
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDetection of sulfenic acid in intact proteins by mass spectrometric techniques: application to serum samples†
M. Sharar a,
Humberto Rodríguez-Sollab,
M. W. Linscheidc and
Maria Montes-Bayón*d
a,
Humberto Rodríguez-Sollab,
M. W. Linscheidc and
Maria Montes-Bayón*d
aDepartment of Chemistry, Humboldt Universität zu Berlin, 10117 Berlin, Germany
bDepartment of Organic and Inorganic Chemistry, University of Oviedo, 33006 Oviedo, Spain
cDepartment of Chemistry, Humboldt Universität zu Berlin, 10117 Berlin, Germany. E-mail: analytik@chemie.hu-berlin.de
dDepartment of Physical and Analytical Chemistry, University of Oviedo, 33006 Oviedo, Spain. E-mail: montesmaria@uniovi.es
First published on 12th September 2017
Abstract
Formation of cysteine sulfenic acid (SA) is considered a transient state of thiol oxidation in living organisms that can be either reduced back or continue to result in the formation of sulfinic and sulfonic acids. As any disturbance in oxidation is correlated to age-related diseases such as cancer and Alzheimer's disease, the detection of SA transient state formed a sensor for such redox-mediated events. Thus, detection of low amounts of SA is critical in order to prevent further oxidative damage of cells and tissues and for this aim specific strategies have to be developed. In this work, detection and quantification of induced SA in human serum albumin is reported by specific capture using alkyne β-ketoester (KE) previously linked to a lanthanide (Ln)-containing chelator (Ln–DOTA, where DOTA is 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid). SA formation was induced by hydrogen peroxide to mimic oxidative conditions produced in living cells by ROS and was detected using molecular and elemental mass spectrometric (MS) techniques. The developed strategy has been further applied to the determination of SA-induced formation in human serum by using affinity chromatography for purification of albumin followed by inductively coupled plasma mass spectrometry (ICP-MS) to monitor the formed SA linked to Ln–DOTA–KE in combination with isotope dilution analysis (IDA) for absolute quantification. Quantitative results showed levels of oxidative damage regarding SA formation in up to 40% of the albumin present.
Introduction
A well-known mechanism of redox signaling that can also conduct to further cell damage involves H2O2-mediated oxidation of cysteine residues within proteins. Cysteine residues exist as a thiolate anion (Cys–S−) at physiological pH and are more susceptible to oxidation compared with protonated cysteine thiol (Cys–SH).1 During redox signaling, H2O2 oxidizes the thiolate anion to the sulfenic form (Cys–SOH, SA), causing allosteric changes within the protein that alter its function. It is estimated that thiolate oxidation in living cells occurs in the nanomolar range of H2O2, whereas higher levels of H2O2 further oxidize thiolate anions to sulfinic (SO2H) or sulfonic (SO3H) species.2–5 Unlike sulfenic modification, sulfinic and sulfonic acids modifications are irreversible and result in permanent protein damage (i.e., oxidative stress).6These formed redox transformations for cysteine residue are called post translational modifications (PTMs); they can modify the function of proteins and have been connected to disease states (such as cancer, Alzheimer's, and Parkinson's diseases).6–8 Therefore, the quantification of cysteine PTMs in biological systems has become essential for understanding many age-related diseases.9–12 In particular, the detection and quantification of transient cysteine SA is a challenging task that requires the use of highly sensitive analytical techniques and suitable “capturing strategies”.
The most common way to detect SA formation in proteins is by “trapping” it through a chemical reaction. Current methods for this objective involve the use of dimedone and other nucleophilic 1,3-dicarbonyl probes that form covalent adducts with cysteine-derived protein SA. As a mechanistic alternative, recent studies describe highly strained cyclic alkynes as concerted traps for sulfenic acids.13 However, in most cases, the formed derivative showed limited ionization when analyzed using electrospray (ESI) or matrix-assisted laser desorption/ionization (MALDI) mass spectrometry (MS). This situation has motivated the development of other strategies, like those based on the use of biotinylated and fluorophore-conjugated analogues, in order to study labelled SA residues.12–15
Nevertheless, for quantitative analysis of SA formation in proteins, alternative detection possibilities have been explored based on trapping of the SA groups with specific molecules that incorporate lanthanide atoms in their structure followed by elemental detection using inductively coupled plasma mass spectrometry (ICP-MS).16,17 Since ketoesters (KE) have been reported to be specific for SA detection, and to exhibit improved reactivity at physiological pH compared to other probes (such as dimedone),14 we developed a MS and ICP-MS detectable compound for SA detection. In the prepared compound, alkyne KE was linked to macrocyclic 1,4,7,10-tetraazacyclododecane-1,4,7-tris(acetic acid)-10-azidopropylethylacetamide(azide-DOTA) which was previously metallated with a lanthanide (Ln). Being specific for SA labelling, the developed Ln–DOTA–KE enabled use of MS and ICP-MS for the detection of SA in model peptides.17
However, the detection of SA in proteins is still a complex subject, in particular, in real samples like serum or whole blood. Here, we report the absolute quantification of SA in human serum albumin (HSA) in samples of serum after trapping the SA using a linear alkyne KE linked to an Ln-containing DOTA. Albumin contains 35 cysteines forming 17 disulfide bridges (–S–S–) with only one free thiol, Cys-34, which comprises 80% of the total free thiols in plasma and can follow the smallest change in the redox environment of a biological system.18–20 Ln–DOTA–KE was used to label the free cysteine (Cys-34) in albumin using Nd as metal to follow with ICP-MS. The use of isotope dilution analysis in conjunction with liquid chromatography-ICP-MS permits quantifying the evolution of the oxidative process in the albumin after a suitable purification process. The accuracy of this technique in addition to the high sensitivity of ICP-MS contributed significantly to quantitative proteomics.
Experimental
Chemicals and materials
Purchased materials were used without further purification. High-purity Milli-Q water was used for samples preparation (Millipore, Bedford, MA, USA). Argon for ICP-MS, 99.99% purity, was delivered by Alphagaz (Madrid, Spain). 1,4,7,10-Tetraazacyclododecane-1,4,7-tris-acetic acid-10(azidopropylethylacetamide) (azide-DOTA) was purchased from Macrocyclics (Dallas, TX, USA).Neodymium(III) chloride hexahydrate, 99.99%, human serum albumin standard and human serum (ERM® certified reference serum proteins) were purchased from Sigma-Aldrich (Madrid, Spain). Amicon® Ultra centrifugal filter units, hydrogen peroxide 30%, potassium dihydrogen phosphate, acetonitrile and tris(hydroxymethyl) amino methane (TRIS) were purchased from Merck KGaA (Darmstadt, Germany). Isotopically enriched (145Nd) neodymium standard was purchased from Euriso-Top GmbH (Saarbrücken, Germany); the isotopic abundances were 0.59% 142Nd, 0.45% 143Nd, 2.61% 144Nd, 86.57% 145Nd, 9.44% 146Nd, 0.22% 148Nd, and 0.11% 150Nd. The HiTrap™ Blue HP column was purchased from GE Healthcare (Uppsala, Sweden).
Instrumentation
Purification for Ln–DOTA and Ln–DOTA–KE was performed using an Agilent 1100 system equipped with a diode array detector; they were purified using a Nucleosil® 100 Å C18 column (5 μm, 200 × 4.6 mm) (Phenomenex, Spain) operating at a flow rate of 0.5 mL min−1. The employed mobile phases were: phase (A) 0.01 M phosphoric acid (H3PO4) in H2O and phase (B) 100% acetonitrile (ACN).The HiTrap™ Blue HP column was used to purify albumin from human serum using an Agilent 1100 system equipped with a diode array detector. Employed binding buffer was 50 mM KH2PO4 (pH = 7.00) and the elution buffer was 50 mM KH2PO4 + 1.5 M KCl (pH = 7.00).
To monitor signals, an electrospray time-of-flight instrument (ultra-high-resolution double-quadrupole time-of-flight mass spectrometer; Impact II, Bruker, Germany) was used. Two different ICP-MS systems were used: an Agilent 7500 ce (Agilent Technologies, Kyoto, Japan) and an iCAP TQ ICP-MS (Thermo Fisher Scientific, Bremen, Germany). Both systems were coupled on-line to size exclusion chromatography (SEC) using a Superdex 200 10/300 GL column (GE Healthcare Bio-Sciences, Sweden) with a mobile phase containing ammonium acetate (50 mM, pH = 6.5). The mobile phase was pumped using a chromatographic pump (Shimadzu LC-10AD, Shimadzu Corporation, Japan) with a flow rate of 0.6 mL min−1, and the injection volume of samples was 20 μL using a six-way peek injection valve.
Preparation of Ln–DOTA–KE
Azide-DOTA was metallated with a poly-isotopic neodymium salt (NdCl3·6H2O). Azide-DOTA was added to the Nd solution (10-fold excess of Nd dissolved in 110 mM sodium acetate buffer, pH = 6.20). The reaction was left for 2 h in darkness with gentle shaking at room temperature. Nd–DOTA was purified using a C18 column and the signals were monitored at 215 nm and 280 nm.Copper catalyzed azide alkyne cycloaddition (CuAAC), click reaction, was carried out to perform the selective reaction between Nd–DOTA and alkyne β-ketoester (KE) (Scheme S1 in ESI †). The reaction was carried out according to the following order: KE 2 (4 mM) was mixed with TEAA buffer (100 mM, pH = 7.00); two-fold excess of Nd–DOTA 1 (8 mM) to KE 2 was added. THPTA (500 μM) and copper(II) sulphate (100 μM) were premixed in 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio and then added to the previous mixture, and finally a solution of sodium ascorbate (5 mM) was added. The reaction was carried out in 500 μL TEAA buffer and left in darkness for 1 h under sonication to yield compound 3.21,22 Compound 3 was then purified using a C18 column and the reaction products were monitored at 200 nm and 280 nm.23 The produced Nd–DOTA–KE 3 was then used for SA labelling 4.
:1 ratio and then added to the previous mixture, and finally a solution of sodium ascorbate (5 mM) was added. The reaction was carried out in 500 μL TEAA buffer and left in darkness for 1 h under sonication to yield compound 3.21,22 Compound 3 was then purified using a C18 column and the reaction products were monitored at 200 nm and 280 nm.23 The produced Nd–DOTA–KE 3 was then used for SA labelling 4.
Synthesis of KE was performed in the Organic Chemistry Department (University of Oviedo) and further details regarding KE synthesis and click reaction conditions can be found elsewhere.17
Labelling of SA with Nd–DOTA–KE
Nd–DOTA–KE was used to label intact albumin first as protein models susceptible to being oxidized in free cysteine to form SA. For this objective, an albumin solution of 90 μM was prepared in TRIS buffer (pH = 8.4). For SA labelling in human serum albumin (HSA), HSA was denatured in 8 M urea and then oxidized with different excess ratios of hydrogen peroxide (H2O2) (5 mM). Finally, 30-fold excess of Nd–DOTA–KE to cysteine was added to the mixture. The labelling reaction was carried out in TRIS buffer (100 mM, pH = 8.4) and the mixture was left for 4 h with gentle shaking at R.T.In the case of the serum sample, the protein was previously purified from the remaining proteins using affinity chromatography on a HiTrap™ Blue HP column and the signal was monitored at 280 nm. Collected albumin fractions were lyophilized and cleaned through 30 kDa amicons in order to remove any traces of salts that might affect further labelling reactions. Labelling of the oxidized HSA was conducted under the same conditions as carried out in the albumin standard. The labelling signal for albumin (Nd–DOTA–KE–SA) was monitored with LC-ESI-MS and SEC-ICP-MS.
Quantification of the labelled SA by Nd-isotope dilution analysis (IDA-SEC-ICP-MS)
The isotopically enriched 145Nd standard was used for post column isotope dilution analysis (ID-SEC-ICP-MS). For this, a continuous flow of the tracer (0.1 mL min−1) was added and mixed with the column eluent using a T piece. The concentration of the spiked solution (145Nd) was first estimated using a natural Nd standard solution. Further calibration using reverse ID-ICP-MS analysis was performed to measure the precise concentration of the spiking solution.24Optimum ratio between Nd standard and spike was experimentally determined (Rm)142Nd/145Nd,25and a 20 μL loop was used for introducing the sample of labelled SA with Nd–DOTA–KE. A 5 ppb solution of 145Nd prepared in 2% HNO3 was finally used. All samples were precisely weighed using a four-digit analytical balance.
Protein electrophoresis
In order to test the purity of the labelled product, separation of the labelled human serum albumin was carried out using sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) according to the Laemmli gel system.26 Proteins were separated by 10% SDS-PAGE under denaturing conditions (boiled for 1 min in 4× SDS sample buffer before loading into the gel) by using a Bio-Rad electrophoresis system.Results and discussion
Characterization of the labelling reagent Nd–DOTA–KE
Metalation of the azide-DOTA with Nd was conducted according to the previously published strategy and using the CuAAC reaction.17 The CuAAC reaction was carried out between Nd–DOTA and alkyne KE to produce the labelling reagent Nd–DOTA–KE which then was purified and used for SA labelling. LC-ESI-MS was used to confirm the structure of the synthetic product of the CuAAC reaction (Nd–DOTA–KE). The obtained result is provided in ESI (Fig. S1†) where the isotopic pattern of Nd can be observed. The calculated mass accuracy for the most abundant isotope was of −0.51 ppm. In addition, the purified Nd–DOTA–KE signal was also monitored with SEC-ICP-MS to obtain the elution time for Nd–DOTA–KE and to address the absence of any species that might have been formed during the synthesis. The Nd chromatogram corresponding to 50 ppb of Nd is shown in ESI (Fig. S2†) where the metal is associated to a single peak at about 31.5 min.Since the labelling reagent (Nd–DOTA–KE) is aimed to be used for quantitative labelling of SA in albumin, its concentration must be determined. For this purpose, ICP-MS analysis of Nd by flow injection analysis (FIA) of the purified Nd–DOTA–KE was carried out followed by a calibration curve of the metal measured under the same conditions (FIA-ICP-MS). By knowing the stoichiometry of the Nd:complex (1:1), the obtained Nd concentration can be transformed into Nd–DOTA–KE concentration. Thus, the final concentration of the synthetic labelling reagent turned out to be 30.3 mM which is in good agreement with the calculated concentration (32 mM). As expected, all Nd in the Nd–DOTA–KE solution was the metal in the complex and no free Nd was present in the solution.
Evaluation of SA labelling in albumin standards and serum samples
A human serum albumin standard was prepared and analyzed using ESI-MS to evaluate different forms of the protein and potential post-translational modifications. The ESI-MS spectrum provided the multiple charge profile of albumin (see ESI, Fig. S3†) and deconvolution of the spectrum resulted in a calculated molecular mass of 66557 Da. This corresponds mainly to the form of cysteinylated albumin (covalently bound cysteine residue). Since only cysteine that is not forming disulfide bridges, and is susceptible to form SA for subsequent labelling with the Ln–DOTA–KE, Cys-34, can be affected by this modification, treatment with a reducing agent is considered necessary to reduce the proportions of the existing disulfides. However, the use of reducing agents (such as β-mercaptoethanol or TCEP) will affect as well the other internal disulfide bridges present in albumin.19,20 Therefore, labelling was attempted directly in albumin from serum, expecting the presence of some traces of the native form of the protein.
For this aim, albumin was first separated from the rest of the serum components using the HiTrap™ Blue HP affinity column and the elution profiles were monitored by UV/Vis. The chromatogram is shown in ESI (Fig. S4†) in which two separated peaks correspond to plasma proteins (fraction A) and albumin (fraction B). The fractions were collected, purified, and the products monitored using gel electrophoresis (10% SDS-PAGE). As can be seen in the inset of Fig. S4,† the results obtained for fraction A showed bands that correspond to α, β, and γ-globulins among others and fraction B exhibited mainly the band corresponding to the purified albumin which appeared at around 70 kDa, confirming the successful isolation of HSA with the proposed set-up.
The collected albumin (Fig. S4† – fraction B) was also analyzed using ESI-MS and in this case, a single chain of 585 amino acids of albumin (unmodified) was obtained as can be seen in Fig. 1 at m/z 66438.27 In this case, also different PTMs and variants such as cysteinylated (Cys–HSA) and glycated (Glc–HSA) formed corresponding to 66557 Da and 66598 Da, respectively. As before, urea was used for denaturing albumin and further labelling for SA was carried out in 100 mM of THAM buffer, pH = 8.4 using 30-fold excess of Nd–DOTA–KE to cysteine unit. Different oxidation conditions were tested and different fold excess of hydrogen peroxide (H2O2) (5 mM) to Cys-34 were used to form the SA. The mixture was left for 4 h with gentle shaking at R.T. The labelling reaction was monitored by ESI-MS and the results are shown in Fig. 2. As observed, a mixture of labelled (marked with a star) and unlabelled HSA was obtained with a mass difference between both species corresponding to the molecular mass of the used labelling reagent (Nd–DOTA–KE–HSA) which has 780 Da. Results are summarized in Table 1. The deconvoluted mass spectrum showed a high peak of native unlabelled HSA with fractions of cysteinylated HSA, along the labelled HSA. Moreover, used labelling reagent (Nd–DOTA–KE) did not show any reactivity towards any amino acid residue other than SA.14
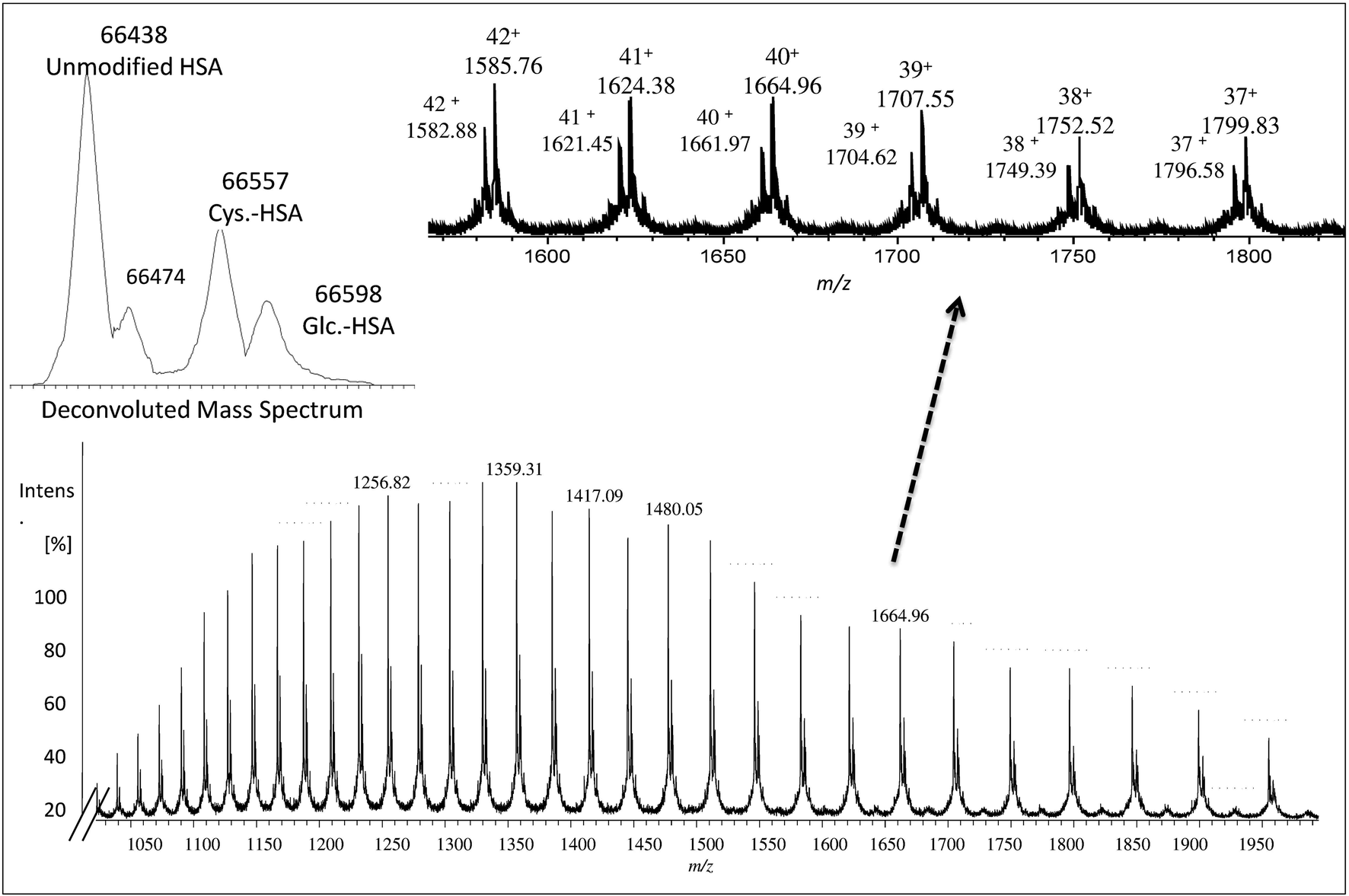 | ||
| Fig. 1 Mass spectrum obtained by ESI-q-TOF mass spectrometry for collected albumin from human serum. The deconvoluted mass spectrum represents different PTMs and variant average molecular weight of albumin in the range of 66438–66600 Da. | ||
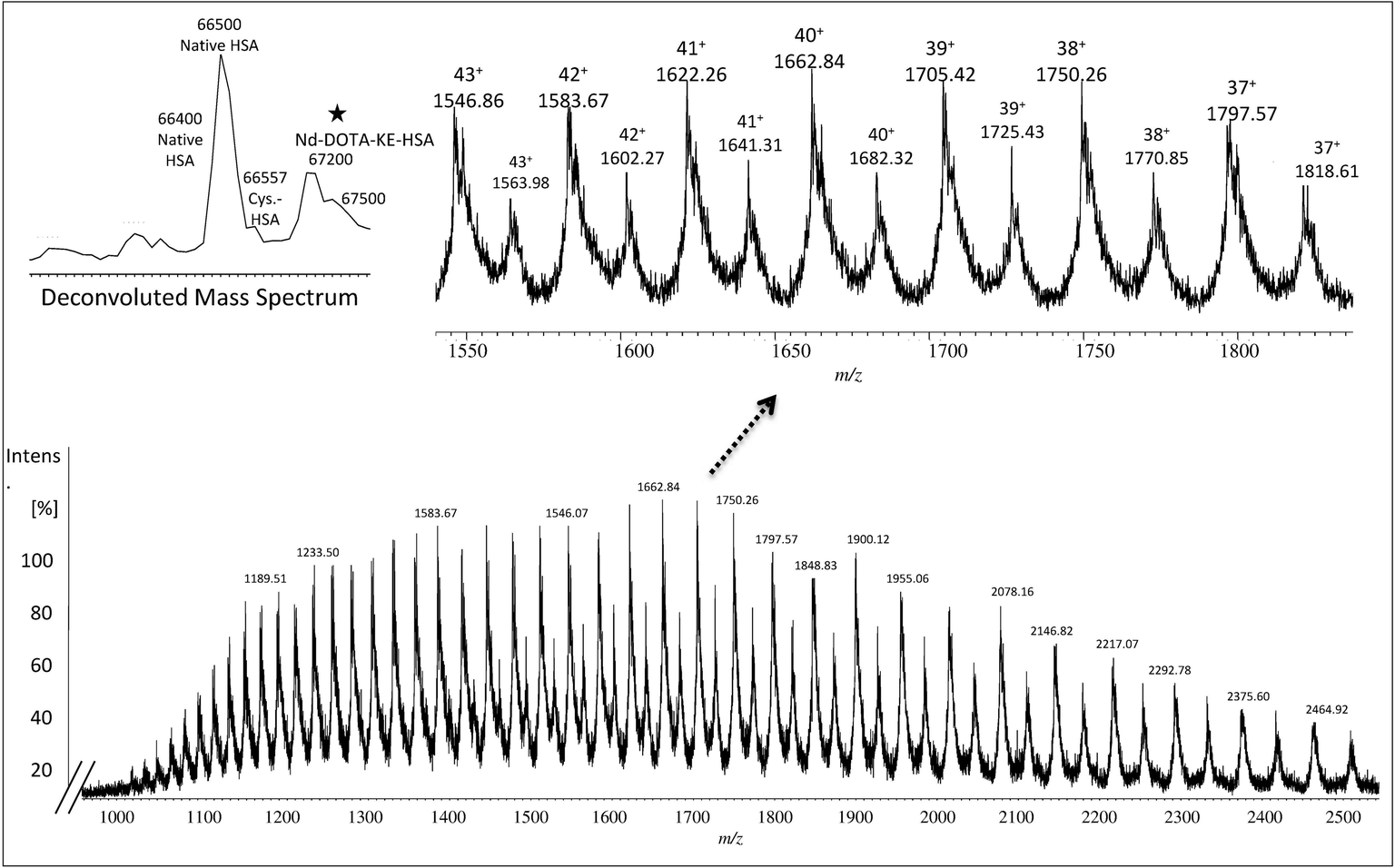 | ||
| Fig. 2 Mass spectrum obtained by ESI-q-TOF mass spectrometry for a labelled sulfenic acid (SA) in albumin with Nd–DOTA–KE. Labelled SA residues with Nd–DOTA–KE are indicated with stars. Deconvoluted mass spectrum represents different PTMs and average molecular weight of labelled albumin with Nd–DOTA–KE (67252 Da) and the remaining unlabelled (66472 Da). | ||
| Charge | Fragmentation pattern of albumin monoisotopic mass | Fragmentation pattern of Nd–DOTA–KE-albumin monoisotopic mass |
|---|---|---|
| 43+ | 1545.86 | 1563.96 |
| 42+ | 1582.65 | 1601.18 |
| 41+ | 1621.23 | 1640.21 |
| 40+ | 1661.73 | 1681.19 |
| 39+ | 1704.31 | 1724.27 |
| 38+ | 1749.14 | 1769.62 |
| 37+ | 1796.38 | 1817.41 |
Analysis of the samples by SDS-PAGE did not show any difference between the bands corresponding to the labelled and unlabelled compounds. As shown in the ESI (Fig. S5†) the slight increase in albumin molecular weight band (C and D) can be attributed to the higher molecular weight resulting from the mixture of labelled and unlabelled albumin (slight differences in band concentrations can be seen as all samples were filtered with 30 kDa filters after labelling to remove excess of reagent and the used denaturing agent).
Finally, the SEC-ICP-MS signal was used for monitoring of both Nd and S simultaneously in the same peak. The instrument was used in the oxygen reaction mode in order to form the oxide of both species, which exhibited higher sensitivity in particular in the case of S analysis. Fig. 3 shows the elution time for the labelled SA with Nd–DOTA–KE (S and Nd signal) as well as presence of the excess labelling compound. It is noteworthy that the first compound (peak A) shows the corresponding S signal for labelled and unlabelled HSA since they cannot be separated under these conditions. However, since the Nd concentration is proportional to the labelled species and the sensitivity of this element in ICP-MS is substantially higher, quantification by the Nd signal will be very advantageous.
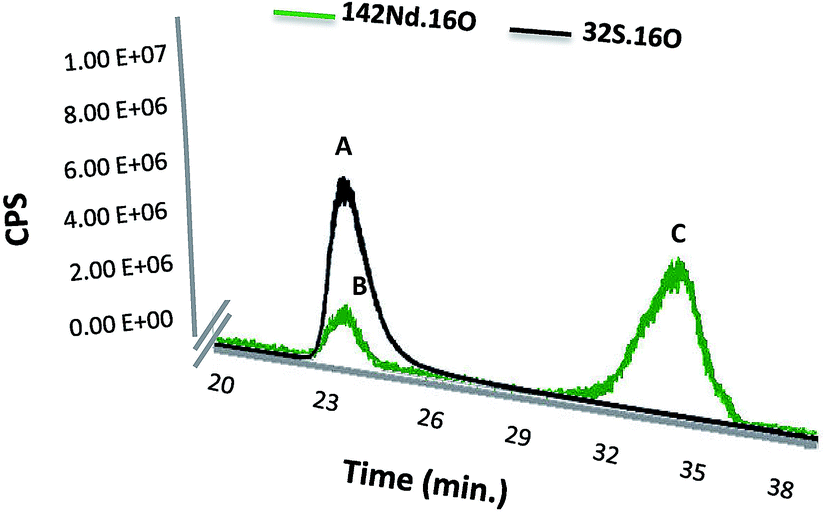 | ||
| Fig. 3 Chromatograms obtained by SEC-ICP-MS for labelled SA in HSA with Nd–DOTA–KE. Monitored isotopes were 142Nd and 32S where both were measured with the oxygen reaction mode. Monitored 32S16O represented in peak A is related to the total HSA in the sample, where monitored 142Nd16O represented in peak B is related to the labelled HSA with Nd–DOTA–KE, and finally peak C represents the excess of the labelling reagent. | ||
Quantification for labelled SA with isotopic dilution analysis (IDA-MS)
Quantification for the labelled SA with Nd–DOTA–KE was done using an isotopically enriched 145Nd isotopic standard. Optimum ratio between 142Nd standard and 145Nd spike was experimentally determined (Rm) 142Nd/145Nd in order to achieve the best precision for the measurement (see ESI Fig. S7†). Calculated Rm ratios (Table 2) were between 0.01–1. Post-column isotope dilution analysis of Nd (ID-ICP-MS) was carried out. For this purpose, a continuous flow of a 145Nd solution (5 ng mL−1) was constantly pumped (0.1 mL min−1) and mixed with the chromatography eluent. The mixed solution was then introduced into the ICP-MS.| Excess of H2O2 to free Cys-34 | Rm |
|---|---|
| Control | 0.054 |
| 2 excess | 0.046 |
| 5 excess | 0.372 |
| 8 excess | 0.545 |
| 10 excess | 0.078 |
This set-up was used to evaluate the formation of SA upon incubation with the oxidizing reagent and after labelling with Nd–DOTA–KE. Fig. 4 shows the quantitative results of SA formation upon exposure of 90 μM of albumin to 0 (control), 0.18 mM, 0.45 mM, 0.72 mM, and 0.9 mM H2O2 which corresponded to 0, 2-fold, 5-fold, 8-fold, and 10-fold excess with respect to the –SH. Labelling percentages were found to be 2.6%, 2.2%, 22.8%, 39.8%, and 4.1%, respectively.
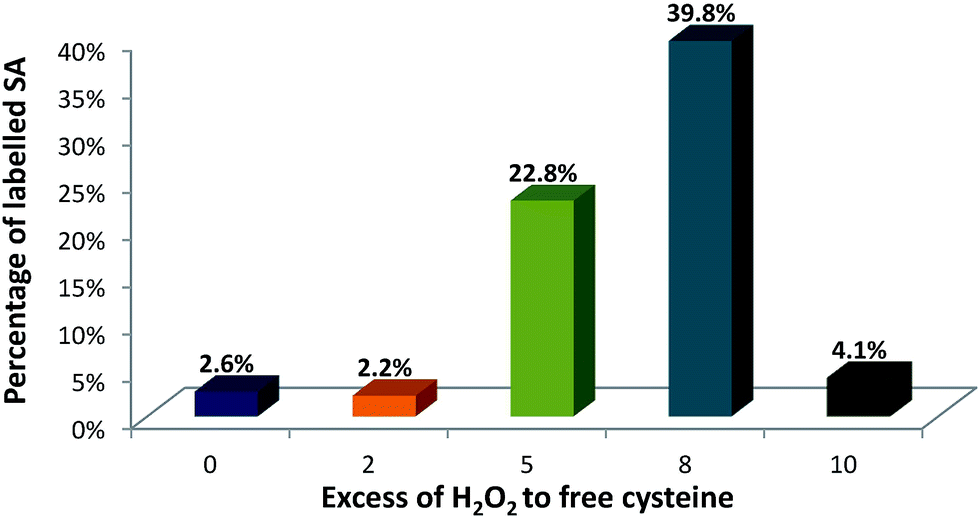 | ||
| Fig. 4 Percentage of labelled SA in HSA with Nd–DOTA–KE with different excess of hydrogen peroxide (H2O2) to free cysteine. Labelling was carried out with 90 μM of HSA, 8 M urea, and 30 excess of Nd–DOTA–KE in tris buffer, pH 8.4 for 4 hours at R.T. | ||
As a relatively stable label, SA can be spontaneously formed in HSA,19 and the control sample (without H2O2 addition) indicated some minor labelling percentage for SA. A similar percentage for SA labelling with Nd–DOTA–KE was detected for the 2-fold excess of H2O2, which might indicate that this concentration is too low for any further formation of SA above the spontaneously existing amount. Formed SA can undergo chemical reactions such as oxidation, reduction, and alkylation. Revised literature regarding the reactivity of initially formed SA from HSA revealed that once formed: (1) the accessibility of sulfenic acid (or bulk of the chemical probe) affects labelling efficiency, and/or (2) some environments may facilitate the chemical reaction between the probe and RSOH.28 In this regard, although there is no data available for the labelling reagent synthesized here, the reported data showed that the trapping of SA with dimedone (upon oxidation with H2O2) shows a constant rate of 0.025 M−1 s−1; meanwhile, the further reaction with H2O2 to form overoxidized species is of 0.4 M−1 s−1. Thus, considering a similar situation with the alkyne KE used here, only a small percentage of the formed SA can be trapped by the labelling probe. Since higher labelling percentages were observed upon the addition of 5-fold and 8-fold excess of H2O2, the high reactivity of the labelling probe for the SA in comparison to other reagents like dimedone seems to be proved here. The result of 10 mM could be ascribed to the excessive oxidation and formation of higher oxidation states such as sulfinic (–SO2H) and sulfonic (–SO3H) acids that cannot be labelled under the studied conditions. Therefore, 8-fold excess of H2O2 was found to be optimum for producing sufficient SA residues.
Conclusions
In this work we have presented a new methodology for sulfenic acid (SA) labelling and quantification in human serum albumin (HSA). Effective and selective labelling for SA was carried out using alkyne β-ketoester (KE) which was coupled to Ln–DOTA forming a new labelling reagent (Ln–DOTA–KE). With the use of the reagent (Ln–DOTA–KE), elemental and molecular mass spectrometry (MS) techniques were used to reveal the labelled SA in HSA in a complementary way.Using a poly-isotopic Ln, the prepared Ln–DOTA–KE allowed quantification of the detected SA using isotopic dilution analysis (IDA-ICP-MS) where an enriched tracer was used for the absolute quantification for labelled SA. Quantitative results can be achieved with the proposed strategy and SA formation in HSA isolated from serum turned out to be as high as 40% which shows the high reactivity of the labelling probe. This feature should permit the determination of smaller quantities of SA, even those spontaneously oxidized with atmospheric oxygen, in future work.
The straightforward methodology and specificity of the prepared reagent for SA detection suggest a promising future for the specific quantification of SA in real samples and tissues.
Conflicts of interest
The authors declare that they have no conflict of interest.Acknowledgements
This work was financially supported by the DFG (German Research Foundation). Sharar, M. acknowledges the SALSA graduate program (School of Analytical sciences in Adlershof) and Bundesanstalt fur Materialforschung und prufung (BAM) for funding through the scholarship.References
- R. A. Bradshaw and E. A. Dennis, Handbook of cell signaling, Academic Press, San Diego, California, 2003 Search PubMed
.
- C. Jacob, G. I. Giles, N. M. Giles and H. Sies, Angew. Chem., Int. Ed., 2003, 42, 4742–4758 CrossRef CAS PubMed
- C. Jacob, I. Knight and P. G. Winyard, Biol. Chem., 2006, 387, 1385–1397 CrossRef CAS PubMed
- C. E. Paulsen and K. S. Carroll, Chem. Rev., 2013, 113, 4633–4679 CrossRef CAS PubMed
- S. Carballal, R. Radi, M. C. Kirk, S. Barnes, B. A. Freeman and B. Alvarez, Biochemistry, 2003, 42, 9906–9914 CrossRef CAS PubMed
- U. Jacob and D. Reichmann, Oxidative stress and redox regulation, Springer, Dordrecht Heidelberg, New York, London, 2013 Search PubMed
- N. L. Anderson and N. G. Anderson, Electrophoresis, 1998, 19, 1853–1861 CrossRef CAS PubMed
- M. Mann and O. N. Jensen, Nat. Biotechnol., 2003, 21, 255–261 CrossRef CAS PubMed
- L. Turell, H. Botti, S. Carballal, R. Radi and B. Alvarez, J. Chromatogr., 2009, 877, 3384–3392 CAS
- K. G. Reddie and K. S. Carroll, Curr. Opin. Chem. Biol., 2008, 12, 746–754 CrossRef CAS PubMed
- W. S. Allison, Acc. Chem. Res., 1976, 9, 293–299 CrossRef CAS
- R. L. Charles, E. Schröder, G. May, P. Free, R. P. Gaffney, R. Wait, S. Begum, R. J. Heads and P. Eaton, Mol. Cell. Proteomics, 2007, 6, 1473–1484 CAS
- T. H. Poole, J. A. Reisz, W. Zhao, L. B. Poole, C. M. Furdui and S. B. King, J. Am. Chem. Soc., 2014, 136, 6167–6170 CrossRef CAS PubMed
- J. Qian, R. Wani, C. Klomsiri, L. B. Poole, A. W. Tsang and C. M. Furdui, Chem. Commun., 2012, 48, 4091–4093 RSC
- N. J. Kettenhofen and M. J. Wood, Chem. Res. Toxicol., 2010, 23, 1633–1646 CrossRef CAS PubMed
- A. H. El-Khatib, D. Esteban-Fernández and M. W. Linscheid, Anal. Chem., 2014, 86, 1943–1948 CrossRef CAS PubMed
- M. Sharar, E. M. Saied, M. C. Rodriguez, C. Arenz, M. Montes-Bayón and M. W. Linscheid, Anal. Bioanal. Chem., 2017, 409, 2015–2027 CrossRef CAS PubMed
- D. S. Rehder and C. R. Borges, Biochemistry, 2010, 49, 7748–7755 CrossRef CAS PubMed
- B. Alvarez, S. Carballal, L. Turell and R. Radi, Methods Enzymol., 2010, 473, 117–136 CAS
- T. Peters, All About Albumin: Biochemistry, Genetics, and Medical applications, Academic Press, San Diego, California, 1996 Search PubMed
- V. Hong, S. I. Presolski, C. Ma and M. G. Finn, Angew. Chem., Int. Ed., 2009, 48, 9879–9883 CrossRef CAS PubMed
- S. I. Presolski, V. P. Hong and M. G. Finn, Curr. Protoc. Chem. Biol., 2011, 3, 153–162 Search PubMed
- S. A. King, J. Armstrong and J. Keller, Org. Synth., 2009, 81, 178–187 Search PubMed
- J. Alonso and P. Gonzalez, Isotope Dilution Mass Spectrometry, Royal Society of Chemistry, Cambridge, 2013 Search PubMed
- J. D. Fassett and P. J. Paulsen, Anal. Chem., 1989, 61, 643A–649A CrossRef CAS
- U. K. Laemmli, Nature, 1970, 227, 680–685 CrossRef CAS PubMed
- UniProt, http://www.uniprot.org/blast/?about=P02768[25-609]%26key=Chain%26id=PRO_0000001068, accessed March 2017.
- V. Gupta and K. S. Carroll, Biochim. Biophys. Acta, Gen. Subj., 2014, 1840, 847–875 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra06241a |
| This journal is © The Royal Society of Chemistry 2017 |