
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tailored theranostic apolipoprotein E3 porphyrin-lipid nanoparticles target glioblastoma†
M. A.
Rajora
ab,
L.
Ding
a,
M.
Valic
ab,
W.
Jiang
a,
M.
Overchuk
ab,
J.
Chen
a and
G.
Zheng
 *abc
*abc
aPrincess Margaret Cancer Centre, University Health Network, 101 College Street, Toronto, Ontario M5G 1L7, Canada. E-mail: gzheng@uhnresearch.ca
bInstitute of Biomaterials and Biomedical Engineering, University of Toronto, 164 College Street, Toronto, Ontario M5S 3G9, Canada
cDepartment of Medical Biophysics, University of Toronto, 101 College Street, Toronto, Ontario M5G 1L7, Canada
First published on 23rd May 2017
Abstract
The development of curative glioblastoma treatments and tumour-specific contrast agents that can overcome the blood–brain barrier (BBB) and infiltrative tumour morphology remains a challenge. Apolipoprotein E3 (apoE3) is a high density lipoprotein apolipoprotein that chaperones the transcytosis of nanoparticles across the BBB, and displays high-affinity binding with the low density lipoprotein receptor (LDLR), a cell-surface receptor overexpressed by glioblastoma cells. This LDLR overexpression and apoE3 binding capacity was exploited for the development of glioblastoma-targeted porphyrin-lipid apoE3 lipid nanoparticles (pyE-LNs) with intrinsic theranostic properties. Size-controlled discoidal and cholesteryl oleate (CO)-loaded spherical pyE-LNs were synthesized through the systematic variation of particle composition, which dictated nanoparticle size and morphology. Composition optimization yielded 30 nm pyE-LNs with stable loading of apoE3 and porphyrin-lipid that simultaneously conferred the nanoparticles with glioblastoma targeting and activatable near-infrared fluorescence imaging functionalities. A 4-fold higher uptake of pyE-LNs by LDLR-expressing U87 glioblastomas cells relative to minimally expressing ldlA7 cells was observed in vitro. This uptake was a result of receptor-mediated endocytosis, which could be inhibited through LDL competition and acetylation of particle apoE3 moieties. ApoE3-dependent delivery of pyE-LN to glioblastomas was also demonstrated in orthotopic U87-GFP tumour-bearing animals. Quantification of CO-loaded pyE-LN biodistribution demonstrated successful selective uptake of porphyrin by malignant tissue, with a 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 tumour:healthy tissue particle specificity. This allowed for the detection of strong, tumour-localized porphyrin fluorescence, which was diminished when apoE3-devoid py-LN particles were administered. Furthermore, this selective uptake yielded cell-specific potent PDT sensitization in vitro, resulting in an 83% reduction in glioblastoma cell viability. These results highlight the promising capacity of pyE-LNs to target porphyrin delivery to glioblastoma tumours for theranostic applications.
:1 tumour:healthy tissue particle specificity. This allowed for the detection of strong, tumour-localized porphyrin fluorescence, which was diminished when apoE3-devoid py-LN particles were administered. Furthermore, this selective uptake yielded cell-specific potent PDT sensitization in vitro, resulting in an 83% reduction in glioblastoma cell viability. These results highlight the promising capacity of pyE-LNs to target porphyrin delivery to glioblastoma tumours for theranostic applications.
Introduction
High grade gliomas, termed glioblastomas, remain a highly lethal disease with a mere 5% five-year survival rate.1 The aggressively invasive, heterogenous nature of these brain tumours prevent current treatment paradigms (surgical tumour resection and adjunctive radio or chemotherapy) from achieving successful remission, leading to inevitable recurrence.1 The development of accurate imaging modalities and curative glioblastoma interventions is challenged by the blood–brain barrier (BBB). Despite the presence of leaky vasculature within solid glioma tumours, the small 12 nm upper-limit size of these pores, in addition to the sustained presence of an intact BBB within the infiltrative tumour component, prevents the extravasation of contrast agents and drugs to all areas of the tumour.2–4 Furthermore, the diffuse infiltration of glioblastoma cells into healthy brain parenchyma not only prevents complete surgical resection of tumours,5 but also requires the use of imaging and therapeutic agents with high glioblastoma cell selectivity for accurate tumour delineation and prevention of off-target toxicity. Consequently, there exists an immediate need for therapeutic and imaging platforms that can cross the BBB and target glioma cells.To this end, apolipoprotein E3 (apoE3) nanoparticles have gained attention as promising glioblastoma drug delivery vehicles. ApoE3 is one of three polymorphisms of the 34 kDa apoE glycoprotein; the most prominent lipoprotein produced within the central nervous system, where as a component of high-density lipoprotein (HDL), it regulates cholesterol and lipid homeostasis.6,7 Recently, apoE3 nanoparticles were shown to cross the BBB via transcytosis and exert neuroprotective effects in animal models of Alzheimer's disease.8–12 Furthermore, the transport of non-proteinaceous gold and polymeric nanoparticles is thought to be chaperoned by endogenous apoE adsorbed onto particle surfaces during their circulation in the blood stream.13,14 This mediation of nanoparticle active transport into the brain is thought to be in part due to the high-avidity binding of apoE3 to the low density lipoprotein receptor (LDLR) expressed in brain endothelium.15,16
The BBB permeability of apoE3 nanoparticles and this high affinity LDLR binding capacity makes this an interesting class of nanoparticles to explore as glioblastoma-targeted drug delivery vehicles. Several tumour types, including gliomas, require increased cholesterol metabolism to support lesion growth.17,18 This results in an upregulation of cell-surface LDLR, which facilitates cellular cholesterol uptake via receptor-mediated endocytosis of lipoproteins. Consequently, LDLR is overexpressed in human glioma cells compared to surrounding normal brain tissue.19 Recently, a limited number of studies exploited this LDLR overexpression for the delivery of apoE3-vitamin D3 nanoparticles and resveratrol, curcumin or DNA-loaded apoE3-HDL to glioblastoma cells.20–23 Though these studies demonstrated effective drug delivery in vitro, more extensive exploration of targeting specificity in vitro and in vivo is needed to further validate the utility of apoE3 nanoparticles for glioblastoma drug delivery.
In this study, we take advantage of the LDLR binding affinity of apoE3 and the overexpression of LDLR in malignant cells to address current needs in glioblastoma therapy and imaging via the development of porphyrin apoE3 lipid nanoparticles (pyE-LNs). Porphyrins are heterocyclic organic molecules that exhibit photophysical properties conducive to cancer fluorescence imaging and photodynamic therapy (PDT).24 Their integration into the shell of nanoparticles as lipid conjugates yields stable supramolecular structures with unique intrinsic multimodal (photoacoustic, near infrared fluorescence, positron emission tomography, magnetic resonance) imaging and phototherapy capabilities.25–28 By integrating porphyrin-lipid and apoE3 within a single nanostructure (Fig. 1), we expanded upon the currently limited repertoire of glioma-targeted apoE3 nanoparticles via the introduction of intrinsic all-in-one theranostic properties. Here, we present the development and characterization of these size-controlled pyE-LNs, and demonstrate their ability to specifically target glioblastoma cells in an apoE3-dependent manner in vitro and in vivo.
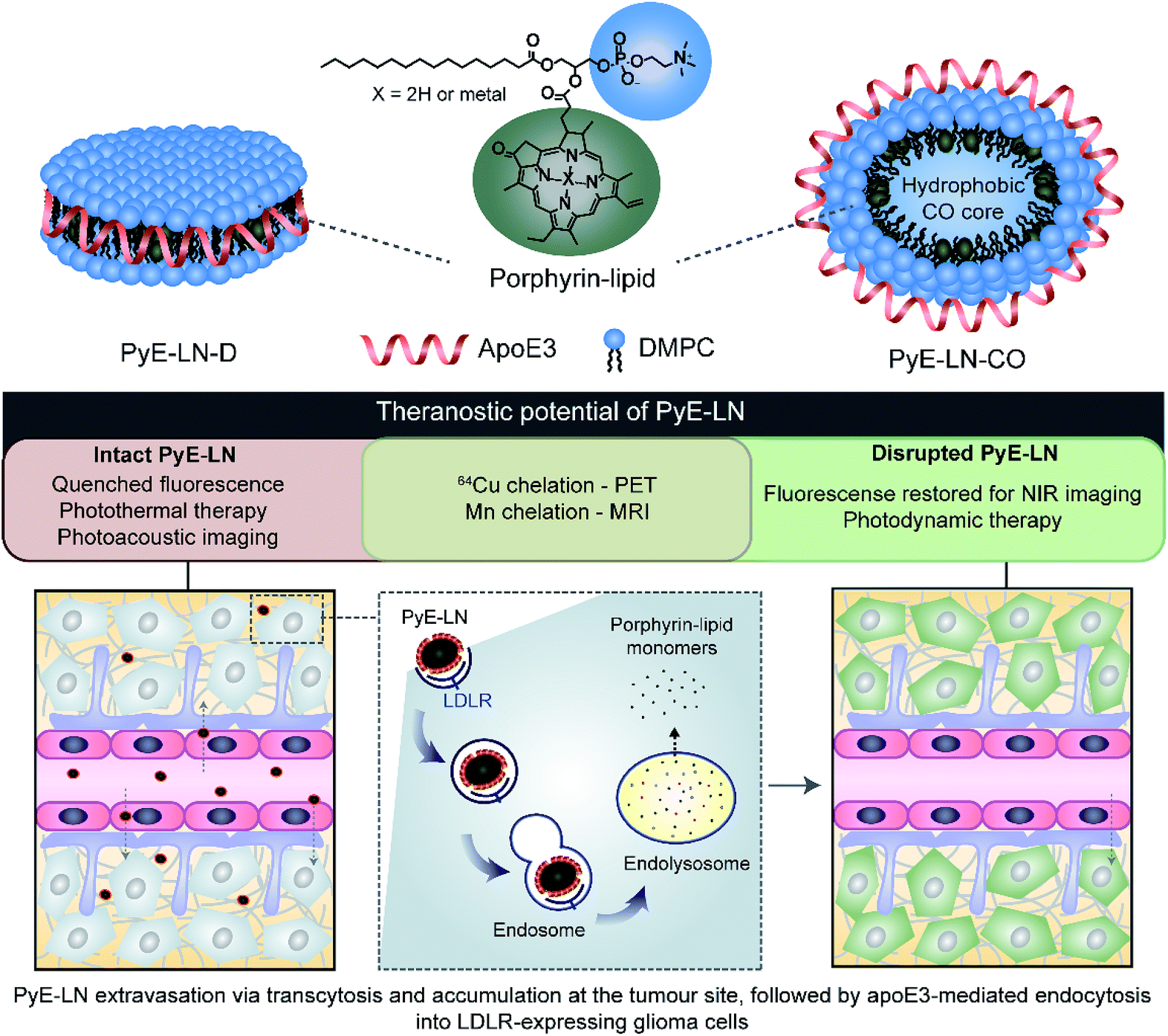 | ||
| Fig. 1 PyE-LN particles were formulated to comprise 1,2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC) and porphyrin-lipid within the particle shell, assembled and size-constrained by apoE3 in either a discoidal or cholesteryl oleate (CO) core-loaded state, which represent two predominant populations of native HDL. It is anticipated that particles will permeate the BBB, accumulate at the tumour site, and undergo apoE3-directed LDLR-targeted uptake by glioma cells. Here, the multifunctional properties of porphyrin-lipid can be utilized for theranostic purposes. Intact particles display quenched fluorescence, giving rise to photoacoustic imaging and photothermal therapy capabilities. Following targeted endolysosomal uptake into glioma cells, particles are disrupted and monomeric porphyrin-lipid is released intracellularly. This dissociation restores porphyrin fluorescence to yield NIR imaging and photodynamic therapy capabilities. | ||
Results and discussion
Tailoring the size and composition of pyE-LN
Native HDL is comprised of a heterogeneous mixture of particle subclasses that range from dense discs to spherical particles core-loaded with cholesteryl esters and triglycerides, and which exhibit differing physiochemical and biological properties.29–33 To account for this variability, pyE-LNs were synthesized as discs (pyE-LN-D), comprising phospholipid, porphyrin-lipid and apoE3, or additionally loaded with cholesterol-oleate (pyE-LN-CO) to adopt a more spherical particle morphology (Fig. 1). Size and composition play important roles in the pharmacokinetic profiles of nanoparticles. Smaller subclasses of HDL exhibit higher serum stability and slower blood clearance relative to larger HDL populations.34,35 Furthermore, smaller nanoparticles permeate through the sub-40 nm extracellular matrix fibre spacing in glioblastoma tumours more effectively than their larger counterparts.2,36–38 As such, in order to promote pyE-LN stability and delivery while simultaneously capitalizing on the theranostic and translational potential of the shell-loaded porphyrin, pyE-LN compositions were tailored to minimize particle size and heterogeneity, and maximize porphyrin-lipid and CO loading.Systematic optimization of the pyE-LNs, illustrated in Fig. 2, was conducted to achieve these design criteria. As the composition, and particularly the protein:lipid ratio, within HDL subclasses is associated with their size,32,39,40 the first step in this optimization process involved the variation of the apoE3:total lipid molar ratio. ApoE3/1,2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC) particles were synthesized by sonicating hydrated DMPC lipid films, yielding aqueous lipid vesicle suspensions to which apoE3 was added to consume and size-constrain the vesicles into proteinaceous discs. Increasing the apoE3:lipid ratio decreased the vesicle population (Fig. 2 and S1†); an important consideration during particle formation as these vesicles, devoid of apoE3, lack targeting moieties and thus effectively increase particle impurity, while also increasing morphological heterogeneity and average particle size. To this end, increasing the apoE3:lipid ratio also decreased particle size, a trend consistent with what is observed for native HDL.32,39,40 Beyond a 1:50 apoE3:lipid molar ratio, protein aggregates were visible by TEM (Fig. S1†). As such, when taking lipid vesicle consumption, lack of protein aggregation, and the formation of small, morphologically-homogenous particles into account, a 1:75 apoE3:lipid molar ratio was selected for further steps in the pyE-LN optimization process. Similar endpoints were also considered when subsequently selecting a 5:1 DMPC:CO molar ratio for the generation of pyE-LN-CO, whereby CO loading into the DMPC film resulted in a disc to ellipse/sphere shape change (Fig. 2 and S2†).
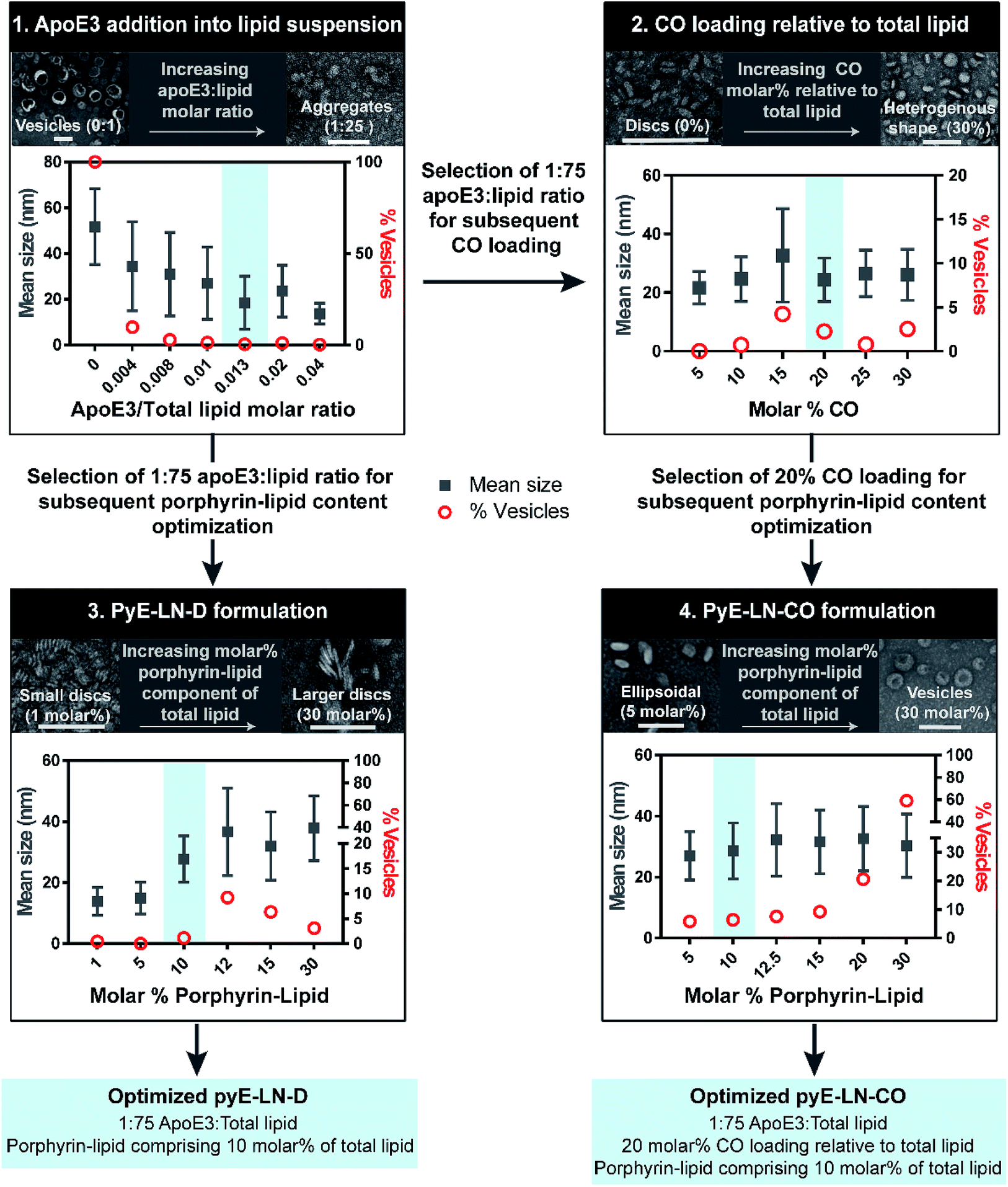 | ||
| Fig. 2 Optimization of pyE-LNs was performed via the systematic modification of particle composition. The molar ratio of DMPC and apoE3 was first varied to formulate discoidal particles which were then loaded with CO. Selection of optimal apoE3:DMPC and CO/DMPC molar ratios was followed by the addition of porphyrin-lipid to the formulations, constituting up to 30 molar% of the total lipid content. Particles were imaged with TEM and analysed for morphology and size via ImageJ. Sizes are displayed as the average ± standard deviation of a minimum of 450 particle measurements from a minimum of three representative fields of view. Representative TEM images and histograms of all particle formulations are presented in Fig. S1–S4.† Scale bars = 100 nm. | ||
Lastly, porphyrin-lipid was added into DMPC or DMPC/CO lipid films to formulate optimized pyE-LN-D and pyE-LN-CO respectively using the total apoE3, lipid and CO compositions selected as described above in steps 1 and 2 of the optimization process (Fig. 2, S3 and S4†). Interestingly, increasing the porphyrin-lipid loading yielded differing trends for the two pyE-LN formulations: disc size increased with increasing porphyrin-lipid loading in pyE-LN-D, while particle size remained constant, but the vesicle population increased with higher loading of porphyrin-lipid into pyE-LN-CO. In both cases, a 90 molar% DMPC/10 molar% porphyrin-lipid composition was selected for optimal size-controlled particle formation. Overall, the results of this optimization process indicated that pyE-LN composition governed particle size and morphology, which could be tuned through the variation of lipid, apoE3 and CO molar ratios.
Characterization of optimized pyE-LN
Following the identification of optimal apoE3, DMPC, porphyrin-lipid and CO compositions, tailored pyE-LNs were reproduced, and their physical chemical properties were characterized. As illustrated in Fig. 3A, a mean particle size of approximately 30 nm was achieved for optimized pyE-LN-D and pyE-LN-CO particles; a size potentially amenable to diffusion through the sub-40 nm spacing between glioblastoma tumour collagen fibrils,37 and consistent with sizes obtained for BBB-permeating apoE3/DMPC nanoparticles previously reported.11,12 Size distributions attained by TEM demonstrated the formation of monodisperse particles, wherein pyE-LN-D samples were composed of discs (Fig. 3 and S3†), while pyE-LN-CO samples were composed predominantly of spherical and more elliptical discoidal particles (Fig. 3 and S4†), a mixture observed consistently in previously reported TEM and cryo-TEM images of reconstituted HDL.41,42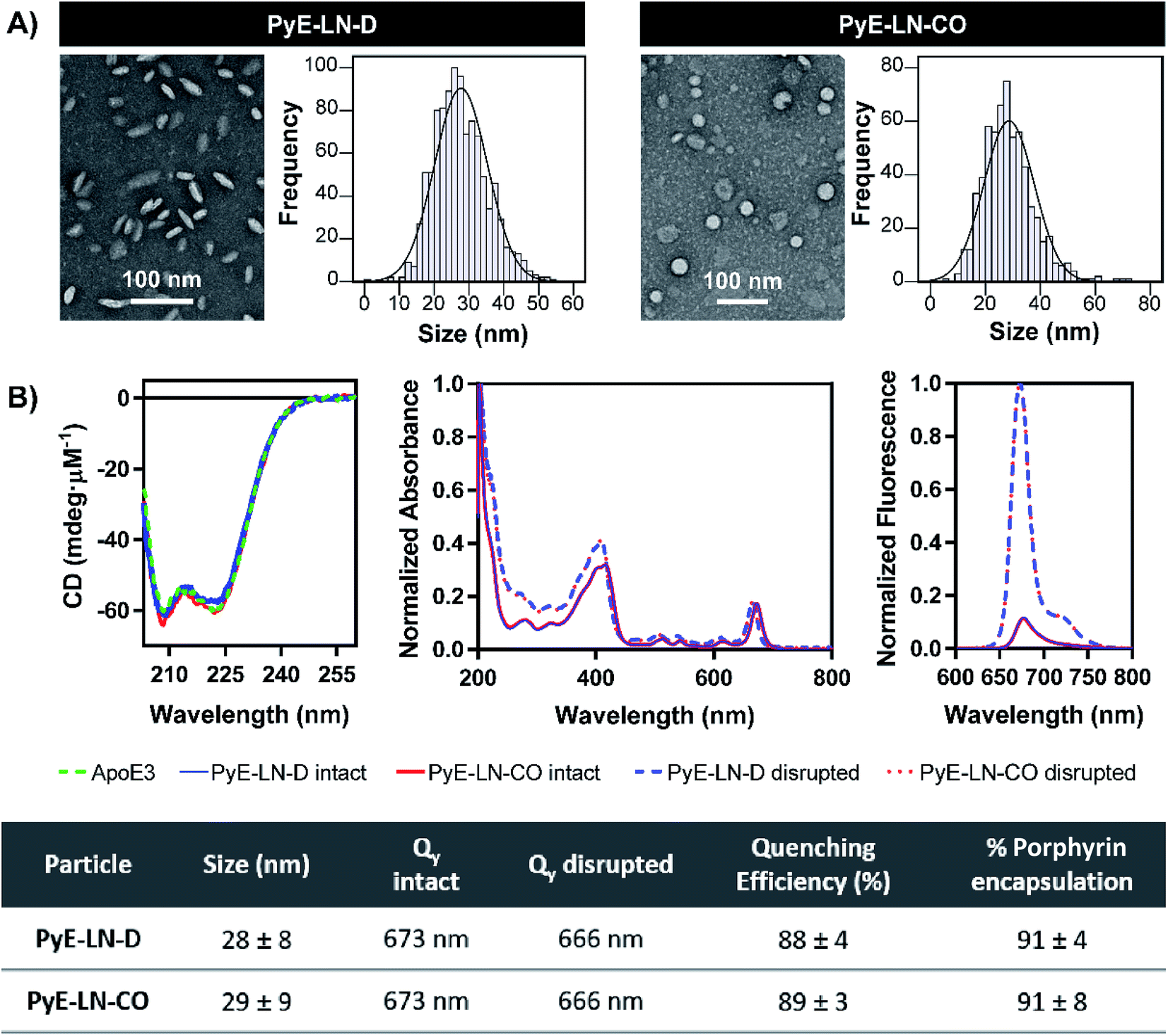 | ||
| Fig. 3 Characterization of the optimized pyE-LN-D and pyE-LN-CO. Particle size and morphology were assessed by TEM (A), while optical properties (B) were assessed using CD spectroscopy (presented as mdeg μM−1 of apoE3), spectrophotometry (intact particles measured in PBS, disrupted in methanol), and spectrofluorometry (410 nm excitation, disrupted particles measured in 1 v/v% Triton X-100, intact particles measured in PBS). Size is presented as an average ± standard deviation of a minimum of 450 ImageJ particle measurements from three sample replicates over three or more fields of view. Quenching efficiency and porphyrin encapsulation are presented as the average ± standard deviation of five samples. Absorbance and fluorescence profiles are normalized to the maximum absorbance and fluorescence intensity respectively in each spectra. | ||
Optical characterization of discoidal and CO-loaded pyE-LN demonstrated successful incorporation of apoE3 and porphyrin-lipid within the particles. Both nanoparticles, as illustrated in Fig. 3B, yielded circular dichroism (CD) spectra with a signature double peak between the 200 and 230 nm ultraviolet light range characteristic of the alpha-helical secondary structure of apoE in reconstituted HDL.43 CD values of −57.4 and −60.9 mdeg μM−1 protein were observed respectively for pyE-LN-D and pyE-LN-CO, similar to that of concentration-equivalent free apoE3 (−60.0 mdeg μM−1), demonstrating that apoE3 was present in the particle suspensions. Native gradient polyacrylamide gel electrophoresis of the pyE-LN samples demonstrated co-localization of protein and porphyrin fluorescence bands and stronger staining of particulate apoE3 bands versus those of free apoE3 (Fig. S5†). These observations illustrated that apoE3 within the particle suspensions was successfully co-assembled alongside porphyrin-lipid. Furthermore, a slight red-shift (7 nm) of the Q-band within the absorbance spectra of the intact particles was observed relative to the Q-band associated with disrupted particles (Fig. 3B). A similar red shift of 4 nm was noted in the fluorescence spectra of the intact particles relative to disrupted particles. Combined with an achievement of high porphyrin loading efficiency (∼90%) and porphyrin fluorescence quenching (∼90%) in intact particles, these observations indicated the presence of strong ground state coupling and the successful, stable assembly of porphyrin-lipid within both discoidal and CO-loaded pyE-LN.44 These assemblies remained stable under storage conditions (4 °C in PBS), whereby no significant difference (n = 3, p > 0.05) in quenching efficiency was observed for 30 days following particle synthesis (Fig. S6†). Particle size and morphology also remained stable over a period of 30 days (Fig. S6†), with no significant increase in size (n = 3, p > 0.05), indicating a lack of instability-induced particle aggregation. Thus, optimization of pyE-LN synthesis generated small (30 nm), monodisperse, stable pyE-LN-D and pyE-LN-CO particles loaded effectively with apoE3 and porphyrin-lipid.
In addition to remaining stable under storage conditions, discoidal and CO-loaded pyE-LNs maintained over 97% of their initial fluorescence quenching efficiencies over a 24 hour period at 37 °C in serum solutions composed of 10% FBS, and approximately 80% of their quenching efficiencies in 50% FBS solutions (Fig. S6†). This serum stability allows the activatable fluorescence of pyE-LN to be feasibly applied for theranostic purposes. As illustrated in Fig. 1, the fluorescence self-quenching of pyE-LN holds theranostic advantages: following the LDLR-targeted apoE3-mediated uptake of particles by glioblastoma cells, it is proposed that particles disassemble within endolysosomes into monomeric porphyrin-lipid with restored fluorescence, which among other applications, can be exploited to delineate tumours via near infrared (NIR) fluorescence imaging. The proposed utility of this target-specific activatable fluorescence was contingent on and conserved by the production of pyE-LNs stable under physiological conditions.
Targeted in vitro glioblastoma cellular uptake of pyE-LN
The proposed targeting ability of pyE-LNs was evaluated in vitro using human U87 glioblastoma and Chinese hamster ovary ldlA7 cells, which exhibited high and low levels of LDLR expression respectively (Fig. S7†). Activation of porphyrin fluorescence upon cellular delivery of the particles enabled the qualitative assessment and comparison of pyE-LN uptake between U87 and ldlA7 cells via fluorescence microscopy (Fig. 4A). Strong porphyrin fluorescence was observed in U87 cells treated with either discoidal or CO-loaded pyE-LN, while negligible fluorescence signal was detected in ldlA7 cells at equivalent exposure times, suggesting that pyE-LN uptake was facilitated by LDLR. This was further confirmed through flow cytometry quantitative measurements of porphyrin uptake in the two cell types exposed to either pyE-LN-D or pyE-LN-CO over a 24 hour period. Porphyrin fluorescence intensity increased qualitatively (Fig. S8†) and quantitatively in a time-dependent manner for discoidal and CO-loaded particles in both U87 and ldlA7 cells (Fig. 4B). However, particle uptake was 3–4-fold and 2–3 fold higher in U87 cells than ldlA7 cells for pyE-LN-CO and pyE-LN-D respectively (n = 4, p < 0.05). Interestingly, despite the two particles having similar average sizes, quenching efficiencies, serum stability, porphyrin loading, and qualitative time-dependent increase in cell uptake by U87 and ldlA7 cells (Fig. S8†), flow cytometry quantification of porphyrin uptake in U87 cells demonstrated a 1.5-fold higher (n = 4, p < 0.05) porphyrin fluorescence intensity following treatment with pyE-LN-CO versus pyE-LN-D at each time-point tested. This increased uptake of the more spherical variant of pyE-LN was consistent with previous findings of enhanced glioblastoma cell uptake of spherical apoE3-HDL particles compared to discoidal apoE3-HDL.45 The tertiary structure and microenvironment of apoE influences its LDLR binding avidity.46,47 The microenvironment of lysine residues and thereby apoE conformation is known to differ between discoidal and spherical apoE3 lipid nanoparticles.48 As such, although apoE3 within pyE-LN-D and pyE-LN-CO bears a similar secondary structure per CD spectroscopy, it is plausible that differences in the tertiary structure of apoE3 in the two pyE-LN variants leads to differential LDLR binding and subsequent uptake by the LDLR-expressing U87 cells. Nevertheless, the qualitative and quantitative observation of increased delivery of both pyE-LNs to U87 cells relative to ldlA7 cells indicates that the particles undergo cell-specific uptake.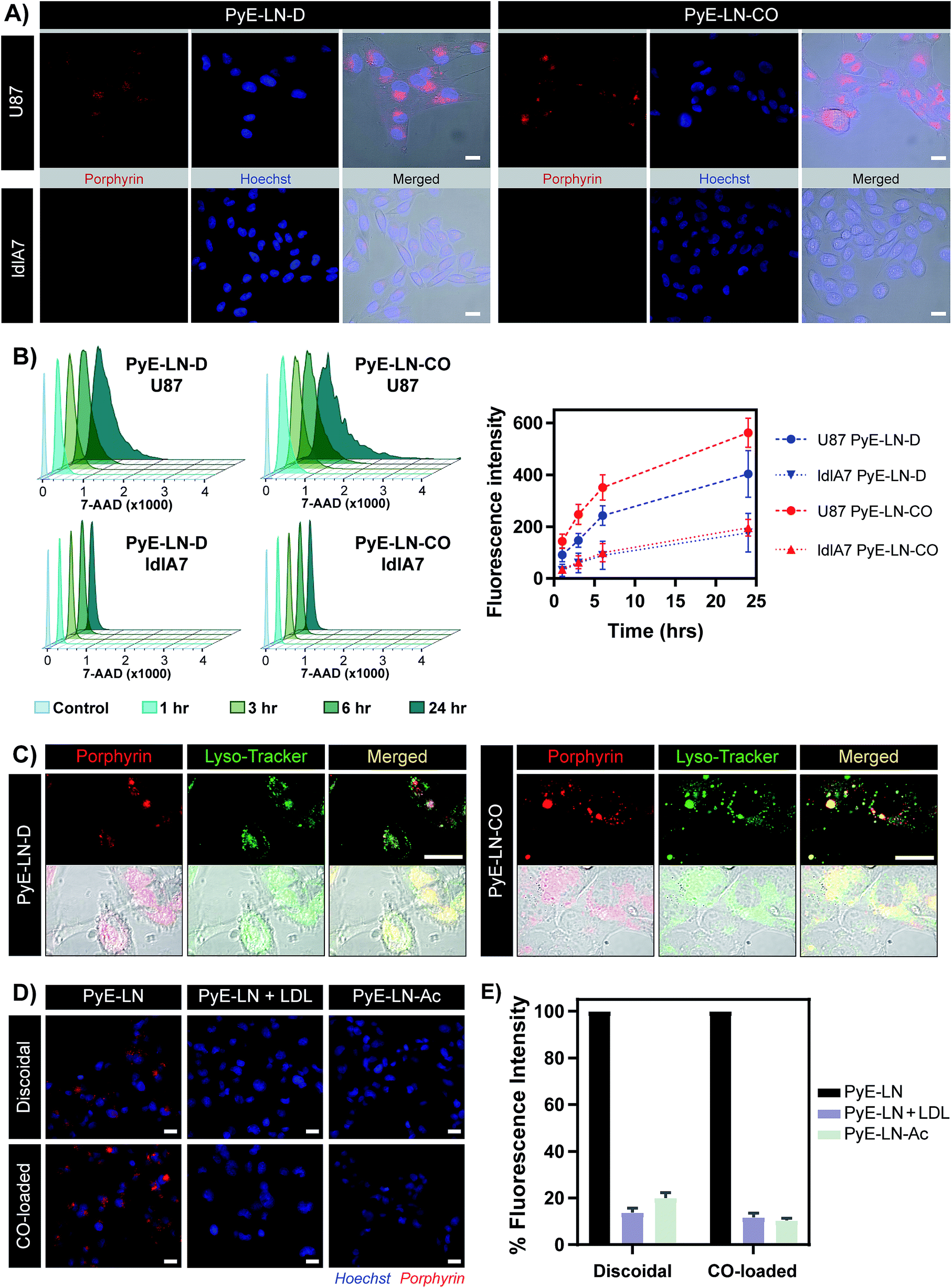 | ||
| Fig. 4 In vitro cell uptake studies of pyE-LN (5 μM porphyrin treatment concentration). (A) Comparison of pyE-LN uptake by U87 (high LDLR expression) and ldlA7 (low LDLR expression) cells with fluorescence microscopy following incubation with particles for 3 hours. (B) Representative flow cytometry histograms and associated quantification of time-dependent porphyrin uptake. At each time point, U87 cells displayed significantly higher porphyrin uptake than ldlA7 cells, while pyE-LN-CO demonstrated higher uptake than pyE-LN-D (p < 0.05, n = 4). (C) Confocal imaging of Lyso-Tracker-stained U87 cells following incubation with particles for 6 hours. (D and E) Treatment of U87 cells with pyE-LN, pyE-LN + 50× excess LDL or acetylated pyE-LN. Fluorescence microscope images (D) and flow cytometry quantification of cell uptake normalized to pyE-LN fluorescence intensity (E) are shown, with the latter demonstrating a significant decrease in absolute porphyrin fluorescence intensity (p < 0.005, n = 3) with LDL competition or particle acetylation for both pyE-LN-D and pyE-LN-CO (representative flow cytometry histograms are shown in Fig. S10†). Scale bars = 20 μm. | ||
Binding of a ligand to LDLR results in its receptor-mediated endocytosis followed by lysosomal fusion and degradation.49,50 Thus, in order to further substantiate whether the U87 cell-specific uptake of pyE-LNs was indeed a result of LDLR-mediated endocytosis, particles were incubated with U87 cells and stained with Lyso-Tracker. Confocal microscopy revealed a punctate pattern of porphyrin uptake that co-localized with the Lyso-Tracker signal (Fig. 4C), indicating that discoidal and CO-loaded pyE-LNs were sequestered into late endosomes or lysosomes, as would be expected following LDLR-mediated endocytosis. This potential delivery mechanism was further elucidated through the successful competitive inhibition of pyE-LN uptake in U87 cells by LDL. The strong fluorescence signal observed in U87 cells following a 3 hour treatment with either pyE-LN-D or pyE-LN-CO was eliminated when a 50-fold excess of LDL was added to the treatment media (Fig. 4D). Flow cytometry quantification revealed that LDL addition significantly reduced pyE-LN-D and pyE-LN-CO uptake in U87 cells by 86 ± 2% and 88 ± 2% respectively (n = 3, p < 0.005). PyE-LNs were thus shown to undergo LDLR-mediated uptake by glioblastoma cells.
Basic residues within the N-terminal domain of apoE3 are known to direct apoE3-LDLR binding, which is attenuated with the chemical modification of these residues.51,52 Consequently, acetylated pyE-LNs (PyE-LN-Ac) were synthesized and assessed in vitro to establish the role played by apoE3 in the glioma cell targeting abilities displayed by pyE-LN. Acetylation of protein moieties was conducted subsequent to particle formation in order to evade apoE3 modification-induced changes in particle self-assembly. Physical chemical properties of pyE-LNs, including particle size, apoE3 secondary structure, apoE3 and porphyrin-lipid incorporation, and quenching efficiencies, were conserved following acetylation (Fig. S5 and S9†). As was observed for LDL competition, acetylation of apoE3 moieties in pyE-LNs decreased the intracellular porphyrin signal observable by fluorescence microscopy following the treatment of U87 cells with pyE-LN-Ac (Fig. 4D). Acetylation significantly (n = 3, p < 0.005) decreased porphyrin uptake in U87 cells by 80 ± 2% and 90 ± 1% for discoidal and CO-loaded particles respectively, indicating that apoE3 mediated the in vitro LDLR-targeting abilities of pyE-LNs.
In vivo glioblastoma tumour homing and theranostic utility
The apoE3-mediated glioblastoma-targeting capacity of pyE-LN was subsequently characterized in vivo using U87-GFP orthotopic tumour-bearing mice. Due to its superior uptake by U87 cells in vitro and longer circulation half-life relative to discoidal particles (Fig. 5A), pyE-LN-CO was used for in vivo studies. By taking advantage of the intrinsic metal chelation abilities of porphyrin centres, pyE-LN-CO was radiolabelled with copper-64, yielding 64Cu-pyE-LN-CO, and allowing for the quantitative assessment of particle biodistribution via gamma counting. Importantly, the intrinsic 64Cu radiolabelling of porphyrin-lipid nanostructures does not necessitate the incorporation of added imaging agents, allowing the photophysical and biological properties of the parent particles to be conserved.53 Accordingly, 64Cu-pyE-LNs were synthesized with high radiolabelling efficiency (100 ± 1.6%) while maintaining the physical chemical properties of unlabelled pyE-LN-CO (Fig. S11†).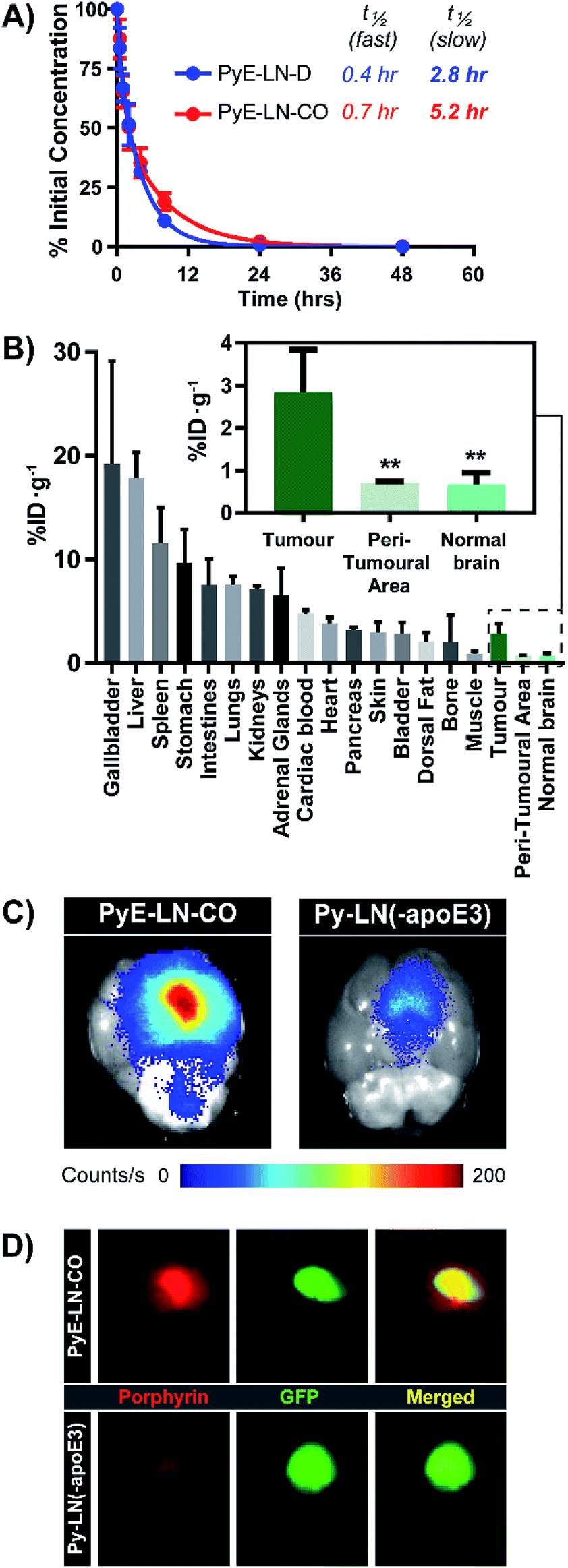 | ||
| Fig. 5 In vivo evaluation of pyE-LN. (A) Blood clearance profiled of pyE-LN formulations, and associated half-lives in healthy mice (n = 5). (B) Porphyrin biodistribution in U87-GFP tumour-bearing mice 6 hours following the administration of 64Cu-pyE-LN-CO (average ± standard deviation, n = 6). Significantly higher porphyrin accumulation (**p < 0.005, n = 6) was observed at the tumour site compared to surrounding tissue. Comparison of ex vivo fluorescence imaging of porphyrin signal intensity (C) and GFP signal co-localization (D) in brains harvested from U87-GFP tumour-bearing mice administered 64Cu-pyE-LN-CO or porphyrin dose equivalent py-LN(-apoE3). | ||
As illustrated in Fig. 5B, 64Cu-pyE-LN-CO accumulated to the greatest extent in the liver, gallbladder, spleen and gastrointestinal tract. This pattern of accumulation is consistent with the high expression of LDLR in the liver, stomach and intestinal tissue, the hepatobiliary clearance of porphyrins, sequestration of nanoparticles by the mononuclear phagocytic system, and with previously reported biodistribution of apoE3/lipid nanoparticles.54–5664Cu-pyE-LN-CO accumulated in tumour tissue at 3 ± 1% injected dose per gram of tissue. This accumulation was highly targeted to glioblastoma tissue, with a 4:1 tumour:peri-tumoral and tumour:contralateral normal brain tissue specificity. To the best of our knowledge, this is the first study to quantitatively establish the in vivo glioblastoma targeting specificity of synthetic apoE3 nanoparticles.
This tumour-homing ability was further evaluated via the activatable NIR fluorescence imaging capacity of pyE-LN-CO. Ex vivo fluorescence imaging of brains harvested from U87-GFP tumour-bearing mice 6 hours following the administration of 64Cu-pyE-LN-CO demonstrated strong porphyrin fluorescence at the tumour site that highly co-localized with U87 GFP signal (Fig. 5C and D). This tumour-specific imaging functionality was compared to that of DMPC/porphyrin-lipid nanoparticles synthesized without apoE3 (py-LN(-apoE3)). These particles displayed a larger size range due to the lack of apoE3 to mediate size control, but optical properties consistent with those of pyE-LN-CO (Fig. S11†). In comparison to pyE-LN-CO, py-LN(-apoE3) yielded weak fluorescence signal at the tumour site with no observable co-localization with U87 GFP fluorescence, indicating that pyE-LN uptake was apoE3-dependent (Fig. 5C and D). Collectively, these results validate the remarkable in vivo glioblastoma targeting capacity of pyE-LN.
To further explore this targeting capacity and demonstrate the theranostic utility of pyE-LN, in vitro PDT was conducted in U87 and ldlA7 cells (Fig. 6). Laser or pyE-LN-CO administration alone yielded no cytotoxic effects against either cell line. In fact, pyE-LN-CO treatment of U87 cells enhanced cell viability by 20%. Cholesterol is an important substrate in sustaining cancer cell proliferation.17 As such, this augmentation of cell viability may be a result of increased cholesterol substrate delivery in the form of pyE-LN-CO, consistent with studies in which lipoprotein cholesterol treatment enhanced cancer cell proliferation.57,58 Nevertheless, co-treatment of U87 cells with pyE-LN-CO and low dose laser light yielded significant and highly potent cytotoxicity against U87 cells, wherein light doses of 1 and 5 J cm−2 yielded 67% and 83% cell death respectively. Such cytotoxic effects were markedly reduced by approximately 3-fold in ldlA7 cells co-treated with pyE-LN-CO and a 5 J cm−2 light dose, consistent with the higher expression of LDLR by U87 cells and the associated 3-fold reduced uptake of pyE-LN-CO by ldlA7 cells observed by flow cytometry (Fig. 4B) relative to U87 cells over a 24 hour treatment period. Thus, in addition to providing strong NIR fluorescence contrast in vivo, pyE-LN also demonstrated potent and specific phototherapeutic effects, making it a targeted and truly theranostic platform, which can be exploited in future studies to enhance in vivo glioblastoma PDT, and positron emission tomography and magnetic resonance imaging tumour contrast.
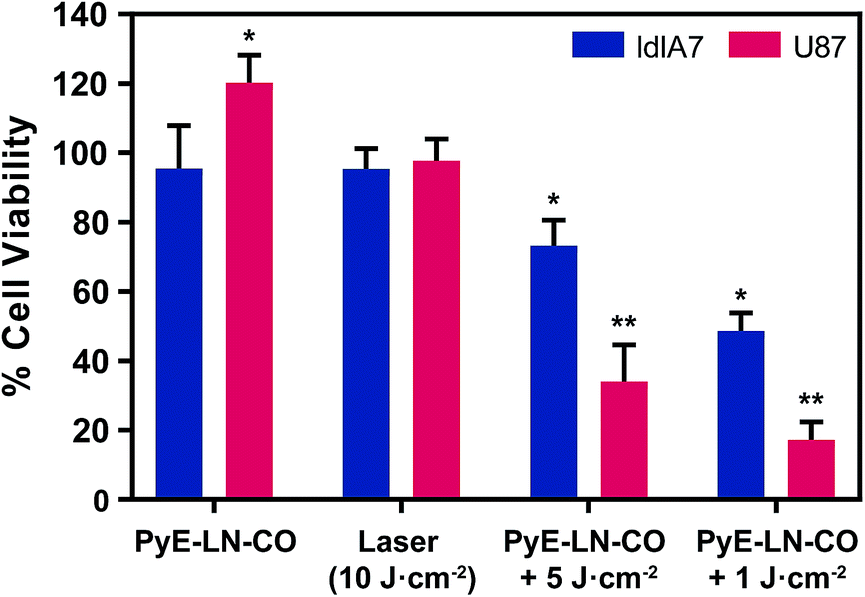 | ||
| Fig. 6 In vitro evaluation of pyE-LN PDT sensitization. Cell viability was normalized to untreated cells and is presented as the average of three replicates ± standard deviation. Cells were treated with py-LN-CO (3 μM), laser (671 nm) or a combination of laser and particle. Significant differences (*p < 0.01, n = 3) were observed between treated and untreated cells, wherein significantly higher toxicity (**p < 0.01, n = 3) was observed in U87 cells versus ldlA7 cells treated with particle and laser. | ||
Conclusions
The lack of curative interventions available for glioblastoma necessitates the development of platforms that can cross the BBB and specifically target malignant cells. PyE-LNs represent a novel class of theranostic vehicles that have the potential to address these needs. PyE-LN compositions could be tailored to generate size-controlled discoidal and spherical/elliptical CO-loaded particles with stable loading of porphyrin-lipid. These particles underwent LDLR-mediated endocytosis for the selective delivery of porphyrin to LDLR-overexpressing glioblastoma cells in vitro. Interestingly, CO-loaded pyE-LN displayed a longer blood clearance half-life and was more effectively uptaken than the discoidal variant by glioblastoma cells, suggesting that pyE-LN morphology influences pharmacokinetics and protein binding. The intrinsic multimodal properties of pyE-LNs were applied in vivo to illustrate the apoE3-dependent tumour-homing abilities of pyE-LN-CO, and in vitro to demonstrate potent PDT sensitizing capabilities. To the best of our knowledge, this is the first report of inherently multifunctional apoE3 nanoparticles with in vivo glioblastoma targeting capabilities. Furthermore, by establishing tumour specificity in vivo, this study validated the utility of apoE3 nanoparticles as glioma drug and contrast agent delivery platforms.Experimental
PyE-LN synthesis
:total lipid, CO/total lipid and porphyrin-lipid/total lipid molar ratios were systematically modified in a 4-step optimization procedure for the synthesis of pyE-LNs (Fig. 2). Established procedures were followed during this optimization study.12,28 Briefly, 0.1–0.15 μmol lipid films were generated by combining appropriate quantities of DMPC (10 mg mL−1 in chloroform; Avanti Polar Lipids), CO (2.5 mg mL−1 in chloroform; Sigma Aldrich) and porphyrin-lipid (4–5 mg mL−1 in chloroform; pyropheophorbide/1-palmitoyl-2-hydroxy-sn-glycero-3-phosphocholine conjugate synthesized as described previously25) stock solutions into a glass vial, as summarized in Table 1. The chloroform was dried under nitrogen gas and then under vacuum to generate lipid films. The dried lipid films were re-hydrated with 1 mL phosphate buffered saline (PBS; 10 mM phosphate, pH 7.4) and subsequently bath-sonicated for 90 min. The resulting lipid solutions were allowed to passively cool to room temperature prior to the drop-wise addition of a 0.4 mg mL−1 apoE3 solution in PBS in a 75:1 total lipid:apoE3 molar ratio for steps 2, 3 and 4 of the optimization process and in a 1:0, 250:1, 125:1, 100:1, 75:1, 50:1 or 25:1 total lipid:apoE3 molar ratio for step 1 of the optimization procedure. The resulting solutions were slowly rotated overnight at 4 °C, after which the particle suspensions were centrifuged at 4 °C, 17000g for 5 min to remove free porphyrin-lipid, DMPC or CO precipitates. The resulting supernatants were filtered through Corning® 0.2 μm syringe filters prior to characterization by TEM.
| Step | DMPC (μmol) | Porphyrin-lipid (μmol) | CO (μmol) |
|---|---|---|---|
| 1 | 0.15 | — | — |
| 2 | 0.15 | — | 0–0.03 |
| 3 | 0.1485 | 0.0015 | — |
| 0.1425 | 0.0075 | ||
| 0.135 | 0.015 | ||
| 0.132 | 0.018 | ||
| 0.1275 | 0.0225 | ||
| 0.105 | 0.045 | ||
| 4 | 0.095 | 0.005 | 0.02 |
| 0.09 | 0.01 | ||
| 0.0875 | 0.0125 | ||
| 0.085 | 0.015 | ||
| 0.08 | 0.02 | ||
| 0.07 | 0.03 |
:1 total lipid:apoE3 molar ratio. The resulting particle suspensions were rotated overnight at 4 °C to allow for gentle mixing, after which free lipid and CO were precipitated via centrifugation at 4 °C, 17000g for 5 min. The resulting supernatants were filtered through Corning® 0.2 μm syringe filters. PyE-LN solutions used for in vitro and in vivo studies were concentrated by 3 and 5–6-fold respectively using 10000 NMWL regenerated cellulose Amicon® Ultra-15 centrifuge filter units prior to sterile filtration through Corning® 0.2 μm syringe filters. Particles were shielded from light and stored at 4 °C prior to use.
000 NMWL regenerated cellulose Amicon® Ultra-15 centrifuge filter unit, which was subsequently centrifuged at 4000g, 4 °C for 20 min. The resulting concentrate was diluted with 10 mL of PBS and centrifuged again. Washes were repeated until no 200–300 nm UV-Vis optical signatures of free imidazole were observed in the wash filtrate (this required 5–7 total washes). The final concentrate was then diluted up to a 1 mL volume with PBS, centrifuged in an Eppendorf tube at 17000g, 4 °C for 5 min prior to sterile filtration through a Corning® 0.2 μm syringe filters. Particles were shielded from light and stored at 4 °C prior to use.
:45:40:5 molar ratio in a glass vial. The chloroform was dried under nitrogen and vacuum to form lipid films, which were then hydrated with PBS. The hydrated films were subjected to 9 freeze/thaw cycles prior to extrusion via a LIPEX Thermobarrel Extruder through a polycarbonate membrane (100 nm pore size) 10 times at 65 °C to yield py-LN(-apoE3). The particles were sterile filtered through a Corning® 0.2 μm syringe filters, shielded from light and stored at 4 °C prior to use.
000 NMWL regenerated cellulose Amicon® centrifugal filter units to remove ammonium acetate buffer and non-chelated 64CuCl2. Two washes of the concentrate containing the 64Cu-labelled pyE-LN-CO were conducted with sterile PBS and the filtrate was recovered to characterize radiolabelling efficiency via gamma-counting. The resulting counts were used to calculated a radiolabelling efficiency of 100 ± 1.6% as follows: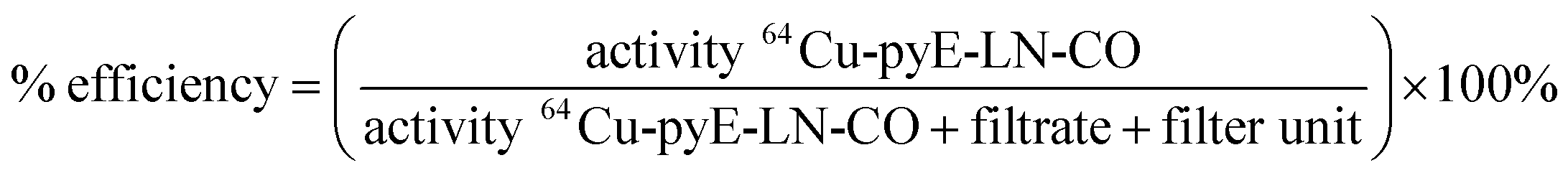
Lastly, the purified 64Cu-pyE-LN-CO nanoparticles were diluted in sterile PBS to yield a final activity concentration of approximately 1.0 mCi mL−1.
PyE-LN characterization
000–150000× using a FEI Techai 20 electron microscope (Nanoscale Biomedical Imaging Facility, Peter Gilgan Centre for Research and Learning, Toronto) equipped with a digital camera. Samples were adsorbed onto charged carbon grids (Electron Microscopy Sciences) and imaged following 2% uranyl acetate negative staining. Average particle sizes were obtained from the measurement of a minimum of 450 particles over three representative fields of view using ImageJ. The percentage of the particles with a vesicular morphology was determined by counting the total number of vesicles and particles over three representative fields of view for each particle formulations. Dynamic light scattering (Zetasizer, Malvern Instruments) was used as a supplementary technique to compare size distributions of acetylated and radiolabelled particles against parent pyE-LNs.
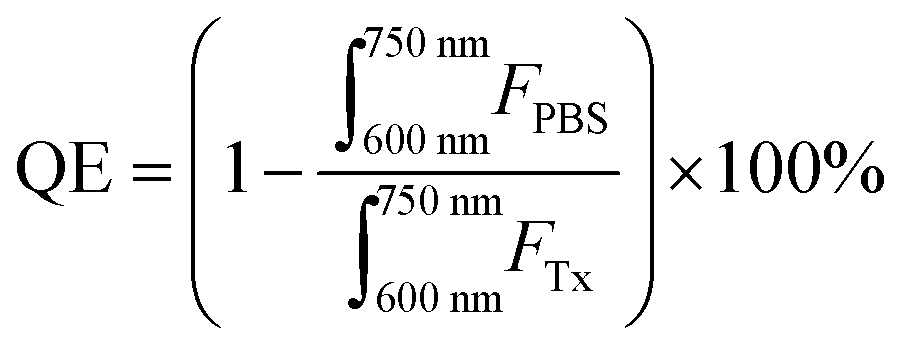
000 M−1 cm−1 at 667 nm in methanol). Fluorescence quenching efficiency was calculated over a 600–750 nm emission range as follows:where, FTx and FPBS represent fluorescence intensities of concentration-equivalent particle solutions in 0.5–1 v/v% Triton X-100 (disrupted particles) and PBS (intact particles) respectively.
PyE-LN cell uptake and in vitro PDT studies
:5000 in 5% BSA/TBST. Five million U87, ldlA7 and HepG2 cells were collected and lysed in lysis buffer, from which protein samples were obtained and loaded into a 10% SDS-PAGE gels, and then transferred to a PVDF membrane (Bio-Rad). The membranes were subsequently blocked with 5% fat-free milk in TBST overnight. Membranes were then incubated with primary antibodies overnight at 4 °C, followed by incubation with 1:2000 diluted HRP-conjugated anti-rabbit antibody (Cell Signaling Technology) for 1 hour at room temperature. Detection of protein bands was performed by a Konica SRX-101A Medical Film Processor. The blots were stripped and re-probed with HRP-conjugated anti β-actin antibody as a loading control.
000 events. Median fluorescence intensity was measured and corrected for background signal from control cells, and histograms were generated using FlowJo software.
In vivo pharmacokinetics and biodistribution of pyE-LNs
All in in vivo studies were approved and conducted in compliance with the University Health Network Animal Resource Centre guidelines.Statistics
Independent samples t-tests were used to compare the means of two groups (equal variances not assumed) with a p < 0.05 denoting statistical significance.Acknowledgements
The authors would like to thank Jenny Lou for technical assistance in conducting biodistribution studies, and Dr Christina MacLaughlin and Danielle Charron for their technical expertise and assistance in acquiring TEM images. The authors would also like to acknowledge their funding sources: Vanier Canada Graduate Scholarship, Canadian Institutes of Health Research, Natural Sciences and Engineering Research Council of Canada, Terry Fox Research Institute, Canadian Cancer Society Research Institute, Canada Foundation for Innovation, Princess Margaret Cancer Foundation, and the Joey and Toby Tanenbaum/Brazillian Ball Chair in Prostate Cancer Research.Notes and references
- Q. T. Ostrom, H. Gittleman, P. Liao, C. Rouse, Y. Chen, J. Dowling, Y. Wolinsky, C. Kruchko and J. Barnholtz-Sloan, Neuro-Oncology, 2014, 16, iv1–iv63 CrossRef PubMed.
- H. Sarin, A. S. Kanevsky, H. Wu, K. R. Brimacombe, S. H. Fung, A. A. Sousa, S. Auh, C. M. Wilson, K. Sharma, M. A. Aronova, R. D. Leapman, G. L. Griffiths and M. D. Hall, J. Transl. Med., 2008, 6, 80 CrossRef PubMed.
- H. Sarin, A. S. Kanevsky, H. Wu, A. A. Sousa, C. M. Wilson, M. A. Aronova, G. L. Griffiths, R. D. Leapman and H. Q. Vo, J. Transl. Med., 2009, 7, 51 CrossRef PubMed.
- D. J. Brat and E. G. Van Meir, Lab. Invest., 2004, 84, 397–405 CrossRef CAS PubMed.
- A. Giese, R. Bjerkvig, M. E. Berens and M. Westphal, J. Clin. Oncol., 2003, 21, 1624–1636 CrossRef CAS PubMed.
- G. Bu, Nat. Rev. Neurosci., 2009, 10, 333–344 CrossRef CAS PubMed.
- D. M. Holtzman, J. Herz and G. Bu, Cold Spring Harbor Perspect. Med., 2012, 2, a006312 Search PubMed.
- K. Michaelis, M. M. Hoffmann, S. Dreis, E. Herbert, R. N. Alyautdin, M. Michaelis, J. Kreuter and K. Langer, J. Pharmacol. Exp. Ther., 2006, 317, 1246–1253 CrossRef CAS PubMed.
- C. Balducci, S. Mancini, S. Minniti, P. La Vitola, M. Zotti, G. Sancini, M. Mauri, A. Cagnotto, L. Colombo, F. Fiordaliso, E. Grigoli, M. Salmona, A. Snellman, M. Haaparanta-Solin, G. Forloni, M. Masserini and F. Re, J. Neurosci., 2014, 34, 14022–14031 CrossRef PubMed.
- A. Zensi, D. Begley, C. Pontikis, C. Legros, L. Mihoreanu, S. Wagner, C. Buchel, H. von Briesen and J. Kreuter, J. Controlled Release, 2009, 137, 78–86 CrossRef CAS PubMed.
- Q. Song, H. Song, J. Xu, J. Huang, M. Hu, X. Gu, J. Chen, G. Zheng, H. Chen and X. Gao, Mol. Pharm., 2016, 13, 3976–3987 CrossRef CAS PubMed.
- Q. Song, M. Huang, L. Yao, X. Wang, X. Gu, J. Chen, J. Chen, J. Huang, Q. Hu, T. Kang, Z. Rong, H. Qi, G. Zheng, H. Chen and X. Gao, ACS Nano, 2014, 8, 2345–2359 CrossRef CAS PubMed.
- M. Schaffler, F. Sousa, A. Wenk, L. Sitia, S. Hirn, C. Schleh, N. Haberl, M. Violatto, M. Canovi, P. Andreozzi, M. Salmona, P. Bigini, W. G. Kreyling and S. Krol, Biomaterials, 2014, 35, 3455–3466 CrossRef CAS PubMed.
- J. Kreuter, J. Microencapsulation, 2013, 30, 49–54 CrossRef CAS PubMed.
- G. Sarkar, G. L. Curran, E. Mahlum, T. Decklever, T. M. Wengenack, A. Blahnik, B. Hoesley, V. J. Lowe, J. F. Poduslo and R. B. Jenkins, PLoS One, 2011, 6, e28881 CAS.
- M. Lucarelli, V. Borrelli, A. Fiori, A. Cucina, F. Granata, R. L. Potenza, S. Scarpa, A. Cavallaro and R. Strom, FEBS Lett., 2002, 3, 19–23 CrossRef.
- P. M. Cruz, H. Mo, W. J. McConathy, N. Sabnis and A. G. Lacko, Front. Pharmacol., 2013, 4, 119 Search PubMed.
- G. R. Villa, J. J. Hulce, C. Zanca, J. Bi, S. Ikegami, G. L. Cahill, Y. Gu, K. M. Lum, K. Masui, H. Yang, X. Rong, C. Hong, K. M. Turner, F. Liu, G. C. Hon, D. Jenkins, M. Martini, A. M. Armando, O. Quehenberger, T. F. Cloughesy, F. B. Furnari, W. K. Cavenee, P. Tontonoz, T. C. Gahman, A. K. Shiau, B. F. Cravatt and P. S. Mischel, Cancer Cell, 2016, 30, 683–693 CrossRef CAS PubMed.
- M. J. Rudling, B. Angelin, C. O. Peterson and V. P. Collins, Cancer Res., 1990, 50, 483–487 CAS.
- N. Maleklou, A. Allameh and B. Kazemi, J. Drug Targeting, 2016, 24, 348–358 CrossRef CAS PubMed.
- S. H. Kim, B. B. Adhikari, S. Cruz, M. P. Schramm, J. A. Vinson and V. Narayanaswami, PLoS One, 2015, 10, e0135130 Search PubMed.
- M. Mousazadeh, A. Palizban, R. Salehi and M. Salehi, J. Drug Targeting, 2007, 15, 226–230 CrossRef CAS PubMed.
- M. Ghosh and R. O. Ryan, Nanomedicine, 2014, 9, 763–771 CrossRef CAS PubMed.
- L. B. Josefsen and R. W. Boyle, Theranostics, 2012, 2, 916–966 CrossRef CAS PubMed.
- J. F. Lovell, C. S. Jin, E. Huynh, H. Jin, C. Kim, J. L. Rubinstein, W. C. W. Chan, W. Cao, L. V. Wang and G. Zheng, Nat. Mater., 2011, 10, 324–332 CrossRef CAS PubMed.
- T. W. Liu, T. D. MacDonald, C. S. Jin, J. M. Gold, R. G. Bristow, B. C. Wilson and G. Zheng, ACS Nano, 2013, 7, 4221–4232 CrossRef CAS PubMed.
- T. D. MacDonald, T. W. Liu and G. Zheng, Angew. Chem., Int. Ed., 2014, 53, 6956–6959 CrossRef CAS PubMed.
- L. Cui, Q. Lin, C. S. Jin, W. Jiang, H. Huang, L. Ding, N. Muhanna, J. C. Irish, F. Wang, J. Chen and G. Zheng, ACS Nano, 2015, 9, 4484–4495 CrossRef CAS PubMed.
- G. H. Rothblat and M. C. Phillips, Curr. Opin. Lipidol., 2010, 21, 229–238 CrossRef CAS PubMed.
- W. S. Davidson, D. L. Sparks, S. Lund-Katz and M. C. Phillips, J. Biol. Chem., 1994, 269, 8959–8965 CAS.
- R. S. Rosenson, H. B. Brewer Jr, M. J. Chapman, S. Fazio, M. M. Hussain, A. Kontush, R. M. Krauss, J. D. Otvos, A. T. Remaley and E. J. Schaefer, Clin. Chem., 2011, 57, 392–410 CAS.
- V. P. Skipski, M. Barclay, R. K. Barclay, V. A. Fetzer, J. J. Good and F. M. Archibald, Biochem. J., 1967, 104, 340–352 CrossRef CAS PubMed.
- B. F. Asztalos, M. de la Llera-Moya, G. E. Dallal, K. V. Horvath, E. J. Schaefer and G. H. Rothblat, J. Lipid Res., 2005, 46, 2246–2253 CrossRef CAS PubMed.
- X. Gao, S. Yuan, S. Jayaraman and O. Gursky, J. Mol. Biol., 2009, 387, 628–638 CrossRef CAS PubMed.
- B. Lamarche, K. D. Uffelman, G. Steiner, P. H. Barrett and G. F. Lewis, J. Lipid Res., 1998, 39, 1162–1172 CAS.
- J. Mo, L. He, B. Ma and T. Chen, ACS Appl. Mater. Interfaces, 2016, 8, 6811–6825 CAS.
- A. Pluen, Y. Boucher, S. Ramanujan, T. D. McKee, T. Gohongi, E. di Tomaso, E. B. Brown, Y. Izumi, R. B. Campbell, D. A. Berk and R. K. Jain, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 4628–4633 CrossRef CAS PubMed.
- H. Kafa, J. T. Wang, N. Rubio, R. Klippstein, P. M. Costa, H. A. Hassan, J. K. Sosabowski, S. S. Bansal, J. E. Preston, N. J. Abbott and K. T. Al-Jamal, J. Controlled Release, 2016, 225, 217–229 CrossRef CAS PubMed.
- H. M. Wilson, B. A. Griffin, C. Watt and E. R. Skinner, Biochem. J., 1992, 284, 477–481 CrossRef CAS PubMed.
- L. I. Gidez, G. J. Miller, M. Burstein, S. Slagle and H. A. Eder, J. Lipid Res., 1982, 23, 1206–1223 CAS.
- J. T. Tansey, T. Y. Thuren, W. G. Jerome, R. R. Hantgan, K. Grant and M. Waite, Biochemistry, 1997, 36, 12227–12234 CrossRef CAS PubMed.
- L. Zhang, J. Song, Y. Newhouse, S. Zhang, K. H. Weisgraber and G. Ren, J. Lipid Res., 2010, 51, 1228–1236 CrossRef CAS PubMed.
- V. Narayanaswami, J. N. Maiorano, P. Dhanasekaran, R. O. Ryan, M. C. Phillips, S. Lund-Katz and W. S. Davidson, J. Biol. Chem., 2004, 279, 14273–14279 CrossRef CAS PubMed.
- B. M. Luby, D. M. Charron, C. M. MacLaughlin and G. Zheng, Adv. Drug Delivery Rev., 2016 DOI:10.1016/j.addr.2016.08.010.
- J.-L. Huang, J. Gan, Q.-X. Song, X. Gu, H.-H. Song, X.-L. Wang, D. Jiang, T. Kang, X.-Y. Feng, X.-G. Jiang, H.-Z. Chen and X.-L. Gao, Die Pharmazie, 2016, 71, 709–714 Search PubMed.
- S. Lund-Katz, S. Wehrli, M. Zaiou, Y. Newhouse, K. H. Weisgraber and M. C. Phillips, J. Lipid Res., 2001, 42, 894–901 CAS.
- V. Gupta, V. Narayanaswami, M. S. Budamagunta, T. Yamamato, J. C. Voss and R. O. Ryan, J. Biol. Chem., 2006, 281, 39294–39299 CrossRef CAS PubMed.
- S. Lund-Katz, K. H. Weisgraber, R. W. Mahley and M. C. Phillips, J. Biol. Chem., 1993, 268, 23008–23015 CAS.
- R. G. Anderson, M. S. Brown and J. L. Goldstein, Cell, 1977, 10, 351–364 CrossRef CAS PubMed.
- J. L. Goldstein, R. G. Anderson and M. S. Brown, Nature, 1979, 279, 679–685 CrossRef CAS PubMed.
- M. Zaiou, K. S. Arnold, Y. M. Newhouse, T. L. Innerarity, K. H. Weisgraber, M. L. Segall, M. C. Phillips and S. Lund-Katz, J. Lipid Res., 2000, 41, 1087–1095 CAS.
- T. L. Innerarity, E. J. Friedlander, S. C. Rall Jr, K. H. Weisgraber and R. W. Mahley, J. Biol. Chem., 1983, 258, 12341–12347 CAS.
- T. W. Liu, T. D. MacDonald, J. Shi, B. C. Wilson and G. Zheng, Angew. Chem., 2012, 51, 13128–13131 CrossRef CAS PubMed.
- LDLR tissue atlas, Human protein atlas, Knut & Alice Wallenberg foundation, http://www.proteinatlas.org/ENSG00000130164-LDLR/tissue, 2017 Search PubMed.
- J. R. Bloomer, J. Gastroenterol. Hepatol., 1998, 13, 324–329 CrossRef CAS PubMed.
- A. J. Versluis, P. C. Rensen, E. T. Rump, T. J. Van Berkel and M. K. Bijsterbosch, Br. J. Cancer, 1998, 78, 1607–1614 CrossRef CAS PubMed.
- C. W. Lu, Y. H. Lo, C. H. Chen, C. Y. Lin, C. H. Tsai, P. J. Chen, Y. F. Yang, C. H. Wang, C. H. Tan, M. F. Hou and S. F. Yuan, Cancer Lett., 2017, 388, 130–138 CrossRef CAS PubMed.
- M. Rotheneder and G. M. Kostner, Int. J. Cancer, 1989, 43, 875–879 CrossRef CAS PubMed.
- K. H. Weisgraber, T. L. Innerarity and R. W. Mahley, J. Biol. Chem., 1978, 253, 9053–9062 CAS.
- A. V. Nichols, R. M. Krauss and T. A. Musliner, Methods Enzymol., 1986, 128, 417–431 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7sc00732a |
| This journal is © The Royal Society of Chemistry 2017 |