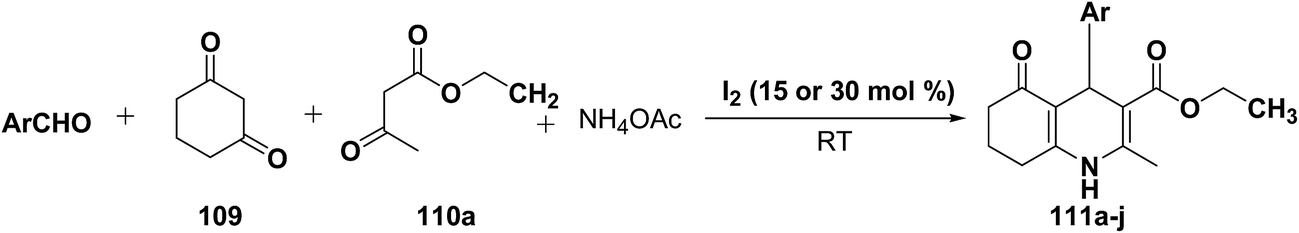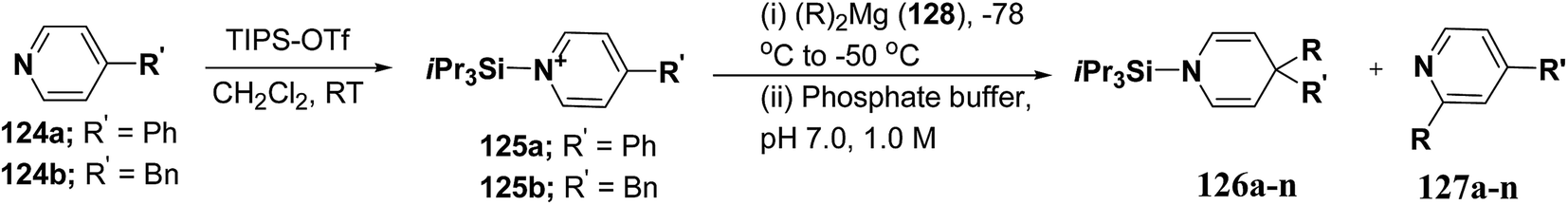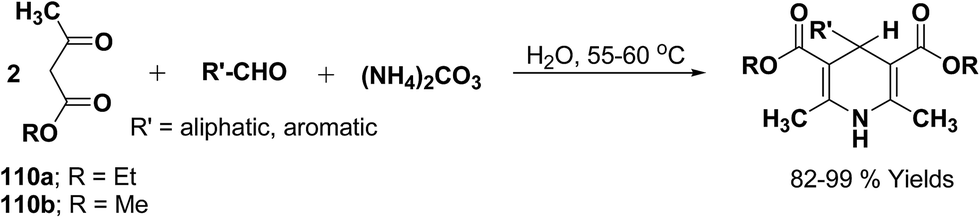DOI:
10.1039/C6RA24823C
(Review Article)
RSC Adv., 2017,
7, 2682-2732
Synthesis, utility and medicinal importance of 1,2- & 1,4-dihydropyridines
Received
6th October 2016
, Accepted 14th November 2016
First published on 12th January 2017
Abstract
Dihydropyridine (DHP) is among the most beneficial scaffolds that have revolutionised pharmaceutical research with unprecedented biological properties. Over the years, metamorphosis of easily accessible 1,2- and 1,4-dihydropyridine (1,4-DHP) intermediates by synthetic chemists has generated several drug molecules and natural products such as alkaloids. The 1,4-dihydropyridine (1,4-DHP) moiety itself is the main fulcrum of several approved drugs. The present review aims to collate the literature of 1,2- and the 1,4-DHPs relevant to synthetic and medicinal chemists. We will describe various methodologies that have been used for the synthesis of this class of compounds, including the strategies which can furnish enantiopure DHPs, either by asymmetric synthesis or by chiral resolution. We will also elaborate the significance of DHPs towards the synthesis of natural products of medicinal merit.
 Vivek K. Sharma | Vivek received his PhD degree in Chemistry in 2014 from University of Delhi, India. Currently he is working as Postdoctoral Fellow at RNA Therapeutics Institute, University of Massachusetts Medical School, USA. His current research interests include development of low-cost microfluidic gene synthesis, to improve in vivo delivery of peptide nucleic acid (PNA), and study the effect of neutral linkage between conformationally locked nucleotides for gene silencing applications. |
 Sunil K. Singh | Sunil K. Singh has obtained his M.Sc and PhD in Chemistry from University of Delhi. He is currently working as Assistant Professor in Chemistry Department at Kirori Mal College (A constituent college), University of Delhi. His current research works include multicomponent synthesis and biocatalysis. He has 17 research publications in his credit. |
1. Introduction
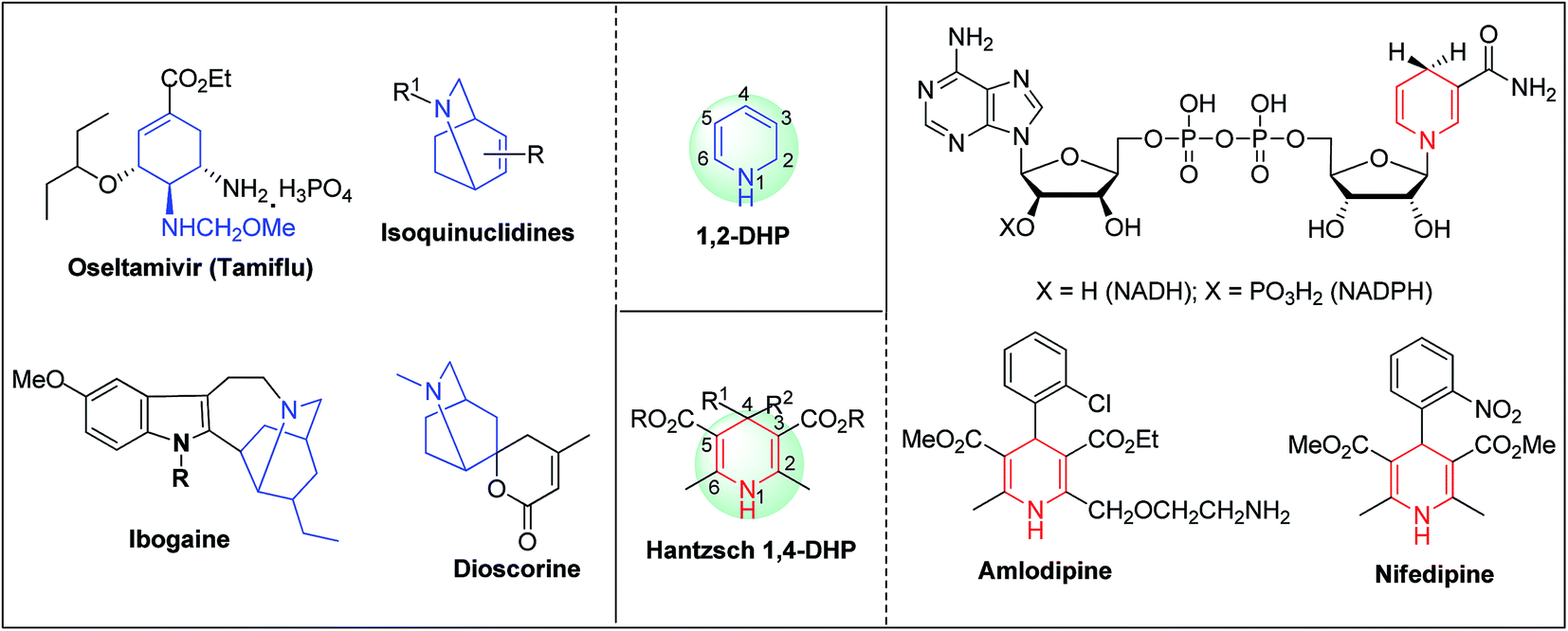
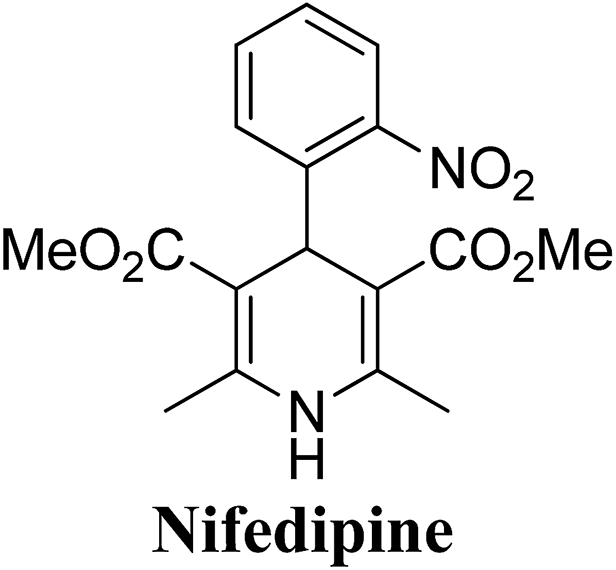
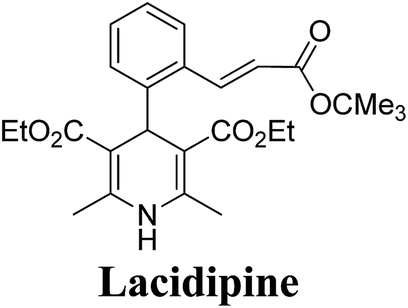
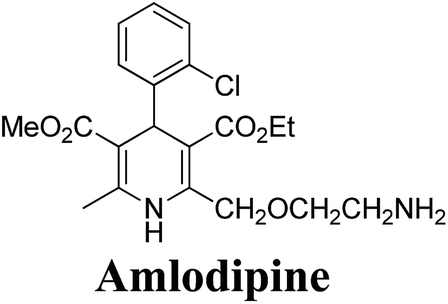
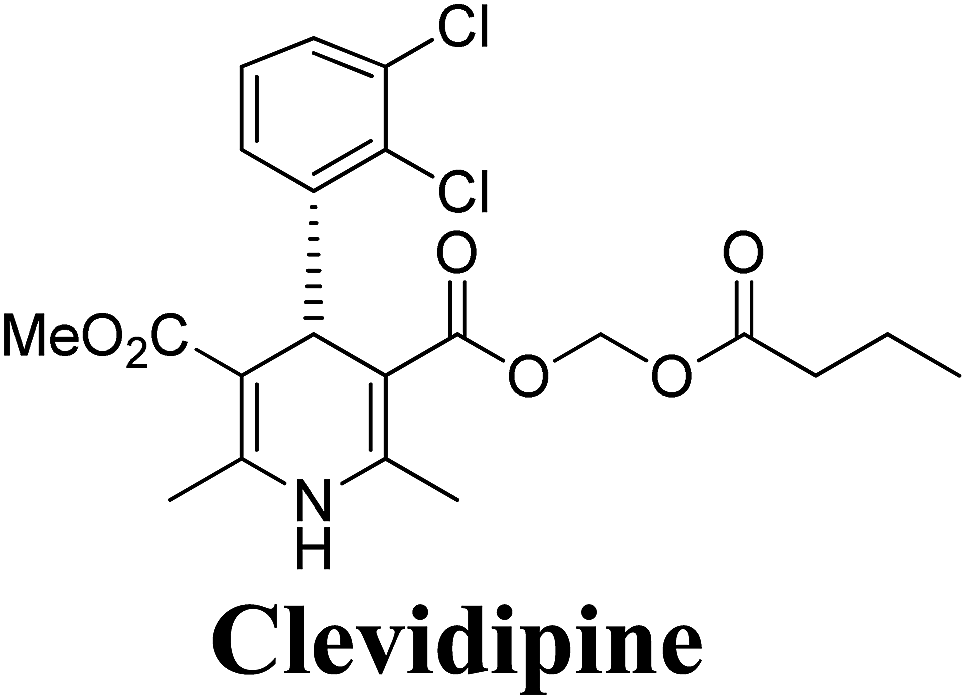
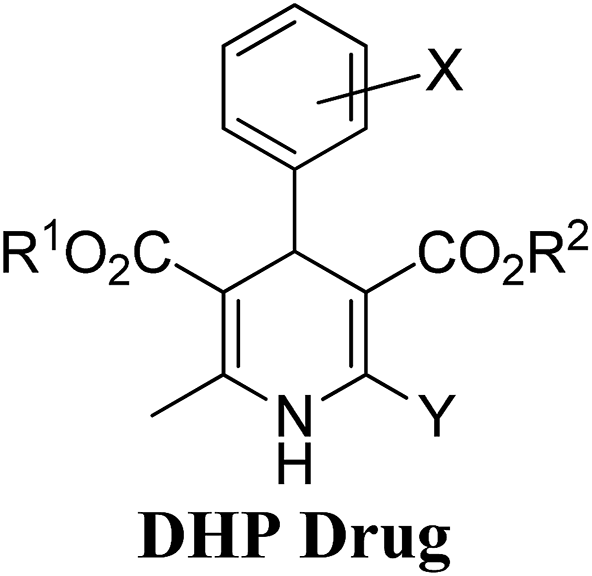
Arthur Hantzsch added one of the most valuable scaffolds to the toolbox of medicinal chemists, reporting the synthesis of dihydropyridine (DHP) in 1882. Among five possible regio-isomers only 1,2- and the 1,4-DHP (Fig. 1) have gained significant attention. The 1,4-DHP scaffold has served as a nucleus for several blockbuster drugs such as nifedipine and amlodipine.1 Close resemblance to nicotinamine adenine dinucleotide (NADH) coenzyme, which has an important role in biological oxidation–reduction reactions, has made the 1,4-DHP core even more lucrative. Perhaps less studied in the past, the potential of 1,2-dihydropyridines has recently been explored as a critical scaffold for the synthesis of alkaloids and other drugs. 1,2-DHPs are now popular as a precursor for the synthesis of the 2-azabicyclo[2.2.2]octanes (isoquinuclidines) ring system present in alkaloids, ibogaine and dioscorine. The anti-influenza drug, oseltamivir phosphate (Tamiflu), is also synthesised from 1,2-DHP via an isoquinuclidine intermediate (Fig. 1).2
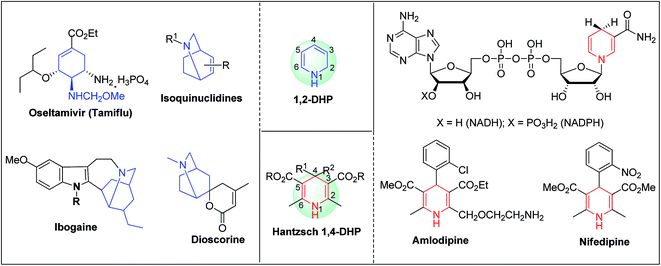 |
| | Fig. 1 Structures of 1,2-DHP, Hantzsch 1,4-DHP, NADH/NADPH and some medicinally important compounds/cores accessible from DHPs. | |
Recently, we reviewed reactions of 1,2- and the 1,4-dihydropyridines.3a The present manuscript aims to highlight the importance of 1,2- and the 1,4-dihydropyridines relevant to both synthetic and medicinal chemists. We will describe various methodologies that have been used for the synthesis of this class of compounds. We then focus on strategies which can furnish enantiopure DHPs, either by asymmetric synthesis or by chiral resolution. We will also spotlight the utility of DHPs towards the synthesis of natural products of medicinal merit. In 2002, Rodolfo Lavilla compiled the synthesis, reactivity, and applications of DHPs in medicinal chemistry.3b
2. Synthesis of dihydropyridines
Substituted dihydropyridine are usually prepared either by the cyclization reactions (Hantzsch ring closure) or by reduction of pyridinium ions. The reviews by Eisner and Kuthan,3c and Stout and Meyers3d covers the chemistry of DHPs from early development till 1982. Beside these, R. Lavilla3b in 2002 and Silva, et al.3e in 2013 also compiled the literature for the synthesis of DHPs.
2.1. Synthesis of 1,2-dihydropyridine
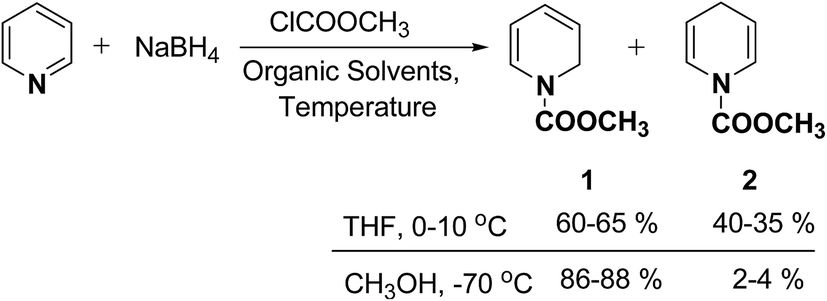
In 1972, Fowler et al.4 reported the synthesis of both N-carbomethoxy-1,2- and 1,4-dihydropyridine by treating a mixture of pyridine and sodium borohydride with methyl chloroformate in various organic solvents like, ether, glyme, THF, methanol and water. They observed that reaction in THF at 0 °C gave a mixture of dihydropyridines containing about 35–40% of 1,4-dihydropyridine along with the 1,2-dihydropyridine. However, the amount of 1,4-isomer can be reduced to 2–4% by performing the reaction in methanol at −70 °C giving 1,2-DHP in 86–88% yields (Scheme 1).
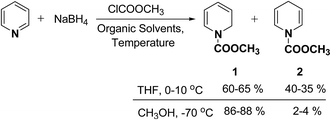 |
| | Scheme 1 Synthesis of 1,2- and 1,4-dihydropyridines 1 and 2 by reduction of pyridine using NaBH4. | |
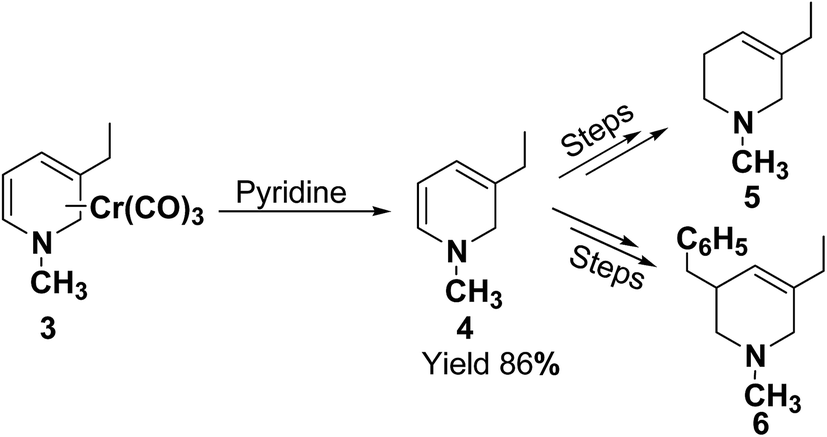
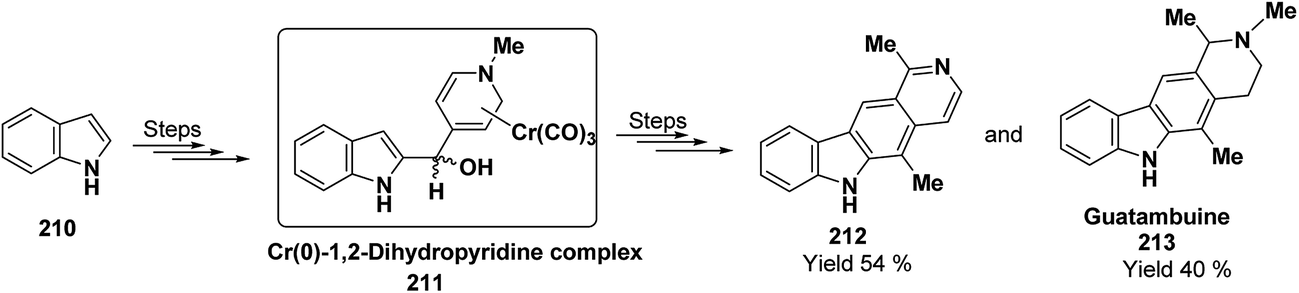
Kutney, et al.5 have described the synthesis of novel and stable chromium complex of N-methyl-3-ethyl-1,2-dihydropyridine. This metal carbonyl complex 3 has been used for the synthesis of essentially pure N-methyl-3-ethyl-1,2-dihydropyridine 4, which can be used as a precursor to synthesize a number of useful heterocyclic compounds such as 5 and 6 (Scheme 2).
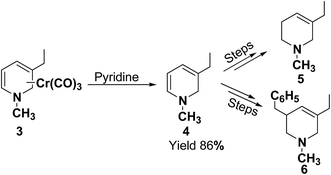 |
| | Scheme 2 Dihydropyridine 4 generated from precursor chromium complex 3. | |
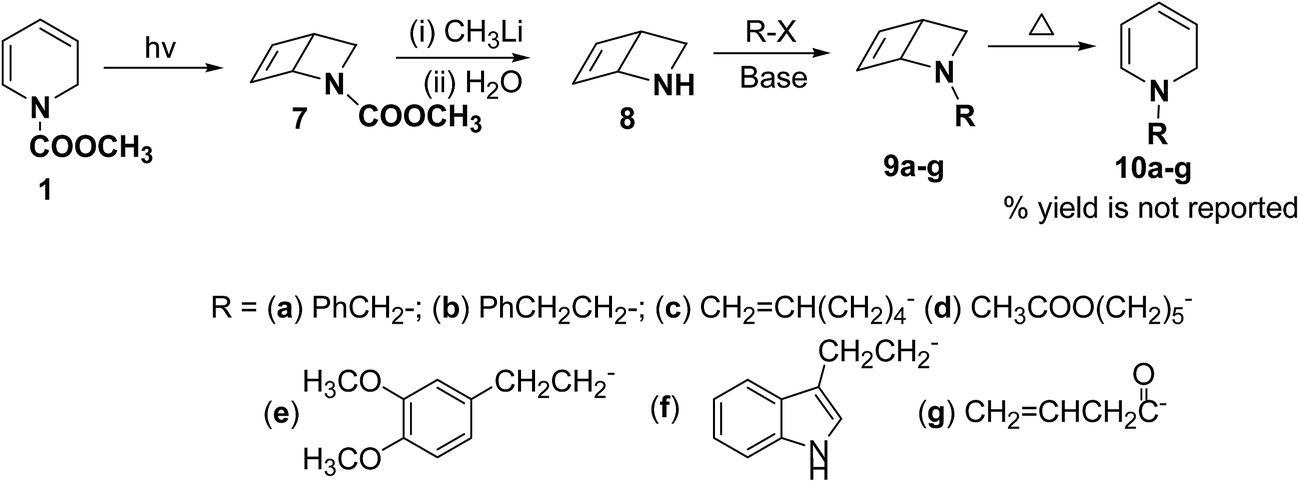
Beeken, et al.6 have developed a synthetic methodology for the synthesis of 1,2-dihydropyridines 10a–g via 2-azabicyclo[2.2.0]hex-5-ene 8 as a synthetic equivalent. The amine 8 was conveniently synthesized by methyl lithium mediated hydrolysis of carbamate derivative of 1,2-dihydropyridine 7 which in turn was photochemically synthesized from compound 1. Alkylation and acylation of amine 8 has lead to the formation of 9a–g which were easily isolated. The alkylated/acylated analogues 9a–g were converted into the corresponding 1,2-dihydropyridines 10a–g under thermal heating condition (Scheme 3).
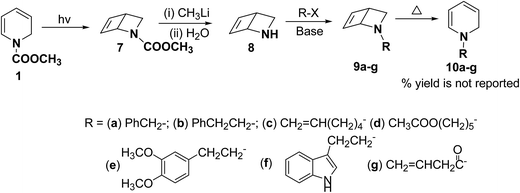 |
| | Scheme 3 Synthesis of N-substituted 1,2-dihydropyridines 10a–g. | |
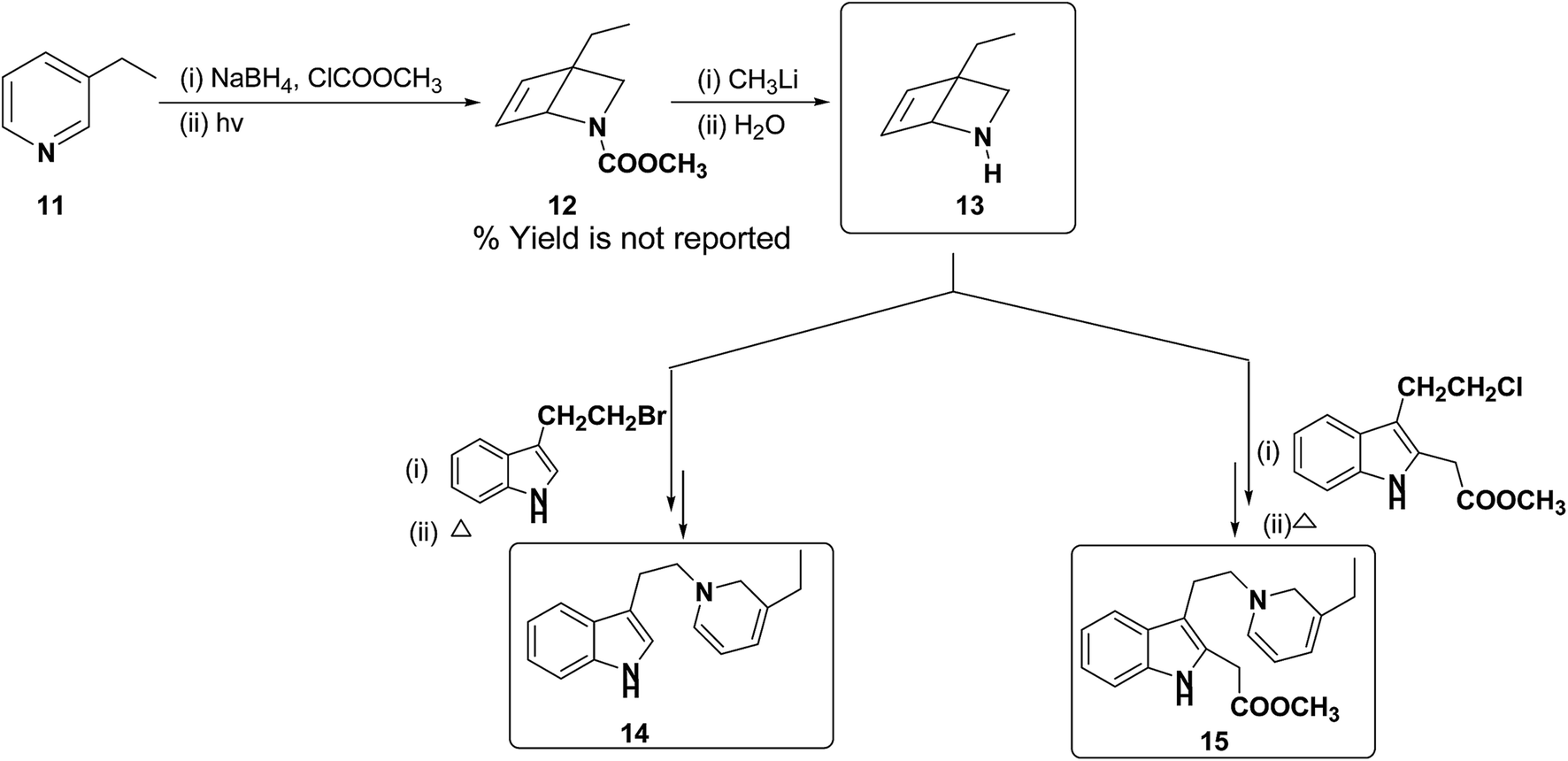
Further, Fowler et al. have successfully prepared dihydropyridines 14 and 15 from 3-ethyl pyridine 11 (Scheme 4). 3-Ethyl pyridine 11 was first converted into N-carboxymethyl amine 12 using ClCOOCH3 and sodium borohydride, which on hydrolysis yield amine 13. This method is significantly important as it can be applied to relatively more complex 1,2-DHP structures which may serve as an intermediate for the synthesis of alkaloids, e.g. compound 15 is an intermediate in a biomimetic synthesis of Strychnos alkaloids.
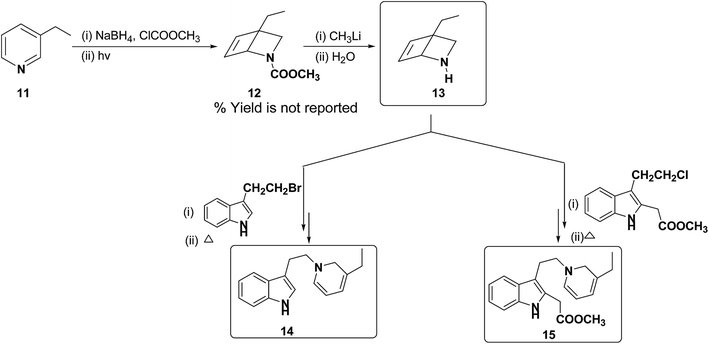 |
| | Scheme 4 Synthesis of 1,2-dihydropyridines 14 and 15 from 3-ethyl pyridine 11. | |
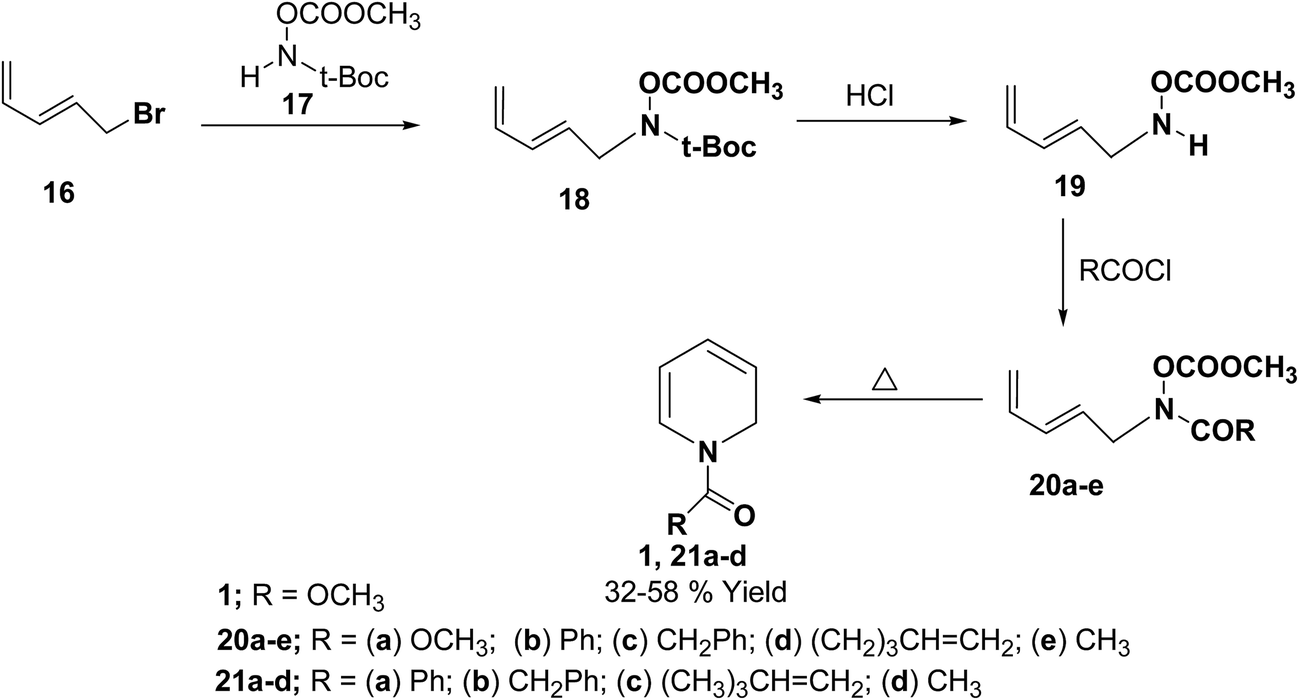
Fowler, et al.7 has synthesized the 1,2-dihydropytidines 1, 21a–d by the thermal cyclisation of hydroxamic acid esters 20. Reaction of the hydroxamic acid ester 17 with 5-bromopenta-1,3-diene 16 gave the compound 18. The removal of t-Boc protecting group from compound 18 followed by acylation with various acid chlorides gave the hydroxamic acid derivatives 20. Evaporation of these hydroxamic acid derivatives through a hot tube gave the 1,2-dihydropyridines 1, 21a–d as the only isolable products in 32 to 58% yields (Scheme 5).
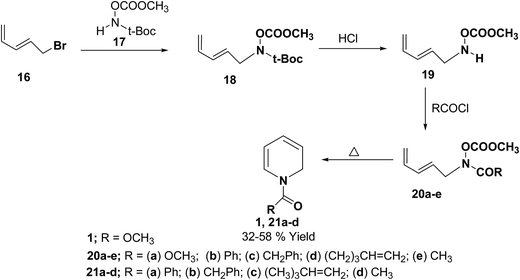 |
| | Scheme 5 Synthesis of 1,2-dihydropyridines 1, 21a–d by thermal cyclisation of hydroxamic acid derivatives. | |
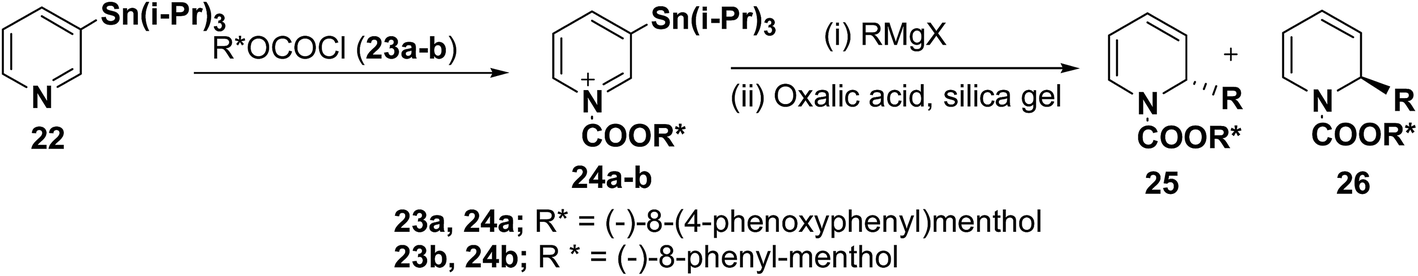
2-Substituted-1,2-dihydropyridine analogues are useful intermediate for the synthesis of natural products such as piperidine, indolizidine, quinolizidine and cis-decahydroquinoline alkaloids. Comins, et al.8 have reported the asymmetric synthesis of 2-substituted-1,2-dihydropyridines 25 & 26 from N-acyl pyridinium salt 24, which was synthesized via the addition of 3-(triisopropylstannyl)pyridine 22 to an enantiopure chloroformate 23a–b derived from (−)-8-(4-phenoxyphenyl)menthol or (−)-8-phenyl-menthol. The reaction of N-acyl pyridinium salt 24 with different Grignard reagents followed by treatment of the reaction mixture with silica gel containing oxalic acid provided chiral 1,2-dihydropyridines 25 and 26 (Scheme 6, Table 1).
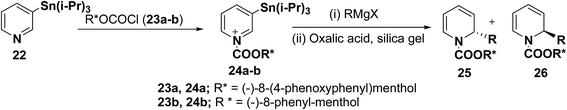 |
| | Scheme 6 Asymmetric synthesis of 1-acyl-α-alkyl-1,2-dihydropyridines 25 and 26. | |
Table 1 Synthesis of 1-acyl-2-alkyl-1,2-dihydropyridines 25 and 26
| Entry |
R*OCOCl |
RMgX |
Product |
Yield% |
de |
| 1 |
23a |
n-PrMgCl |
25a |
72 |
82 |
| 2 |
23b |
n-PrMgCl |
25b |
81 |
78 |
| 3 |
23a |
n-HexMgCl |
25c |
81 |
91 |
| 4 |
23a |
PhCH2MgCl |
25d |
58 |
76 |
| 5 |
23a |
VinylMgBr |
25e |
71 |
90 |
| 6 |
23a |
PhMgCl |
25f |
85 |
89 |
| 7 |
23b |
PhMgCl |
25g |
87 |
84 |
| 8 |
23a |
p-MePhMgBr |
25h |
86 |
92 |
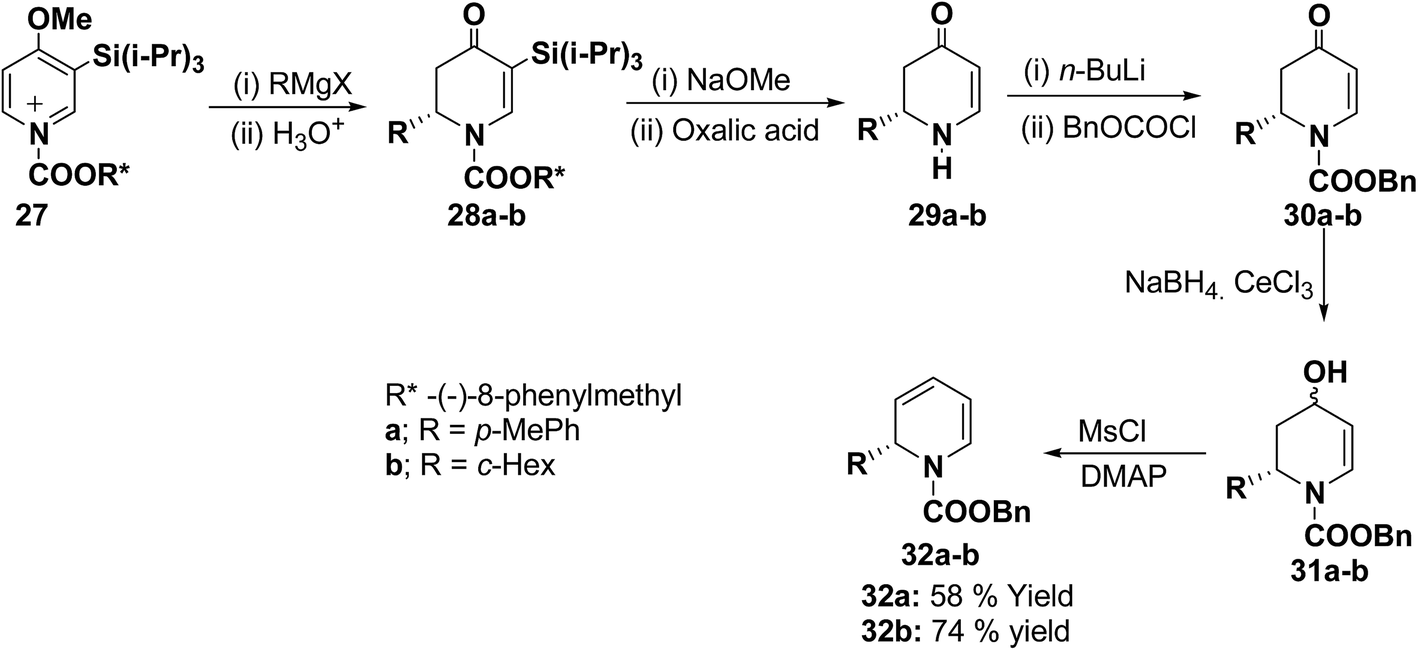
The presence of the chiral auxiliary in 25 may find application in the several transformations. Another asymmetric synthesis was developed for the synthesis of 1,2-dihydropyridines as shown in Scheme 7. The chiral salt 27 prepared in situ from 4-methoxy-3-(triisopropylsilyl)pyridine and (−)-8-phenylmethyl chloroformate, was treated with Grignard reagent (RMgX) to give tetrahydropyridone 28a–b in high yield and high diastereomeric excess (de). The chiral auxiliary and the triisopropyl silyl groups were removed from purified diastereomers 28 with sodium methoxide/methanol and oxalic acid to give enantiopure dihydropyridones 29a (88% yield) and 29b (81% yield) via one pot reaction. Deprotonation of 29 with n-BuLi and addition of benzyl chloroformate provided 30 in almost quantitative yield. Reduction of 30 to alcohols and subsequent dehydration gave the desired enantiomerically pure 1-(benzyloxycarbonyl)-1,2-dihydropyridine 32a and 32b in 58 and 74% yields, respectively (Scheme 7).
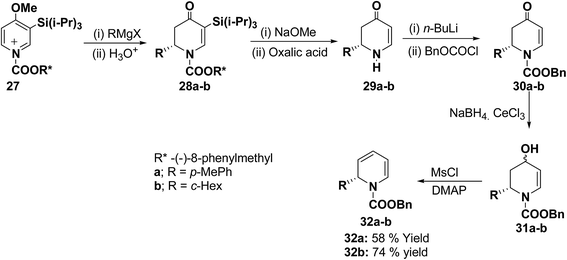 |
| | Scheme 7 A highly enantioselective synthesis of 1,2-dihydropyridines 32a–b. | |
Donohoe, et al.9 has used Birch conditions or sodium naphthalene in THF for the partial reduction of electron deficient pyridines for the synthesis of 1,2-dihydropyridines 34. Reductive alkylation of electron deficient pyridine diester 33 under Birch type conditions (Scheme 8, method A, quenching the reaction with an electrophile followed by a proton source) gave excellent yield of the corresponding monoalkylated dihydropyridine 34. The partial reduction using sodium and naphthalene in THF (Scheme 8, method B) was also accomplished, thus avoiding the use of liquid ammonia. Moreover, both sets of reducing conditions were compatible with a range of electrophiles (Table 2).
 |
| | Scheme 8 Reduction of electron deficient pyridine 33: preparation of 1,2-dihydropyridines 34. | |
Table 2 Reduction of pyridine 33 by using method A or B and their alkylationa
| Entry |
R–X |
R |
Method |
Yield (%) |
| Method A: Na (3.5 equiv.), NH3/THF, −78 °C, then R–X (3.5 equiv.), then NH4Cl after 5–30 s (time delay depends upon the electrophile). Method B: Na (3.5 equiv.), naphthalene (5 equiv.), THF, −78 °C, then R–X (3.5 equiv.), then NH4Cl after 5–30 s (time delay depends upon the electrophile). b = formed as a mixture of diastereomers. c = this compound will aromatize in approximately 24 h at room temperature and should be stored in the freezer. |
| 1 |
MeI |
Me |
A |
99 |
| 2 |
MeI |
Me |
B |
86 |
| 3 |
EtI |
Et |
A |
98 |
| 4 |
EtI |
Et |
B |
78 |
| 5 |
iBuI |
iBu |
A |
93 |
| 6 |
Epibromohydrin |
 |
B |
84 |
| 7 |
Allyl-Br |
Allylc |
A |
90 |
| 8 |
I(CH2)3Cl |
(CH2)3Cl |
A |
96 |
| 9 |
I(CH2)3Cl |
(CH2)3Cl |
B |
96 |
| 10 |
I(CH2)4Cl |
(CH2)4Cl |
A |
99 |
| 11 |
I(CH2)4Cl |
(CH2)4Cl |
B |
91 |
| 12 |
I(CH2)5Cl |
(CH2)5Cl |
A |
97 |
| 13 |
I(CH2)5Cl |
(CH2)5Cl |
B |
94 |
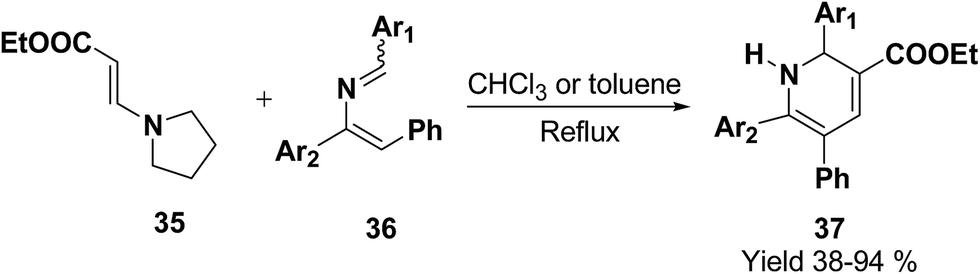
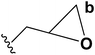
Palacios, et al.10 has reported the synthesis of 1,2-dihydropyridine 37 using enamines 35 and 2-azadienes 36 (readily prepared by aza-Wittig reactions). The compounds and 36 under reflux in toluene form a broad variety of substituted dihydropyridines 37 in a regiospecific manner (Scheme 9).
 |
| | Scheme 9 Regioselective synthesis of 1,2-dihydropyridine. | |
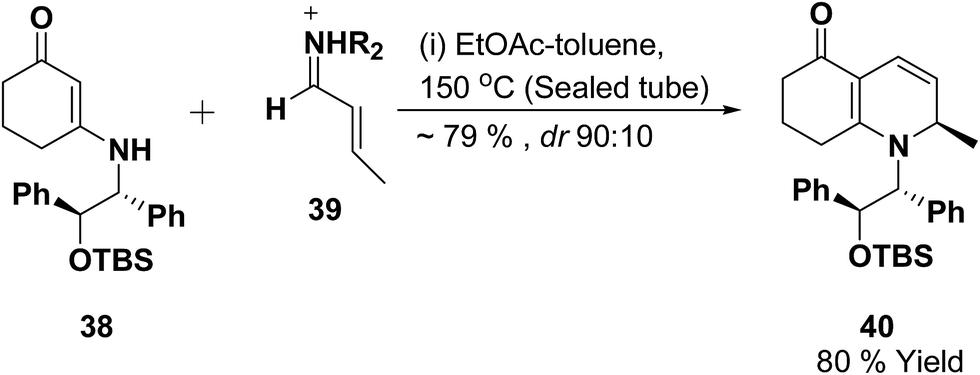
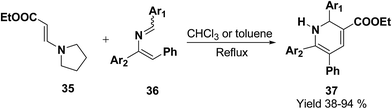
Vinylogous amides 38 undergo [3 + 3] cycloaddition reaction with α,β-unsaturated iminium ions 39 to yield 1,2-dihydropyridines 40. Intramolecular and stereoselective versions of this process have been successfully developed (Scheme 10).11
 |
| | Scheme 10 1,2-Dihydropyridine via [3 + 3] cycloaddition reaction. | |
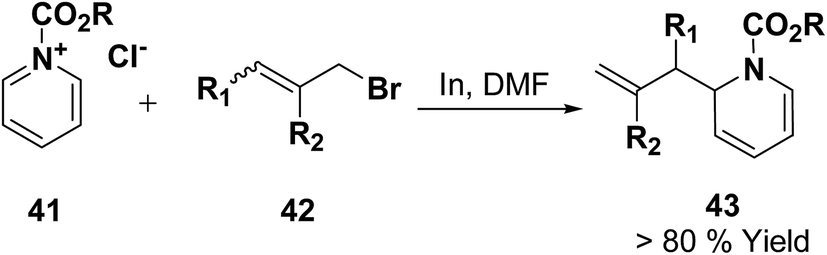
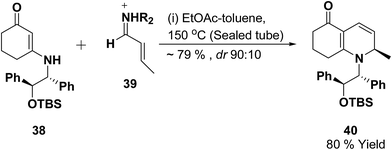
Loh, et al.12 has reported the indium-promoted allylations of N-acetylpyridinium salt 41 with allyl bromide 42 in DMF, to furnish 1,2-dihydropyridines 43 regioselectively in good yields (Scheme 11).
 |
| | Scheme 11 Indium catalysed synthesis of 1,2-dihydropyridine. | |
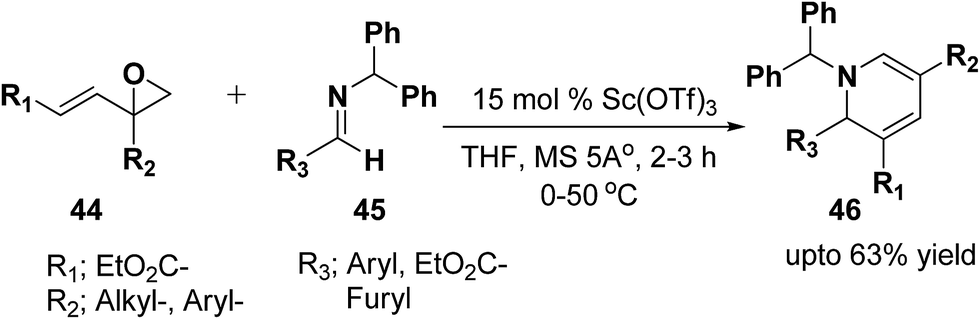
Brunner, et al.13 has synthesized the functionalized 1,2-dihydropyridine in moderate to good yield by using vinyloxiranes as dienolates in imino-aldol reactions. Under the optimized reaction condition the reaction of vinylepoxide 44 (1.5 equiv.) with benzhydril protected aldimine 45 (1.0 equiv.) in the presence of catalytic amount of Sc(OTf)3 (15.0 mmol) lead to the formation of stable 1,2-dihydropyridine 46 in upto 63% yield (Scheme 12). Further it has been illustrated by the author that the reaction proceeds via the vinylogous imino-aldol type reaction.
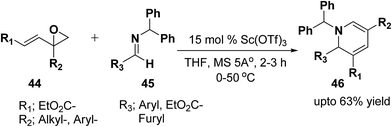 |
| | Scheme 12 Vinylogous imino-aldol reaction with vinyloxiranes: synthesis if 1,2-dihydropyridines. | |
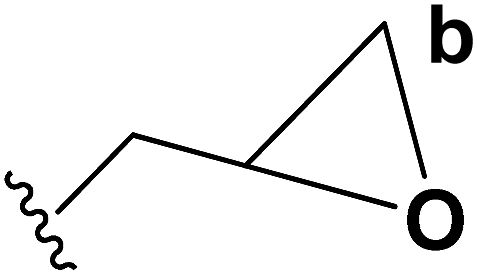
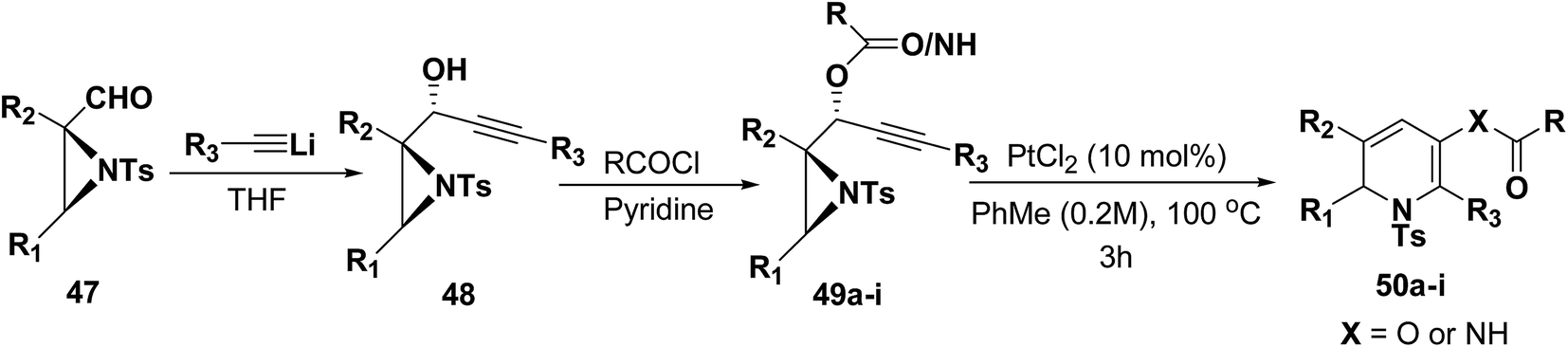
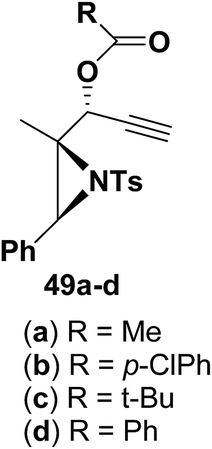
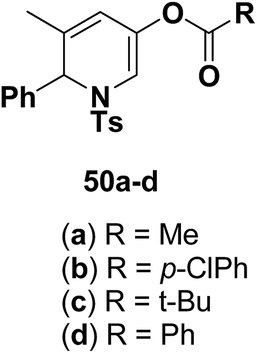
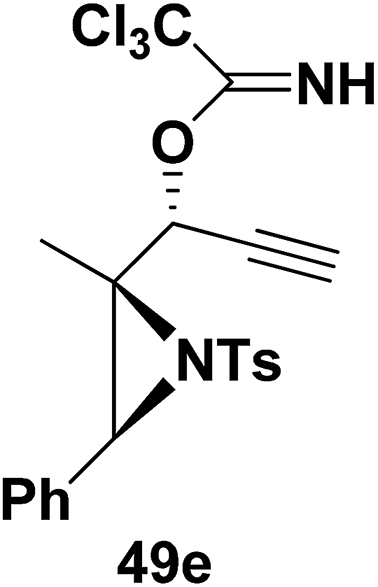
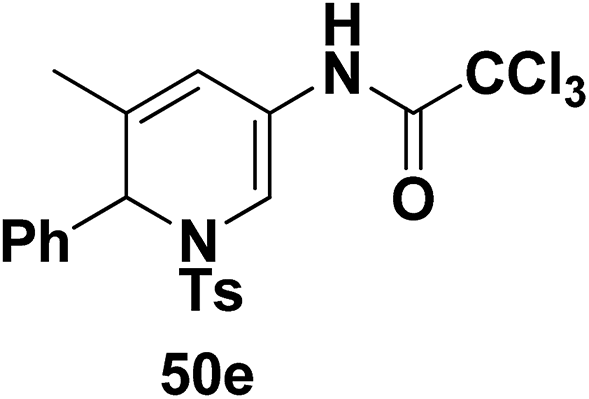
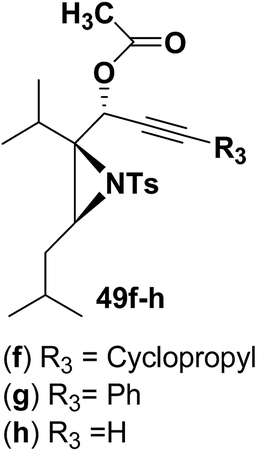
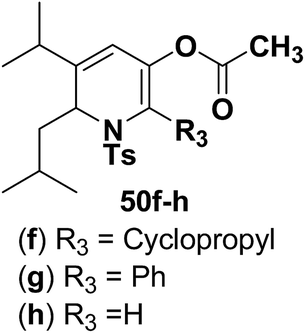
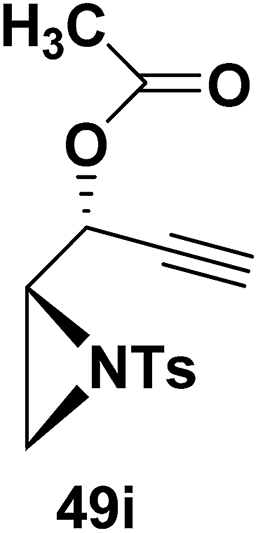
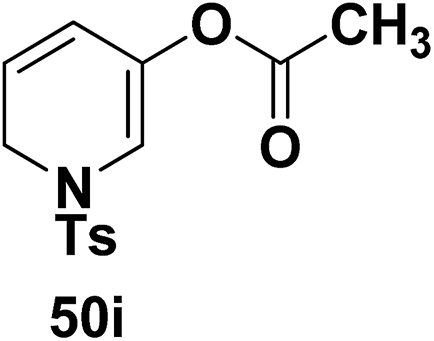
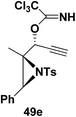
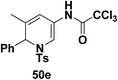
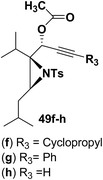
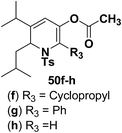
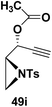
Motamed, et al.14 has reported the first Pt(II) catalyzed cycloisomerisation of aziridinyl propargylic esters 49a–i to afford 1,2-dihydropyridines 50a–i (Scheme 13). The aziridinyl propargylic ester substrates (Table 3) were prepared by acylation of the corresponding aziridine propargylic alcohols 48, which were synthesized from aziridinyl aldehyde 47 via a highly diastereoselective (dr > 95%) 1,2-addition of the corresponding alkyllithium or Grignard reagent. The aziridinyl propargylic esters 49a–i on cycloisomerisation using Pt(II) catalysis (10 mol% of PtCl2, 0.2 M in toluene, 100 °C, 3 h) afforded the corresponding 1,2-DHP products 50a–i in moderate to good yields (Table 3).
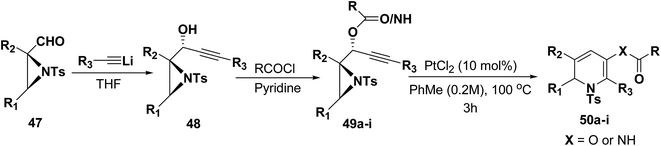 |
| | Scheme 13 Pt(II) catalyzed synthesis of 1,2-dihydropyridines. | |
Table 3 Pt(II)-Catalyzed synthesis of 1,2-dihydropyridinesa
| Entry |
Substrate |
Product |
Yieldb (%) |
| Standard conditions: 10 mol% of PtCl2, 0.2 M in toluene at 100 °C over 3 h. Isolated yields after column chromatography. |
| 1 |
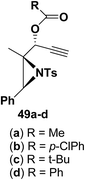 |
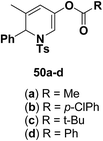 |
(50a) 76%, (50b) 72%, (50c) 76%, (50d) 76% |
| 2 |
 |
 |
(50e) 56% |
| 3 |
 |
 |
(50f) 62%, (50g) 69%, (50h) 70% |
| 4 |
 |
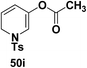 |
(50i) 76% |
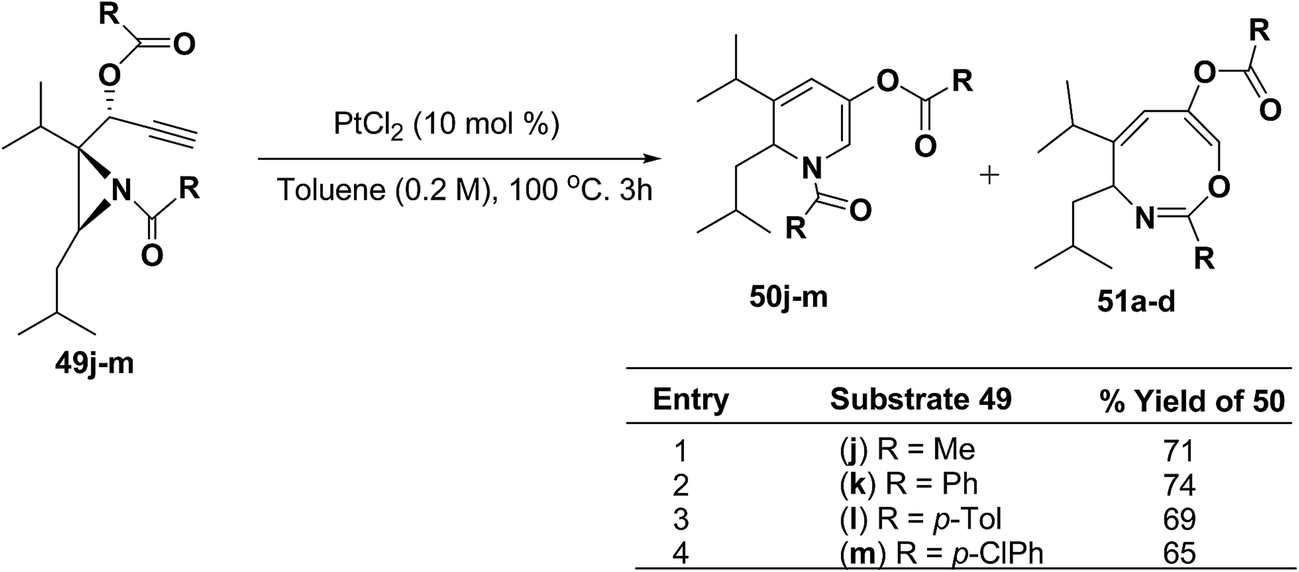
To explore the scope of the substrates that participate in the reaction, other substituents at the aziridine nitrogen were explored. The N-acyl substrates 49j–m (Scheme 14) were subjected to the Pt(II) catalyzed reaction to afford the 1,2-dihydropyridines 50j–m in 65–74% yield along with the another heterocycle 51a–d as by product, which were not isolated.
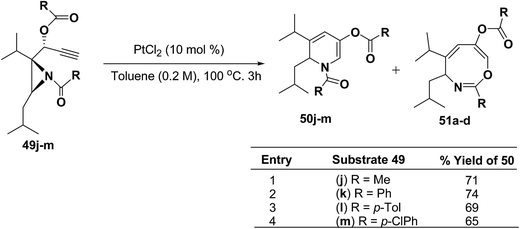 |
| | Scheme 14 Pt(II)-Catalyzed cycloisomerization of acylated aziridines substrates to 1,2-dihydropyridines. | |
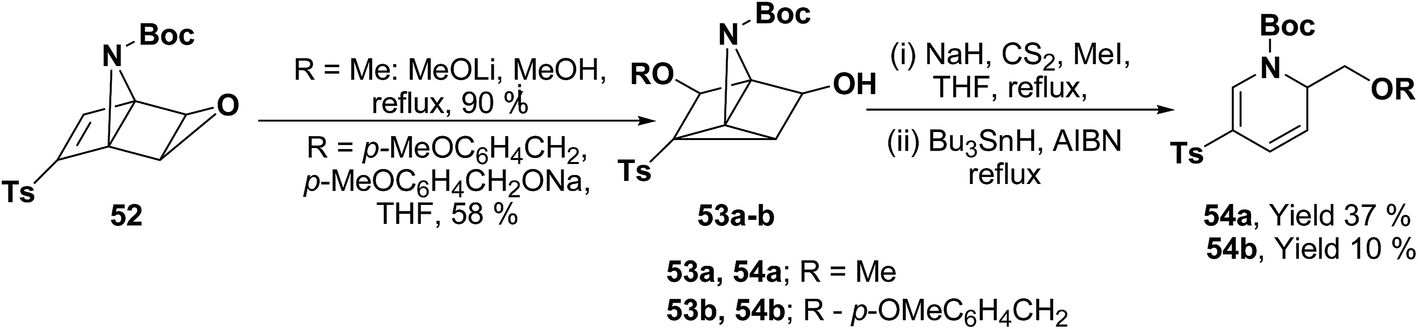
Hodgson, et al.15 have developed a new methodology of radical deoxygenation process on 3-azatricyclo[2.2.1.02,6]heptan-5-ols 53 using tributyltin hydride to afford the 1,2-dihydropyridines. 3-Azatricyclo[2.2.1.02,6]heptan-5-ols 54a–b were synthesized from the corresponding epoxide 52 (readily available from cycloaddition of the N-Boc pyrrole and tosyl ethyne, followed by the epoxidation of the resulting bicyclic diene), which on radical deoxygenation (via the xanthate) using Bu3SnH (2.0 equiv.) gave the 1,2-dihydropyridines 54a–b in 37 and 10% yields, respectively (Scheme 15).
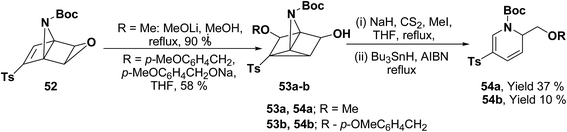 |
| | Scheme 15 Bu3SnH catalyzed radical deoxygenation on xanthate: synthesis of 1,2-dihydropyridines. | |
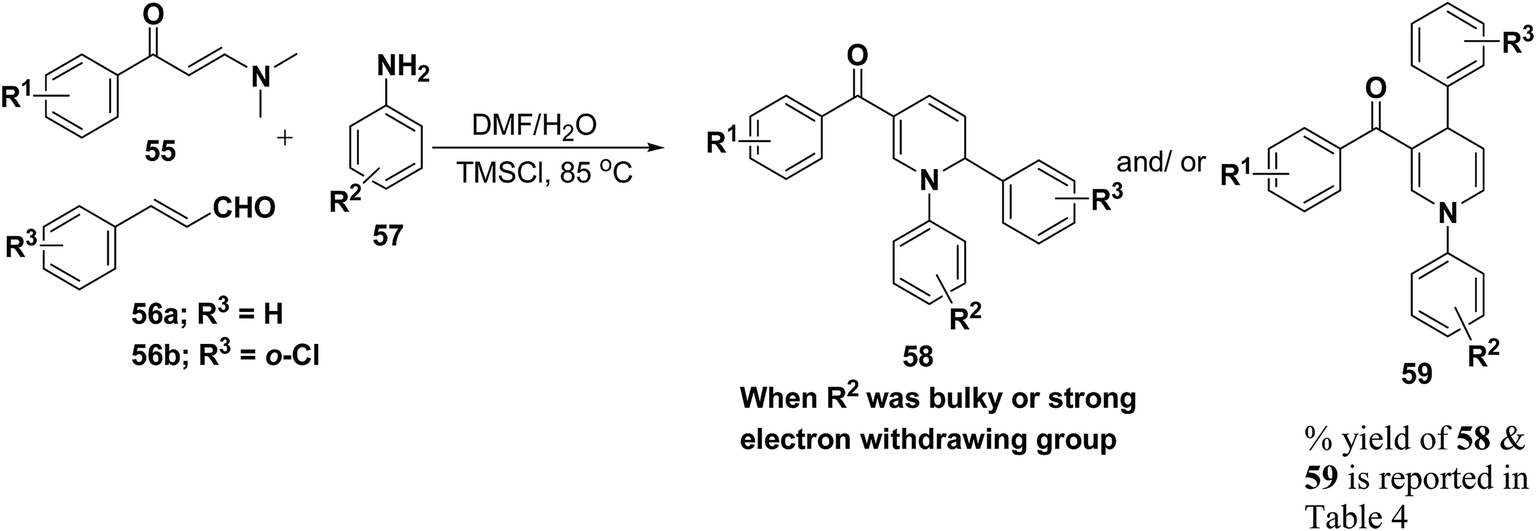
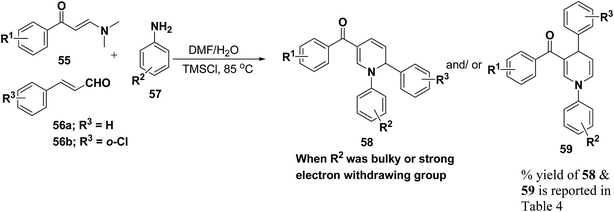
Wan, et al.16 have reported the three component sequential reaction enaminones 55, α,β-unsaturated aldehydes 56a–b and amines 57a–k to afford 1,2-dihydropyridines 58 in highly regioselective manner. This novel regioselectivity has been assigned to both steric and electronic effects originating from the amine partner (Scheme 16).
 |
| | Scheme 16 Regioselective synthesis of 1,2-dihydropyridines. | |
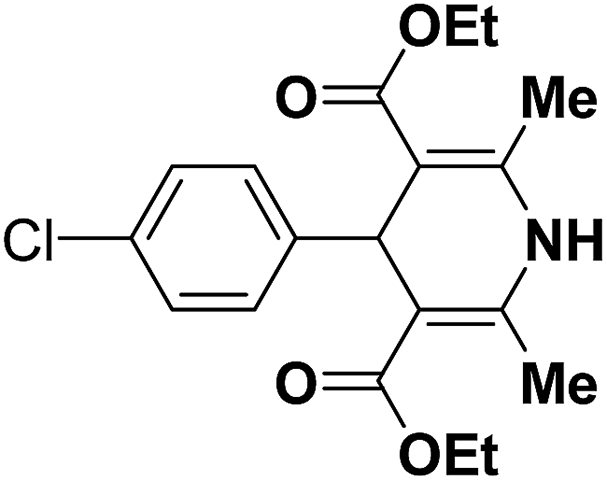
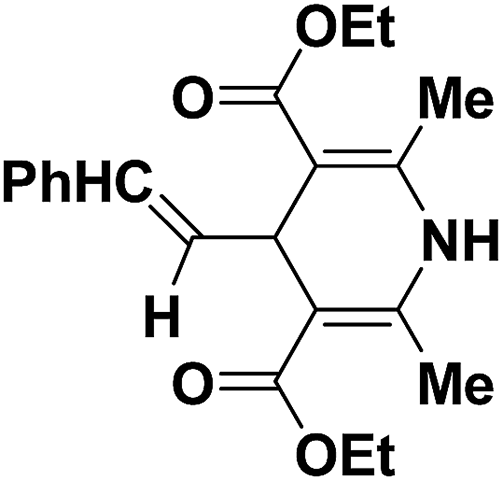
To examine the factors contributing to the selectivity, a series of reactions by taking p-nitroaniline and ortho-substituted anilines, have been carried out with enaminones and α,β-unsaturated aldehydes. The results are highlighted in Table 4. As shown in Table 4 both 1,2-DHPs 58 and/or 1,4-DHPs 59 could be furnished depending on the properties of the substituent on phenyl ring of the anilines. p-Nitroaniline mainly offered 1,2-DHPs 58 as a major product, while with different ortho-anilines indicate that the regioselectivity was affected by both the size and the electronic profiles of the ortho groups. The reaction of o-haloanilines, fluoro- and bromoaniline gave 1,4-DHP 59c, 59d and 59h as the major product (entries 3, 4, and 8), while o-chloroaniline gave 1,2-DHP 58e, 58f and 58g as the only isolated product (entries 5–7). Integrating the 1,2- and/or 1,4-DHP furnished in the 2-iodoaniline, 2-methylaniline, and 2,4,6-trimethylaniline entries (entries 9–11), it is evident that the bulky ortho group is important, but not the only inducing factor for 1,2-DHP formation. Further it was observed that the sequence of adding the reactants did not make a visible difference in the regioselectivity of the reactions.
Table 4 Regioselective synthesis of 1,2-DHPs and 1,4-DHPsa
| Entry |
R1 |
R3 |
R2 (57a–k) |
% yieldb of 58 |
% yieldb of 59 |
| General conditions: 0.3 mmol of enaminones, 0.35 mmol of aldehyde, 0.3 mmol of amine, 0.3 mmol of TMSCl mixed in 1 mL of solvents (0.5 mL H2O + 0.5 mL of DMF), stirred at 85 °C for 10 h. Isolated yield based on the corresponding enaminone. nd = not determined. |
| 1 |
H |
H |
(57a) p-NO2 |
(58a) 63 |
nd |
| 2 |
p-OCH3 |
H |
(57a) p-NO2 |
(58b) 14 |
(59c) 53 |
| 3 |
H |
H |
(57b) o-F |
(58c) ndc |
(59c) 70 |
| 4 |
p-OCH3 |
H |
(57b) o-F |
(58d) nd |
(59d) 52 |
| 5 |
H |
H |
(57c) o-Cl |
(58e) 59 |
(59e) nd |
| 6 |
o-Cl |
o-Cl |
(57d) H |
(58f) 52 |
(59f) nd |
| 7 |
o-Cl |
H |
(57c) o-Cl |
(58g) 60 |
(59g) nd |
| 8 |
H |
H |
(57e) o-Br |
(58h) <10 |
(59h) 46 |
| 9 |
H |
H |
(57d) o-I |
(58i) 27 |
(59i) 51 |
| 10 |
H |
H |
(57g) o-CH3 |
(58j) nd |
(59j) 65 |
| 11 |
H |
H |
(57h) 2,4,6-trimethyl |
(58k) <10 |
(59k) 51 |
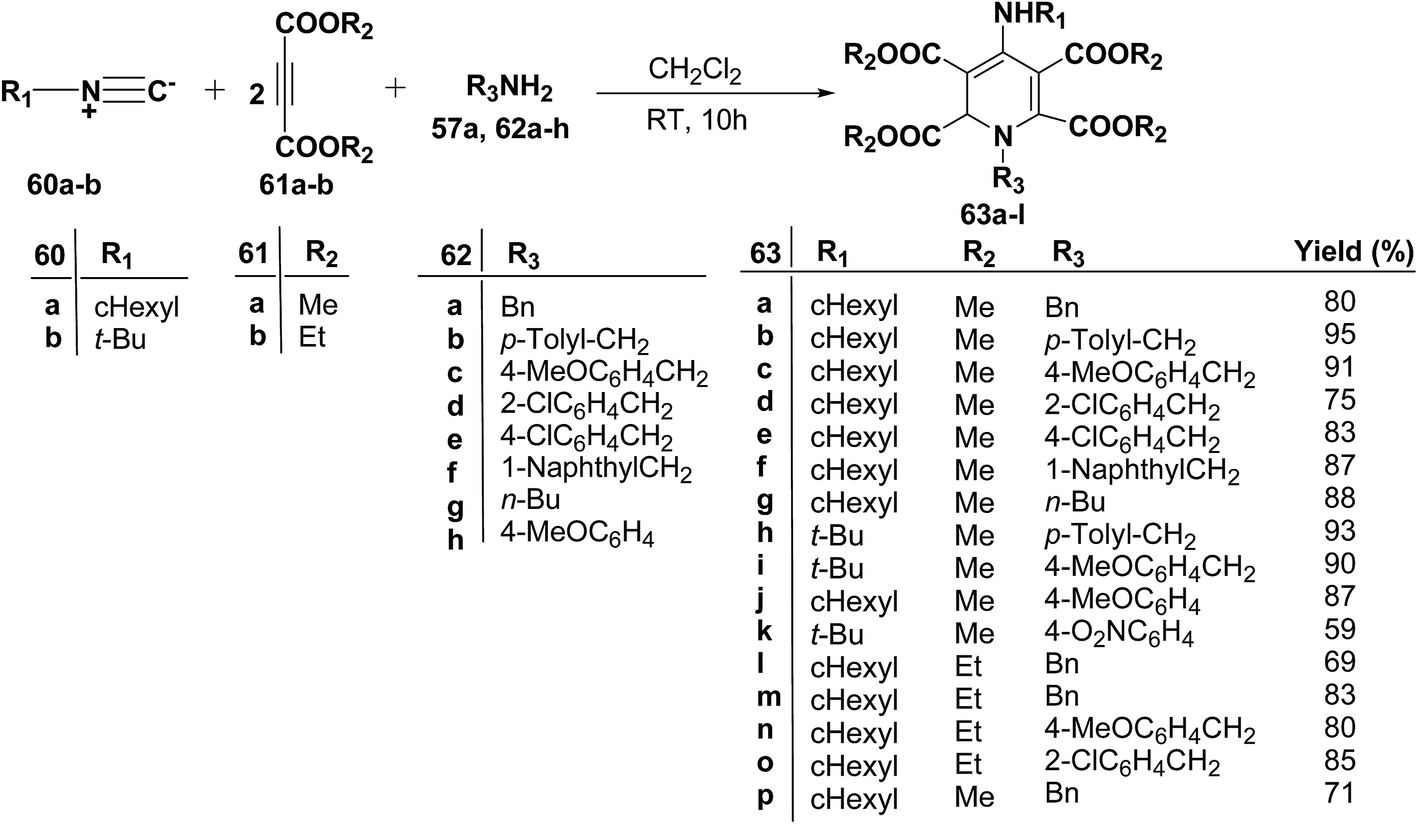
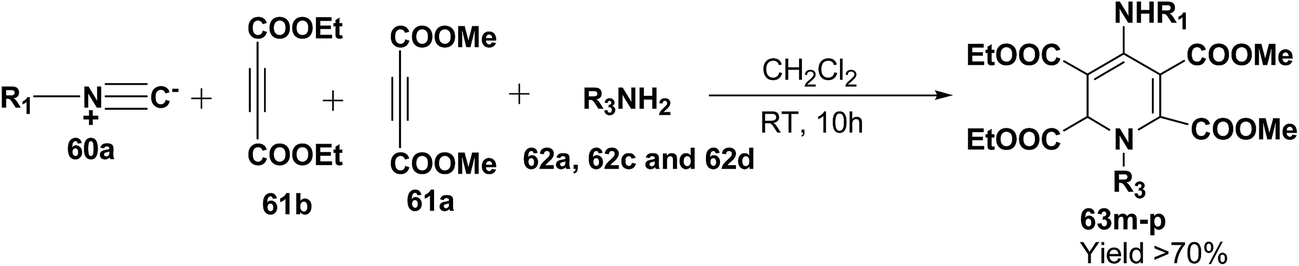
Yavari, et al.17 has reported one pot synthesis of highly functionalized 1,2-dihydropyridines from alkyl isocyanide 60a–b, acetylenic esters 61a–b and primary alkylamine (62a–i). The reaction of alkyl isocyanides 60a–b, dimethyl acetylenedicarboxylate (61a), and primary amines 57a, 62a–h, proceeded smoothly in CH2Cl2 at room temperature and produced tetramethyl 4-(alkylamino)-1-alkyl(aryl)-1,2-dihydropyridine-2,3,5,6-tetracarboxylates 63a–k in good yields after purification (Scheme 17). Similarly, taking two equivalent of diethyl acetylenedicarboxylate 61b in above reaction gives 63l in 69% yield. A wide range of structurally varied primary amines were employed in this cyclocondensation reaction. Addition of a solution of equimolar amounts of primary amine 62a, 62c & 62d and 61a to a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of cyclohexyl isocyanide (60a), and diethyl acetylenedicarboxylate (61b) in CH2Cl2 at rt, produced 5,6-diethyl 2,3-dimethyl 4-(cyclohexylamino)-1-(arylmethyl)-1,2-dihydropyridine-2,3,5,6-tetracarboxylates 63m–o which contain two different ester groups (Scheme 18). Similarly reaction of 62a and 61b in a 1:1 mixture of 60a & 61a yields 63p. The formation of a single product when two different acetylenic esters are used is presumably controlled by the sequence in which the reaction is carried out.
:1 mixture of cyclohexyl isocyanide (60a), and diethyl acetylenedicarboxylate (61b) in CH2Cl2 at rt, produced 5,6-diethyl 2,3-dimethyl 4-(cyclohexylamino)-1-(arylmethyl)-1,2-dihydropyridine-2,3,5,6-tetracarboxylates 63m–o which contain two different ester groups (Scheme 18). Similarly reaction of 62a and 61b in a 1:1 mixture of 60a & 61a yields 63p. The formation of a single product when two different acetylenic esters are used is presumably controlled by the sequence in which the reaction is carried out.
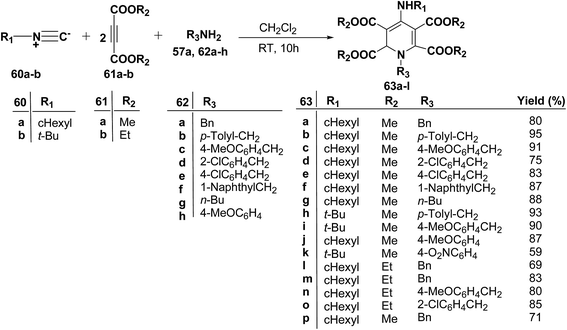 |
| | Scheme 17 Three component one-pot synthesis of functionalized 1,2-dihydropyridines 63a–l. | |
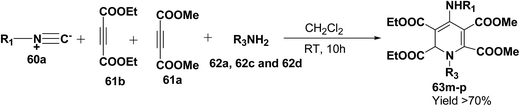 |
| | Scheme 18 Four component one-pot synthesis of functionalized 1,2-dihydropyridines 63m–p. | |
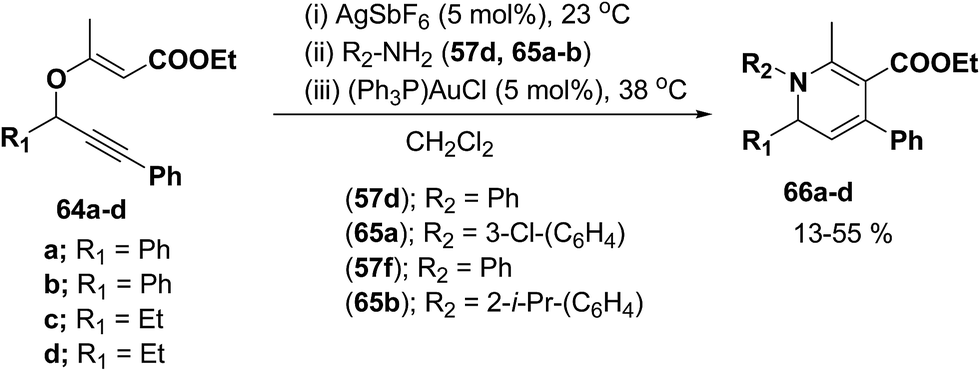
Binder and Kirsch et al.18 has synthesized 1,2-dihydropyridines 66a–d from propargyl vinyl ethers 64a–d through a sequence of propargyl-Claisen rearrangement, condensation, and heterocyclization (Scheme 19). The scope of this protocol was quite limited, only four examples of 1,2-dihydropyridines with low to moderate yields are known.
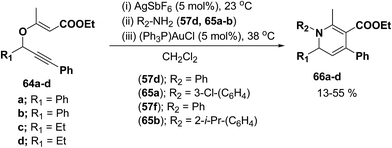 |
| | Scheme 19 Synthesis of 1,2-dihydropyridines 66a–d from propargyl ethers. | |
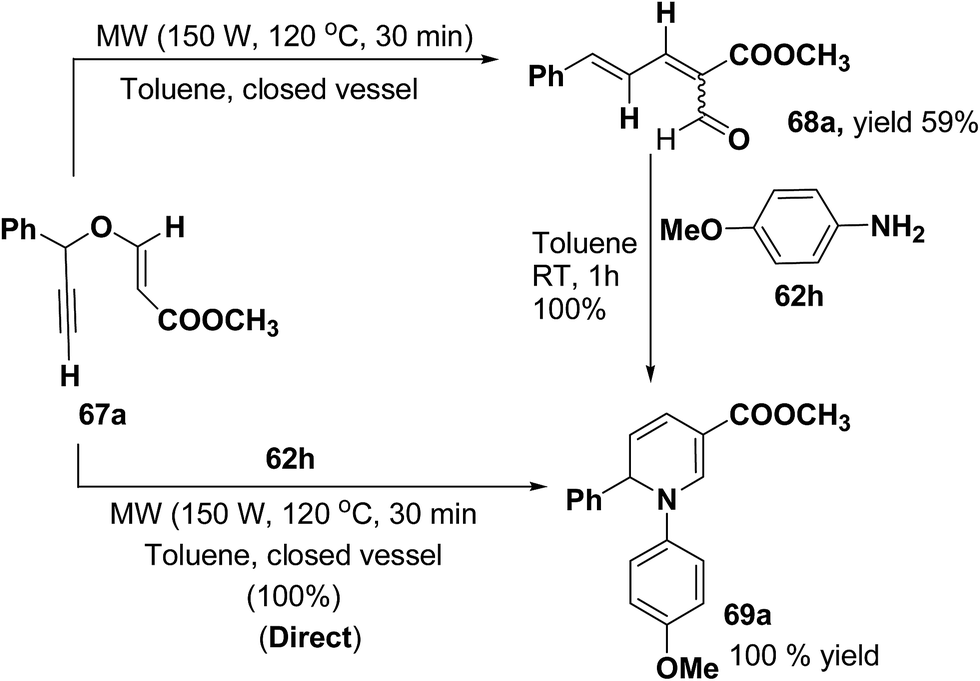
Tejedor, et al.19 has developed a convenient domino access to substituted alkyl 1,2-dihydropyridine-3-carboxylates from propargyl enol ethers and primary amine. After some experimental work, it has been found that microwave irradiation of a solution of 67a in toluene (150 W, 120 °C, 30 min) afforded the corresponding dienal 68a, which could be isolated as a mixture of E/Z (1:1) isomers in 59% yield after flash-chromatographic purification (Scheme 20). Subsequent treatment of dienal 68a with p-anisidine (62h; 1.0 equiv.) at room temperature in toluene for 1 h afforded 1,2-dihydropyridine 69a in quantitative yield. The direct microwave irradiation of 67a with 62h in toluene (150 W, 120 °C, 30 min) afforded the corresponding 1,2-dihydropyridine 69a in almost quantitative yield.
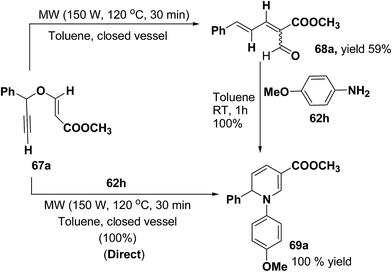 |
| | Scheme 20 Two step versus domino reaction: synthesis of 1,2-dihydropyridine 69a. | |
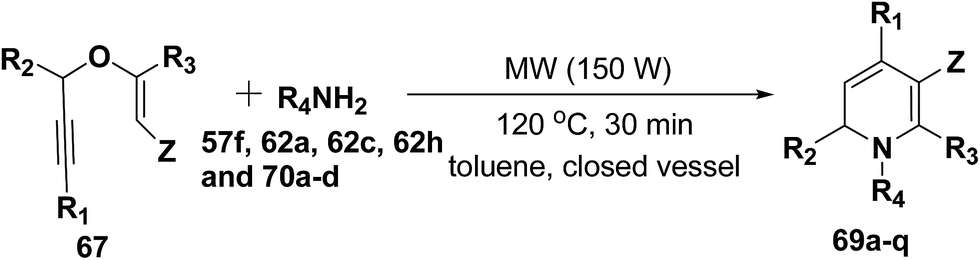
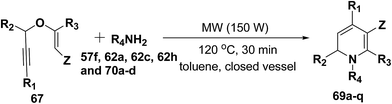
After the development of one step domino process the scope of this reaction has been extended with regard to the propargylic component and amine (Table 5). In general, the reaction presented a broad spectrum for the amine although aromatic amines gave better yields than aliphatic amines (compare entries 1, 15 and 16 with entries 10–14). The effect of diastereo induction by the amine component was studied with the chiral amine 70c, which in turn yields chiral 1,2-dihydropyridine 69m with a significant 50% de (entry 13).
Table 5 Domino synthesis of 1,2-dihydropyridines 69a–q from propargyl enol ethers 67 and primary aminesa
|
| Entry |
R1 |
R2 |
R3 |
67 |
R4NH2 |
69 |
Yield (%) |
| Propargyl vinyl ether 67 (1.0 equiv.), primary amine 62h (1.1 equiv.) in toluene (5 mL). Z = CO2CH3. 300 W, 150 °C, 2 h. 300 W, 150 °C, 3 h, Z = CO2CH2CH3. Z = SO2Tol. Ad = adamantyl. 50% de. PMB = p-methoxybenzyl. (R)-1-Phenylprop-2-yn-1-ol was used to prepare enantiopure (R)-69q; product 69q obtained as a racemic mixture. |
| 1 |
H |
Ph |
H |
a |
pMeOC6H4 (62h) |
a |
100 |
| 2 |
H |
H |
H |
b |
pMeOC6H4 (62h) |
b |
51b |
| 3 |
H |
Me |
H |
c |
pMeOC6H4 (62h) |
c |
87 |
| 4 |
h |
nPent |
H |
d |
pMeOC6H4 (62h) |
d |
71 |
| 5 |
Ph |
Ph |
H |
e |
pMeOC6H4 (62h) |
e |
95 |
| 6 |
cHex |
Ph |
H |
f |
pMeOC6H4 (62h) |
f |
55 |
| 7 |
H |
Ph |
Me |
g |
pMeOC6H4 (62h) |
g |
24c |
| 8 |
H |
Me |
Me |
h |
pMeOC6H4 (62h) |
h |
17c |
| 9 |
H |
Ph |
H |
i |
pMeOC6H4 (62h) |
i |
80d |
| 10 |
H |
Ph |
H |
a |
Bn (62a) |
j |
83 |
| 11 |
H |
Ph |
H |
a |
Allyl (70a) |
k |
72 |
| 12 |
H |
Ph |
H |
a |
Ade (70b) |
l |
87 |
| 13 |
H |
Ph |
H |
a |
(S)PhCHMe (70c) |
m |
83f |
| 14 |
H |
Ph |
H |
a |
PMBg (62c) |
n |
78 |
| 15 |
H |
Ph |
H |
a |
Ph (57f) |
o |
93 |
| 16 |
H |
Ph |
H |
a |
4-Cl-C6H4 (70d) |
p |
88 |
| 17 |
H |
Ph |
H |
a |
pMeOC6H4 (62h) |
q |
100h |
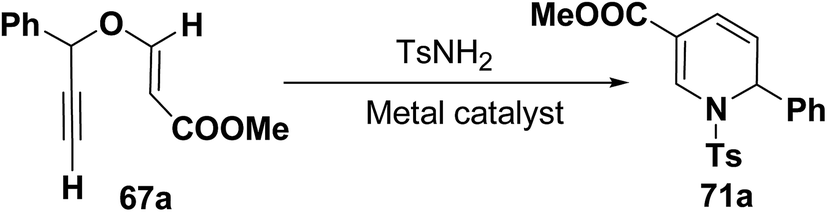
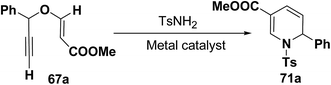
Wei, et al.20 has reported the synthesis of 1,2-dihydropyridines from propargyl vinyl ethers and allenic vinyl ethers by gold-catalyzed Claisen rearrangement and 6π-aza-electrocyclization. No reaction occurred with AuCl3 or AuPPh3Cl/AgOTf (5 mol% each) in CH2Cl2 at room temperature (entries 1 and 2, Table 6). 1,2-Dihydropyridine was obtained in 84% yield when AuPPh3Cl/AgBF4 (5 mol%) was used (entry 3). It was interesting result that 92% yield was realized by using AuPPh3Cl/AgSbF6 (5 mol%) at room temperature (entry 4). Ag(I) could serve to abstract the chloride from AuPPh3Cl to form a more electrophilic catalyst. On the contrary, when Au(PPh3)Cl or AgSbF6 was used alone (entries 5 and 6), the tandem reaction did not take place at all. On the other hand, other transition metal catalysts such as PtCl2 or PdCl2(CN)2 did not promote any transformation (entries 7 and 8). The investigation on the solvent effect showed that the best choice of the solvent was CH2Cl2 (entries 9–11). However, the yield decreased when the amount of TsNH2 was reduced from 2.0 to 1.2 equivalents (entry 12). Thus the use of 5 mol% of AuPPh3Cl/AgSbF6, with 2.0 equivalents of TsNH2 in CH2Cl2 at room temperature constituted the optimal reaction conditions.
Table 6 Optimization of reaction conditionsa
|
| Entry |
Catalyst (mol%) |
Solution |
TsNH2 (mol%) |
Yield (%) |
| Reactions were conducted with 0.4 mmol of 1a in 3 mL of solvent at room temperature. No reaction. Most of the material decomposed. |
| 1 |
AuCl3 |
DCM |
200 |
NRb |
| 2 |
PPh3AuCl/AgOTf |
DCM |
200 |
NRb |
| 3 |
PPh3AuCl/AgBF4 |
DCM |
200 |
84 |
| 4 |
PPh3AuCl/AgSbF6 |
DCM |
200 |
92 |
| 5 |
PPh3AuCl |
DCM |
200 |
NRb |
| 6 |
AgSbF6 |
DCM |
200 |
NRb |
| 7 |
PdCl2(CN)2 |
DCM |
200 |
NRb |
| 8 |
PtCl2 |
DCM |
200 |
NRb |
| 9 |
PPh3AuCl/AgSbF6 |
Toluene |
200 |
Tracec |
| 10 |
PPh3AuCl/AgSbF6 |
CH3CN |
200 |
NRb |
| 11 |
PPh3AuCl/AgSbF6 |
DCE |
200 |
77 |
| 12 |
PPh3AuCl/AgSbF6 |
DCM |
120 |
80 |
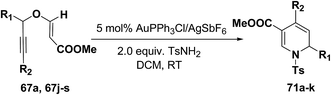
The scope of the reaction has been explored in gold catalysed tandem reaction by studying a wide variety of substrates (Table 7). Substrates containing electron-rich or electron-deficient aryl groups at the propargylic position gave good to excellent yields of desired 1,2-dihydropyridines (entries 2–9).
Table 7 Tandem synthesis of 1,2-dihydropyridines from propargyl vinyl ethers and TsNH2a
|
| Entry |
R1 |
R2 |
67 |
Product |
Yieldb (%) |
| Reactions were conducted with 0.4 mmol of 67 in 3 mL of solvent. Isolated yield after column chromatography. |
| 1 |
C6H5 |
H |
67a |
71a |
92 |
| 2 |
2-Me-C6H4 |
H |
67j |
71b |
90 |
| 3 |
4-Me-C6H4 |
H |
67k |
71c |
91 |
| 4 |
3-Me-C6H4 |
H |
67l |
71d |
80 |
| 5 |
3,4-Di-Me-C6H3 |
H |
67m |
71e |
78 |
| 6 |
2-MeO-C6H4 |
H |
67n |
71f |
89 |
| 7 |
4-CI-C6H4 |
H |
67o |
71g |
85 |
| 8 |
2-CI-C6H4 |
H |
67p |
71h |
84 |
| 9 |
2-Naphthyl |
H |
67q |
71i |
88 |
| 10 |
n-Hexyl |
H |
67r |
71j |
82 |
| 11 |
C6H5 |
n-Pentyl |
67s |
71k |
60 |
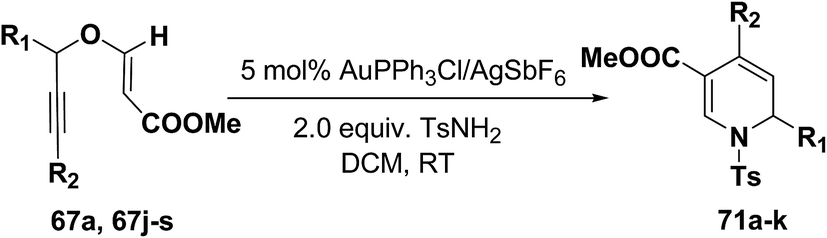
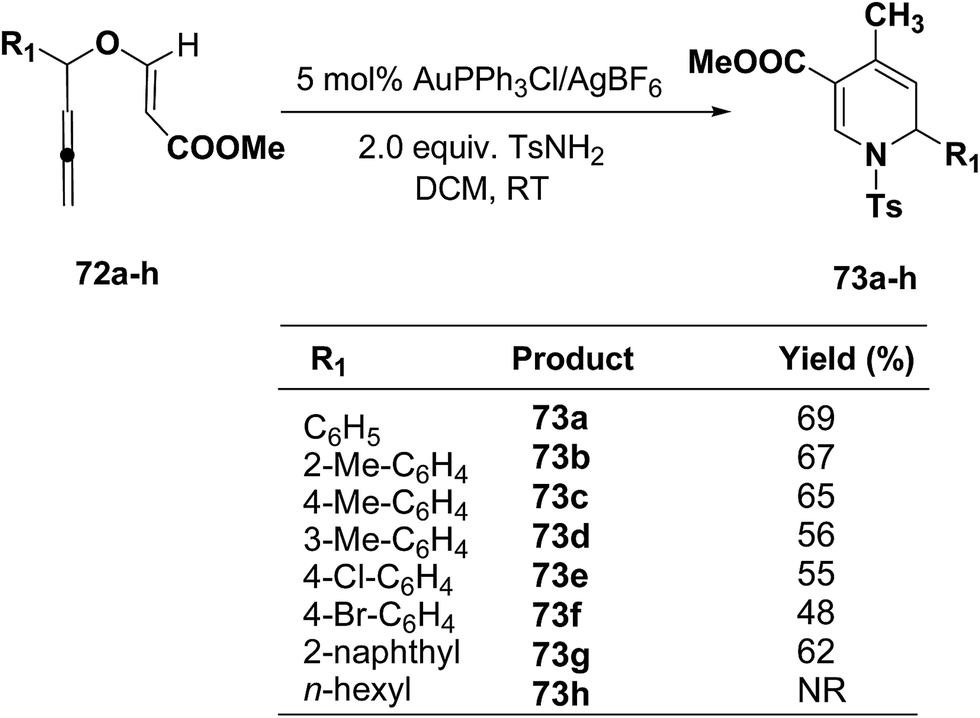
The scope of this reaction has been extended by Wei, et al. on allenic vinyl ethers 72a–h and it has been found that 5 mol% AuPPh3Cl/AgBF4 in presence of 2.0 equiv. TsNH2 gave the moderate to good yield of diversified 1,2-dihydropyridines 73a–h (Scheme 21).
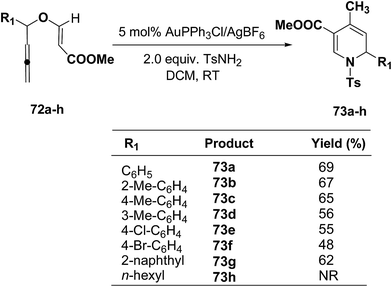 |
| | Scheme 21 Tandem synthesis of 1,2-dihydropyridines 73a–h from allenic vinyl ethers and TsNH2. | |
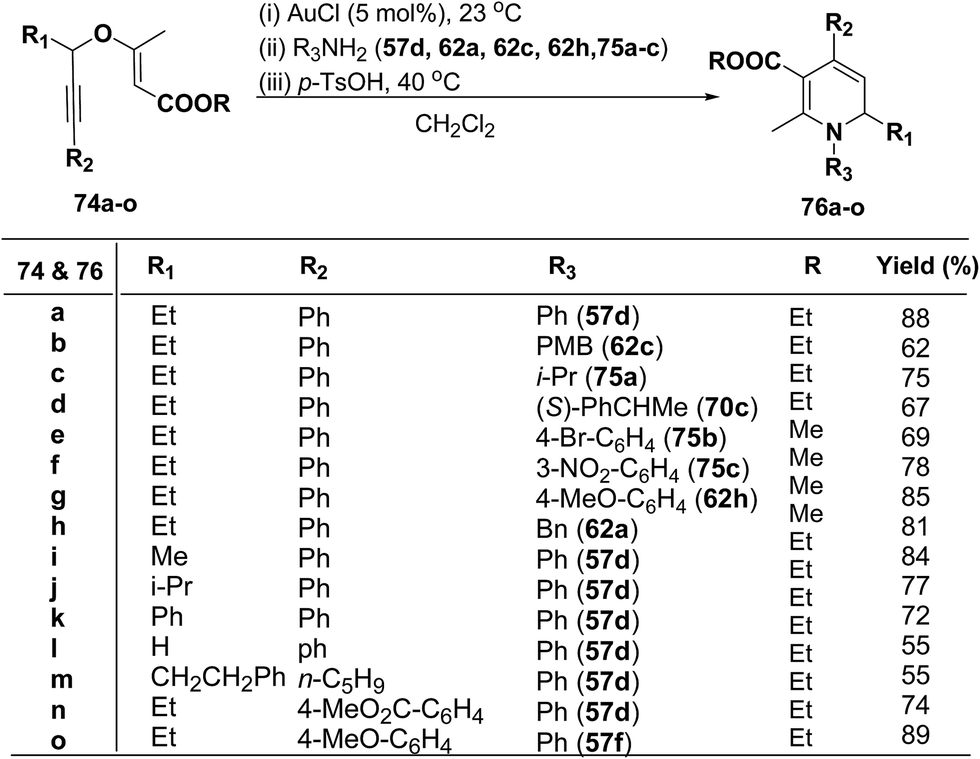
1,2-Dihydropyridines 76a–o were synthesized in good to excellent yields by using a standardized protocol of Kirsch et al.21 with internal alkynes 74a–o and used a sequential addition of reagents and catalysts to obtain reproducibly high yields [(i) AuCl (5 mol%), 23 °C; (ii) R3NH2 (57d, 62a, 62c, 62h and 75a–c); (iii) p-TsOH (20 mol%), 40 °C, CH2Cl2] (Scheme 22). The formation of 1,2-dihydropyridines 76a–o tolerated substitution of R1 and R2 with both aryl and alkyl groups (Scheme 22). Compound 76l bearing no substituent in the 2-position (R1 = H) was obtained in only moderate yield.
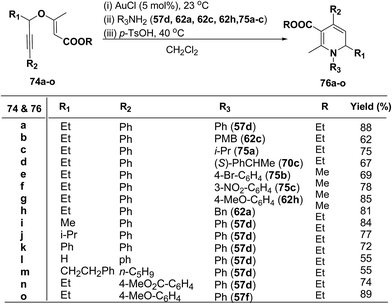 |
| | Scheme 22 The conversion of 74a–o in 1,2-dihydropyridines 76a–o. | |
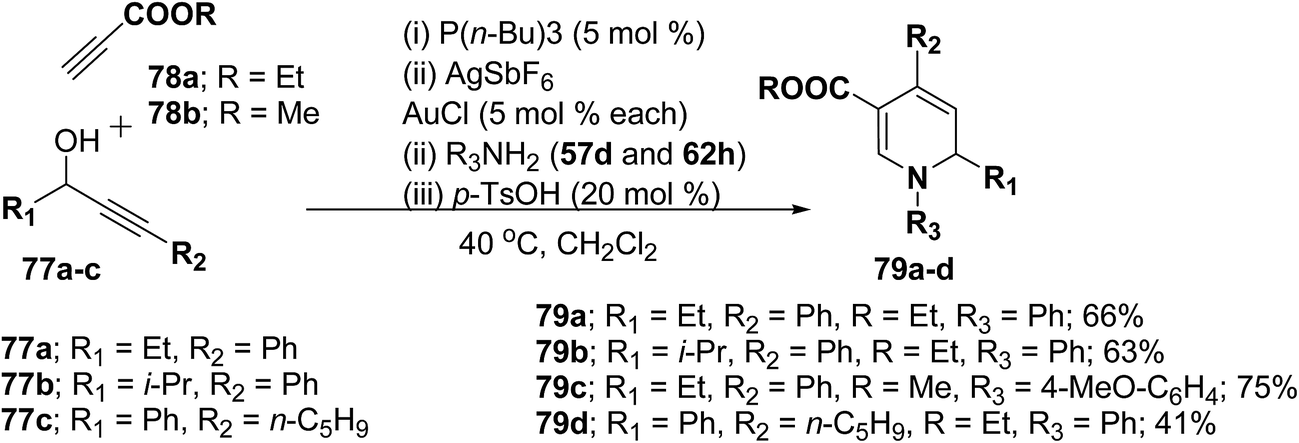
Further this one-pot procedure is extended by an additional step that is the formation of the propargyl vinyl ethers. When propargylic alcohols 77a–c were reacted with 1.0 equiv. of ethyl propiolate 78a/methyl propiolate 78b and 5 mol% of P(n-Bu)3, the Michael-addition remained an efficient process. Subsequent addition of both AuCl and AgSbF6 (5 mol% each) furnished the allenylcarbonyl compounds. Notably, only the use of both catalysts together provided good and reproducible results; neither one was able to catalyze the reaction on its own. The sequence was terminated by condensation with 1.5 equiv. of an amine and 0.2 equiv. of p-TsOH at 40 °C for 15 h to give the desired products in good yields (Scheme 23). It is of particular note that this one-pot procedure presents the possibility to access 1,2-dihydropyridines 79a–d even in cases when the required propargyl vinyl ether is not accessible due to poor stability.
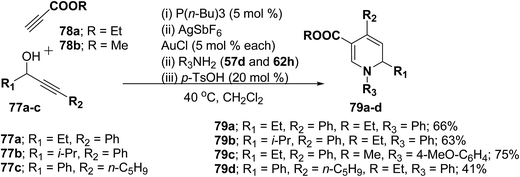 |
| | Scheme 23 One pot synthesis of 1,2-dihydropyridines 79a–d starting from propargylic alcohols 77a–c. | |
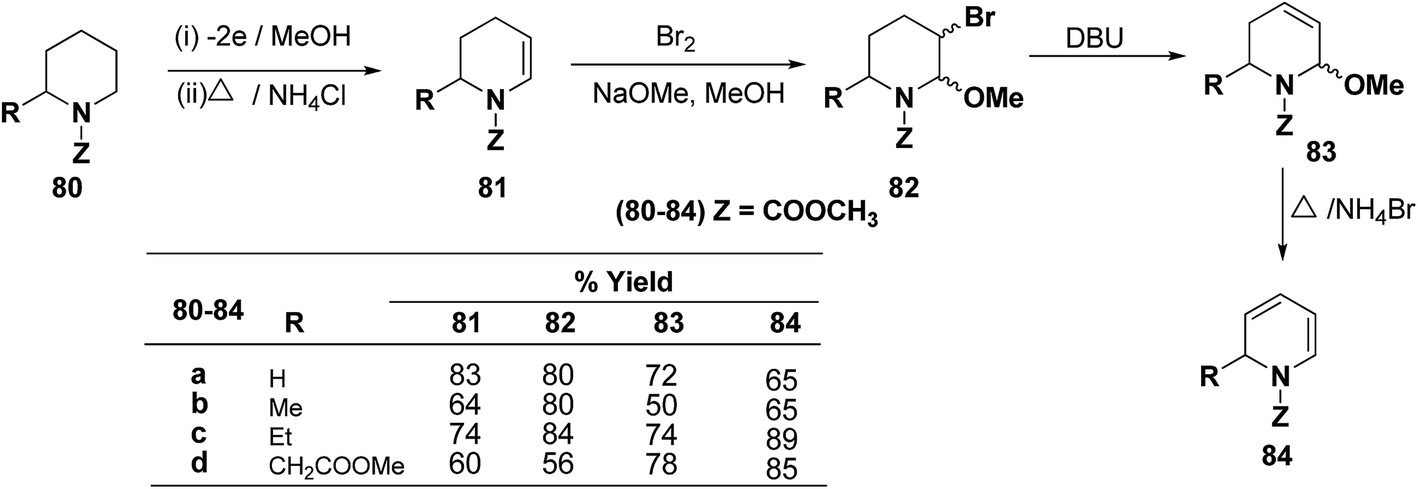
Shono, et al.22 have developed a convenient and regioselective method for the synthesis of 1,2-dihydropyridines, especially those possessing substituents at certain positions of the pyridine nucleus. 2-Substituted 1-(methoxycarbonyl)-1,2-dihydropyridines have been synthesized from piperidines electrochemically (Scheme 24).
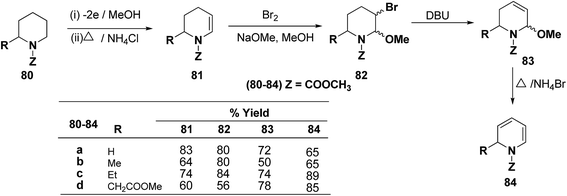 |
| | Scheme 24 Electrochemical synthesis of 1,2-dihydropyridines 84a–d. | |
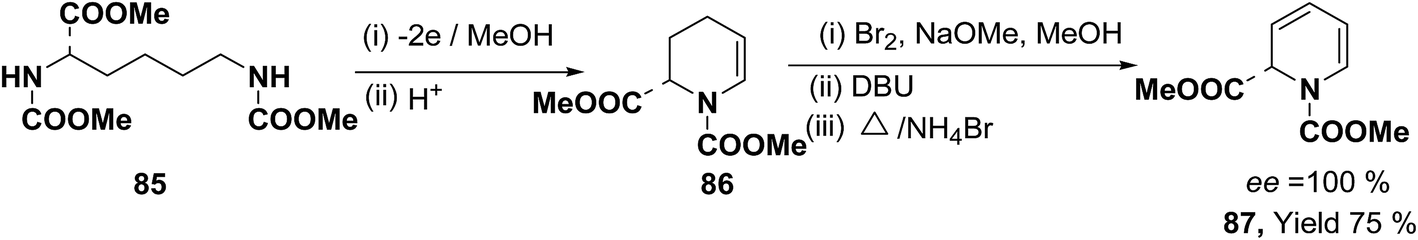
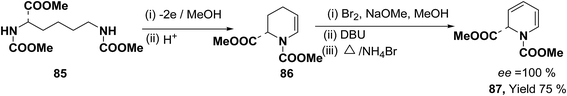
Optically active 1,2-dihydropyridine 87 has been synthesized starting from L-lysine derivative 85 by electrochemically in two steps via the synthesis of tetrahydropyridine 86 in 75% yield, ee ∼ 100% (Scheme 25).
 |
| | Scheme 25 Electrochemical synthesis of enantiomerically pure 1,2-dihydropyridine 87. | |
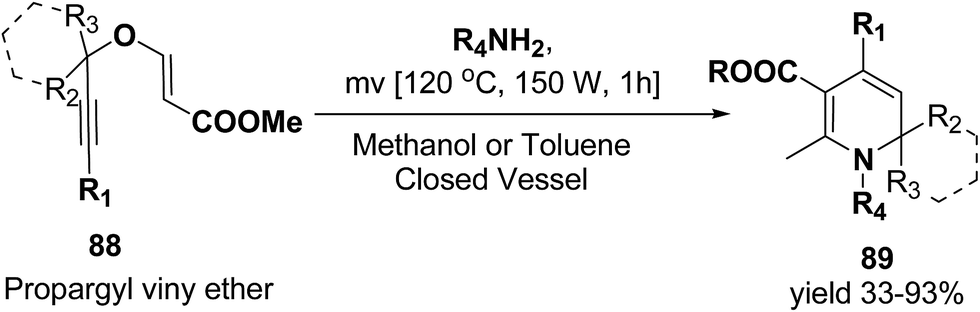
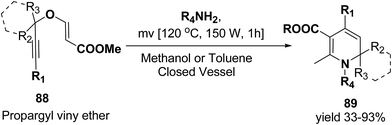
Tejedor, et al.23 has developed a general and practical metal-free protocol for the synthesis of 1,2-dihydropyridines (Scheme 26) with variety of structural/functional diversity at the ring and featuring mono, double, or spiro substitution at the sp3 hybridized carbon position. The protocol contains a microwave-assisted domino reaction of propargyl vinyl ether 88 (secondary or tertiary) and a primary amine (aliphatic or aromatic) in toluene or methanol.
 |
| | Scheme 26 Microwave-assisted domino synthesis of 1,2-dihydropyridines 89 from propargyl vinyl ether 88. | |
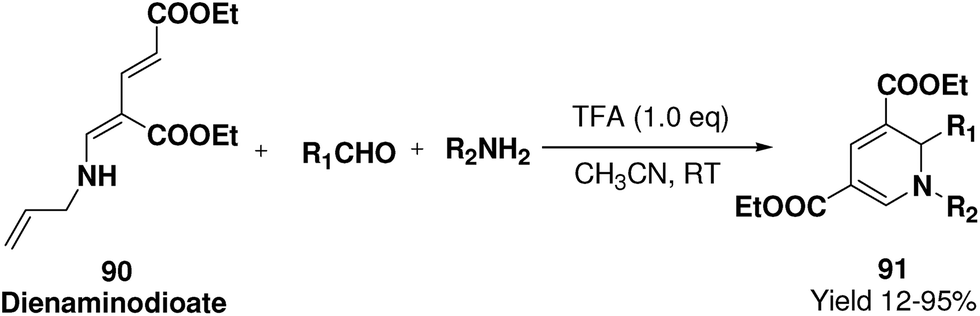
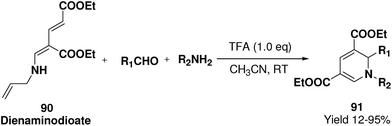
Challa, et al.24 has developed a convenient synthesis of 1,2-dihydropyridine (91) from dienaminodioate 90 and an imine (generated in situ) mediated by CF3COOH in a one-pot cascade synthesis. The advantages associated with this transformation include conditions that are metal-free, room temperature, undistilled solvent, and expeditious in excellent yields. Out of the several acid catalyst used for the optimisation of the reaction condition trifluoroacetic acid was found to me excellent, which yield 1,2-dihydropyridine in excellent yield (Scheme 27). The substrate scope has been demonstrated with various aromatic, heteroaromatic, unsaturated aldehydes, and anilines, benzylic amines in impressive yields.
 |
| | Scheme 27 Cascade synthesis of 1,2-dihydropyridine 91 by variation of aldehydes and amines. | |
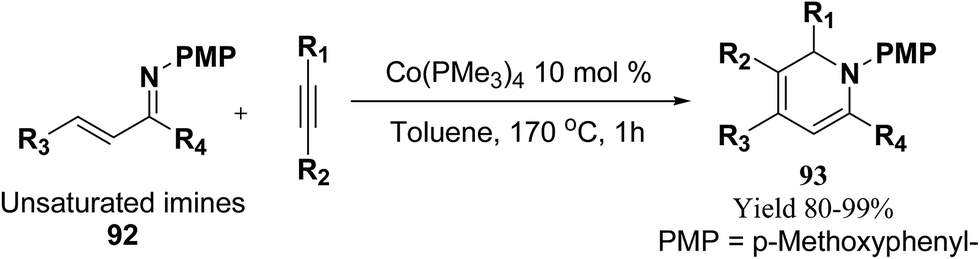
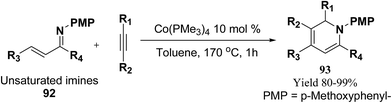
Fallon, et al.25 has developed a convenient one-pot synthesis of highly substituted 1,2-dihydropyridines 93 from unsaturated imines 92 and alkynes via C–H activation/6π-electrocyclisation pathway (Scheme 28). The reaction proceeds with high regioselectivity, and the author has disclosed the first example of isolated 1,2-dihydropyridines lacking substitution at 2 position. A low valent cobalt complex without reducing agents or additives provides C–H activation.
 |
| | Scheme 28 Annulation reaction between unsaturated imines and alkynes for the formation of 1,2-dihydropyridines 93. | |
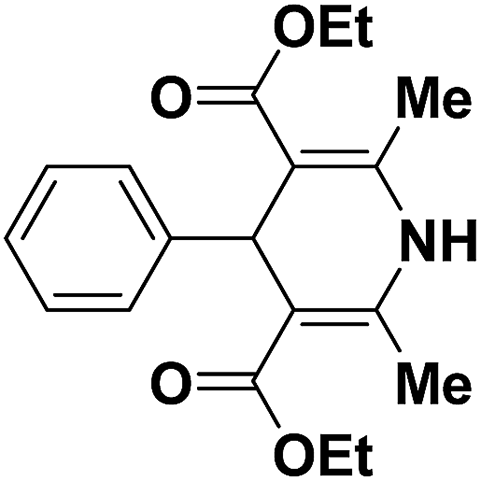
2.2. Synthesis of 1,4-dihydropyridines
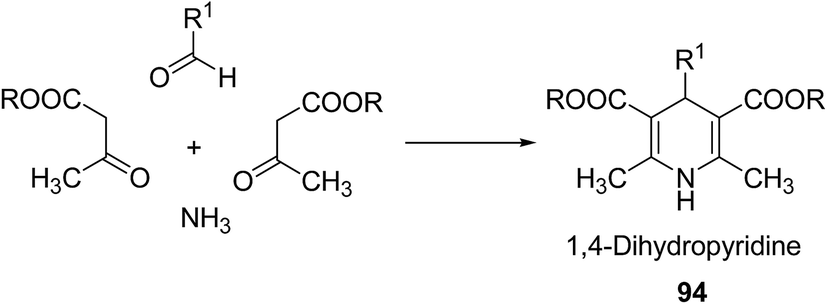
Over the years many outstanding discoveries have been made in the field of drug development and research with multifaceted drugs having great potential. 1,4-Dihydropyridine is one such privileged structure which has revolutionized the pharmaceutical industry. It is a well explored scaffold which binds to multiple receptors, possesses a wide variety of biological features and also has its basis in biologically active natural products. The first synthesis of 1,4-dihydropyridines was reported by Arthur Hantzsch in 1882.26 He observed that in the process of synthesizing pyridine by a one pot three-component condensation reaction of acetoacetic ester, aldehyde and ammonia, an intermediate 1,4-dihydropyridine (1,4-DHP) 94 is formed, which could be isolated easily. Since then this reaction has been successfully employed in the synthesis of 1,4-DHPs and bears his name as “Hantzsch dihydropyridine synthesis” (Scheme 29).
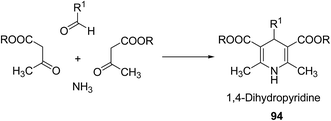 |
| | Scheme 29 Hantzsch dihydropyridine synthesis. | |
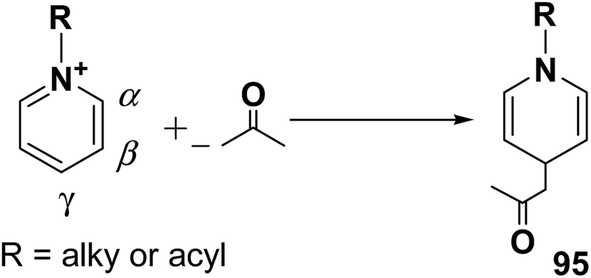
The addition of nucleophiles to α- and γ- positions of N-substituted pyridinium salts gives both 1,2- and 1,4-dihydropyridines. The regioselectivity of the addition reaction depends on the hardness of the nucleophile: hard nucleophiles preferentially attack at C-2 whereas soft ones attack at C-4 position of the pyridinium salts. The addition is also believed to proceed at both the α- and γ-carbon centres under kinetic control and only at the γ-carbon under thermodynamic control.27 The Kröhnke procedure28–30 and sodium dithionite reduction31–33 are examples of reactions involving addition of nucleophiles at γ-position. The Kröhnke procedure,28–30 consisting of base-catalysed reaction of ketones with alkyl or N-acyl pyridinium salts, was investigated years ago (Scheme 30). It is of particular synthetic interest since it constitutes a useful method of forming new carbon–carbon bonds to give γ-substituted 1,4-dihydropyridines 95.22
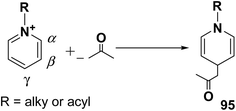 |
| | Scheme 30 An example of Kröhnke procedure. | |
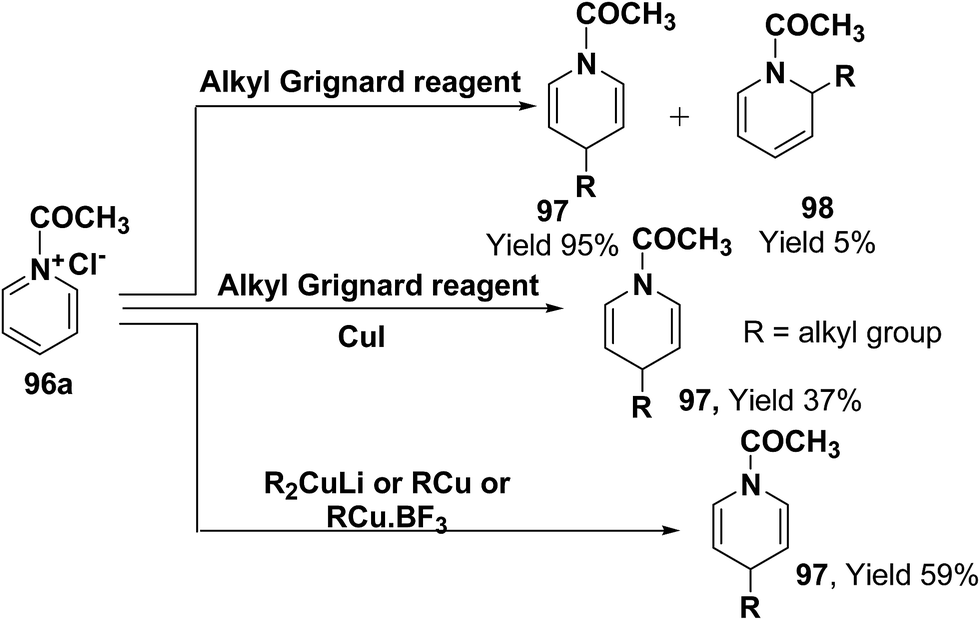
Comins, et al.34 and Yamaguchi, et al.35 have reported the reaction of 1-acetylpyridinium chloride with alkyl Grignard reagent, which lead to the formation of 1,4-dihydropyridines 97 and 1,2-dihydropyridines 98.34a However, when a catalytic amount of CuI is present, the addition is regiospecific and resulted in the exclusive formation of 1,4-dihydropyridines 97. Stoichiometric organocopper reagent (e.g. R2CuLi, RCu, RCu·BF3) also give 1,4-addition product and resulted into the formation of 1,4-dihydropyridines 97 (Scheme 31).36
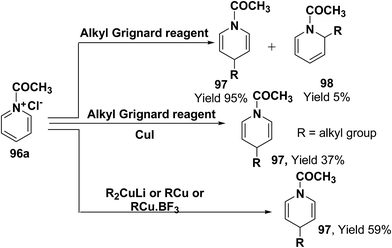 |
| | Scheme 31 Grignard and organocopper reagents catalyzed exclusive synthesis of 1,4-dihydropyridine 97. | |
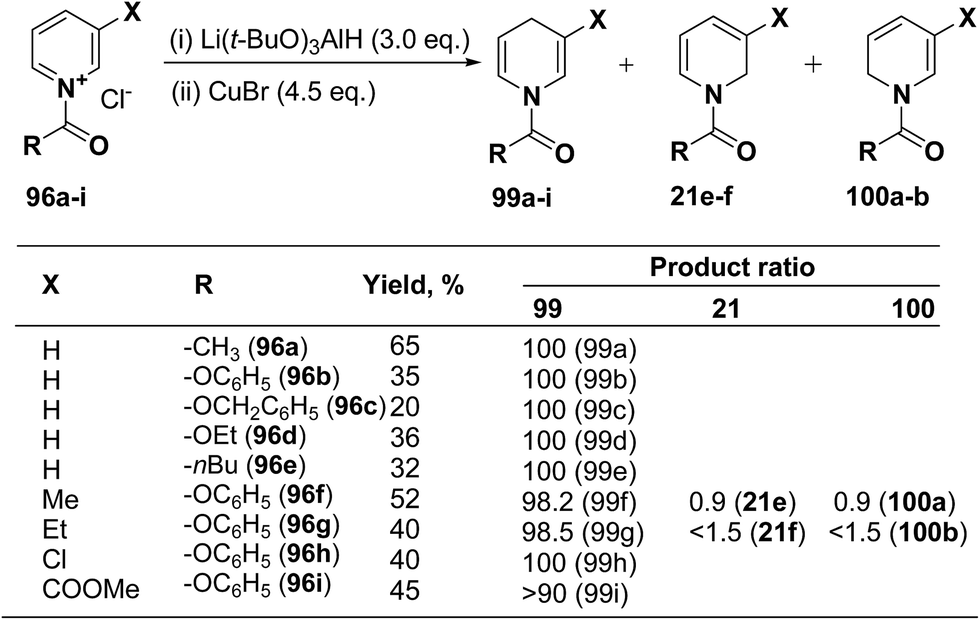
Several hydride reagents e.g. NaBH4,3 NaBH3CN, (PPh3)2CuBH4,37 (Ph2MeP)3CuBH4,38 Semmelhack's “NaCuH2”39 and Semmelhack's “LiCuH2”39 has been used for the regioselective synthesis of 1,4-dihydropyridines from 1-(phenoxycarbony1)pyridinium chloride 99a (generate in situ by the reaction between pyridine and phenyl chloriformate), but none of these procedure resulted into the regioselective formation of 1,4-DHP. Comins, et al.40 has used a copper hydrido reagent prepared from lithium tri-tert-butoxyaluminum hydride (3.0 equiv.) and copper bromide (4.5 equiv.) as a reducing agent for the regioselective reduction of 1-(phenoxycarbony1)pyridinium chloride 96a in almost quantitative yield (Scheme 32). Further this procedure has been extended to the variety of the 1-(alkoxy/aryloxy/acyloxycarbony1)pyridinium chlorides 99b–i (generated in situ by the reaction between substituted pyridine and corresponding alkyl/phenyl chloroformate or acid chloride). This convenient one-pot preparation of 1-(alkoxycarbonyl)-1,4-dihydropyridines by using the Comins' copper hydride complements the two-step procedure developed by Fowler.4
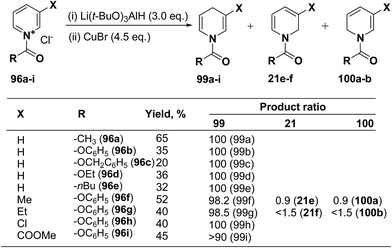 |
| | Scheme 32 Comins copper hydride catalyzed regioselective synthesis of 1,4-dihydropyridines 99a–i. | |
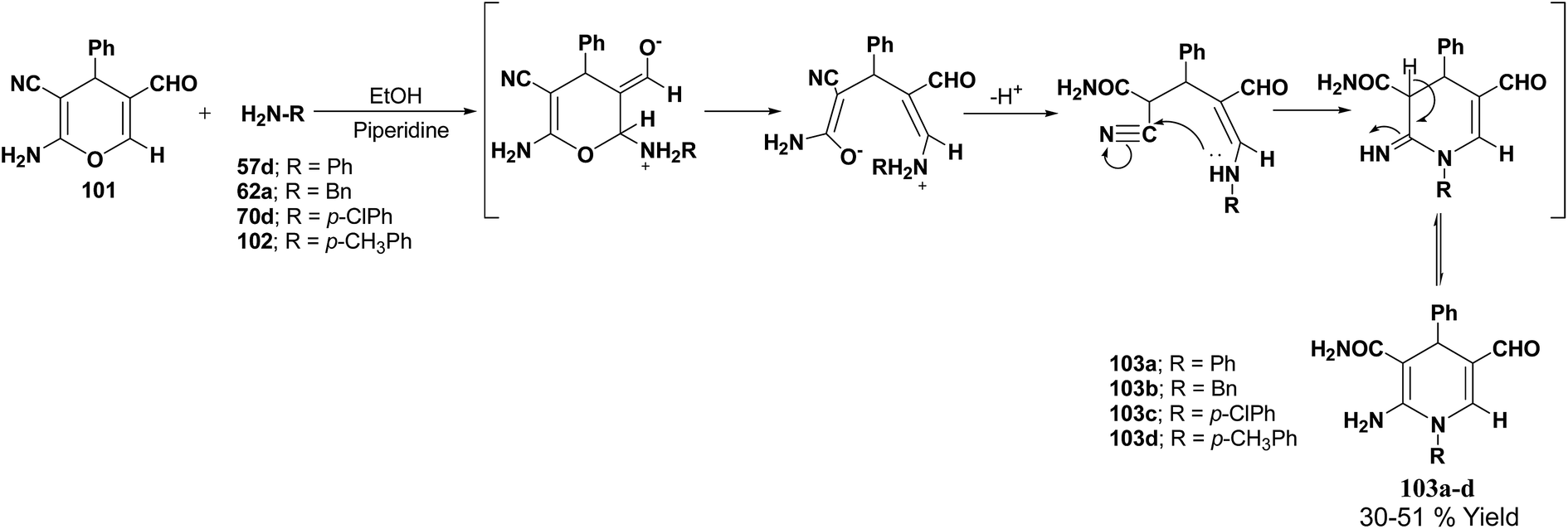
De Lucas, et al.41 have developed a novel methodology for the synthesis of N-substituted 1,4-dihydropyridines 103a–d by the reaction of 2-amino-5-formyl-4H-pyran 101 with primary amines 57d, 62a, 70d and 102 in moderate yield (Scheme 33). The formation of 1,4-dihydropyridines involves cleavage of the 4H-pyran ring by nucleophilic attack of the respective amine and subsequent 6-exo-dig cyclisation.
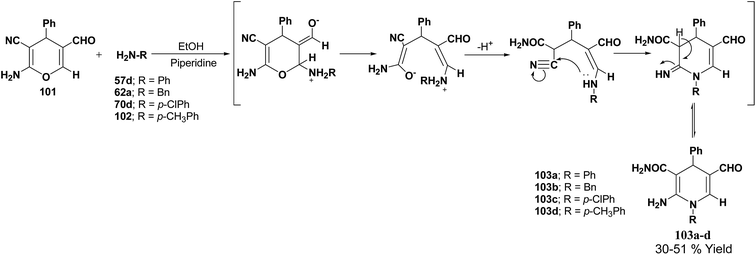 |
| | Scheme 33 Synthesis of 1,4-dihydropyridines 103a–d from 2-amino-5-formyl-4H-pyran 101. | |
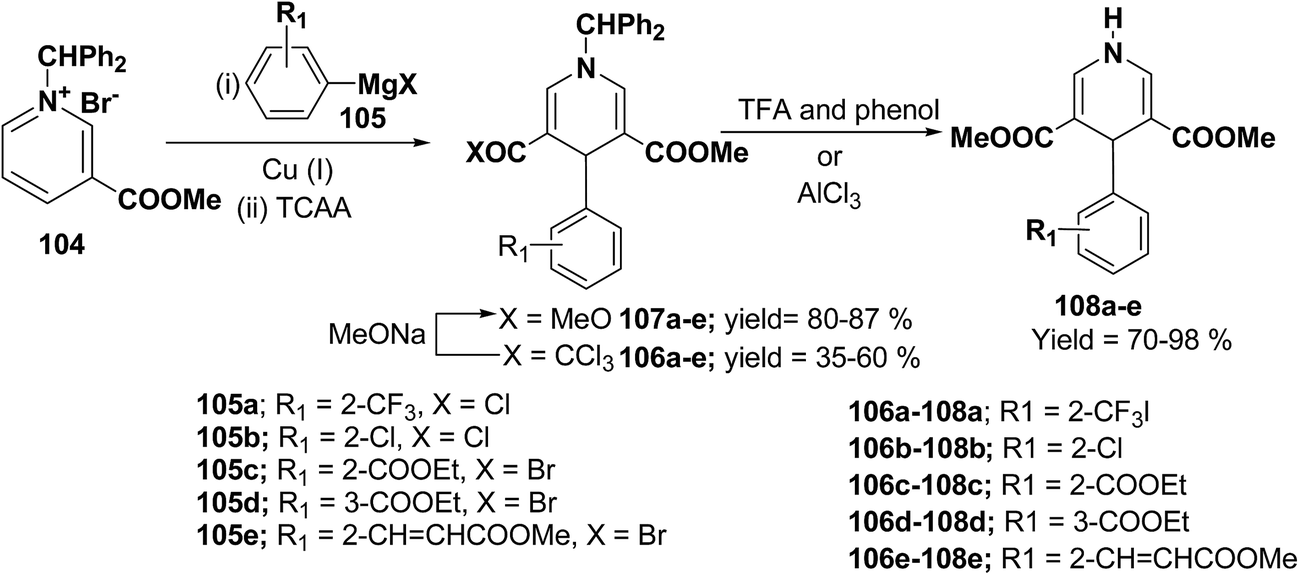
Bennasar, et al.42 have developed a regioselective synthesis of 4-functionalized 3,5-diacyl-4-aryl-1,4-dihydropyridines 108a–e by the copper-mediated addition of functionalized arylmagnesium reagent 105a–e to the N-benzhydrilpyridinium salt 104, followed by acylation with trichloroacetic anhydride. The subsequent haloform reaction on 106a–e yields 107a–e, which on N-deprotection gives 108a–e (Scheme 34).
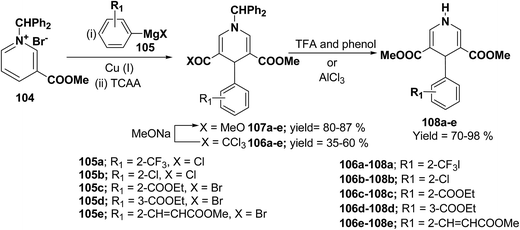 |
| | Scheme 34 Synthesis of 4-functionalized 3,5-diacyl-4-aryl-1,4-dihydropyridines 108a–e. | |
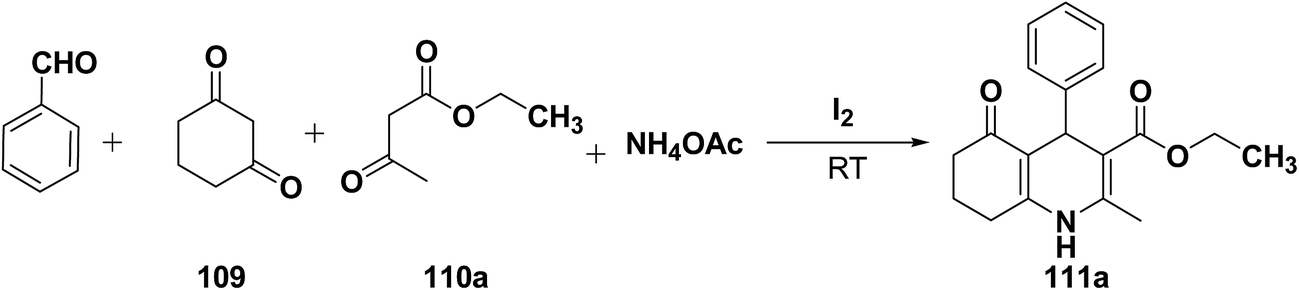
Yao, et al.43 have developed a simple, inexpensive and efficient one-pot synthesis of 1,4-dihydropyridine derivatives with excellent product yields at room temperature using catalytic amount of molecular iodine. Initially, benzaldehyde, 1,3-cyclohexanedione 109, ethyl acetoacetate 110a and ammonium acetate were stirred at room temperature in a few mL of ethanol. After 4 hours, only 56% of product 111a was realized after recrystallization of the crude product from ethanol (entry 1, Table 8). To improve the product yields and to optimize the reaction condition, molecular iodine was used in catalytic amount (15 mol%) and a reaction was carried out under similar reaction conditions to give the product 111a in 99% yield (entry 2). The increase in the quantity of molecular iodine from 15 to 30 mol% not only reduced the reaction time from 4 to 2.5 h, but also enhanced the product yield from 56% to 99% (entry 3). Similarly, using 50 mol% of iodine as catalyst the reaction time further reduced to 1.5 h along with a decrease in the yield of the product 111a (70%) (entry 4).
Table 8 Optimizing the reaction conditionsa
A range of aryl and alkyl aldehydes were subjected to react with 109, 110a and NH4OAc in the presence of either 15 or 30 mol% of iodine to generate corresponding 1,4-DHP 111b. The results are summarized in Table 9, e.g. with 30 mol% of iodine as catalyst, p-chlorobenzaldehyde is converted into the product in 6 h with 99% yield, whereas the same reaction takes place within 40 min with 15 mol% of iodine without significant loss of yield (Table 9, entries 9 & 10). In general, the yields are little less with 15 mol% of catalyst and the reaction times are also short. Both aliphatic and aromatic aldehydes react equally good to give the products with excellent yields. The aryl group substituted with different groups and same groups located at different positions of the aromatic ring has not shown much effect on the formation of the final product.
Table 9 Iodine catalyzed synthesis of 1,4-dihydropyridine derivatives through Hantzsch reaction
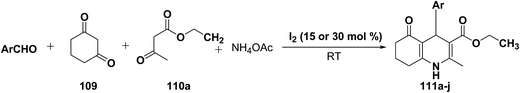
|
| Entry |
105 |
Ar |
Iodine (mol%) |
Time (h) |
Yielda (%) |
| Crude isolated yield. |
| 1 |
111a |
C6H5 |
30 |
2.5 h |
99 |
| 2 |
111a |
15 |
30 min |
93 |
| 3 |
111b |
p-Me-C6H4 |
30 |
1.5 h |
90 |
| 4 |
111b |
15 |
35 min |
99 |
| 5 |
111c |
p-Me-C6H4 |
30 |
4 h |
97 |
| 6 |
111c |
15 |
40 min |
87 |
| 7 |
111d |
p-F-C6H4 |
30 |
3.5 h |
91 |
| 8 |
111d |
15 |
35 min |
87 |
| 9 |
111e |
p-CI-C6H4 |
30 |
6 h |
99 |
| 10 |
111e |
15 |
40 min |
90 |
| 11 |
111f |
p-OH-C6H4 |
30 |
2.5 h |
99 |
| 12 |
111f |
15 |
45 min |
92 |
| 13 |
111g |
o-NO2-C6H4 |
30 |
1.5 h |
94 |
| 14 |
111g |
15 |
25 min |
91 |
| 15 |
111h |
m-NO2-C6H4 |
30 |
1.5 h |
99 |
| 16 |
111h |
15 |
25 min |
92 |
| 17 |
111i |
p-NO2-C6H4 |
30 |
3 h |
99 |
| 18 |
111i |
15 |
25 min |
85 |
| 19 |
111j |
Isopropyl |
30 |
5 h |
99 |
| 20 |
111j |
15 |
40 min |
99 |
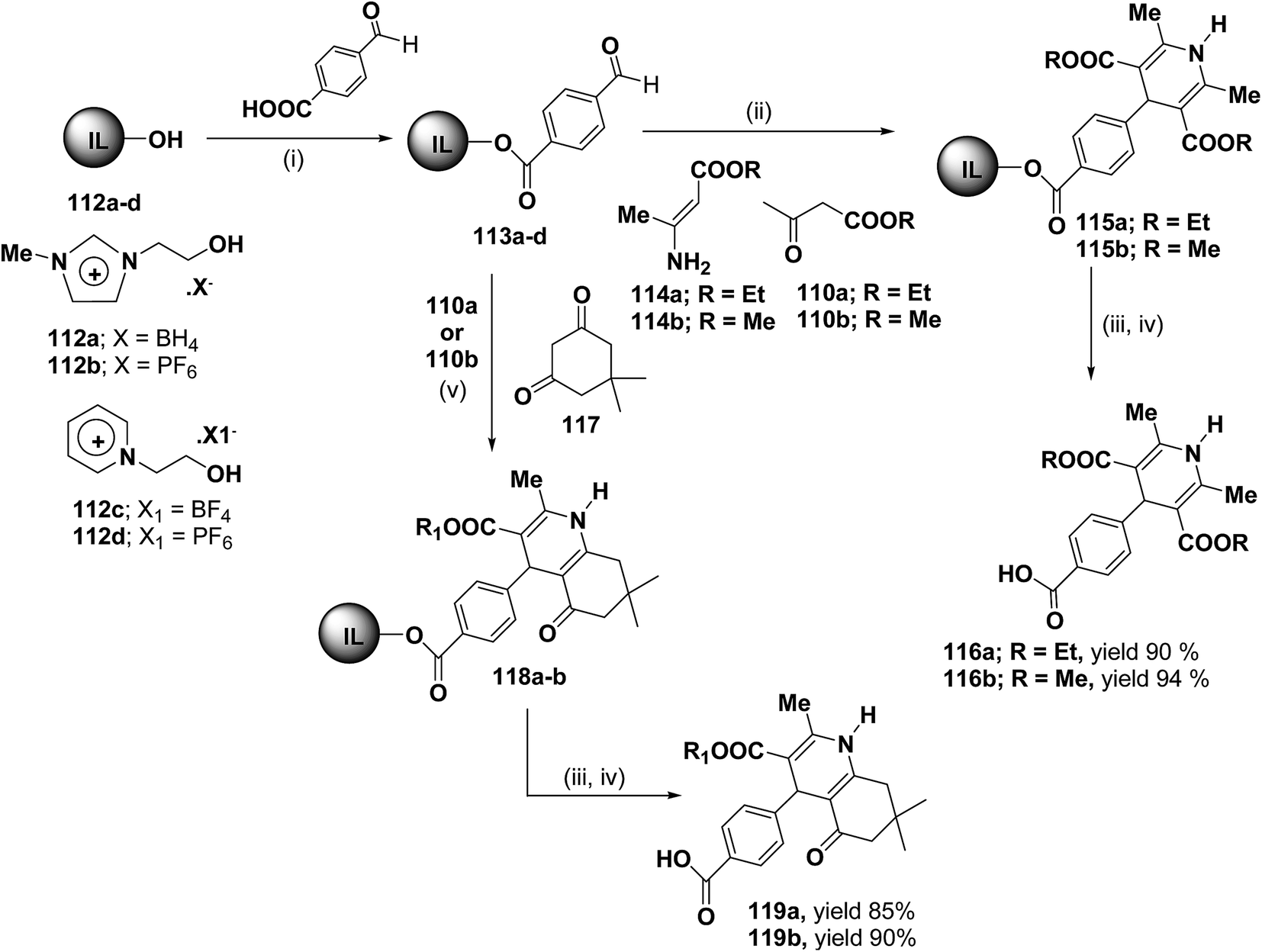
Bazureaua, et al.44 have developed a three component one pot microwave dielectric heating assisted liquid phase synthesis of 1,4-dihydropyridines using task specific ionic liquid as a soluble solid support. They synthesized the ILP bound aldehydes 113a–d in high yields by the esterification of PEG-ILPs 112a–d with 4-formylbenzoic acid in dry MeCN with dicyclohexylcarbodiimide (DCC) and 5% of dimethylamino pyridine (DMAP) as catalyst. The ILP bound 1,4-dihydropyridines 115 has been synthesized in an excellent yields via two methods. Method A involves the condensation of aldehydes 113a–d and 1.0 equiv. of β-ketoester ethyl acetoacetate 110a or methyl acetoacetate 110b and 1.0 equiv. of aminocrotonate 114 under microwave irradiation (120 °C, 150 W, 50% power level, time exposure: 10 min), in the method B the IL-phase bound 1,4-DHP 115 was prepared by the condensation from β-ketoester 110a or 110b (2.0 equiv.) and NH4OAc (2.0 equiv.) using the same microwave reaction conditions (120 °C, 10 min) (Scheme 35). IL-phase, the bound products 115a–b were subjected to cleavage by: (i) transesterification with 30% of MeONa in refluxed MeOH during 18 h, (ii) saponification with 60% of LiOH in THF at room temperature, followed by controlled acidification with a solution of 3.0 M HCl to obtain the desired 1,4-DHP 116a–b in an good yield. Similarly 118a–b was synthesized by the condensation of 113a–d with 110a or 110b (1.0 equiv.) and 117 (1.0 equiv.) and NH4OAc (2.0 equiv.) under the same microwave radiation condition. 118a–b on transesterification followed by saponification yielded 119a–b.
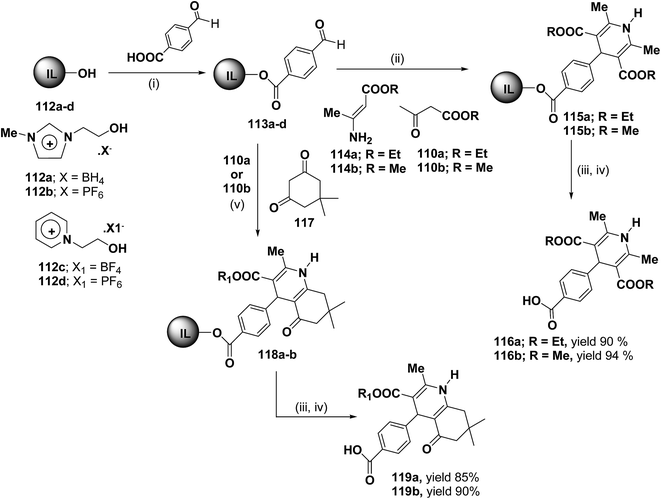 |
| | Scheme 35 Ionic liquid supported synthesis of 1,4-DHPs. Reagents and conditions (i) DCC (1.0 equiv.), DMAP (5%), dry MeCN, rt, 24 h; (ii) method A: 110 (1.0 equiv.), 114 (1.0 equiv.), mw: 120 °C (power level: 50%, 150 W), 10 min or method B: 110 (2.0 equiv.), NH4OAc (2.0 equiv.), mw: 120 °C (power level: 50%, 150 W); (iii) MeONa 30%, MeOH, reflux, 18 h; (iv) LiOH 60%, THF, rt, 20 h then 3 M HCl; (v) 110 (1.0 equiv.), 117 (1.0 equiv.), NH4OAc (2.0 equiv.), mw: 120 °C (power level: 50%, 150 W). | |
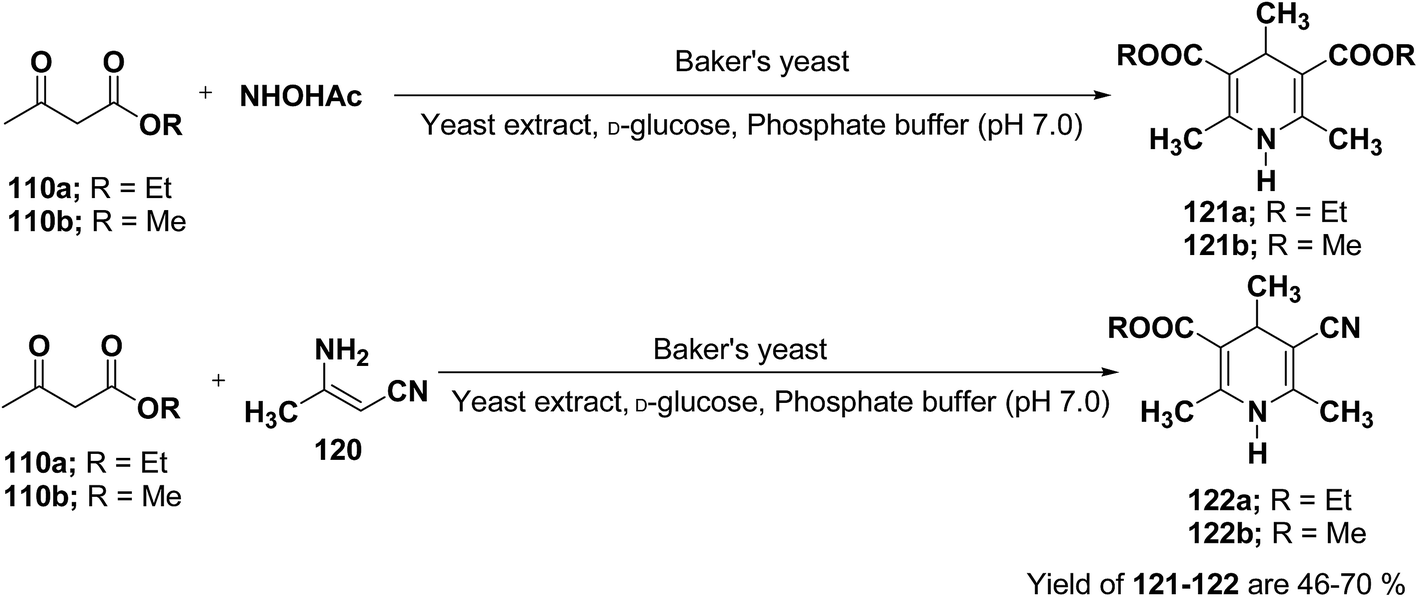
J. H. Lee45 has described an efficient method for the synthesis of 1,4-DHP under mild reaction condition using Baker's yeast. In a typical procedure a solution of 100 mL of pH 7.0 phosphate buffer, 5.0 g of D-glucose, and 2.0 g of yeast extract was warmed at 35 °C. Further 5.0 g of dry active Baker's yeast was added to this solution and the mixture was stirred at 30 °C for 30 min, after which, acetoacetic ester 110a or 110b (1.0 mmol) and ammonium acetate or 3-amino crotonitrile 120 were added. The mixture was stirred at room temperature for 24 h and then extracted with diethyl ether. The organic layer was dried and concentrated in vacuo and the resulting crude products were recrystallized using ether–n-hexane to afford pure products 121a–b and 122a–b in 46–70% yields (Scheme 36).
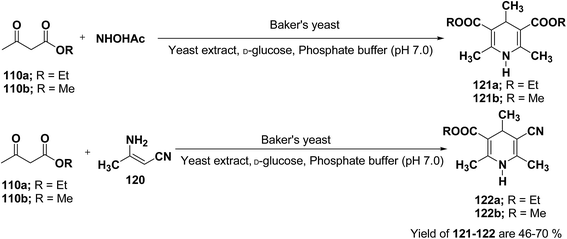 |
| | Scheme 36 Baker's yeast catalyzed synthesis of 1,4-DHPs 121 and 122. | |
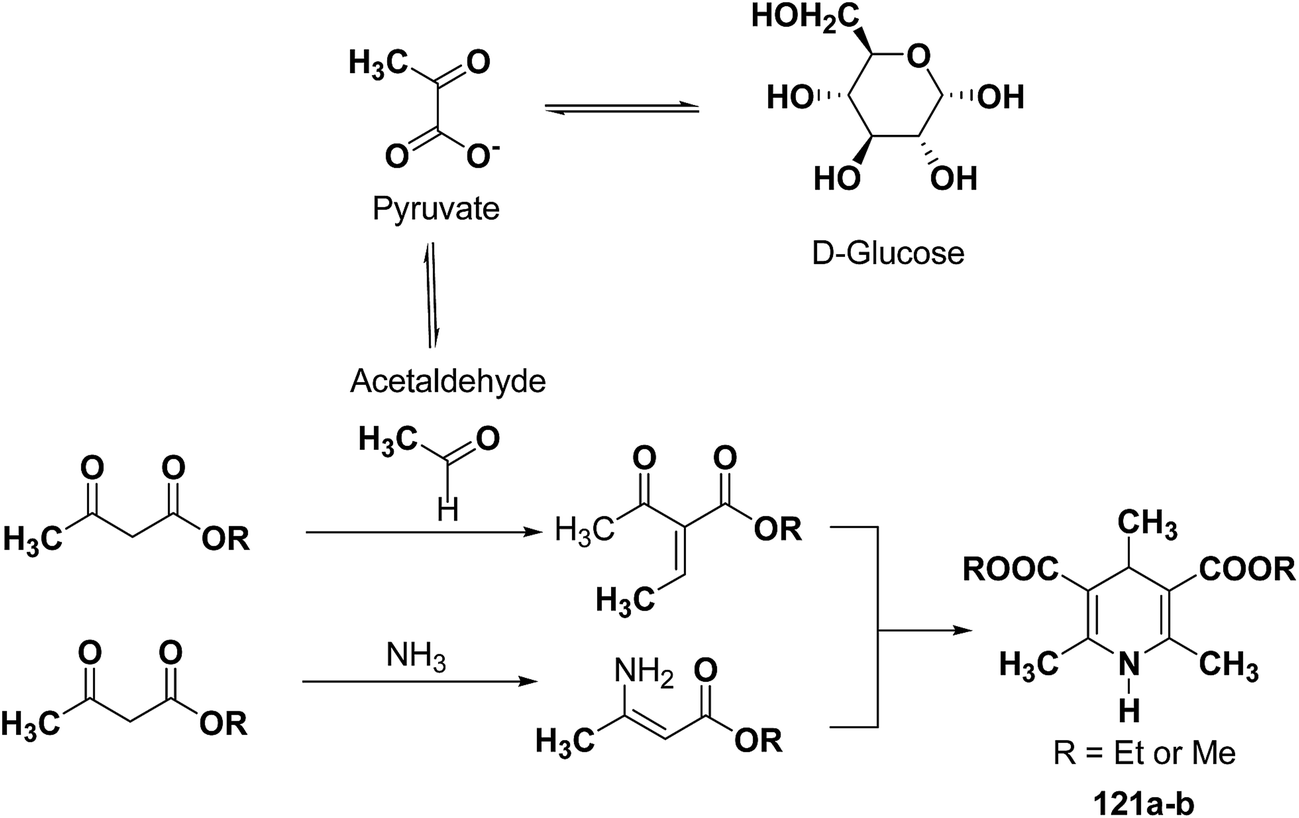
The glycolytic pathway from D-glucose to pyruvate is one of the most universal metabolic pathway known. In yeast, glycolysis is supposed to be the main pathway for the catabolism of glucose. According to the classical concept of glycolysis, metabolic acetaldehyde, resulting in the formation of acetoin, should be released from pyruvate in aerobic conditions. It is assumed that this acetaldehyde is involved in this Hantzsch-type reaction (Scheme 37).
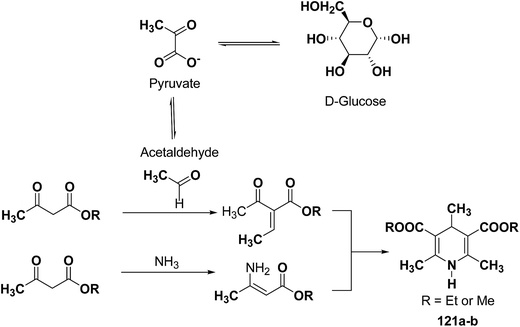 |
| | Scheme 37 Proposed mechanism for the baker's yeast catalysed synthesis of 1-4-DHPs. | |
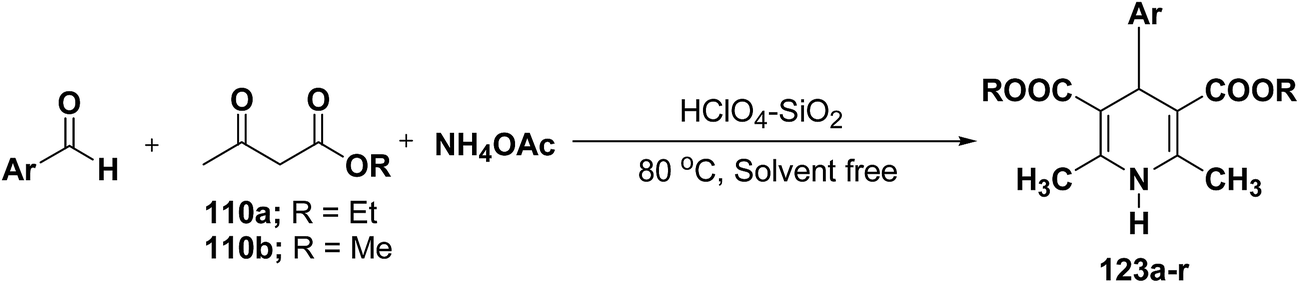
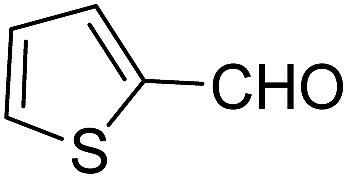
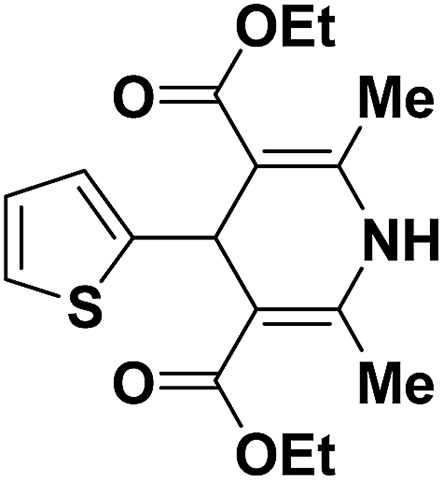
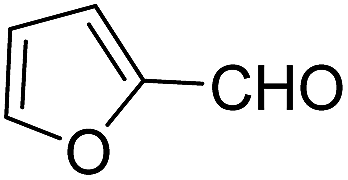
Sridhar, et al.46 have reported an efficient and convenient procedure for the one-pot synthesis 1,4-dihydropyridine derivatives 123a–r in excellent yields (Scheme 38) by the condensation of β-dicarbonyl compounds 110a or 110b, aldehydes and ammonium acetate, by using heterogeneous catalyst (HClO4–SiO2) under solvent-free conditions. The experimental procedure has been found very simple, convenient, and has the ability to tolerate a variety of other functional groups on phenyl ring of the aromatic aldehydes such as methoxy, nitro, hydroxyl and halides under the reaction conditions. Further the optimized reaction condition has been examined to prove the universality of this catalyst application. Various aromatic, aliphatic and heterocyclic aldehydes were selected to undergo the Hantzsch condensation in the presence of heterogeneous catalyst (HClO4–SiO2). The results of this study are summarized in (Table 10).
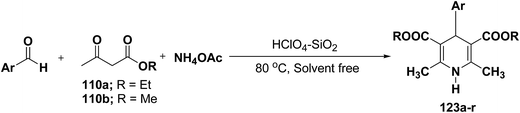 |
| | Scheme 38 Heterogeneous catalyst (HClO4–SiO2) catalyzed synthesis 1,4-DHPs 123. | |
Table 10 One-pot synthesis of 1,4-DHP under solvent free conditions using HClO4–SiO2a
| Entry |
(Ar) 123 |
110 |
Reaction time (min.) |
% yield |
| The structures of the products were determined from their spectroscopic (IR, NMR and MS) data. |
| 1 |
C6H5 (123a) |
110b |
20 |
95 |
| 2 |
p-CH3-C6H4 (123b) |
110b |
25 |
90 |
| 3 |
p-CH3O-C6H4 (123c) |
110b |
28 |
92 |
| 4 |
p-Cl-C6H4 (123d) |
110b |
20 |
89 |
| 5 |
p-OH-C6H4 (123e) |
110b |
26 |
90 |
| 6 |
o-NO2-C6H4 (123f) |
110b |
35 |
92 |
| 7 |
o-Cl-C6H4 (123g) |
110b |
26 |
87 |
| 8 |
p-NO2-C6H4 (123h) |
110a |
33 |
92 |
| 9 |
o-CH3O-C6H4 (123i) |
110a |
42 |
90 |
| 10 |
p-Br-C6H4 (123j) |
110a |
40 |
92 |
| 11 |
o-Br-C6H4 (123k) |
110a |
40 |
89 |
| 12 |
2-Furyl (123l) |
110a |
52 |
86 |
| 13 |
2-Thienyl (123m) |
110a |
56 |
90 |
| 14 |
3-Pyridyl (123n) |
110a |
55 |
82 |
| 15 |
C6H5 (123o) |
110b |
20 |
94 |
| 16 |
p-CH3-C6H4 (123p) |
110b |
25 |
92 |
| 17 |
o-CH3O-C6H4 (123q) |
110b |
25 |
90 |
| 18 |
o-NO2-C6H4 (123r) |
110b |
22 |
91 |
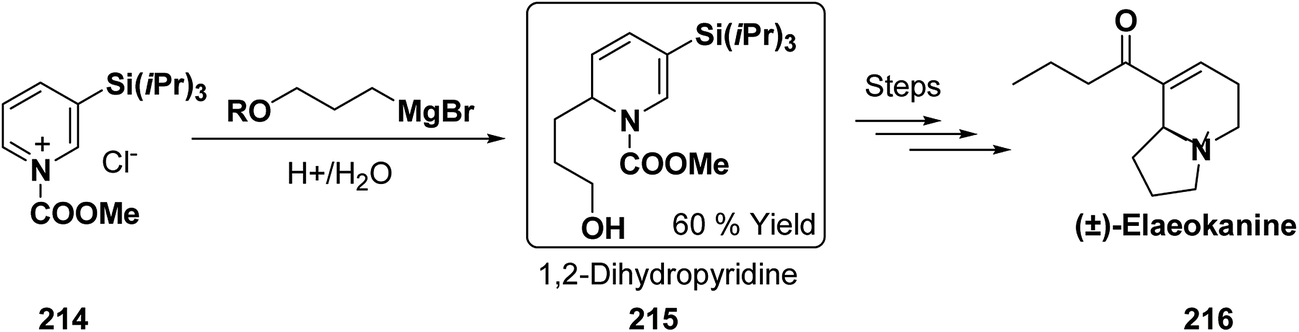
Wanner, et al.47 have synthesized N-silyl protected 1,4-DHP 126a–n by the addition reaction of dialkylmagnesium reagents (R2Mg) to N-triisopropylsilylpyridinium ions 125a–b (Scheme 39). The N-triisopropylsilylpyridinium ions 125a–b were generated in situ by the reaction of 4-substituted pyridines 124a–b and triisopropylpyridinium-triflate (TIPS-OTf). The regioselectivity of 4-addition product of this reaction was quite satisfactory (Table 11, entries 1, 2, 3, 6, 7, 9, 10, 11 and 12).
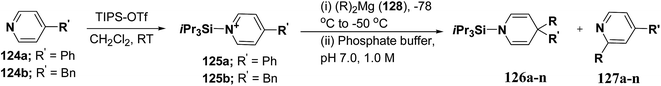 |
| | Scheme 39 Synthesis of 1,4-DHPs 126a–n via addition reaction of Grignard reagent to N-triisopropylsilylpyridinium ions 125a–b. | |
Table 11 Addition of various dialkylmagnesium reagents to N-triisopropylsilylpyridinium ions 125a–b
| Entry |
125 |
R2Mg (128)a |
Product |
% yieldb |
Product ratio 126/127/125 (%)c |
| After addition of the organomagnesium compound R2Mg to the N-silylpyridinium ion 125 at −78 °C the mixture was slowly warmed to −50 °C. Isolated yield. According to 1H NMR of the crude reaction product. Sum of 127 and non oxidized 1,2-addition product, which were both present. Yield 63%, ratio 86/2/12 when addition performed by warming the mixture from −78 °C to room temperature. Yield 29%, ratio 32/0/68 when addition performed by warming the mixture from −78 °C to room temperature. |
| 1 |
125a |
Et2Mg |
126a, 127a |
78 |
90/4/6 |
| 2 |
125a |
nBu2Mg |
126b, 127b |
82 |
100/0/0 |
| 3 |
125a |
Bn2Mg |
126c, 127c |
92 |
95/2/3 |
| 4 |
125a |
Ph2Mg |
126d, 127d |
0 |
5/95d/0 |
| 5 |
125a |
Me2Mg |
126e, 127e |
0 |
0/59/41 |
| 6 |
125a |
iPr2Mg |
126f, 127f |
91 |
99/0/1 |
| 7 |
125a |
tBu2Mg |
126g, 127g |
56 |
80/3/17e |
| 8 |
125a |
Allyl2Mg |
126h, 127h |
20 |
20/78d/2 |
| 9 |
125b |
Et2Mg |
126i, 127i |
73 |
86/2/12 |
| 10 |
125b |
nBu2Mg |
126j, 127j |
54 |
54/6/40 |
| 11 |
125b |
Bn2Mg |
126k, 127k |
85 |
95/0/5 |
| 12 |
125b |
iPr2Mg |
126l, 127l |
78 |
100/0/0 |
| 13 |
125b |
tBu2Mg |
126m, 127m |
5 |
6/0/94f |
| 14 |
125b |
Allyl2Mg |
126n, 127n |
12 |
21/70/9 |
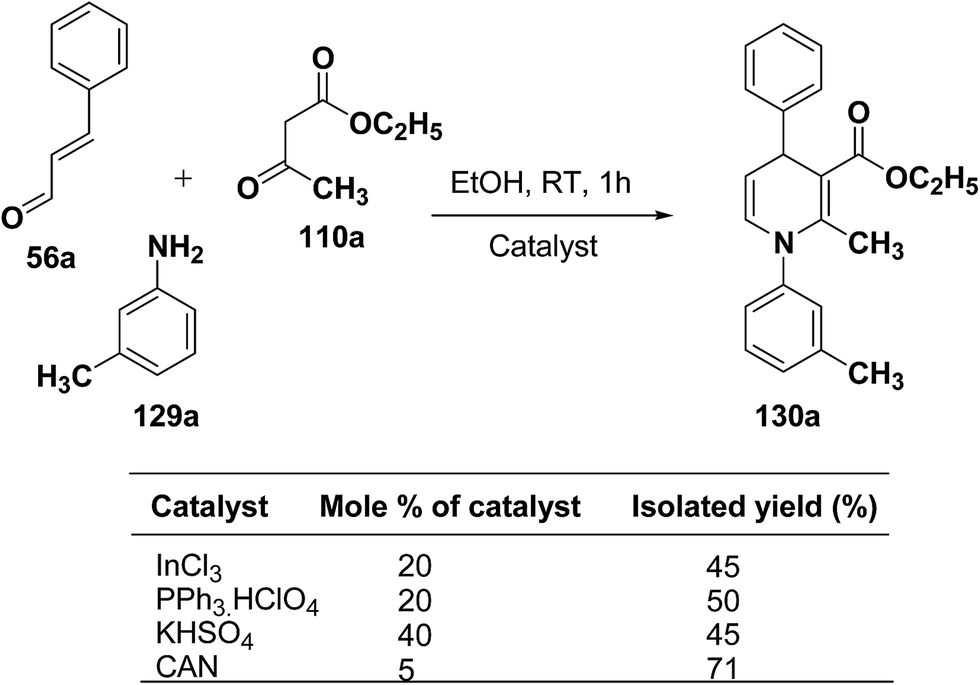
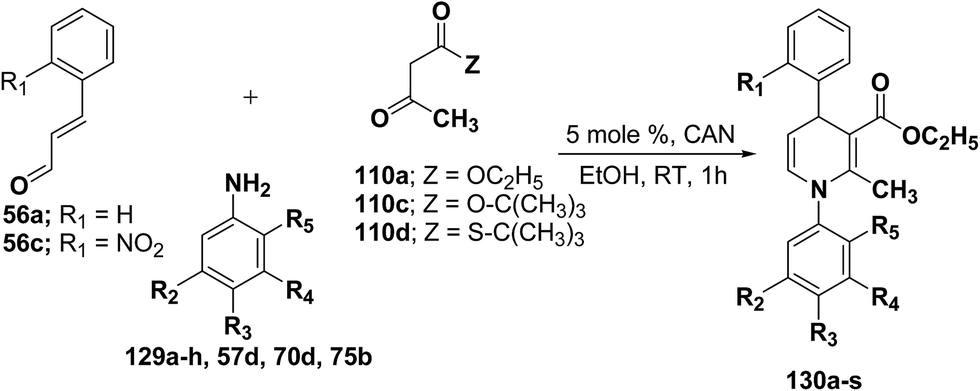
Perumal, et al.48–50 has carried out an initial study in search for the suitable catalyst and solvent for the synthesis of 1,4-DHP using cinnamaldehyde 56a, m-toluidine 129a and ethyl acetoacetate 110a. Author first assayed indium trichloride,44 triphenylphosphonium perchlorate,45 and potassium hydrogen sulfate as catalysts,46 which have been successfully employed in related condensation reactions, but the yields obtained were only moderate, even with high catalyst loadings. The best results were obtained with cerium ammonium nitrate (CAN) in ethanol, which gave the desired dihydropyridine derivative in 71% isolated yield (Scheme 40). Further Perumal, et al.51 have developed cerium ammonium nitrate (CAN) catalyzed three-component domino reaction between aromatic amines 57d, 70d & 129a–h, α,β-unsaturated aldehydes 56a & 56c and alkyl acetoacetate 110a, 110c & 110d to afford the 1,4-dihydropyridines 130a–s in moderate to good yields (Scheme 41, Table 12).
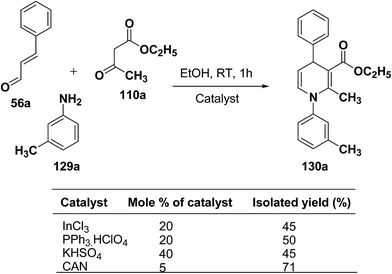 |
| | Scheme 40 Screening of the catalysts for the three-component domino reaction between 56a, 129a and 110a. | |
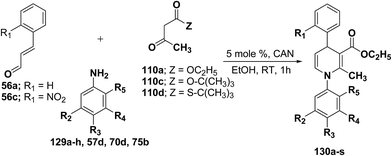 |
| | Scheme 41 Three-component domino reaction between 56, 110 and 129a–h, 57d & 70d. | |
Table 12 Scope and yields of the CAN-catalyzed synthesis of 1,4-dihydropyridines 130a–s
| Entry |
Compound |
110 |
56 |
R2 |
R3 |
R4 |
R5 |
Amine |
Time (h) |
Yield (%) |
| Isolated as an 1.1:1 rotamer mixture. |
| 1 |
130a |
110a |
56a |
CH3 |
H |
H |
H |
129a |
1 |
74 |
| 2 |
130b |
110a |
56a |
H |
H |
H |
H |
57d |
1 |
71 |
| 3 |
130c |
110a |
56a |
H |
CH3 |
H |
H |
129b |
1 |
70 |
| 4 |
130d |
110a |
56a |
H |
F |
H |
H |
129c |
1 |
76 |
| 5 |
130e |
110a |
56a |
H |
Cl |
H |
H |
70d |
1 |
74 |
| 6 |
130f |
110a |
56a |
CH3 |
CH3 |
H |
H |
129d |
1 |
70 |
| 7 |
130g |
110a |
56a |
Cl |
H |
H |
H |
129e |
1 |
72 |
| 8 |
130h |
110a |
56a |
H |
Br |
H |
H |
75b |
1 |
74 |
| 9 |
130i |
110a |
56a |
CH3 |
H |
CH3 |
H |
129f |
1 |
72 |
| 10 |
130j |
110a |
56a |
OCH3 |
H |
H |
H |
129g |
1 |
65 |
| 11 |
130k |
110a |
56c |
H |
H |
H |
H |
57d |
1 |
50 |
| 12 |
130la |
110a |
56a |
H |
CH3 |
H |
CH3 |
129h |
1 |
72 |
| 13 |
130m |
110c |
56a |
H |
H |
H |
H |
57d |
2 |
52 |
| 14 |
130n |
110c |
56a |
H |
CH3 |
H |
H |
129b |
2 |
61 |
| 15 |
130o |
110c |
56a |
H |
Cl |
H |
H |
70d |
2 |
65 |
| 16 |
130p |
110d |
56a |
H |
H |
H |
H |
57d |
1 |
61 |
| 17 |
130q |
110d |
56a |
H |
F |
H |
H |
129c |
1 |
62 |
| 18 |
130r |
110d |
56a |
H |
Cl |
H |
H |
70d |
1 |
63 |
| 19 |
130s |
110d |
56a |
H |
CH3 |
H |
H |
129b |
1 |
61 |
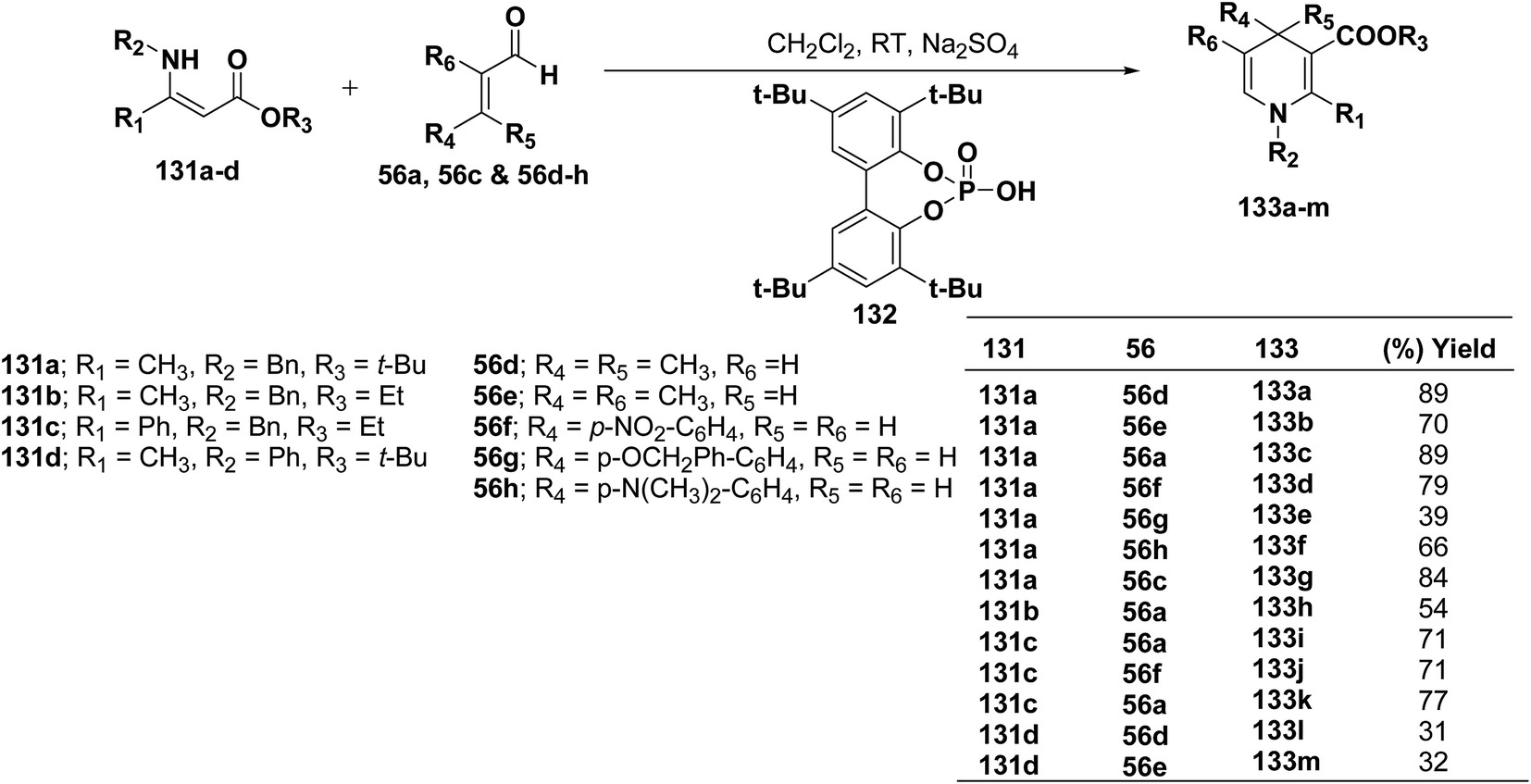
Renaud, et al.52 has developed Brønsted acid 132 catalyzed addition of β-enaminoacrylates 131a–d to α,β-unsaturated aldehydes 56a, 56c and 56d–h to afford the corresponding N-substituted 1,4-dihydropyridines 133a–m in 31–89% yields (Scheme 42).
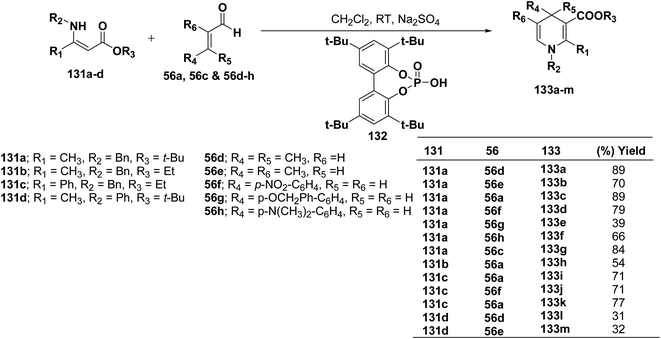 |
| | Scheme 42 Synthesis of 1,4-dihydropyridines 133a–m catalyzed by Brønsted acid 132. | |
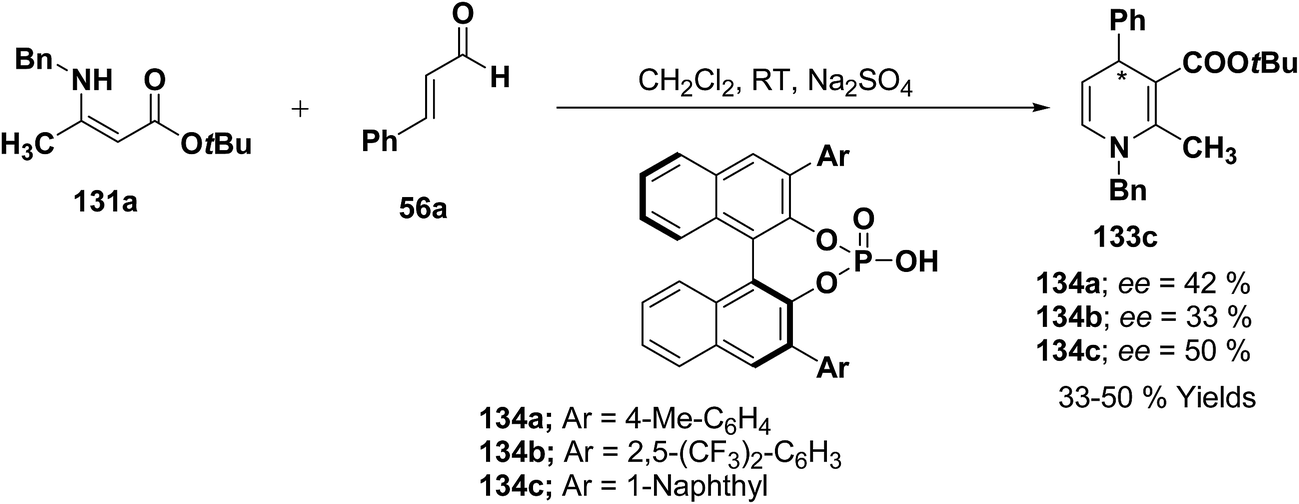
Further author have also reported the enantioselective synthesis of 1,4-dihydropyridine 133c starting from 131a and 56a using three different chiral Brønsted acids 134a–c in ee 33–50% (Scheme 43).
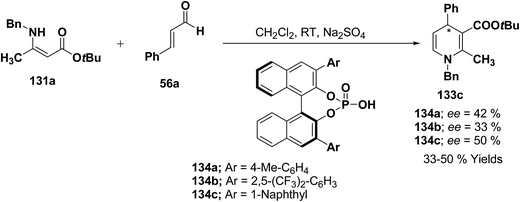 |
| | Scheme 43 Enantioselective synthesis of 1,4-dihydropyridine 133c catalyzed by chiral Brønsted acids 134a–c. | |
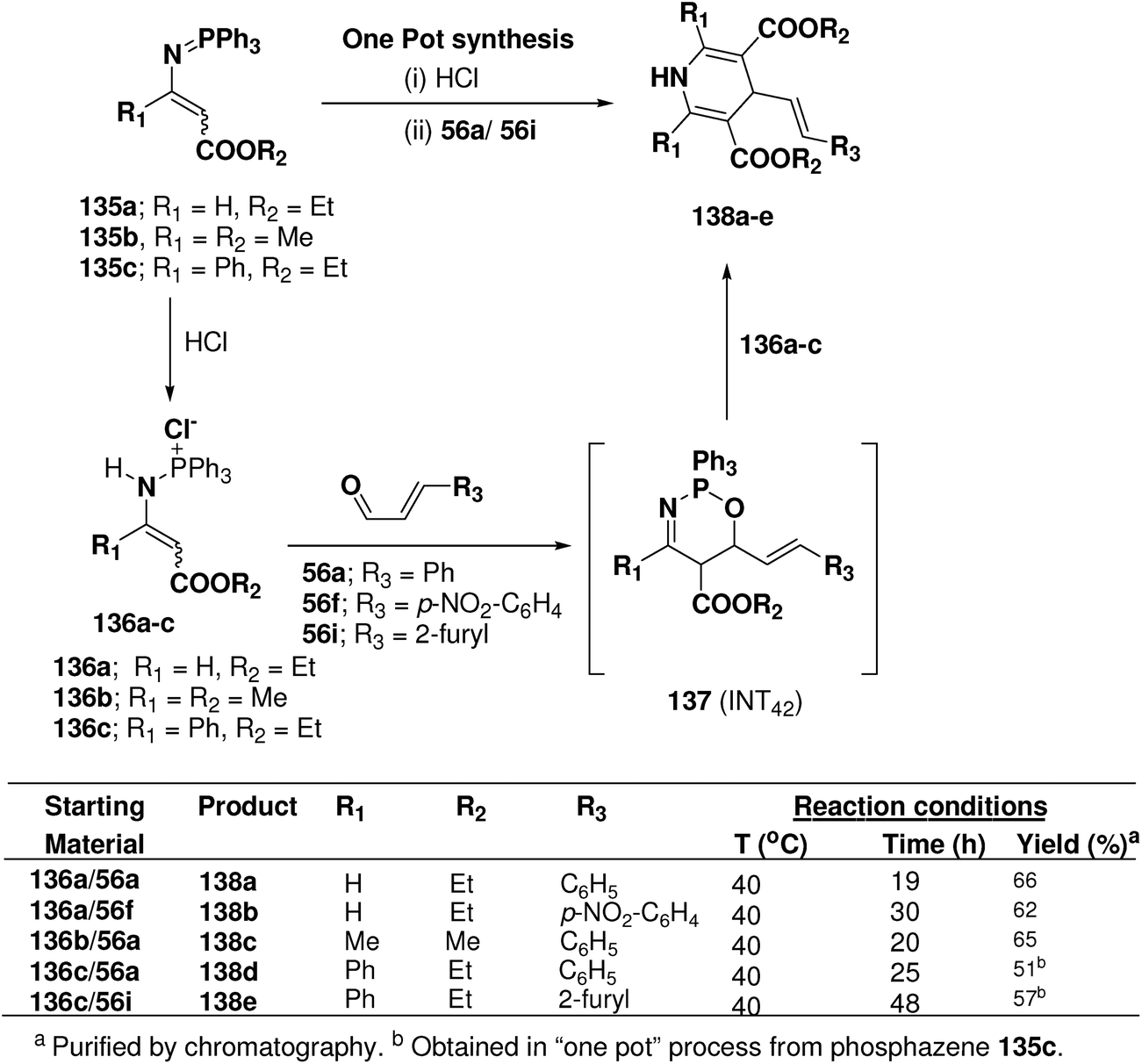
Palacios, et al.53 explored the reactivity of enaminophosphonium salts 136a–c derived from phosphazenes 135a–c with α,β-unsaturated aldehydes 56a, 56f and 56i (Scheme 44). The reaction of enaminophosphonium salt 135a, prepared by hydrochloric acid treatment of N-vinylic phosphazene 135a, with cinnamaldehyde 56a or p-nitrocinnamaldehyde 56f in refluxing CH2Cl2 led to the formation of 4-(2-phenylethenyl)-3,5-diethoxycarbonyl-1,4-dihydropyridine 138a and 4-[2-(4-nitrophenyl)ethenyl]-3,5-diethoxycarbonyl-1,4-dihydropyridine 138b in 66 and 62% yields, respectively. The formation of these dihydropyridines 138 could be explained by means of a formal [4 + 2]-cyclization process involving an initial 1,4-addition of the γ-carbon atom of the enaminophosphonium salt 136a to the carbonyl group of the aldehydes 56a to give the cycloadduct intermediate 1,2,5-oxaazaphosphoranes 137 (INT42) followed by regioselective attack of a second molecule of the phosphonium salt 130a (Scheme 44). 1,4-DHPs 138d and 138e were also obtained in “one pot” process from phosphazene 135c in 51 and 57% yields, respectively.
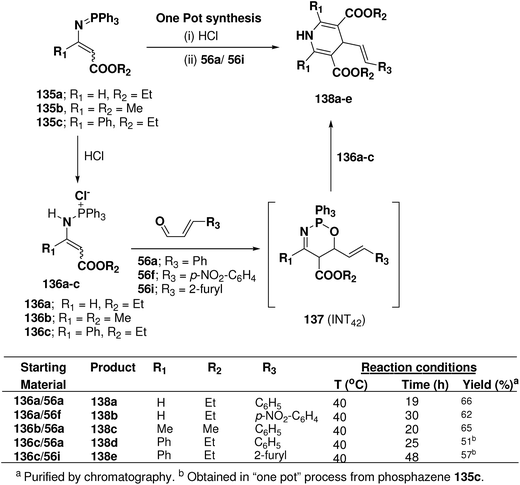 |
| | Scheme 44 Reaction of enamino-phosphonium salts 136 with α,β-unsaturated aldehydes 55a, 56f and 56i: preparation of symmetrical dihydropyridines 138a–e. | |
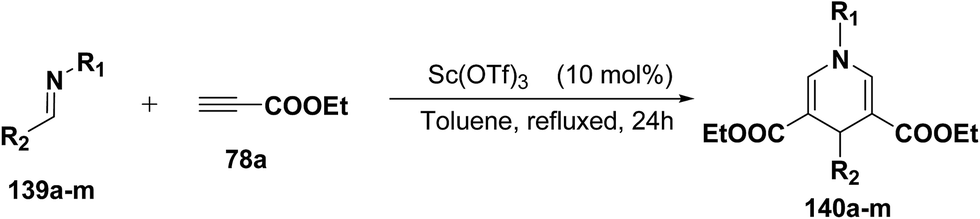
Fukuzawa, et al.54 have developed a procedure for the synthesis of N-substituted-1,4-dihydropyridines 140a–m in moderate to good yields by scandium(III) triflate catalyzed reaction of imines 139a–m with ethyl propiolate 78a (2.5 equiv.) in toluene or benzotrifluoride (BTF) under refluxed conditions (Table 13).
Table 13 The reaction of imines 139a–m with 78aa
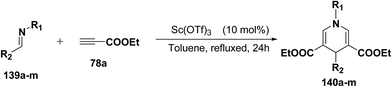
|
| Entry |
Imine |
R1 |
R2 |
Product 140 |
Yieldb (%) |
| 133 (0.5 mmol), 78a (1.25 mmol), Sc(OTf) (0.05 mmol) in toluene (5 mL) at reflux for 24 h. Isolated yield. GC yield. BTF was used as a solvent instead of toluene. |
| 1 |
139a |
c-Hex |
Ph |
140a |
22c |
| 2 |
139b |
t-Bu |
Ph |
140b |
18 |
| 3d |
139c |
Bn |
Ph |
140c |
28 |
| 4 |
139d |
CHPh2 |
Ph |
140d |
35 |
| 5 |
139e |
Ph |
Ph |
140e |
45c |
| 6 |
139f |
p-MeOC6H4 |
Ph |
140f |
75c |
| 7d |
139g |
p-MeC6H4 |
Ph |
140g |
62c |
| 8d |
139h |
2,6-MeC6H3 |
Ph |
140h |
77 |
| 9 |
139i |
p-MeOC6H4 |
p-MeC6H4 |
140i |
78 |
| 10 |
139j |
p-MeOC6H4 |
p-CI-C6H4 |
140j |
54 |
| 11 |
139k |
p-MeOC6H4 |
p-FC6H4 |
140k |
47 |
| 12 |
139l |
p-MeOC6H4 |
p-NO2C6H4 |
140l |
34 |
| 13 |
139m |
p-MeOC6H4 |
PhCH2CH2 |
140m |
0 |
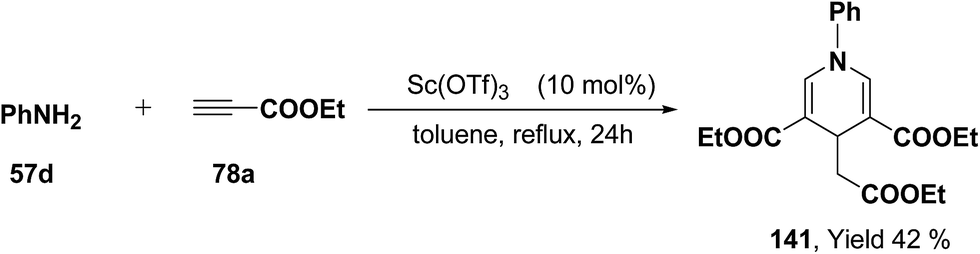
Further, it has also been reported by the author's that the Sc(OTf)3 catalyzed reaction of aniline 57d and ethyl propiolate 78a (3.2 equiv.) yielded another 1,4-dihydropyridine 141 bearing three ester groups in 42% yield under the same reaction conditions (Scheme 45).
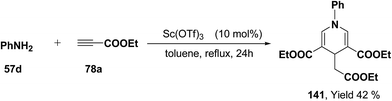 |
| | Scheme 45 Sc(OTf)3 catalyzed reaction of aniline 57d and 78a: preparation of 1,4-DHP 141. | |
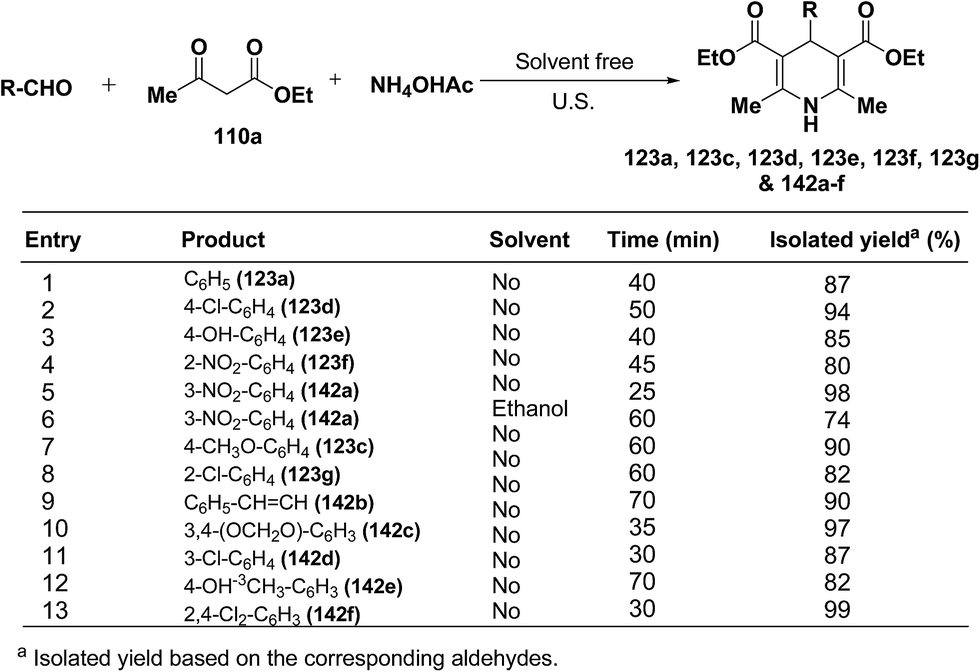
Wang, et al.55 has developed a three-component one pot condensation of aldehydes, ethyl acetoacetate 110a and ammonium acetate to afford the 1,4-dihydropyridines 123a, 123c, 123d, 123e, 123f, 123g and 142a–f in 82–99% yields under ultrasound irradiation without solvent and catalyst at room temperature (Scheme 46). The main advantages of this procedure are mild reaction condition, shorter reaction time and high product yield.
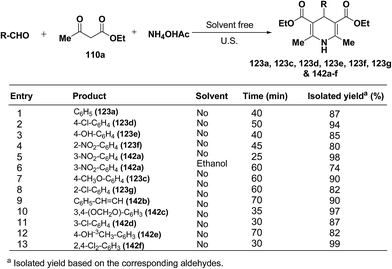 |
| | Scheme 46 Synthesis of 1,4-dihydropyridines under ultrasound at room temperature. | |
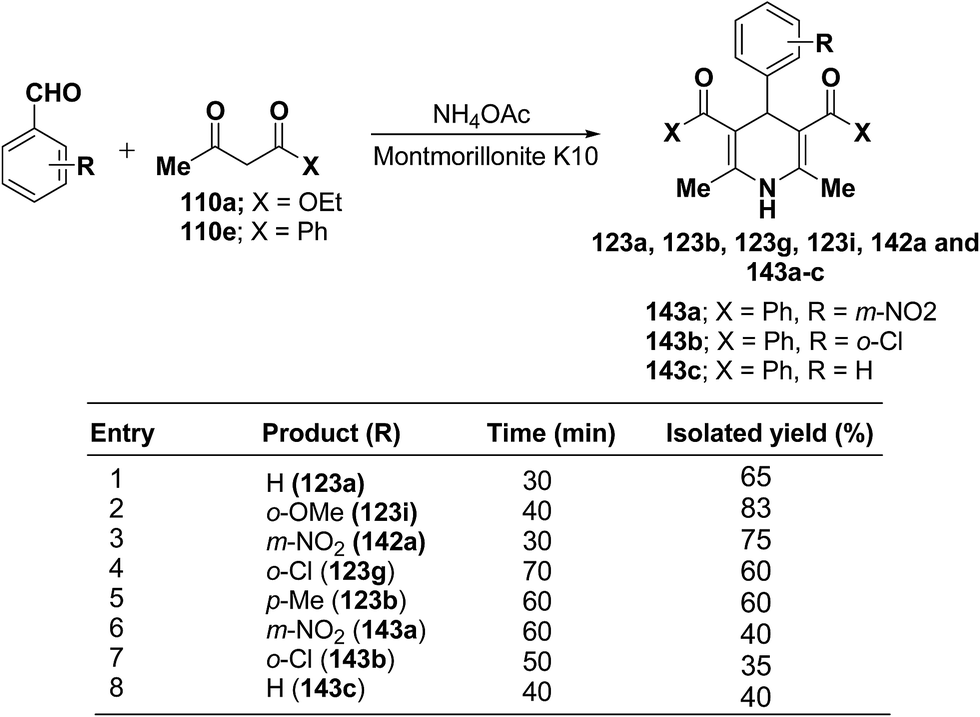
Zonouz, et al.56 have developed a three-component one pot condensation of aldehydes ethylacetoacetate 110a/phenylacetylacetone 110e and NH4OAc in the presence of a solid catalyst (montmorillonite K10 clay) to afford 1,4-dihydropyridines 123a, 123b, 123g, 123i, 142a and 143a–c in good yields (Scheme 47). This procedure has advantages such as short reaction time, high yield and simple reaction workup conditions.
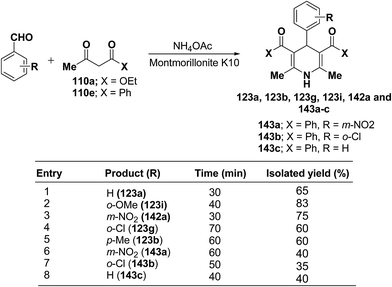 |
| | Scheme 47 Synthesis of 1,4-dihydropyridines catalyzed by montmorillonite K10 clay. | |
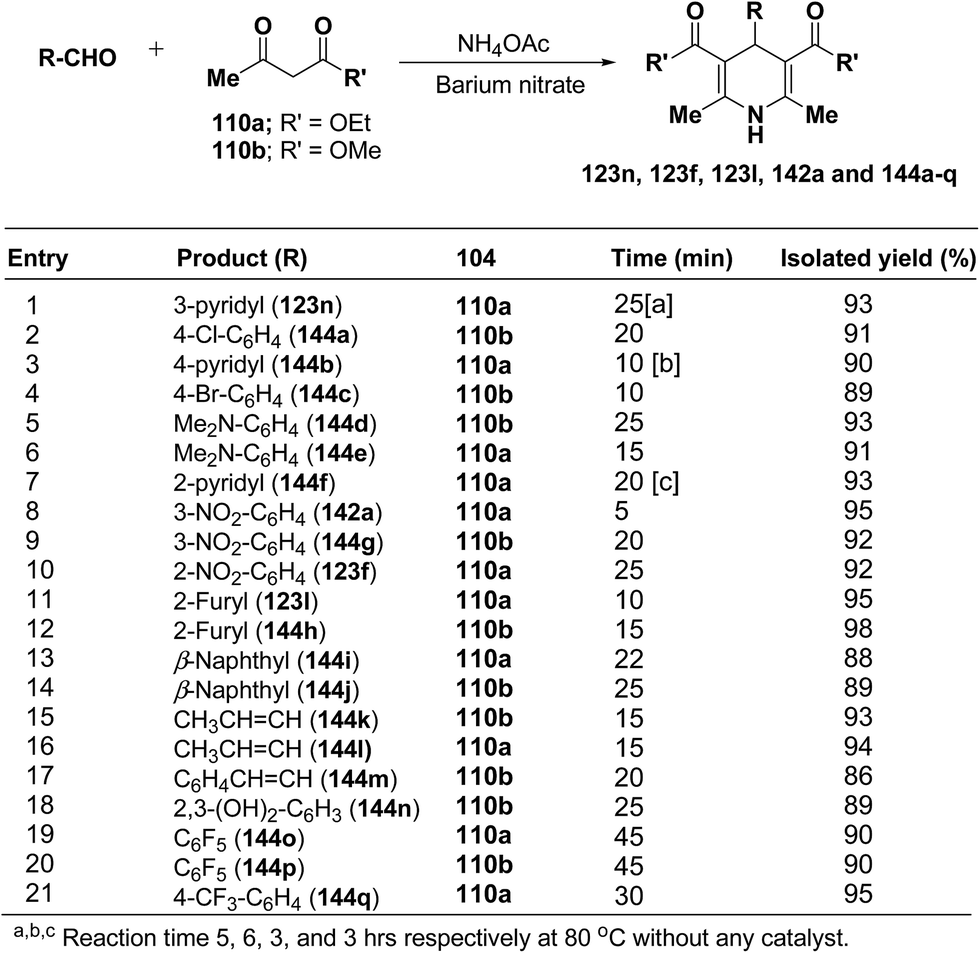
Rawat, et al.57 have developed an efficient methodology for the three-component one pot condensation of aldehydes, alkylacetoacetate 110a/110b and NH4OAc using barium nitrate as a efficient catalyst to afford 1,4-dihydropyridines 123n, 123f, 123l, 142a and 144a–q (Scheme 48).
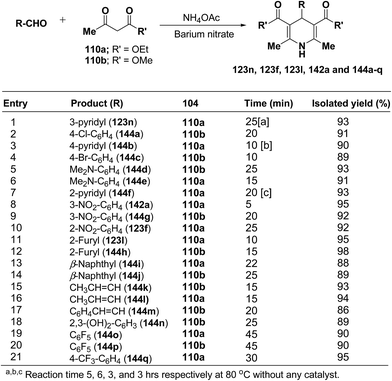 |
| | Scheme 48 Synthesis of 1,4-dihydropyridines in presence of 5 mol% of the barium nitrate. | |
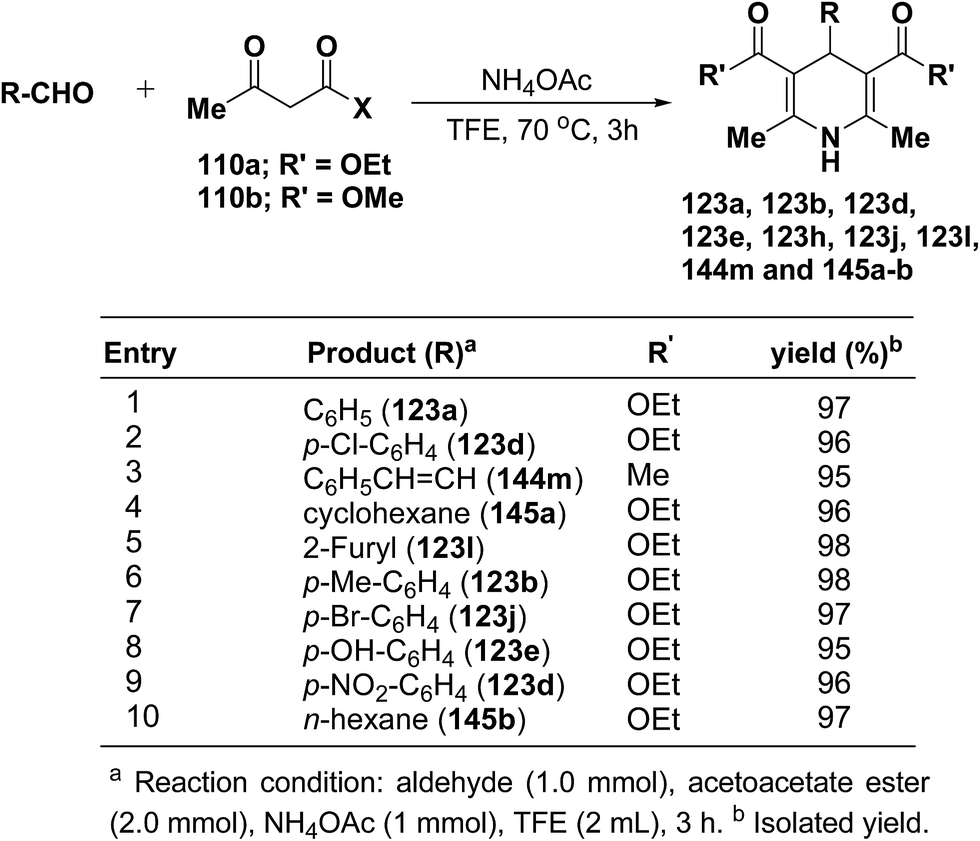
Heydari, et al.58 has synthesized 1,4-dihydropyridines 123a, 123b, 123d, 123e, 123h, 123j, 123l, 144m and 145a–b in excellent yields by the reaction of aldehydes, alkyl acetoacetate 110a–b, and ammonium acetate (Scheme 49) at 70 °C in trifluoroethanol (TFE). The advantage of this reaction was the solvent (TFE), which can be readily separated from reaction products and recovered in excellent purity for reuse.
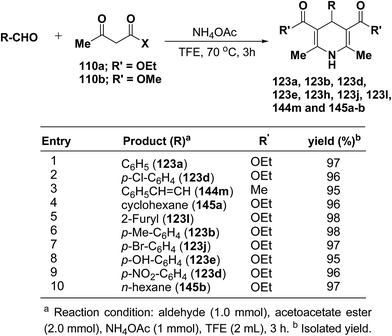 |
| | Scheme 49 Synthesis of 1,4-dihydropyridines through Hantzsch reaction in TFE. | |
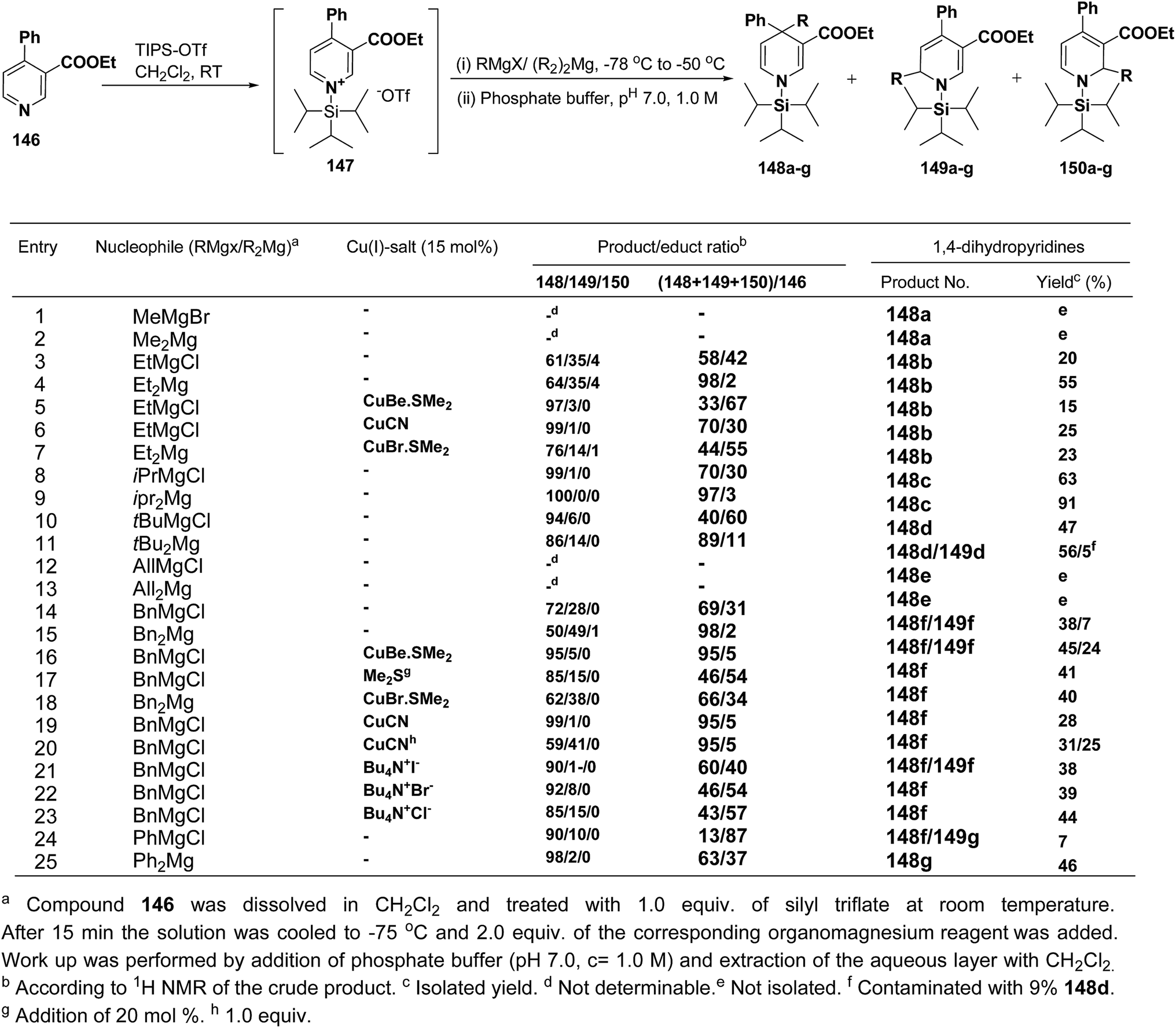
Wanner, et al.59 have developed a methodology for the synthesis of 4,4-disubstituted 1,4-dihydronicotinates 148a–g by the regioselective addition of Grignard reagents RMgX or R2Mg to nicotinic acid ester 146, activated with triisopropylsilyl triflate 147 (Scheme 50). The regioselectivity of this reaction, where 4-unsubstituted and 4-substituted pyridine derivatives were employed as starting materials, was examined. Depending on the structure of the organomagnesium reagent varying ratios of 1,2- (149a–g), 1,4- (148a–g), and 1,6-regioisomers (150a–g) were obtained but in all the cases 1,4-addition products were predominating one.
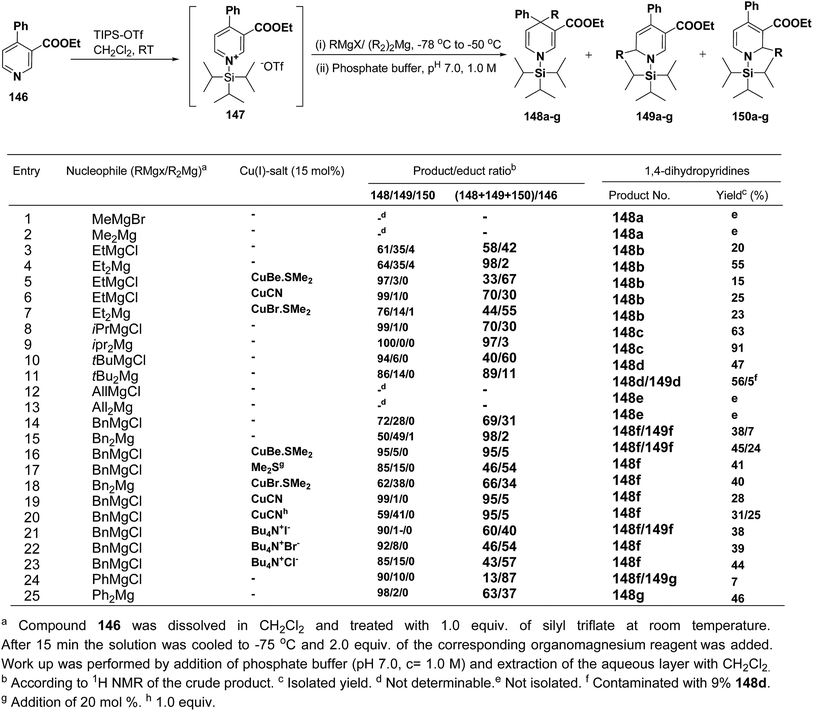 |
| | Scheme 50 Addition of various organomagnesium reagents to N-triisopropylsilylpyridinium ions 147: preparation of 1,4-dihydropyridines. | |
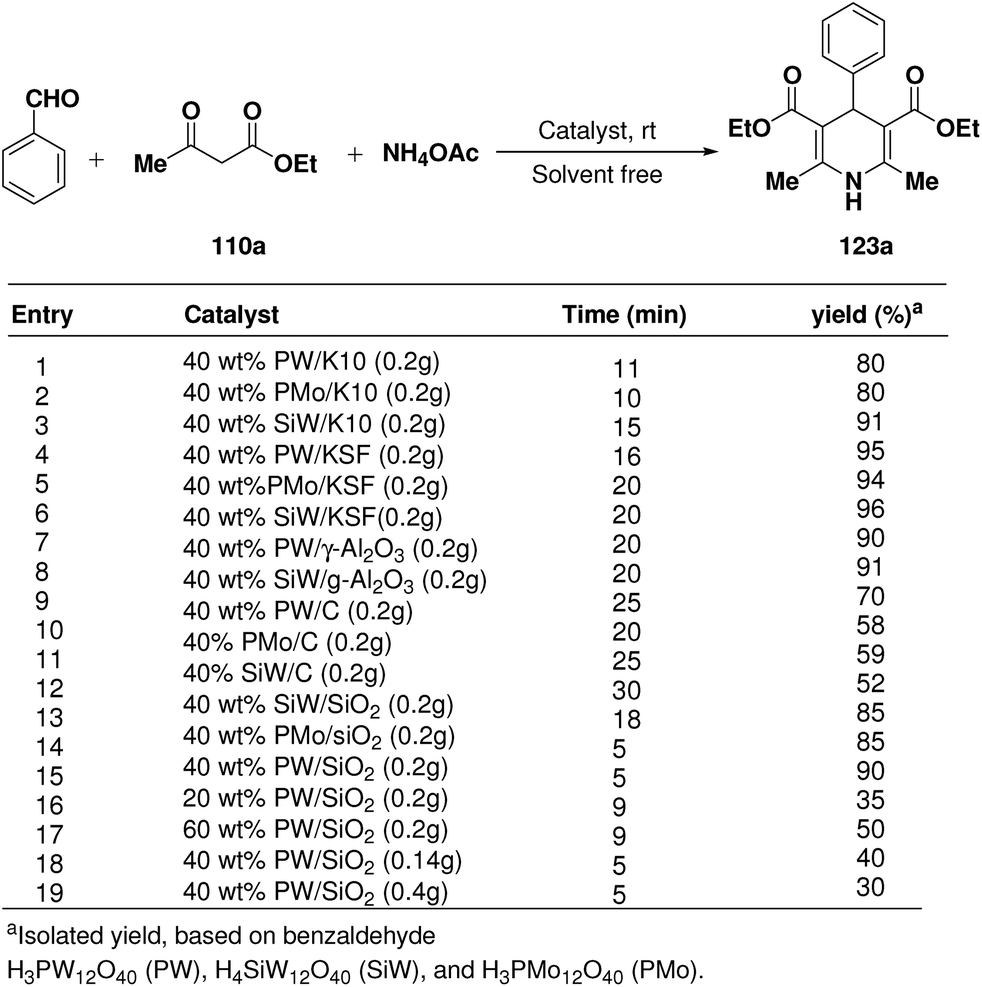
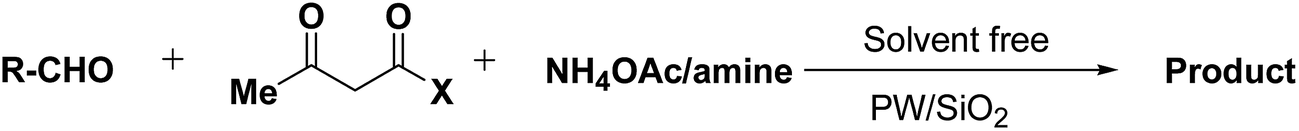
Rafiee, et al.60 have prepared 12-tungstophosphoric acid (PW) supported on different metal oxides and their catalytic performances have been evaluated in the three component condensation of benzaldehyde, ethyl acetoacetate 110a and ammonium acetate to afford the corresponding 1,4-dihydropyridine 123a (Scheme 51). A high catalytic activity was found over silica supported PW. Effect of PW loading, catalyst loading and solvent has been studied by the author to find out the best reaction condition. 40% PW onto SiO2 (0.2 g) under solvent-free condition has been found to be the best catalyst for the synthesis of 1,4-dihydropyridines.
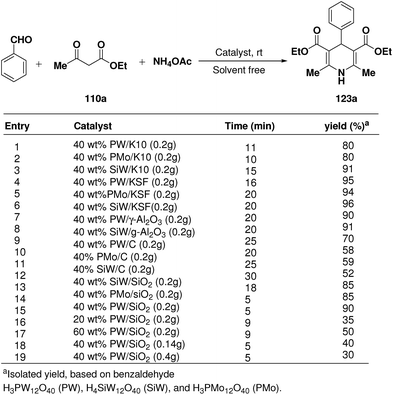 |
| | Scheme 51 Effect of different catalyst on the yield of 1,4-dihydropyridine. | |
A series of 4-aryl, N-alkyl, and N-aryl substituted 1,4-dihydropyridines 123a, 123b, 123m, 123l, 123d, 142b and 151a–f have been synthesized using different aldehydes in good to excellent yield in short reaction times (Table 14).
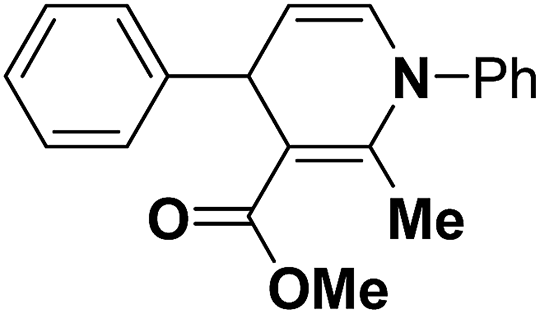
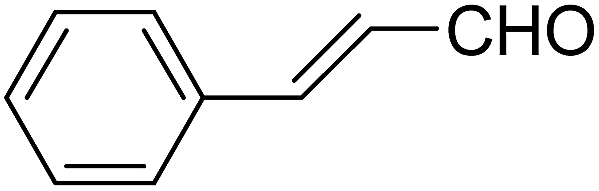
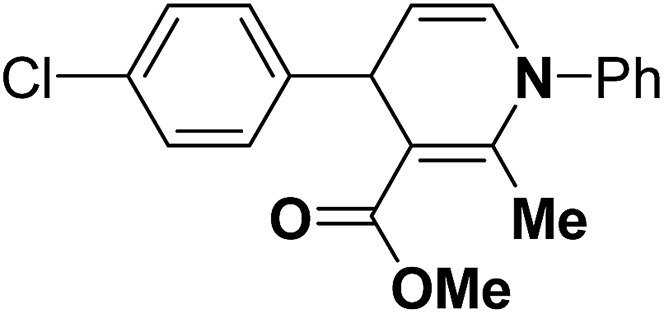
Table 14 Synthesis of Hantzsch 1,4-dihydropyridines by the condensation of aldehydes, β-dicarbonyl compounds and ammonium acetate/amines using PW/SiO2 as catalyst
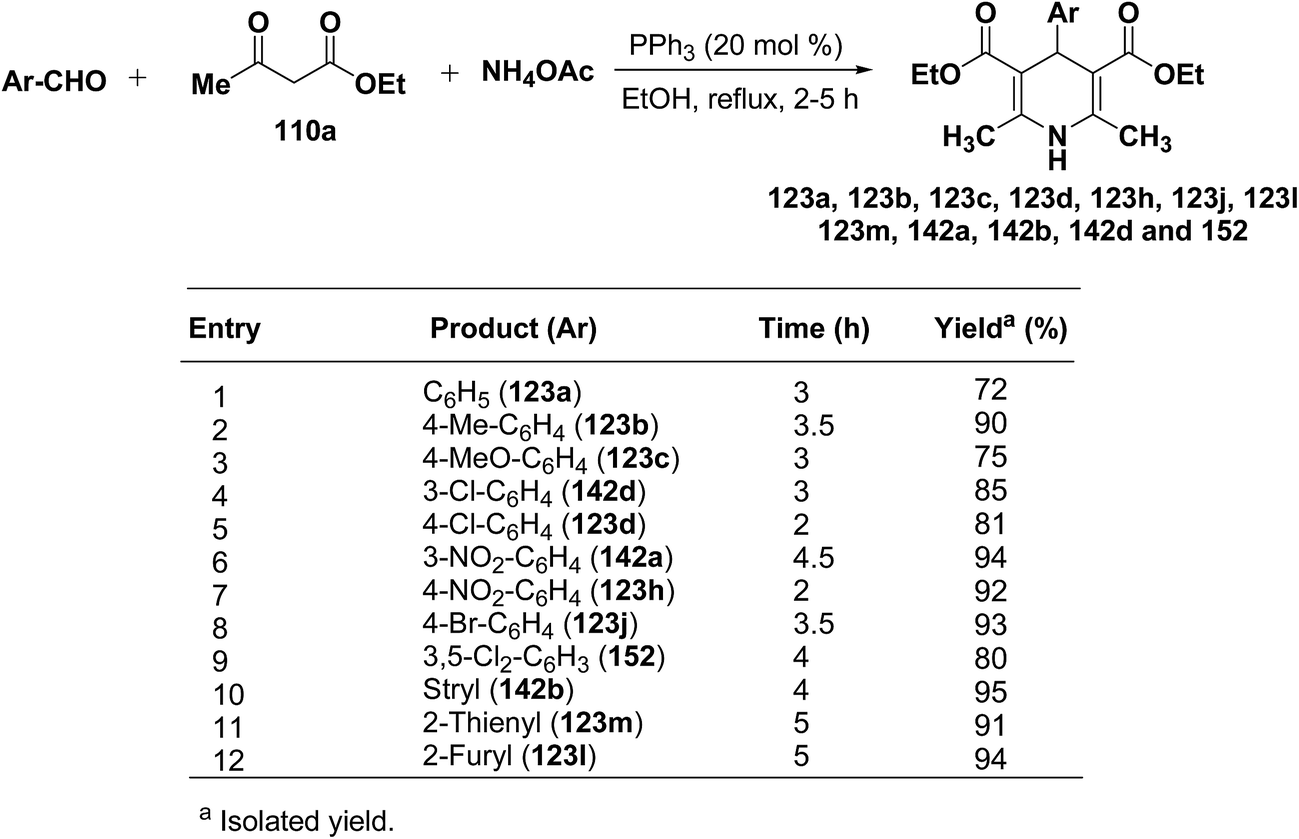
Debache, et al.61 have developed triphenylphosphine-catalyzed Hantzsch three-component reaction of an aromatic aldehydes, ethyl acetoacetate 110a and ammonium acetate to afford Hantzsch 1,4-dihydropyridines 123a, 123b, 123c, 123d, 123h, 123j, 123l, 123m, 142a, 142b, 142d and 152 in good to excellent yields (Scheme 52).
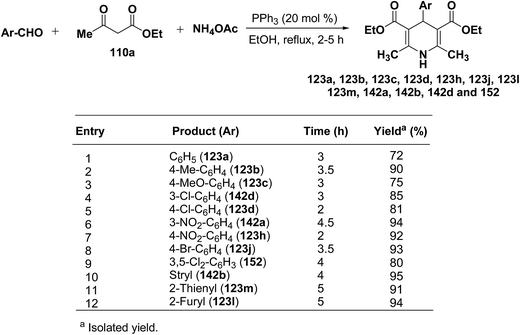 |
| | Scheme 52 Triphenylphosphine-catalyzed Hantzsch synthesis of 1,4-dihydropyridines. | |
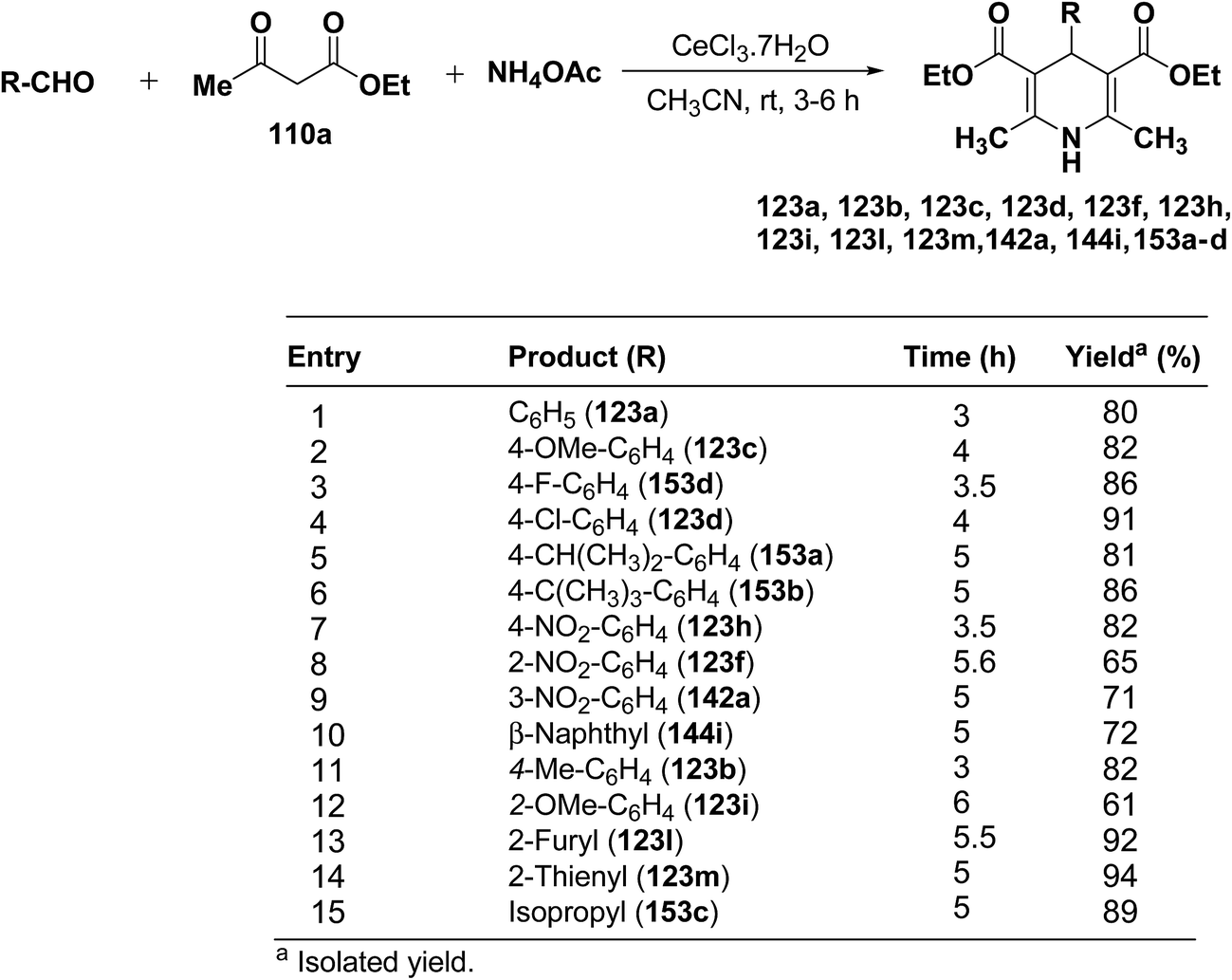
Yadav, et al.62 have developed an efficient one-pot synthesis of 1,4-dihydropyridines 123a, 123b, 123c, 123d, 123f, 123h, 123i, 123l, 123m, 142a, 144i, 153a–d in good to excellent yields via the CeCl3·7H2O-catalyzed Hantzsch three-component reaction of an aromatic aldehydes, ethyl acetoacetate 110a and ammonium acetate (Scheme 53).
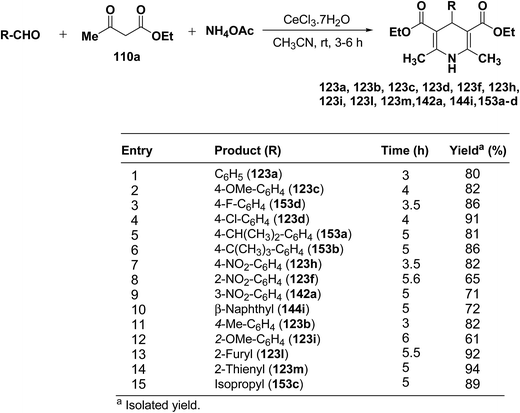 |
| | Scheme 53 CeCl3·7H2O-catalyzed synthesis of Hantzsch 1,4-dihydropyridines. | |
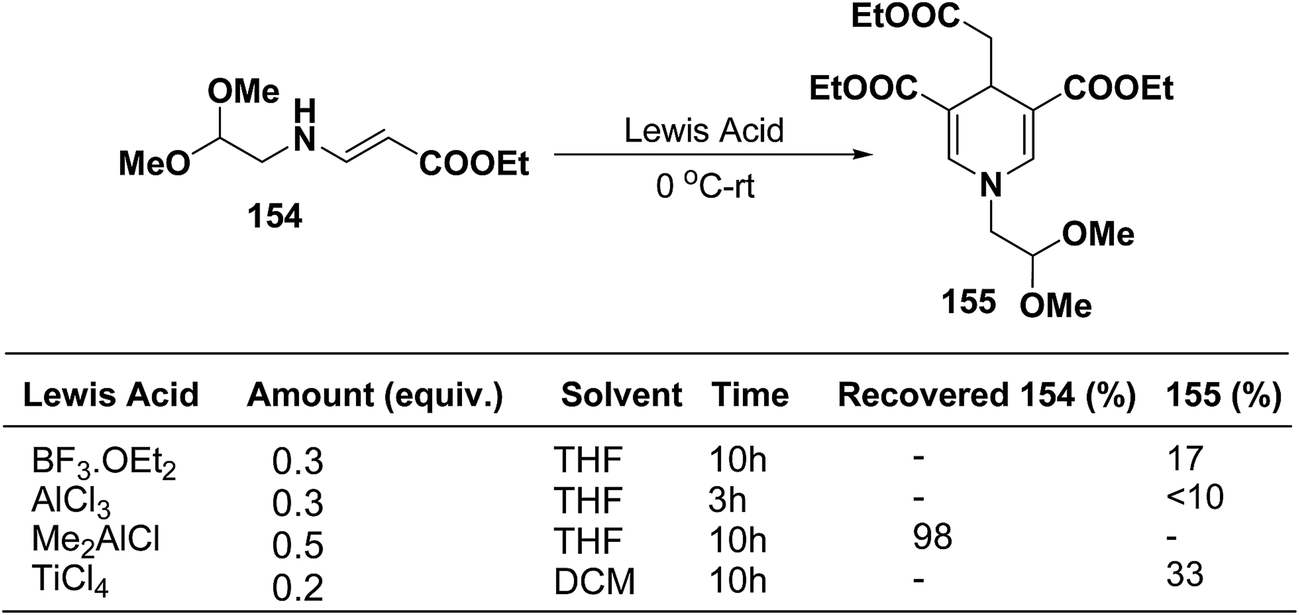
Ajavakom, et al.63 have screened several Lewis acid catalysts such as BF3·OEt2, AlCl3, and TiCl4 on β-amino acrylates 154, for the synthesis of 1,4-dihydropyridine 155 (Scheme 54). The yield of product 155 obtained from these initial screening of the Lewis acid catalyst were not satisfactory, which may be likely due to the decomposition of the acetal group in β-amino acrylate 154 under acidic conditions. Out of the several screened Lewis acid catalyst TiCl4 in dichloromethane was found to be best catalyst for the synthesis of 1,4-dihydropyridine 155.
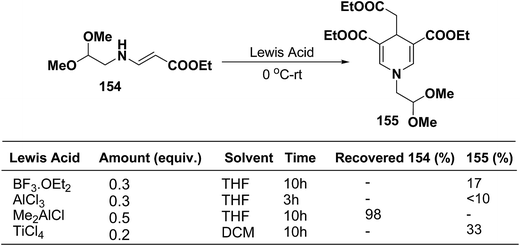 |
| | Scheme 54 Optimization of Lewis acid catalysts for the synthesis of 1,4-dihydropyridine 155. | |
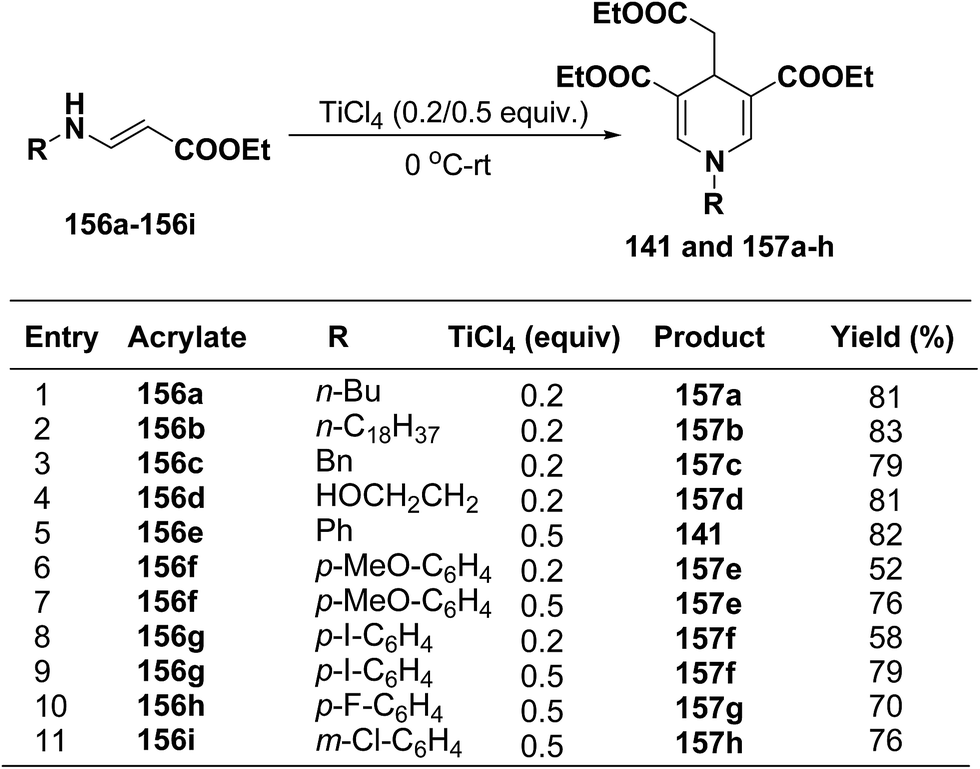
Several 1,4-dihydropyridines derivatives 141 and 157a–h has been synthesized from β-amino acrylates 156a–i using TiCl4 as a Lewis acid catalyst (0.2/0.5 equiv.) at 0 °C – rt (Scheme 55).
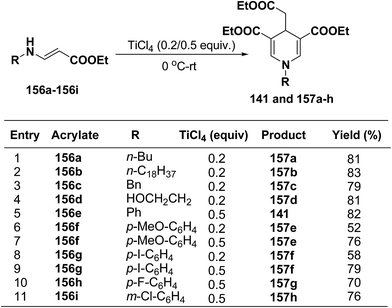 |
| | Scheme 55 TiCl4 catalysed synthesis of 1,4-dihydropyridines 141 and 157a–h. | |
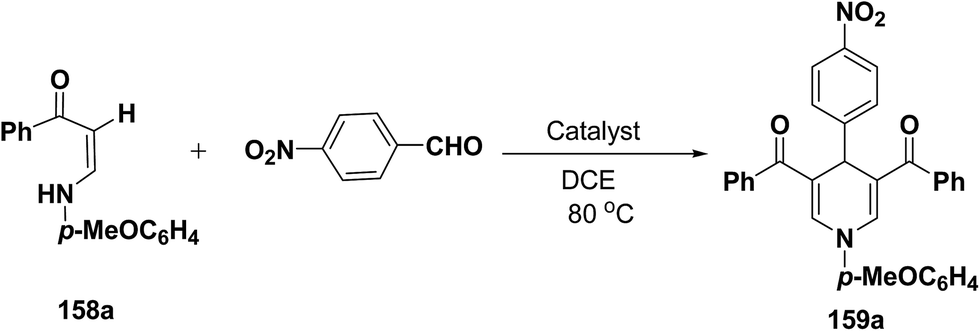
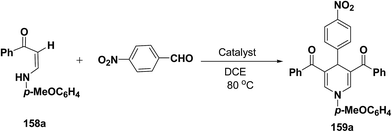
Li, et al.64 have screened several experimental conditions to synthesize 1,4-dihydropyridines by the condensation of (Z)-3-(4-methoxyphenylamino)-1-phenylprop-2-ene-1-one (158a) with p-nitrobenzaldehyde in the presence of catalytic amount of Lewis acids. The reaction of 158a with p-nitrobenzaldehyde in the presence of a catalytic amount of TsOH·H2O, TFA or NaAuCl4·2H2O yielded the desired 1,4-DHP 159a in 71–79% yield (Table 15, entries 2–4). Increase in the aldehyde equivalent from 0.75 to 1.0 not only lessened the reaction time, but also enhanced the yield of the desired product (Table 15, entry 2 vs. 6). The high yield of the 1,4-DHP 159 was achieved in the presence of TsOH·H2O as a catalyst (85–87%; Table 15, entries 6 and 7).
Table 15 Optimization of catalyst for the synthesis of 1,4-dihydropyridine 159a
|
| Entry |
p-NO2-C6H4-CHO (eq.) |
Catalyst (mol%) |
Time (h) |
% yield of 159aa |
| Isolated yields. NR = no reaction. |
| 1 |
0.75 |
Sc(OTf)3 (5) |
12 |
34 |
| 2 |
0.75 |
TsOH·H2O (10) |
3 |
75 |
| 3 |
0.75 |
TFA (10) |
5 |
71 |
| 4 |
1.0 |
NaAuCl4·2H2O (5) |
1 |
79 |
| 5 |
1.0 |
TFA (5) |
20 |
91 |
| 6 |
1.0 |
TsOH·H2O (10) |
1 |
85 |
| 7 |
1.0 |
TsOH·H2O (5) |
1 |
87 |
| 8 |
1.0 |
FeCl3·6H2O (10) |
3 |
48 |
| 9 |
1.0 |
— |
10 |
NRb |
Tamaddon, et al.65 have developed a green, efficient one-pot synthesis of various 4-alkyl/aryl-1,4-dihydropyridines in an excellent yields via the Hantzsch three-component reaction of an aromatic/aliphatic aldehydes, alkyl acetoacetate 110a/110b and ammonium carbonate in water (Scheme 56).
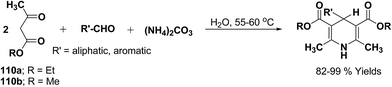 |
| | Scheme 56 Synthesis of 1,4-dihydropyridines using ammonium carbonate in water. | |
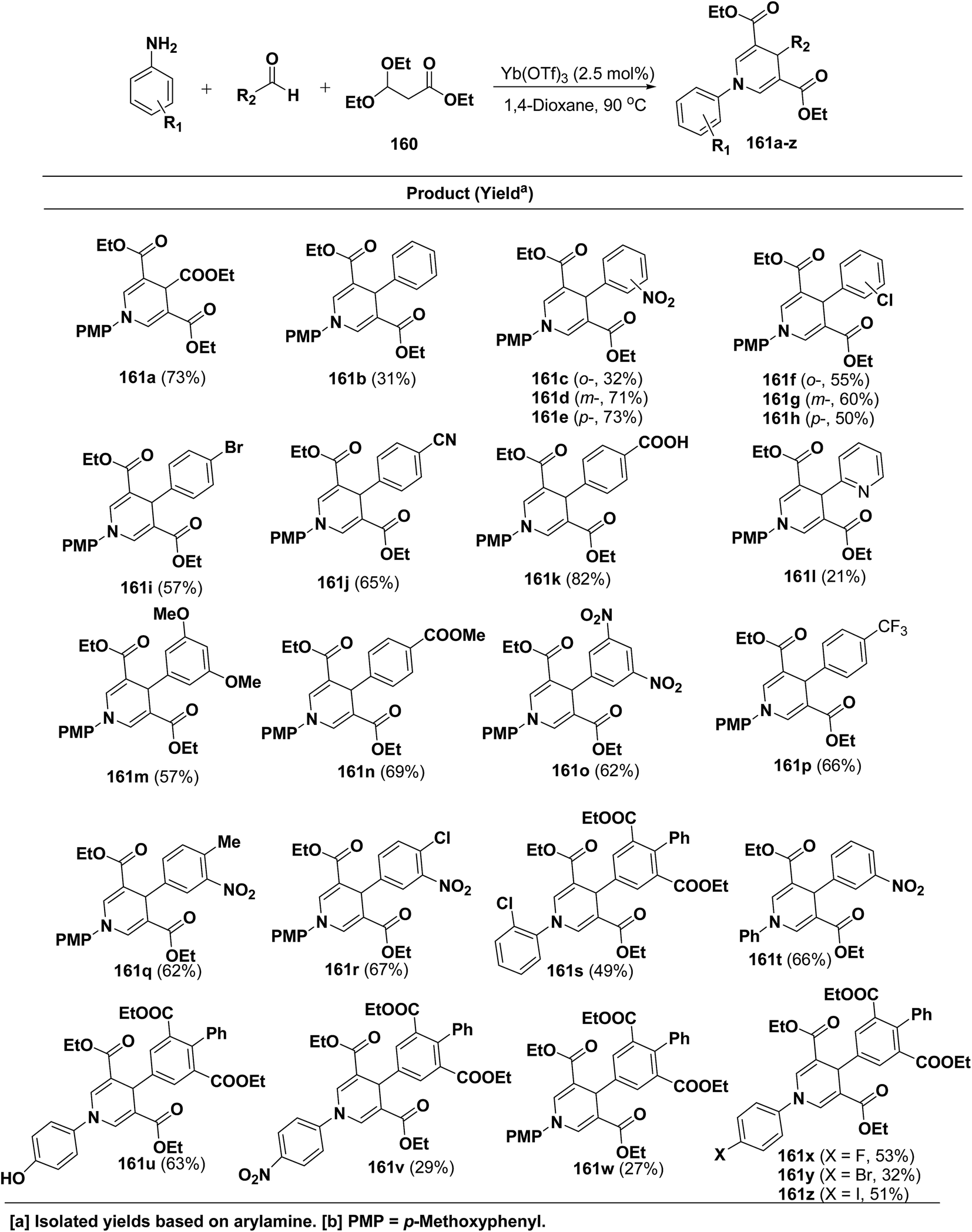
Shimizu, et al.66 have developed an efficient one-pot synthesis of 1,4-dihydropyridines 161a–z in moderate to good yields via the Yb(OTf)3-catalyzed Hantzsch three-component reaction of an aromatic aldehydes, 3,3-dimethoxy propionate 160 and aromatic amines in 1,4-dioxane at 90 °C (Scheme 57).
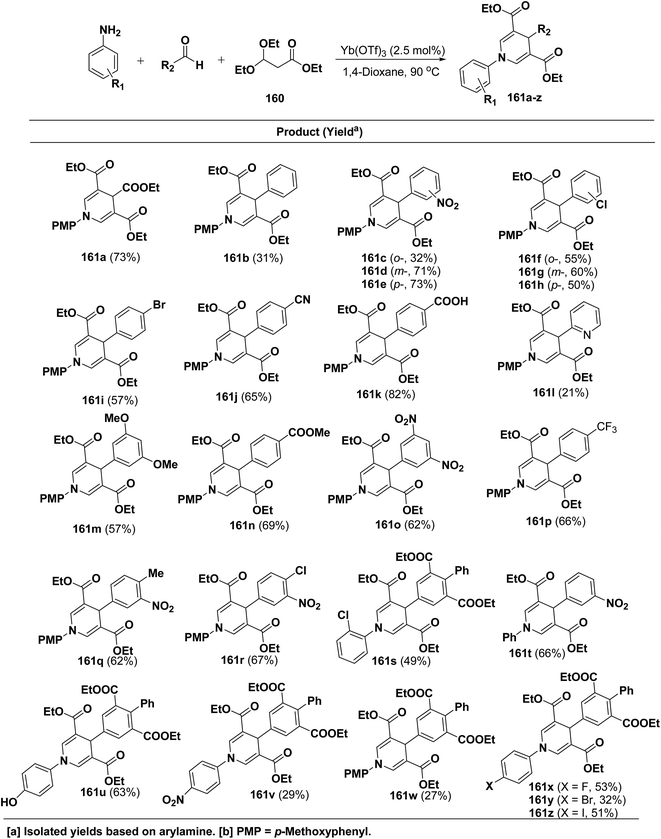 |
| | Scheme 57 Yb(OTf)3 catalyzed one-pot synthesis of 1,4-DHPs 161a–z. | |
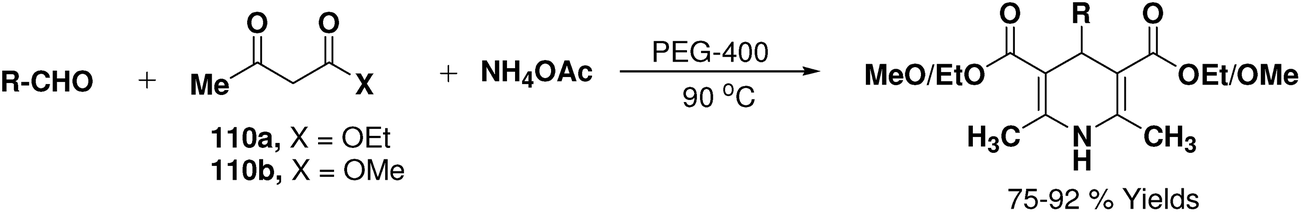
Wang, et al.67 have developed an efficient one-pot synthesis of 1,4-dihydropyridines in an excellent yields via the Hantzsch three-component reaction of an aromatic/aliphatic aldehydes, alkyl acetoacetate 110a/110b and ammonium acetate in PEG-400 as solvent at 90 °C (Scheme 58). PEG-400 facilitates the reactants converting into corresponding products smoothly in excellent yields because of its good immiscibility with a number of organic reagents. Moreover, PEG-400 is inexpensive, thermally stable, nonvolatile, and nontoxic.
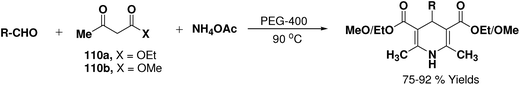 |
| | Scheme 58 Synthesis of 1,4-dihydropyridines in PEG-400. | |
2.3. Synthesis of enantiopure 1,4-dihydropyridines
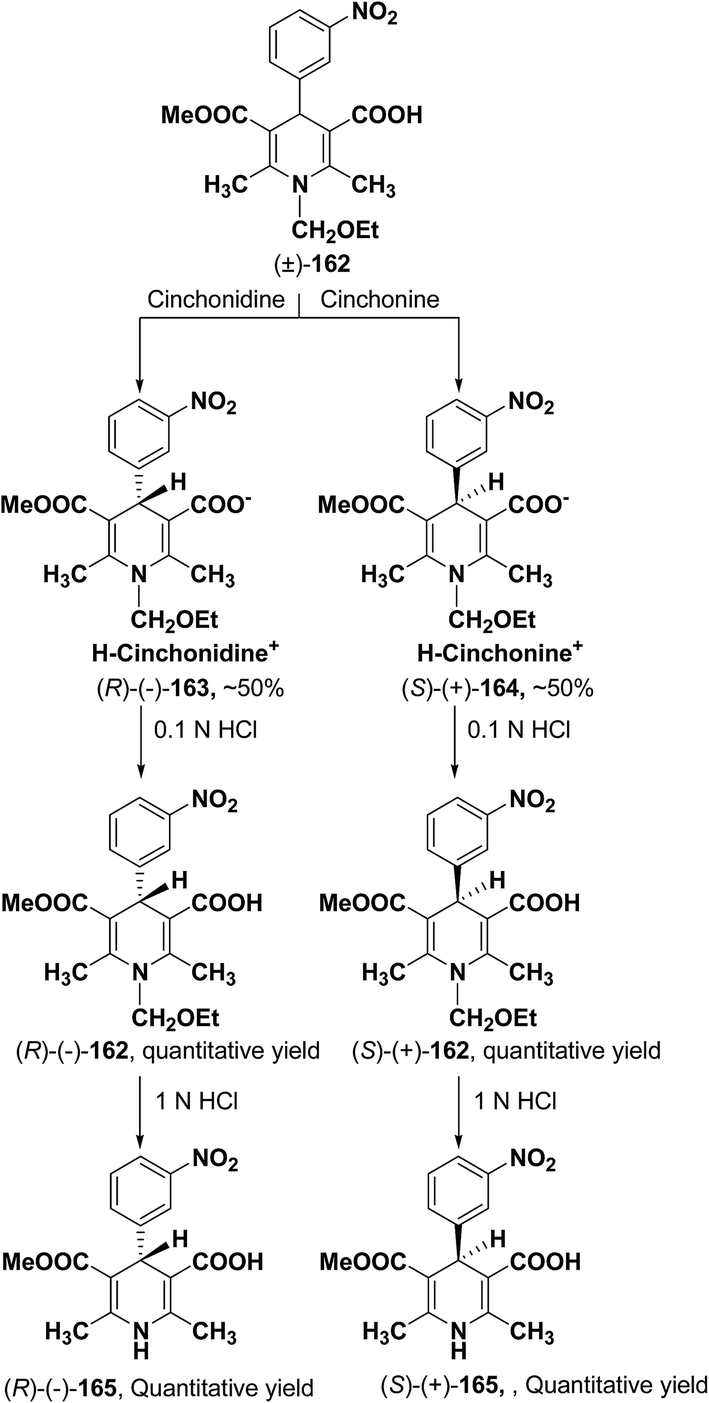
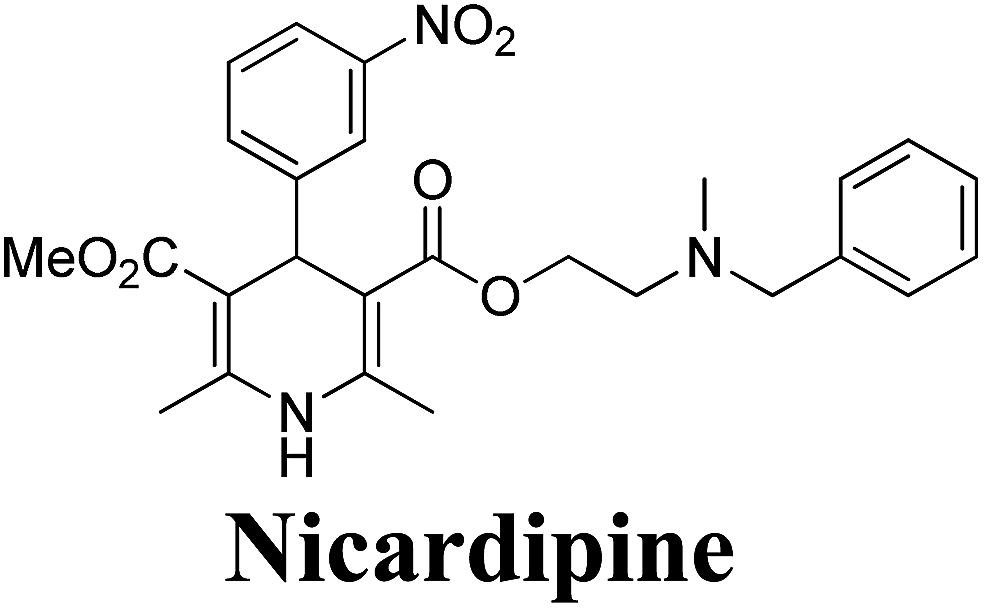
2.3.1. Resolution of racemic 1,4-dihydropyridine carboxylic acids via diastereomeric salt formation. Chiral crystallization with the use of resolving agents to form diastereomeric salts is a widely used technique for resolving chiral compounds. Cinchonidine, cinchonine and quinidine are the most commonly used natural chiral amine bases applied which are used to resolve racemic 1,4-dihydropyridinecarboxylic acid via forming their diastereomeric salts. It has been observed that these alkaloids behave as quasi-mirror images and are used in a complementary way to obtain both enantiomers of 1,4-dihydropyridines in a pure form. The method involves treatment of racemic acid with a chiral base which leads to the formation of diastereomeric salts (Scheme 59). The enantiomer is then obtained in pure acid form by repeated crystallization followed by acidification. The other enantiomer is recovered from the mother liquor and resolved using other suitable chiral base reagent by going through the same procedure. The first resolution of racemic dihydropyridinecarboxylic acids through this method was carried out by Shibanuma, et al.68 to synthesize nicardipine from 1-ethoxymethyl-5-methoxycarbonyl-2,6-dimethyl-4-(m-nitrophenyl)-1,4-dihydropyridine-3-carboxylic acid (±)-162 using cinchonine and cinchonidine (Scheme 59). Various other 1,4-dihydropyridine derivatives have been synthesized similarly through optical resolution of racemic mixture of 1-ethoxymethylated dihydropyridines and by subsequently removing the ethoxymethyl group.69
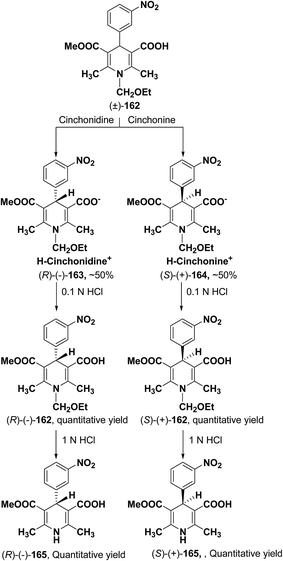 |
| | Scheme 59 Synthesis of enantiopure 1,4-dihydropyridines 165 via diastereomeric salt formation method. | |
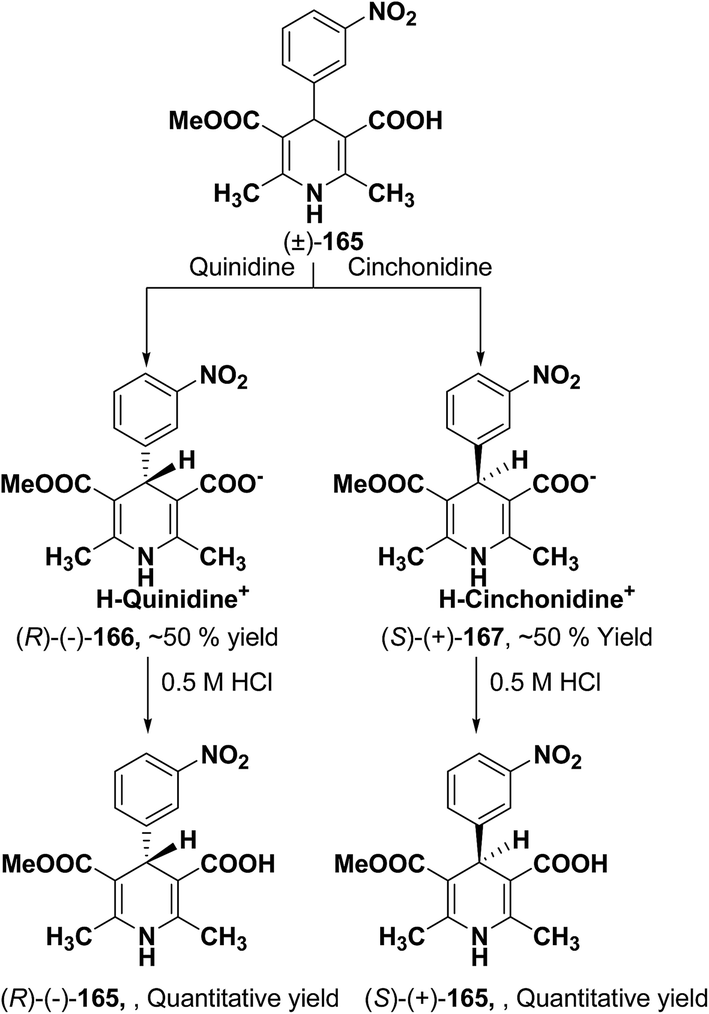
Synthesis of optically-active 1,4-dihydropyridine derivatives from 1-ethoxymethylated carboxylic acid 162 involves protection and deprotection approach for NH group in 1,4-DHP ring. Use of unprotected racemic acid for direct resolution has also been investigated in detail. Zhang, et al.70 treated racemic acid (±)-165 with cinchonidine to form diastereomeric salt (S)-(+)-167 (Scheme 60). (S)-(+)-167 enantiomer was formed by repeated crystallization and further treatment with hydrochloric acid. When quinidine was used (R)-(−)-166 was formed.
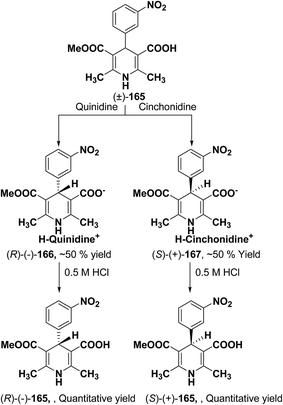 |
| | Scheme 60 Synthesis of chiral 1,4-dihydropyridine 165 via diastereomeric salt formation method. | |
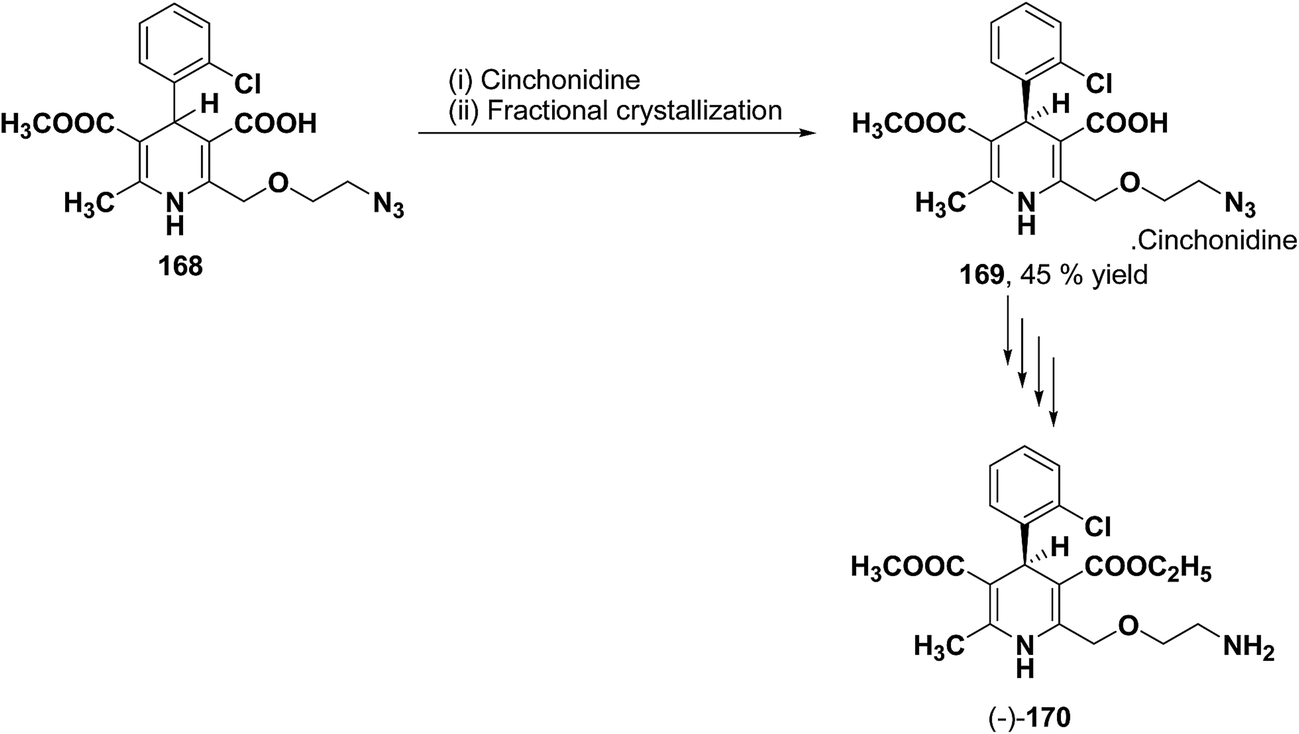
The separation of (R)- and (S)-amlodipine 170 has been achieved by treatment of racemic acid 168 with cinchonidine in methanol. Dilution with water formed a crystalline precipitate which was recrystallized to yield diastereomerically pure cinchonidine salt 169, which was further converted to enantiomerically pure (R)- and (S)-amlodipine in several synthetic steps (Scheme 61).71
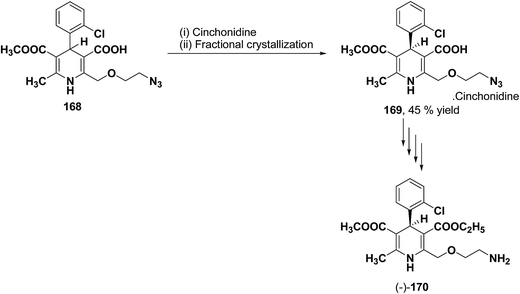 |
| | Scheme 61 Separation of (R)- and (S)-amlodipine 170 by treatment of racemic acid 168 with cinchonidine in methanol. | |
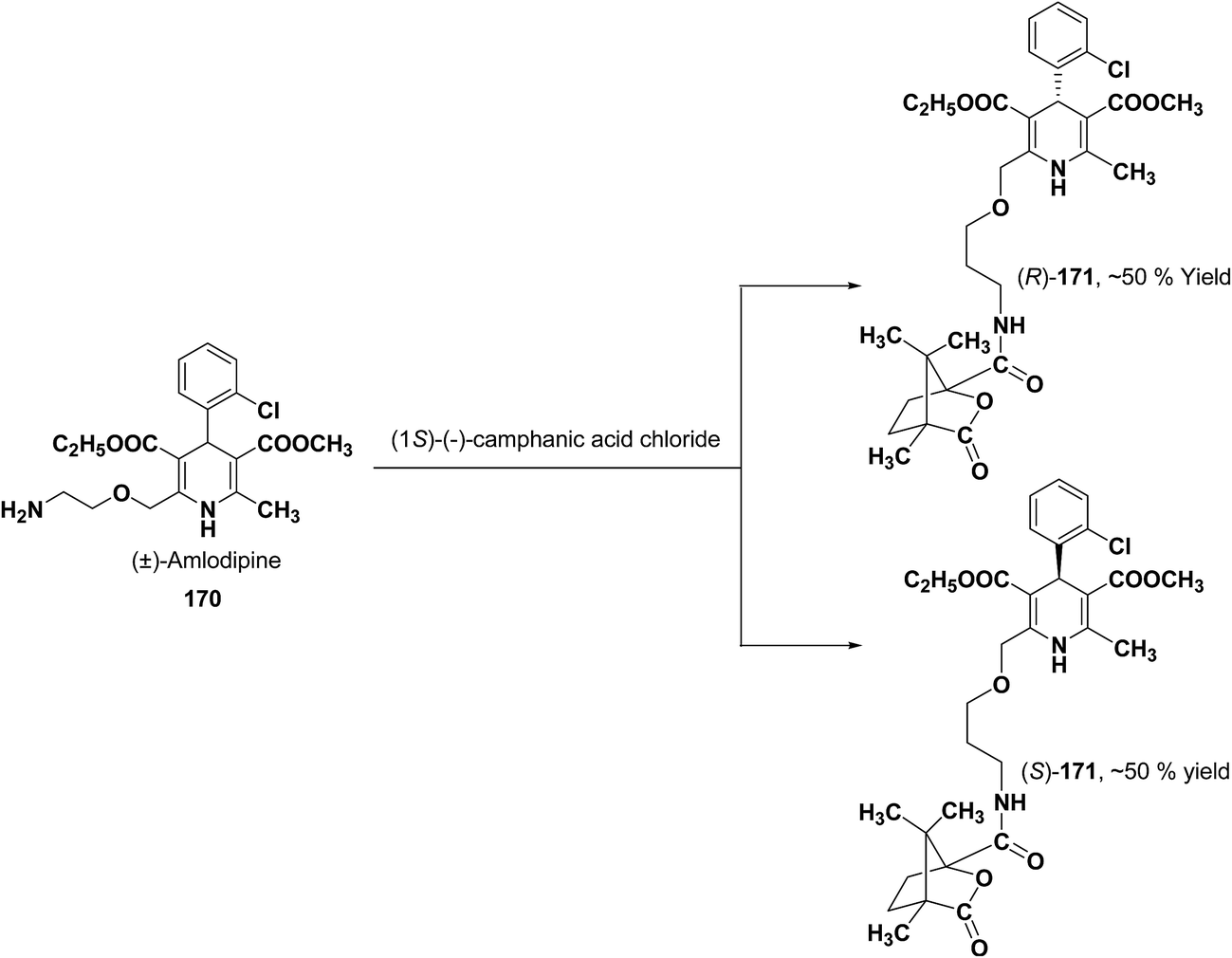
Various chiral acids have been used for the separation of racemic basic dihydropyridines. Racemic amlodipine 170 has been resolved using (1S)-(−)-camphanic acid chloride (Scheme 62) by Goldmann, et al.72 The diastereomeric amides 171 obtained were easily separated chromatographically on Chiralcel OD column followed by crystallization from DMF/water.
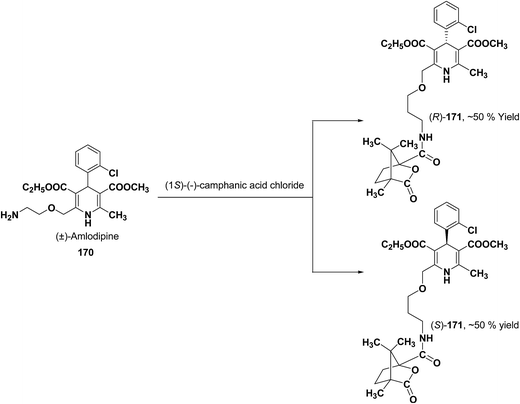 |
| | Scheme 62 Resolution of racemic amlodipine 170 using (1S)-(−)-camphanic acid chloride. | |
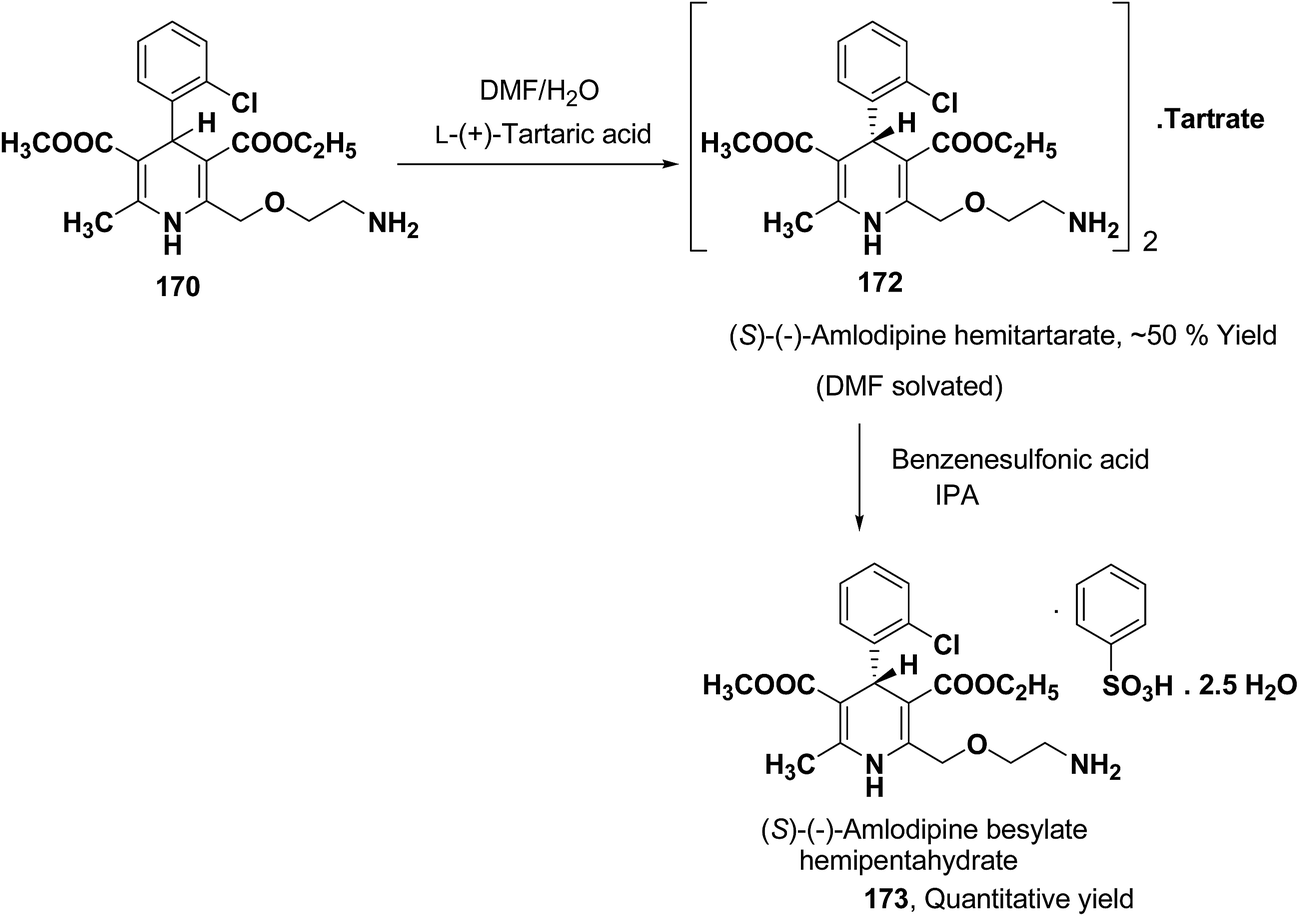
Another remarkable method to get R-(+) and S-(−) amlodipine 170 is through L or D-tartaric acid respectively in DMSO.73 It has been reported that (R)-amlodipine tartrate crystallizes out preferentially when naturally occurring L-tartaric acid is used with racemic amlodipine in DMSO. Recently it has been described that (S)-isomer of amlodipine could also be obtained with L-tartaric acid just by changing the solvent system (Scheme 63). By using DMF/H2O (85:15 ratio) desired (S)-(−)-172 crystallized out with 99% purity.74 By reacting 172 with benzenesulfonic acid, active pharmaceutical ingredient (S)-(−)-amlodipine besylate hemipentahydrate salt 173 was obtained.
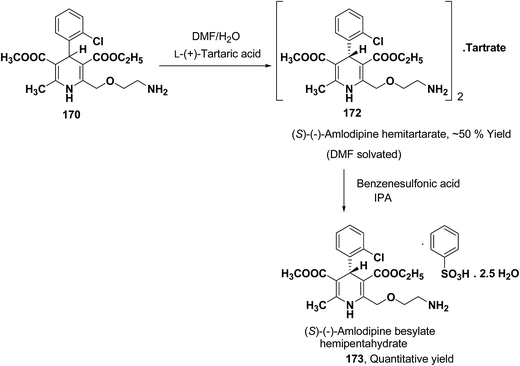 |
| | Scheme 63 Resolution of R-(+) and S-(−) amlodipine 170 via diastereomeric salt formation with L or D-tartaric acid, respectively in DMSO. | |
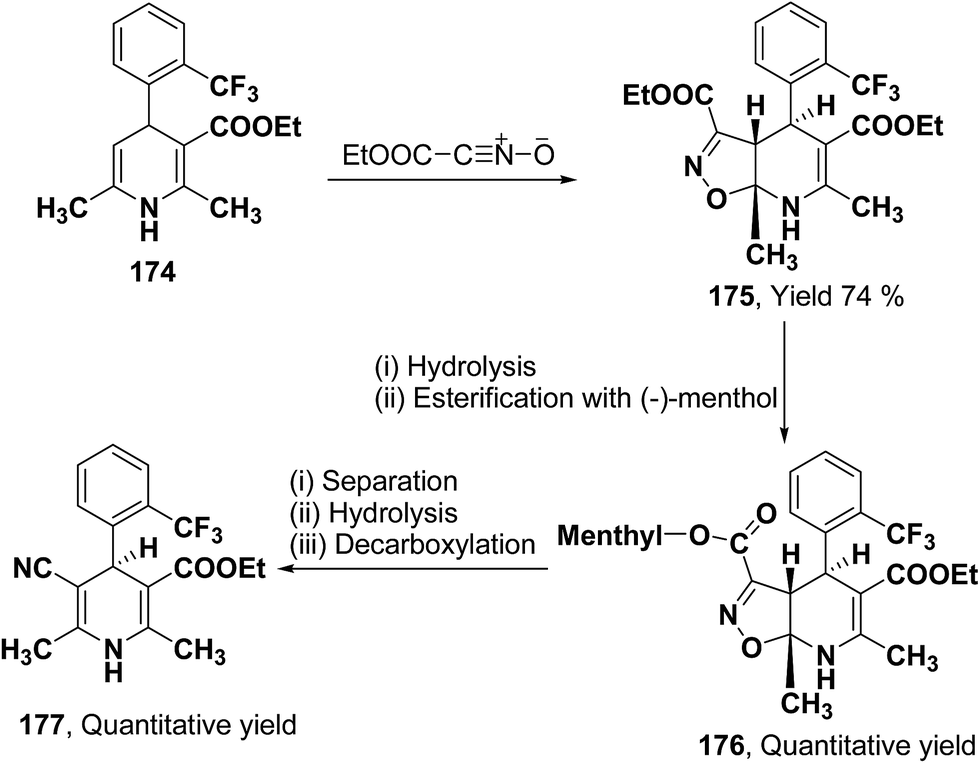
2.3.2. Separation of racemic 1,4-dihydropyridines by using chiral auxiliary. This synthetic strategy utilizes chiral auxiliary for the synthesis of optically active 1,4-dihydropyridines by forming diastereomeric esters which can be separated and eventually the chiral auxiliary can be regioselectively removed. Synthesis of 5-cyano-1,4-dihydropyridines from isoxazolo[5,4-b]pyridines have been achieved by Taylor, et al.75 The monoester derivative 174 was reacted with nitrile oxide to produce isoxazolo[5,4-b]pyridine 175 in 74% yield (Scheme 64). Its saponification formed an acid which was then re-esterified with (−)-menthol. The resulting diastereomeric esters 176 were separated through flash chromatography, which on hydrolysis and decarboxylation yielded the desired 5-cyano-1,4-dihydropyridine-3-carboxylic acid 177.
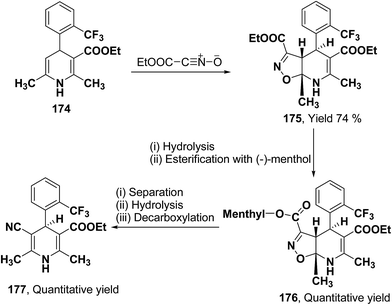 |
| | Scheme 64 Separation of 174 using nitrile oxide as a chiral auxiliary. | |
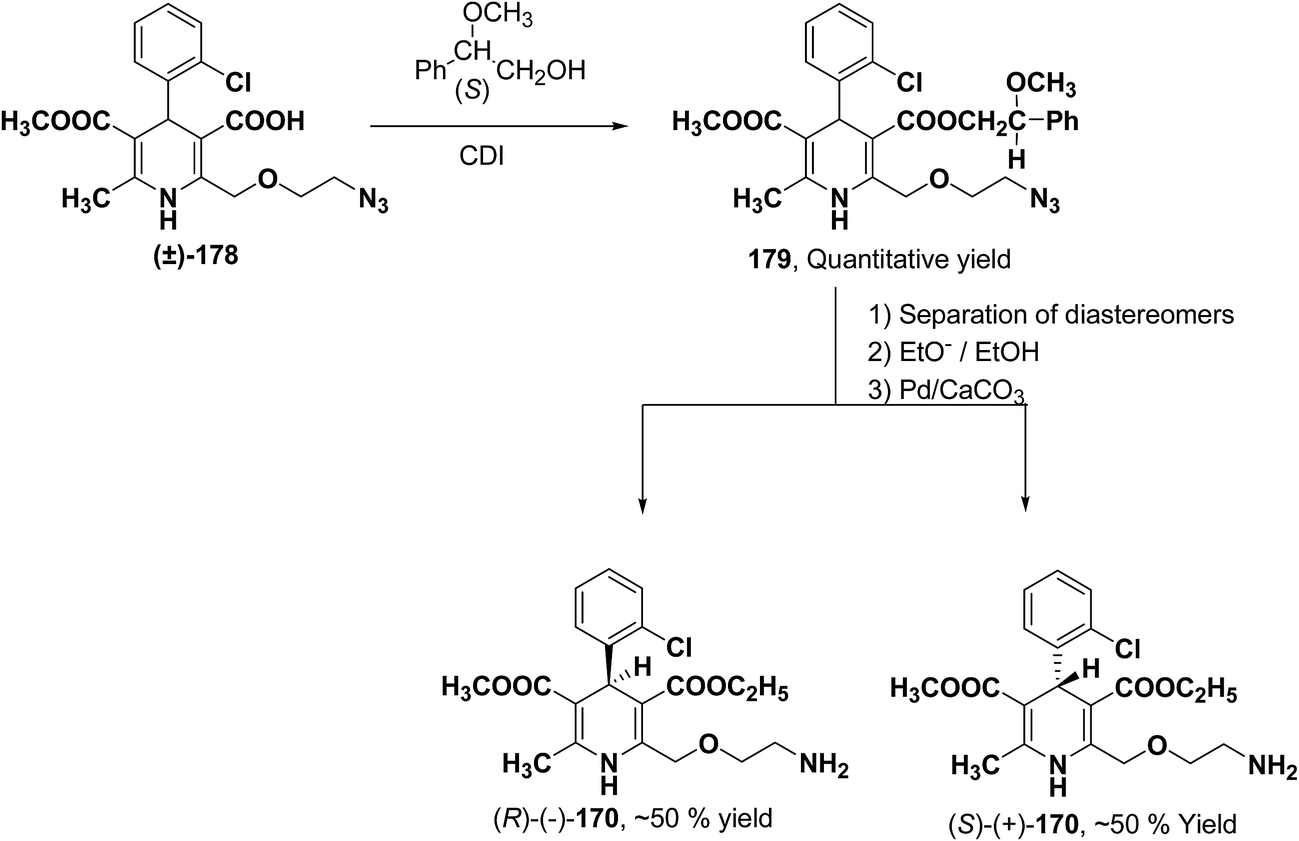
Arrowsmith, et al.76 esterified (±)-178 using optically pure (S)-(+)-2-phenylethanol to give diastereomeric mixture 179. It was chromatographically separated on silica gel to form pure diastereomers and further treatment with sodium ethoxide and finally reduction of azide group by palladium on calcium carbonate yielded pure enantiomers (R)-(−)-amlodipine and (S)-(+)-amlodipine 170 (Scheme 65).
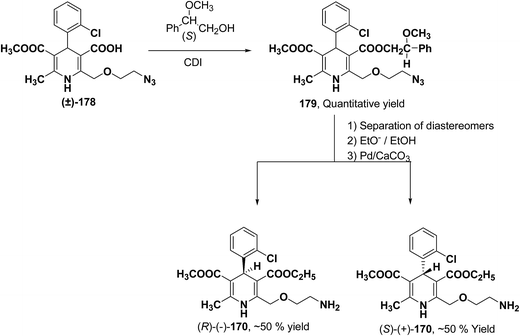 |
| | Scheme 65 Resolution of 170 from 178 using optically pure (S)-(+)-2-phenylethanol. | |
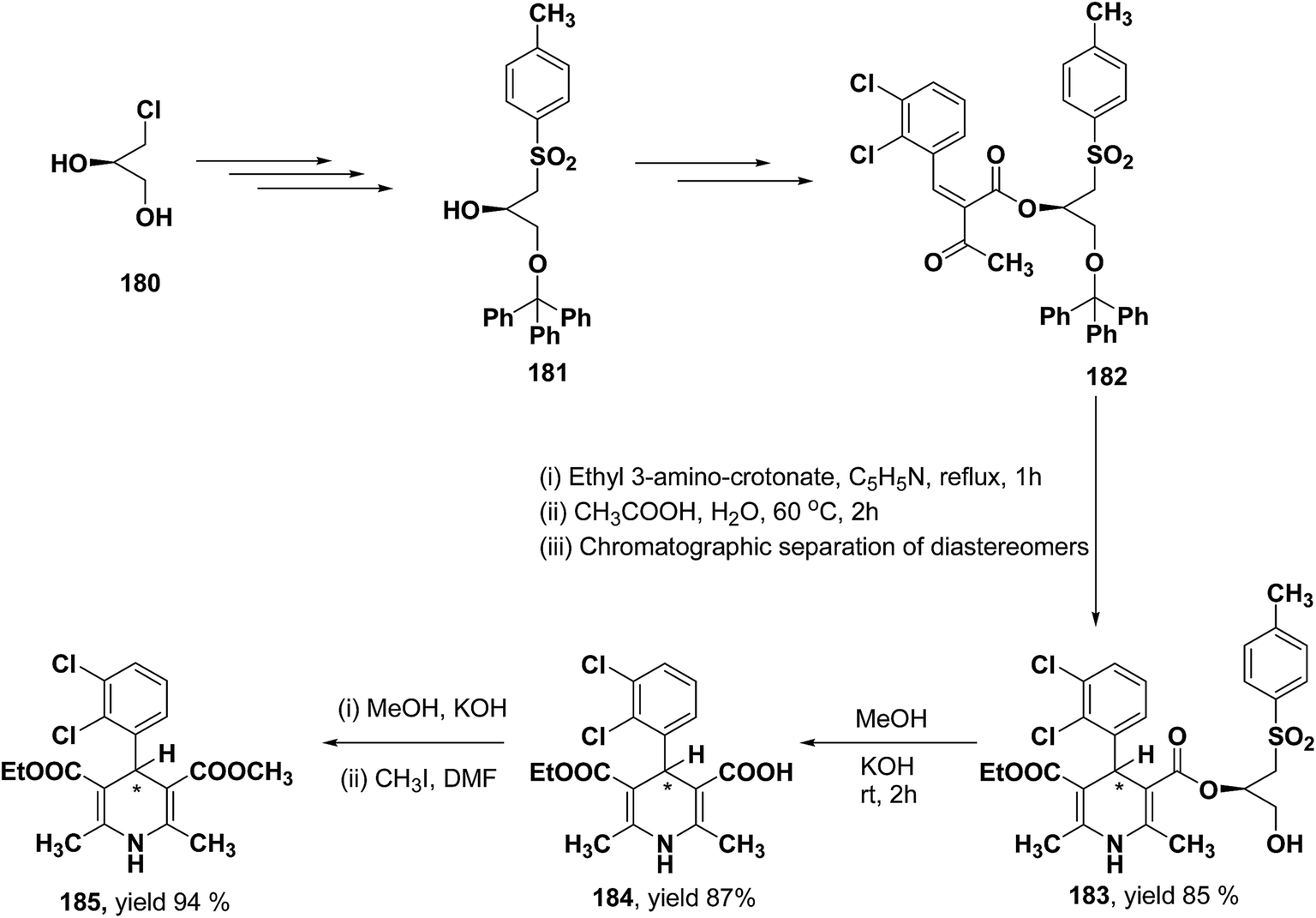
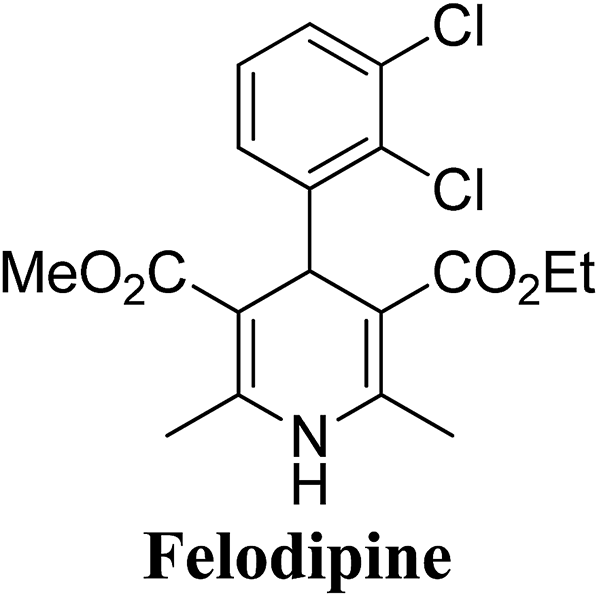
Lamm, et al.77 disclosed the formation of pure enantiomers of felodipine 185, a calcium channel antagonist by chromatographic separation of diastereomeric esters 183 prepared from (R)-1-(p-toluenesulfonyl)-3-tritylpropan-2-ol 181 (Scheme 66). This chiral auxiliary was removed easily by β-elimination reaction using potassium hydroxide in methanol at room temperature. Esterification of 184 with iodomethane successfully yielded (R)- and (S)-enantiomers of felodipine 185.
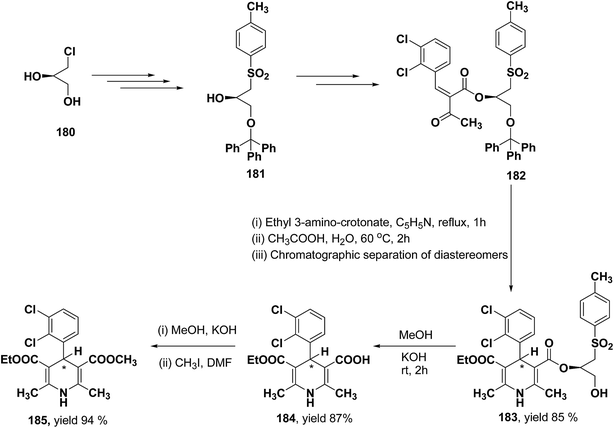 |
| | Scheme 66 Formation of enantiopure 185 using chiral auxiliary. | |
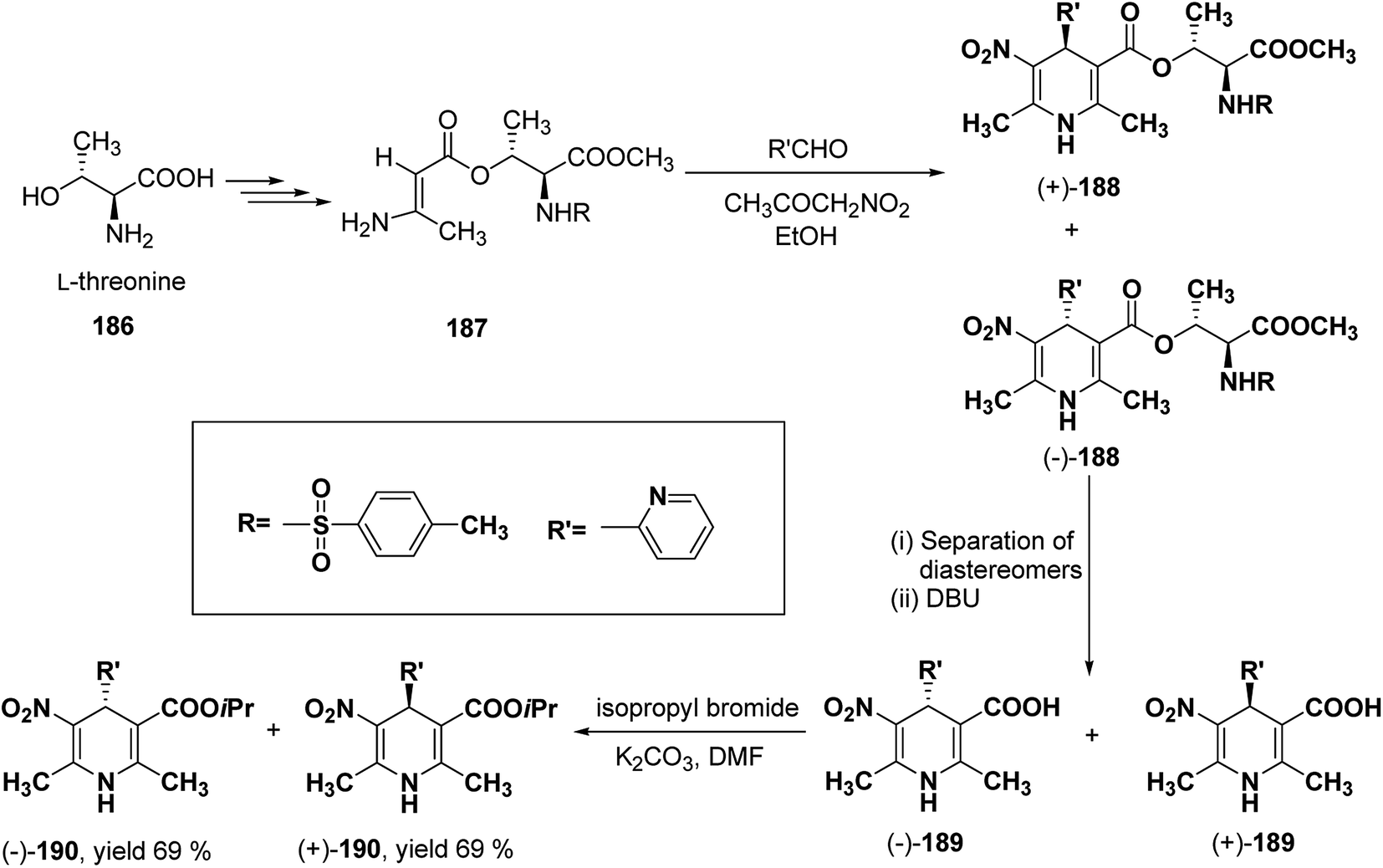
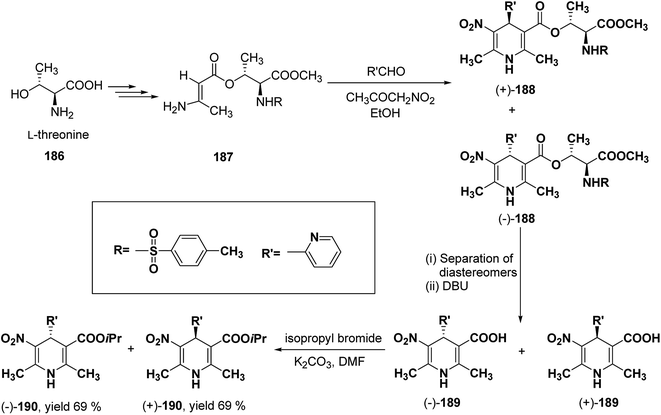
Racemic isopropyl 1,4-dihydro-2,6-dimethyl-3-nitro-4-pyridinylpyridine-5-carboxylate isomers with in vitro calcium channel modulating activities have been described by Vo, et al.78 Out of them the most promising 4-(2-pyridinyl) compound was resolved into pure enantiomers with the help of L-threonine 186 (Scheme 67). Through a multistep synthesis L-threonine was converted to β-aminocrotonate derivative 187, which through Hantzsch condensation afforded the mixture of 1,4-DHP diastereomers 188 separable by silica gel column chromatography. This was followed by removal of chiral auxiliary using DBU to yield 189 and desired enantiopure (−)-190 and (+)-190 were synthesized using isopropyl bromide and potassium carbonate in DMF.
 |
| | Scheme 67 Resolution of racemic isopropyl 1,4-dihydro-2,6-dimethyl-3-nitro-4-pyridinylpyridine-5-carboxylate using L-threonine. | |
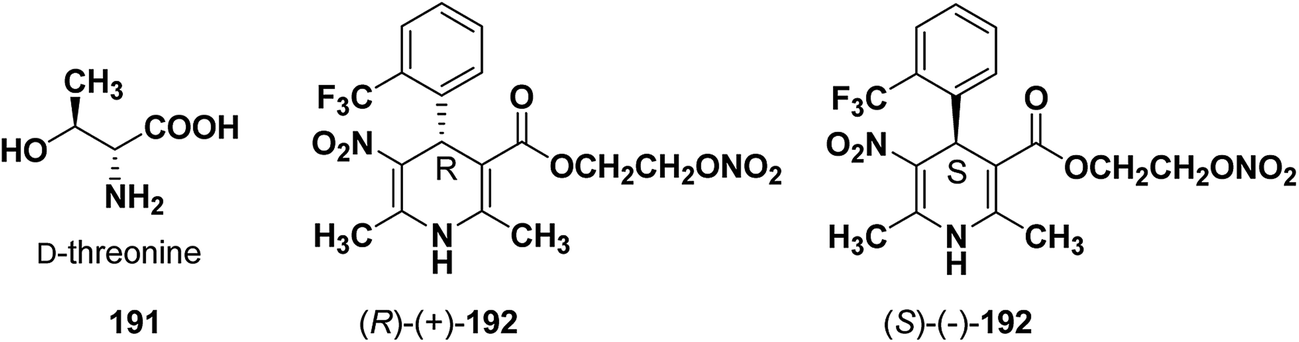
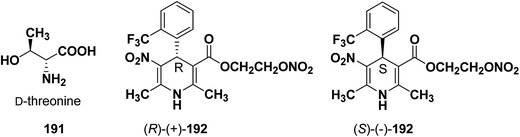
Similarly synthesis of (S)-(−)- and (R)-(+)-enantiomers of 2-nitrooxyethyl 1,4-dihydro-2,6-dimethyl-3-nitro-4-(2-trifluoromethylphenyl)pyridine-5-carboxylate79 192 exhibiting dual cardioselective agonist/smooth muscle selective antagonist activity80 were synthesized utilizing D-threonine 191 as chiral auxiliary (Fig. 2). In both the cases the chiral esterifying group was easily removed by non-nucleophilic base 1,8-diazabicyclo[5.4.0]-undec-7-ene (DBU).
 |
| | Fig. 2 Structures of D-threonine 191 and 2-nitrooxyethyl 1,4-dihydro-2,6-dimethyl-3-nitro-4-(2-trifluoromethylphenyl)pyridine-5-carboxylate 192. | |
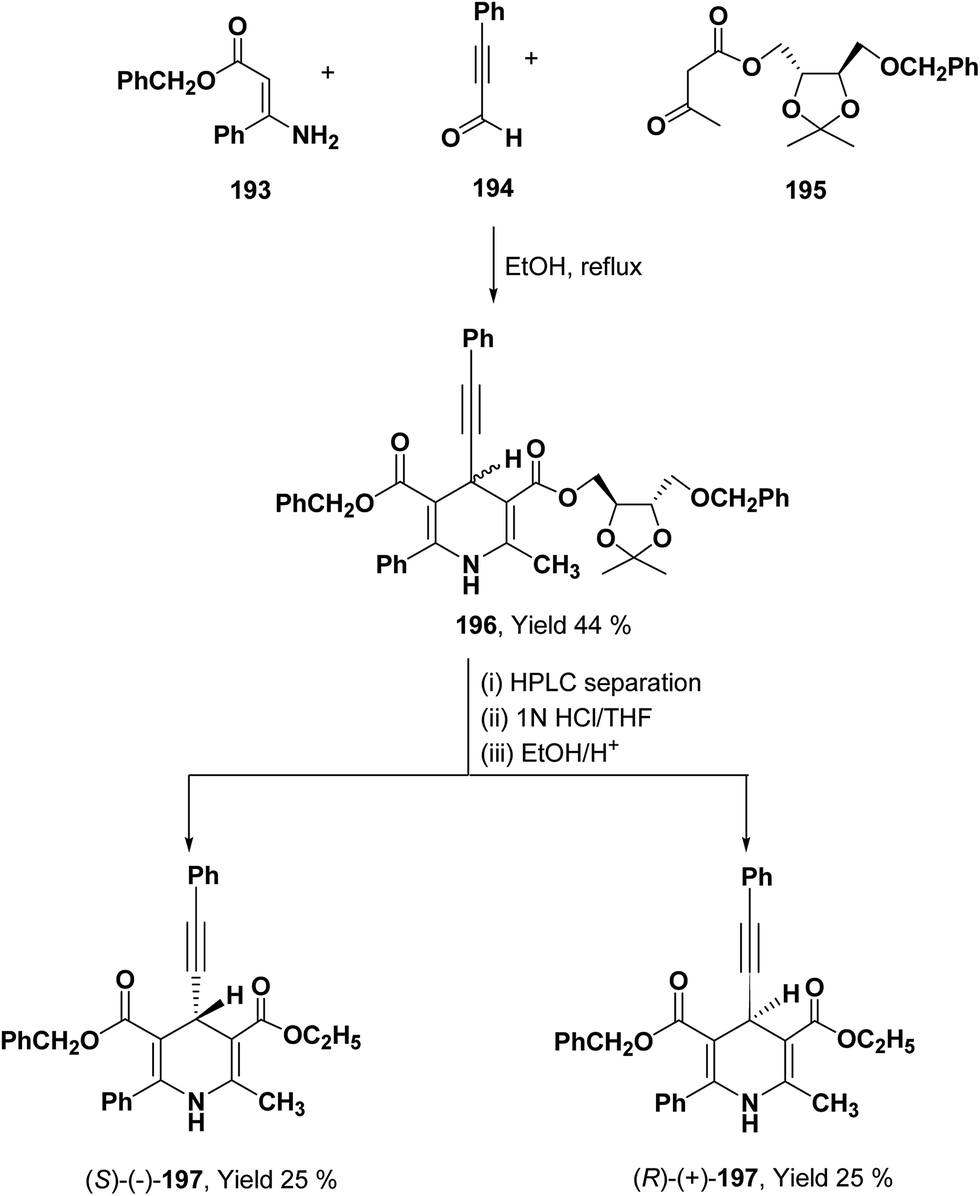
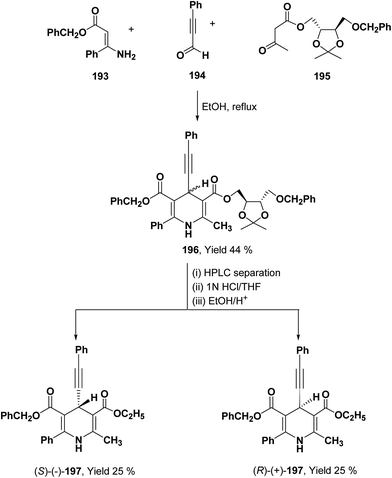
To synthesize chiral β-ketoester 195, (4R,5R)-(−)-2,3-O-isopropylidine-D-threitol was used as chiral auxiliary (Scheme 68).81 Using 195 as chiral β-ketoester, phenylpropargyl aldehyde 194 and benzyl 3-amino-3-phenyl-2-propenoate 193, 1,4-DHP diastereomers 196 were obtained which were resolved through HPLC. 2,2-Dimethyl-1,3-dioxolane-4-methanol and 2,3-O-isopropylidine-D-threitol moieties on the chiral ester were replaced by ethyl group through deprotection of diol by acidic treatment followed by transesterification in ethanol to afford 197.
 |
| | Scheme 68 Use of (4R,5R)-(−)-2,3-O-isopropylidine-D-threitol was used as chiral auxiliary in the separation of 1,4-DHP. | |
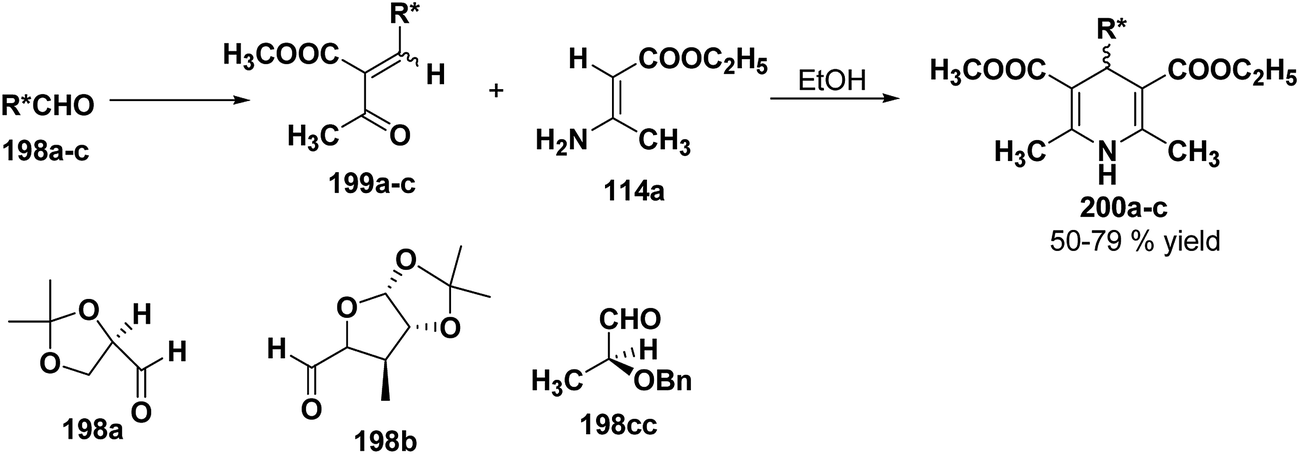
2.3.3. Synthesis of enantiomerically pure 1,4-DHPs using chiral aldehydes. Synthetic methodology to synthesize 4-alkyl-3,5-dialkoxycarbonyl-2,6-dimethyl-1,4-dihydropyridines 200a–c has been developed by utilizing chiral aldehydes 198a–c (Scheme 69).82 Enantiomerically pure 1,4-DHPs 200a–c were synthesized by Michael addition of ethyl aminocrotonate 114a to chiral α-acetylacrylates 199a–c. The synthesis of α-acetylacrylates 199a–c was carried out by knoevenagel condensation of methylacetyl acetate and the corresponding chiral aldehyde.
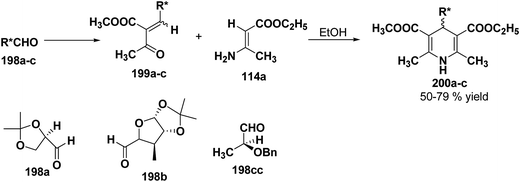 |
| | Scheme 69 Synthesis of chiral 1,4-DHPs 200a–c using chiral aldehydes. | |
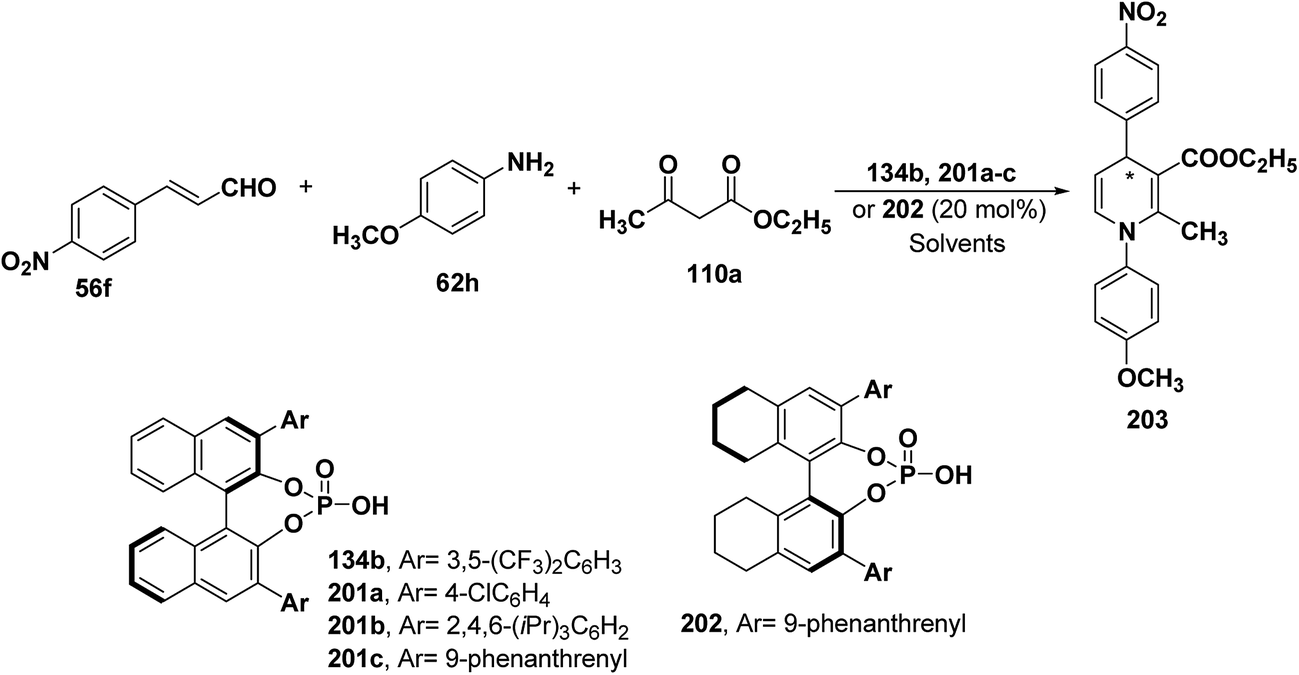
2.3.4. Organocatalytic asymmetric synthesis to form enantiomerically enriched dihydropyridines. Chiral phosphonic acids 134b, 201a–c and 202 have been recognized as promising catalysts for asymmetric synthesis of 1,4-dihydropyridines (Scheme 70). A three-component cyclization of an α,β-unsaturated aldehyde 56f, a primary amine 62h and ethyl acetoacetate 110a in presence of chiral phosphonic acids 134b, 201a–c and 202 have been explored to generate chiral 1,4-dihydropyridines 203 with excellent ee upto 98% (Scheme 70).83 The screening of phosphonic acids 134b, 201a–c and 202 showed that increasing size of substituents at 3,3′-positions of the chiral catalyst and increase in the reaction temperature leads to enhanced enantioselectivities (Table 16). It was concluded that in PhCN at 50 °C phosphonic acids 201c and 202 afforded 203 with 89% and 90% ee, respectively. Decreasing the amount of 202 from 20 mol% to 10 mol% led to an improved ee of 203 up to 92%.
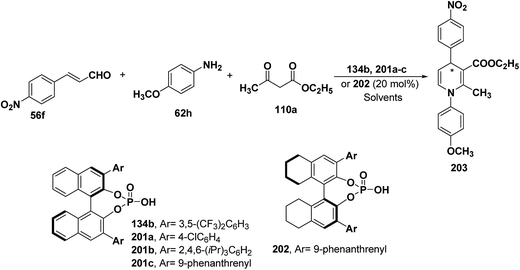 |
| | Scheme 70 Chiral phosphonic acids catalyzed synthesis of 1,4-DHP 203. | |
Table 16 Effect of phosphonic acids 134b, 201a–c and 202 and increase in the reaction temperature on the enantioselectivity of 203
| Catalyst |
Solvent |
Temperature (°C) |
Yield (%) |
ee (%) |
Product |
| 201a |
CHCl3 |
25 |
72 |
19 |
(S)-203 |
| 134b |
CHCl3 |
25 |
85 |
19 |
(S)-203 |
| 201b |
CHCl3 |
25 |
62 |
31 |
(S)-203 |
| 201c |
CHCl3 |
25 |
67 |
73 |
(S)-203 |
| 201c |
CHCl3 |
40 |
80 |
80 |
(S)-203 |
| 201c |
CHCl3 |
50 |
85 |
82 |
(S)-203 |
| 201c |
PhCN |
50 |
82 |
89 |
(S)-203 |
| 202 |
CHCl3 |
25 |
69 |
64 |
(R)-203 |
| 202 |
CHCl3 |
50 |
86 |
72 |
(R)-203 |
| 202 |
PhCN |
50 |
83 |
90 |
(R)-203 |
| 202 (10 mol%) |
PhCN |
50 |
82 |
92 |
(R)-203 |
Yamamoto, et al.84 described an efficient asymmetric synthesis of 1,4-dihydropyridine derivatives. The key step of this reaction was the stereoselective Michael addition using t-butyl ester of L-valine 207 as a chiral auxiliary to achieve good ee of 209 (>95%) and moderate yield (Scheme 71). With this method, (+)-4-(3-chlorophenyl)-6-dimethoxymethyl-2-methyl-1,4-dihydropyridine-3,5-dicarboxylic acid cinnamyl ester (+)-209 was obtained and was characterized as a promising N-type calcium channel blocker with improved selectivity over L-type compared to its (−)- and racemic isomers.
 |
| | Scheme 71 Stereoselective synthesis of 1,4-dihydropyridine 209. | |
3. Utility of dihydropyridines in the synthesis of natural products
3.1. Utility of 1,2-dihydropyridines in the synthesis of natural products
Kutney, et al. have synthesized two pyridocarbazole alkaloids, olivacine 212 and guatambuine 213 by utilizing the tricarbonylchromium complex 211 of the suitable 1,2-dihydropyridine, which was synthesized from indole 210 (Scheme 72).85
 |
| | Scheme 72 Synthesis of pyridocarbazole alkaloids. | |
The synthesis of (±)-elaeokanine A 216 has been achieved in a regioselective manner from 1-methoxycarbonyl salt of 3-(iPr)3 silyl pyridine 214 by Comins and Myoung.86 The 1-methoxycarbonyl salt of 3-(iPr)3 silyl pyridine was first converted into the 1,2-dihydropyridine derivative 215, which was then converted into the (±)-elaeokanine A into several number of synthetic steps (Scheme 73).
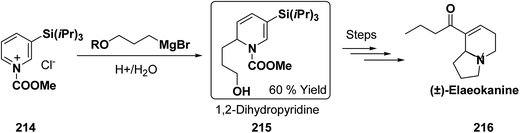 |
| | Scheme 73 Synthesis of (±)-elaeokanine A using 1,2-dihydropyridine derivative. | |
2-Substituted-1,2-dihydropyridine and piperidines has been synthesized in regio- and stereoselective manner from unsubstituted pyridinium salt. The approach relies on stereoselective formation of (E)-isomer of N-pyridinium imidate 218 from amide 217 in which the nitrogen lone pair is oriented at the proper position to direct the addition of Grignard reagent 219 at the 2-position. This approach has been used for the synthesis of 2-substituted-1,2-dihydropyridine 220 and R-(−)-coniine 221 (Scheme 74).87
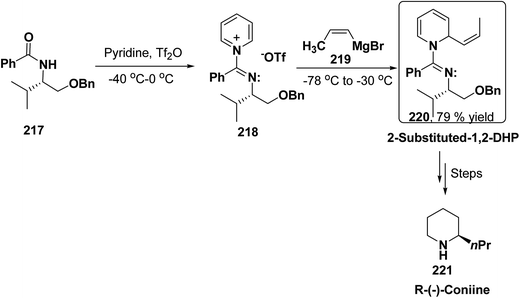 |
| | Scheme 74 Synthesis of R-(−)-coniine. | |
2,3-Disubstituted-1,2-dihydropyridines 224 and 225 have been synthesized from the 3-substituted pyridinium salts 222 and 223, respectively (Scheme 75). This methodology has been applied for the synthesis of (−)-L-733061 (226) and (−)-CP-99994 (227), two members of a new class of highly potent, nonpeptide, substance P antagonists.88
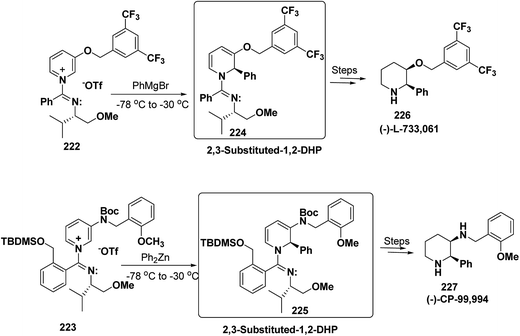 |
| | Scheme 75 Synthesis of 2,3-disubstituted-1,2-DHP: synthesis of (−)-L-733, 061 and (−)-CP-99, 994. | |
A simple synthesis of the fused tetrahydro-imidazopyridine 229 was accomplished via selective addition of protected guanidine 228 to N-carbomethoxy-1,2-dihydropyridine 1 in the presence of bromine at room temperature. Base-mediated semicleavage of the aminal gave 4-substituted 2-aminoimidazole 230 (Scheme 76). With this new method, natural marine metabolite 3-amino-1-(2-aminoimidazol-4-yl)-prop-1-ene (marine C6N4 2-aminoimidazole alkaloids) 231 and its derivatives may be prepared starting from pyridine.89
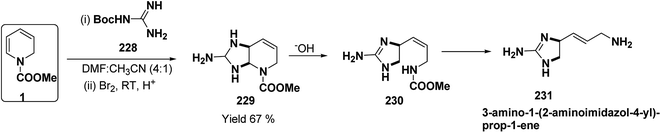 |
| | Scheme 76 Synthesis of marine C6N4 2-aminoimidazole alkaloids. | |
The asymmetric synthesis of 2,6-disubstituted 3-piperidinols having a 2,3-cis and 2,6-trans relative stereochemistry has been accomplished from 2-substituted 1,2-dihydropyridine 233 (synthesized from unsubstituted pyridinium salt 232) via a one-pot, highly diastereoselective epoxidation-nucleophilic addition with a heteroatom nucleophile or an organometallic reagent. This methodology was applied to the expedient asymmetric synthesis of (+)-julifloridine 234 in four steps (Scheme 77).90
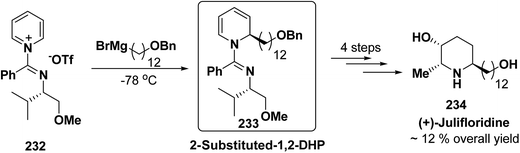 |
| | Scheme 77 Synthesis of (+)-julifloridine from 1,2-dihydropyridine. | |
Marine metabolites pyrrole-2-aminoimidazole oroidine, hymenidin and clathrodine have been synthesized via one pot oxidative bromine mediated addition of 2-aminopyrimidine 237 to the N-acyl-1,2-dihydropyridine 236 to form compound 238. The N-acyl-1,2-dihydropyridine was synthesized by the reaction of pyrrole-2-carbonyl chloride 235 with pyridine followed by the reduction using sodium borohydride. The compound 238 was treated with hydroxylamine hydrochloride and resulted into the formation of amide 239 and 240. The treatment of amide 239 and 240 with trifluoroacetic acid in dichloromethane resulted into the formation of oroidine 241 and hymenidine 242 or clathrodine 243 in good yields (Scheme 78).91
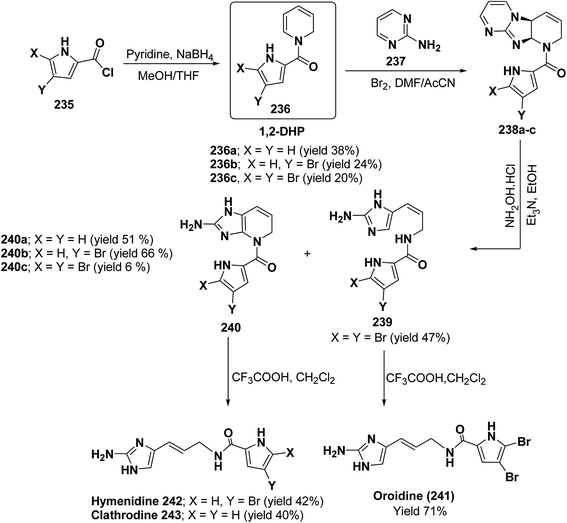 |
| | Scheme 78 Synthesis of oroidine, hymenidine and clathrodine using 1,2-dihydropyridine as an intermediate. | |
Stereoselective synthesis of L-picolic acid and (2S,3S)-3-hydroxypipecolic acid were achieved from a chiral 1,2-dihydropyridine intermediate 244. L-Picolic acid 248 has been obtained via reduction of 244 to 245, removal of chiral auxiliary yields 246, oxidation yields 247 and basic hydrolysis of 247 yields L-picolic acid 248. (2S,3S)-3-Hydroxypipecolic acid 253 was obtained in five steps (i) tandem hetero Diels–Alder reaction of 244 with oxygen to yield 249, (ii) alane reduction to yield 250, (iii) N and O-protection to afford 251, (iv) reduction of double bond to yield 252, and (v) sharpless oxidation to yield 253 (Scheme 79). L-Picolic acid is a naturally occurring non-proteinogenic α-amino acid and many biological alkaloids contain this moiety as a key structural unit or are derived from it. (2S,3S)-3-Hydroxypipecolic acid is found in febrifugine a potent antimalarial agent. Both L-picolic acid and (2S,3S)-3-hydroxypipecolic acid are conformationally constrained amino acids relevant to the study of peptide structure and drug design.92
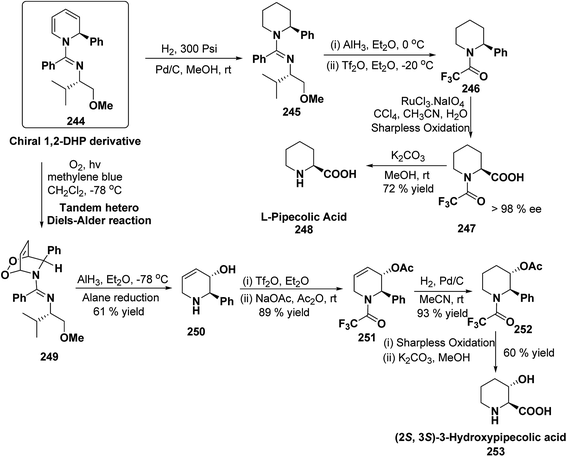 |
| | Scheme 79 Synthesis of L-picolic acid and (2S,3S)-3-hydroxypipecolic acid. | |
3.2. Utility of 1,4-dihydropyridine in the synthesis of natural products
Yohimbines are pentacyclic indole alkaloids containing five chiral centres. A convergent six steps synthesis of (±)-pseudo-yohimbines has been accomplished from methyl-β-(β-pyridyl)acrylate 254 (Scheme 80). Methyl-β-(β-pyridyl)acrylate 254 was converted into pyridinium salt 256 using tryptophyl bromide 255, which was then converted into 1,4-dihydropyridine derivative 257. The 1,4-dihydropyridine 257 was converted into (±)-pseudo-yohimbines 258 in several steps.93
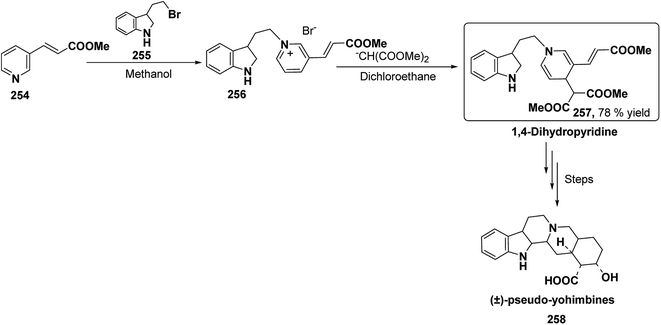 |
| | Scheme 80 Synthesis of (±)-pseudo-yohimbines. | |
Indoloquinolizine alkaloid, deplancheine has been synthesized starting from pyridinium salt 256. The pyridinium salt 256 was reduced to 1,4-dihydropyridine derivative 259 using sodium dithionate. The treatment of 1,4-dihydropyridine with HCl·MeOH resulted into the formation of cyclised product 260. The chloride salt 261 was synthesized using HCl·H2O from 260, which was reduced selectively to yield (±)-deplancheine 262 in good yield (Scheme 81).94
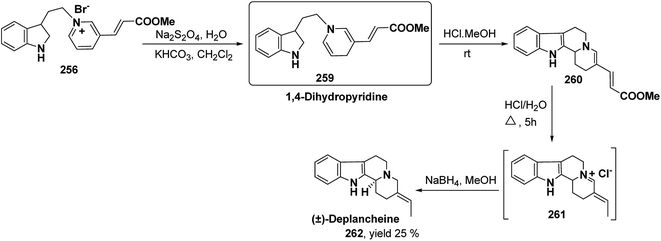 |
| | Scheme 81 Synthesis of (±)-deplancheine. | |
Vinoxine is amino indole alkaloid and its first total synthesis is based on the intramolecular cyclisation of suitably substituted 1,4-dihydropyridine–indole conjugate 265. The dihydripyridine–indole conjugate was synthesized by the reaction between methyl 2-(1H-indol-1-yl)acetate 263 and pyridinium salt 264 in the presence of LDA in THF. The intramolecular cyclization reaction on compound 265 was carried out using C6H6·HCl to afford 266. The chloride salt 267 of 266 was synthesized using 4 N HCl followed by the esterification and selective reduction afford the vinoxine 268b in good yield (Scheme 82).95
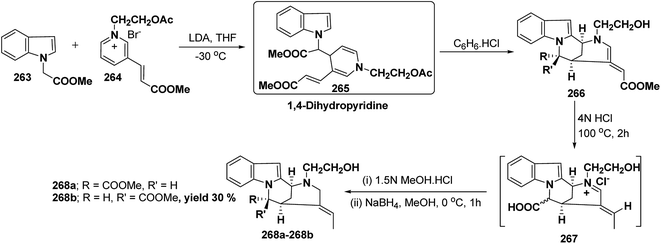 |
| | Scheme 82 Synthesis of vinoxine 268b. | |
Bosch, et al.96 has reported the synthesis of vinoxine analogue bearing C-16 methoxycarbonyl substituent, 19,20-dihydro-16-epivinoxine 275. The synthesis of 19,20-dihydro-16-epivinoxine 275 has been achieved from 3-ethyl-4-pyridine carboxylate 269 via the synthesis of 1,4-dihydropyridine analogue 274 (Scheme 83).
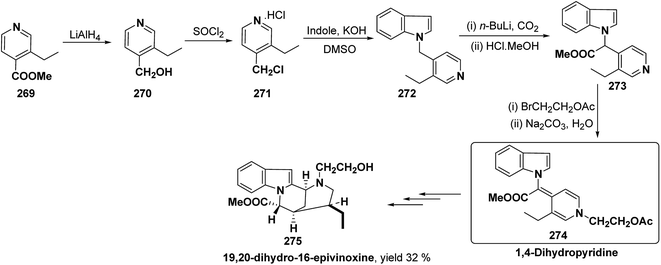 |
| | Scheme 83 Synthesis of 19,20-dihydro-16-epivinoxine 275. | |
An efficient route for the synthesis of tetracyclic ABCD ring substructures of Strychnos alkaloids has been carried out by synthesizing the 1,4-dihydropyridine 277 derivative from indole-2-carboxylate 276 and 264. The compound 1,4-dihydropyridine 277 was converted into disulfane analogue 278 in six steps, which was converted into pentacyclic compound 279 by generating the carbanion of compound 278 using DMTSF (dimethyl(methylthio)sulfonium fluoroborate). The resulting pentacyclic compound has been converted into tubifolidine 280 by treating 279 with RANEY® Ni. Tubifoline 281 and 19,20-dihydroakummicine 282 were synthesized by first N-protection with methyl carboxylate and desulferisation to yield 281 followed by treatment with sodium methoxide and photolysis, respectively (Scheme 84).97
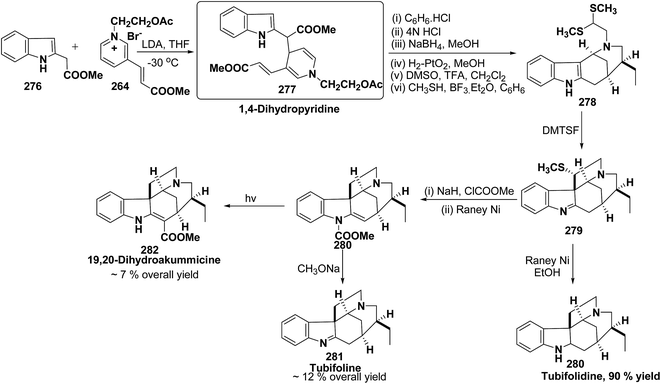 |
| | Scheme 84 Synthesis of tubifolidine 280, tubifoline 281 and 19,20-dihydroakummicine 282. | |
Ervitsine 289 is a minor 2-acylindole alkaloid isolated in 1977 from Pandaka boiteau. The first total synthesis of ervitsine 289 through a straightforward, biomimetic sequence involving only three separate synthetic steps (Scheme 85) has been reported by Bennasar, et al.98 The key intermediate was the iminium cation 286. The iminium cation 286 was synthesized from 1,4-dihydropyridine derivative 285 using Me2N+![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2I−. The 1,4-dihydropyridine derivative 285 was synthesized using 2-acetyl indole 283 and pyridinium salt 284. The iminium cation 286 would undergo regioselective cyclization to the bridged tetracyclic system 287, which was converted into N-methyl ervitsine 289a and ervitsine 289b into two synthetic steps first by treatment with mCPBA to yield 288 followed by borohydride reduction. A short four-step synthesis of N-methyl ervitsine involving the nucleophilic addition of acetyl indole to pyridinium salt, with subsequent C6H5SeBr-promoted cyclization of 1,4-dihydropyridine instead of Me2N+CH2I− promoted cyclization has been further reported by Bennasar, et al.99
CH2I−. The 1,4-dihydropyridine derivative 285 was synthesized using 2-acetyl indole 283 and pyridinium salt 284. The iminium cation 286 would undergo regioselective cyclization to the bridged tetracyclic system 287, which was converted into N-methyl ervitsine 289a and ervitsine 289b into two synthetic steps first by treatment with mCPBA to yield 288 followed by borohydride reduction. A short four-step synthesis of N-methyl ervitsine involving the nucleophilic addition of acetyl indole to pyridinium salt, with subsequent C6H5SeBr-promoted cyclization of 1,4-dihydropyridine instead of Me2N+CH2I− promoted cyclization has been further reported by Bennasar, et al.99
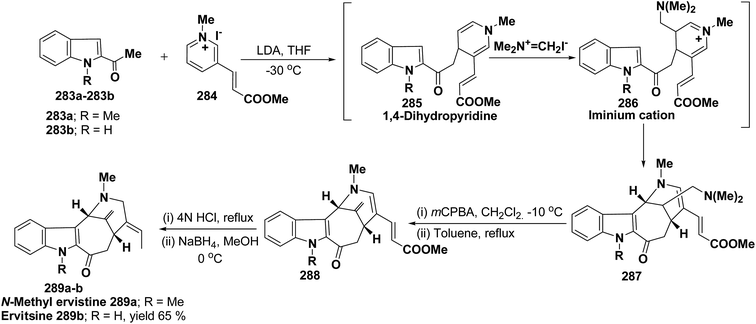 |
| | Scheme 85 Synthesis of N-methyl ervitsine 289a and ervitsine 289b. | |
The first total synthesis of (±)-2,7-dihydropleiocarpamine 295 has been reported by Bennasar, et al.100 via the synthesis of 1,4-dihydropyridine 291 as an intermediate (Scheme 86). The 1,4-dihydropyridine 291 was synthesized from nucleophilic addition of 263 on pyridinium salt 290 in the presence of LDA in THF. The key step is the photocyclization of the tetracyclic chloroacetamide 294 to give the required six membered ring via closure of C-6/C-7 bond formed by diradical coupling, which was not achieved by electrophilic cyclization.
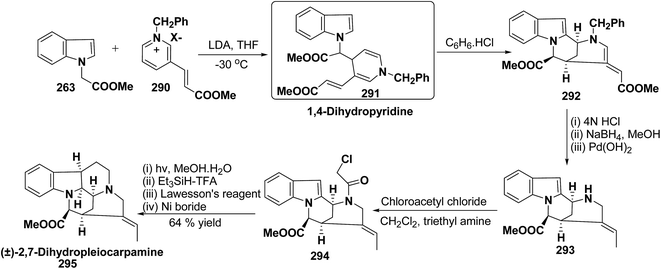 |
| | Scheme 86 Synthesis of (±)-2,7-dihydropleiocarpamine 295. | |
The ervatamine alkaloids (19,20-dehydroervatamine 307, 20-epiervatamine 308 and ervatamine 309) constitute a group of 2-acylindole alkaloids. Bennasar, et al.101 has reported the total synthesis of 19,20-dehydroervatamine and 20-epiervatamine, whereas ervatamine was synthesized from 19,20-dehydroervatamine via catalytic hydrogenation. The intermediate 1,4-dihydropyridine 299 formed via the nucleophilic attack of N-benzyl-2-acetyl indole 296 to the γ-position of the pyridinium salt 297. The 1,4-dihydropyridine 298 was functionalized to give 3,5-diacyl-1,4-dihydropyridine 299, which was reduced to tetrahydropyridine 300, and was further converted to cis-fused pentasubstituted piperidines (19,20-dehydroervatamine 307, 20-epiervatamine 308 and ervatamine 309) in number of steps by using suitable reagents (Scheme 87).
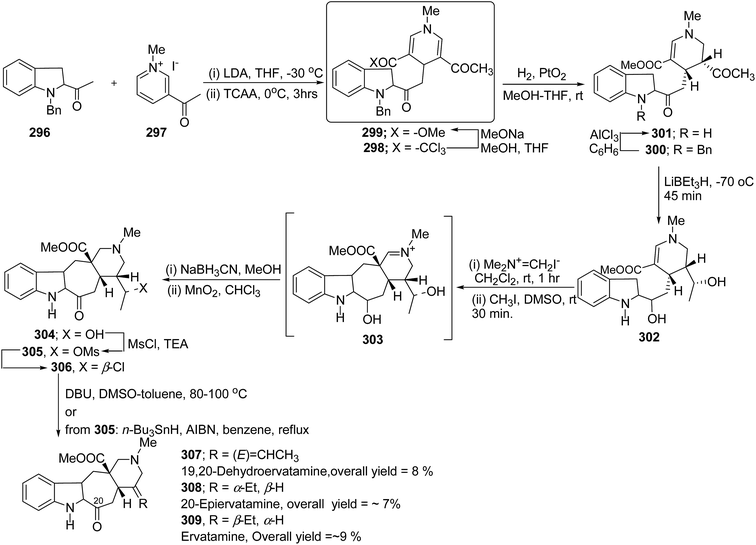 |
| | Scheme 87 Synthesis of 19,20-dehydroervatamine 307, 20-epiervatamine 308 and ervatamine 309. | |
Bennasar, et al.102 has reported a general route for the synthesis of tetracyclic ring system of silicine–methuenine alkaloids 311 and 312. The key step in the synthesis was the selective reduction of the intermediate 299 by using PtO2 followed by debenzylation to afford 301, which on cyclisation using trimethylsilyl polyphosphate PPSE yielded 310 (Scheme 88). The compound 310 was further converted into tetracyclic ring system of silicine–methuenine alkaloids 311 and 312 in number of steps. The 1,4-dihydropyridine intermediate 299 was synthesized according to the literature procedure reported by Bennasar, et al.101
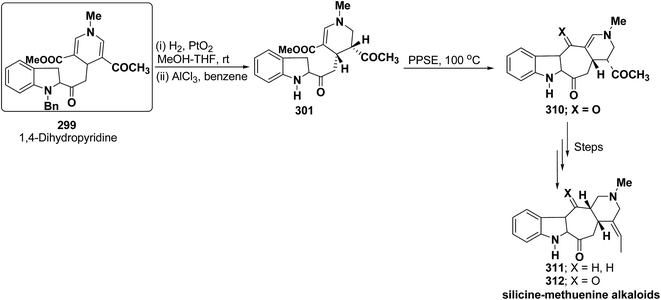 |
| | Scheme 88 Synthesis of silicine–methuenine alkaloids 311 and 312. | |
Bennasar, et al.103 has reported a general route for the synthesis of 6-oxo-16-episilicine 320 (a silicine–methuenine alkaloid), involving the nucleophilic addition of an acetylindole enolate 296 to the pyridinium salt 284 to yield 1,4-dihydropyridine 313 as an intermediate. The 1,4-dihydropyridine 313 was functionalized to give 3,5-diacyl-1,4-dihydropyridine 314, which was reduced to tetrahydropyridine 315, and was subjected to PPSE-induced cyclisation to yield 316. Hydrolysis of compound 316 using MeOH·H2O, HCl followed by decarboxylation using 2,2′-dithiobis-(pyridine-N-oxide) yielded 317, which on N-debenzylation yielded 318. The debenzylated compound 318 on reduction using NaBH3CN yields 319, which on oxidation with MnO2 yield 6-oxo-16-episilicine 320 (Scheme 89).
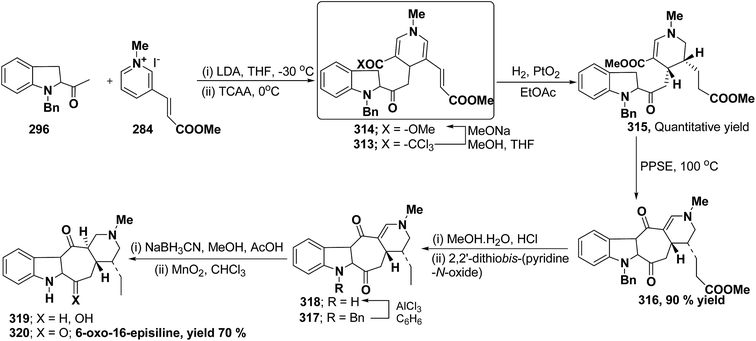 |
| | Scheme 89 Synthesis of 6-oxo-16-episilicine 320. | |
Pyridoacridines are a family of alkaloids based on 1H-pyrido[4,3,2,mn]acridine skeleton. The synthesis of pyridoacridine ring 329 of alkaloid amphimedine 330 was achieved by condensing tbutyl-protected 2-lithio-1,4-DHP 324 with 3,4-disubstituted cyclobutanediones 325 to yield 326 followed by thermolysis to yield 327. The conversion of 327 in aniline derivative 328 was achieved using hydroxylamine hydrochloride and oxidation with t-BuOOH in presence of KOH yielded dihydroquinoline hydroquinine ring system 329. The Boc-protected 2-lithio-1,4-DHP was synthesized by the reaction between the Grignard reagent of N-(2-bromophenyl)-2,5-dimethylpyrrole 321 with N-carboxyphenylpyridinium chloride (Scheme 90).104
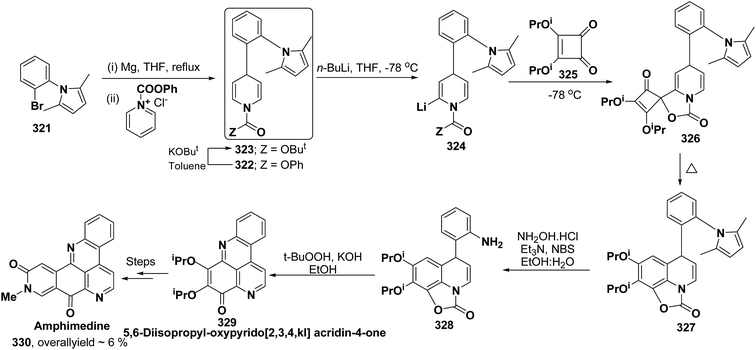 |
| | Scheme 90 Synthesis of 5,6-diisopropyl-oxypyrido[2,3,4,kl]acridin-4-one 330. | |
Akagerine 338 is a tetracyclic indole alkaloid. A formal stereoselective synthesis of akagerine 338 had been reported via pentacyclic dilactam 337.105 The intermediate dilactam 337 was synthesized starting from 1,4-dihydropyridine 333, which was synthesized from the nucleophilic attack of the N-acetyl indole 331 on pyridinium salt 332. Compound 333 was converted into 334 using sulfonic acid. The hydroxyl group of the compound 334 was removed via first triflation followed by reduction to afford compound 335. The key step is the formation of piperidine ring of compound 336, which was accomplished by cyclization of thionium ion generated by Pummerer rearrangement using TFAA in presence of 2,6-di(t-butyl)pyridine. Dilactam 337 was obtained by the treatment of compound 336 with tributyltin hydride in benzene (Scheme 91).106
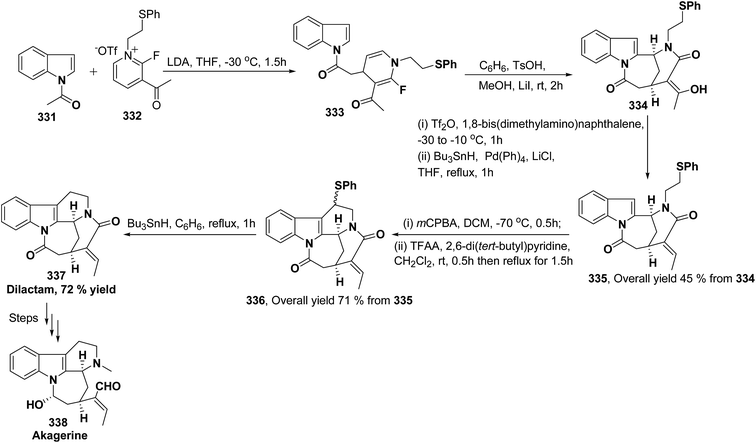 |
| | Scheme 91 Synthesis of dilactam 337. | |
Formal synthesis of (±)-geissoschizine had been carried out using 1,4-dihydropyridine derivative 340.107 The 1,4-dihydropyridine derivative 340 was synthesized from nucleophilic attack of the N-acetyl indole 331 on pyridinium salt 339. Compound 340 was converted into 341 using sulfonic acid in benzene, which on treatment with HCl followed by borohydride reduction yielded 342a. Compound 342a was first converted into sulfone 342b, followed by treatment with sodium methoxide resulted into the formation of compound 343, which on desulferisation using tributyltin hydride and AlBn give compound 344 (Scheme 92). Compound 344 has been converted into geissoschizine 345 by Yamada, et al.108
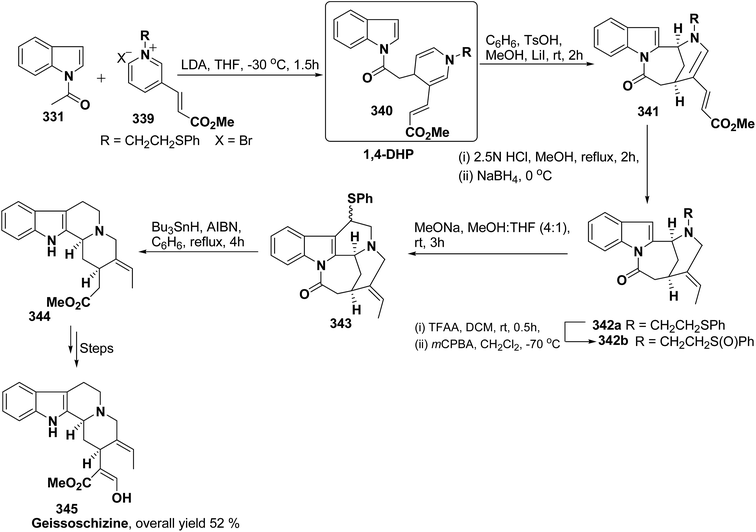 |
| | Scheme 92 Synthesis of geissoschizine 345. | |
A concise total synthesis of (±)-camptothecin and natural (+)-camptothecin has been reported starting from 2-fluoro-1,4-dihydropyridine adduct 351.109 The 1,4-DHP adduct 351 was achieved from compound 346 in three steps. 2-Fluoro-1,4-dihydropyridine adduct 351 on oxidation with DDQ resulted into the formation of compound 352, which on treatment with hydrogen atom donor tris(trimethylsilyl)silane-AIBN give compound 353 (Scheme 93). Formation of the pyran ring in compounds 354 and 20-dioxycamptothecin 355 takes place by the treatment of compound 353 with DIBAL and borohydride reduction. 20-Dioxycamptothecin 355 was further used for the synthesis of (±)-camptothecin and natural (+)-camptothecin via the literature procedure.110
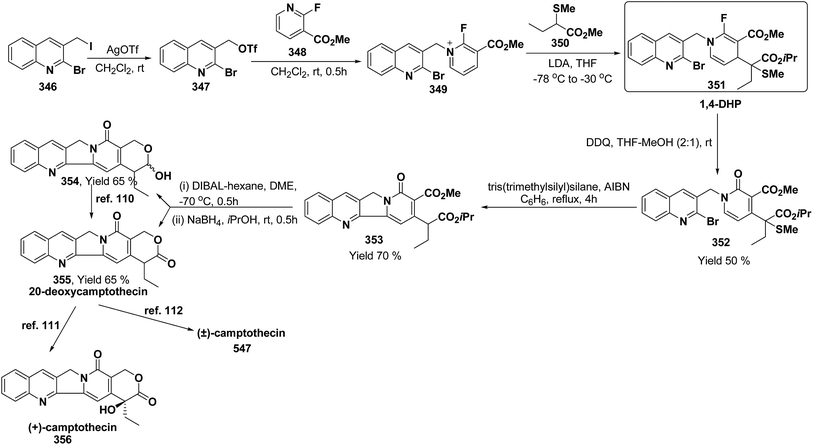 |
| | Scheme 93 Synthesis of (±)-camptothecin and natural (+)-camptothecin. | |
The synthesis of natural (+)-camptothecin 356 has been reported by the nucleophilic addition of a chiral auxiliary 357 to the pyridinium salt 349 (Scheme 94). The remaining synthetic steps are similar as used in Scheme 93.109
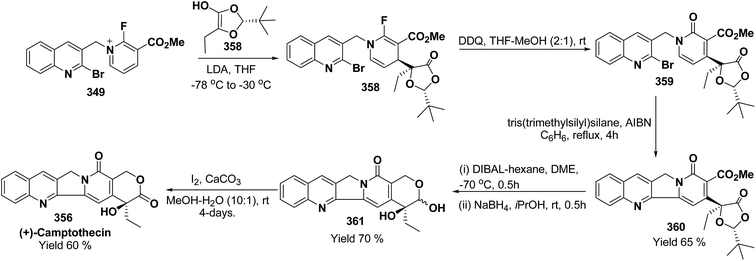 |
| | Scheme 94 Synthesis of (+)-camptothecin 356. | |
Putkonen, et al.111 has reported the total synthesis of (±)-tangutorine 368 and modified the first synthetic procedure reported by Berner, et al.112 For the synthesis of (±)-tangutorine 368, desired quinoline derivative 362 was reacted with tryptophyl bromide 255 to afford pyridinium salt 363. Since the total yield of the tangutorine skeleton was relatively low due to the different stereoisomers formed in the Fry reaction.112 The total synthesis of indole alkaloid (±)-tangutorine 368 has been carried out using dithionite reduction of pyridinium salt 363 to afford the 1,4-dihydropyridine intermediate 365. Cyclization of compound 364 in HCl/MeOH to compound 365 and reduction with sodium borohydride in glacial acetic acid for overnight yielded a mixture of isomers of compound 366. (±)-Tangutorine 368 was afforded after dehydration of compound 366 and finally reduction of the ester group in compound 367 (Scheme 95).
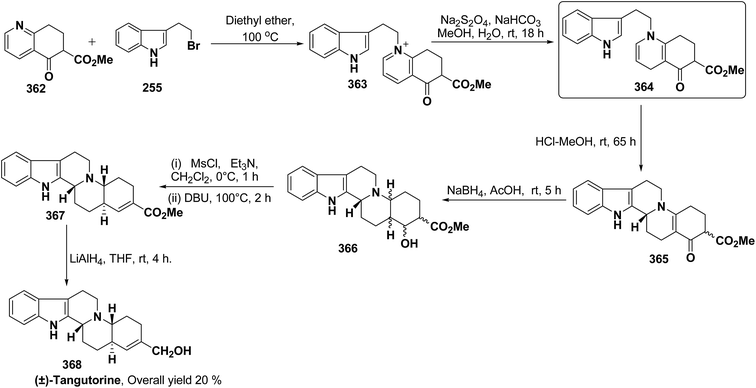 |
| | Scheme 95 Synthesis of (±)-tangutorine 368. | |
The harman-1,4-dihydropyridines 373a–b, which constitutes the original proposed structures for the indole alkaloid lyadine 375 and lyaline 376 have been synthesized by Bennasar, et al.113 (Scheme 96). The harman-1,4-dihydropyridines 373a–b were synthesized starting from harman 369 and pyridine 371. The nucleophilic addition reaction of Grignard reagent of harman 370 with pyridinium salt 372 prepared from pyridine 371. The harman-1,4-dihydropyridine 373a was converted into lyadine 375 in two synthetic steps first by N-deacetylation followed by borohydride reduction, whereas lyaline 376 was obtained from the N-deacetylation of compound 373b.
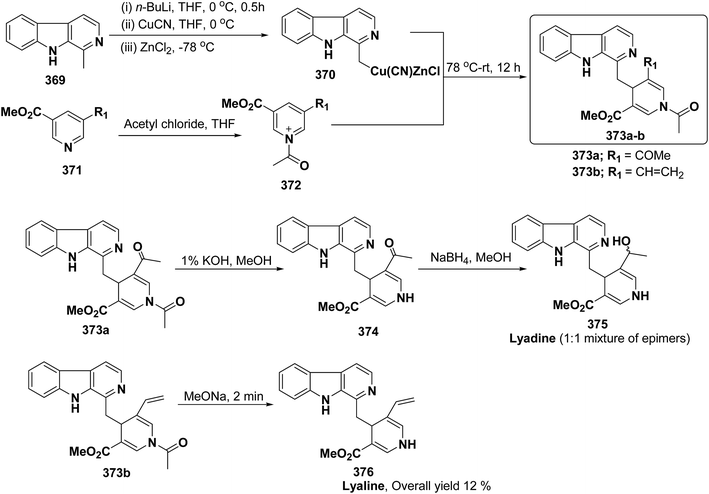 |
| | Scheme 96 Synthesis of lyadine 375 and lyaline 376. | |
Amat, et al.114 has proposed a model for the synthesis of Strychnos alkaloid e.g. dihydroakuammicine 380 could be obtained starting from a suitable functionalized enantiopure 2-(1,4-dihydro-4-pyridylmethyl)indole 377 (Scheme 97).
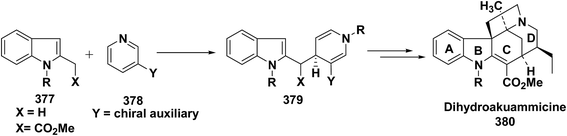 |
| | Scheme 97 Proposed procedure for the synthesis of dihydroakuammicine 380. | |
The nucleophilic addition of indole acetic ester 381 and N-alkyl pyridinium salt 382a–b followed by acid cyclization gave a mixture of expected tetracyclic compound 383a–b along with regioisomer 384a–b (Scheme 98). It is probably the bulkiness of chiral auxiliary hinders the approach of the nucleophile to the adjacent 4-position of pyridine ring.
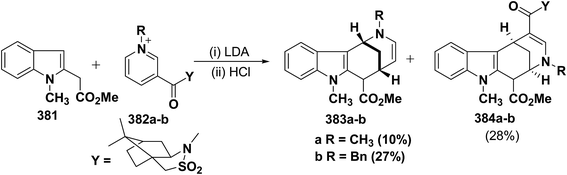 |
| | Scheme 98 Synthesis of tetracyclic precursors 383 and 384 of dihydroakuammicine. | |
The strategy for the synthesis of tetracyclic precursors ABCD of dihydroakuammicine using chiral auxiliary suffers two major drawbacks (i) low facial stereoselectivity due to inefficient coordination of nucleophile with the chiral auxiliary and (ii) low regioselectivity due to bulkiness of chiral auxiliary.114
4. Medicinal importance of dihydropyridines
The first synthesis of 1,4-dihydropyridine (1,4-DHP) nucleus was reported in 1881 by A. Hantzsch.26a It took 80 years to test these compounds for their biological activity, and finally Bossert and Vater at Bayer AG got the Hantzsch type 1,4-DHP possessing outstanding coronary vasodilator activity.115 Nifedipine, the 2-nitrophenyl derivative (Table 17) was then introduced for the treatment of coronary diseases.116 Due to the resemblance with NADH and also interesting biological activities, subsequently several other 1,4-DHPs (Table 17) have been introduced for clinical use as hypertensive agents that have a worldwide market of several billion dollars.117 Amlodipine (Norvasc), with sales of more than $ 5 billion in recent years is among the top-five best selling drugs. DHP's have also been explored as anti-inflammatory,118 anti-tumor,119 anti-tubercular,120 anti-convulsant activity121 etc. 1,4-DHP drugs can be divided into three generations depending on their pharmacologic and pharmacokinetic profiles.122 Because of the short duration and rapid onset of vasodilator action, nifedipine activated sympathetic tone.123 Therefore, in the quest of better therapeutic standard, the first-generation drug, nifedipine has been extensively modified. The second generation drugs exert less sympathetic reflex by designing slow-release formulations of preparations of the short-acting drugs.124 The third generation drugs exhibit more stable pharmacokinetics than the second-generation. These are less cardioselective and, hence well tolerated in patients with heart failure.125 Now second and third generation 1,4-DHPs are being explored for the treatment of hypertension, angina, hypertrophic cardiomyopathy,126 post-hemorrhagic cerebral vasospasm127 and pulmonary hypertension, Raynaud's phenomenon.128
Table 17 Commercially important 1,4-DHP's
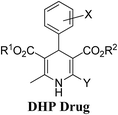
|
Brand name |
X |
R1 |
R2 |
Y |
| 1st generation |
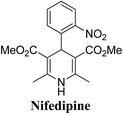 |
Procardia, Adalat |
2-NO2 |
–Me |
–Me |
Me |
|
| 2nd generation |
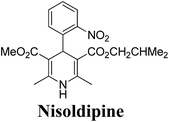 |
Baymycard, Sular, Syscor |
2-NO2 |
–Me |
–CH2CHMe2 |
Me |
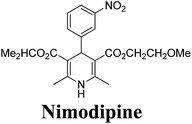 |
Nimotop |
3-NO2 |
–CHMe2 |
–CH2CH2OMe |
Me |
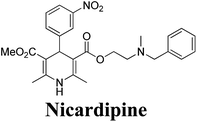 |
Cardene, Carden SR |
3-NO2 |
–Me |
–CH2CH2N(Me)CH2Ph |
Me |
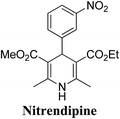 |
Cardif, Nitrepin, Baylotensin |
3-NO2 |
–Me |
–Et |
Me |
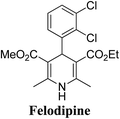 |
Plendil |
2,3-Di-Cl |
–Me |
–Et |
Me |
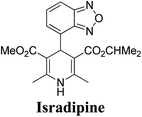 |
DynaCirc, DynaCirc CR |
 |
–Me |
–CHMe2 |
Me |
|
| 3rd generation |
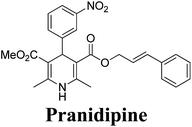 |
Acalas |
3-NO2 |
–Me |
–CH2CHCHPh |
Me |
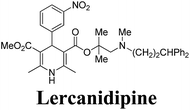 |
Zanidip |
3-NO2 |
–Me |
–C(Me)2CH2N(Me)CH2CH2CHPh2 |
Me |
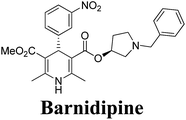 |
HypoCa |
3-NO2 |
–Me |
 |
Me |
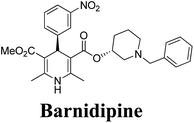 |
Coniel |
3-NO2 |
–Me |
 |
Me |
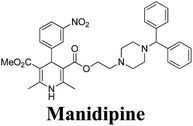 |
Calslot, Madipine |
3-NO2 |
–Me |
 |
Me |
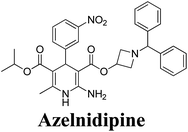 |
Calblock |
3-NO2 |
–CHMe2 |
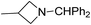 |
NH2 |
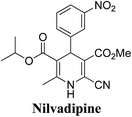 |
Nilvadil |
3-NO2 |
–CHMe2 |
–Me |
CN |
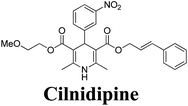 |
Atelec, Cinalong, Siscard |
3-NO2 |
–CH2CH2OMe |
–CH2CHCHPh |
Me |
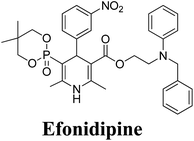 |
Landel |
3-NO2 |
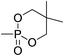 |
–CH2CH2N(Ph)CH2Ph |
Me |
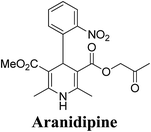 |
Sapresta |
2-NO2 |
–Me |
–CH2COCH3 |
Me |
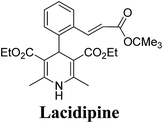 |
Motens, Lacipil |
2-CHCHCOOCMe3 |
–Et |
–Et |
Me |
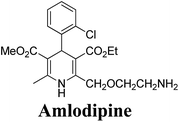 |
Norvasc, Amlodin |
2-Cl |
–Me |
–Et |
CH2OCH2CH2NH2 |
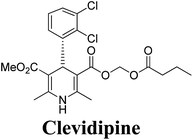 |
Cleviprex |
2,3-Di-Cl |
–Me |
–CH2OCOnPr |
Me |
5. Conclusion
Dihydropyridine (DHP) scaffold has certainly revolutionalised the pharmaceutical research with its unprecedented biological properties. We have reviewed various methods for the synthesis of 1,2- and 1,4-dihydropyridine including the strategies which can furnish enantiopure DHPs, either by asymmetric synthesis or by chiral resolution. DHPs accept wide range of reactions that has triggered the synthesis of different class of compounds of great medicinal value, including natural products. Alkaloids deplancheine, tangutorine, dihydroakummicine, olivacine, (±)-guatambuine, L-pipecolic acid, (±)-geissoschizine, (±)-akagerine, lyaline, lyadine, harman-dihydropyrimidine, camptothecin, 20-deoxycamptothecin, akuammiline alkaloids precursor, silicine–methuenine alkaloids, vinoxine, (±)-2,7-dihydropleiocarpamine, tubifoline & tubifolidine, ervitsine and several other alkaloids have been synthesized following two simple reaction methodologies of DHPs. First, both 1,2 and 1,4-dihydropyridines give 1,3-dipolar addition reaction and readily undergo [2 + 2] cycloaddition reactions with dienophiles. Second, reaction of enamine functional group in dihydropyridines with electrophile (x+) to form iminium salt, which undergoes α-attack of nucleophiles in a regio- and stereoselective manner (non-biomimetic oxidation) to give substituted tetrahydropyridine derivatives.
We have collated vast amount of literature regarding the strategies for the synthesis of DHPs. However, the continuously growing field of DHPs demands more diversity, tethering/merging of new moieties with DHP scaffold. Therefore this is still a vibrant and challenging area for medicinal chemists to explore new synthetic methodologies or sync different methodologies together to develop new DHP-drugs. Many DHP-based drugs have been blockbuster of their time; therefore, we have also highlighted commercial value of previously approved DHP-based drugs in brief.
Acknowledgements
One of the author SKS thank University Grants Commission (UGC), India for providing the UGC startup Grant, No. F.30-109/2015, FD Diary No. 7235.
References
- Q. Huang, Y. Li, C. Sheng, Y. Dou, M. Zheng, Z. Zhu and J. Wang, J. Hypertens., 2015, 33(1), e94 CrossRef PubMed.
- H. Nakano, K. Osone, M. Takeshita, E. Kwon, C. Seki, H. Matsuyama, N. Takano and Y. Kohari, Chem. Commun., 2010, 46, 4827–4829 RSC.
-
(a) S. K. Singh and V. K. Sharma, Curr. Org. Chem., 2014, 18, 1159–1207 CrossRef CAS;
(b) R. Lavilla, J. Chem. Soc., Perkin Trans. 1, 2002, 1141–1156 RSC;
(c) U. Eisner and J. Kuthan, Chem. Rev., 1972, 72, 1–43 CrossRef CAS;
(d) D. M. Stout and A. I. Meyers, Chem. Rev., 1982, 82, 223–243 CrossRef CAS;
(e) E. M. P. Silva, P. A. M. M. Varandas and A. M. S. Silva, Synthesis, 2013, 45, 3053–3089 CrossRef CAS.
- F. W. Fowler, J. Org. Chem., 1972, 37, 1321–1323 CrossRef.
-
(a) C. A. Bear, W. R. Cullen, J. P. Kutney, V. E. Ridaura, J. Trotter and A. Zanarotti, J. Am. Chem. Soc., 1973, 95, 3058–3060 CrossRef CAS;
(b) J. P. Kutney, R. Greenhouse and V. E. Riduara, J. Am. Chem. Soc., 1974, 96, 7364–7365 CrossRef CAS.
- P. Beeken, J. N. Bonfiglio, I. Hasan, J. J. Piwinski, B. Weinstein, K. A. Zoo and F. W. Fowler, J. Am. Chem. Soc., 1979, 101, 6677–6682 CrossRef CAS.
-
(a) Y.-S. Cheng, A. Lupo and F. W. Fowler, J. Am. Chem. Soc., 1983, 105, 7696–7703 CrossRef CAS;
(b) M. J. Wyle and F. W. Fowler, J. Org. Chem., 1984, 49, 4025–4029 CrossRef CAS.
- D. L. Comins, H. Hong and J. M. Salvador, J. Org. Chem., 1991, 56, 7197–7199 CrossRef CAS.
- T. J. Donohoe, A. J. McRiner and P. Sheldrake, Org. Lett., 2000, 2, 3861–3863 CrossRef CAS PubMed.
-
(a) F. Palacios, C. Alonso, G. Rubiales and J. M. Ezpeleta, Eur. J. Org. Chem., 2001, 2115–2122 CrossRef CAS;
(b) F. Palacios, E. Herrán and G. Rubiales, J. Org. Chem., 1999, 64, 6239–6246 CrossRef CAS.
-
(a) H. M. Sklenicka, R. P. Hsung, L.-L. Wei, M. J. McLaughlin, A. I. Gerasyuto and S. J. Degen, Org. Lett., 2000, 2, 1161–1164 CrossRef CAS PubMed;
(b) L.-L. Wei, R. P. Hsung, H. M. Sklenicka and A. I. Gerasyuto, Angew. Chem., Int. Ed., 2001, 40, 1516–1518 CrossRef CAS;
(c) J. K. F. Geirsson and J. F. Johannesdottir, J. Org. Chem., 1996, 61, 7320–7325 CrossRef CAS PubMed.
- T.-P. Loh, P.-L. Lye, R.-B. Wang and K.-Y. Sim, Tetrahedron Lett., 2000, 41, 7779–7783 CrossRef CAS.
- B. Brunner, N. Stogaitis and M. Lautens, Org. Lett., 2006, 8, 3473–3476 CrossRef CAS PubMed.
- M. Motamed, E. M. Bunnelle, S. W. Singaram and R. Sarpong, Org. Lett., 2007, 9, 2167–2170 CrossRef CAS PubMed.
- D. M. Hodgson, M. L. Jones, C. R. Maxwell, A. R. Cowely, A. L. Thompson, O. Ichihara and I. R. Matthews, Tetrahedron, 2009, 65, 7825–7836 CrossRef CAS.
- J.-P. Wan, S.-F. Gan, G.-L. Sun and Y.-J. Pan, J. Org. Chem., 2009, 74, 2862–2865 CrossRef CAS PubMed.
- I. Yavari, M. J. Bayat, M. Sirouspour and S. Souri, Tetrahedron, 2010, 66, 7995–7999 CrossRef CAS.
- J. T. Binder and S. F. Kirsch, Org. Lett., 2006, 8, 2151–2153 CrossRef CAS PubMed.
- D. Tejedor, G. Méndez-Abt and F. García-Tellado, Chem.–Eur. J., 2010, 16, 428–431 CrossRef CAS PubMed.
- H. Wei, Y. Wang, B. Yue and P.-F. Xu, Adv. Synth. Catal., 2010, 352, 2450–2454 CrossRef CAS.
- T. Harschneck and S. F. Kirsch, J. Org. Chem., 2011, 76, 2145–2156 CrossRef CAS PubMed.
- T. Shono, Y. Matsumara, O. Onomura and Y. Yamada, Tetrahedron Lett., 1987, 28, 4073–4074 CrossRef CAS.
-
(a) D. Tejedor, L. Cotos, G. Méndez-Abt and F. García-Tellado, J. Org. Chem., 2014, 79, 10655–10661 CrossRef CAS PubMed;
(b) D. Tejedor, M. C. Prieto-Ramírez, M. Ingold, M. Chicón and F. García-Tellado, Org. Lett., 2016, 18, 2770–2773 CrossRef CAS PubMed.
- C. Challa, M. John and R. S. Lankalapalli, Tetrahedron Lett., 2013, 54, 3810–3812 CrossRef CAS.
- B. J. Fallon, J. B. Garsi, E. Derat, M. Amatore, C. Aubert and M. Petit, ACS Catal., 2015, 5, 7493–7497 CrossRef CAS.
-
(a) A. Hantzsch, Ber. Dtsch. Chem. Ges., 1881, 14, 1637–1638 CrossRef;
(b) A. Hantzsch, Justus Liebigs Ann. Chem., 1882, 1, 215–219 Search PubMed.
- M.-L. Bennasar, R. Lavilla, M. Alvarez and J. Bosch, Heterocycles, 1988, 27, 789–824 CrossRef CAS.
- F. Kröhnke, Angew. Chem., 1953, 65, 605–628 CrossRef.
- F. Kröhnke, K. Ellgast and E. Bertram, Liebigs Ann. Chem., 1956, 600, 176–210 CrossRef.
- H. Ahlbrecht and F. Kröhnke, Liebigs Ann. Chem., 1967, 704, 133–139 CrossRef CAS.
- D. Mauzerall and F. H. Westheimer, J. Am. Chem. Soc., 1955, 77, 2261–2264 CrossRef CAS.
- R. F. Hutton and F. H. Westheimer, Tetrahedron, 1958, 3, 73–77 CrossRef CAS.
- H. Minato, S. Fujie, K. Okuma and M. Kobayashi, Chem. Lett., 1977, 1091–1094 CrossRef CAS.
-
(a) D. L. Comins and A. H. Abdullah, J. Org. Chem., 1982, 47, 4315–4319 CrossRef CAS;
(b) D. L. Comins and N. B. Mantlo, J. Heterocycl. Chem., 1983, 20, 1239–1243 CrossRef CAS;
(c) D. L. Comins, A. H. Abdullah and R. K. Smith, Tetrahedron Lett., 1983, 24, 2711–2714 CrossRef CAS;
(d) D. L. Comins, Tetrahedron Lett., 1983, 24, 2807–2810 CrossRef CAS;
(e) D. L. Comins and N. B. Mantlo, Tetrahedron Lett., 1983, 24, 3683–3686 CrossRef CAS;
(f) D. L. Comins, E. D. Stroud and J. J. Herrick, Heterocycles, 1984, 22, 151–157 CrossRef CAS.
- R. Yamaguchi, Y. Nakazono and M. Kawanisi, Tetrahedron Lett., 1983, 24, 1801–1804 CrossRef CAS.
-
(a) E. Piers and M. Soucy, Can. J. Chem., 1974, 52, 3563–3564 CrossRef CAS;
(b) K. Akiba, Y. Iseki and M. Wada, Tetrahedron Lett., 1982, 23, 429–432 CrossRef CAS;
(c) D. D. Weller, G. R. Luellen and D. L. Weller, J. Org. Chem., 1983, 48, 3061–3067 CrossRef CAS;
(d) D. D. Weller, E. P. Stirchak and D. L. Weller, J. Org. Chem., 1983, 48, 4597–4605 CrossRef CAS.
- G. W. J. Fleet, C. J. Fuller and P. J. C. Harding, Tetrahedron Lett., 1978, 19, 1437–1440 CrossRef.
-
(a) J. C. Bommer and K. W. Morse, J. Chem. Soc., Chem. Commun., 1977, 137–138 RSC;
(b) J. C. Bommer and K. W. Morse, Inorg. Chem., 1980, 19, 587–593 CrossRef CAS.
- M. F. Semmelhack, R. D. Stauffer and A. Yamashita, J. Org. Chem., 1977, 42, 3180–3188 CrossRef CAS.
- D. L. Comins and A. H. Abdullah, J. Org. Chem., 1984, 49, 3392–3394 CrossRef CAS.
- A. I. De Lucas, J. Fernández-Gadea, N. Martín and C. Seoane, Tetrahedron, 2001, 57, 5591–5595 CrossRef CAS.
- M.-L. Bennasar, T. Roca, M. Monerris, C. Juan and J. Bosch, Tetrahedron, 2002, 58, 8099–8106 CrossRef CAS.
- S. Ko, M. N. V. Sastry, C. Lin and C.-F. Yao, Tetrahedron Lett., 2005, 46, 5771–5774 CrossRef CAS.
- J.-C. Legeay, J. J. V. Eynde and J. P. Bazureaua, Tetrahedron, 2005, 61, 12386–12397 CrossRef CAS.
- J. H. Lee, Tetrahedron Lett., 2005, 46, 7329–7330 CrossRef CAS.
- M. Meheswara, V. Siddaiah, Y. K. Rao, Y.-M. Tzeng and C. Sridhar, J. Mol. Catal. A: Chem., 2006, 260, 179–180 CrossRef.
- J. Bräckow and K. T. Wanner, Tetrahedron, 2006, 62, 2395–2404 CrossRef.
- G. Babu and P. T. Perumal, Aldrichimica Acta, 2000, 33, 16–22 CAS.
-
(a) R. Nagarajan, S. Chitra and P. T. Perumal, Tetrahedron, 2001, 57, 3419–3423 CrossRef CAS;
(b) M. Anniyappan, D. Muralidharan and P. T. Perumal, Tetrahedron Lett., 2003, 44, 3653–3657 CrossRef CAS.
- R. S. Kumar, R. Nagarajan and P. T. Perumal, Synthesis, 2004, 949–959 CAS.
- V. Sridharan, P. T. Perumal, C. Avendano and J. C. Menendez, Tetrahedron, 2007, 63, 4407–4413 CrossRef CAS.
- J. Moreau, A. Duboc, C. Hubert, J.-P. Hurvois and J.-L. Renaud, Tetrahedron Lett., 2007, 48, 8647–8650 CrossRef CAS.
- F. Palacios, E. Herrán, C. Alonso and G. Rubiales, ARKIVOC, 2007, iv, 397–407 Search PubMed.
- S. Kikuchi, M. Iwai, H. Murayama and S. Fukuzawa, Tetrahedron Lett., 2008, 49, 114–116 CrossRef CAS.
- S.-X. Wang, Z. Y. Li, J.-C. Zhang and J.-T. Li, Ultrason. Sonochem., 2008, 15, 677–680 CrossRef CAS PubMed.
- A. M. Zonouz and S. B. Hosseini, Synth. Commun., 2008, 38, 290–296 CrossRef CAS.
- M. Sharma, N. Aggarwal and D. S. Rawat, J. Heterocycl. Chem., 2008, 45, 737–739 CrossRef CAS.
- A. Heydari, S. Khaksar, M. Tajbakhsh and H. R. Bijanzadeh, J. Fluorine Chem., 2009, 130, 609–614 CrossRef CAS.
- C. A. Sperger and K. T. Wanner, Tetrahedron, 2009, 65, 5824–5833 CrossRef CAS.
- E. Rafiee, S. Eavani, S. Rashidzadeh and M. Joshaghani, Inorg. Chim. Acta, 2009, 362, 3555–3562 CrossRef CAS.
- A. Debache, W. Ghalem, R. Boulcina, A. Belfaitah, S. Rhouati and B. Carboni, Tetrahedron Lett., 2009, 50, 5248–5250 CrossRef CAS.
- G. Sabitha, K. Arundhathi, K. Sudhakar, B. S. Sastry and J. S. Yadav, Synth. Commun., 2009, 39, 2843–2851 CrossRef CAS.
- T. Sirijindalert, K. Hansuthirakul, P. Rashatasakhon, M. Sukwattanasinitt and A. Ajavakom, Tetrahedron, 2010, 66, 5161–5167 CrossRef CAS.
- J. Yang, C. Wang, X. Xie, H. Li and Y. Li, Eur. J. Org. Chem., 2010, 4189–4193 CrossRef CAS.
- F. Tamaddon, Z. Razmi and A. A. Jafari, Tetrahedron Lett., 2010, 51, 1187–1189 CrossRef CAS.
- S. Sueki, R. Takei, J. Abe and I. Shimizu, Tetrahedron Lett., 2011, 52, 4473–4477 CrossRef CAS.
- X. Wang, H. Gong, Z. Quan, L. Li and H. Ye, Synth. Commun., 2011, 41, 3251–3258 CrossRef CAS.
- T. Shibanuma, M. Iwanani, K. Okuda, T. Takenaka and M. Murakami, Chem. Pharm. Bull., 1980, 28, 2809–2812 CrossRef CAS PubMed.
- T. Tamazawa, H. Arima, T. Kojima, Y. Isomura, M. Okada, S. Fujita, T. Furuya, T. Takenaka, O. Inagaki and M. Terai, J. Med. Chem., 1986, 29, 2504–2511 CrossRef PubMed.
- B.-L. Zhang, W. He, X. Shi, M.-L. Huan, Q.-J. Huang and S.-Y. Zhou, Bioorg. Med. Chem. Lett., 2010, 20, 805–808 CrossRef CAS PubMed.
- J. W. Young, US Pat., 2002, vol. 6, 448, pp. 275B2.
- S. Goldmann, J. Stolefuss and L. Born, J. Med. Chem., 1992, 35, 3341–3344 CrossRef CAS PubMed.
-
(a) Z. Xitian, US Pat., 2003, vol. 6, 646, p. 131;
(b) R. Joshi and M. Gurjar, US Pat., 2005, vol. 7, 148, p. 358;
(c) P. Spargo, US Pat., 2000, vol. 6, 046, p. 338.
- D. M. Gotrane, R. D. Deshmukh, P. V. Ranade, S. P. Sonawane, B. M. Bhawal, M. M. Gharpure and M. K. Gurjar, Org. Process Res. Dev., 2010, 14, 640–643 CrossRef CAS.
- M. D. Taylor, R. J. Himmelsbach, B. E. Kornberg, J. Quin, E. Lunney and A. Michel, J. Org. Chem., 1989, 54, 5585–5590 CrossRef CAS.
- J. E. Arrowsmith, S. F. Campbell, P. E. Cross, J. K. Stubbs, R. A. Burges, D. G. Gardiner and K. J. Blackburn, J. Med. Chem., 1986, 29, 1696–1702 CrossRef CAS PubMed.
- B. Lamm, R. Simonsson and S. Sundell, Tetrahedron Lett., 1989, 30, 6423–6426 CrossRef CAS.
- D. Vo, W. C. Matowe, M. Ramesh, N. Iqbal, M. W. Wolowyk, S. E. Howlett and E. E. Knaus, J. Med. Chem., 1995, 38, 2851–2859 CrossRef CAS PubMed.
- R. Shan, S. E. Howlett and E. E. Knaus, J. Med. Chem., 2002, 45, 955–961 CrossRef CAS PubMed.
- R. Shan and E. E. Knaus, Bioorg. Med. Chem. Lett., 1999, 9, 2613–2614 CrossRef CAS PubMed.
- J. Jiang, A.-H. Li, S.-Y. Jang, L. Chang, N. Melman, S. Moro, X. Ji, E. B. Lobkovsky, J. C. Clardy and K. A. Jacobson, J. Med. Chem., 1999, 42, 3055–3065 CrossRef CAS PubMed.
- N. Martín, A. Martinez-Grau, C. Seoane, J. L. Marco, A. Albert and F. H. Cano, Tetrahedron: Asymmetry, 1995, 6, 877–880 CrossRef.
- J. Jiang, J. Yu, X.-X. Sun, Q.-Q. Rao and L.-Z. Gong, Angew. Chem., Int. Ed., 2008, 47, 2458–2462 CrossRef CAS PubMed.
- T. Yamamoto, S. Ohno, S. Niwa, M. Tokumasu, M. Hagihara, H. Koganei, S. Fujita, T. Takeda, Y. Saitou, S. Iwayama, A. Takahara, S. Iwata and M. Shoji, Bioorg. Med. Chem. Lett., 2011, 21, 3317–3319 CrossRef CAS PubMed.
- J. P. Kutney, M. Noda, N. G. Lewis, B. Monteiro, D. Mostowicz and B. R. Worth, Can. J. Chem., 1982, 60, 2426–2430 CrossRef CAS.
- D. L. Comins and Y. C. Myoung, J. Org. Chem., 1990, 55, 292–298 CrossRef CAS.
- A. B. Charette, M. Grenon, A. Lemire, M. Pourashraf and J. Martel, J. Am. Chem. Soc., 2001, 123, 11829–11830 CrossRef CAS PubMed.
- A. Lemire, M. Grenon, M. Pourashraf and A. B. Charette, Org. Lett., 2004, 6, 3517–3520 CrossRef CAS PubMed.
- R. Abou-Jneid, S. Ghoulami, M.-T. Martin, E. T. H. Dau, N. Travert and A. Al-Mourabit, Org. Lett., 2004, 6, 3933–3936 CrossRef CAS PubMed.
- A. Lemire and A. B. Charette, Org. Lett., 2005, 7, 2747–2750 CrossRef CAS PubMed.
- C. Schroif-Gregoire, N. Travert, A. Zaparucha and A. Al-Mourabit, Org. Lett., 2006, 8, 2961–2964 CrossRef CAS PubMed.
- A. Lemire and A. B. Charette, J. Org. Chem., 2010, 75, 2077–2080 CrossRef CAS PubMed.
- E. Wenkert, T. D. J. Halls, G. Kunesch, K. Orito, R. L. Stephens, W. A. Temple and J. S. Yadav, J. Am. Chem. Soc., 1979, 101, 5370–5376 CrossRef CAS.
- R. Besselievre, B.-P. Cosson, B. C. Das and H.-P. Husson, Tetrahedron Lett., 1980, 21, 63–66 CrossRef CAS.
- J. Bosch, M.-L. Bennasar, E. Zulaica and M. Feliz, Tetrahedron Lett., 1984, 25, 3119–3122 CrossRef CAS.
- J. Bosch, M.-L. Bennasar and E. Zulaica, J. Org. Chem., 1986, 51, 2289–2297 CrossRef CAS.
- M. Alvarez, M. Salas, A. de Veciana, R. Lavilla and J. Bosch, Tetrahedron Lett., 1990, 31, 5089–5091 CrossRef CAS.
- M.-L. Bennasar, B. Vidal and J. Bosch, J. Am. Chem. Soc., 1993, 115, 5340–5341 CrossRef CAS.
- M.-L. Bennasar, B. Vidal and J. Bosch, J. Chem. Soc., Chem. Commun., 1995, 125–126 RSC.
- M.-L. Bennasar, E. Zulaica, J.-M. Jiménez and J. Bosch, J. Org. Chem., 1993, 58, 7756–7767 CrossRef CAS.
-
(a) M.-L. Bennasar, B. Vidal and J. Bosch, J. Org. Chem., 1996, 61, 1916–1917 CrossRef CAS;
(b) M.-L. Bennasar, B. Vidal and J. Bosch, J. Org. Chem., 1997, 62, 3597–3609 CrossRef CAS.
- M.-L. Bennasar, B. Vidal, A. Lázaro, R. Kumar and J. Bosch, Tetrahedron Lett., 1996, 37, 3541–3544 CrossRef CAS.
- M.-L. Bennasar, B. Vidal and J. Bosch, Chem. Commun., 1996, 2755–2756 RSC.
- D. Zhang, I. Llorente and L. S. Liebesking, J. Org. Chem., 1997, 62, 4330–4338 CrossRef CAS PubMed.
- W. Benson and E. Winterfeldt, Heterocycles, 1981, 15, 935–941 CrossRef CAS.
- M.-L. Bennasar, B. Vidal, B. A. Sufi and J. Bosch, Chem. Commun., 1998, 2639–2640 RSC.
- M.-L. Bennasar, J.-M. Jimenez, B. Vidal, B. A. Sufi and J. Bosch, J. Org. Chem., 1999, 64, 9605–9612 CrossRef CAS.
- K. Yamada, K. Aoki, T. Kato, D. Uemura and T. E. E. Van, J. Chem. Soc., Chem. Commun., 1974, 908–909 RSC.
- M.-L. Bennasar, E. Zulaica, C. Juan, Y. Alonso and J. Bosch, J. Org. Chem., 2002, 67, 7465–7474 CrossRef PubMed.
-
(a) R. T. Brown, L. Jianli and C. A. M. Santos, Tetrahedron Lett., 2000, 41, 859–862 CrossRef CAS;
(b) W. Shen, G. A. Coburn, W. G. Bornmann and S. J. Danishefsky, J. Org. Chem., 1993, 58, 611–617 CrossRef CAS;
(c) K. Tagami, N. Nakazawa, S. Sano and Y. Nagao, Heterocycles, 2000, 53, 771–775 CrossRef CAS.
- T. Putkonen, A. Tolvanen and R. Jokela, Tetrahedron Lett., 2001, 42, 6593–6594 CrossRef CAS.
- M. Berner, A. Tolvanen and R. Jokela, Tetrahedron Lett., 1999, 40, 7119–7122 CrossRef CAS.
- M.-L. Bennasar, T. Roca and M. Monernis, J. Org. Chem., 2004, 69, 752–756 CrossRef CAS PubMed.
- M. Amat, M.-D. Coll, N. Llor, C. Escolano, E. Molins, C. Miravitlles and J. Bosch, Tetrahedron: Asymmetry, 2003, 14, 1691–1699 CrossRef CAS.
- F. Bossert and W. Vater, Med. Res. Rev., 1989, 9, 291–324 CrossRef CAS PubMed.
- F. Bossert and W. Vater, Naturwissenschaften, 1971, 58, 578 CrossRef CAS PubMed.
-
(a) H. Nakayama and Y. Kanaoka, Heterocycles, 1996, 42, 901–909 CrossRef CAS;
(b) F. Bossert, H. Meyer and E. Wehinger, Angew. Chem., Int. Ed., 1981, 20, 762–769 CrossRef.
- H. Komoda, T. Inoue and K. Node, Clin. Exp. Hypertens., 2010, 32, 121–128 CrossRef CAS PubMed.
- R. Boer and R. Gekeler, Drugs Future, 1995, 20, 499–509 Search PubMed.
-
(a) G. A. Wächter, M. C. Davis, A. R. Martin and S. G. Franzblau, J. Med. Chem., 1998, 41, 2436–2438 CrossRef PubMed;
(b) B. Desai, D. Sureja, Y. Naliapara, A. Shah and A. K. Saxena, Bioorg. Med. Chem., 2001, 9, 1993–1998 CrossRef CAS PubMed.
- J. M. Tusell, S. Barrón and J. Serratosa, Brain Res., 1993, 622, 99–104 CrossRef CAS PubMed.
- P. Ioan, E. Carosati, M. Micucci, G. Cruciani, F. Broccatelli, B. S. Zhorov, A. Chiarini and R. Budriesi, Curr. Med. Chem., 2011, 18, 4901–4922 CrossRef CAS PubMed.
- F. H. Leenen, Am. J. Hypertens., 1996, 9, 97S–104S CrossRef CAS PubMed.
- D. D. Freedman and D. D. Waters, Drugs, 1987, 34, 578–598 CrossRef CAS PubMed.
- T. F. Lüscher and F. Cosentino, Drugs, 1998, 55, 509–517 CrossRef.
- T. Tokushima, T. Utsunomiya, T. Ogawa, K. Kidoh, Y. Ohtsubo, T. Ryu, K. Yoshida, T. Ogata, S. Tsuji and S. Matsuo, Basic Res. Cardiol., 1996, 91, 329–336 CrossRef CAS PubMed.
- D. Tomassoni, A. Lanari, G. Silvestrelli, E. Traini and F. Amenta, Clin. Exp. Hypertens., 2008, 30, 744–766 CrossRef CAS PubMed.
- A. Herrick, Curr. Treat. Options Cardiovasc. Med., 2008, 10, 146–155 CrossRef PubMed.
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a