
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Rh(III)-catalyzed double C–H activation of aldehyde hydrazones: a route for functionalized 1H-indazole synthesis†
Pan
Xu
a,
Guoqiang
Wang
b,
Zhongkai
Wu
a,
Shuhua
li
*b and
Chengjian
Zhu
*ac
aState Key Laboratory of Coordination Chemistry, School of Chemistry and Chemical Engineering, Nanjing University, Nanjing 210093, P. R. China. E-mail: cjzhu@nju.edu.cn
bKey Laboratory of Mesoscopic Chemistry of Ministry of Education, Institute of Theoretical and Computational Chemistry School of Chemistry and Chemical Engineering, Nanjing University, Nanjing, 210093, P. R. China. E-mail: shuhua@nju.edu.cn
cState Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, Shanghai 200032, P. R. China
First published on 7th October 2016
Abstract
A novel and straightforward strategy for functionalized 1H-indazoles is realized by the Rh(III)-catalyzed double C–H activation and C–H/C–H cross coupling of readily available aldehyde phenylhydrazones. The reaction is scalable and various 1H-indazoles could be afforded in moderate to high yields with good functional-group compatibility. Mechanism experiments and DFT calculations suggest the distinctive Rh(III)-catalyzed C–H/C–H cross coupling reaction underwent a cascade C(aryl)–H bond metalation, C(aldhyde)–H bond insertion and reductive elimination process.
Introduction
Hydrazones are particularly attractive as building blocks in synthetic organic chemistry because of their easy availability and versatile reactivity.1 As well as enjoying the features of carbonyl compounds, the most unique property of aldehyde hydrazone is its ability to react with active electrophiles as a neutral acyl anion equivalent, which represents an important method for the C(sp2)–H functionalization of aldehyde hydrazones.2 However, the demand of limited strong electrophilic reagents such as acyl chloride and the subsequent problem in terms of functional-group tolerance have restricted its widespread application. Hence the development of effective and general methodologies for the C(sp2)–H functionalization of aldehyde hydrazones is highly desired. A recent breakthrough in this area is the transition metal (Cu, Pd) or visible light catalytic strategies for the C(sp2)–H functionalization of aldehyde hydrazones (Scheme 1a).3 Mechanism experiments as well as theoretical calculations in our previous report3b support an electron transfer induced aminyl radical-polar crossover (ARPC) process, in which the key activating unit, the N–N bond, plays a crucial role. From another point of view, the N–N bond, a Lewis basic functional unit, often appears as a directing group in transition metal-catalyzed C–H activation reactions. To complement these radical-type strategies, we have recently initiated a project to exploit the directing group strategy for the C(sp2)–H bond functionalization of aldehyde hydrazones.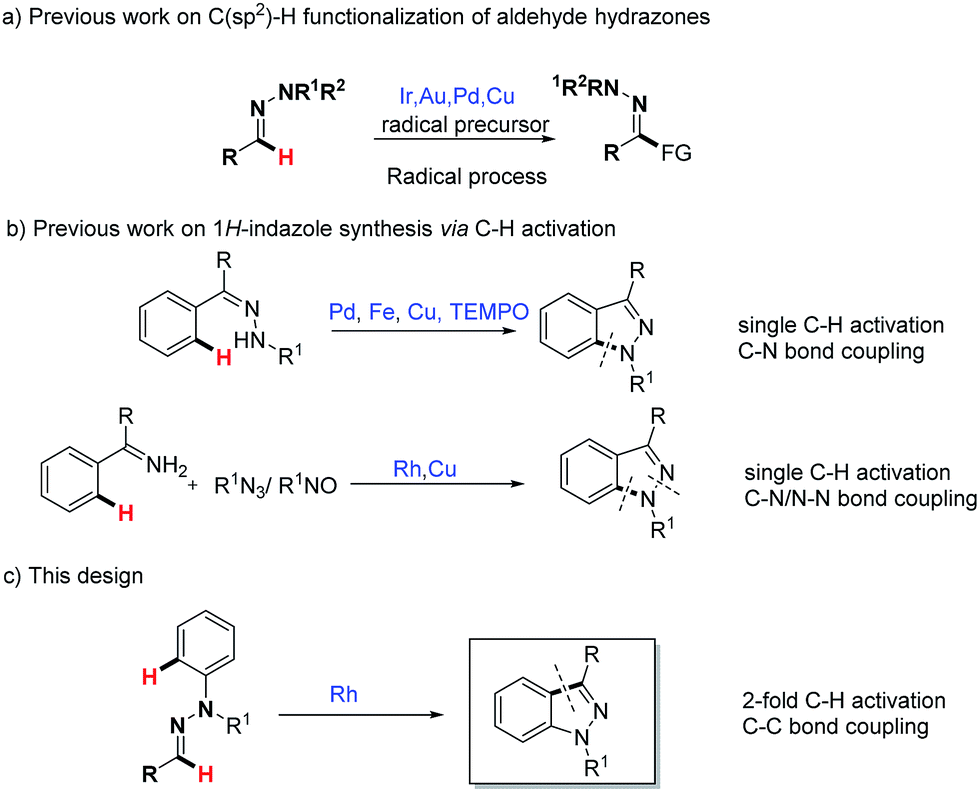 | ||
| Scheme 1 Strategy design for 1H-indazole synthesis from aldehyde hydrazones. | ||
1H-Indazole, a privileged pharmacophore in pharmaceuticals, is widely incorporated in multiple drugs such as anti-HIV, anti-inflammatory and anti-cancer drugs.4 The efficient synthesis of functionalized 1H-indazole has particularly attracted the increased attention of organic synthetic chemists.5,6 In the past few years, transition metal-catalyzed C–H activation has emerged with significant advantages toward the synthesis of a diverse array of heterocycles.7 Among them, Pd-,6a Fe-,6b Cu-6c,e and Rh-6d,f catalyzed C–H activation strategies have been applied to the synthesis of 1H-indazoles (Scheme 1b), which complement conventional approaches. Despite progress, it is noteworthy here that previous wisdom unanimously focused on the 1H-indazole synthesis via C–N bond formation. Given the importance of 1H-indazole synthesis and our recent ongoing interest in the C(sp2)–H bond functionalization of aldehyde hydrazones,6b,c we wondered here whether functionalized 1H-indazole could be directly synthesized from readily available aldehyde phenylhydrazones through a Rh(III)-catalyzed cascade 2-fold C–H activation and oxidative C–C bond formation (Scheme 1c). Importantly, different from previous 1H-indazole syntheses, such as the synthetic method, which is based on the challenging C–C bond construction, this method has its own particular advantages in the synthesis of certain distinctive and significant 1H-indazole scaffolds. In our strategy design, the N–N bond is initially used as a directing group in the Rh(III)-catalyzed C(aryl)–H bond activation step. Then the resulting active metallacyclic intermediate could serve as a potential reactive linkage that is capable of the ultimate C(N)–H bond functionalization. Difficulties in this design need to be considered: (1) the regioselectivity in the presence of two different aromatic rings (C-aryl and N-aryl) is challenging; and (2) the stability of the N–N bond under the reaction condition, as in previous Rh(III)-catalyzed hydrazine or other N–N group directed C–H activations, the N–N bond tends to cleave as an internal oxidant.8 To the best of our knowledge, the Rh(III)-catalyzed C(sp2)–H functionalization of aldehyde hydrazones is unprecedented, thus representing an unexplored research topic.
Results and discussion
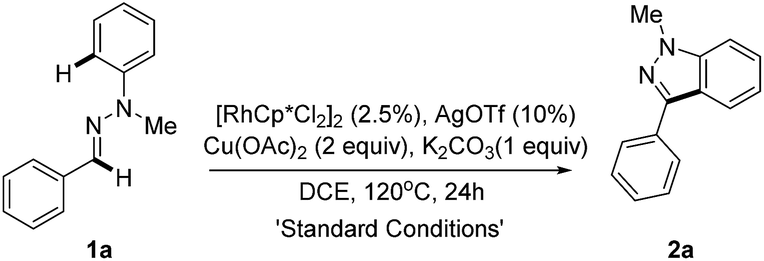
We initially selected the readily accessible hydrazone 1a as the model substrate to investigate our hypothesis about the Rh(III)-catalyzed intramolecular oxidative C–H/C–H bond cross coupling. Various optimization studies revealed that the desired product 1H-indazole 2a could be achieved in 80% yield with (RhCp*Cl2)2/AgOTf as the catalyst precursor, Cu(OAc)2 as the oxidant and K2CO3 as the base at 120 °C in 1,2-dichloroethane (Table 1 and ESI†). Control experiments were performed to test the role of each reactant. Hydrazone tends to decompose under pH-acidic reaction conditions. Thus, K2CO3 is added to neutralize the in situ generated acetic acid (entry 2). As expected, rhodium plays a core role in the transformation (entry 3). Catalytic Ag salt is not necessary, yet could promote the reaction with higher efficiency (entry 4). A lower loading of Cu(OAc)2 would induce a lower yield (entry 5) and an attempt to make the reaction greener with O2 as the sole oxidant failed (entry 6). The temperature effect was also examined and it was found that a higher temperature did not increase the reaction efficiency (entry 7), while lowering the reaction temperature decreased the yield (entry 8).|
|
|||
|---|---|---|---|
| Entry | Variation from the “standard conditions” | Conv. of 1ab (%) | Yieldb (%) |
| a The reactions were run on a 0.20 mmol scale in 0.5 mL of DCE. b Yields determined by 1NMR spectroscopy using N-(4-methoxyphenyl)acetamide as the internal standard. c Yield of isolated products. Cp* = 1,2,3,4,5-pentamethylcyclopentadiene, and DCE = 1,2-dichloroethane. | |||
| 1 | None | 84 | 81(80)c |
| 2 | Without K2CO3 | 5 | Trace |
| 3 | Without [RhCp*Cl2]2 | 0 | 0 |
| 4 | Without AgOTf | 55 | 50 |
| 5 | 1.5 equiv. of Cu(OAc)2 | 50 | 42 |
| 6 | O2 and without Cu(OAc)2 | 5 | 0 |
| 7 | 135 °C instead of 120 °C | 85 | 80 |
| 8 | 100 °C instead of 120 °C | 66 | 65 |
With the optimized reaction conditions in hand, the substrate scope of the reaction was next systematically explored. The representative examples are shown in Table 2. Substrates with different N-alkyl (Me, Et and Bn) or aryl groups all furnished the corresponding 1H-indazoles 2a–d with good to excellent yields (78–95%), whereas N–H or N–Ac aldehyde hydrazones failed to give the desired product, which is probably a result of the operation of electronic effect. In addition, we found that O-aryl oxime derivatives cannot be transferred into benzisoxazoles under the reaction conditions. Various electron-donating (methoxy, methyl, dimethylamine) and electron-withdrawing (fluoro, chloro, bromo, trifluoromethyl, ester) groups substituted at the aldehyde phenyl rings were readily tolerated (2e–p). However, the position of the substituted group at the aryl group has an obvious effect on this reaction. Only para (2e–k) and meta (2l–o) substituted aldehyde hydrazones could proceed well, giving the corresponding products in good yields while ortho substituted aldehyde hydrazones proved unreactive with only ortho-fluoro hydrazone 1p furnishing the desired product in a low yield (2p). This unforeseen steric effect prompted us to rethink the mechanism of this Rh(III)-catalyzed oxidative C–H/C–H cross coupling reaction.9 Importantly, hetero-aldehyde derived aldehyde hydrazones such as furan and thiophene also exhibit good reactivity, providing the expected products in moderate to good yields (2q–s).
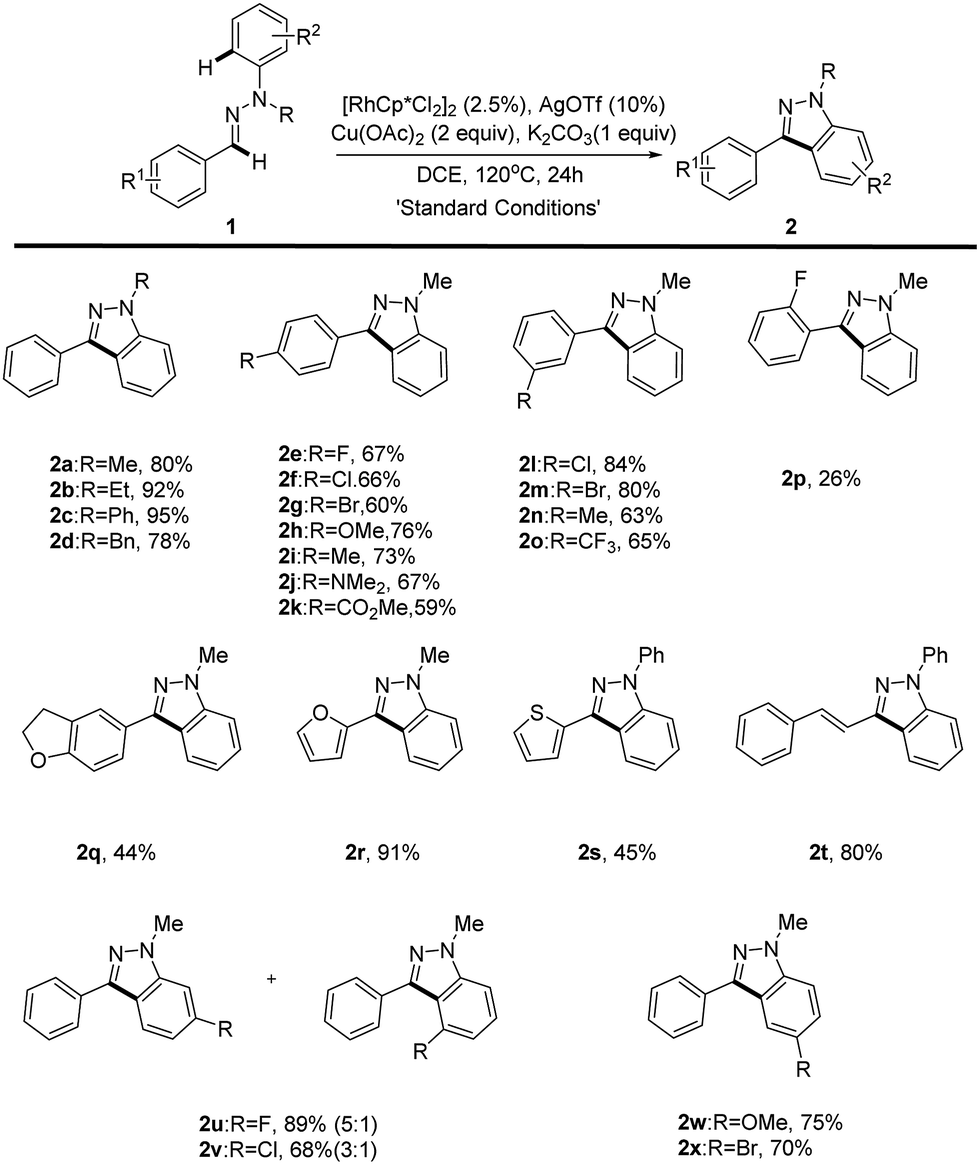
Unfortunately, the aliphatic aldehyde-derived or alkyl-substituted alkenyl aldehyde hydrazones decomposed and failed to give the desired product under our reaction conditions. The broad substrate scope of this reaction was further illustrated by altering the substitution patterns on the aryl-hydrazine unit (2u–x). When meta-substituted substrates underwent the oxidative coupling reaction, the C–H activation occurred at both positions with moderate regioselectivities (2u–v).
Remarkably, a gram-scale reaction of 1a could still be transformed into the product 2a in a good yield (73%) under standard reaction conditions, thus making our strategy for 1H-indazole synthesis more attractive and robust (Scheme 2).
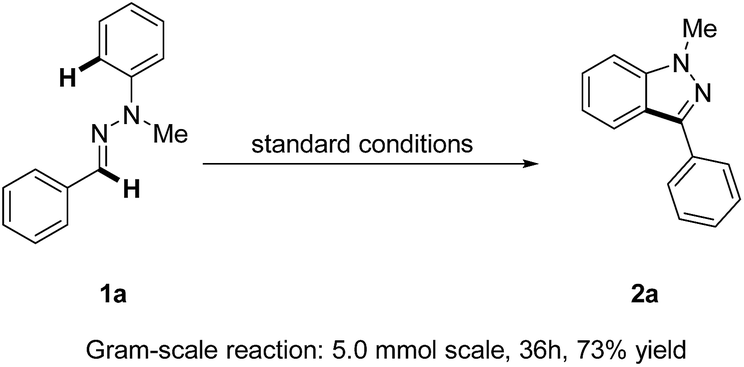 | ||
| Scheme 2 Gram-scale synthesis of 1H-indazole 2a. | ||
To further demonstrate the unique advantages of our reaction, we applied our synthetic strategy to the rapid synthesis of certain significant and bioactive 1H-indazole scaffolds (Table 3), which show special 5-HT4/5-HT3 receptor antagonist activity.10 With readily available tetrahydroquinoline and benzo[b][1,4]oxazine as starting materials, a simple four-step sequence including N-nitrosation, reduction, condensation with aldehyde and Rh(III)-catalyzed intramolecular oxidative C–H/C–H cross coupling could concisely furnish the desired 1H-indazole scaffolds in moderate overall yields. It is worth mentioning that this kind of 1H-indazole scaffold could not be accessed via previous reported synthetic strategies. The X-ray single-crystal structure analysis of the 1H-indazole 4f is shown in Table 3.
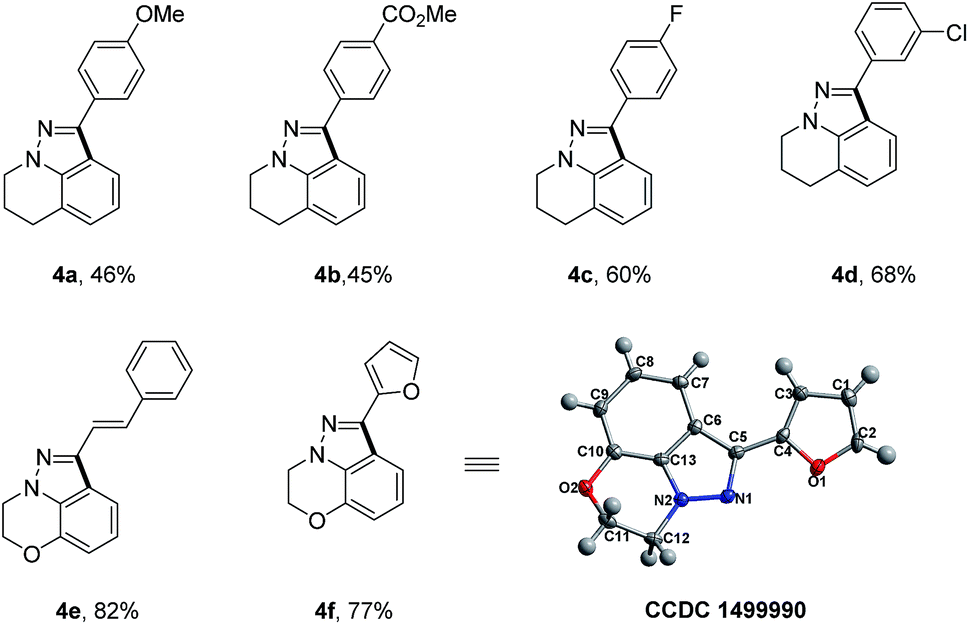
To gain insight into this Rh(III)-catalyzed oxidative C–H/C–H cross coupling reaction, a deuterium labeling experiment with 1j-d3 was carried out (Scheme 3). Deuterium losses both in the product and reclaimed substrate were observed, suggesting the process involved an initial reversible C–H bond metalation. In addition, the identification of C–H bond deuteration at the ortho-position of the C-aryl rings (both product and substrate) provides an evidence for the initial two competitive coordination sites with the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–N system as the directing group. However, valuable and convincible KIE data for this intramolecular double C–H activation process can be obtained with difficultly via experiments to study the rate-limiting process.
N–N system as the directing group. However, valuable and convincible KIE data for this intramolecular double C–H activation process can be obtained with difficultly via experiments to study the rate-limiting process.
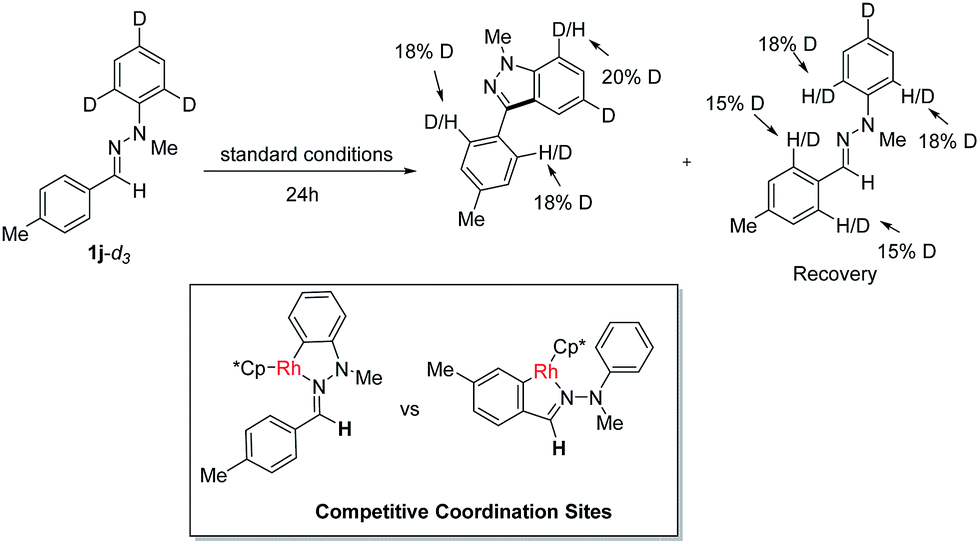 | ||
| Scheme 3 Deuterium labeling experiment. | ||
Based on the preliminary studies, the mechanism of this Rh(III)-catalyzed oxidative C–H/C–H cross coupling is proposed in Scheme 4. In the presence of catalytic AgOTf and an equivalent of Cu(OAc)2, Rh(III)Cp*(OAc)2I is generated in situ as an active catalyst. The transformation is initiated through the coordination of the nitrogen atom of the CN bond to the cationic Rh center and subsequent C(aryl)–H activation, generating the five-membered rhodacyclic complex II.8 Then the active intermediate serves as a reactive linkage for the second C(sp2)–H bond insertion,11 resulting in the six-membered rhodacyclic complex III (path 1). Final reductive elimination will give the desired 1H-indazole 2a and Rh(I), which can be oxidized by Cu(II) to regenerate the active Rh(III) catalyst. However, another mechanism, which involves CN bond insertion12 rather than C–H bond insertion, is also possible at this stage (path 2). The resulting polar C(aryl)–Rh bond in the first step can serve as a nucleophilic aryl source and undergo nucleophilic addition to the CN bond, forming the N–Rh species IV. Under the basic conditions, the N–Rh species then undergoes β-H elimination to furnish the desired product 2a.
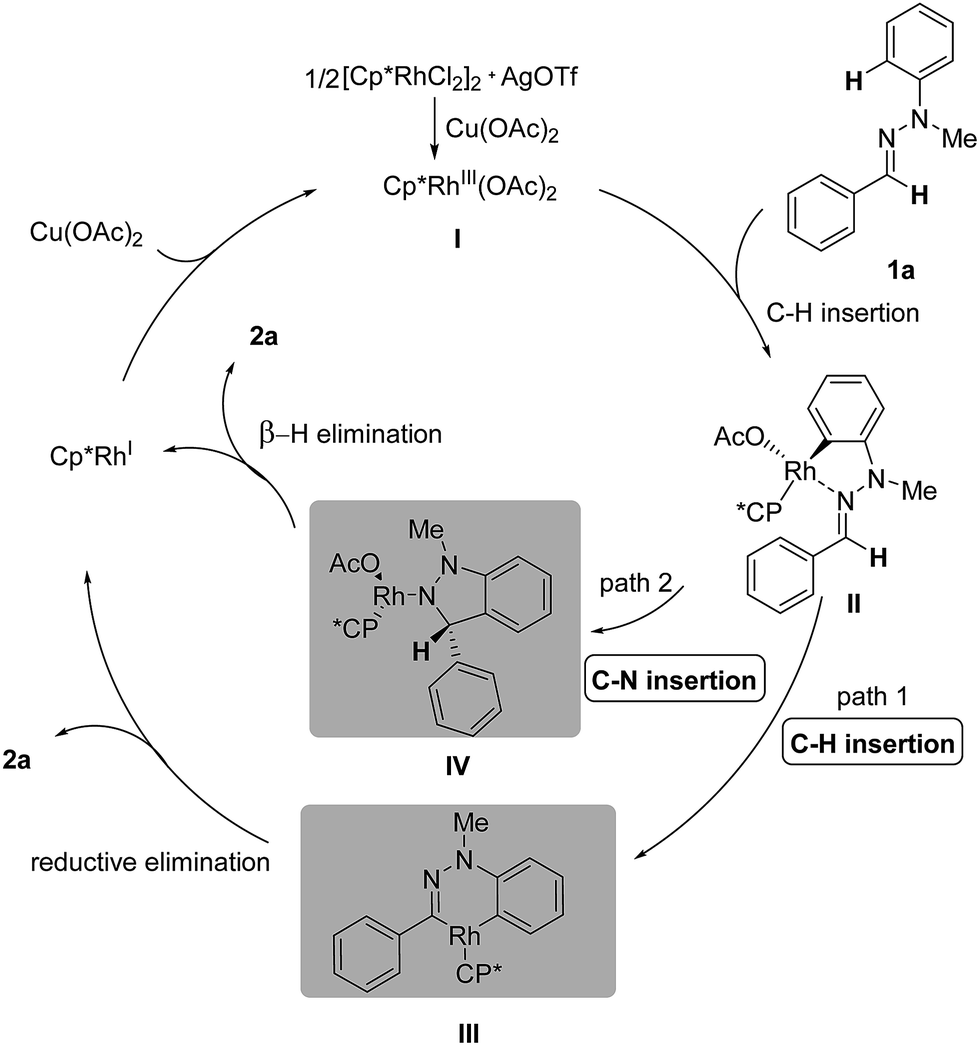 | ||
| Scheme 4 Proposed mechanism. | ||
Furthermore, density functional theory (DFT) calculations were performed to investigate the detailed mechanism of this Rh(III)-catalyzed oxidative C–H/C–H cross coupling reaction; two possible reaction pathways are proposed in Scheme 4, and the free energy profiles are shown in Fig. 1 (see ESI† for details). The active catalyst I and hydrazone 1a were chosen as the model system. First, the coordination of the nitrogen atom of the CN bond in 1a to the Rh center and subsequent C(aryl)–H activation8 generates the five-membered rhodacyclic intermediate IIviaTS1, with a barrier of 33.3 kcal mol−1. The formation of intermediate II is endothermic by 5.4 kcal mol−1, which is consistent with the deuterium labeling experiment (a reversible C–H bond metalation). Then, the N–N bond rotation of intermediate II forms the rotation isomer II′viaTS2, with a barrier of 22.4 kcal mol−1. Subsequently, the second C(aldehyde)–H bond activation of the II′ intermediate assisted by an acetate yields the 16-electron six-membered rhodacyclic complex IIIviaTS3 (with a barrier of 26.4 kcal mol−1), releasing an acetic acid molecule. Finally, the reductive elimination of the intermediate III gives the desired 1H indazole (2a) and Rh(I) intermediate (Cp*Rh(I)) viaTS4, with a barrier of 32.3 kcal mol−1 (path 1, black lines). The generation of Cp*Rh(I) and 2a is endothermic by 19.8 kcal mol−1 (with respect to the active catalyst I and reactant 1a). This result is consistent with the observed fact that the stoichiometric amount of oxidant (Cu(OAc)2) and base (K2CO3) were required for promoting this reaction. In addition to path 1, path 2 which involves the nucleophilic addition of the C–Rh bond to the CN bond of species II11 is also possible at this stage (as shown in Scheme 4, path 2). However, all attempts that tried to locate the transition state of the intramolecular nucleophilic addition (or CN insertion) failed. Nevertheless, we performed a relaxed potential energy scan by fixing the C–C distance at a series of values to estimate the approximate barrier (as shown in Fig. S1†). The generation of the CN bond insertion intermediate IV is endothermic by 48.8 kcal mol−1 (path 2, red lines), which suggests that path 2 can be excluded. Therefore, path 1 which involves the C(aryl)–H metalation/C(aldehyde)–H insertion/reductive elimination sequence is more responsible for the Rh(III)-catalyzed C–H/C–H cross coupling reaction.
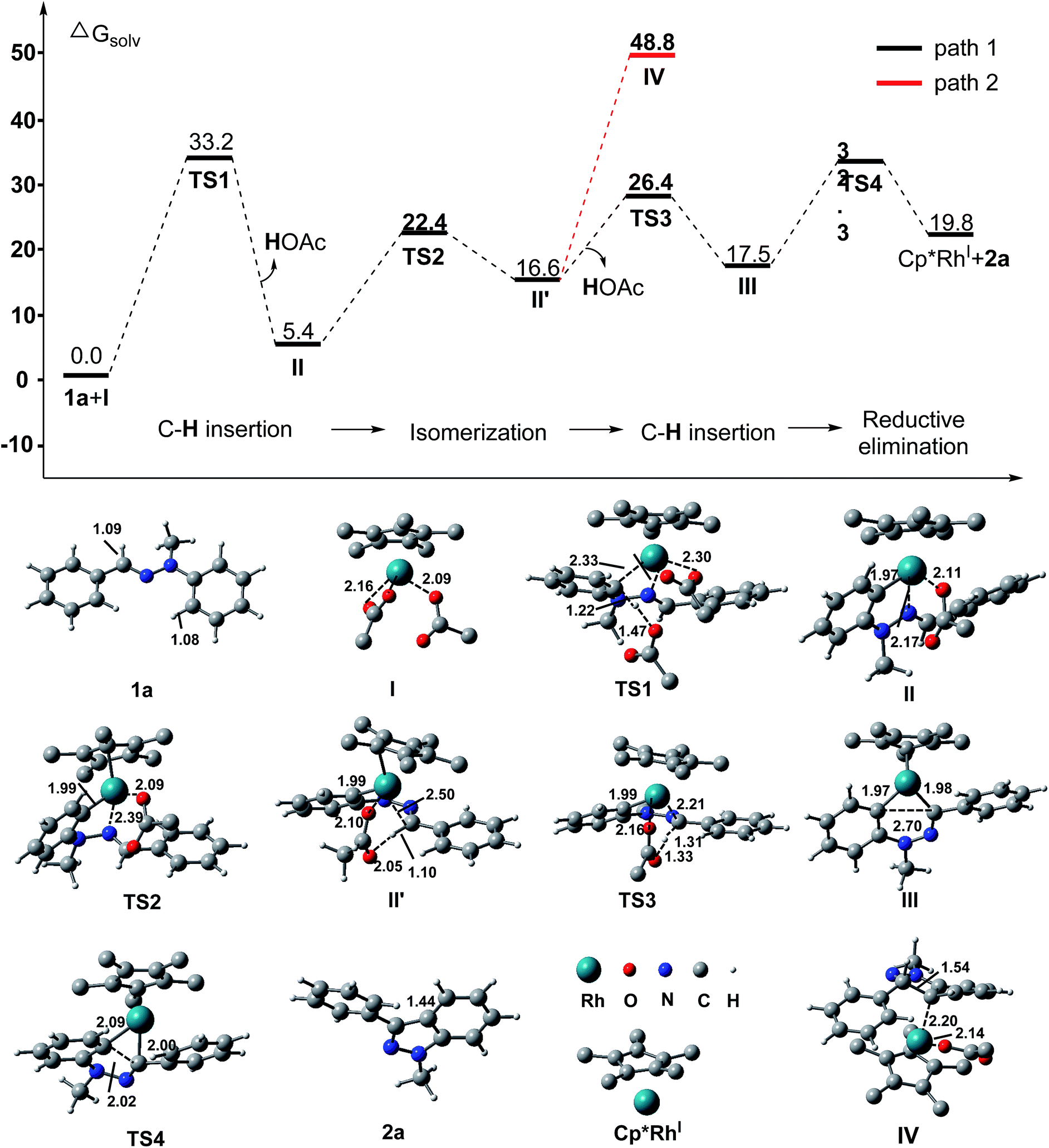 | ||
| Fig. 1 Computed Gibbs free energy (in kcal mol−1) profile for the reaction between the hydrazone 1a and active catalyst I in the solvent (1,2-dichloroethane). Distances are in Å. | ||
Conclusions
In conclusion, an intramolecule directing group strategy has been successfully applied for the C(aldehyde)–H functionalization of aldehyde hydrazones for the first time. Highly functionalized 1H-indazoles could be directly accessed from easily available aldehyde phenylhydrazones via Rh(III)-catalyzed oxidative C–H/C–H cross coupling. The reaction is scalable and various 1H-indazoles can be afforded in moderate to high yields with good functional-group compatibility. A mechanism study indicates this distinctive C–H/C–H cross coupling was enabled by a C(aryl)–H metalation/C(aldhyde)–H insertion/reductive elimination sequence. We believe this new strategy for the C(sp2)–H functionalization of aldehyde hydrazones will be extended to the concise synthesis of other important heterocyclic skeletons.Acknowledgements
We gratefully acknowledge the National Natural Science Foundation of China (21474048, 21372114 and 21672099).Notes and references
- R. Lazny and A. Nodzewska, Chem. Rev., 2010, 110, 1386 CrossRef CAS PubMed.
- R. Brehme, D. Enders, R. Fernandez and J. M. Lassaletta, Eur. J. Org. Chem., 2007, 5629 CrossRef CAS.
- (a) E. Pair, N. Monteiro, D. Bouyssi and O. Baudoin, Angew. Chem., Int. Ed., 2013, 52, 5346–5349 CrossRef CAS PubMed; (b) P. Xu, G. Wang, Y. Zhu, W. Li, Y. Cheng, S. Li and C. Zhu, Angew. Chem., Int. Ed., 2016, 55, 2939 CrossRef CAS PubMed; (c) P. Xu, Z. Wu, N. Zhou and C. Zhu, Org. Lett., 2016, 18, 1143 CrossRef CAS PubMed; (d) A. Prieto, R. Melot, D. Bouyssi and N. Monteiro, Angew. Chem., Int. Ed., 2016, 55, 1885 CrossRef CAS PubMed; (e) J. Xie, T. Zhang, F. Chen, N. Mehrkens, F. Rominger, M. Rudolph and A. S. Hashmi, Angew. Chem., Int. Ed., 2016, 55, 2934 CrossRef CAS PubMed.
- (a) R. S. Turnbull, J. Can. Dent. Assoc., 1995, 61, 127 CAS; (b) J. D. Rodgers, B. L. Johnson, H. Wang, R. A. Greenberg, S. Erickson-Viitanen, R. M. Klabe, B. C. Cordova, M. M. Rayner, G. N. Lam and C. H. Chang, Bioorg. Med. Chem. Lett., 1996, 6, 2919 CrossRef CAS; (c) J. H. Sun, C. A. Teleha, J. S. Yan, J. D. Rodgers and D. A. Nugiel, J. Org. Chem., 1997, 62, 5627 CrossRef CAS; (d) A. Jennings and M. Tennant, J. Chem. Inf. Model., 2007, 47, 1829 CrossRef CAS PubMed; (e) J. Magano, M. Waldo, D. Greene and E. Nord, Org. Process Res. Dev., 2008, 12, 877 CrossRef CAS; (f) A. V. Dolzhenko and W. K. Chui, Heterocycles, 2008, 75, 1575 CrossRef CAS.
- For selected reports on 1H-indazole syntheses: (a) S. Caron and E. Vazquez, Synthesis, 1999, 588 CrossRef CAS; (b) W. Stadlbauer, Sci. Synth., 2002, 12, 227 CAS; (c) F. Crestey, V. Collot, S. Stiebing and S. Rault, Tetrahedron, 2006, 62, 7772 CrossRef CAS; (d) K. Lukin, M. C. Hsu, D. Fernando and M. R. Leanna, J. Org. Chem., 2006, 71, 8166 CrossRef CAS PubMed; (e) T. Jin and Y. Yamamoto, Angew. Chem., Int. Ed., 2007, 46, 3323 ( Angew. Chem. , 2007 , 119 , 3387 ) CrossRef CAS PubMed; (f) J. Hu, H. Xu, P. Nie, X. Xie, Z. Nie and Y. Rao, Chem.–Eur. J., 2014, 20, 3932 CrossRef CAS PubMed; (g) M. Tang, Y. Kong, B. Chu and D. Feng, Adv. Synth. Catal., 2016, 358, 926 CrossRef CAS.
- For recent reports on 1H-indazole syntheses via C–H activation: (a) K. Inamoto, T. Saito, M. Katsuno, T. Sakamoto and K. Hiroya, Org. Lett., 2007, 9, 2931 CrossRef CAS PubMed; (b) T. Zhang and W. Bao, J. Org. Chem., 2013, 78, 1317 CrossRef CAS PubMed; (c) X. Li, L. He, H. Chen, W. Wu and H. Jiang, J. Org. Chem., 2013, 78, 3636 CrossRef CAS PubMed; (d) D. G. Yu, M. Suri and F. Glorius, J. Am. Chem. Soc., 2013, 135, 8802 CrossRef CAS PubMed; (e) J. Peng, Z. Xie, M. Chen, J. Wang and Q. Zhu, Org. Lett., 2014, 16, 4702 CrossRef CAS PubMed; (f) Q. Wang and X. Li, Org. Lett., 2016, 18, 2102 CrossRef CAS PubMed.
- For selected reviews: (a) C. Sun, B. Li and Z. Shi, Chem. Rev., 2011, 111, 1293 CrossRef CAS PubMed; (b) C. Liu, H. Zhang, W. Shi and A. Lei, Chem. Rev., 2011, 111, 1780 CrossRef CAS PubMed; (c) D. A. Colby, A. S. Tsai, R. G. Bergman and J. A. Ellman, Acc. Chem. Res., 2012, 45, 814 CrossRef CAS PubMed; (d) K. M. Engle, T.-S. Mei, M. Wasa and J.-Q. Yu, Acc. Chem. Res., 2012, 45, 788 CrossRef CAS PubMed; (e) N. Kuhl, M. N. Hopkinson, J. Wencel-Delord and F. Glorius, Angew. Chem., Int. Ed., 2012, 51, 10236 CrossRef CAS PubMed; (f) S. A. Girard, T. Knauber and C. Li, Angew. Chem., Int. Ed., 2014, 53, 74 CrossRef CAS PubMed; (g) L. Ackermann, Acc. Chem. Res., 2014, 47, 281 CrossRef PubMed; (h) X. Guo, D. Gu, Z. Wu and W. Zhang, Chem. Rev., 2015, 115, 1622 CrossRef CAS PubMed; (i) G. Song and X. Li, Acc. Chem. Res., 2015, 48, 1007 CrossRef CAS PubMed.
- For selected reports with an N–N bond as an internal oxidant: (a) B. Liu, Y. Fan, Y. Gao, C. Sun, C. Xu and J. Zhu, J. Am. Chem. Soc., 2013, 135, 468–473 CrossRef CAS PubMed; (b) B. Liu, C. Song, C. Sun, S. Zhou and J. Zhu, J. Am. Chem. Soc., 2013, 135, 16625 CrossRef CAS PubMed; (c) D. Zhao, Z. Shi and F. Glorius, Angew. Chem., Int. Ed., 2013, 52, 12426 CrossRef CAS PubMed.
- We have performed the computational investigations to investigate the influence of steric effects on this reaction, please see Table S1.†.
- (a) E. Luc, G. Claudie, A. Michel, B. R. Philippe, PCT Int. Appl., WO 9920633 A1 19990429, 1999; (b) E. Luc, A. Michel, PCT Int. Appl., WO 9811112 A1 19980319, 1998.
- (a) N. Kuhl, M. N. Hopkinson and F. Glorius, Angew. Chem., Int. Ed., 2012, 51, 8230 ( Angew. Chem. , 2012 , 124 , 8354 ) CrossRef CAS PubMed; (b) J. Delord, C. Nimphius, H. Wang and F. Glorius, Angew. Chem., Int. Ed., 2012, 51, 13001 CrossRef PubMed; (c) K. Morimoto, M. Itoh, K. Hirano, T. Satoh, Y. Shibata, K. Tanaka and M. Miura, Angew. Chem., Int. Ed., 2012, 51, 5359 CrossRef CAS PubMed.
- (a) Y. Li, B. Li, W. Wang, W. Huang, X. Zhang, K. Chen and Z. Shi, Angew. Chem., Int. Ed., 2011, 50, 2115 CrossRef CAS PubMed; (b) A. S. Tsai, M. E. Tauchert, R. G. Bergman and J. A. Ellman, J. Am. Chem. Soc., 2011, 133, 1248 CrossRef CAS PubMed; (c) A. Wangweerawong, R. G. Bergman and J. A. Ellman, J. Am. Chem. Soc., 2014, 136, 8520 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1499990. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6sc03888c |
| This journal is © The Royal Society of Chemistry 2017 |