
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThermoresponsive poly(2-oxazoline)s, polypeptoids, and polypeptides
Richard
Hoogenboom
*a and
Helmut
Schlaad
*b
aSupramolecular Chemistry Group, Department of Organic and Macromolecular Chemistry, Ghent University, Krijgslaan 281 S4, B-9000 Ghent, Belgium. E-mail: richard.hoogenboom@ugent.be
bInstitute of Chemistry, University of Potsdam, Karl-Liebknecht-Str. 24-25, 14476 Potsdam, Germany. E-mail: schlaad@uni-potsdam.de
First published on 5th September 2016
Abstract
This review covers the recent advances in the emerging field of thermoresponsive polyamides or polymeric amides, i.e., poly(2-oxazoline)s, polypeptoids, and polypeptides, with a specific focus on structure–thermoresponsive property relationships, self-assembly, and applications.
 Richard Hoogenboom | Richard Hoogenboom (1978) studied chemical engineering at the Eindhoven University of Technology (the Netherlands). In 2005, he obtained his Ph.D. under the supervision of Ulrich S. Schubert and continued working as a project leader for the Dutch Polymer Institute. After postdoctoral training with Martin Moeller and Roeland Nolte, he was appointed as associate professor at Ghent University in 2010 and in October 2014 he was promoted to full professor. His research interests include stimuli-responsive polymers, supramolecular polymers, and poly(2-oxazoline)s. He has published more than 300 scientific articles and is currently associate editor for European Polymer Journal and Australian Journal of Chemistry. Prof. Hoogenboom is the recipient of the inaugural Polymer Chemistry Lectureship (2015) and the fifth PI IUPAC award (2016). |
 Helmut Schlaad | Helmut Schlaad (1967) studied chemistry at the University of Mainz (Germany) and earned a doctoral degree in Physical Chemistry, under Axel H. E. Müller in 1997. After one year post-doctoral fellowship with Rudolf Faust at the University of Massachusetts in Lowell (USA), he moved to the Max Planck Institute of Colloids and Interfaces in Potsdam (Germany). He finished habilitation in 2004, mentored by Markus Antonietti, and became professor in 2014 at the University of Potsdam. His research interests are directed towards polymer synthesis, bio-sourced polymers, smart functional materials, and bio-inspired polymer structures. He is currently an associate editor for Polymer International (Wiley). |
1. Introduction
Natural systems are governed by adaptive and responsive behavior to survive, which is mostly driven by conformational changes and catalytic actions of proteins. These perfectly defined polyamide structures take on defined folded structures to get very sophisticated response behavior. Inspired by such systems, polymer scientists have developed a wide range of synthetic responsive polymer materials that respond to a wide range of stimuli (pH, temperature, ionic strength, molecules, etc.) with various changes in the polymer materials, including phase transition, color change, and shape transformation.1Thermoresponsive synthetic polymers that undergo a temperature induced solubility phase transition in aqueous solutions have received significant interest as mild temperature changes provide an easy way to trigger the solubility.2–8 Furthermore, such systems are highly appealing for development of drug delivery systems if the transition temperature is close to body temperature, allowing to prepare formulations that are soluble at room temperature and gel upon injection, polymeric sensors as well as switchable surfaces.9–14 Two different types of thermoresponsive polymers exist, namely those that undergo a demixing phase transition upon heating and those that demix upon cooling.
Polymers that are fully soluble at low temperatures and phase separate upon heating are so-called lower critical solution temperature (LCST) polymers, whereby the LCST represents the lowest phase transition temperature in the entire binodal phase diagram. Most commonly, the cloud point temperature (Tcp) is reported as the phase separation temperature at a specific concentration, determined by turbidimetry. The LCST phase transition is driven by the entropy-loss due to interaction of water molecules with the polymer and upon heating this entropy-loss becomes dominant eventually leading to dehydration of the polymer and phase separation.
Polymers with opposite behavior are known as upper critical solution temperature (UCST) polymers and such behavior is much more rare as it is mostly based on the enthalpic attraction between the polymer chains.5 Such enthalpic attraction is based on supramolecular interactions, such as electrostatic interactions or hydrogen bonding. A second type of UCST polymer phase transition is more commonly observed in alcohol–water mixtures, which is driven by the non-ideal mixing of the solvent mixture leading to a significant drop in solvent polarity upon heating, thereby increasing the polymer solubility.7
Within this review, we provide an overview of recent work on synthetic thermoresponsive polyamides, or polymeric amides, that closely resemble the (random coil) structure of proteins, namely poly(2-oxazoline)s, polypeptoids, and polypeptides (Fig. 1). Synthetic polypeptides are analogues of natural peptides that are prepared by ring-opening polymerization of the corresponding amino acid N-carboxyanhydrides,15 which are obviously not limited to the use of natural amino acids but allow the introduction of other functionalities to tune the thermoresponsive behavior. Polypeptoids are the tertiary amide analogues of polypeptides that can also be prepared by similar methods. Poly(2-oxazoline)s, prepared by living cationic ring-opening polymerization of 2-oxazolines,16,17 are polymers having tertiary amides units too, albeit with a slightly different configuration as only the nitrogen is part of the main chain and the carbonyl is part of the side chain. These three classes of polymes have in common that they have a hydrophilic main chain (partially) consisting of the repeating amide groups. As such, the introduction of slightly hydrophobic side chains allows accurate tuning of the hydrophilic–hydrophobic balance to obtain thermoresponsive polymers.
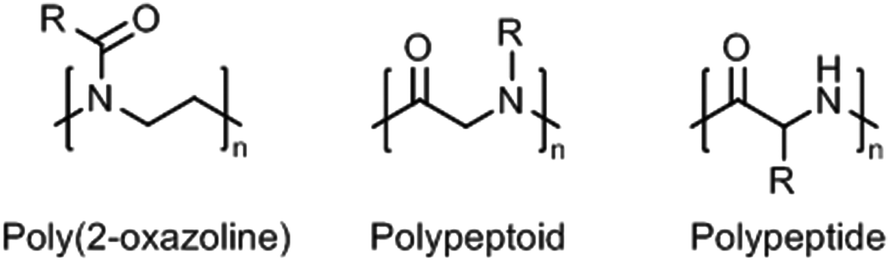 | ||
| Fig. 1 General chemical structures of (partially) main-chain polyamides: poly(2-oxazoline), polypeptoid, and polypeptide; R = organic substituent (usually alkyl). | ||
In the following, the recent developments in the areas of thermoresponsive poly(2-oxazoline)s (section 2.1), polypeptoids (section 2.2), and polypeptides (section 2.3) will be discussed mostly focusing on work from the past five years.
2. Thermoresponsive polyamides or polymeric amides
2.1 Poly(2-oxazoline)s
Poly(2-oxazoline)s are a synthetic class of polyamides, or polymeric amides, that are obtained by (living) cationic ring-opening polymerization of 2-oxazoline monomers yielding the corresponding ring-opened polymers that have a tertiary amide structure of which just the nitrogen is incorporated in the polymer backbone.17–21 The 2-substituent of the monomer can be varied allowing accurate control over the hydrophilic–hydrophobic balance of the resulting poly(2-oxazoline)s.22–25 If the hydrophilic tertiary amide bond of the polymer backbone is complimented with small hydrophobic side chains, this leads to thermoresponsive poly(2-oxazoline)s with lower critical solution temperature (LCST) behavior. In this regard, the boundaries are set by poly(2-methyl-2-oxazoline), which is very hydrophilic and does not show LCST behavior in water, and by poly(2-butyl-2-oxazoline), in which the hydrophobic butyl side chains dominate the behavior making the polymer insoluble in water. Obviously, larger hydrophobic aliphatic or aromatic side chains also result in hydrophobic poly(2-oxazoline)s. Poly(2-oxazoline) homopolymers bearing side chains with intermediate hydrophobicity will lead to thermoresponsive polymers with LCST behavior in water. An overview of the poly(2-oxazoline) homopolymers that have been reported to exhibit LCST behavior is shown in Fig. 2.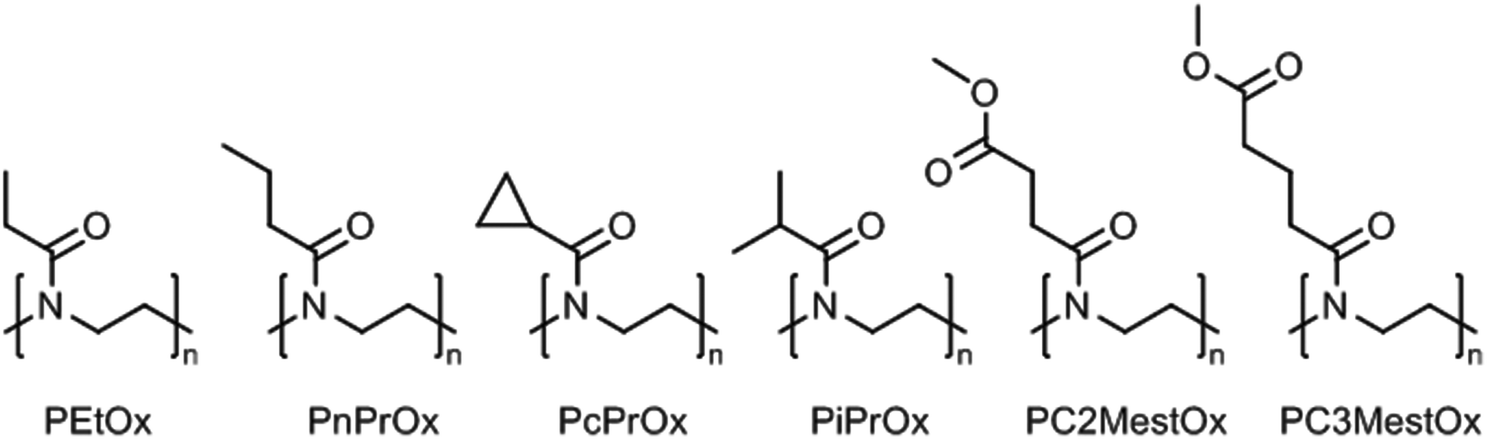 | ||
| Fig. 2 Thermoresponsive poly(2-oxazoline) homopolymers with the abbreviated names. | ||
Poly(2-isopropyl-2-oxazoline) (PiPrOx; Fig. 2) was first reported to be thermoresponsive by Uyama and Kobayashi in 1992.40 As the LCST of PiPrOx is close to body temperature, being 26 °C to 34 °C depending on polymer molar mass indicative of type 1 Flory–Huggins behavior,41 this polymer has developed into the most studied and most popular thermoresponsive poly(2-oxazoline) derivative.42–44 Similar to PEtOx, Filippov and coworkers demonstrated for PiPrOx that the Tcp decreases for star-shaped polymers compared to linear analogues due to enhanced local polymer concentration and polymer–polymer interactions.45 PiPrOx comb-shaped polymers were demonstrated by Jordan to have Tcp's of 28 °C to 31 °C with side chain graft lengths down to DP 4 units indicating that the high local concentration induces the thermoresponsive behavior since linear PiPrOx with DP 17 has been reported to have a Tcp of 73 °C.42,46 Furthermore, the Tcp of PiPrOx has been demonstrated to strongly depend on the end-group, especially for shorter polymer chains where the effect of the end-group is more dominant. Winnik et al. reported that the Tcp of PiPrOx with DP 57 decreased from 48.1 °C with methyl and hydroxyl end-groups to 32.5 °C with n-octadecyl and hydroxyl end-groups and 31.6 °C with two n-octadecyl end-groups (all at 0.1 mg mL−1 in water).47 A detailed investigation by high sensitivity differential scanning calorimetry revealed that both polymers with one and two n-octadecyl end-groups organized into micellar aggregates, thereby leading to similar enhanced polymer–polymer interactions and similar Tcp values. Similar results were reported by Jordan who demonstrated that the Tcp of PiPrOx with DP 27 decreased from 47 °C to 28 °C and 32 °C by introduction of one or two n-nonyl end-groups, respectively (all at 20 mg mL−1), tentatively ascribed to micellization.48 Furthermore, it was demonstrated that introduction of short hydrophilic PMeOx outer blocks with DP 3 increased the Tcp to 53 °C while introducing short hydrophobic poly(2-n-nonyl-2-oxazoline) outer blocks with DP 1 or DP 2 decreased the Tcp to 15 °C and 11 °C.
Even though all the early reports on the LCST behavior of PiPrOx demonstrated fully reversible phase transitions without significant hysteresis, Schlaad and coworkers were the first to report irreversible crystallization driven self-assembly of PiPrOx upon continued heating above the Tcp.49–51 In 2012, Winnik and coworkers demonstrated by a combination of optical spectroscopy and solution vibrational spectroscopy and molecular dynamics simulations that heating of PiPrOx aqueous solution leads to an irreversible change in the chain conformation to a more regular all-trans conformation (Fig. 3).52 Despite this change in chain conformation, the LCST phase transition remains fully reversible upon repetitive heating-cooling cycles. However, prolonged heating leads to crystallization facilitated by the more regular chain conformation. This hypothesis was more recently confirmed by the work of Wu based on temperature variable 1H NMR, Fourier-transform infrared and Raman spectroscopy, including two-dimensional correlation spectroscopy.53
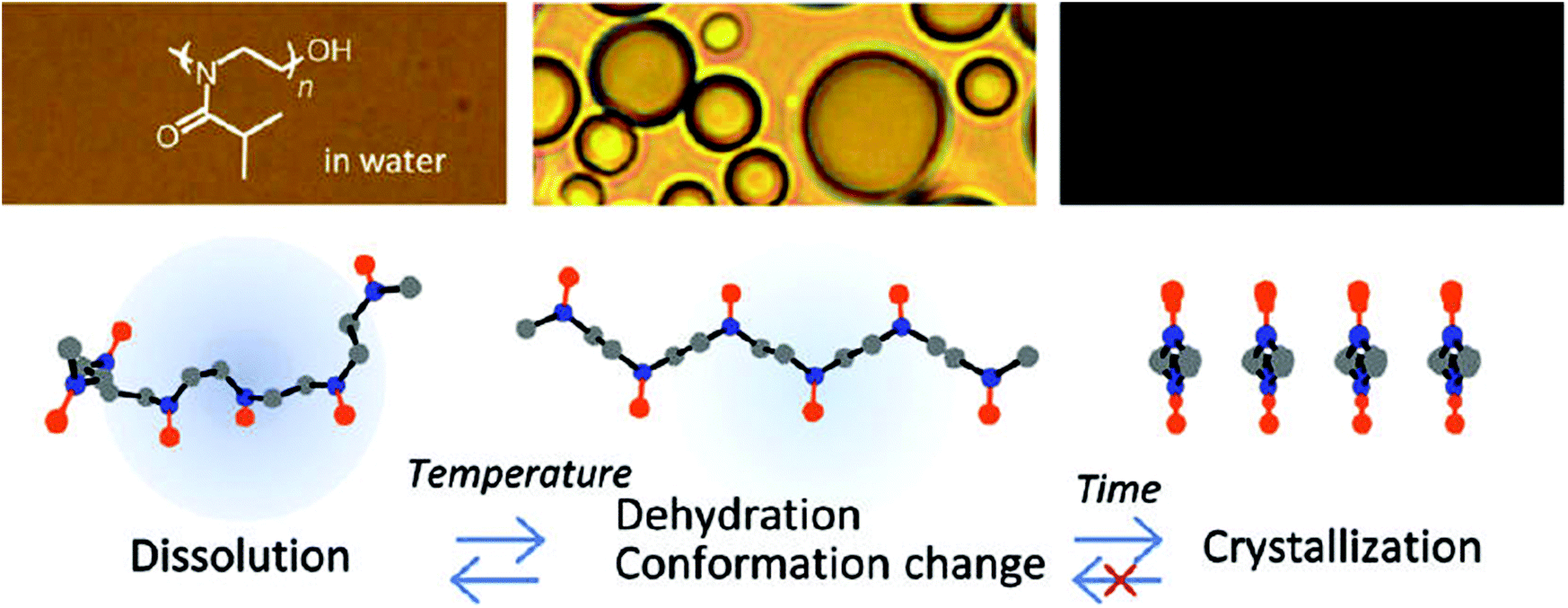 | ||
| Fig. 3 Proposed mechanism for the irreversible crystallization of PiPrOx upon annealing of aqueous solutions above the cloud point temperature. Reprinted with permission from ref. 52. Copyright 2012 American Chemical Society. | ||
Only 15 years after the first report on the LCST behavior of PiPrOx, Kataoka and coworkers reported that poly(2-n-propyl-2-oxazoline) (PnPrOx; Fig. 2) also exhibits LCST behavior with a Tcp around 25 °C.54 The Tcp of PnPrOx revealed much smaller molar mass and concentration dependence than PEtOx as may be attributed to its more hydrophobic character that facilitates dehydration.28 The third possible C3 side chain, namely cyclopropyl, was reported by Schubert et al. to lead to poly(2-cyclopropyl-2-oxazoline) (PcPrOx; Fig. 2), which is also thermoresponsive and has a Tcp that is intermediate to PiPrOx and PnPrOx.55 For both PnPrOx and PcPrOx it has been demonstrated that incorporation as side chains in comb polymers leads to a decrease in Tcp.46,55
Poly(2-oxazoline)s with methyl ester side chains were first reported by Litt in 1968,56 but it was only reported in 2015 by Hoogenboom that such polymers exhibit thermoresponsive LCST behavior in aqueous solution.57 In fact, poly(2-methoxycarbonylethyl-2-oxazoline) (PC2MestOx; Fig. 2) has very similar solution behavior as PEtOx with a Tcp around 100 °C for a polymer with DP 100 while poly(2-methoxycarbonylpropyl-2-oxazoline) (PC3MestOx; Fig. 2) has very similar behavior as PnPrOx with a Tcp around 25 °C.
In recent years, some more exotic comonomers have been explored to tune the Tcp of poly(2-oxazoline)s in aqueous solution. Volet and coworkers reported copolymers of PMeOx and poly(2-(5-azidopentyl)-2-oxazoline) that have Tcp values in between 0 °C and 100 °C depending on the comonomer composition.71 Schubert et al. reported the synthesis of copolymers of PEtOx and poly(2-(4-Boc-aminobutyl)-2-oxazoline) (Boc = tert-butyloxycarbonyl) that after removal of the Boc group yielded copolymers of PEtOx and poly(2-(4-aminobutyl)-2-oxazoline).72 Even though the amino group is more hydrophilic in water due to protonation leading to fully water-soluble copolymers that do not show LCST behavior in neutral water, it was demonstrated that at pH 14, where the amino-group is in its free base form, the copolymers had Tcp values in between 25 °C and 65 °C, depending on the copolymer composition and the polymer concentration. Copolymers of PEtOx, PnPrOx, PC2MestOx and PC3MestOx were also shown to have tunable Tcp values in between 25 °C and 100 °C.57 Interestingly, copolymers of PEtOx and PC3MestOx revealed a linear relationship between composition and Tcp while copolymers of PnPrOx and PC2MestOx cover exactly the same range of Tcp values, but with a strong non-linear correlation. This difference in behavior is not yet understood, but may be related to the size of the side chains and their exposure to the aqueous phase. Hydrolysis of PcPrOx-PC2MestOx and PnPrOx-PC2MestOx copolymers was demonstrated to lead to dual responsive polymers that are sensitive to both temperature and pH resulting from (de)protonation of the carboxylic acid units.73,74 Similarly, direct amidation of the PC2MestOx with ethylene diamine led to an amine functional copolymer showing temperature and pH sensitivity based on (de)protonation of the amino groups.73,75 Schlaad and coworkers reported dual responsive poly(2-oxazoline) micelles containing carboxylic acid or amine groups in the core, prepared by post-polymerization modification of core-crosslinked micelles using thiol–yne radical coupling.32 These ionically modified crosslinked micelles were demonstrated to not only respond to changes in temperature and pH, but their Tcp values were also strongly dependent on the presence of salts following the Hofmeister series.37
These latter examples already indicated that post-polymerization modification is another popular strategy to control the Tcp of poly(2-oxazoline)s. The popularity of this strategy is based on the possibility to modify the side chains of (co)polymers without affecting the polymer chain length and molar mass distribution allowing direct interpretation of the effect of the side chain on the Tcp. Schlaad et al. introduced poly(2-butenyl-2-oxazoline) (PButenOx) as a robust scaffold for post-polymerization modification using radical thiol–ene chemistry.76,77 This methodology was then applied for post-polymerization modification of PiPrOx-PButenOx copolymers to tune the Tcp providing a versatile platform in which the Tcp could be tuned by copolymer composition as well as by post-polymerization modification.78 As expected, introduction of hydrophobic side chains such as acetyl-protected thioglucose and 1-octanethiol led to a decrease in Tcp, whereas introduction of hydrophilic side chains such as 2-mercaptoethanol and 1-thioglycerol increased the Tcp. Similarly, deprotection of the acetyl-glucose side chains led to significant increase in Tcp. Schubert and coworkers reported a very similar strategy for the post-polymerization modification of PEtOx-poly(2-(9-decenyl)-2-oxazoline) (PEtOx-PDecenOx) copolymers.79 However, the presence of the long hydrophobic alkyl side chain led to a decrease in Tcp when unprotected thioglucose was attached to the terminal double bonds by thiol–ene coupling.80 This difference in behavior between the PButenOx and PDecenOx can be ascribed to the formation of hydrophobic pockets that favor hydrogen bonding between the glucose and the amide groups of the polymer backbone in the case of PDecenOx,81 thereby enhancing the polymer–polymer interactions and decreasing the solubility. A final example of tuning the Tcp by post-polymerization modification of poly(2-oxazoline)s was reported by Meier and Hoogenboom et al. making use of Passerini and Ugi multicomponent side chain modification of an acid-containing copolymer obtained by hydrolysis of PEtOx-PC3MestOx.82 By variation of the small molecule reagents for these multicomponent reactions, the Tcp of the acid-functionalized copolymer (Tcp = 78 °C) could be lowered to 15 °C while intermediate transition temperatures could also be achieved.
A double thermoresponsive PEtOx-b-PnPrOx block copolymer was reported by Hoogenboom and Kjoniksen et al.86 The thermoresponsive behavior of this block copolymer was studied by a combination of static and dynamic light scattering and turbidimetry revealing that below the Tcp of both blocks the polymer is present as unimers and in loose aggregates (Fig. 4). When passing the Tcp of the PnPrOx block, large aggregates are formed that upon further heating undergo a transition to defined micellar aggregates with a PnPrOx core and a PEtOx corona. Further heating beyond the Tcp of PEtOx led to the formation of large macroscopic aggregates as the entire polymer becomes insoluble in water. A related study on PEtOx-b-PiPrOx block copolymers was reported by Sato and Winnik et al.87 Upon heating the block copolymer solution in water to 50 °C, which is in between the Tcp of both individual blocks, the initial formation of star-like micelles was observed that further aggregate to induce macroscopic phase segregation. This is in contrast to the PEtOx-b-PnPrOx block copolymer micelles that remained stable in time, which may be ascribed to the smaller difference in Tcp of PEtOx and PiPrOx leading to more cooperative behavior compared to PEtOx and PnPrOx. Similarly, PiPrOx-b-PNIPAM block copolymers were also reported to undergo one cooperative phase separation rather than individual collapse of the blocks.88
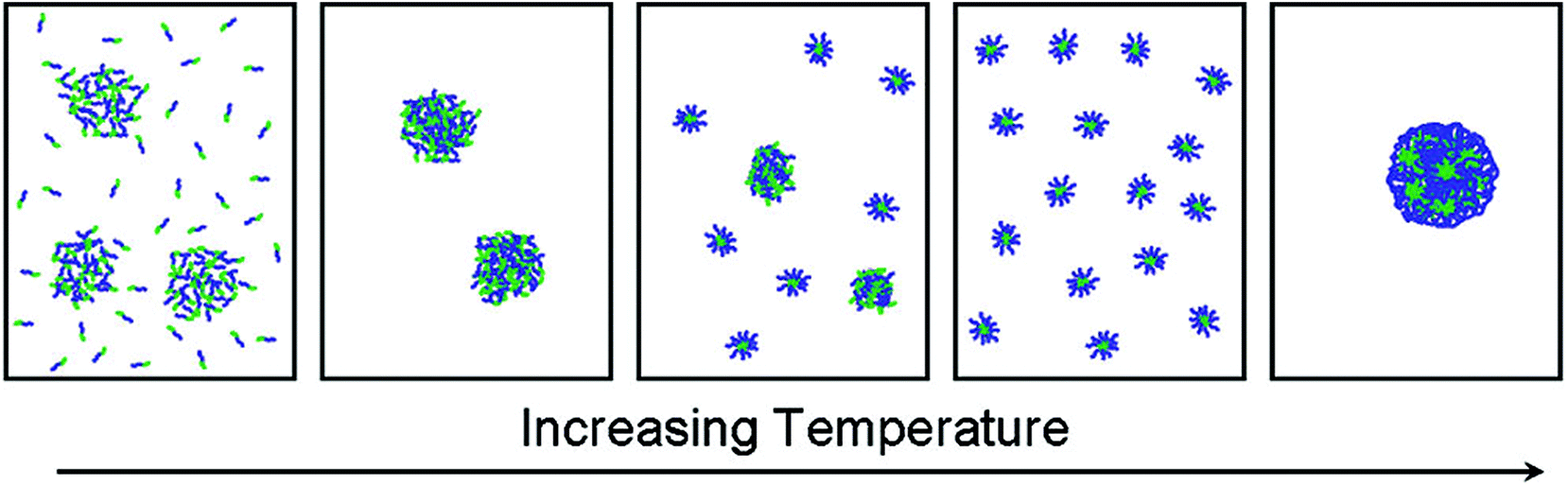 | ||
| Fig. 4 Schematic representation of the temperature induced self-assembly of PEtOx-b-PnPrOx going from a mixture of unimers and loose aggregates below Tcp to large aggregates that fall apart in discrete and defined micelles with a PnPrOx core and PEtOx corona until finally macroscopic aggregation when passing the Tcp of PEtOx. Reprinted with permission from ref. 86. Copyright 2012 American Chemical Society. | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 wt% ethanol–water solvent mixtures, more and more ethanol was required to get such behavior if the side chain length was increased up to PNonOx.89 A broad screening of the effect of poly(2-oxazoline) side chain on the LCST and UCST behavior in ethanol–water solvent mixtures revealed a general correlation between hydrophobicity and required amount of ethanol to gain UCST behavior (Fig. 5). Besides control over the UCST behavior by variation of the 2-oxazoline monomer structure, it can also be controlled by copolymerization of different monomers as was demonstrated for PEtOx-NonOx random copolymers.69
:50 wt% ethanol–water solvent mixtures, more and more ethanol was required to get such behavior if the side chain length was increased up to PNonOx.89 A broad screening of the effect of poly(2-oxazoline) side chain on the LCST and UCST behavior in ethanol–water solvent mixtures revealed a general correlation between hydrophobicity and required amount of ethanol to gain UCST behavior (Fig. 5). Besides control over the UCST behavior by variation of the 2-oxazoline monomer structure, it can also be controlled by copolymerization of different monomers as was demonstrated for PEtOx-NonOx random copolymers.69
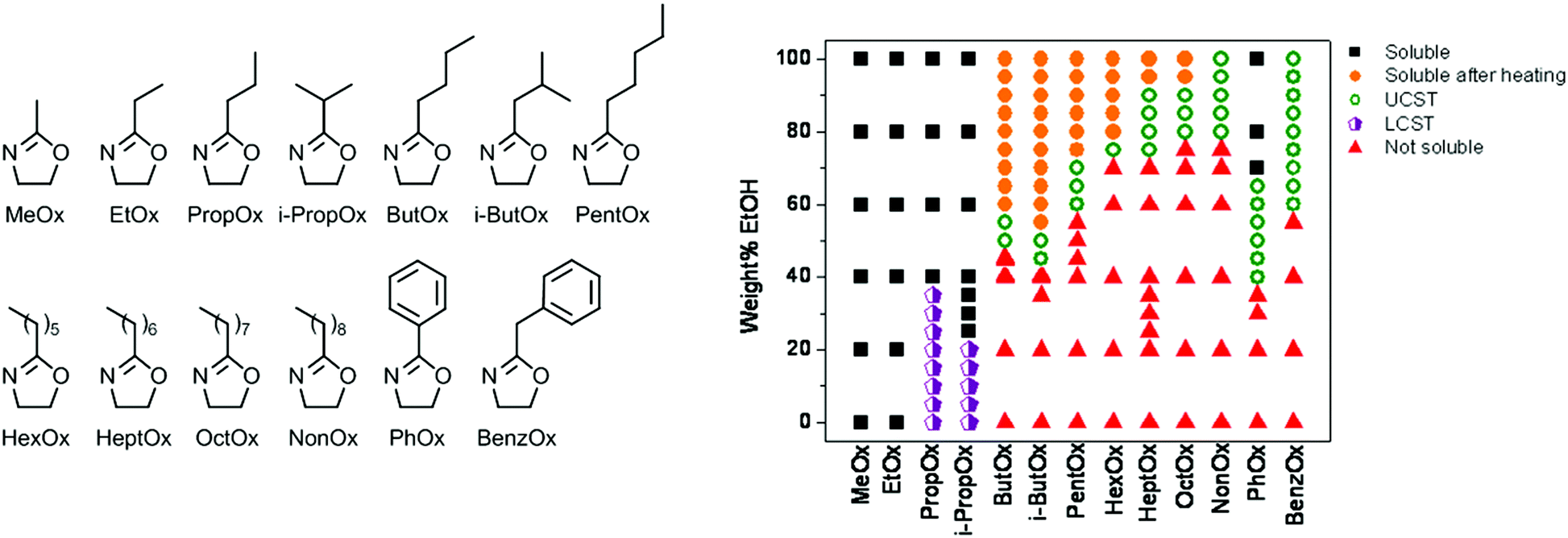 | ||
| Fig. 5 Screening of solubility behavior of a wide set of poly(2-oxazoline)s (monomer structures shown) (left) in different ethanol–water solvent mixtures (right). Reprinted with permission from ref. 89. Copyright 2010 MDPI AG. | ||
Annealing of poly(2-isobutyl-2-oxazoline) (PiBuOx) and PNonOx below their UCST in ethanol–water solvent mixtures was demonstrated by Schlaad and coworkers to lead to isothermal crystallization leading to nanosized hierarchically organized structures.90 Furthermore, the UCST behavior of PPhOx in ethanol–water solvent mixtures was exploited by Schubert et al. for the preparation of thermoresponsive micelles consisting of PMeOx or PEtOx as hydrophilic part and PPhOx as UCST-switchable part.91 The resulting micelles that are formed below the UCST phase transition temperature dissolve into unimoleculaly dissolved polymer chains upon heating. Another example of UCST-switchable micelles was reported by Hoogenboom et al. based on copolymers of PNonOx and PPhOx.92 By variation of the solvent system, one single copolymer was found to form UCST switchable micelles with either PNonOx as switchable core or PPhOx as switchable core.
In a related context, Osada and Kataoka et al. reported a triblock copolymer consisting of PEtOx, PnPrOx as middle thermoresponsive block and polylysine as cationically charged block.94 The polylysine could be exploited for complexation with DNA resulting in cylindrical micelle self-assembled structures. Increasing the temperature of the solution then led to collapse of the PnPrOx block onto the DNA-polylysine core, thereby providing a barrier to stabilize the polyplex on the one hand and to protect the DNA against degradation on the other hand. This novel strategy was demonstrated to lead to enhanced transfection efficiency.
The formation of defined mesoglobules upon increasing the temperature of an aqueous solution of PiPrOx was utilized by Rangelov et al. as template for the construction of hollow capsules.95 The collapsed mesoglobules of PiPrOx were utilized as seeds for the radical polymerization of NIPAM and a crosslinker above the Tcp of both PiPrOx and PNIPAM. Subsequent lowering of the temperature below the Tcp of both polymers allowed extraction of the non-crosslinked PiPrOx core by diffusion through the cross-linked PNIPAM layer leading to hollow thermoresponsive PNIPAM capsules.
A last example where the thermoresponsive behavior of a poly(2-oxazoline) homopolymer was exploited to control assembly processes was reported by Hoogenboom and De Geest et al. for layer-by-layer (LBL) assembly of PnPrOx and tannic acid.96 It was demonstrated that LBL assembly of alternating layers of PnPrOx and tannic acid led to much faster growth of the multilayer film thickness when performed above the Tcp of PnPrOx due to adsorption of mesoglobules rather than individual PnPrOx chains as is the case below the Tcp.
Since the LCST phase transition of poly(2-oxazoline)s is accompanied by a transition from a hydrophilic to a hydrophobic polymer, this feature can be utilized to obtain temperature control over adsorption and desorption processes. Claesson et al. demonstrated that the adsorption of a block copolymer of PiPrOx as themoresponsive block and poly(3-acrylamidopropyltrimethylammonium chloride) as charged block to adsorb onto a negatively charged silica substrate is indeed temperature dependent.97 Upon increasing the temperature, the PiPrOx is partially dehydrated and becomes more hydrophobic which favors the adsorption process leading to an increase in adsorbed mass. This process was found to be reversible and decreasing the temperature led to partial desorption of the block copolymer. The design of such a adsorption–desorption system can also be reversed by attaching the thermoresponsive polymer to a substrate. Dworak and coworkers demonstrated that a PiPrOx or PEtOx-PNonOx thermoresponsive polymer modified glass substrate can be used for temperature controlled cell-culture surfaces.98 Cells can be grown and cultured at 37 °C on the collapsed and dehydrated poly(2-oxazoline) layer. After growth of the cells, it was demonstrated that the cell sheet can easily be removed by simply lowering the temperature below the Tcp of the poly(2-oxazoline). The hydration and swelling of the polymer layer leads to spontaneous detachment of the cell layer. In an improved system, the PiPrOx polymer layer was modified by deposition of PiPrOx crystallites that were formed by annealing of PiPrOx above its Tcp.99 This modified system enhanced proliferation of the human dermal fibroblast cells while facilitating their detachment from the substrate. A very recent example of temperature controlled adsorption and desorption with thermoresponsive poly(2-oxazoline)s was reported by Maskos and Bertin et al.100 PiPrOx coated rhodamine labeled polyorganosiloxane were prepared and their interaction with serum proteins was studied. It was demonstrated that the adsorption of serum proteins is significantly enhanced upon heating beyond the Tcp while subsequent cooling below the Tcp led to release of the proteins indicating the reversible nature of this process.
A final application area where thermoresponsive poly(2-oxazoline)s are receiving significant attention is as sensors and molecular logic gates. The most straightforward sensor with a thermoresponsive polymer is as temperature sensor, whereby the polymer phase transition induces clouding of the solution which can be the output signal. Hoogenboom has recently shown that PEtOx-PNonOx copolymers are ideal for this application as the exact phase transition temperature that is ‘sensed’ can be accurately tuned by addition of different amounts of α-cyclodextrins that form host guest complexes with the nonyl side chains, thereby altering the hydrophilic–hydrophobic balance of the copolymer and hence the Tcp.101,102 Furthermore, it was demonstrated that introducing a large content of hydrophobic PNonOx leads to a widening of the phase transition hysteresis in presence of α-cyclodextrins, that is upon heating the clouding of the solution takes place at 50 °C while upon cooling the solution only turns transparent when cooled below 10 °C.103 This hysteresis originates from the supramolecular host–guest complexation that results in the formation of a meta-stable soluble phase. Once collapsed, reformation of the host–guest complexation only takes place at the original phase transition temperature of the non-complexed polymer. It was shown that this large hysteresis can be exploited as memory function for the molecular thermometer. A solution of the copolymer and α-cyclodextrin will remember whether it has been heated beyond 50 °C or not for more than 1.5 months at room temperature.
A similar sensor concept was developed by Jang, although employing a much more sophisticated fluorescence output signal.104 PiPrOx was prepared with a tetraphenylethene end-group that translated the polymer phase transition in a fluorescent output signal. This system acts as a sensor for the polymer concentration as well as the presence of γ-cyclodextrin. Moreover, the polymer γ-cyclodextrin host guest complex acts as temperature sensor as collapse of the polymer upon heating releases the γ-cyclodextrin to the aqueous solution, thereby inducing a change in fluorescence of the tetraphenylethene end-group. Very recently, Jang and coworkers reported PiPrOx end-modified with blue (pyrene), green (boron-dipyrromethene) and red (porphyrin) emissive dyes.105 Even though the same PiPrOx polymer was used, the Tcp values were tuned by variation of the polymer chain length and by making star-shaped polymers. As such, the three emissive polymers all had different Tcps and each of the individual polymers acts as a temperature sensor. However, by careful optimization of the concentration of the three polymers in one solution it was possible to design systems with different emission color in response to variations in temperature as shown in Fig. 6.
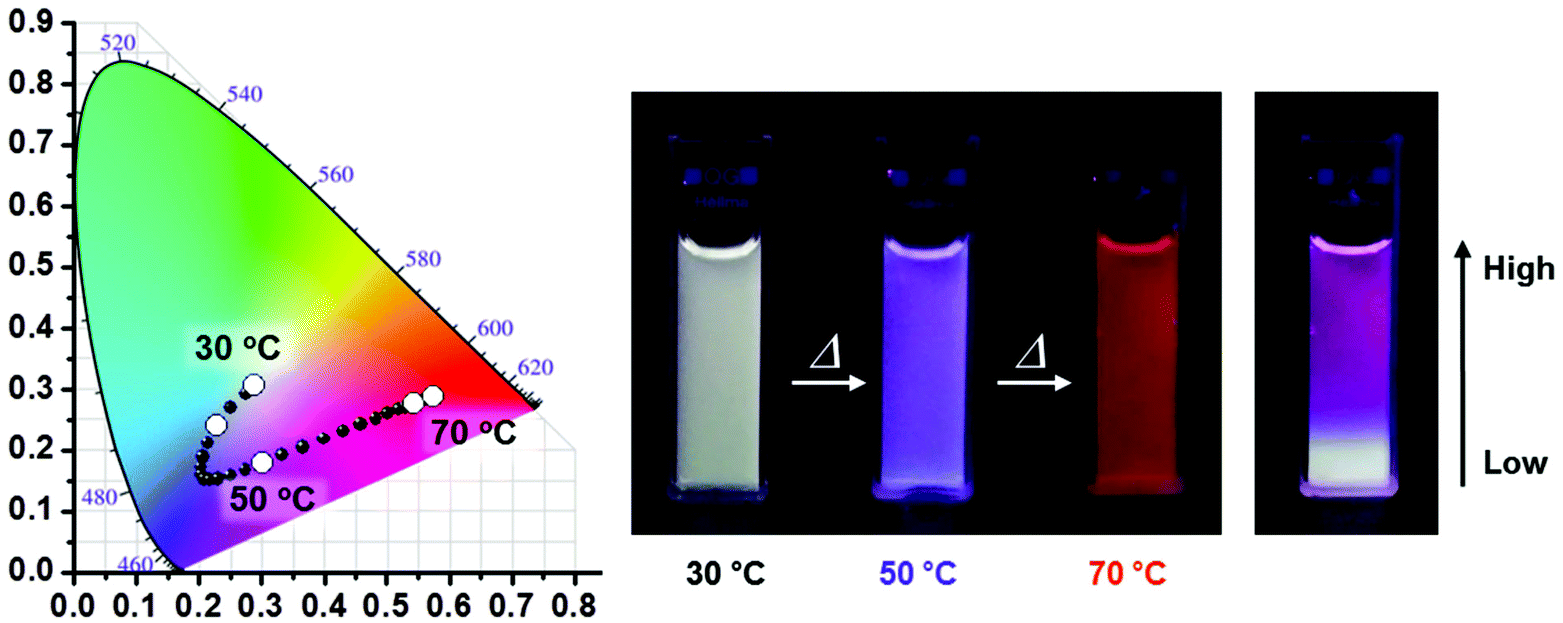 | ||
| Fig. 6 Temperature-dependent fluorescence emission changes of three-component mixtures of PiPrOx modified with pyrene (blue), boron-dipyrromethene (green) and porphyrin (red). The right picture show the solution with a temperature gradient from bottom to top. Reprinted with permission from ref. 105. Copyright 2016 WILEY-VCH Verlag GmbH & Co. KGaA. | ||
The final two examples describe the use of multiresponsive poly(2-oxazoline)s for molecular logic gate operations. Hoogenboom et al. reported the preparation of poly(2-oxazoline) coated gold nanoparticles that aggregate and change color from red to purple upon increasing the temperature above the Tcp of the polymer coating and in presence of sodium chloride.106 It was demonstrated that the system operates as AND logic gate and that only a color change occurs when both triggers are present. Variation of the poly(2-oxazoline) from PiPrOx to PEtOx allowed tuning of the temperature required for the input. A triple responsive poly(2-oxazoline) was developed by Ju and Jang as AND-OR logic gates with the solution being opaque or transparent as output signal.107 PiPrOx was prepared having two azobenzene end-groups, whereby the PiPrOx induces thermoresponsivity while photoisomerization of the azobenzene governs host–guest complexation with either α-cyclodextrin in the trans-form and with β-cyclodextrin in the cis-form. The AND-OR logic gate operation is based on photoirradiation for isomerization of the azobenzene, presence of cyclodextrin and the temperature as input signals.
2.2 Polypeptoids
Polypeptoids, i.e., poly(N-alkyl glycine)s,8 in aqueous solution can exhibit lower critical solution (LCST) behavior, which they share with other tertiary amides like poly(2-oxazoline)s (see previous section) and also poly(meth)acrylamides.2,3Peptoid homopolymers with methyl (C1, sarcosine) and ethyl (C2) side chains are readily soluble in water while all other polypeptoids with longer alkyl side chains (C3 and longer) were found to be insoluble.108 Afterwards, it has been demonstrated that amorphous samples of polypeptoids with C3 side chains, i.e., n-propyl, allyl, and i-propyl (Fig. 7), can be dissolved in water at 20–40 g L−1 and show LCST behavior; poly(N-propargylglycine), however, remains insoluble in water.109,110 The cloud point temperatures (Tcp) were found to increase in the order n-propyl (15–25 °C) < allyl (27–54 °C) < i-propyl (47–58 °C), depending on the chain length and polymer concentration (classical Flory–Huggins type 1 behavior). The phase transitions were reversible only for shorter time scales of a few hours. Long-term annealing of dilute aqueous solutions of poly(N-n-propylglycine) and poly(N-allylglycine) resulted in the formation of crystalline precipitates (melting at 188–198 °C and 157–165 °C, respectively) with complex morphologies (cf. crystallization of PEtOx or PiPrOx in hot water, see above).109
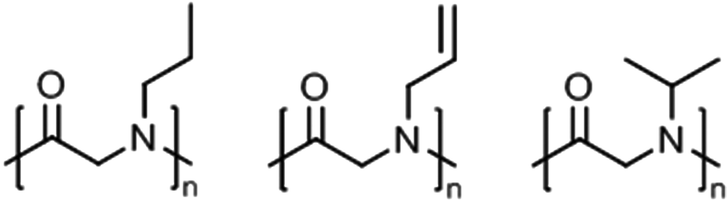 | ||
| Fig. 7 Thermoresponsive polypeptoid homopolymers. | ||
It is worth to mention that thermoresponsive polypeptoid homopolymer fiber mats could be prepared by electrospinning of blends of semicrystalline poly(N-n-propylglycine) and high molar mass poly(ethylene oxide) (PEO).111 Annealing of the electrospun fibers at about 100 °C and subsequent washing with water selectively removed the amorphous PEO fraction to give stable crystalline poly(N-n-propylglycine) fibers.
Thermo-induced aggregation and crystallization (see above) was observed for poly(N-n-propylglycine)-polysarcosine diblock copolypeptoids in water (double hydrophilic at T < Tcp and amphiphilic at T > Tcp).112 Interestingly, the morphology of initially spherical aggregates at T > Tcp was not retained during crystallization of the hydrophobic poly(N-n-propylglycine) core, resulting in the formation of larger complex assemblies (100–500 nm in size) with flower-like, ellipsoidal, or irregular shapes (Fig. 8). Evidently, the crystallization of the hydrophobic core is not applicable for the stabilization of aggregates against dilution, as for instance desirable in drug delivery applications. Also, the incorporation of hydrophobic molecules or drugs into the crystallized compartment might be difficult.
 | ||
| Fig. 8 Time-dependant evolution of aggregate structures, as visualized by cryogenic scanning electron microscopy (scale bar = 500 nm), in 1 wt% aqueous solutions of poly(N-n-propylglycine)70-block-polysarcosine23 at 48 °C (T > Tcp). Reprinted (modified) with permission from ref. 112. Copyright 2016 American Chemical Society. | ||
The Tcp values could be adjusted for statistical copolymers with hydrophilic units, usually sarcosine or N-ethylglycine, and hydrophobic units, usually butyl or higher alkyl substituted glycines, at various ratios. Examples include poly[sarcosine-ran-(N-butylglycine)] with tunable Tcp values in the range of 27–71 °C (at 0.3 wt% in water; sarcosine content increasing from 42 to 73 mol%)113,114 and poly[(N-ethylglycine)-stat-(N-butylglycine)] 20–60 °C (at 0.1 wt% in water).115 For the latter case, a distinct impact of the architecture, linear vs. cyclic, on Tcp could be recognized. The phase transition of the cyclic copolypeptoids was shifted to lower temperature, by five degrees or less, as compared to the linear analogue with the same composition, which was explained in terms of lower entropic loss. Also, the Tcp was found to decrease upon the addition of sodium salts, the degree of depression being in line with the Hofmeister series, i.e., sulphate > chloride > iodide. Such a salting-in/out effect was also observed for poly[sarcosine-ran-(N-butylglycine)].114
Interestingly, when comparing linear norbornyl-poly[(N-ethylglycine)-ran-(N-butylglycine)] and polynorbornene-graft-poly[(N-ethylglycine)-ran-(N-butylglycine)] bottlebrushes (prepared by ring-opening metathesis polymerization, ROMP), the polymer architecture seemed to have no impact on the Tcp.116 However, the phase behavior was strongly dependent on the thermal history of the samples and the presence of inorganic salts.
ABC block copolypeptoids, i.e., poly(N-allylglycine)-block-polysarcosine-block-poly(N-n-decylglycine), were found to undergo sol-to-gel transitions with increasing temperature in aqueous solutions at 2.5–10 wt% (Fig. 9).117 The gelation temperature (Tgel) and mechanical properties (storage modulus, G′, and Young's modulus, E) of the hydrogel could be tuned in the range of Tgel = 26–60 °C, G′ = 0.2–780 Pa, and E = 0.5–2346 Pa, by varying the copolypeptoid composition and polymer concentration. The hydrogels are injectable through a 24 gauge (0.635 mm) syringe, maintain their shape upon contact with surfaces, and exhibit minimal cytotoxicity.
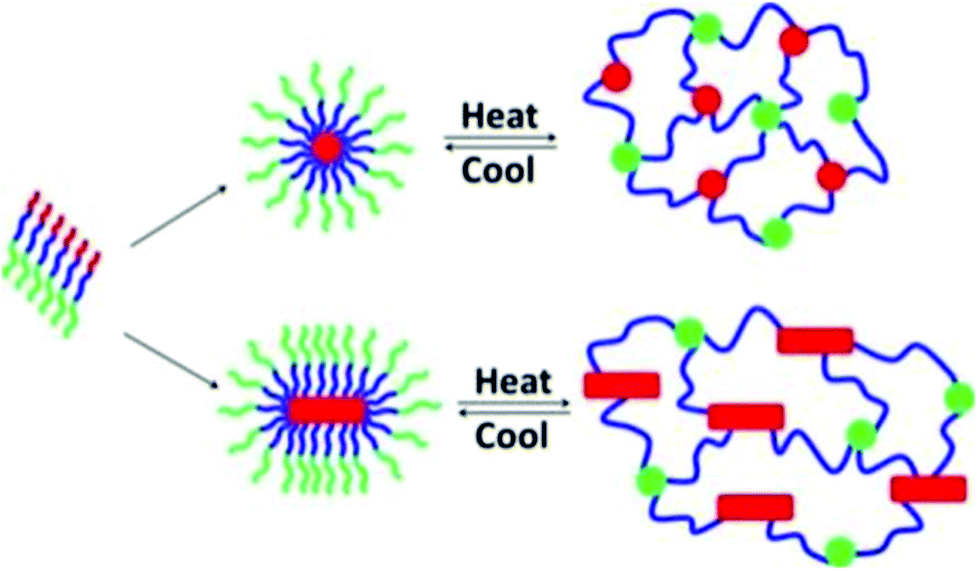 | ||
| Fig. 9 Schematic representation of the gelation mechanism of aqueous solutions of ABC triblock copolypeptoid; red = poly(N-allylglycine), green = polysarcosine, blue = poly(N-n-decylglycine). Reprinted with permission from ref. 117. Copyright 2016 American Chemical Society. | ||
2.3 Polypeptides
Synthetic polypeptides118,119 based on naturally occuring amino acids do not exhibit thermoresponsive solution behavior, except elastin-mimetic peptide sequences or elastin-like polypeptides (ELPs)120 made by genetic engineering techniques (not considered here). Thermoresponsive polypeptide materials have been obtained by grafting hydrophilic side chains, usually tertiary amine, oligo(ethylene glycol) or poly[oligo(ethylene glycol) methacrylate], onto a hydrophobic poly(γ-substituted L-glutamate) or side chain modified poly(L-cysteine) and poly(L-lysine).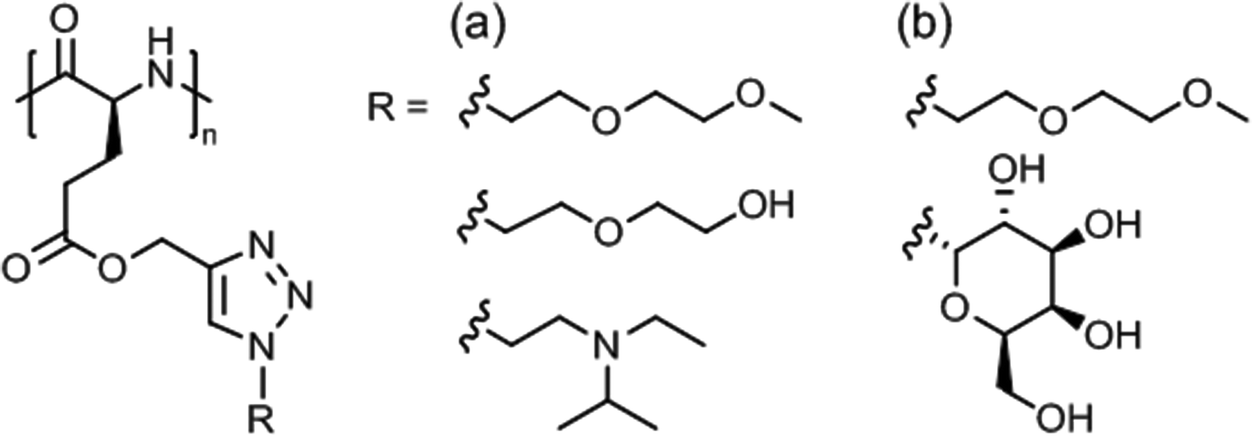 | ||
| Fig. 10 Themoresponsive poly(L-glutamate)s with (a) diethylene glycol/tertiary amine side chains and (b) diethylene glycol/galactose side chains. | ||
Aqueous solutions of diethylene glycol/galactose functionalized poly(L-glutamate)s (Fig. 10b) exhibited Tcp values in the range of 15–80 °C, the Tcp values increasing linearly with galactose content (0–24%).123 These glycopolypeptides were designed for use in temperature controlled biological response, here, the specific recognition of lectins via selective carbohydrate-protein interactions. Lectin binding appeared to occur only at a temperature below Tcp and was suppressed at above Tcp.
A systematic study on the pH- and thermo-responsiveness of tertiary amine functionalized poly(L-glutamate)s (Fig. 11) revealed that the Tcp depended delicately on the alkyl spacer (between triazole and amine) and the N-substituted groups.124 Polypeptides with moderate N-substituted amine group (e.g., diethylamine, pyrolidine, and piperidine) or branched spacer displayed more likely an LCST-type phase transition (Tcp ∼ 15–50 °C), tuned by pH variation, than polypeptides with (lower substituted) amine, methylamine, dimethylamine, and morpholine groups or (higher substituted) diisopropylamine and hexamethylene groups. Interestingly, the diisopropylamine substituted poly(L-glutamate) exhibited different behaviors in acidified water (no phase transition)124 and in aqueous phosphate buffer (Tcp = 38–66 °C at pH 5.0–6.2),121 which might be attributed to a salting out effect.
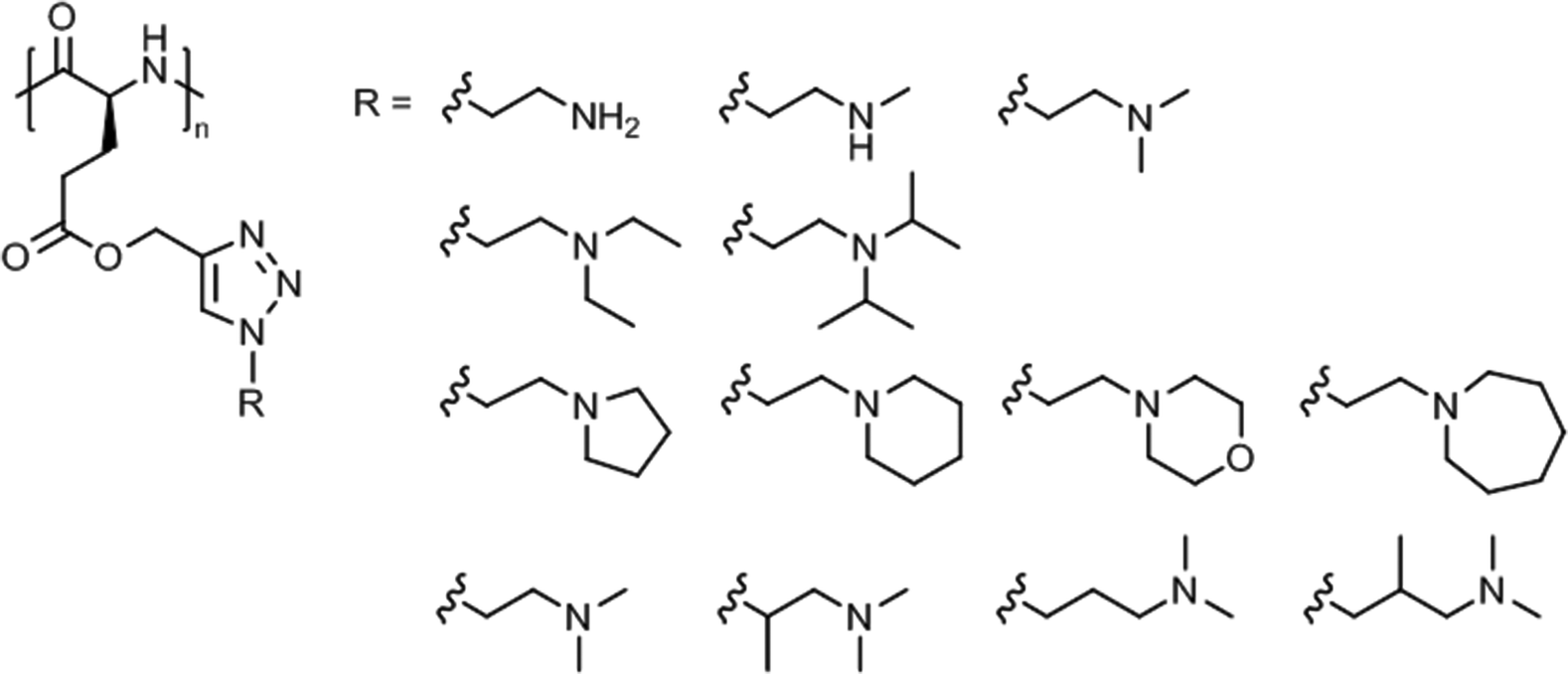 | ||
| Fig. 11 Thermo- and pH-responsive poly(L-glutamate)s with amine side chains. | ||
Tang et al. introduced very special types of thermoresponsive helical poly(γ-benzyl-L-glutamate)s bearing alkyl/oligoethylene glycol triazolium side chains (Fig. 12a)125 or alkyl imidazolium side chains (Fig. 12b),126 which can be regarded as poly(ionic liquid)s.127,128 The first polypeptide exhibited LCST behavior in deionized water and the Tcp could be varied in the range of 40–75 °C, depending on the alkyl/oligoethylene glycol substitution (alkyl = butyl, hexyl, dodecyl) and polymer concentration (0.1–0.6 wt%).125 The Tcp's were also affected considerably by the presence of salt (NaBF4), i.e., ΔT = −5 °C to −25 °C, due to salting out effect. The most pronounced effect, however, was found for the permanently charged polypeptide with butyl/oligoethylene glycol side chains at 0.6 wt% NaBF4.
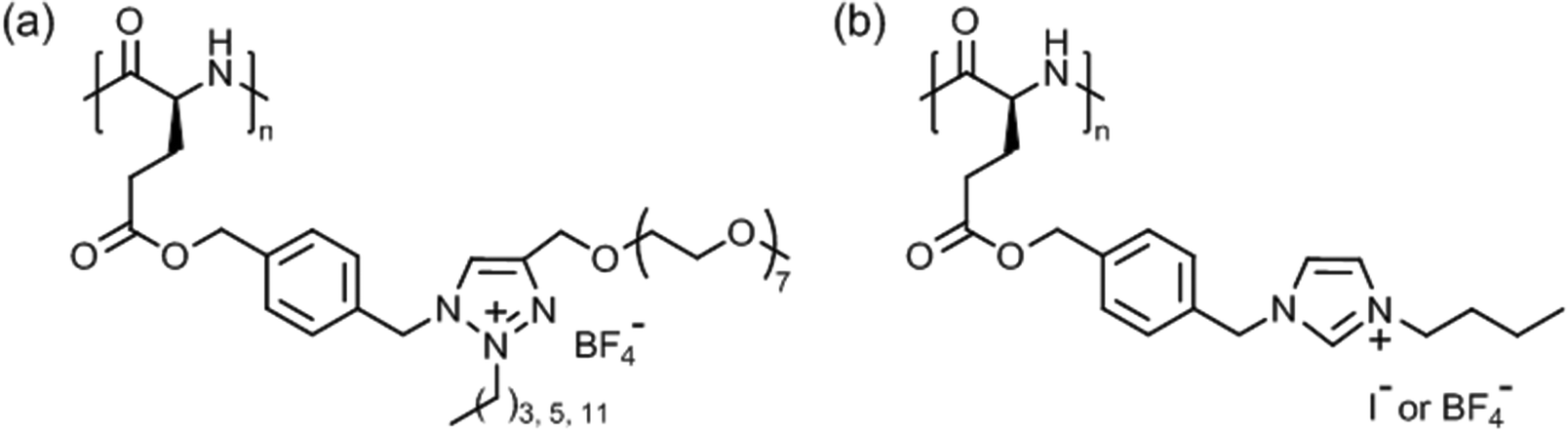 | ||
| Fig. 12 Thermoresponsive poly(γ-benzyl-L-glutamate)s with (a) alkyl/oligoethylene glycol triazolium and (b) n-butyl imidazolium side chains. | ||
The second polypeptide with n-butyl imidazolium side chains, however, showed UCST behavior in deionized water.126 Depending on the counter ion, I− or BF4−, the transition or clearing temperature was 35 °C or 69 °C, respectively, at 0.1 wt%; no phase transition was observed for polypeptides with alkyl = methyl/Cl−, I−, BF4− and n-butyl/Cl−. The transition temperature was unaffected by the polymer chain length but was significantly affected by salts, and it increased in the presence of NaI and NaBF4 and decreased in the presence of NaCl (cf. Hofmeister series). This effect was ascribed to electrostatic interactions and anionic exchange reactions.
Water-soluble random copoly(L-glutamate)s with benzyl and triethylene glycol pendants (Fig. 13) were found to exhibit thermoresponsive properties.129 Depending on the composition, i.e., ratio of hydrophobic benzyl groups vs. hydrophilic triethylene glycol groups, the Tcp could be tuned between 22 °C (57% hydrophilic units) and 53 °C (90% hydrophilic units) (at 0.2 wt% in water). Furthermore, the helicity of copolypeptide chains increased from 65% to 90% with increasing triethylene glycol content.
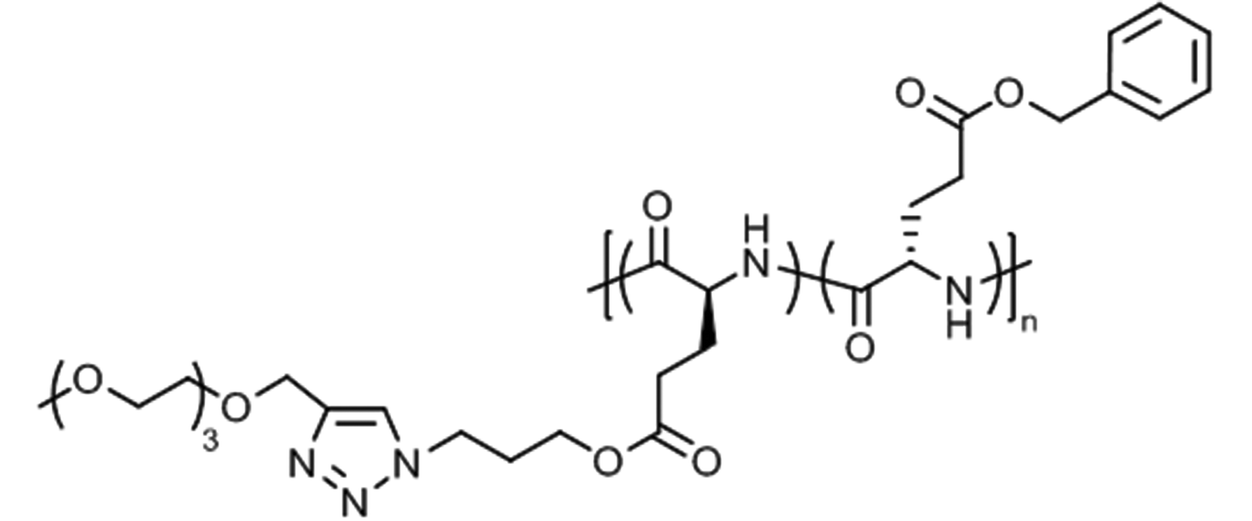 | ||
| Fig. 13 Thermoresponsive copoly(L-glutamate) with benzyl and triethylene glycol side chains. | ||
Linear poly(ethylene glycol)45-block-poly[γ-(methoxy diethylene glycol)-L-glutamate]43 (Fig. 14a) was soluble in water (or 100 mM NaCl solution) at room temperature but formed wormlike micelles at a temperature above Tcp = 53 °C.130 Aggregation occurred due to the temperature-induced dehydration of the polypeptide block without affecting its α-helical conformation. Extension of the thermal annealing time (12 hours at 80 °C) drove the secondary structure transformation of the polypeptide block from α-helix to β-sheet, which accounted for a transition from wormlike micelles into nanoribbons measuring about 70 nm in width and several micrometers in length.
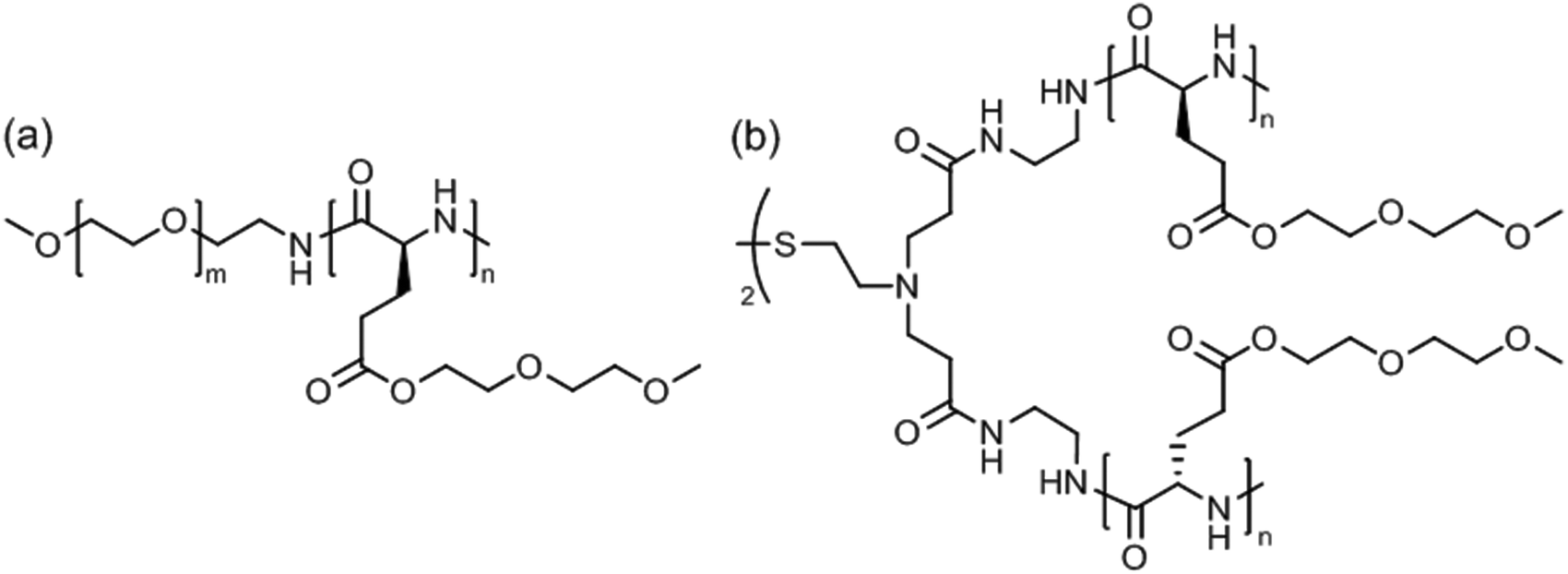 | ||
| Fig. 14 Thermoresponsive poly[γ-(methoxy diethylene glycol)-L-glutamate]-based (a) block copolymer and (b) 4-arm star polymer. | ||
Star-shaped poly[γ-(methoxy diethylene glycol)-L-glutamate] with a disulfide-containing core (apparent molar mass, Mn = 15.5 or 33.8 kDa) (Fig. 14b) exhibited dual thermo- and redox-responsiveness.131 Addition of dithiothreitol to the aggregates in water reduced the disulfide bonds, cutting the 4-arm star polypeptide into 2-arm star (linear) fragments, which however caused a reduction of the size of aggregates from about 330 nm to 180 nm. The aggregate size gradually decreases upon heating (20–50 °C) due to the collapse of the polypeptide side chains; the process could be reversed by cooling. Interestingly, 3–4 wt% aggregate solutions formed a hydrogel at room temperature, which could be dissolved upon heating to above Tcp ∼ 40 °C.
The graft copolymer (or molecular bottlebrush) poly(L-glutamate)-graft-poly[2-(2-methoxyethoxy)ethyl methacrylate] (molar mass, Mn = 221 kDa) (Fig. 15a) exhibited thermoresponsive LCST behavior in aqueous solution.132 The phase transition was rather broad in pure water (Tcp ∼ 27 °C) but sharp in saline solution (Tcp = 22–23 °C, 0.1–0.9 wt% NaCl). It was stated, based on circular dichroism spectroscopic measurements, that the helicity of polypeptide chains (DP ∼ 45) was almost 100% at 25 °C and also at 60 °C. Aggregates (assumed to be spheres with helical polypeptide core and thermoresponsive polymethacrylate shell) were formed in water, measuring 150 nm in diameter at 25 °C and 60 nm at 60 °C.
 | ||
| Fig. 15 Thermoresponsive “hairy-rod” graft copolymers with poly(L-glutamate) backbone and poly[methoxy oligo(ethylene glycol) methacrylate] side chains, prepared by (a) grafting from and (b) grafting onto. | ||
Also the “hairy-rod” polypeptides consisting of a poly(L-glutamate)40 backbone and poly[(methoxy diethylene glycol methacrylate)-ran-(methoxy triethylene glycol methacrylate)] side chains (grafting ratio ∼90%; molar mass, Mn = 195–227 kDa) (Fig. 15b) displayed thermoresponsive properties.133 The Tcp values, measured at 2 wt% in physiological saline solution (0.9 wt% NaCl), were found to increase from 20 °C to 41 °C with increasing hydrophilicity of the side chains, i.e., increasing amount of methoxy triethylene glycol methacrylate. In the same line, the hydrodynamic size of the aggregates formed in water increased from 170 nm to 310 nm. All polypeptide copolymers adopted 100% α-helical conformation, except the most hydrophilic one bearing poly(methoxy triethylene glycol methacrylate) side chains, 72%.
Klok et al. synthesized surface-tethered helical poly[γ-(oligoethylene glycol)-L-glutamate) brushes.134 The polypeptide chains did not undergo any changes in the secondary structure between 10 °C and 70 °C, but revealed a significant dehydration upon heating from 10 °C to 40 °C. Interestingly, the film thickness remained unchanged due to the shape-persistant nature of the polypeptide brushes (Fig. 16).
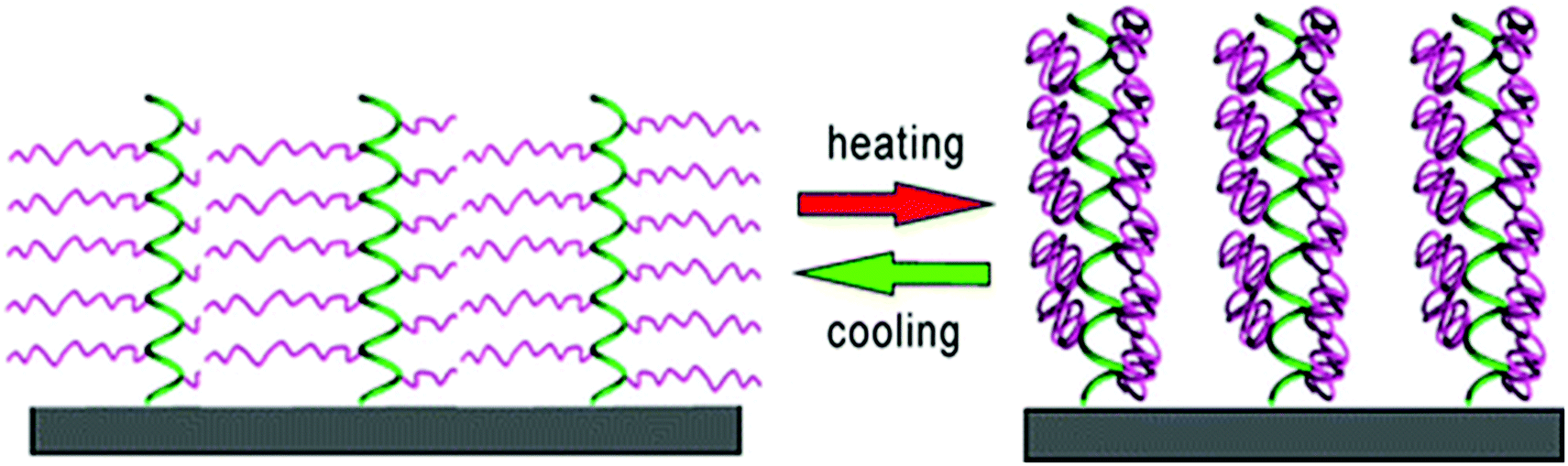 | ||
| Fig. 16 Thermo-induced hydration/dehydration of polypeptide brushes; green = helical poly(L-glutamate) backbone, purple = thermoresponsive oligoethylene glycol side chains. Reprinted with permission from ref. 134. Copyright 2015 American Chemical Society. | ||
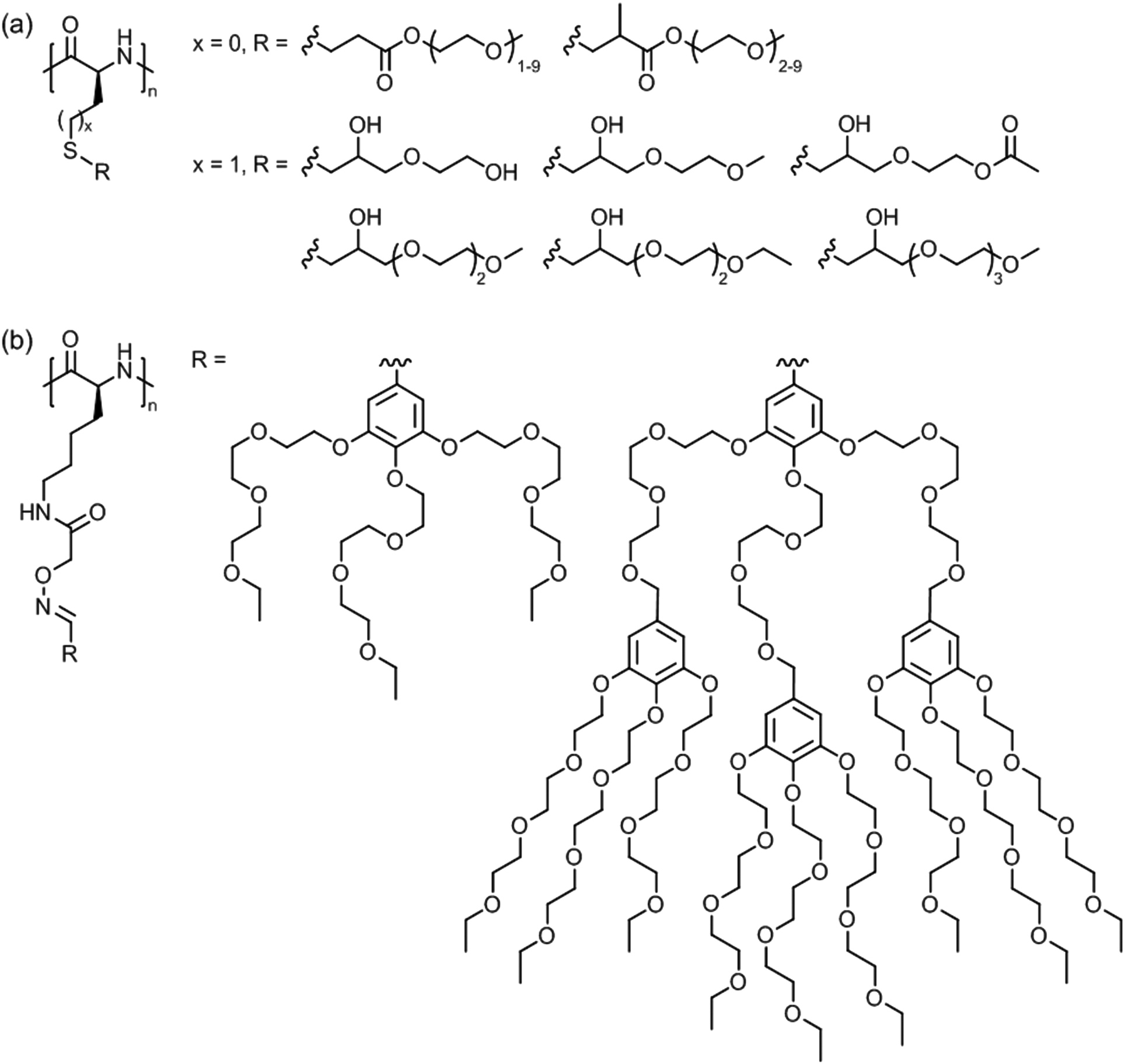 | ||
| Fig. 17 Thermoresponsive polymers based on side chain modified (a) poly(L-cysteine) (x = 0) and poly(L-homocysteine) (x = 1) and (b) poly(L-lysine). | ||
Poly(L-lysine)s bearing triethylene glycol dendrons of generation 1 and 2 (Fig. 17b) exhibited LCST behavior in pH 7 buffered aqueous solutions.137 At 0.1 wt%, the cloud point temperatures were in the range of 30–37 °C depending on the polypeptide chain length and dendron generation.
3. Summary
Poly(2-oxazoline)s, polypeptoids, and polypeptides are interesting materials especially for use in biomedical applications, which not least is due to their good (bio-) compatibility and degradability. Recent efforts were focussed to implement external stimuli-responsiveness, for instance to temperature, pH, etc., to these polymers for the generation of advanced smart materials.Thermoresponsive poly(2-oxazoline)s are known for almost three decades while thermoresponsive polypeptoids and polypeptides have just emerged during the last five years (however, thermoresponsive elastin-like polypeptides and pH-responsive polypeptides are known for much longer). Accordingly, the research in thermoresponsive materials based on poly(2-oxazoline)s is far more established and yet more applications have been developed, for instance as biomedical devices, sensors, or molecular logic gates, as compared to polypeptoids and polypeptides. Applications of thermoresponsive polypeptoids and polypeptides are still scarce, which with the rapid research developments should change in the very near future.
Key parameter for the control of the thermoresponsive properties of a polymer, no matter if it is a poly(2-oxazoline), polypeptoid, polypeptide, or any other kind of material, is the hydrophilic–hydrophobic balance. This balance can be tuned by the proper choice of side chains or by the copolymerization of hydrophilic and hydrophobic monomers. Also the blending of homopolymers might be a suitable approach to tune the thermoresponsive behavior.
Yet a plethora of thermoresponsive polyamides have been prepared based of this concept of hydrophilic–hydrophobic balance (or by trial and error), though the self-assembly behavior, structure formation, and applications need to be further explored.
Acknowledgements
Katharina Zesch is thanked for proofreading of the manuscript.References
- M. A. Cohen Stuart, W. T. S. Huck, J. Genzer, M. Muller, C. Ober, M. Stamm, G. B. Sukhorukov, I. Szleifer, V. V. Tsukruk, M. Urban, F. Winnik, S. Zauscher, I. Luzinov and S. Minko, Nat. Mater., 2010, 9, 101–113 CrossRef CAS PubMed.
- I. Dimitrov, B. Trzebicka, A. H. E. Müller, A. Dworak and C. B. Tsvetanov, Prog. Polym. Sci., 2007, 32, 1275–1343 CrossRef CAS.
- V. Aseyev, H. Tenhu and F. Winnik, Adv. Polym. Sci., 2011, 242, 29–89 CrossRef CAS.
- C. Weber, R. Hoogenboom and U. S. Schubert, Prog. Polym. Sci., 2012, 37, 686–714 CrossRef CAS.
- J. Seuring and S. Agarwal, Macromol. Rapid Commun., 2012, 33, 1898–1920 CrossRef CAS PubMed.
- G. Vancoillie, D. Frank and R. Hoogenboom, Prog. Polym. Sci., 2014, 39, 1074–1095 CrossRef CAS.
- Q. Zhang and R. Hoogenboom, Prog. Polym. Sci., 2015, 48, 122–142 CrossRef CAS.
- N. Gangloff, J. Ulbricht, T. Lorson, H. Schlaad and R. Luxenhofer, Chem. Rev., 2016, 116, 1753–1802 CrossRef CAS PubMed.
- H. Schlaad, C. Diehl, A. Gress, M. Meyer, A. L. Demirel, Y. Nur and A. Bertin, Macromol. Rapid Commun., 2010, 31, 511–525 CrossRef CAS PubMed.
- M. A. Ward and T. K. Georgiou, Polymers, 2011, 3, 1215–1242 CrossRef CAS.
- C. Pietsch, U. S. Schubert and R. Hoogenboom, Chem. Commun., 2011, 47, 8750–8765 RSC.
- M. R. Islam, Z. Z. Lu, X. Li, A. K. Sarker, L. Hu, P. Choi, X. Li, N. Hakobyan and M. J. Serpe, Anal. Chim. Acta, 2013, 789, 17–32 CrossRef CAS PubMed.
- J. K. Chen and C. J. Chang, Materials, 2014, 7, 805–875 CrossRef CAS.
- A. Gandhi, A. Paul, S. O. Sen and K. K. Sen, Asian J. Pharm. Sci., 2015, 10, 99–107 CrossRef.
- H. R. Kricheldorf, Angew. Chem., Int. Ed., 2006, 45, 5752–5784 CrossRef CAS PubMed.
- K. Aoi and M. Okada, Prog. Polym. Sci., 1996, 21, 151–208 CrossRef CAS.
- B. Verbraeken, K. Lava and R. Hoogenboom, in Encyclopedia of Polymer Science and Technology, John Wiley & Sons, Inc., 2014 Search PubMed.
- D. A. Tomalia and D. P. Sheetz, J. Polym. Sci., Part A: Polym. Chem., 1966, 4, 2253–2265 CrossRef CAS.
- W. Seeliger, E. Aufderhaar, W. Diepers, R. Feinauer, R. Nehring, W. Thier and H. Hellmann, Angew. Chem., Int. Ed., 1966, 5, 875–888 CrossRef CAS PubMed.
- T. Kagiya, S. Narisawa, T. Maeda and K. Fukui, J. Polym. Sci., Part B: Polym. Lett., 1966, 4, 441–445 CrossRef CAS.
- T. G. Bassiri, A. Levy and M. Litt, J. Polym. Sci., Part B: Polym. Lett., 1967, 5, 871–879 CrossRef.
- R. Hoogenboom, M. W. M. Fijten, H. M. L. Thijs, B. M. van Lankvelt and U. S. Schubert, Des. Monomers Polym., 2005, 8, 659–671 CrossRef CAS.
- E. Rossegger, V. Schenk and F. Wiesbrock, Polymers, 2013, 5, 956–1011 CrossRef.
- B. Guillerm, S. Monge, V. Lapinte and J.-J. Robin, Macromol. Rapid Commun., 2012, 33, 1600–1612 CrossRef CAS PubMed.
- K. Lava, B. Verbraeken and R. Hoogenboom, Eur. Polym. J., 2015, 65, 98–111 CrossRef CAS.
- P. Y. Lin, C. Clash, E. M. Pearce, T. K. Kwei and M. A. Aponte, J. Polym. Sci., Part B: Polym. Phys., 1988, 26, 603–619 CrossRef CAS.
- D. Christova, R. Velichkova, W. Loos, E. J. Goethals and F. Du Prez, Polymer, 2003, 44, 2255–2261 CrossRef CAS.
- R. Hoogenboom, H. M. L. Thijs, M. J. H. C. Jochems, B. M. van Lankvelt, M. W. M. Fijten and U. S. Schubert, Chem. Commun., 2008, 5758–5760 RSC.
- A. Kowalczuk, J. Kronek, K. Bosowska, B. Trzebicka and A. Dworak, Polym. Int., 2011, 60, 1001–1009 CrossRef CAS.
- M. A. Cortez, W. T. Godbey, Y. Fang, M. E. Payne, B. J. Cafferty, K. A. Kosakowska and S. M. Grayson, J. Am. Chem. Soc., 2015, 137, 6541–6549 CrossRef CAS PubMed.
- J. Xu, J. Ye and S. Liu, Macromolecules, 2007, 40, 9103–9110 CrossRef CAS.
- N. Ten Brummelhuis and H. Schlaad, Polym. Chem., 2011, 2, 1180–1184 RSC.
- R. Chapman, P. J. M. Bouten, R. Hoogenboom, K. A. Jolliffe and S. Perrier, Chem. Commun., 2013, 49, 6522–6524 RSC.
- C. Weber, S. Rogers, A. Vollrath, S. Hoeppener, T. Rudolph, N. Fritz, R. Hoogenboom and U. S. Schubert, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 139–148 CrossRef CAS.
- M. M. Bloksma, D. J. Bakker, C. Weber, R. Hoogenboom and U. S. Schubert, Macromol. Rapid Commun., 2010, 31, 724–728 CrossRef CAS PubMed.
- P. T. Güner and A. L. Demirel, J. Phys. Chem. B, 2012, 116, 14510–14514 CrossRef PubMed.
- N. ten Brummelhuis, C. Secker and H. Schlaad, Macromol. Rapid Commun., 2012, 33, 1690–1694 CrossRef CAS PubMed.
- J.-F. Lutz, Ö. Akdemir and A. Hoth, J. Am. Chem. Soc., 2006, 128, 13046–13047 CrossRef CAS PubMed.
- P. T. Güner, A. Miko, F. F. Schweinberger and A. L. Demirel, Polym. Chem., 2012, 3, 322–324 RSC.
- H. Uyama and S. Kobayashi, Chem. Lett., 1992, 21, 1643–1646 CrossRef.
- J. Zhao, R. Hoogenboom, G. Van Assche and B. Van Mele, Macromolecules, 2010, 43, 6853–6860 CrossRef CAS.
- C. Diab, Y. Akiyama, K. Kataoka and F. M. Winnik, Macromolecules, 2004, 37, 2556–2562 CrossRef CAS.
- J.-S. Park, Y. Akiyama, F. M. Winnik and K. Kataoka, Macromolecules, 2004, 37, 6786–6792 CrossRef CAS.
- S. Salzinger, S. Huber, S. Jaksch, P. Busch, R. Jordan and C. M. Papadakis, Colloid Polym. Sci., 2012, 290, 385–400 CAS.
- A. I. Amirova, M. M. Dudkina, A. V. Tenkovtsev and A. P. Filippov, Colloid Polym. Sci., 2015, 293, 239–248 CAS.
- N. Zhang, R. Luxenhofer and R. Jordan, Macromol. Chem. Phys., 2012, 213, 973–981 CrossRef CAS.
- R. Obeid, F. Tanaka and F. M. Winnik, Macromolecules, 2009, 42, 5818–5828 CrossRef CAS.
- S. Huber, N. Hutter and R. Jordan, Colloid Polym. Sci., 2008, 286, 1653–1661 CAS.
- M. Meyer, M. Antonietti and H. Schlaad, Soft Matter, 2007, 3, 430–431 RSC.
- A. L. Demirel, M. Meyer and H. Schlaad, Angew. Chem., Int. Ed., 2007, 46, 8622–8624 CrossRef CAS PubMed.
- C. Diehl, P. Cernoch, I. Zenke, H. Runge, R. Pitschke, J. Hartmann, B. Tiersch and H. Schlaad, Soft Matter, 2010, 6, 3784–3788 RSC.
- Y. Katsumoto, A. Tsuchiizu, X. P. Qiu and F. M. Winnik, Macromolecules, 2012, 45, 3531–3541 CrossRef CAS.
- T. Li, H. Tang and P. Wu, Langmuir, 2015, 31, 6870–6878 CrossRef CAS PubMed.
- J.-S. Park and K. Kataoka, Macromolecules, 2007, 40, 3599–3609 CrossRef CAS.
- M. M. Bloksma, C. Weber, I. Y. Perevyazko, A. Kuse, A. Baumgärtel, A. Vollrath, R. Hoogenboom and U. S. Schubert, Macromolecules, 2011, 44, 4057–4064 CrossRef CAS.
- A. Levy and M. Litt, J. Polym. Sci., 1968, 6, 1883–1894 CrossRef CAS.
- P. J. M. Bouten, K. Lava, J. C. M. van Hest and R. Hoogenboom, Polymers, 2015, 7, 1998–2008 CrossRef CAS.
- N. S. Ieong, M. Hasan, D. J. Phillips, Y. Saaka, R. K. O'Reilly and M. I. Gibson, Polym. Chem., 2012, 3, 794–799 RSC.
- Q. L. Zhang, L. Voorhaar, S. K. Filippov, B. F. Yesil and R. Hoogenboom, J. Phys. Chem. B, 2016, 120, 4635–4643 CrossRef CAS PubMed.
- E. Djokpé and W. Vogt, Macromol. Chem. Phys., 2001, 202, 750–757 CrossRef.
- L. Starovoytova, J. Spěváček, L. Hanyková and M. Ilavský, Polymer, 2004, 45, 5905–5911 CrossRef CAS.
- T. J. Li, H. Tang and P. Y. Wu, Soft Matter, 2015, 11, 3046–3055 RSC.
- W. Zhang, X. Chen and M. Zhang, Polym. Bull., 2014, 71, 243–260 CrossRef CAS.
- B. Trzebicka, E. Haladjova, Ł. Otulakowski, N. Oleszko, W. Wałach, M. Libera, S. Rangelov and A. Dworak, Polymer, 2015, 68, 65–73 CrossRef CAS.
- A. El Asmar, O. Gimello, G. Morandi, D. Le Cerf, V. Lapinte and F. Burel, Macromolecules, 2016, 49, 4307–4315 CrossRef CAS.
- J.-S. Park and K. Kataoka, Macromolecules, 2006, 39, 6622–6630 CrossRef CAS.
- M. Glassner, K. Lava, V. R. de la Rosa and R. Hoogenboom, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 3118–3122 CrossRef CAS.
- S. Huber and R. Jordan, Colloid Polym. Sci., 2008, 286, 395–402 CAS.
- H. M. L. Lambermont-Thijs, R. Hoogenboom, C.-A. Fustin, C. Bomal-D'Haese, J.-F. Gohy and U. S. Schubert, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 515–522 CrossRef CAS.
- N. Zhang, R. Luxenhofer and R. Jordan, Macromol. Chem. Phys., 2012, 213, 1963–1969 CrossRef CAS.
- G. Le Fer, C. Amiel and G. Volet, Eur. Polym. J., 2015, 71, 523–533 CrossRef CAS.
- M. Hartlieb, D. Pretzel, C. Engert, M. Hentschel, K. Kempe, M. Gottschaldt and U. S. Schubert, Biomacromolecules, 2014, 15, 1970–1978 CrossRef CAS PubMed.
- J. C. Rueda, M. Asmad, V. Ruiz, H. Komber, S. Zschoche and B. Voit, Des. Monomers Polym., 2015, 18, 761–769 CrossRef CAS.
- M. A. Boerman, H. L. van der Laan, J. Bender, R. Hoogenboom, J. A. Jansen, S. C. Leeuwenburgh and J. C. M. Van Hest, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 1573–1582 CrossRef CAS.
- M. A. Mees and R. Hoogenboom, Macromolecules, 2015, 48, 3531–3538 CrossRef CAS.
- A. Gress, A. Völkel and H. Schlaad, Macromolecules, 2007, 40, 7928–7933 CrossRef CAS.
- C. Diehl and H. Schlaad, Chem. – Eur. J., 2009, 15, 11469–11472 CrossRef CAS PubMed.
- C. Diehl and H. Schlaad, Macromol. Biosci., 2009, 9, 157–161 CrossRef CAS PubMed.
- K. Kempe, R. Hoogenboom and U. S. Schubert, Macromol. Rapid Commun., 2011, 32, 1484–1489 CrossRef CAS PubMed.
- K. Kempe, T. Neuwirth, J. Czaplewska, M. Gottschaldt, R. Hoogenboom and U. S. Schubert, Polym. Chem., 2011, 2, 1737–1743 RSC.
- T. R. Dargaville, K. Lava, B. Verbraeken and R. Hoogenboom, Macromolecules, 2016, 49, 4774–4783 CrossRef CAS.
- A. Sehlinger, B. Verbraeken, M. A. R. Meier and R. Hoogenboom, Polym. Chem., 2015, 6, 3828–3836 RSC.
- M. Hruby, S. K. Filippov, J. Panek, M. Novakova, H. Mackova, J. Kucka, D. Vetvicka and K. Ulbrich, Macromol. Biosci., 2010, 10, 916–924 CrossRef CAS PubMed.
- J. H. Kim, E. Lee, J. S. Park, K. Kataoka and W. D. Jang, Chem. Commun., 2012, 48, 3662–3664 RSC.
- E. V. Korchagina, X. P. Qiu and F. M. Winnik, Macromolecules, 2013, 46, 2341–2351 CrossRef CAS.
- L. T. T. Trinh, H. M. L. Lambermont-Thijs, U. S. Schubert, R. Hoogenboom and A. L. Kjoniksen, Macromolecules, 2012, 45, 4337–4345 CrossRef CAS.
- R. Takahashi, T. Sato, K. Terao, X. P. Qiu and F. M. Winnik, Macromolecules, 2012, 45, 6111–6119 CrossRef CAS.
- R. Takahashi, X. P. Qiu, N. Xue, T. Sato, K. Terao and F. M. Winnik, Macromolecules, 2014, 47, 6900–6910 CrossRef CAS.
- H. M. L. Lambermont-Thijs, H. P. C. Van Kuringen, J. P. W. Van der Put, U. S. Schubert and R. Hoogenboom, Polymers, 2010, 2, 188–199 CrossRef CAS.
- C. Diehl, I. Dambowsky, R. Hoogenboom and H. Schlaad, Macromol. Rapid Commun., 2011, 32, 1753–1758 CrossRef CAS PubMed.
- R. Hoogenboom, H. M. L. Thijs, M. W. M. Fijten, B. M. van Lankvelt and U. S. Schubert, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 416–422 CrossRef CAS.
- R. Hoogenboom, H. M. L. Lambermont-Thijs, M. J. H. C. Jochems, S. Hoeppener, C. Guerlain, C.-A. Fustin, J.-F. Gohy and U. S. Schubert, Soft Matter, 2009, 5, 3590–3592 RSC.
- O. Sedlacek, P. Cernoch, J. Kucka, R. Konefal, P. Stepanek, M. Vetrik, T. P. Lodge and M. Hruby, Langmuir, 2016, 32, 6115–6122 CrossRef CAS PubMed.
- S. Osawa, K. Osada, S. Hiki, A. Dirisala, T. Ishii and K. Kataoka, Biomacromolecules, 2016, 17, 354–361 CrossRef CAS PubMed.
- N. Toncheva, C. Tsvetanov, S. Rangelov, B. Trzebicka and A. Dworak, Polymer, 2013, 54, 5166–5173 CrossRef CAS.
- A. Antunes, M. Dierendonck, G. Vancoillie, J. P. Remon, R. Hoogenboom and B. G. De Geest, Chem. Commun., 2013, 49, 9663–9665 RSC.
- J. X. An, A. Dedinaite, F. M. Winnik, X. P. Qiu and P. M. Claesson, Langmuir, 2014, 30, 4333–4341 CrossRef CAS PubMed.
- A. Dworak, A. Utrata-Wesolek, N. Oleszko, W. Walach, B. Trzebicka, J. Aniol, A. L. Sieron, A. Klama-Baryla and M. Kawecki, J. Mater. Sci.: Mater. Med., 2014, 25, 1149–1163 CrossRef CAS PubMed.
- N. Oleszko, W. Walach, A. Utrata-Wesolek, A. Kowalczuk, B. Trzebicka, A. Klama-Baryla, D. Hoff-Lenczewska, M. Kawecki, M. Lesiak, A. L. Sieron and A. Dworak, Biomacromolecules, 2015, 16, 2805–2813 CrossRef CAS PubMed.
- O. Koshkina, T. Lang, R. Thiermann, D. Docter, R. H. Stauber, C. Secker, H. Schlaad, S. Weidner, B. Mohr, M. Maskos and A. Bertin, Langmuir, 2015, 31, 8873–8881 CrossRef CAS PubMed.
- V. R. de la Rosa, W. M. Nau and R. Hoogenboom, Org. Biomol. Chem., 2015, 13, 3048–3057 CAS.
- V. R. de la Rosa, W. M. Nau and R. Hoogenboom, Int. J. Mol. Sci., 2015, 16, 7428–7444 CrossRef CAS PubMed.
- V. R. de la Rosa and R. Hoogenboom, Chem. – Eur. J., 2015, 21, 1302–1311 CrossRef CAS PubMed.
- J. H. Kim, D. Yim and W. D. Jang, Chem. Commun., 2016, 52, 4152–4155 RSC.
- J. H. Kim, Y. Jung, D. Lee and W. D. Jang, Adv. Mater., 2016, 28, 3499–3503 CrossRef CAS PubMed.
- V. R. de la Rosa, Z. Y. Zhang, B. G. De Geest and R. Hoogenboom, Adv. Funct. Mater., 2015, 25, 2511–2519 CrossRef CAS.
- J. H. Kim, E. Koo, S. Y. Ju and W. D. Jang, Macromolecules, 2015, 48, 4951–4956 CrossRef CAS.
- C. Fetsch, A. Grossmann, L. Holz, J. F. Nawroth and R. Luxenhofer, Macromolecules, 2011, 44, 6746–6758 CrossRef CAS.
- J. W. Robinson, C. Secker, S. Weidner and H. Schlaad, Macromolecules, 2013, 46, 580–587 CrossRef CAS.
- J. W. Robinson and H. Schlaad, Chem. Commun., 2012, 48, 7835–7837 RSC.
- M. W. Thielke, C. Secker, H. Schlaad and P. Theato, Macromol. Rapid Commun., 2016, 37, 100–104 CrossRef CAS PubMed.
- C. Secker, A. Völkel, B. Tiersch, J. Koetz and H. Schlaad, Macromolecules, 2016, 49, 979–985 CrossRef CAS.
- X. Tao, Y. Deng, Z. Shen and J. Ling, Macromolecules, 2014, 47, 6173–6180 CrossRef CAS.
- X. Tao, J. Du, Y. Wang and J. Ling, Polym. Chem., 2015, 6, 3164–3174 RSC.
- S. H. Lahasky, X. Hu and D. Zhang, ACS Macro Lett., 2012, 1, 580–584 CrossRef CAS.
- S. H. Lahasky, L. Lu, W. A. Huberty, J. Cao, L. Guo, J. C. Garno and D. Zhang, Polym. Chem., 2014, 5, 1418–1426 RSC.
- S. Xuan, C.-U. Lee, C. Chen, A. B. Doyle, Y. Zhang, L. Guo, V. T. John, D. Hayes and D. Zhang, Chem. Mater., 2016, 28, 727–737 CrossRef CAS PubMed.
- T. J. Deming, Prog. Polym. Sci., 2007, 32, 858–875 CrossRef CAS.
- N. Hadjichristidis, H. Iatrou, M. Pitsikalis and G. Sakellariou, Chem. Rev., 2009, 109, 5528–5578 CrossRef CAS PubMed.
- E. R. Wright and V. P. Conticello, Adv. Drug Delivery Rev., 2002, 54, 1057–1073 CrossRef CAS PubMed.
- C. M. Chopko, E. L. Lowden, A. C. Engler, L. G. Griffith and P. T. Hammond, ACS Macro Lett., 2012, 1, 727–731 CrossRef CAS PubMed.
- Y. Wu, Y. Deng, Q. Yuan, Y. Ling and H. Tang, J. Appl. Polym. Sci., 2014, 131, 1097–4628 Search PubMed.
- A. Kapetanakis and A. Heise, Eur. Polym. J., 2015, 69, 483–489 CrossRef CAS.
- C. Xiao, Y. Cheng, Y. Zhang, J. Ding, C. He, X. Zhuang and X. Chen, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 671–679 CrossRef CAS.
- Y. Xu, M. Zhu, M. Li, Y. Ling and H. Tang, Polym. Chem., 2016, 7, 1922–1930 RSC.
- Y. Deng, Y. Xu, X. Wang, Q. Yuan, Y. Ling and H. Tang, Macromol. Rapid Commun., 2015, 36, 453–458 CrossRef CAS PubMed.
- J. Y. Yuan and M. Antonietti, Polymer, 2011, 52, 1469–1482 CrossRef CAS.
- Y. J. Men, H. Schlaad and J. Y. Yuan, ACS Macro Lett., 2013, 2, 456–459 CrossRef CAS.
- Y. Yang, Y. Wu, R. Li, Y. Ling and H. Tang, Aust. J. Chem., 2016, 69, 112–118 CAS.
- J. Shen, C. Chen, W. Fu, L. Shi and Z. Li, Langmuir, 2013, 29, 6271–6278 CrossRef CAS PubMed.
- D.-L. Liu, X. Chang and C.-M. Dong, Chem. Commun., 2013, 49, 1229–1231 RSC.
- J. X. Ding, C. S. Xiao, Z. H. Tang, X. L. Zhuang and X. S. Chen, Macromol. Biosci., 2011, 11, 192–198 CrossRef CAS PubMed.
- J. Ding, L. Zhao, D. Li, C. Xiao, X. Zhuang and X. Chen, Polym. Chem., 2013, 4, 3345–3356 RSC.
- Y. Shen, S. Desseaux, B. Aden, B. S. Lokitz, S. M. Kilbey, Z. Li and H.-A. Klok, Macromolecules, 2015, 48, 2399–2406 CrossRef CAS.
- X. Fu, Y. Shen, W. Fu and Z. Li, Macromolecules, 2013, 46, 3753–3760 CrossRef CAS.
- E. G. Gharakhanian and T. J. Deming, J. Phys. Chem. B, 2016, 120, 6096–6101 CrossRef CAS PubMed.
- J. Yan, K. Liu, W. Li, H. Shi and A. Zhang, Macromolecules, 2016, 49, 510–517 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2017 |