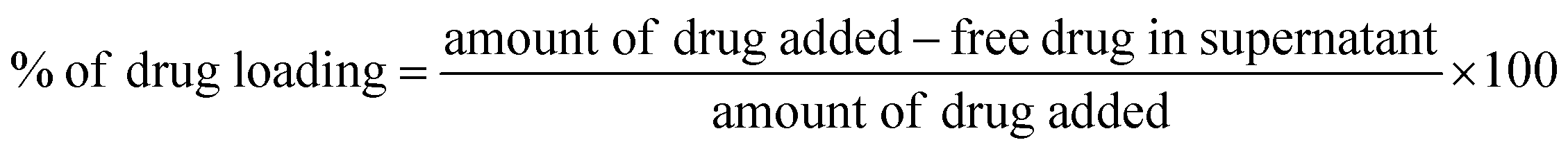DOI:
10.1039/C7RA06615E
(Paper)
RSC Adv., 2017,
7, 46271-46285
Magneto-chemotherapy for cervical cancer treatment with camptothecin loaded Fe3O4 functionalized β-cyclodextrin nanovehicle†
Received
13th June 2017
, Accepted 4th August 2017
First published on 28th September 2017
Abstract
We portray a novel way to deal with the synthesis of iron oxide magnetic nanoparticle incorporated β-cyclodextrin (β-CD) nanocarrier stabilized by ethylenediamine tetra acetic acid (EDTA), which obtained remarkable biocompatibility and biodegradability. The functionality, crystalline nature, morphology and thermal decomposition nature of Fe3O4, β-CD–EDTA–Fe3O4 and β-CD–EDTA–Fe3O4/CPT nanocarriers were confirmed by Fourier transform infrared spectroscopy (FT-IR), X-ray powder diffraction (XRD), scanning electron microscopy (SEM) and thermogravimetric analysis (TGA). The mechanistic action of camptothecin (CPT) on cancer cells was studies relying to incorporation with the topoisomerase (topo I) and DNA complex formation. This prevented DNA re-ligation, causing DNA damage which resulted in apoptosis in cervical cancer (HeLa) cells. We noticed a higher number of cells arrested in the G1 phase with a decreasing cell count in the S phase and the G2/M phase at a lower concentration of β-CD–EDTA–Fe3O4/CPT. Consequently, via conjugation with Fe magnetic nanoparticles (MNPs), the molecular recognition between β-CD and target the cancerous cells make this nanovehicle a promising candidate for the non-invasive treatment of HeLa cells.
1. Introduction
In cancer chemotherapy, a major difficulty is to design highly effective drug vehicles that can deliver bioactive agents specifically to cells or organs and reduce or eliminate unwanted side effects. Recently, innovative magnetic cargo releasing carriers have been developed for medicinal applications.1,2 MNPs have been examined for medicinal uses, such as cell labeling/cell partition, gene delivery, and as contrast agents for magnetic resonance imaging (MRI).3 MNPs with medical applications are principally made from magnetic iron oxide (Fe3O4), along with general attributes of nanoparticles, are biocompatible and can be relaxed to synthesize4 chemically stable and magnetically responsive. Fe3O4 nanoparticles are the main magnetic nanomaterial approved for medical applications by the FDA (US Food and Drug Administration).5,6 Magnetic iron oxide-assisted drug carriers can significantly hinder tumor multiplication and initiate apoptosis of cancer cells in dosage- and time-variable methods. These effects may be associated with bioactive compounds released by the magnetic nanomaterials and the in vivo influence on the metabolic processes of magnetic iron oxide once transported into the diseased cells.7–9
The expenditure of conventional drugs from natural and chemical synthesis for tumor treatment has drawbacks such as the quick discharge of their therapeutic effect, short blood circulation, and non-target specificity that can damage normal cells, causing serious side effects.10,11 Currently, improved drugs that incorporate nanoparticle-based remedies are emerging tools in nanopharmacology. Nanoparticle-based treatment provides controlled drug release, improved cell uptake, and target specificity.12,13 Furthermore, the anti-cancer agents cause minimal damage to normal cells, yet target and destroy cancer-causing cells readily.14 The highly significant features of nanoparticles used in medicine are particle size, molecular weight, pH, ionic strength, and charge of the particles.15 In addition, site-specific delivery need to incorporate one or more targeting molecules to reach the cancer cells without disrupting the delivery mechanism.16 Polymers commonly used for preparation of nanoparticles as drug carriers include grafts, block copolymers, micelles, poly ester–amides, dendrimers and some hybrid materials.13,16–18 In our present work, β-CD was functionalized with magnetic iron oxide by cross-linking of EDTA. The multifunctional qualities of β-CD in biomedical applications make this molecule attractive for chemotherapeutic applications. The specific property of CDs is their capability to enhance drug delivery through biological membranes.19 The most significant property of CDs is their geometric structure, valuable in enhancing host–guest interactions with various types of moieties with appropriate polarity and size.20 In addition, simulation studies have long affirmed the hypothetical concept of probable application of CD-encapsulated drugs for the deliver of anesthesia and bioactive agents to treat diseases of the nervous system.21,22 Research into the possibility of incorporating CD with metals utilizing cross-linkage with cross-linkers, for instance, glutaraldehyde (GLA) and epichlorohydrin (EPI) to produce CD polymers (CDP) have been reported.23 However, both GLA and EPI are recognized to be harmful and carcinogenic to both human and animals.24 EDTA is efficient chelating operators that produce nontoxic, stable chelates with metals.25
CPT is frequently used anti-cancer drug that works effectively on numerous types of cancer, including ovarian and colorectal tumors. The alkaloid compound of CPT was initially isolated from a Chinese tree, Camptotheca acumincta, in the early 1960s.26 CPT is recognized as a selective topoisomerase I inhibitor.27 Emerging studies proposed that the potential of CPT to act as anti-tumor agent might be attributed to its ability to inhibit nitric oxide (NO) biosynthesis.28 As a DNA topoisomerase I inhibitor, camptothecin forms a stable ternary topoisomerase I-DNA cleavable complex, which initiates an apoptotic signaling pathway, ultimately resulting in cell death.29 Meanwhile, a main drawback of CPT is that it has a reduced therapeutic efficacy due to its poor solubility in aqueous media30 and active lactone ring instability at physiological pH.30 As chemotherapy is one of the emerging treatment methods to treat cancer, various nano-carriers are continuously being formulated and designed to increase the solubility of camptothecin. The solubility of chemical drugs is essential for complete therapeutic effect and bioavailability at the targeted location. Poor solubility greatly reduces the anti-cancer capabilities of some chemotherapy analogs, such as CPT, and limits their use to only certain cancers. To rectify this problem, multiple analogs of CPT with improved lactone stability and aqueous solubility have been developed. Formulations of CPT have been developed through conjugation with various polymers, including polyethylene glycol (PEG),31 CD copolymer,32 poly(L-glutamic acid),33 and chitosan.34 Studies to enhance the anti-cancer potential of CPT through the formation of water-soluble analogs are continuously conducted. The main purpose of this research was to achieve a soluble form of CPT analogs by conjugation with β-CD and iron nanoparticles (Fe3O4) cross-linked using EDTA, to significantly increase the efficiency of CPT as an anti-cancer drug.
2. Materials and methods
2.1. Materials
Ferrous sulfate hepta-hydrate (FeSO4·7H2O), ferric chloride hexa-hydrate (FeCl3·6H2O), liq. ammonia, β-cyclodextrin, ethylenediaminetetraaceticacid (EDTA), disodium hydrogen phosphate (Na2HPO4·H2O), polyethylene glycol (PEG), camptothecin (CPT) were purchased from Sigma Aldrich (Mumbai, India). Solvents such as ethanol (C2H5OH) were purchased from Himedia Laboratories Pvt. Ltd, Chennai, India. Annexin V-FITC apoptosis detection kit and mitochondrial membrane potential detection kit were purchased from BD Bioscience (BD Pharmingen™, USA). 3-(4,5-Dimethyl-thiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), Dulbecco's Modified Eagle Medium (DMEM) 1640 medium, fetal bovine serum (FBS), penicillin (100 U mL−1), streptomycin (100 μg mL−1), 0.25% trypsin–EDTA were acquired from Naccalai, (Kyoto, Japan). Caspase-3 colorimetric assay kit was obtained from R&D Systems Co. (Minneapolis, USA), and propidium iodide flow cytometry kit for cell cycle analysis was obtained from Abcam (ab139418).
2.2. Synthesis of magnetite nanoparticles
Magnetite nanoparticles were prepared as previously reported based on the controlled chemical co-precipitation35 of Fe2+ and Fe3+ (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 ratio) in an ammoniacal medium at 80 °C under a nitrogen atmosphere. In a representative preparation, 0.02 M of ferrous sulfate (FeSO4·7H2O) and 0.04 M of FeCl3·6H2O were liquefied in 200 mL of DD water. The components were stirred and heated to 80 °C under a nitrogen atmosphere. A volume of 12 mL of a 25% ammonia solution was added into the reaction round bottom flask and stirred for 20 min to permit settle down the formed of the iron oxide magnetic nanoparticles (FMNPs). After 20 min, the temperature of the product was reduced to 27 °C (room temperature) and the resulting magnetite nanoparticles were centrifuged. The nanoparticles were cleaned with DD water for five times. The solution pH was brought to neutral by adding dilute HCl, and the FMNPs were rewashed with DD water for additional analysis.
:2 ratio) in an ammoniacal medium at 80 °C under a nitrogen atmosphere. In a representative preparation, 0.02 M of ferrous sulfate (FeSO4·7H2O) and 0.04 M of FeCl3·6H2O were liquefied in 200 mL of DD water. The components were stirred and heated to 80 °C under a nitrogen atmosphere. A volume of 12 mL of a 25% ammonia solution was added into the reaction round bottom flask and stirred for 20 min to permit settle down the formed of the iron oxide magnetic nanoparticles (FMNPs). After 20 min, the temperature of the product was reduced to 27 °C (room temperature) and the resulting magnetite nanoparticles were centrifuged. The nanoparticles were cleaned with DD water for five times. The solution pH was brought to neutral by adding dilute HCl, and the FMNPs were rewashed with DD water for additional analysis.
2.2.1. Synthesis of EDTA–β-CD. EDTA–β-CD polymers were synthesized by reacting β-CD with EDTA as a cross-linker and disodium hydrogen phosphate (MSP) as a catalyst. β-CD (4 g, 3.5 mmol), EDTA (6 g, 20.4 mmol), MSP (Na2HPO4·7H2O, 2.68 g, 10 mmol), and 20 mL of deionized water were mixed in a round-bottomed flask and stirred in an oil bath for 1 h at a 100 °C. Polyethylene glycol 200 (PEG-200, 0.5 g, 2.5 mmol) as a dispersant was added drop-wise to increase the solubilizing of β-CD in water. The mixture was transferred into a petri dish (diameter 160 mm) and heated in an oven at 155 °C for 10 h. After cooling at RT, the resulting condensation polymer product was ground and soaked with 500 mL of deionized water, and then suction filtered and rinsed with a large amount of 0.1 M HCl, deionized water, 0.1 M NaOH, again deionized water, and methanol, to remove the unreacted materials and catalyst. The final product was dried in a vacuum at 60 °C overnight.36
2.2.2. Synthesis of β-CD–EDTA with Fe3O4. Initially, 0.03 g of β-CD–EDTA was liquefied with 10 mL of double distilled water. A 0.03 g sample of magnetic nanoparticles was dissolved in 2.0 mL of Phosphate Buffer Solution (PBS) pH 6.8 solution. Then the magnetic nanoparticle-containing solution was mixed with the β-CD–EDTA solution with incessant stirring at 1000 rpm for 3 h, followed by centrifuging at 4000 rpm for 15 min. Finally, the iron functionalized carrier was dried at 38 °C for 24 h.
2.2.3. Preparation of drug loaded magnetic nanoparticles. The β-CD–EDTA modified with Fe3O4 nanoparticles encapsulated with camptothecin was prepared in a PBS/ethanol system by co-lyophilization technique. A 20 mg aliquot of β-CD–EDTA modified with Fe3O4 and 1.0 mg of CPT was dissolved in the mixed solvent system, i.e., 2 mL of PBS and 2 mL of ethanol. The system was left to equilibrate under constant stirring for 18 h at 50 °C in the dark condition. After the organic solvent was completely removed under vacuum condition, the postponement was filtered. Finally, the filtrate containing β-CD–EDTA modified with Fe3O4 (CPT@CD) was lyophilized37 and this work was shortly presented in Poorani et al. 2017.38
2.3. Characterization studies
2.3.1. FT-IR analysis. Tablets were formed from the compression of 200 mg KBR with a minimum amount of magnetic nanoparticles, one tablet without drugs β-CD–EDTA and one tablet with CPT@β-CD–EDTA. These tablets were scanned on a Fourier Transform Infrared Spectrometer (Spectrum GX-1, Perkin Elmer, USA) in the spectral region of 4000–400 cm−1.
2.3.2. Scanning electron microscope. The morphological and surface appearance of the synthesized nanocarrier (i.e., before and after the drug loading) were examined by scanning electron microscopy (VEGA3SB, TESCAN, Czech Republic). One drop of the nanoparticle suspension was coated on a glass surface and dried at 37 °C. The glass plate was mounted onto an aluminum stub using double-sided carbon adhesive tape and covered with gold by an ion sputterer unit. The coating was attained at 25 mA for minimum 60 s. Scanning was performed under high vacuum and ambient temperature with a beam voltage of 5–30 kV.
2.3.3. Thermo gravimetric analysis. The thermal permanence of the materials was characterized by TGA (PerkinElmer) under nitrogen with a flow rate of 20 mL min−1. The materials were heated in a platinum pan from 30–800 °C.
2.3.4. Particle size analysis. Mean particle size (diameter, nm ± S.D.) and PDI (polydispersity index) of the samples were investigated by Beckman Coulter, Delsa™ Nano C, USA. Measurements were taken in triplicate at a 90° angle at 25 °C under suitable dilution conditions.
2.3.5. Zeta potential measurement. Zeta potential of nanoparticle dispersions was measured in mV by Beckman Coulter, Delsa™ Nano C in triplicate to determine the surface charge and the potential physical stability of the Nano system. Zeta potential of nanoparticles was measured in aqueous dispersion. Measurements were analyzed in triplicates at a 120° angle at 25 °C.
2.3.6. Drug loading efficiency. The drug concentration of the prepared drug loaded supernatant was determined by UV spectrophotometer (Shimadzu-1600, Japan). The CPT loading content was calculated by the following equation.
2.3.7. In vitro drug release studies. In vitro release profiles of CPT from drug-loaded nanoparticles were investigated for 100 min in the PBS solution (pH 7.0) and acidic medium (pH 2.4). The nanoparticles (10 mg) and 5 mL of release medium were put into a dialysis tube (MWCO: 12000 Da, Sigma Aldrich, Mumbai, India). The dialysis tube was placed in 50 mL of double distilled water at 37 °C and stirred continuously at 500 rpm. At specific time intervals, 2 mL of solution was withdrawn from the outer compartment and replaced with fresh double distilled water (2 mL). The concentration of the released CPT was analyzed by UV spectrophotometer at λmax 260 nm. The analysis was accomplished in three times of each sample, and the work has been previously reported in one of our earlier publications.38
2.4. Biological characterization
2.4.1. Cell culture.
HeLa. Human cervix adenocarcinoma cells were procured from the Laboratory of Vaccine and Immunotherapy (LIVES) Institute of Biosciences (IBS), Universiti Putra Malaysia (UPM). HeLa cell lines were grown adherently using Dulbecco's Modified Eagle Medium (DMEM) media supplemented with 10% fetal calf serum, 100 U mL−1 penicillin, and 100 μg mL−1 streptomycin at 37 °C and 5% CO2. The cells were seeded in 96-well plates at a concentration of 1 × 104 in 100 μL of cell culture medium. Cells were incubated for 24 h to reach about 90% confluence.
2.4.2. Treatment with CPT/β-CD–EDTA–Fe3O4 (CPT–CEF). The final stock solution of each compound was made by dissolving the compounds in 10% DMSO and cell culture media. Multiple concentrations were made through serial dilutions utilizing the cell culture medium while maintaining the DMSO concentration below 1% (v/v) in all the analysis.
2.4.3. MTT viability assay. The tetrazolium salt 3-[4,5-dimethylthiazol-2-yl]-2-5-diphenyltetrazolium bromide (MTT) assay was performed to determine cell viability and overall cytotoxicity with different concentration of CPT–CEF. Cell viability of HeLa cancer cell lines in response to treatment with various concentrations of CEF–CET, CPT, CEF, and Fe3O4 were determined using MTT as described by Mosmann (1983).39,40 Cells were plated at a density of 1 × 104 cells per well in 96-well plate and incubated at 37 °C for 24 h under 5% CO2 for cell attachment. Treatments were then made with the compound(s) mentioned above. The concentrations used were 250, 125, 62.5, 31.25, 15.62, 7.81, 3.91, 1.95 μg mL−1. The final concentration of DMSO was maintained below 1% v/v. Treated cells were then tested after 24, 48, and 72 h of incubation. For each dosage, three replicates were performed. Negative controls were added to cell culture media only. The MTT assay was performed post-incubation. To initiate the assay, 20 μL of MTT (5 mg mL−1) was added into each well following with incubation for 3 h at 37 °C. Post incubation, the supernatant was carefully discarded and 100 μL of DMSO was added to each well to solubilize the formazan product. The absorbance was measured using a microplate absorbance plate reader (Sunrise™-Tecan Trading AG, Switzerland) at 570 nm with a reference wavelength of 630 nm. Cell viability is presented as the ratio of the absorbance of treated cells to that of blank controls. The IC50 value of CPT–CEF was determined and this concentration was utilized for the subsequent assays.
2.4.4. Annexin V/PI assay. Detection of apoptosis was conducted using the Annexin V-FITC apoptosis detection kit according to manufacturer's protocol. Briefly, cells were plated at a density of 3 × 105 per well in a 6-well plate and treated with different concentrations of CPT–CEF based on the IC50 concentration derived from MTT assay. After 48 h of incubation, all cells were collected, pooled, and washed with PBS. Cells were then resuspended in 1× binding buffer at a concentration of 1 × 106 cells per mL and 100 μL of the solution (1 × 105 cells) was transferred to a 5 mL culture tube. A volume of 5 μL of FITC Annexin V and 5 μL PI were added to the culture tube and incubated for 15 min at RT in the dark. A volume of 400 μL of 1× binding buffer was again added to each tube and then examined using BD FACSARIA flow cytometer (BD Biosciences, San Jose, CA, USA).
2.4.5. Mitochondrial depolarization assay (JC-1). Mitochondrial depolarization was determined using mitochondrial membrane potential detection kit. The cell treatment procedure is maintained as Annexin V/PI assay treatment. Following treatment, 1 mL of each cell suspension was transferred into a sterile 15 mL polystyrene centrifuge tube. Cells were then centrifuged at 400 × g for 5 min at RT and the supernatant was then discarded. A volume of 0.5 mL of JC-1 working solution was added to the tubes and incubated for 10–15 min at 37 °C in a CO2 incubator. Cells were then washed twice with 1× assay buffer and centrifuged 400 × g for 5 min. Cells were finally resuspended in 0.5 mL 1× assay buffer and analyzed using flow cytometry.
2.4.6. Cell cycle analysis. HeLa cells were treated and incubated for 48 h as explained previously. Cell cycle analysis was carried out using propidium iodide flow cytometry kit. Cells (3 × 105 per well) were grown in 6-well plates and then treated with multiple concentrations of CPT–CEF for 48 h. Post treatment, the culture media was removed and cells were rinsed with PBS. Trypsin was used to dissociate the cells. All culture media and PBS rinse were collected and pooled. Cells were then centrifuged at 500 × g for 5 min and pelleted. The supernatant was then removed followed by washing with 1× PBS and centrifugation again at 500 × g for 5 min. Fixation was then performed using 66% ethanol on the ice and stored at 4 °C for at least 2 h. The cells were centrifuged at 500 × g for 5 minutes and then washed with 1 mL 1× PBS and centrifuged again. The cells were gently resuspended in 200 μL 1× propidium iodide + RNAse staining solution. After incubation for 30 min in the dark at 37 °C, cells were analyzed for DNA content using a BD FACSARIA flow cytometer (BD Biosciences, San Jose, CA, USA). The percentage of cells distribution at each cell cycle phases were evaluated as described previously.41 The cell cycle distribution was shown as the percentage of cells containing 2n (G1 phase), 4n (G2 and M phases), and 4n > 3 > 2n DNA amount (S phase) judged by PI staining.
2.4.7. Caspase-3 activity. About 1 × 105 cells per well were cultured overnight in 6-well plates, then treated with various concentrations of CPT–CEF. Cells without any treatment were used as the control. Caspase-3 activity was analyzed according to the manufacturer's protocol (R&D Systems Co. Minneapolis, USA). Briefly, post treatment, cells were harvested and lysed in 50 μL lysis buffer on ice for 10 min and then centrifuged at 10000 × g for 1 min. Following this, the caspase-3 substrate was added into 50 μL of the supernatant in reaction buffer. Samples were then subsequently incubated in 96-well flat bottom microplate at 37 °C for 2 h. The amount of released p-nitroaniline (pNA) was measured with a microplate reader (Sunrise™-Tecan Trading AG, Switzerland) at 405 nm wavelength. Background readings were determined from control wells with culture medium in absence of cells and. Protein concentration was determined using Pierce 660 nm Protein Assay Reagent.
2.4.8. AO/PI staining. The morphological changes in HeLa cells, CPT–CEF-treated HeLa cells were characterized using acridine orange (AO) and propidium iodide (PI) double staining according to the method described by Hajiaghaalipour et al.42 with minor modification. Briefly, 1 × 105 HeLa cells per mL were plated in a 6-well plate and treated with the IC50 concentration of CPT–CEF. The plate was incubated in an atmosphere of 5% CO2 at 37 °C for 48 h. The cells were then trypsinized with trypsin–EDTA, washed twice with PBS and centrifuged at 1800 rpm for 5 min to remove the remaining media. To begin the assay, a fluorescent dye (AO/PI) containing AO (50 μg mL−1) and PI (50 μg mL−1) was freshly prepared and added to the cellular pellet. The freshly stained cells were immediately observed under UV-fluorescence microscopy.
3. Results
3.1. Structure analysis
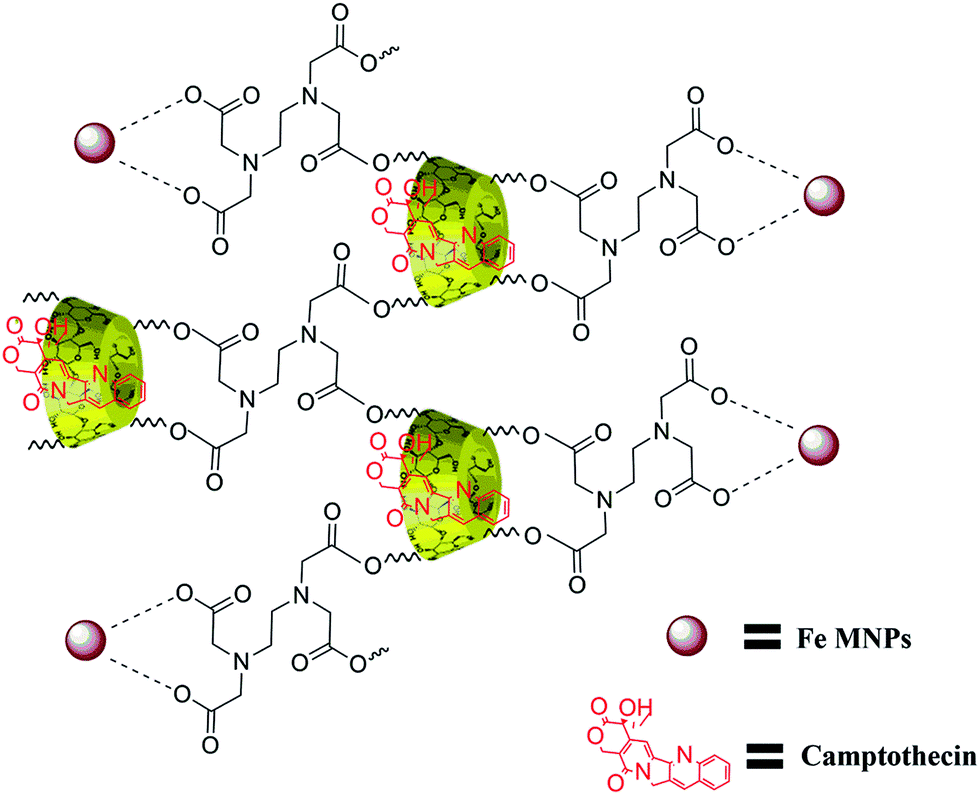
The structure of β-CD cross-linked EDTA was verified with 1H and 13C NMR spectroscopic analysis. The formation of CD–EDTA was under poly condensation process of EDTA with β-CD. Through condensation, the carboxylic acid groups of EDTA were esterified with –CH2OH group of β-CD, because the reaction was conducted on aqueous medium.43 The β-CD cross-linked EDTA image of 1H and 13C are shown in Fig. S1 (ESI) and Fig. S2,† respectively, and the data of 1H were 3.21 (CH2–CH2, EDTA), 4.76 (N–CH2–CO, EDTA), 4.38 (CH–CH–O, CD), 4.09 (CH–CH–O, CD), 4.51 (CH–CH2–O, CD), 3.03 (CH–CH–O, CD), 3.58 (CH–CH–OH, CD), 3.75 (CH–CH–OH, CD), 4.77 (O–CH–O, CD), 3.6 (CH–CH–OH, CD) and 13C were 54.66 (CH2–CH2, EDTA), 60.52 (NCH2CO, EDTA), 70.37 (CH2OH, CD), 72.63 (CH2OCO, CD), 72.99 (CHCHOH, CD), 73.64 (CHCHOH, CD), 82.11 (OCHCH2, CD), 102.55 (OCHO, CD), 171.24 (OCOCH2, CD). The graphical picture of CPT drug loaded β-CD–EDTA–Fe3O4 nanocarrier is given in Fig. 1.
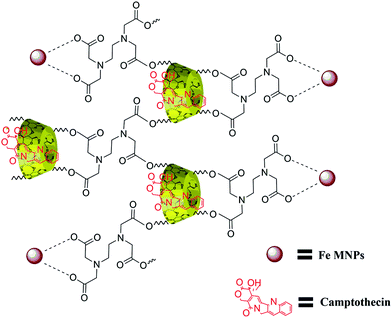 |
| | Fig. 1 Schematic illustration of CPT drug loaded β-CD–EDTA–Fe3O4 nanocarrier. | |
3.2. XRD analysis
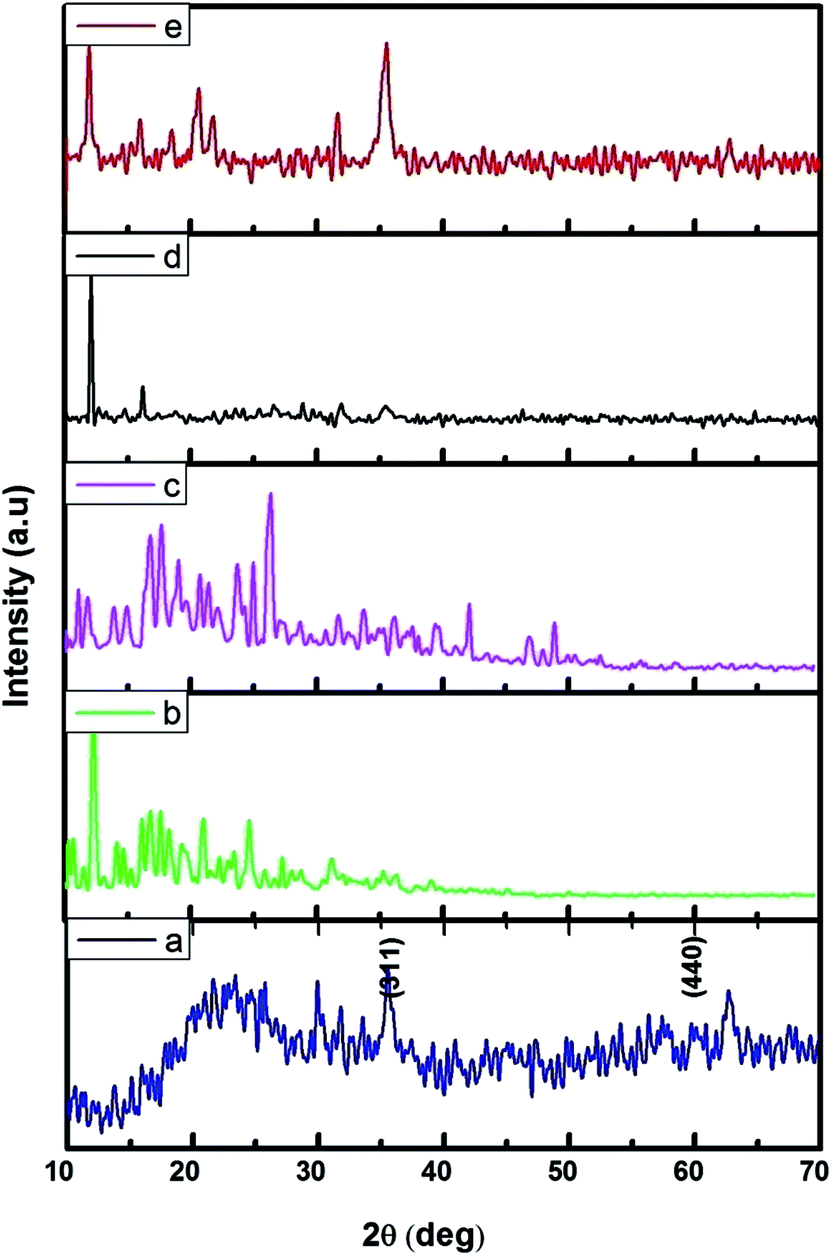
The FMNPs functionalized β-CD nanocarriers were prepared and examined using XRD and the results were presented in Fig. 2. Fig. 2a confirms the formation of FMNPs, and the peaks observed at 2θ = 35.57° and 62.76° are ascribed for (311) and (440) diffraction pattern of cubic Fe3O4 with a space group of Fd![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m. These diffraction lines are in agreement with JCPDS card file no. 65-3107. The estimated d spacing value was 2.5241 Å. Here the broad hump in the lower 2θ region that highlighted the presence of shapeless construction in the samples or from the background of the glass substrate. The XRD pattern of β-CD showed its characteristic peaks with crystalline nature and it is cross-linked with EDTA enhancing their crystalline structure. The functionalized Fe MNs sample on β-CD cross-linked EDTA indicated an observable reduction of crystalline nature of β-CD–EDTA–Fe3O4 composites. The decreased crystalline nature can be understood in the light of decorated FMN particles on the external of the carrier.44 Once the encapsulation of CPT drug on the carrier was achieved, the crystalline nature was improved to a commendable range and this is theoretically due to the regular arrangement of carrier and drug.
m. These diffraction lines are in agreement with JCPDS card file no. 65-3107. The estimated d spacing value was 2.5241 Å. Here the broad hump in the lower 2θ region that highlighted the presence of shapeless construction in the samples or from the background of the glass substrate. The XRD pattern of β-CD showed its characteristic peaks with crystalline nature and it is cross-linked with EDTA enhancing their crystalline structure. The functionalized Fe MNs sample on β-CD cross-linked EDTA indicated an observable reduction of crystalline nature of β-CD–EDTA–Fe3O4 composites. The decreased crystalline nature can be understood in the light of decorated FMN particles on the external of the carrier.44 Once the encapsulation of CPT drug on the carrier was achieved, the crystalline nature was improved to a commendable range and this is theoretically due to the regular arrangement of carrier and drug.
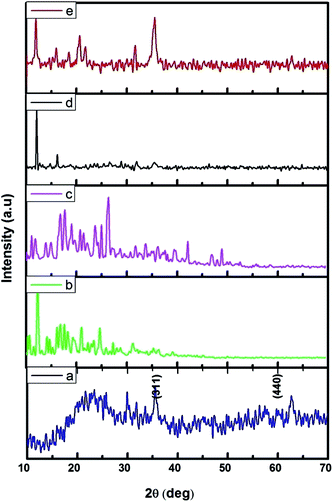 |
| | Fig. 2 XRD pattern of (a) Fe3O4; (b) β-CD; (c) β-CD–EDTA; (d) β-CD–EDTA–Fe3O4; (e) β-CD–EDTA–Fe3O4/CPT nano carrier. | |
3.3. Raman analysis
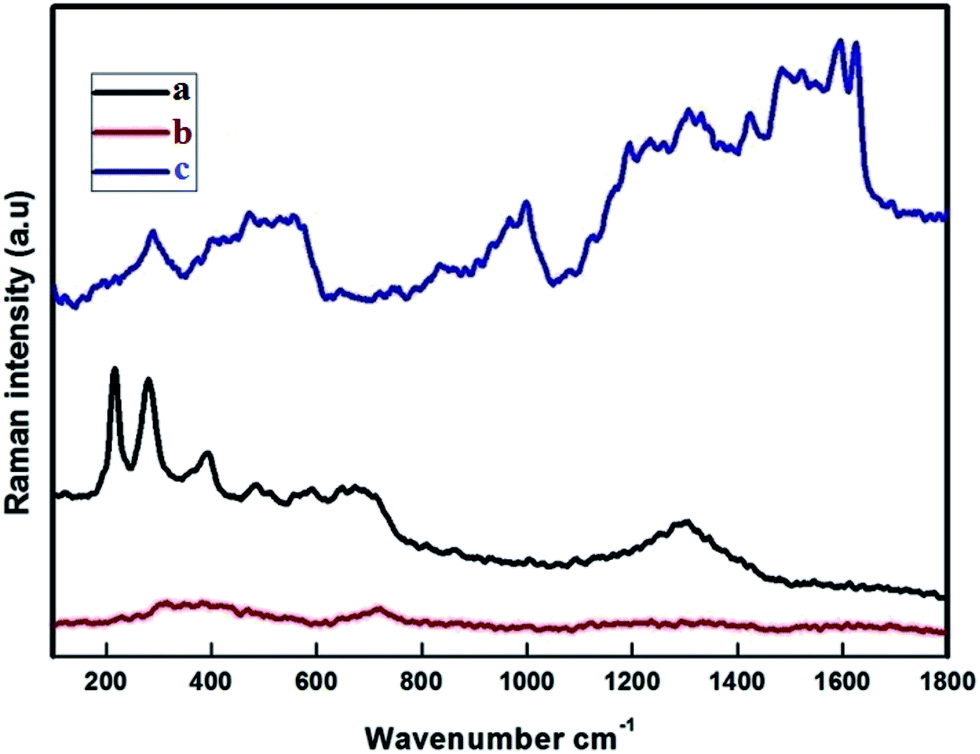
The Raman spectra were recorded at RT with a linear laser excitation of 514 nm (Fig. 3). To understand the characteristic properties, the materials were investigated in the series of 100 to 1800 cm−1. The dominant peaks for FMNPs are at 215, 276, 398, 487, 654 and 1300 cm−1 and agree with previous literature.45 No significant peak was noticed for the CEF sample. The peaks around 1612, 1582 cm−1 correspond to CPT when if forms composites with CEF, as is the circumstance for the peaks at 1300, 398 and 276 cm−1 which agree with FMNPs. These results confirmed the functionalization of Fe on the carriers and encapsulation of CPT drug on the carriers. The data were well correlated with the XRD pattern used to classify the occurrence of FMNPs. For the bare CD and CD with EDTA were not measurable with the high power laser but when bound with FMNPs it has strong binding, hence it is reproduced in the measurements.
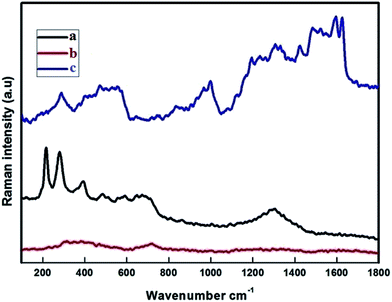 |
| | Fig. 3 Raman band of (a) Fe3O4 (FMN); (b) β-CD–EDTA–Fe3O4 (CEF); (c) β-CD–EDTA–Fe3O4/CPT (CEF–CPT). | |
3.4. Scanning electron microscope
SEM analysis (Fig. 4a–d) provides a surface morphology of the as-synthesized Fe3O4 nanoparticles, β-CD–EDTA, β-CD–EDTA–Fe3O4 nano carriers and β-CD–EDTA–Fe3O4/CPT drug loaded nano carrier. Fig. 4a showed that, the spherical shaped Fe3O4 nanoparticles and sparse distribution with agglomerated clusters. β-CD–EDTA composites appeared as arrangements of network structure with holes (Fig. 4b). When FMN was incorporated into β-CD–EDTA the resulting β-CD–EDTA–Fe3O4 tagged complex showed a drastic change in the shape and morphological features of the original FMNPs and β-CD–EDTA–Fe3O4, which tended to be amorphous as reflected in XRD (Fig. 4c). The variation in the surface properties of the inclusion complex was indicative of the occurrence of a fresh solid phase, which might be due to the molecular encapsulation of CPT in the β-CD–EDTA–Fe3O4. The images confirmed that both individual components, β-CD–EDTA–Fe3O4 and β-CD–EDTA–Fe3O4/CPT, had nanometer range size with a disjointed composition. After the incorporation was performed, the distinct drug particles with solid dense structure were observed.
 |
| | Fig. 4 SEM images of (a) Fe3O4; (b) β-CD–EDTA; (c) β-CD–EDTA–Fe3O4; (d) β-CD–EDTA–Fe3O4/CPT nano carrier. | |
3.5. Thermal gravimetric analysis
TGA curves of β-CD–EDTA, β-CD–EDTA–Fe3O4, and β-CD–EDTA–Fe3O4/CPT nanocarrier are shown in (Fig. S3–S5†). In Fig. S3,† the first stage 32–246 °C, this step includes an initial mass loss around 100 °C, which represents the volatilization of outstanding water molecules, followed by β-CD degradation. Second weight loss stage at temperature range of 245–384 °C can be arrange to the thermal degradation of EDTA and associated to the degradation of free β-CD molecules, while the third weight loss stage at temperature range of 383–794 °C can be accredited to the degradation of carbonaceous residues which in turn resulted from β-CD molecule degradation interacting with the iron oxide nanoparticles surfaces via carbonyl group at the rim. Second weight loss stage of β-CD–EDTA at temperature period of 230–384 °C was 41%, but the second stage of β-CD–EDTA–Fe3O4 (Fig. S4†) sample shows only 23%. It was due to the accumulation of magnetic iron oxide nanoparticles strongly binding with β-CD–EDTA composites and the development of carbonyl groups of the EDTA and Fe complex. The drug loaded β-CD–EDTA–Fe3O4/CPT nano carrier (Fig. S5†) weight loss ranges over only two stages associated with other two samples. The second weight loss stage was extended 256 °C to 796 °C and the weight loss was 52% compare with β-CD–EDTA, β-CD–EDTA–Fe3O4. It may be that the available functional assembly of the β-CD–EDTA–Fe3O4 composites was occupied and bound with the CPT drug.
3.6. Drug loading efficiency
The impact of cyclodextrin-based nano carriers and their therapeutics will likely accelerate in coming years. However, as these products move out of the laboratory and into the clinics, various federal agencies like FDA and US patents have to struggle in order to encourage the development of these products. Since Fe3O4 NPs are capable of preferentially target and invade cancerous cells, use of these particles as an anticancer drug carrier would be ideal for tumor-specific drug delivery. Toward this end, we investigated the drug-loading capacity of these multifunctional NPs, which are already decorated with well-known molecular drug carrier β-cyclodextrin. The encapsulation of CPT drug was successfully loaded by co-lyophilization technique on the cyclodextrin-based nanocarriers. The choice of drug loading method is based on the drug and carriers. The drug loading efficiency of β-CD–EDTA–Fe3O4 polymer composite was shown in (Fig. S6†). The absorption intensity decrease compared with the absorbance of free CPT indicated that the concentration of CPT decreased due to the encapsulation of CPT on β-CD–EDTA–Fe3O4 carrier. From UV-absorbance maximum, drug loading efficiency was calculated and 87% of the CPT drug was encapsulated on the carriers within 3 h. When the cavity groups in the carrier increased, the encapsulation also increased. The affinity between the β-CD and its guest depends on its association constant. During various guest molecules, adamantane and its derivatives have the strongest binding capacity with β-CD in aqueous solution, which could form an exact 1:1 inclusion complex with β-CD. The specific molecular recognition offers the possibility of developing a new trigger. The drug encapsulation accomplished in this study was higher than previously reported.46 This is also projected in the XRD data by means of high crystallinity nature once the drug is loaded carrier i.e., CEF–CPT (Fig. 2).
3.7. In vitro drug release studies
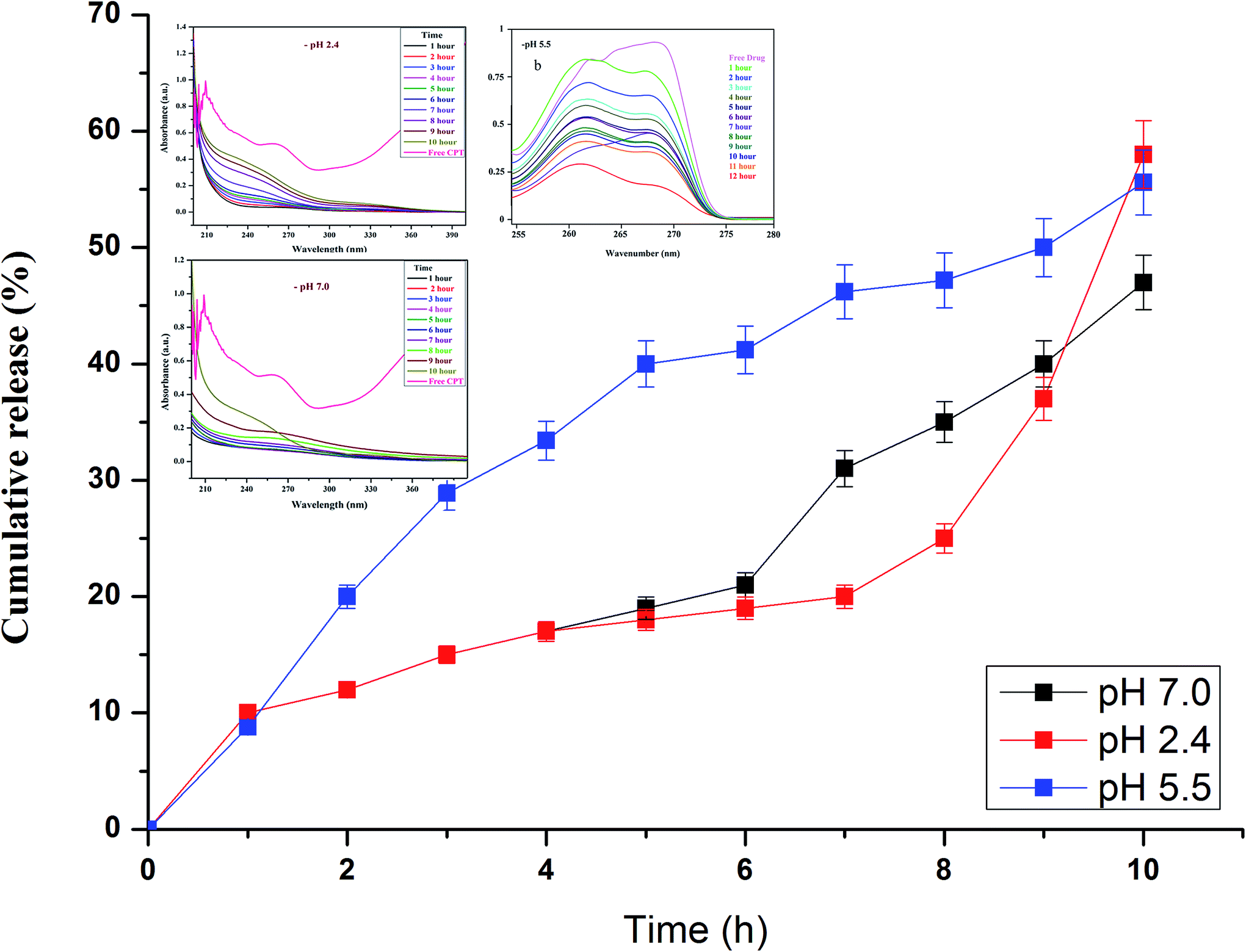
Drug release studies were performed at biologically relevant pH as previously detailed elsewhere.38 In vitro CPT drug discharge summary from the β-CD–EDTA–Fe3O4 carriers was assessed using the dialysis technique at pH 2.4, pH 5.5 and pH 7.0 at 37 °C. From the Fig. 5a and b, nearly 65%, 61% and 58% of the CPT was released during 10 h at pH 2.4, pH 5.5 and 7.0, respectively. The cumulative release curves showed that the release of β-CD–EDTA–Fe3O4–CPT nanoparticles was acid- and hydrogen bond formation responsive. At pH 7.0, the release of CPT was about 58% over a period of 10 h, indicating that β-CD–EDTA–Fe3O4–CPT nano carriers (Fig. 5b) remained stable in the physiological condition. When pH was changed to 2.4 (Fig. 5a), CPT was released more rapidly from the β-CD–EDTA–Fe3O4/CPT nano carriers than pH 7.0. When treated with the acidic condition, the release rate was remarkably increased under pH 2.4 conditions. These results were reliable with the detail that CPT degraded much more quickly under pH 2.4. The absorbance value increased with reverence to the time as the CPT drug was delivery from the carrier. From this study, we confirmed the drug was fruitful released from the β-CD–EDTA–Fe3O4 carrier at pH 2.4 and pH 7.0. The main key advantages of using cyclodextrin functionalized iron nanoparticles for drug carriers is the permanence of cross-linked EDTA agent. This high stability results in a much slower drug release rate than the rate of vascular circulation and cell binding, permitting for the specific release of the nanoparticles and steady release of the loaded drug at the tumor site. The UV absorption peak is shifted to shorter wavelengths with a rise in the presence of drug and dilution of the carriers, accompanied by the increase in absorbance. Similar behaviors of CD with various drugs by UV-Vis spectroscopy have been reported in the research works.47,48
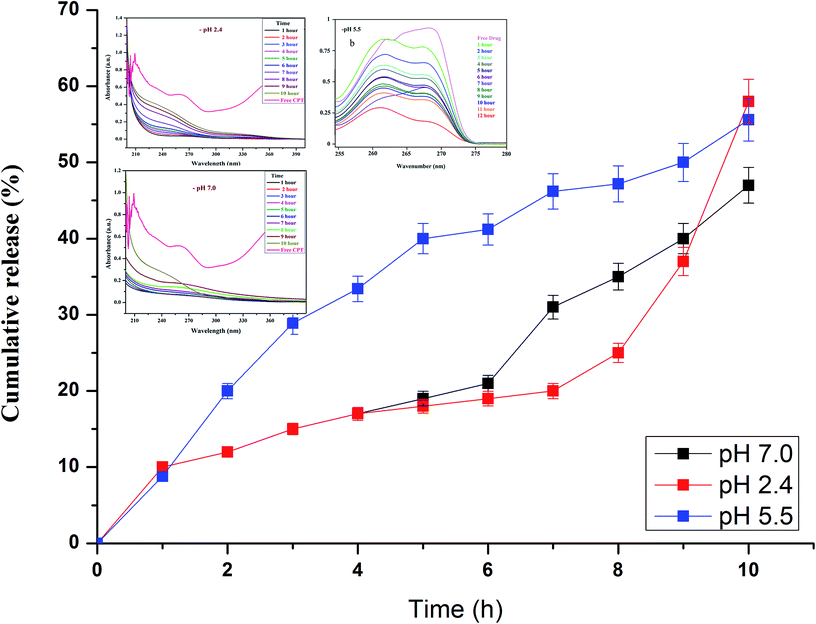 |
| | Fig. 5 In vitro drug release analysis of β-CD–EDTA–Fe3O4/CPT carrier at pH 2.4 (a), at pH 5.5 (b), and at pH 7.0 (c). | |
3.8. The effect of β-CD–EDTA–Fe3O4/CPT on HeLa cell viability
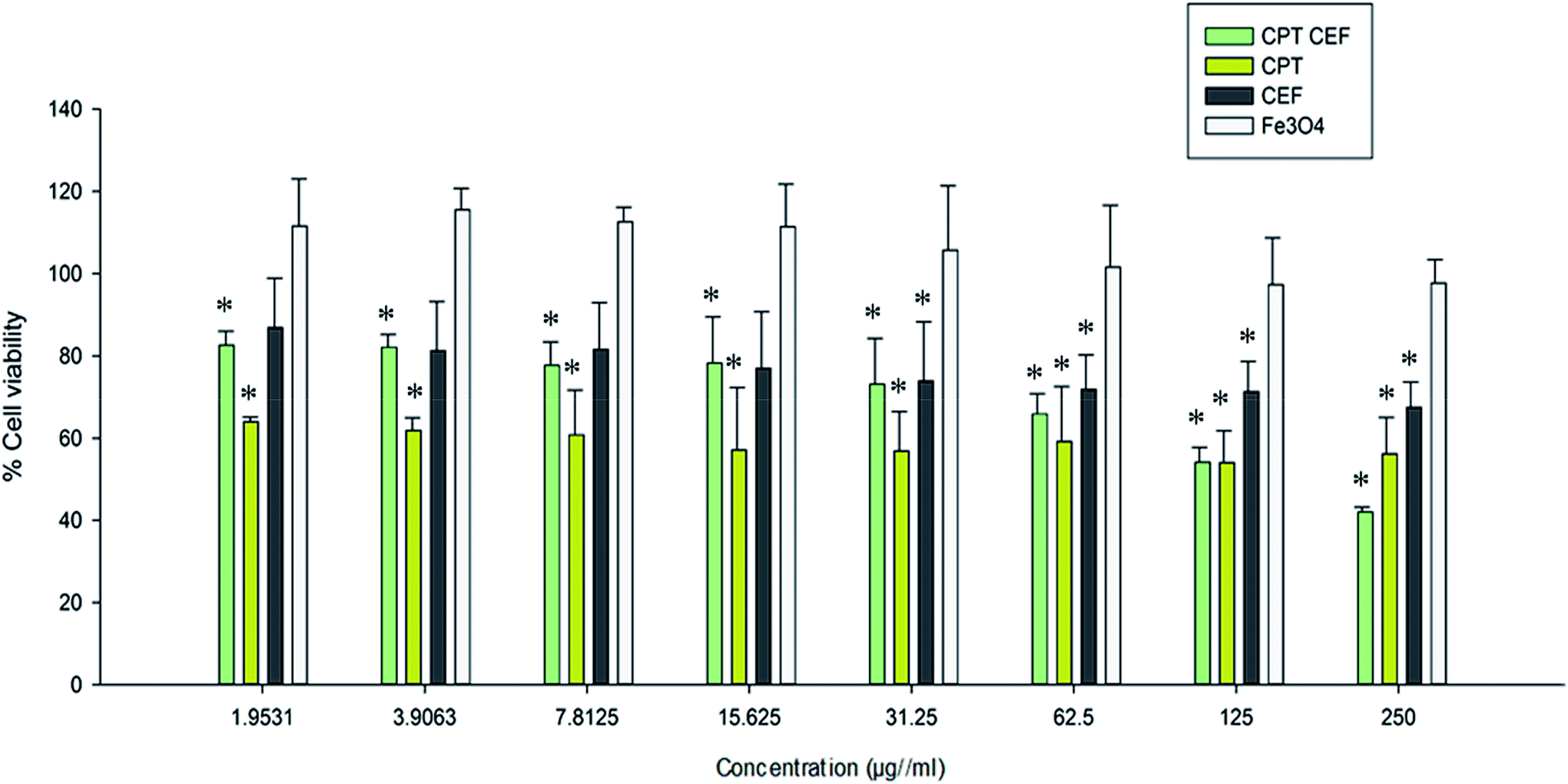
MTT assay was employed to show the effect of β-CD–EDTA–Fe3O4/CPT (CPT–CEF) on the viability of HeLa cells. MTT assays reflect the impact of β-CD–EDTA–Fe3O4/CPT on the mitochondrial activity of HeLa cells, thus reflecting cell cytotoxicity. Cells were treated with various concentrations of free CPT, Fe3O4, β-CD–EDTA–Fe3O4 (CEF) and β-CD–EDTA–Fe3O4/CPT (CPT–CEF) at three different time points of 24, 48, and 72 h (results not shown). The MTT assay result at 48 h treatment (Fig. 6) showed a concentration dependent decrease in cell viability of HeLa when compared to the untreated cells (Fig. 6) thus indicating the ability of β-CD–EDTA–Fe3O4/CPT to reduce cell viability. Significant cell viability decrease was observed at a β-CD–EDTA–Fe3O4/CPT concentration of 200 μg mL−1. The effective β-CD–EDTA–Fe3O4/CPT concentration for 50% inhibition (IC50) of HeLa cell growth after 48 h was 245 μg mL−1. Treatment with β-CD–EDTA–Fe3O4 alone showed mild cytotoxicity toward the viability of HeLa cells.
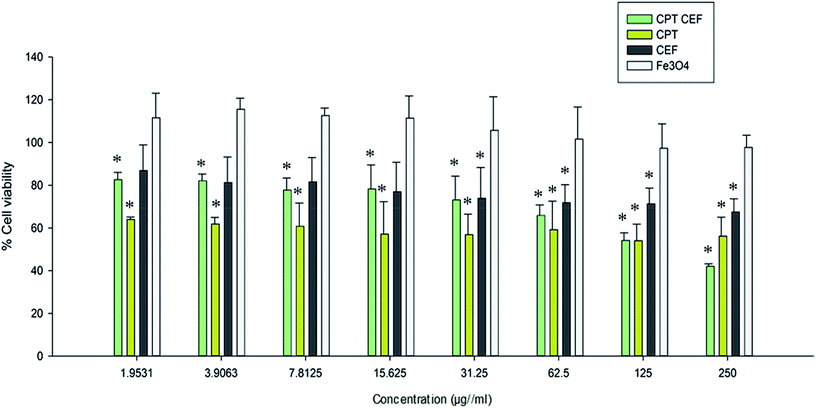 |
| | Fig. 6 The effect of CPT–CEF on cell viability in HeLa cancer cells. Cells were treated with various concentrations of CPT–CEF, CPT, CEF, and Fe3O4 for 48 h and cell viability was determined using an MTT assay. CPT–CEF reduced cell viability in HeLa cells in a dose-dependent manner. *P < 0.05 are significant. Statistical analysis was performed by ANOVA. | |
3.9. β-CD–EDTA–Fe3O4/CPT causes morphological changes in HeLa cancer cell
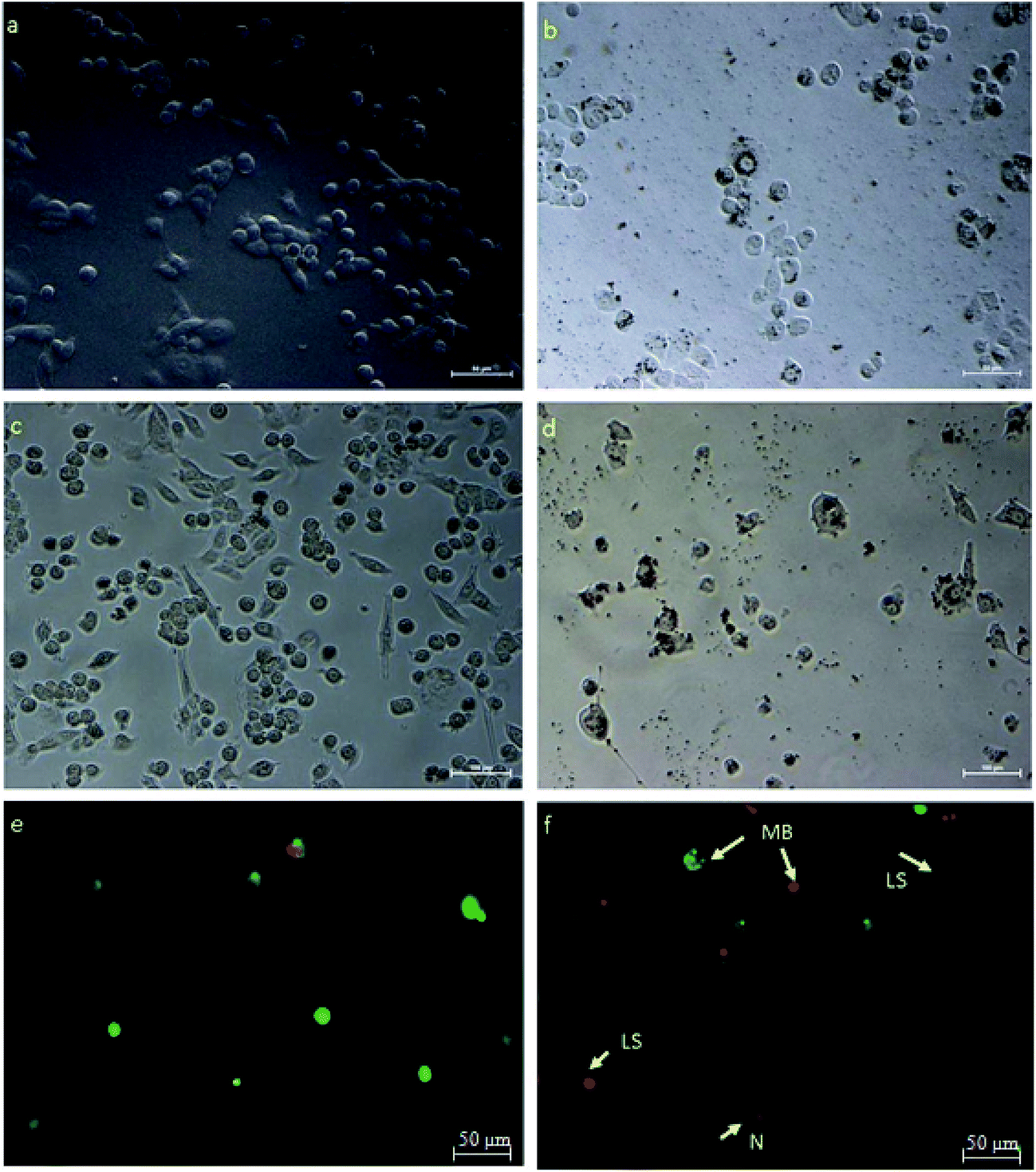
HeLa cells were treated with an IC50 concentration of β-CD–EDTA–Fe3O4/CPT derived from the MTT assay and the changes in morphology of the treated cells were observed under phase contrast microscope. The Fig. 7 showed that, the changes in IC50 concentration-treated HeLa cells (Fig. 7a) in comparison with untreated cells for 24 h (Fig. 7b). At 24 h, minimum cell membrane damage was observed in the β-CD–EDTA–Fe3O4/CPT treated cells. At 48 h incubation period, control cells (Fig. 7c) still maintained intact cell membranes and with minimum cell detachment. However, β-CD–EDTA–Fe3O4/CPT treated cells for 48 h (Fig. 7d) lost cell membrane structure completely and nuclear fragmentation was clearly evident.
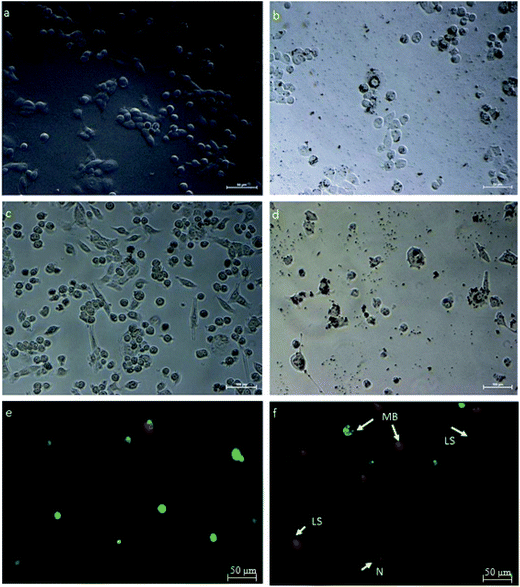 |
| | Fig. 7 HeLa cells treated with β-CD–EDTA–Fe3O4/CPT viewed under phase contrast microscope to show changes in cell morphology at 24 h, and 48 h time points. (a) (Untreated cells at 24 h), (b) (IC50 treated cells for 24 h), (c) (untreated cells at 48 h) and (d) (IC50 treated cells for 48 h). (e & f) Shows fluorescent micrographs of AO/PI double-stained human cervical cancer cells to an IC50 concentration of β-CD–EDTA–Fe3O4/CPT for 48 h. Cell apoptosis was assayed by AO/PI staining to detect chromosomal condensation (CS), Late Stage apoptosis (LS), N (Necrosis) and membrane blebbing (MB) as shown in the micrograph. | |
3.10. Membrane blebbing detected using AO/PI staining assay
Acridine orange (AO)/propidium iodide (PI) double staining were employed to indicate the mode of cell death of HeLa cells treated with an IC50 concentration of β-CD–EDTA–Fe3O4/CPT for 48 h. AO/PI double staining morphological analysis shows viable cells from apoptotic and necrotic cells. AO stains viable cells while PI penetrates cell membranes of dying or dead cells while being excluded by viable cells. Viable cells with an intact nucleus and DNA shows spherical and green nuclei. Nuclei of cells undergoing apoptosis are stained green yet fragmented. Late apoptotic and necrotic cells were stained in a mixture of orange and red.49 Based on these results (Fig. 7f), we conclude that the IC50 concentration of β-CD–EDTA–Fe3O4/CPT leads to an early apoptosis indications, such as cell shrinkage (CS), plasma membrane blebbing (MB), and chromatin condensation (CC). Late stage apoptotic (LA) features are also detected through the intercalation between AO (bright green) and PI reflecting fragmented DNA as well as nuclear fragmentation. The untreated cells are mostly stained green and remained intact (Fig. 7e). Based on the result of AO/PI staining we concluded that β-CD–EDTA–Fe3O4/CPT at a concentration of 245 μg mL−1 has a significant impact on the cell membrane and nuclear membrane of HeLa cells.
3.11. β-CD–EDTA–Fe3O4/CPT induces apoptosis in HeLa cancer cell
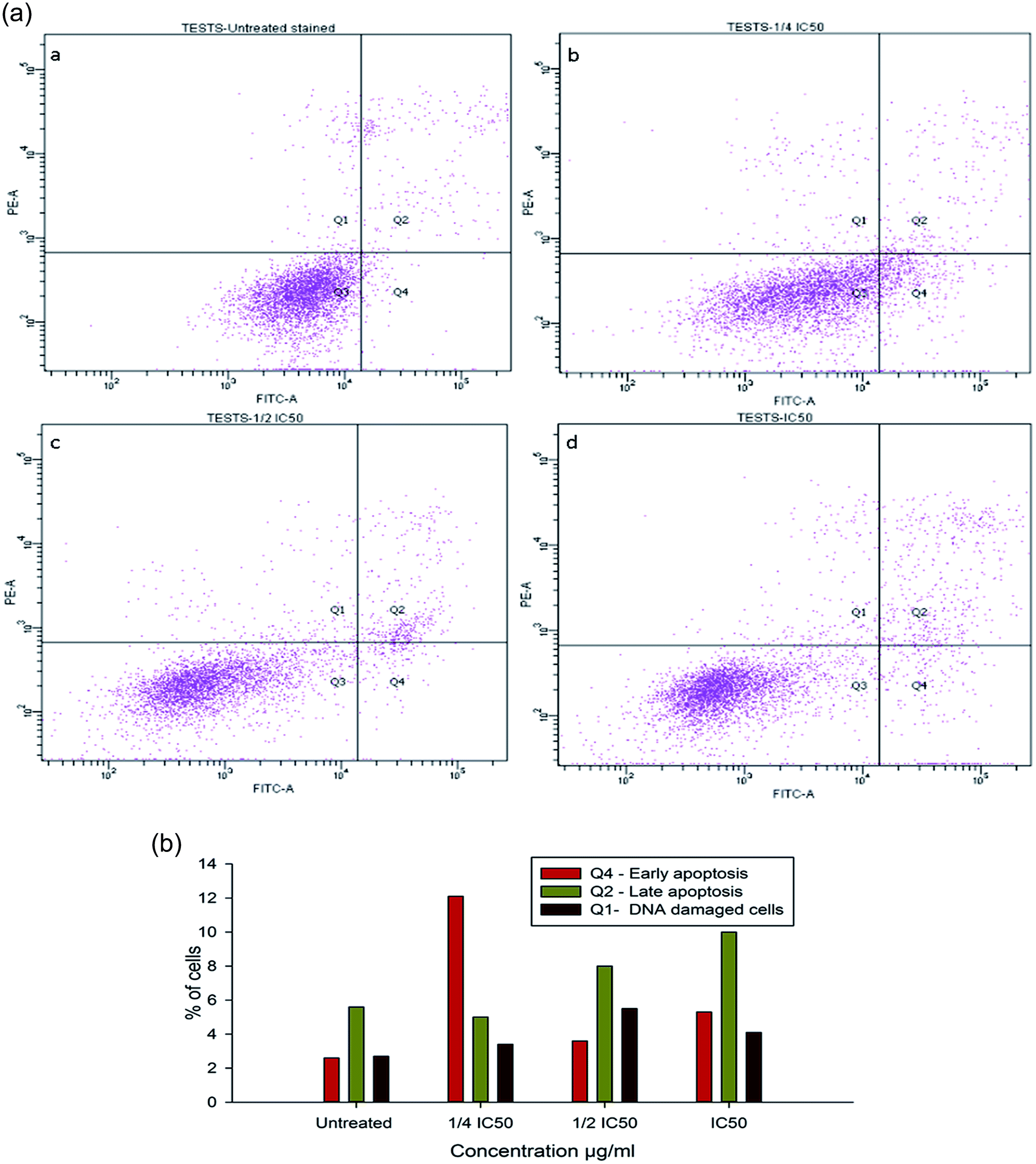
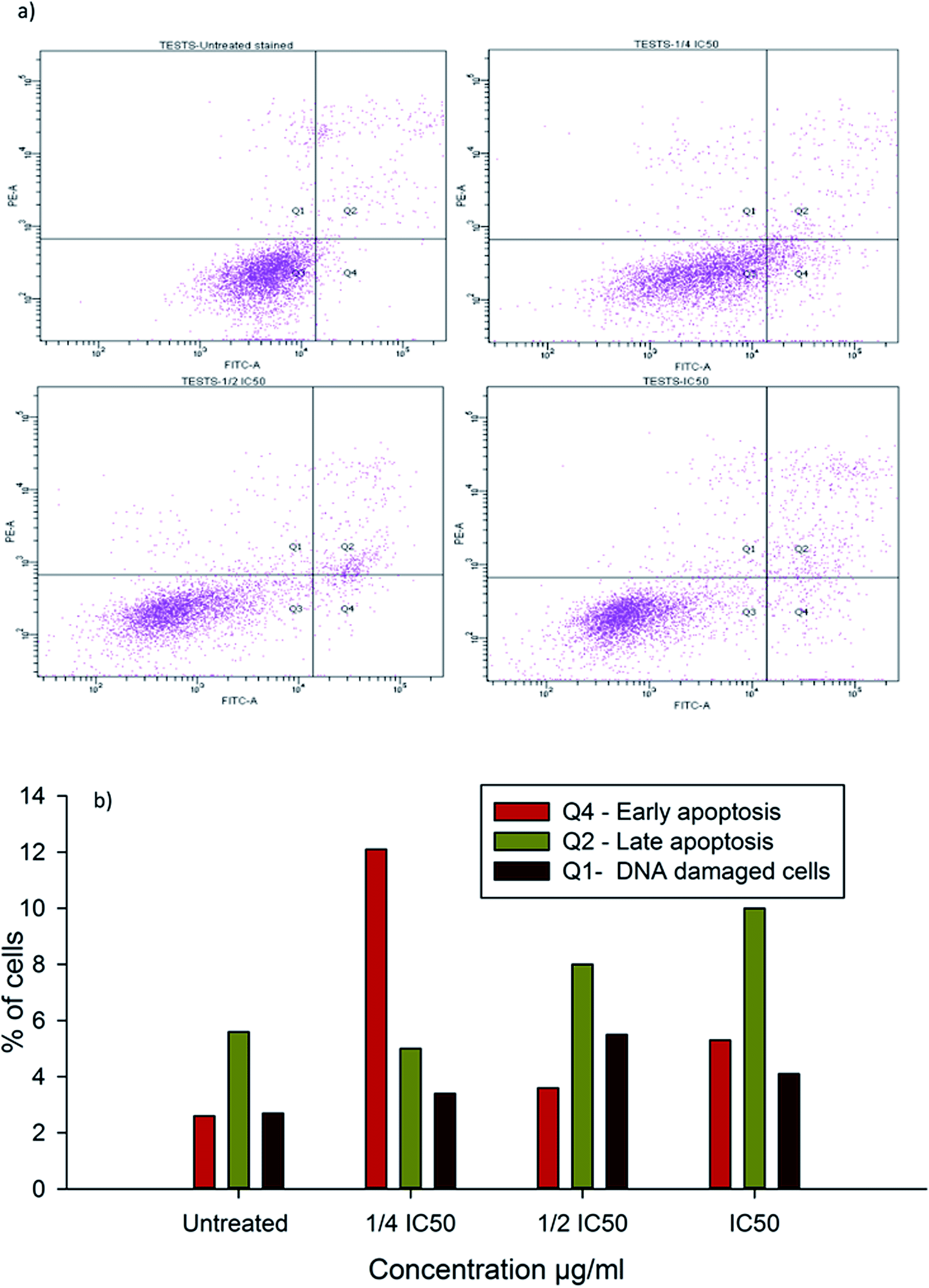
To evaluate the possibility of β-CD–EDTA–Fe3O4/CPT (CPT–CEF) induced cell proliferation inhibition leading to cell apoptosis, the effect of β-CD–EDTA–Fe3O4/CPT on cell apoptosis was studied using Annexin V/PI assay. HeLa cells were exposed to the following inhibition concentration of IC50, 1/2IC50 and 3/4IC50 concentrations of CPT–CEF for 48 h, and subsequently evaluated for apoptosis occurrence by flow cytometry using FITC-conjugated Annexin V (FL1-H) and PI (FL2-H) double staining Fig. 8. The data show an increasing trend in the percentage of both early (Annexin V positive, PI negative) and late (Annexin V positive, PI positive) apoptosis of HeLa cells in a concentration-dependent manner. These results suggest that CPT–CEF has successfully retained the apoptosis inducing potential of CPT. The data clearly shows an increasing trend in the early stage apoptosis and late stage apoptosis as the concentration of β-CD–EDTA–Fe3O4/CPT was gradually doubled. The IC50 concentration of β-CD–EDTA–Fe3O4/CPT has significantly induced early apoptosis in treated HeLa cells compared to the untreated cells.
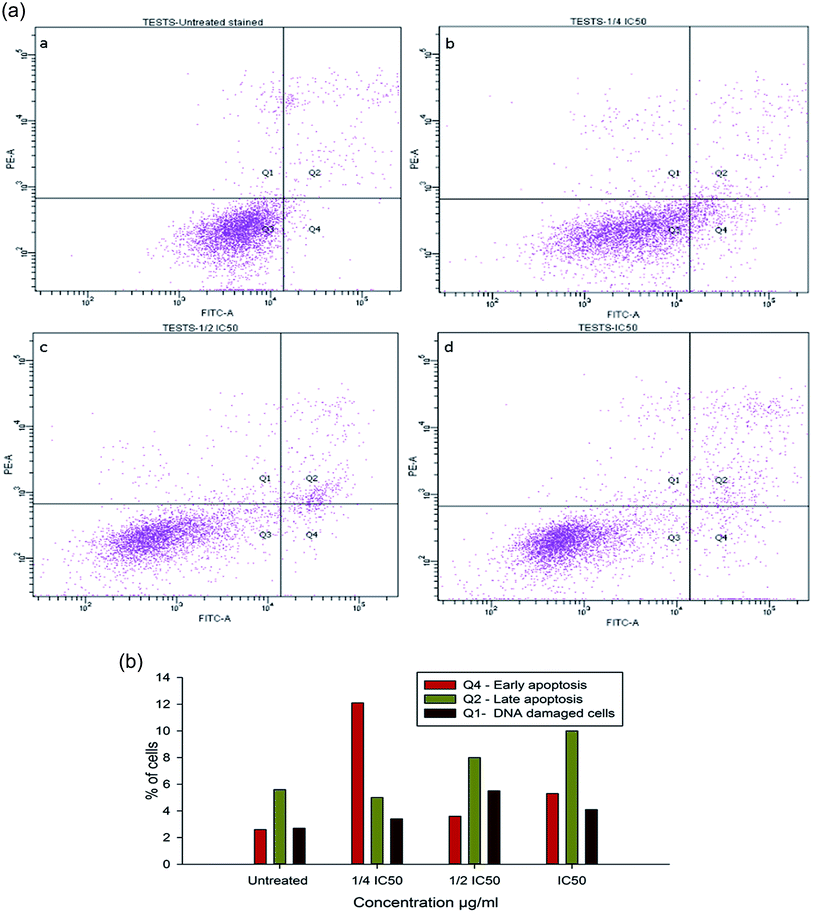 |
| | Fig. 8 (a) Annexin V/PI assay data on the apoptosis inducing effect of β-CD–EDTA–Fe3O4/CPT on HeLa cervical cancer cells with concentrations of β-CD–EDTA–Fe3O4/CPT concentration at (a) untreated, (b) 1/4IC50, (c) 1/2IC50 and (d) IC50 treated HeLa cells for 48 h, and viability was assessed by Annexin V/PI assay. (b) Shows the percentage of cells at different stages of apoptosis. | |
3.12. β-CD–EDTA–Fe3O4/CPT alters the mitochondrial membrane potential in HeLa cell
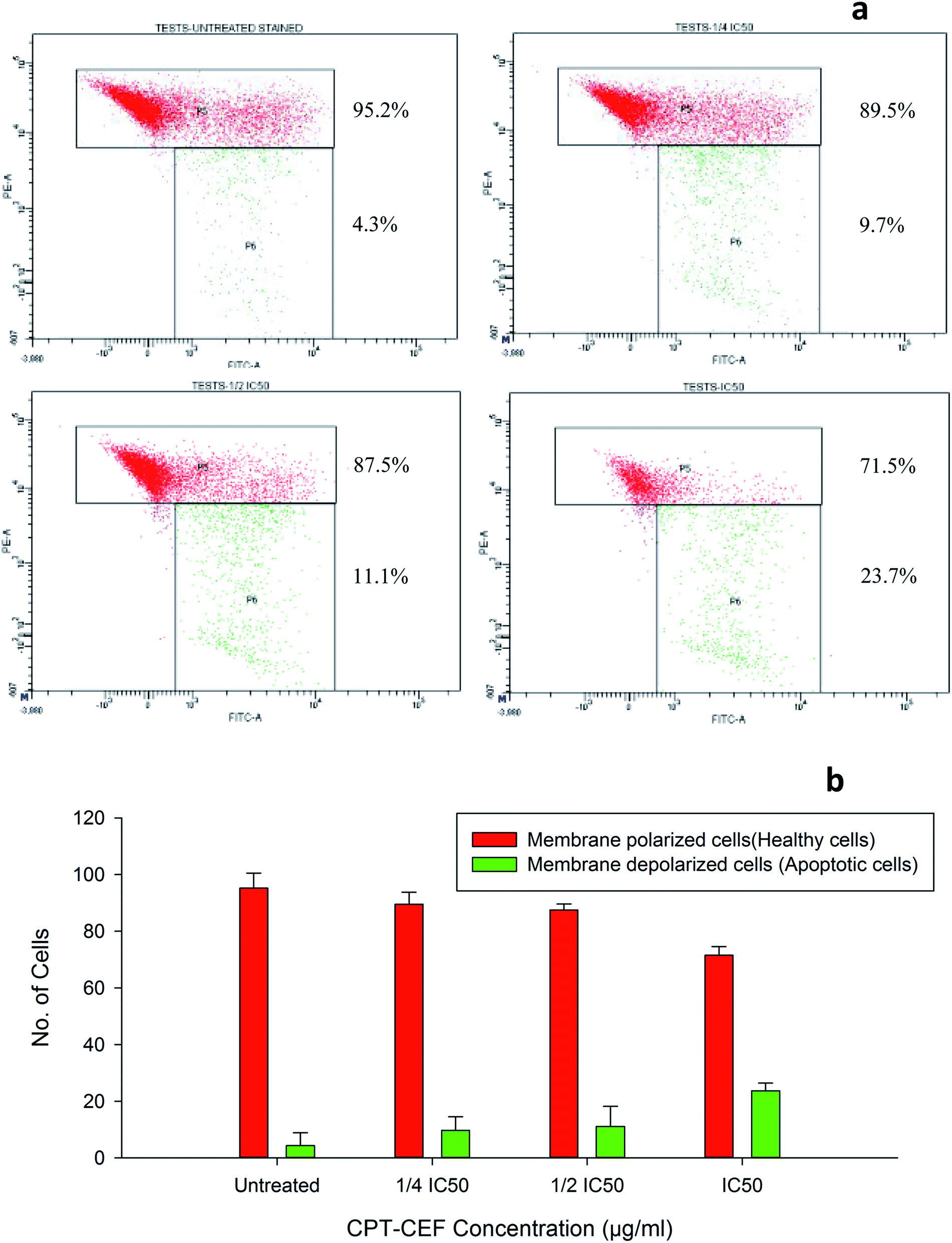
To evaluate whether β-CD–EDTA–Fe3O4/CPT causes alteration to the mitochondrial membrane potential, ΔΨM of cervical cancer cells, JC-1 was employed. At higher potential JC-1 aggregates fluoresce red and at lower potential remains as monomers in the cytoplasm and fluoresce green as it loses the ability to form an aggregate in the mitochondria.50 In this study, HeLa cells were exposed to IC50, 1/2IC50 and 3/4IC50 concentrations of β-CD–EDTA–Fe3O4/CPT for 48 h, and analyzed by flow cytometer. The data shows a substantial escalation in the percentage of mitochondrial membrane depolarized cells in a concentration-dependent manner (Fig. 9). During apoptosis, cells membranes are damaged thus causing an alteration in the ΔΨM. JC-1 staining shows that the at an IC50 concentration of β-CD–EDTA–Fe3O4/CPT significantly enhanced mitochondrial membrane depolarization up to 24% relative to the ΔΨM observed in untreated cells Fig. 9d. These data suggest that β-CD–EDTA–Fe3O4/CPT induced apoptosis accompanied by the alterations in the mitochondrial membrane potential.
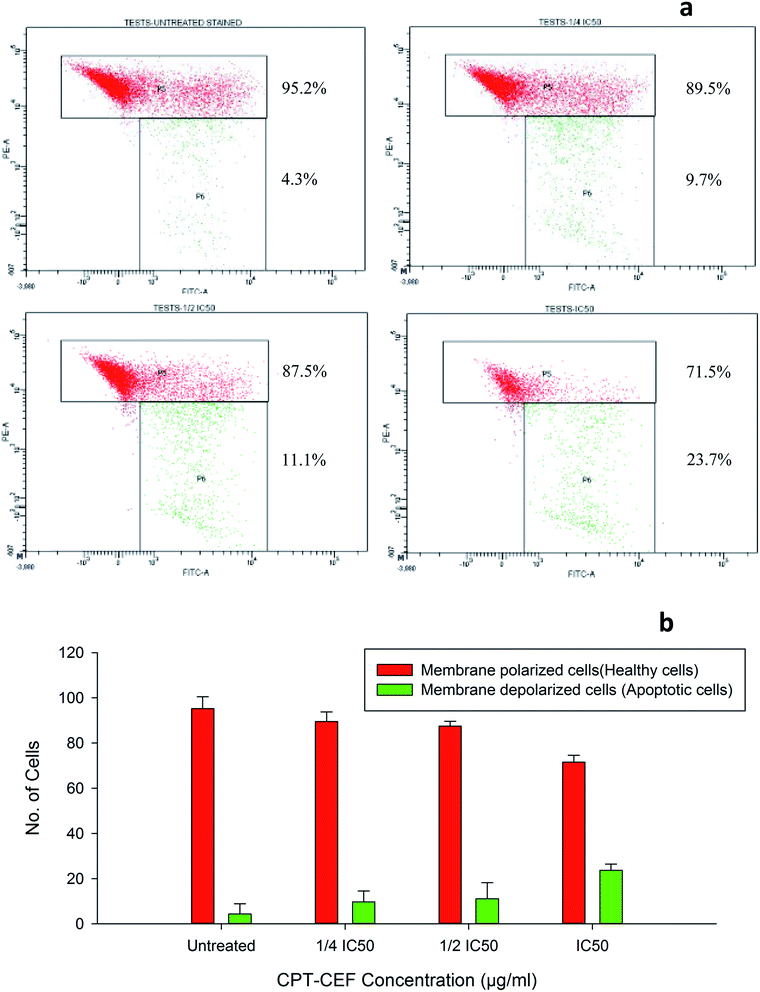 |
| | Fig. 9 (a) The mitochondrial depolarization caused by β-CD–EDTA–Fe3O4/CPT in a concentration dependent manner (a) untreated, (b) 1/4IC50 treated cells, (c) 1/2IC50 treated cells and (d) IC50 treated HeLa cells. The P5 block represents the healthy cells (cell membrane polarized) and P6 block represents the apoptotic cells (cell membrane depolarized). (b) Shows the graphical representation of the trend in membrane depolarization of HeLa cells in response to β-CD–EDTA–Fe3O4/CPT treatment. | |
3.13. β-CD–EDTA–Fe3O4/CPT induces caspase-3 protein expression in HeLa cancer cell
Caspase-3 known as CPP32/Yama/apopain is a key mediator of apoptosis.51 Caspase cascade leading to the release of caspase-3 occurs due to the release of cytochrome c from the mitochondria into the cytosol.52 To further substantiate the existence of caspase-3 in CPT–CEF treated cells colorimetric assay was performed for caspase-3 using its specific substrate poly-ADP ribose polymerase that contains the amino acid motif DEVD conjugated to the color reporter molecule p-nitroaniline (pNA). The role of caspase-3 in the CPT–CEF induced apoptosis was investigated with 1/4IC50, 1/2IC50, and IC50 concentrations. As shown in Fig. S7,† HeLa cells treated with β-CD–EDTA–Fe3O4/CPT for 48 h increased caspase-3 expression in a dose-dependent mode in comparison to the control group. The level of caspase-3 induction in β-CD–EDTA–Fe3O4/CPT treated HeLa cell lines is more than 2-fold greater when compared to the untreated cells.
3.14. Cell cycle arrest in β-CD–EDTA–Fe3O4/CPT treated HeLa cancer cell
Inhibition of cell cycle progression with HeLa cells treated with β-CD–EDTA–Fe3O4/CPT was evaluated with different concentrations. β-CD–EDTA–Fe3O4/CPT treatment at increasing concentration showed an increasing trend in the number of HeLa cells at the G1 phase and decreasing trend in the S-phase and G2/M phase. CPT is commonly known to prompt cell cycle arrest at G2/M phase53 and interestingly, in this study, the results showed a different trend Fig. 10. We noticed a significant increase of cell arrest in the G1 phase while decreasing cell count in S phase and G2/M phase at lower treatment concentration of β-CD–EDTA–Fe3O4/CPT. G2/M phase cell cycle arrest was noticed only at an IC50 concentration of β-CD–EDTA–Fe3O4/CPT. The impact of different concentrations of CPT on the stages of cell cycle arrest has been discussed previously in which at low concentration CPT is found to cause cell cycle arrest at the G2/M phase and shifted to S phase cell cycle arrest at higher concentration of CPT. An increasing trend of cell cycle arrest in the G2/M phase is visible at IC50 concentration.54
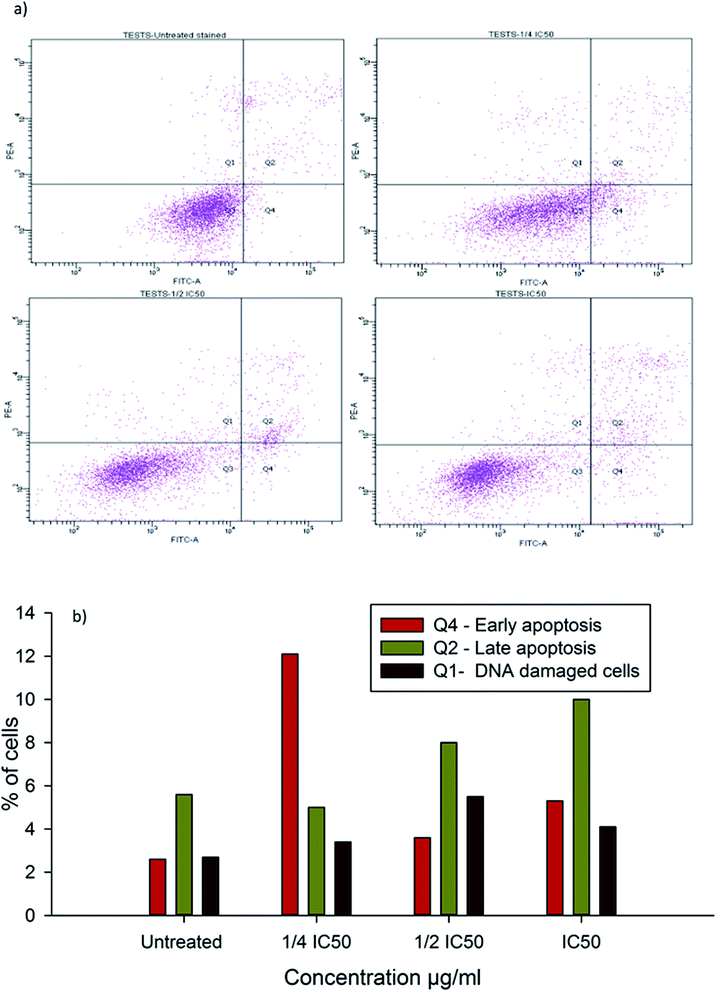 |
| | Fig. 10 (a) Shows the cell cycle arrest caused by CPT–CEF in a concentration dependent manner (a) untreated, (b) 1/4IC50 treated cells, (c) 1/2IC50 treated cells and (d) IC50 treated HeLa cells. (b) Shows the graphical representation of the trend in cell cycle arrest of HeLa cells in response to CPT–CEF treatment. | |
4. Discussion
Cervical cancer is among the major cancers affecting females worldwide and, despite advancement in chemotherapy, it usually causes death, partially due to the failure of chemotherapy.55 The stability and solubility of CPT have greatly diminished CPT's anti-cancer value in present cancer treatments and has diminished its potential to be an anti-cancer agent for other major cancers, such as cervical cancer. This restriction is the driving factor for research within nanotechnology to synthesize an effective nanocarrier for CPT that would enhance its solubility and stability and also improve its aptitude to be an anti-cancer agent for other cancers. β-CD–EDTA–Fe3O4/CPT were developed with these objectives. β-CD–EDTA–Fe3O4 is a nanocarrier that is formulated to encapsulate CPT thus increasing its stability and solubility in aqueous environments. The development of MNPs functionalized CD through the cross linker was examined their crystalline nature, surface morphology, thermal stability and encapsulation, and in vitro drug release of CPT drug on the carrier. The poly-condensation reaction of a secondary alcohol of CD and carboxylic acid group of EDTA was confirmed by 1H and 13C NMR spectroscopy. The SEM morphological examination demonstrates the spherical and smooth surfaces of Fe MNS and CD–EDTA, CD–EDTA–Fe, drug loaded CD–EDTA–Fe has different morphologies. This is due to the modifying parent moiety. The conjugation of Fe on the EDTA linked β-CD increased the size of the carrier compared to non-conjugated β-CD–EDTA composite. Encapsulation of the CPT on the carrier increased the carrier size by the inclusion of CPT drug on the carrier. The conjugated Fe MNs could enhance biomedical application as targeted drug vehicle, a carrier for MRI substances, etc. Fe MNs functionalization affords an essential magnetic treatment method, which increases its usefulness and brings external magnetic force treatment closer to reality.56 The formulation of the magnetic nano-drug delivery vehicle is an expanding area of research. The nanocarrier conjugated with Fe MNs and encapsulation of CPT drug was tested on HeLa cancerous cell line. We characterized the apoptotic pathway induced by water soluble β-CD–EDTA–Fe3O4/CPT in HeLa cells, which share many features common to CPT-induced apoptosis. We identified apoptosis signals, such as upstream caspase-like protease activation and mitochondrial membrane depolarization. The cytotoxicity assay conducted on HeLa cells showed that at an IC50 concentration of 245 μg mL−1, β-CD–EDTA–Fe3O4/CPT were found to be more effective at causing apoptosis in comparison to CPT only. At this concentration, the amount of CPT in the β-CD–EDTA–Fe3O4/CPT formulation is about 7.5% of CPT making the final concentration of CPT IC50 concentration derived from the MTT assay used in this study at a low range of 18.3 μg mL−1 of CPT per 245 μg mL−1 of β-CD–EDTA–Fe3O4/CPT. The solubility showed that β-CD–EDTA–Fe3O4/CPT is soluble in aqueous solution thus improving the efficiency of CPT on the HeLa cancer cells. Also, a multiple cell lines comparison is also made possible by conducting CPT–CEF treatment on HT29 colon and A549 lung cancer cell lines.38 The data showed that the effective CPT–CEF concentration for 50% inhibition (IC50) of HT29 cell growth after 48 h was 133.5 μg mL−1 and of 85 μg mL−1 for achieving 50% inhibition (IC50) in A549 cell growth after 48 h of treatment. This data showed a clear indication of the potential of CPT–CEF to greatly affect A549 cells in terms of cell viability in comparison to colon and lung cancer cell lines.
The JC-1 analysis results suggest that CPT–CEF has the ability to induce apoptosis through the mitochondrial pathway. Mitochondrial membrane potential plays a role in the apoptotic pathway of HeLa cell line treated with β-CD–EDTA–Fe3O4/CPT. Only 23.7% of cells membranes were found to be depolarized at an IC50 concentration as the concentration of β-CD–EDTA–Fe3O4/CPT was doubled the shift in the ΔΨM was reflected as result of β-CD–EDTA–Fe3O4/CPT treatment. This correlates with previous findings that state that CPT alters the mitochondrial membrane potential in treated HeLa cells, which triggers the intrinsic pathway of apoptosis.57 Annexin V results showed an increasing trend in the percentage of cells entering the apoptotic stages. An increased amount of cells in the early and late stage apoptosis were seen at β-CD–EDTA–Fe3O4/CPT treatment IC50 in comparison to the untreated cells. CPT activates the caspase cascade reaction via the mitochondrial apoptotic pathway, leading to cell death.58 Interestingly a 2-fold increase in the percentage of caspase-3 induction was detected at IC50 concentration. Caspases are produced and retained in the cytoplasm as proenzymes and only undergo proteolytic activation when triggered by events such as apoptosis.59 A major shift is observed in cell cycle analysis as cell cycle arrest is noted at G0/G1 phase. We assume this shift might be due to the synergistic effect contributed by the nano-carrier encapsulating camptothecin.
5. Conclusion
In this present work, the new anti-cancer carrier has been developed of using β-CD-modified iron magnetic nanoparticles, showed selective binding to the cancerous cell. Well-dispersed iron oxide magnetic nanoparticles with 168.5 nm diameter and spherical shape were synthesized by co-precipitation method. The morphological analysis of the FMN (Fe3O4 Magnetic Nanoparticles) and the β-CD carrier was carried out by SEM technique. The functionality and thermal decomposition nature of Fe3O4, β-CD–EDTA–Fe3O4, and β-CD–EDTA–Fe3O4/CPT nanocomposites were endorsed by FTIR and TGA studies. To the best of our knowledge, this is the first time that magnetic iron oxide NPs functionalized β-CD nanocarrier system has been developed for camptothecin delivery to HeLa cancer cell line. By taking advantage of the superparamagnetic possessions of the FNM core and the molecular recognition between β-CD and target the cells, these β-CD loaded FMNPs make promising candidates for the non-invasive detection of the cancer cells. We have thoroughly studied the mechanism of action of CPT–CEF by analyzing the nuclear, mitochondrial membrane potential, the activity of caspase-like proteases, and cytosolic changes related with apoptosis in HeLa cells.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
M. Rajan (MR) is thankful to the University Grants Commission (UGC), Government of India, for financial support under the plan of “UGC-MRP” (F. No. 43-187/2014) (SR) and MR acknowledges a major financial support from the Department of Science and Technology, Science and Engineering Research Board (Ref: YSS/2015/001532; New Delhi, India). This project was supported by Universiti Putra Malaysia and funded by Grant Putra-IPS No. 9470200 and Grant Putra-rPM No. 9436400. The authors also extend their appreciation to the International Scientific Partnership Program ISPP at King Saud University for funding this research work through ISPP# 0062.
References
- I. Safarik and M. Safarikova, Chem. Pap., 2009, 63, 497–505 CrossRef CAS.
- E. Peng, F. Wang and J. M. Xue, J. Mater. Chem. B, 2015, 3, 2241–2276 RSC.
- J. Wang, X. Wang, Y. Song, J. Wang, C. Zhang, C. Chang, J. Yan, L. Qiu, M. Wu and Z. Guo, Chem. Sci., 2013, 4, 2605–2612 RSC.
- E. Boros, A. M. Bowen, L. Josephson, N. Vasdevbe and J. P. Holland, Chem. Sci., 2015, 6, 225 RSC.
- T. Neuberger, B. Schöpf, H. Hofmann, M. Hofmann and B. von Rechenberg, J. Magn. Magn. Mater., 2005, 293, 483–496 CrossRef CAS.
- T. T. Thuy, S. Maenosono and N. T. K. Thanh, Magnetic Nanoparticles: From Fabrication to Clinical Applications, CRC Press, 2012, pp. 99–126 Search PubMed.
- F. K. Storm, W. H. Harrison, R. S. Elliott and D. L. Morton, Cancer Res., 1979, 39, 2245–2251 CAS.
- D. A. Richards, A. Maruani and V. Chudasama, Chem. Sci., 2017, 8, 63–77 RSC.
- M. A. Agotegaray, A. E. Campelo, R. D. Zysler, F. Gumilar, C. Bras, A. Gandini, A. Minetti, V. L. Massheimer and V. L. Lassalle, Biomater. Sci., 2017, 5, 772–783 RSC.
- M. Rajan, M. Murugan, D. Ponnamma, S. Kishor Kumar and A. M. Murugan, Biomed. Pharmacother., 2016, 83, 201–211 CrossRef CAS PubMed.
- M. Jeyaraj, R. A. Praphakar, C. Rajendran, D. Ponnamma, S. Kishor Kumar, A. M. Murugan and M. Rajan, RSC Adv., 2016, 6, 51310–51319 RSC.
- M. Rajan, V. Raj, A. A. Al-Arfaj and A. Murugan, Int. J. Pharm., 2013, 453, 514–522 CrossRef CAS PubMed.
- M. Rajan and V. Raj, Carbohydr. Polym., 2013, 98, 951–958 CrossRef CAS PubMed.
- X. Tian, K. H. Baeka and I. Shin, Chem. Sci., 2013, 4, 947–956 RSC.
- Z. Cao, H. Wu, J. Dong and Q. Wang, ACS Appl. Mater. Interfaces, 2016, 8, 28888–28896 CAS.
- T. K. Nguyen, R. Selvanayagam, K. K. K. Ho, R. Chen, S. K. Kutty, S. A. Rice, N. Kumar, N. Barraud, H. T. T. Duong and C. Boyer, Chem. Sci., 2016, 7, 1016–1027 RSC.
- S. Kaur, C. Prasad, B. Balakrishnan and R. Banerjee, Biomater. Sci., 2015, 3, 955–987 RSC.
- M. Guo, C. Que, C. Wang, X. Liu, H. Yan and K. Liu, Biomaterials, 2011, 32(1), 185–194 CrossRef CAS PubMed.
- H. Wei and Y. Cui-yun, Biomater. Sci., 2015, 3, 1050–1060 RSC.
- A. Kulkarni, R. VerHeul, K. DeFrees, C. J. Collins, R. A. Schuldt, A. Vlahu and D. H. Thompson, Biomater. Sci., 2013, 1, 1029 RSC.
- Q. Lin, Y. Yang, Q. Hu, Z. Guo, T. Liu, J. Xu, J. Wu, T. B. Kirk, D. Ma and W. Xue, Acta Biomater., 2016, 49, 456–471 CrossRef PubMed.
- T. Xu, J. Zhang, H. Chi and F. Cao, Acta Biomater., 2016, 36, 152–163 CrossRef CAS PubMed.
- L. D. Wilson, D. Y. Pratt and J. A. Kozinski, J. Colloid Interface Sci., 2013, 393, 271 CrossRef CAS PubMed.
- E. Zeiger, B. Gollapudi and P. Spencer, Mutat. Res., Rev. Mutat. Res., 2005, 589, 136–151 CrossRef CAS PubMed.
- F. Zhao, E. Repo, M. Sillanpää, Y. Meng, D. Yin and W. Z. Tang, Ind. Eng. Chem. Res., 2015, 54, 1271–1281 CrossRef CAS.
- M. Li, W. Tang, F. Zeng, L. Lou and T. You, Bioorg. Med. Chem. Lett., 2008, 18, 6441–6443 CrossRef CAS PubMed.
- Z. Xu, M. Hou, X. Shi, Y. E. Gao, P. Xue, S. Liu and Y. Kang, Biomater. Sci., 2017, 5, 444–454 RSC.
- V. Sharma, T. A. Lansdell, S. Peddibhotla and J. J. Tepe, Chem. Biol., 2004, 11, 1689–1699 CrossRef CAS PubMed.
- V. Sharma, T. A. Lansdell, S. Peddibhotla and J. J. Tepe, Chem. Biol., 2004, 11, 1689–1699 CrossRef CAS PubMed.
- Z. L. Song, H. L. Chen, Y. H. Wang, M. Goto, W. J. Gao, P. L. Cheng, S. L. Morris-Natschke, Y. Qian Liu, G. Xiang Zhu, M. Juan Wang and K. Hsiung Lee, Bioorg. Med. Chem. Lett., 2015, 25, 2690–2693 CrossRef CAS PubMed.
- W. Song, Z. Tang, D. Zhang, M. Li, J. Gu and X. Chen, Chem. Sci., 2016, 7, 728–736 RSC.
- S. Swaminathan, L. Pastero, L. Serpe, F. Trotta, P. Vavia, D. Aquilano, M. Trotta, G. Zara and R. Cavalli, Eur. J. Pharm. Biopharm., 2010, 74, 193–201 CrossRef CAS PubMed.
- R. Bhatt, P. D. Vries, J. Tulinsky, G. Bellamy, B. Baker, J. W. Singer and P. Klein, J. Med. Chem., 2003, 46, 190–193 CrossRef CAS PubMed.
- H. H. Shuai, C. Y. Yang, H. C. Harn, R. L. York, T. C. Liao, W. S. Chen, J. A. Yeha and C. M. Cheng, Chem. Sci., 2013, 4, 3058–3067 RSC.
- B. Sivakumar, R. Girija Aswathy, Y. Nagaoka, M. Suzuki, T. Fukuda, Y. Yoshida, T. Maekawa and D. N. Sakthikumar, Langmuir, 2013, 29, 3453–3466 CrossRef CAS PubMed.
- F. Zhao, R. Eveliina, Y. Dulin, M. Yong, J. Shila and S. Mika, Environ. Sci. Technol., 2015, 49, 10570–10580 CrossRef CAS PubMed.
- F. Du, H. Meng, K. Xu, Y. Xu, P. Luo, Y. Luo and W. Luet, Colloids Surf., B, 2014, 113, 230–236 CrossRef CAS PubMed.
- P. Krishnan, M. Rajan, S. Kumari, S. Sakinah, S. Padma Priya, F. Amira, L. Danjuma, M. Pooi Ling, S. Fakurazi, P. Arulselvan, A. Higuchi, R. Arumugam, A. A. Alarfaj, M. A. Munusamy, R. Awang Hamat, G. Benelli, K. Murugan and S. Suresh Kumar, Sci. Rep., 2017, 7, 10962 CrossRef PubMed.
- T. Mosmann, J. Immunol. Methods, 1983, 65, 55–63 CrossRef CAS PubMed.
- H. A. Azim, Transl Oncol., 2016, 9, 114–123 CrossRef PubMed.
- G. Shaghayegh, A. M. Alabsi, R. A. Saeed, A. Manaf Ali, V. K. Vincent-Chong and R. B. Zain, Cytotechnology, 2016, 68, 1999–2013 CrossRef CAS PubMed.
- F. Hajiaghaalipour, M. S. Kanthimathi, J. Sanusi and J. Rajarajeswaran, Food Chem., 2015, 169, 401–410 CrossRef CAS PubMed.
- D. Zhao, L. Zhao, C. Zhu, Z. Tian and X. Shen, Carbohydr. Polym., 2009, 78, 125–130 CrossRef CAS.
- F. Zhao, E. Repo, M. Sillanpaaa, Y. Meng, D. Yin and Y. Z. Tang, Ind. Eng. Chem. Res., 2015, 54, 1271–1281 CrossRef CAS.
- P. C. Panta and C. P. Bergmann, 4th International conference and exhibition on materials science and engineering, Florida, USA, 2015 Search PubMed.
- K. Wang, Y. Liu, C. Li, S. X. Cheng, R. X. Zhuo and X. Z. Zhang, ACS Macro Lett., 2013, 2, 201–205 CrossRef CAS.
- Y. Liu, G. S. Chen, Y. Chen and J. Lin, Bioorg. Med. Chem., 2005, 13, 4037–4042 CrossRef CAS PubMed.
- H. Wang, L. Tang, C. Tu, Z. Song, Q. Yin, L. Yin, Z. Zhang and J. Cheng, Biomacromolecules, 2013, 14(10), 3706–3712 CrossRef CAS PubMed.
- S. A. AbdGhafar, M. Ismail, L. S. Yazan, S. Fakurazi, N. Ismail, K. W. Chan and P. M. Tahir, J. Evidence-Based Complementary Altern. Med., 2013, 549705, DOI:10.1155/2013/549705.
- N. Sen, B. B. Das, A. Ganguly, T. Mukherjee, G. Tripathi, S. Bandyopadhyay, S. Rakshit, T. Sen and H. K. Majumder, Cell Death Differ., 2004, 11, 924–936 CrossRef CAS PubMed.
- H. Zou, W. J. Henzel, X. Liu, A. Lutschg and X. Wang, Cell, 1997, 90, 405–413 CrossRef CAS PubMed.
- J. Sun, R. Sheng, T. Luo, Z. Wang, H. Li and A. Cao, J. Mater. Chem. B, 2016, 4, 4696–4706 RSC.
- R. Minelli, R. Cavalli, L. Ellis, P. Pettazzoni, F. Trotta, E. Ciamporcero, G. Barrera, R. Fantozzi, C. Dianzani and R. Pili, Eur. J. Pharm. Sci., 2012, 47, 686–694 CrossRef CAS PubMed.
- X. Wang, M. Tanaka, S. Krstin, H. S. Peixoto, C. C. Moura and M. Wink, Eur. J. Pharmacol., 2016, 789, 265–274 CrossRef CAS PubMed.
- M. V. Grau, J. R. Rees and J. A. Baron, Basic Clin. Pharmacol. Toxicol., 2006, 98, 281–287 CrossRef CAS PubMed.
- Y. W. Jun, J. W. Seo and J. Cheon, Acc. Chem. Res., 2007, 41, 179 CrossRef PubMed.
- U. Jeong, X. Teng, Y. Wang, H. Yang and Y. Xia, Adv. Mater., 2007, 19, 33 CrossRef CAS.
- C. W. Zeng, X. J. Zhang, K. Y. Lin, H. Ye, S. Y. Feng, H. Zhang and Y. Q. Chen, Mol. Pharmacol., 2012, 81, 578–586 CrossRef CAS PubMed.
- Y. Zhang, W. Zhang, J. Chen, X. Yang, P. Ma, B. Zhang, B. Liu, J. Ni and R. Wang, Peptides, 2015, 63, 143–149 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra06615e |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *a,
Poorani Krishnanb,
Periyakaruppan Pradeepkumara,
Mayandi Jeyanthinathc,
Murugaraj Jeyarajd,
Mok Pooi Lingef,
Palanisamy Arulselvang,
Akon Higuchihij,
Murugan A. Munusamyj,
Ramitha Arumugamk,
Giovanni Benelliln,
Kadarkarai Muruganmo and
S. Suresh Kumar*be
*a,
Poorani Krishnanb,
Periyakaruppan Pradeepkumara,
Mayandi Jeyanthinathc,
Murugaraj Jeyarajd,
Mok Pooi Lingef,
Palanisamy Arulselvang,
Akon Higuchihij,
Murugan A. Munusamyj,
Ramitha Arumugamk,
Giovanni Benelliln,
Kadarkarai Muruganmo and
S. Suresh Kumar*be