
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of bioactive and stabilized cyclic peptides by macrocyclization using C(sp3)–H activation†
Jian
Tang
,
Yadong
He
,
Hongfei
Chen
,
Wangjian
Sheng
and
Huan
Wang
 *
*
State Key Laboratory of Coordination Chemistry, School of Chemistry and Chemical Engineering, Nanjing University, Nanjing 210093, China. E-mail: wanghuan@nju.edu.cn
First published on 19th April 2017
Abstract
Cyclic peptides have attracted increasing attention in recent years due to their ability to inhibit protein–protein interactions. Current strategies to prepare cyclic peptides often rely on functional amino acid side chains or the incorporation of unnatural amino acids, thus limiting their structural diversity. Here, we describe the development of a highly versatile peptide macrocyclization strategy through a palladium-catalyzed C(sp3)–H activation and the synthesis of cyclic peptides featuring unique hydrocarbon linkages between the β-carbon of amino acids and the aromatic side chains of Phe and Trp. We demonstrate that such peptides exhibit improved biological properties compared to their acyclic counterparts. Finally, we applied this method in the synthesis of the natural product celogentin C.
Introduction
Peptides and peptidomimics are attracting increasing attention in both the chemical and pharmaceutical communities as a rich source of drug candidates and/or biological tools.1 Since the clinical employment of gramicidin S,2 hundreds of peptides have been identified from natural sources, and a number of them have advanced to the clinic.3 One major driving force for the growing interest in these substances is their potency to interrupt or control biological events mediated by protein–protein interactions (PPIs), which often involve binding contacts spread over a surface.4–9 However, native peptides generally suffer from poor pharmacological properties and lack of structural diversity.10 To address these limitations, various peptide modification strategies have been developed, among which macrocyclization is one of the most prominent methods. Compared to their acyclic counterparts, cyclic peptides generally have highly defined conformations, enhanced cell permeability, thermostability and resistance to proteolytic degradation.11–13Classic methods to generate cyclic peptides often follow a one-component strategy, including head-to-tail lactamization,14–16 internal disulfide17 or thioether formation,18 ring-closing olefin metathesis (RCM)19–22 and catalyzed cycloaddition of azides to alkynes23 (Fig. 1a). A number of two-component cyclization procedures rely on external stapling agents, which usually utilize nucleophiles from lysine and cysteine residues. As a representative example, macrocyclization between lysine residues through nitrogen arylation by a perfluoroaryl linker is reported by Pentelute and co-workers.24,25 Recently, several three-component macrocyclization methods were reported by Yudin and co-workers in the synthesis of aziridine- and oxadiazole-containing peptide macrocycles.26,27
 | ||
| Fig. 1 Peptide macrocyclization strategies and functionalization of the β-carbon of amino acids at the N-terminus of peptides via site-selective C(sp3)–H activation. (a) Current peptide macrocyclization strategies. (b) Intermolecular arylation of peptides at the N-terminus via C(sp3)–H activation. (c) Intramolecular macrocyclization of peptides via C(sp3)–H activation. Phth = phthaloyl. | ||
Despite the success achieved in this field, limitations remain to be overcome. Peptides shorter than five amino acids are generally difficult to cyclize by lactamization and often lead to oligomerization.14,15 For RCM and azide/alkyne cycloaddition, incorporation of a pair of unnatural amino acids at specific sites is usually required, making the preparation of precursor peptides challenging.23,28 The use of external stapling agents helps expand the structural diversity of cyclic peptides; however, only amino acids with reactive side chains, such as lysine and cysteine (relative abundance ≈ 1%), can be involved in these cyclization reactions.29,30 Such requirement excludes amino acids with chemically inert aliphatic side chains, which are highly abundant in naturally occurring peptides/proteins, as macrocyclization sites and thus significantly limits the diversity of peptidomimics generated from these protocols. Therefore, strategies that could activate and involve these “inert” amino acids (e.g. at their β positions) in macrocyclization are highly desirable.
C–C coupling assisted by metal catalysis through direct C–H activation has now become a fundamental strategy to introduce functionalities to inert C–H bonds31–35 and has found applications in the synthesis of cyclic peptide natural products.36,37 In our pursuit to generate peptidomimics with novel architecture for biological studies, we envisioned that peptide macrocyclization by activation of the β-carbon C–H bond of amino acids would help overcome the requirement for reactive amino acid side chains, since almost all amino acids contain a β-carbon (Gly as the only exception). Recently, Yu and co-workers reported their inspiring work in the Pd-catalyzed intermolecular arylation of a C(sp3)–H bond of N-terminal amino acid in oligopeptides without the installation of directing groups (Fig. 1b).38 We postulated that this method would allow peptide macrocyclization through intramolecular arylation with β-carbons of amino acids as stapling sites (Fig. 1c).
In this work, we synthesized a variety of cyclic peptides containing unique Cβ–Ar crosslinks between the β-carbon of amino acids (e.g., Ala, Val) and the aromatic ring of Phe/Trp through a Pd-catalyzed C(sp3)–H activation process. This method is highly versatile and compatible with the standard Fmoc SPPS procedure, resulting in cyclic peptides of different sizes. The impact of peptide conformation on the efficiency of macrocyclization is also investigated. Moreover, we demonstrated that peptidomimics generated from this method possess improved stability compared to their linear peptide precursors. In biological studies, cyclic RGD peptide generated by this method exhibited potent binding affinity to integrin. We also successfully applied this methodology to the synthesis of a peptide natural product containing β-carbon-to-Trp indole linkages.
Results and discussion
Investigation of Pd-catalyzed macrocyclization
To apply the Pd-catalyzed β-carbon arylation method to peptide macrocyclization, we started our initial attempt on a tetrapeptide 1a with the sequence of Phth-Ala-Gly-Gly-(m-I-Phe)-OMe (Phth = phthaloyl, m-I-Phe = meta-iodophenylalanine), where the N- and C-termini of the peptide were protected by phthaloyl and methyl group, respectively (Fig. 2, Scheme S1†). This sequence was chosen because it contains minimal side chains and m-I-Phe provides direct access to peptides containing modified phenylalanine moieties. The installation of protecting groups was meant to minimize the undesired coordination to the Pd(II) catalyst. After optimization of reaction conditions (Table S1†), it was found that with 1a (0.05 mmol), Pd(OAc)2 (5 mol%) as the catalyst, AgOAc (1.2 equiv.) as the additive, in dichloroethane (DCE) at 100 °C, the macrocyclization proceeded smoothly, and the desired product 2a was isolated in 62% yield. The resulting cyclic peptide 2a was characterized by analytical HPLC, high-resolution mass spectrometry (HRMS) and NMR analysis (Fig. 2 and S1†). After cyclization, the Ala βH peak shifted significantly (Fig. 2a) in the 1H NMR spectrum, and the newly formed macrocyclic linkage was further assigned by a diagnostic heteronuclear multiple bond correlation (HMBC) between the Ala βH and the Phe C3 (Fig. 2b). No dimerization products were observed by either HPLC or MS analysis.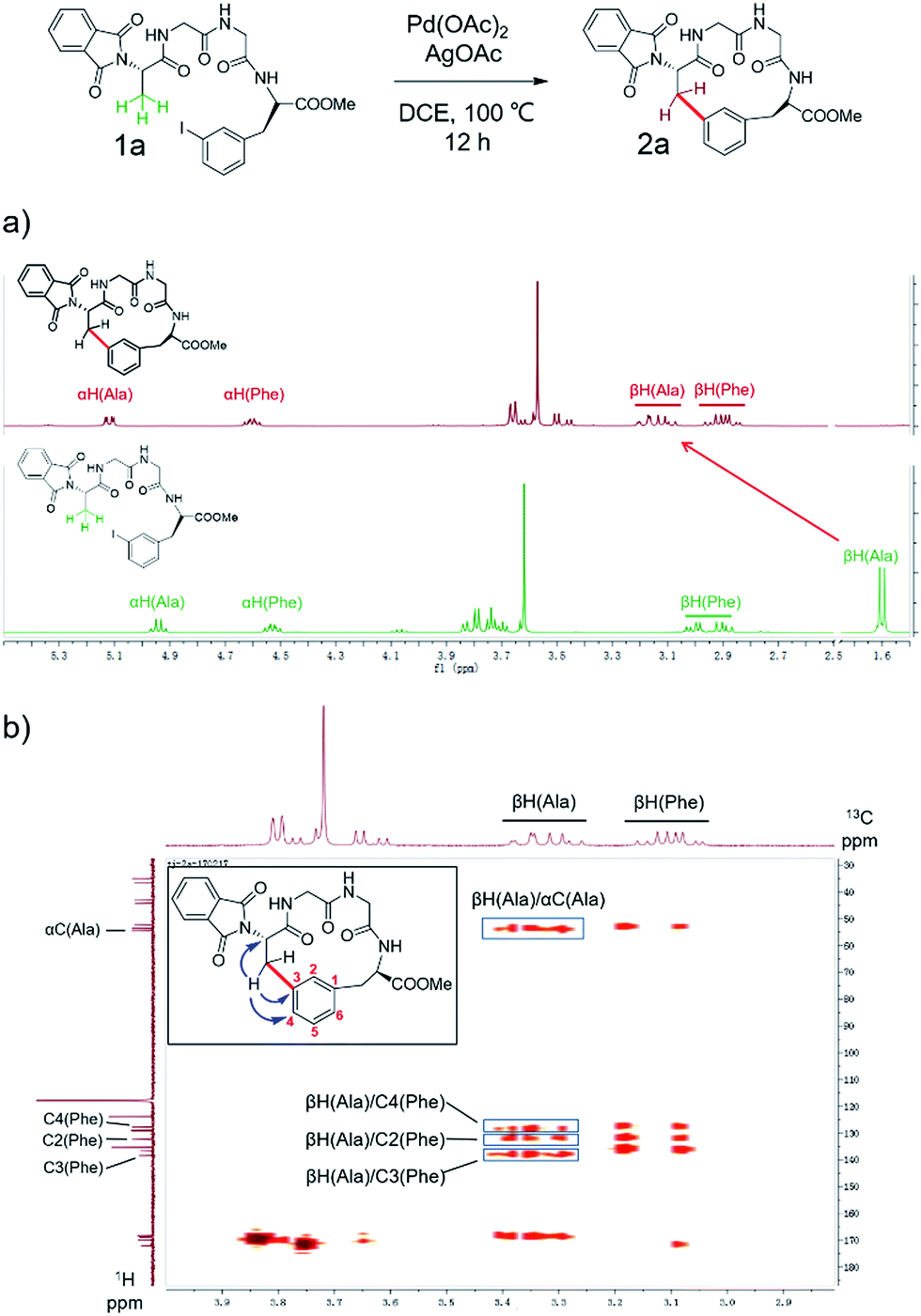 | ||
| Fig. 2 Pd-catalyzed cyclization of peptide substrate 1a Phth-Ala-Gly-Gly-(m-I-Phe)-OMe and structural characterization of the resulting cyclic peptide 2a by NMR spectroscopy. (a) 1H NMR spectra of peptides 1a and 2a. Arrows indicate the resonances of the Ala Cβ protons before (bottom) and after cyclization (top). (b) Key HMBC correlations used to assign the linkage between Ala Cβ and Phe C3 in 2a are indicated by arrows. Full-scale spectra are included in ESI.† | ||
With optimized reaction conditions, we next evaluated the efficiency of this macrocyclization strategy on a variety of tetrapeptide sequences. The incorporation of aliphatic amino acids at position 2 (the second residue to the N-terminus), including Leu, Ile and β-branched Val (2b–2d), did not interrupt the cyclization process, and all afforded cyclic peptides in good isolated yields (53–58%, Table 1). In addition, the crosslinking occurred solely at the β-carbon of the N-terminal Ala, highlighting the regioselectivity of this methodology (see ESI† for structural characterization). Phe-containing peptide 1e and pentapeptide 1f cyclized efficiently under these conditions as well (Table 1).
|
|
|||
|---|---|---|---|
| Entry | Linear peptide | Cyclic peptide | Isolated yield (%) |
| 1a | Phth-Ala-Gly-Gly-m-I-Phe-OMe | 2a | 62 |
| 1b | Phth-Ala-Leu-Gly-m-I-Phe-OMe | 2b | 55 |
| 1c | Phth-Ala-Val-Gly-m-I-Phe-OMe | 2c | 58 |
| 1d | Phth-Ala-Ile-Gly-m-I-Phe-OMe | 2d | 53 |
| 1e | Phth-Ala-Phe-Gly-m-I-Phe-OMe | 2e | 58 |
| 1f | Phth-Ala-Gly-Gly-Ala-m-I-Phe-OMe | 2f | 62 |
| 1g | Phth-Aib-Gly-Gly-m-I-Phe-OMe | 2g | 52 |
| 1h | Phth-Phe-Gly-Gly-m-I-Phe-OMe | 2h | 19 |
| 1i | Phth-Ala-Ser(tBu)-Gly-m-I-Phe-OMe | 2i | 60 |
| 1j | Phth-Ala-Lys(Boc)-Gly-m-I-Phe-OMe | 2j | 65 |
| 1k | Phth-Ala-Asp(OMe)-Gly-m-I-Phe-OMe | 2k | 65 |
| 1l | Phth-Ala-Gly-Gly-m-I-Phe-Gly-Resin | 2l | 21 |
| 1m | Phth-Ala-Gly-Pro-m-I-Phe-OMe | 2m | 87 |
| 1n | Phth-Ala-Gly-Pro-Ala-m-I-Phe-OMe | 2n | 63 |
| 1o | Phth-Ala-Gly-Pro-Ala-p-I-Phe-OMe | 2o | 50 |
| 1p | Phth-Ala-Arg(Pbf)-Gly-Asp(OMe)-m-I-Phe-OMe | 2p | 23 |
Next, we examined peptides with N-terminal residues other than Ala. Linear peptides 1g and 1h with N-terminal 2-aminioisobutyric acid (Aib) and Phe, respectively, were therefore synthesized and subjected to macrocyclization under standard conditions. Results showed that the C–H activation of βH (Aib) in peptide 1g was successful by yielding cyclic peptide 2g in 52% yield. The crosslink in 2g showed low diastereoselectivity (d.r. 60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :40), as determined by 1H NMR and LC-MS analysis (Fig. S2a†). On the other hand, the activation of βH (Phe) in peptide 1h was much less efficient and resulted in a cyclization yield of only 19%, whereas a significant amount of deiodination product was observed by LC-MS, indicating the challenge of crosslinking at the βC of a bulky Phe residue. In contrast to diastereoselectivity observed in peptide 2g, NMR and LC-MS analysis indicated that the resulting cyclic peptide 2h was a single diastereomer (Fig. S2b†). The vicinal coupling between Hα and Hβ of Phe1 of the cyclic peptide 2h was determined to be 12.0 Hz by 1H NMR, suggesting an antiperiplanar arrangement of these two protons and therefore an R configuration of Phe1 Cβ in peptide 2h.
:40), as determined by 1H NMR and LC-MS analysis (Fig. S2a†). On the other hand, the activation of βH (Phe) in peptide 1h was much less efficient and resulted in a cyclization yield of only 19%, whereas a significant amount of deiodination product was observed by LC-MS, indicating the challenge of crosslinking at the βC of a bulky Phe residue. In contrast to diastereoselectivity observed in peptide 2g, NMR and LC-MS analysis indicated that the resulting cyclic peptide 2h was a single diastereomer (Fig. S2b†). The vicinal coupling between Hα and Hβ of Phe1 of the cyclic peptide 2h was determined to be 12.0 Hz by 1H NMR, suggesting an antiperiplanar arrangement of these two protons and therefore an R configuration of Phe1 Cβ in peptide 2h.
To explore the feasibility of performing peptide macrocyclization in solid phase, we first inserted Ser, Lys and Asp residues with side chains masked by standard Fmoc SPPS protecting groups (1i–1k). Results showed that these bulky protecting groups at position 2 were compatible with cyclization conditions without lowering the macrocyclization yields (Table 1). Encouraged by these results, using 2-chlorotrityl chloride resin and following standard Fmoc SPPS procedure, we synthesized peptide 1l with the sequence resin-Gly-(m-I-Phe)-Gly-Gly-Ala-Phth (Table 1 and Scheme S2†). On-resin macrocyclization was achieved with Pd catalyst in DMF at 100 °C for 16 h. The resulting cyclic peptide was cleaved from the resin by TFA and purified by RP-HPLC. NMR and HRMS analysis confirmed that cyclic peptide 2l was successfully synthesized; however, the isolation yield was lower than solution-phase reactions (Table 1). The synthesis of 2l was the first example in which I-Phe is not positioned at the C-terminus of the oligopeptide during cyclization, demonstrating the potential of such method in building in-sequence rings. Interestingly, when para-and ortho-I-Phe was incorporated at the C-terminus of tetrapeptides, none were cyclized successfully, and only deiodination products were detected by MS analysis (Table S2†). Overall, Pd-catalyzed coupling through C(sp3)–H activation was shown to be highly robust and efficient in the preparation of cyclic peptides with novel Cβ–Ar crosslinks (Scheme S3†).
The fact that tetrapeptides with para- or ortho-I-Phe could not be cyclized prompted us to investigate the principle underlining these observations. We reasoned that the cyclic tetrapeptides resulting from macrocyclization were highly constrained, and therefore, conformational organization of linear precursor peptides must allow two reactive ends to coordinate with the catalyst in close spatial proximity before ring closure. We then synthesized peptide 1m with a Pro residue at position 3, which introduced a conformational turn into the linear precursor tetrapeptide. To our delight, the cyclization of peptide 1m proceeded extremely well with almost full conversion as monitored by HPLC with an isolation yield of 87%, indicating the impact of peptide conformation on cyclization efficiency. Pro-containing pentapeptide 1n also cyclized efficiently. Interestingly, pentapeptide 1o (Table 1), which contains a Pro residue and a para-I-Phe, cyclized smoothly with a yield of 50% (Scheme S3†). This result suggested that the failure of tetrapeptides containing of para- or ortho-I-Phe to cyclize was most likely due to conformational bias, instead of inherent reactivity. The insertion of a Pro residue, a conformational turn-inducing element, might relieve the constraint of the cyclic pentapeptide and facilitate Cβ–Ar crosslinking.
Biological evaluation of cyclic peptides
To evaluate the potential of the Cβ–Ar cyclized peptides for biological applications, we synthesized a novel cyclic RGD peptide 2p following this procedure (Fig. 3). We first examined the proteolytic stability of 2p in comparison with its linear counterpart 1p. Peptides 1p and 2p were incubated with endoprotease AspN, which hydrolyzes peptide bonds on the N-terminal side of Asp residue. Results showed that in the presence of AspN, cyclic peptide 2p remained intact for up to 5 h, whereas its linear counterpart 1p degraded quickly with a half-life of 50 min and was fully consumed in 5 h, as determined by HPLC and MS analysis (Fig. 3). Similar results were observed when peptides 1j′ and 2j′, which were resulted from deprotected peptides 1j and 2j, were incubated with protease trypsin (Fig. 3), where peptide 1j′ had a half-life of only 21 min, and up to 95% cyclic peptide 2j′ remained intact after 5 h. These results demonstrate that the Cβ–Ar linkage in cyclic peptides significantly improved their stability against proteolytic degradation.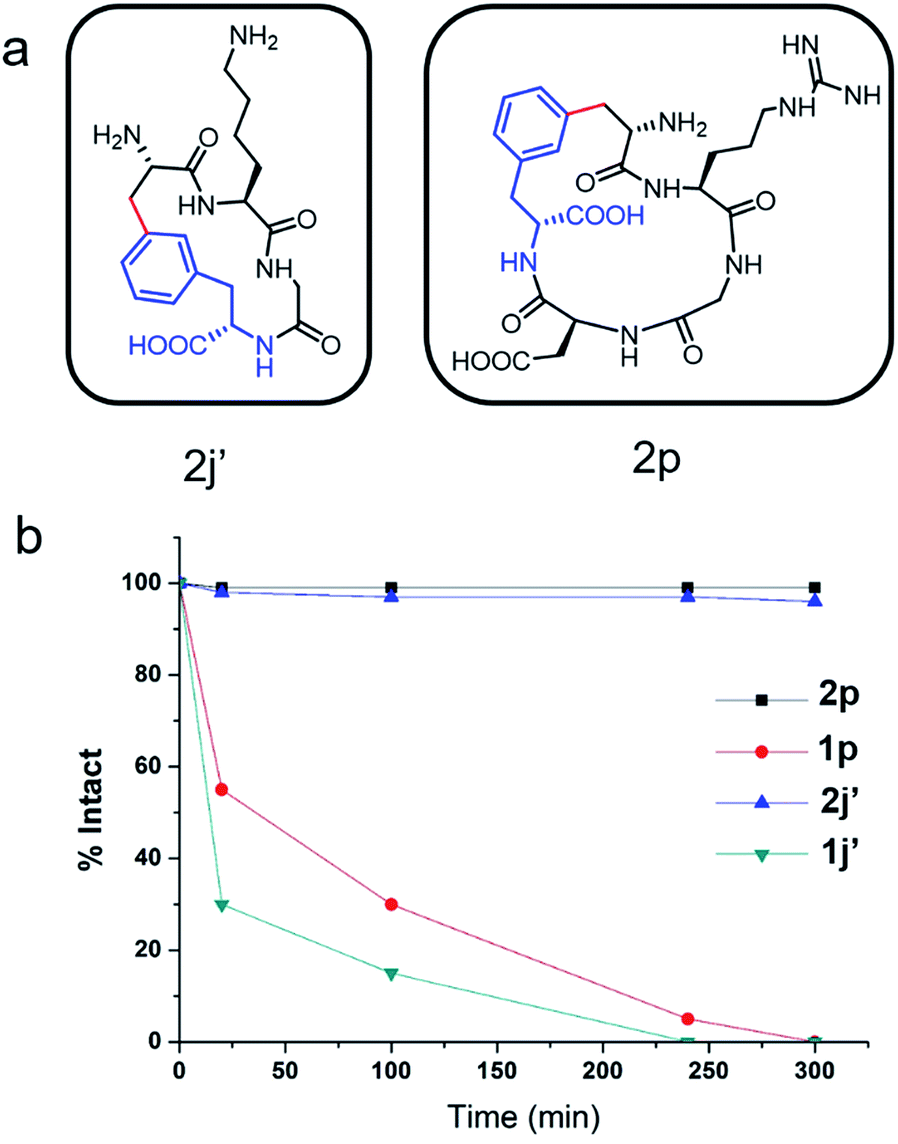 | ||
| Fig. 3 Cyclic peptides exhibited enhanced resistance towards proteolytic degradation. (a) Chemical structures of cyclic peptides 2j′ and 2p. (b) Proteolytic degradation assay of stapled peptides 2j′ and 2p and their linear counterparts 1g and 1m. Assay conditions: 50 mM Tris buffer, pH 7.4, 50 μM peptide substrates, 0.1 mg ml−1 protease AspN or trypsin, at 25 °C. | ||
Next, we examined the binding of cyclic RGD peptide 2p to U87MG cells, which is a glioblastoma cell line overexpressing the αvβ3 integrin. The RGD sequence is well known for its selective binding to integrins when incorporated in proper cyclic structures.39 Fluorescein isothiocyanate (FITC) was then conjugated to the N-terminal amine of 2p to yield fluorescent-labelled 2p-FITC. Linear peptide 1p was also labelled with FITC as a direct comparison. As a positive control, we employed a commercially available cyclo-(RGDfK) and conjugated a FITC moiety onto the Lys amine. U87MG cells were then incubated with 1p-FITC, 2p-FITC and cyclo-(RGDfK)-FITC for 90 min and analyzed by confocal microscopy. As expected, cyclo-(RGDfK)-FITC elicited strong fluorescence staining of cells, whereas the linear peptide conjugate 1p-FITC yielded low fluorescence, suggesting its weak binding to integrins (Fig. 4a and b). Interestingly, cyclic peptide 2p-FITC caused slightly stronger fluorescence staining than cyclo-(RGDfK)-FITC, indicating that the cyclic structure introduced by a Cβ–Ar linkage in 2p significantly enhanced the integrin binding of the RGD sequence. These results nicely demonstrated the applicability of cyclic peptides with Cβ–Ar linkages in biological systems.
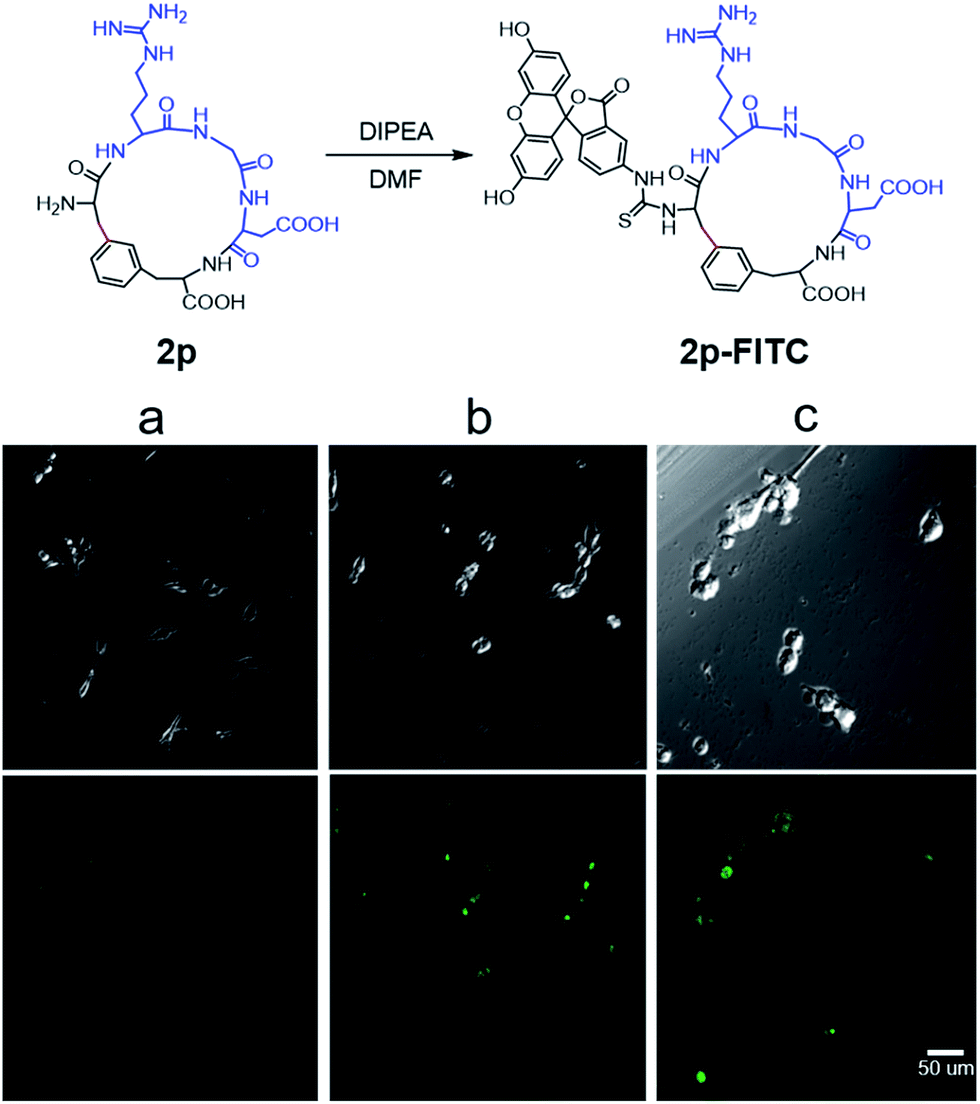 | ||
| Fig. 4 Synthesis of FITC-labelled cyclic RGD peptide 2p and its integrin binding assays analyzed by confocal microscopy. (a) Linear peptide 1p-FITC, (b) cyclo-(RGDfK)-FITC, (c) cyclic peptide 2p-FITC. All peptides were used at 2 μM concentration for the cell binding studies. The RGD motif is in blue, and the Cβ–Ar bond is in red. Scale bar: 50 μm. | ||
Application in natural product synthesis
Finally, we applied this macrocyclization methodology to natural product synthesis. Several peptide natural products containing Cβ–Ar linkages have been identified in recent years. For example, streptide is a recently characterized ribosomally synthesized and posttranslationally modified peptide (RiPP) featuring a C–C crosslink between Trp C5 and Lys Cβ (Fig. 5a).40 Celogentin C, a nonribosomal peptide possessing inhibitory activity against tubulin polymerization, contains a direct linkage of Trp C6 to Leu Cβ (Fig. 5a).41 Towards the total synthesis of celogentin C, Chen and co-workers employed a Pd-catalyzed strategy to construct the Leu1-Trp C–C bond with the assistance of a quinolone directing group.36,42 We postulated that our C–H activation macrocyclization strategy would allow the construction of the celogentin C-ring A from a precursor peptide containing 6-I-Trp and avoid the introduction and removal of directing groups (Fig. 5c). Accordingly, we synthesized a 6-I-Trp containing precursor peptide 1q (Table 1) and subjected it to macrocyclization.36 Gratifyingly, peptide 1q cyclized smoothly under coupling conditions, affording the desired cyclic peptide 2o in 32% isolated yield. Structural characterization of 2q was conducted by HPLC, HRMS and NMR analysis. Product 2q appeared as a sharp single peak in analytical HPLC, suggesting a single diastereoisomer (Fig. S3†). A correlation between Trp C6 and Leu1 βH was observed in HMBC analysis, indicating the direct Leu1 βC-to-Trp C6 linkage in 2q (Fig. S4†). The large vicinal coupling (Jαβ(Leu1) = 12.0 Hz) and the absence of correlation in NOESY analysis between Hα and Hβ of βs-Leu1 strongly suggested an antiperiplanar arrangement of these two protons (Fig. S5†), meaning that the configuration of Leu1 Cβ in 2q is R and thus identical to the Leu2 Cβ of the natural product celogentin C.41 Further derivatizations of 2q yielded compound 2r, the ring A of celogentin C. The 1H NMR and 13C NMR spectra of compound 2r is in very good agreement with the same compound reported by Chen's group, which further supports the correct configuration of Leu1 Cβ.36 This remarkable stereoselective 6-indolation of Leu Cβ in cyclic peptide 2q suggested that analogous to the palladium complex formed with directing groups,36 the amide bond N,N-biscoordinated complex generated during peptide cyclization promoted the formation of the trans-palladacycle intermediate (Fig. S6†).37 In addition, the successful formation of the βC-Trp C6 linkage further expands the scope of this methodology to iodine-substituted indoles and β-branched amino acids.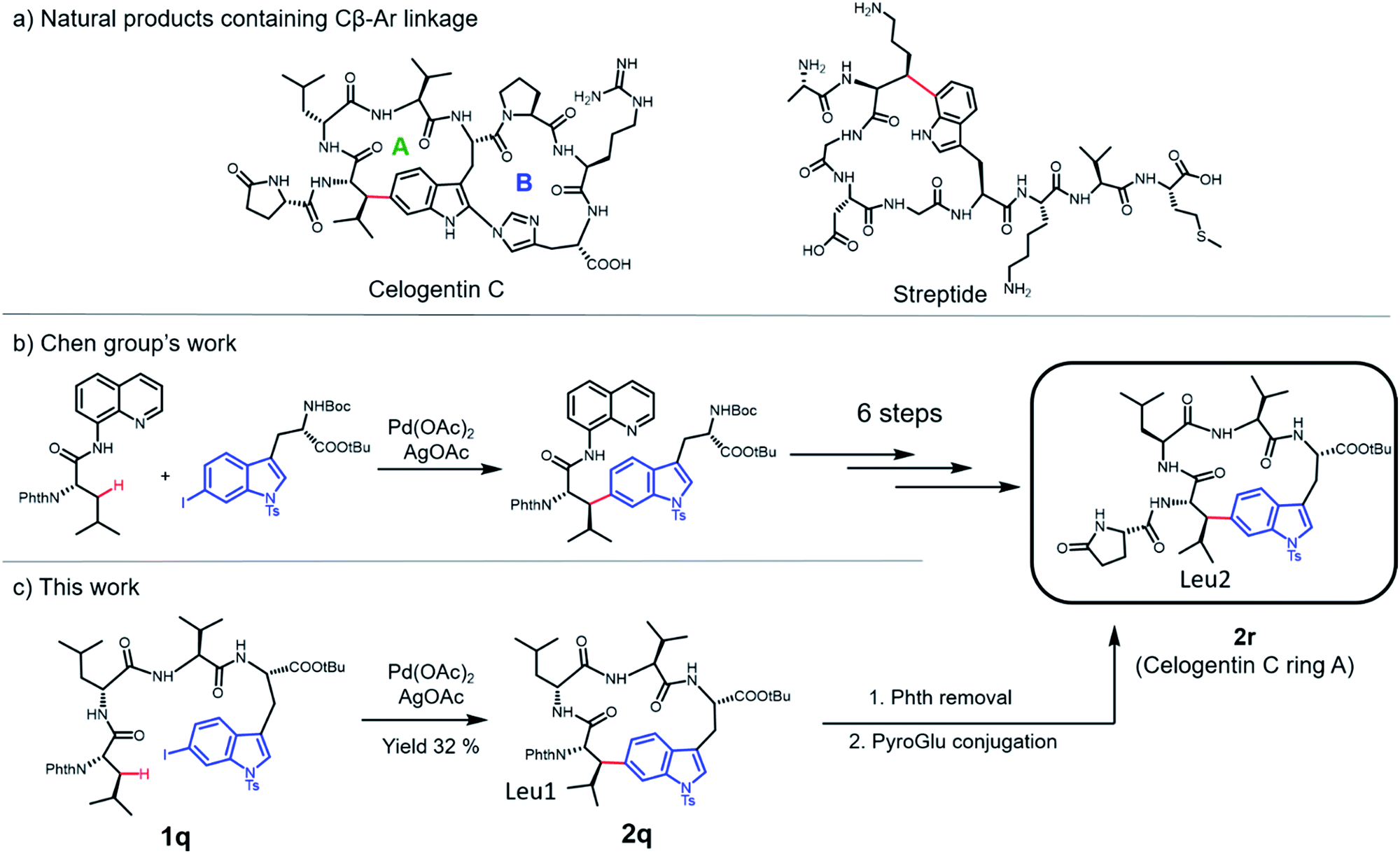 | ||
| Fig. 5 Strategies for the synthesis of natural products containing Cβ–Ar linkages. (a) Examples of natural products containing Cβ-indole crosslinks. (b) Key step for Cβ-indole linkage formation in the total synthesis of celogentin C through directing-group-assisted C–H activation. (c) Macrocyclization strategy by C–H arylation in this work. | ||
Conclusions
In this work, we have developed a robust procedure to generate cyclic peptides with Cβ–Ar linkages through Pd-catalyzed C(sp3)–H activation, which proceeds smoothly in both solution-phase and solid-phase peptide synthesis. This method allows a diverse set of amino acids to serve as peptide stapling sites and therefore could greatly expand the structural diversity of cyclic peptide libraries. This method has a broad scope of peptide substrates, including tetra- and pentapeptides, and it has been demonstrated as a powerful tool in natural product synthesis by introducing a Cβ–Ar bond with high regio- and stereoselectivity. In addition, we demonstrated that a RGD cyclic peptide generated through this method has improved proteolytic stability and higher binding affinity to integrin than a commercially available cyclo-(RGDfK). The generation of natural products and peptidomimics of biological importance through this strategy is in progress in our laboratory.43Acknowledgements
The authors acknowledge the financial support from the National Natural Science Foundation of China (21402091) and the Thousand Talents Program. We would like to thank Prof. Zhuangzhi Shi (NJU) for helpful discussion at the initial stage of the project and Mr Xiao Xiao for confocal microscopy analysis.Notes and references
- S. R. Gracia, K. Gaus and N. Sewald, Future Med. Chem., 2009, 1, 1289–1310 CrossRef CAS PubMed.
- G. F. Gause and M. G. Brazhnikova, Nature, 1944, 154, 703 CrossRef.
- V. Marx, Chem. Eng. News, 2005, 83, 17–24 Search PubMed.
- A. Sandomenico, S. M. Monti, M. Sabatella, A. De Capua, L. Tornatore, N. Doti, F. Viparelli, N. A. Dathan, C. Pedone, M. Ruvo and D. Marasco, Chem. Biol. Drug Des., 2009, 73, 483–493 CAS.
- M. C. Souroujon and D. Mochly-Rosen, Nat. Biotechnol., 1998, 16, 919–924 CrossRef CAS PubMed.
- P. Chene, ChemMedChem, 2006, 1, 400–411 CrossRef CAS PubMed.
- W. L. DeLano, M. H. Ultsch, A. M. de Vos and J. A. Wells, Science, 2000, 287, 1279–1283 CrossRef CAS PubMed.
- W. L. Lian, P. Upadhyaya, C. A. Rhodes, Y. S. Liu and D. H. Pei, J. Am. Chem. Soc., 2013, 135, 11990–11995 CrossRef CAS PubMed.
- A. Czarna, B. Beck, S. Srivastava, G. M. Popowicz, S. Wolf, Y. J. Huang, M. Bista, T. A. Holak and A. Domling, Angew. Chem., Int. Ed., 2010, 49, 5352–5356 CrossRef CAS PubMed.
- D. J. Craik, D. P. Fairlie, S. Liras and D. Price, Chem. Biol. Drug Des., 2013, 81, 136–147 CAS.
- G. H. Bird, N. Madani, A. F. Perry, A. M. Princiotto, J. G. Supko, X. He, E. Gavathiotis, J. G. Sodroski and L. D. Walensky, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 14093–14098 CrossRef CAS PubMed.
- Z. Szewczuk, B. F. Gibbs, S. Y. Yue, E. O. Purisima and Y. Konishi, Biochemistry, 1992, 31, 9132–9140 CrossRef CAS PubMed.
- E. M. Driggers, S. P. Hale, J. Lee and N. K. Terrett, Nat. Rev. Drug Discovery, 2008, 7, 608–624 CrossRef CAS PubMed.
- J. S. Davies, J. Pept. Sci., 2003, 9, 471–501 CrossRef CAS PubMed.
- A. Thakkar, T. B. Trinh and D. Pei, ACS Comb. Sci., 2013, 15, 120–129 CrossRef CAS PubMed.
- G. K. Nguyen, X. Hemu, J. P. Quek and J. P. Tam, Angew. Chem., Int. Ed., 2016, 55, 12802–12806 CrossRef CAS PubMed.
- M. Gongora-Benitez, J. Tulla-Puche and F. Albericio, Chem. Rev., 2014, 114, 901–926 CrossRef CAS PubMed.
- H. Jo, N. Meinhardt, Y. Wu, S. Kulkarni, X. Hu, K. E. Low, P. L. Davies, W. F. DeGrado and D. C. Greenbaum, J. Am. Chem. Soc., 2012, 134, 17704–17713 CrossRef CAS PubMed.
- H. E. Blackwell, J. D. Sadowsky, R. J. Howard, J. N. Sampson, J. A. Chao, W. E. Steinmetz, D. J. O'Leary and R. H. Grubbs, J. Org. Chem., 2001, 66, 5291–5302 CrossRef CAS PubMed.
- H. E. Blackwell and R. H. Grubbs, Angew. Chem., Int. Ed., 1998, 37, 3281–3284 CrossRef CAS.
- L. D. Walensky, A. L. Kung, I. Escher, T. J. Malia, S. Barbuto, R. D. Wright, G. Wagner, G. L. Verdine and S. J. Korsmeyer, Science, 2004, 305, 1466–1470 CrossRef CAS PubMed.
- C. E. Schafmeister, J. Po and G. L. Verdine, J. Am. Chem. Soc., 2000, 122, 5891–5892 CrossRef CAS.
- R. Dharanipragada, Future Med. Chem., 2013, 5, 831–849 CrossRef CAS PubMed.
- A. M. Spokoyny, Y. Zou, J. J. Ling, H. Yu, Y. S. Lin and B. L. Pentelute, J. Am. Chem. Soc., 2013, 135, 5946–5949 CrossRef CAS PubMed.
- G. Lautrette, F. Touti, H. G. Lee, P. Dai and B. L. Pentelute, J. Am. Chem. Soc., 2016, 138, 8340–8343 CrossRef CAS PubMed.
- R. Hili, V. Rai and A. K. Yudin, J. Am. Chem. Soc., 2010, 132, 2889–2891 CrossRef CAS PubMed.
- J. R. Frost, C. C. Scully and A. K. Yudin, Nat. Chem., 2016, 8, 1105–1111 CrossRef CAS PubMed.
- C. J. White and A. K. Yudin, Nat. Chem., 2011, 3, 509–524 CrossRef CAS PubMed.
- E. V. Vinogradova, C. Zhang, A. M. Spokoyny, B. L. Pentelute and S. L. Buchwald, Nature, 2015, 526, 687–691 CrossRef CAS PubMed.
- L. Mendive-Tapia, S. Preciado, J. Garcia, R. Ramon, N. Kielland, F. Albericio and R. Lavilla, Nat. Commun., 2015, 6, 7160 CrossRef PubMed.
- L. Ackermann, Chem. Rev., 2011, 111, 1315–1345 CrossRef CAS PubMed.
- R. Y. Zhu, M. E. Farmer, Y. Q. Chen and J. Q. Yu, Angew. Chem., Int. Ed., 2016, 55, 10578–10599 CrossRef CAS PubMed.
- M. Gulias and J. L. Mascarenas, Angew. Chem., Int. Ed., 2016, 55, 11000–11019 CrossRef CAS PubMed.
- J. Yamaguchi, A. D. Yamaguchi and K. Itami, Angew. Chem., Int. Ed., 2012, 51, 8960–9009 CrossRef CAS PubMed.
- O. Baudoin, Chem. Soc. Rev., 2011, 40, 4902–4911 RSC.
- Y. Feng and G. Chen, Angew. Chem., Int. Ed., 2010, 49, 958–961 CrossRef CAS PubMed.
- A. F. M. Noisier and M. A. Brimble, Chem. Rev., 2014, 114, 8775–8806 CrossRef CAS PubMed.
- W. Gong, G. Zhang, T. Liu, R. Giri and J. Q. Yu, J. Am. Chem. Soc., 2014, 136, 16940–16946 CrossRef CAS PubMed.
- C. Mas-Moruno, F. Rechenmacher and H. Kessler, Anti-Cancer Agents Med. Chem., 2010, 10, 753–768 CrossRef CAS PubMed.
- K. R. Schramma, L. B. Bushin and M. R. Seyedsayamdost, Nat. Chem., 2015, 7, 431–437 CrossRef CAS PubMed.
- J. Kobayashi, H. Suzuki, K. Shimbo, K. Takeya and H. Morita, J. Org. Chem., 2001, 66, 6626–6633 CrossRef CAS PubMed.
- G. Rouquet and N. Chatani, Angew. Chem., Int. Ed., 2013, 52, 11726–11743 CrossRef CAS PubMed.
- Note: while submitting this manuscript, Albericio et al. reported a similar strategy for the preparation of cyclic peptides through C–H activation (Angew. Chem., Int. Ed., 2017, 56, 314–318). Whereas the disclosed data is in accord with our findings, yields are significantly improved under our cyclization conditions. Moreover, the biological properties of the cyclic peptides are evaluated for the first time along with fapplication of this methodology in natural product synthesis.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6sc05530c |
| This journal is © The Royal Society of Chemistry 2017 |