
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEffect of intercalation and chromophore arrangement on the linear and nonlinear optical properties of model aminopyridine push–pull molecules†
Filip
Bureš
*a,
Daniel
Cvejn
a,
Klára
Melánová
b,
Ludvík
Beneš
c,
Jan
Svoboda
b,
Vítězslav
Zima
b,
Oldřich
Pytela
a,
Tomáš
Mikysek
d,
Zdeňka
Růžičková
e,
I. V.
Kityk
f,
Artur
Wojciechowski
f and
Nasser
AlZayed
f
aInstitute of Organic Chemistry and Technology, Faculty of Chemical Technology, University of Pardubice, Studentská 573, Pardubice, 53210, Czech Republic. E-mail: filip.bures@upce.cz
bInstitute of Macromolecular Chemistry, Academy of Science of the Czech Republic, Heyrovsky sq. 2, 162 06 Prague 6, Czech Republic
cJoint Laboratory of Solid State Chemistry, Faculty of Chemical Technology, University of Pardubice, 532 10 Pardubice, Czech Republic
dDepartment of Analytical Chemistry, Faculty of Chemical Technology, University of Pardubice, 532 10 Pardubice, Czech Republic
eDepartment of Inorganic and General Chemistry, Faculty of Chemical Technology, University of Pardubice, 532 10 Pardubice, Czech Republic
fElectrical Engineering Department, Czestochowa University of Technology, Armii Krajowej 17, Czestochowa, 42201, Poland
First published on 2nd December 2015
Abstract
Three push–pull aminopyridine derivatives having D–π–A, D–(π–A)2, and D–(π–A)3 arrangements were examined as model organic chromophores capable of intercalation into inorganic layered materials (alpha modification of zirconium hydrogen phosphate, zirconium 4-sulfophenylphosphonate, and gamma modification of titanium hydrogen phosphate). The fundamental properties of these dyes, their methylated analogues as well as their intercalates were studied by X-ray analysis, electrochemistry, UV/Vis absorption spectra, TGA, IR spectra, SHG, and were completed by DFT calculations. The synthesis of tripodal tris(pyridin-4-yl)amine is given for the first time. The HOMO–LUMO gap, the position of the longest-wavelength absorption maxima, and the dipole moment of aminopyridines can easily be tuned by attaching/removing pyridin-4-yl electron withdrawing units and their quaternization (pyridine vs. pyridinium acceptors). Their intercalation proved to be feasible affording novel inorganic–organic hybrid materials. The intercalation is accompanied by protonation of the guest, which enhances its ICT and strongly anchors the aminopyridines into the confined space of the layered host. Moreover, this process results in ordering of the organic chromophores and also brings improved thermal and chemical robustness. As a result, the measured SHG efficiencies of the intercalates are larger than those observed for the pure organic push–pull chromophores. Hence, the methodology of intercalation turned out to be very useful strategy for property tuning of NLO-active organic molecules.
Introduction
Owing to their easy and modular synthesis as well as peculiar properties such as distinct color, dipolar character, well-defined and tunable structure, and supramolecular arrangement, organic push–pull molecules have become an extensively explored, and burgeoning area of organic and materials chemistry.1 In such D–π–A systems direct intramolecular charge-transfer (ICT) from the electron donor (D) to the acceptor (A) takes place through the π-system and renders the molecule unique linear and nonlinear optical (NLO) properties. Hence, organic push–pull chromophores with large nonlinearities are of substantial interest for all-optical signal processing applications such as optical triggering, light frequency transducers, optical memories, modulators and deflectors.2 On the molecular level, the rational design and property tuning in D–π–A systems rely mostly on modulation of the donating/withdrawing behavior of the D and A parts and extension of the π-system or production of D–π metal–ligand charge transfer complexes.1–3 At the same time, there exist fundamental limits for enhancement of the second-order hyperpolarizabilities of organic chromophores. These limits are mostly determined by energy of the transition from the ground to the first excited states and by the number of effective conjugated electrons.4 However and as a rule, enhanced hyperpolarizability decreases effective energy gap leading to lower transparency. Based on such property tuning, admirable number of organic push–pull molecules with large intrinsic (hyper)polarizabilities were developed.5 However, it turned out to be rather difficult to translate their high molecular optical nonlinearity to macroscopic nonlinear- and electro-optic activity.6 The traditional approach to overcome this issue involves incorporation of the chromophore into a polymeric backbone (either covalently bound or doped), dendrimers, organic glasses, films as well as host–guest systems in which layered inorganic compounds might serve as the host matrix. The host matrix of the latter inorganic–organic hybrid systems comprises clays (LAPONITE®, saponite, montmorillonite, hydrotalcite, kaoline etc.),7 silicates,8 and layered metal oxalates and MPS3 (M = Mn, Cd, Zn).9 Formerly, organic NLOphores capable of intercalation were limited to simple and linear D–π–A systems such as nitroanilines or azobenzenes but, more recently, it was found that large heterocyclic molecules such as (di)azines, porphyrines and some branched chromophores undergo intercalation into a layered material as well.10 The incorporation of organic dyes into a restricted space of an ordered inorganic matrix represents important strategy for achieving materials with the following features and properties:• the periodical ordering of the inorganic matrix, which can induce preferential orientation of the dye
• reduction of random 3D-orientation of the dye into a 2D-arrangement
• planarization of the π-system of the dye
• confined environment with reduced internal motion of the dye enhances its photophysical properties
• encapsulation of less stable organic molecules by a very robust and transparent inorganic carrier
• increased chemical and thermal stability.
Hence, intercalation may open an additional reserve to further enhance and optimize the second-order hyperpolarizabilities without substantial decrease of the energy gap. Based on our recent contribution on intercalation of tripodal push–pull molecule bearing pyridin-4-yl terminus (tris[4-(pyridin-4-yl)phenyl]amine, TPPA),11 we report herein combination of both micro/macroscopic approaches mentioned above. In this respect, three model push–pull molecules with central amino donor and peripheral pyridine acceptor(s) were designed and synthesized. These molecules (4-AminoPYridines, Scheme 1) differ in the number of terminal acceptors and overall molecular arrangement – D–π–A (APY1), D–(π–A)2 (APY2), and D–(π–A)3 (APY3). Due to the presence of peripheral basic pyridin-4-yl moieties, the supramolecular arrangement of APY1–3 can be controlled via intercalation into acidic layered inorganic materials. Protonation of the peripheral pyridine rings will generate pyridinium moieties, which are (i) stronger electron acceptors (ICT enhancement), (ii) strongly anchors the whole chromophore between the host layers, and (iii) organizes the dye molecules in bulk (Fig. 1).
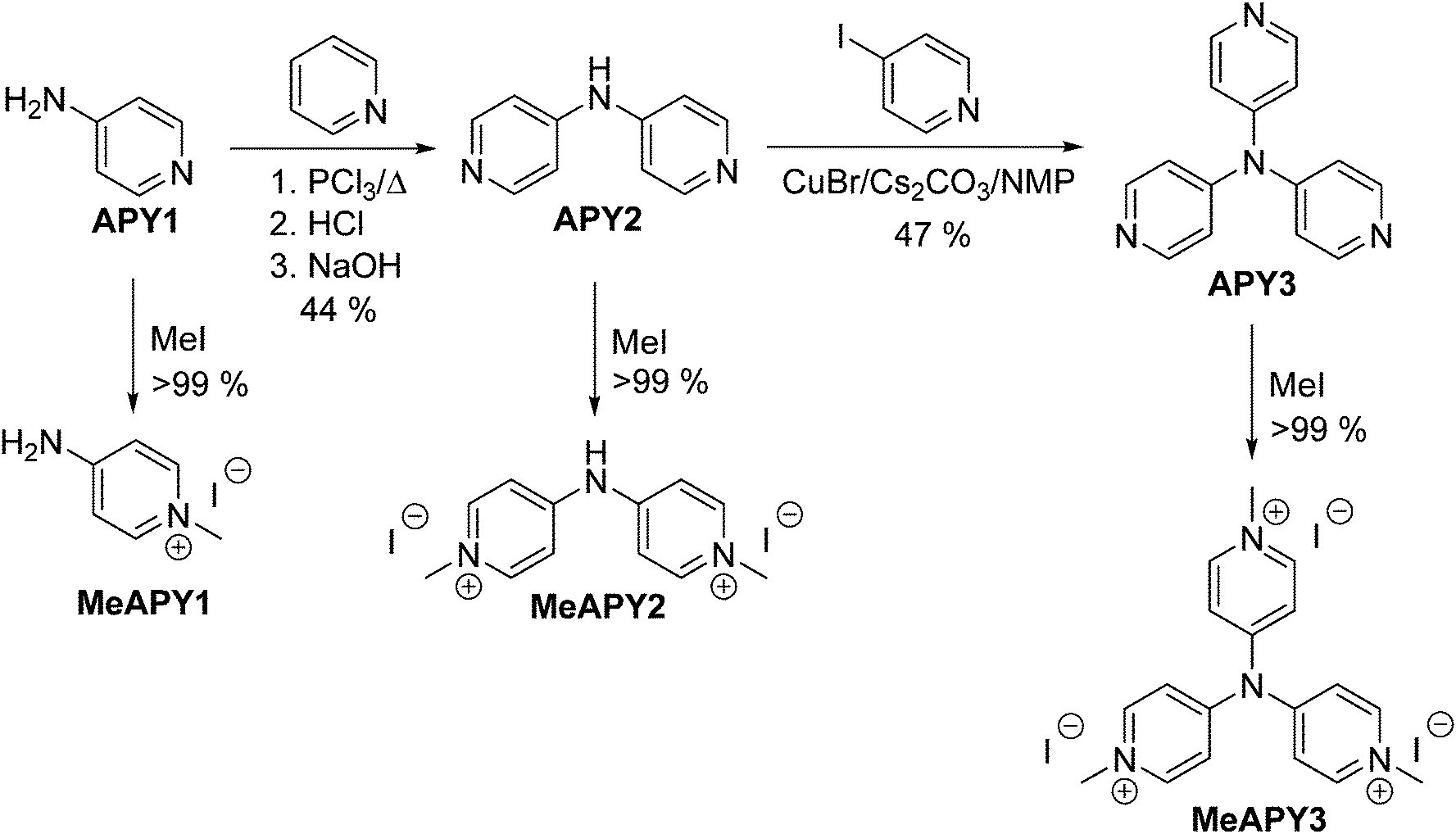 | ||
| Scheme 1 Synthesis of aminopyridines (APY) and aminopyridiniums (MeAPY). | ||
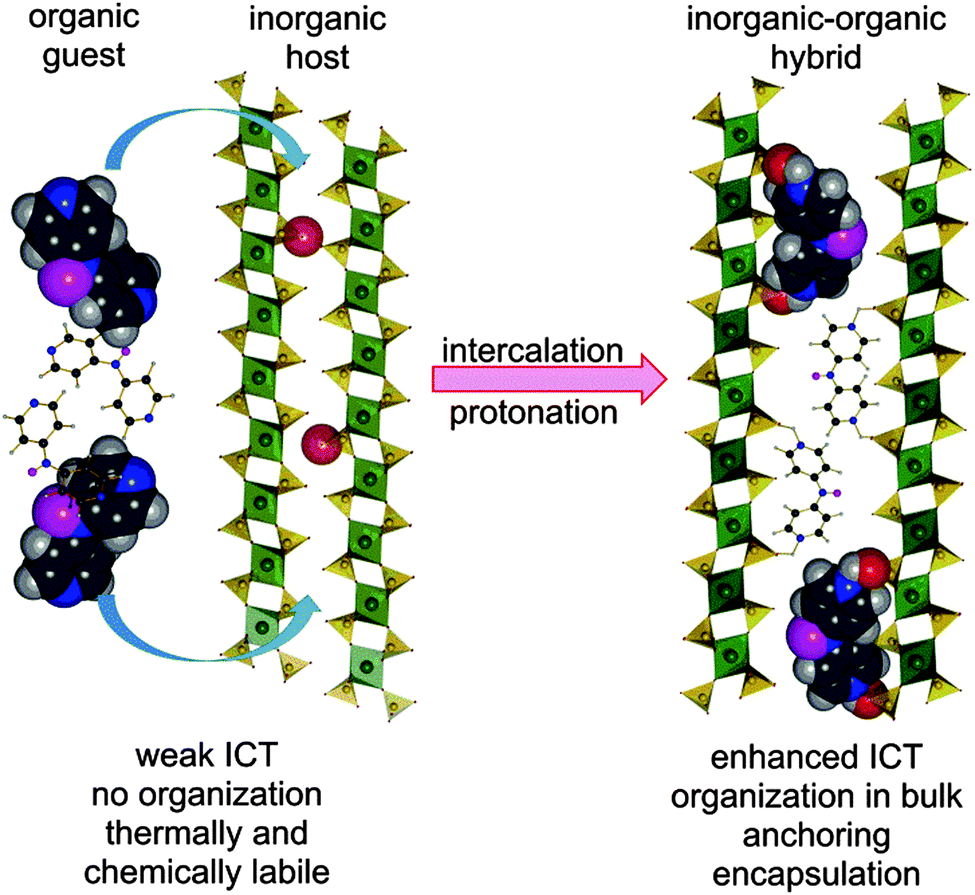 | ||
| Fig. 1 Schematic representation of the intercalation process of APY2. | ||
Aminopyridines APY1–3 were intercalated into α-modification of zirconium hydrogen phosphate (Zr(HPO4)2·2H2O), zirconium 4-sulfophenylphosphonate Zr(HO3SC6H4PO3)2·2H2O and γ-modification of titanium hydrogen phosphate (Ti(PO4)(H2PO4)·2H2O). It has recently been verified that commercially available APY1 undergo intercalation into α-modification of titanium hydrogen phosphate (Ti(HPO4)2·2H2O) and generates significant NLO response on condition of its protonation with maleic acid.12 While APY2 is increasingly being used for construction of metal–organic frameworks (MOF),13APY3, despite it features simple molecular structure, has hypothetically been envisaged as lately as in 2010.14 To the best of our knowledge, we report herein its first successful preparation. All aminopyridines APY1–3 and their intercalates were characterized experimentally by X-ray diffraction, TGA, IR, electrochemistry, UV/Vis spectroscopy as well as theoretically by DFT calculations. Their second order susceptibilities (SHG efficiencies) were examined as well.
Results and discussion
Synthesis of aminopyridines
The synthesis of APY compounds and their quaternized analogues MeAPY is shown in Scheme 1. Dipyridylamine APY2 was synthesized via one-pot pyridine chlorination with PCl3 and subsequent nucleophilic aromatic substitution with commercially available APY1.15 Introduction of the third pyridine branch was initially attempted via Buchwald–Hartwig reaction16 with 4-iodopyridine yielding no desired product. However, by adopting copper(I)-catalyzed strategy of Sarges et al.17 and upon optimization of the reaction condition, APY3 was prepared in 47% isolated yield. Electron withdrawing behavior of azines can easily be improved via quaternization.18 Hence, in order to mimic the anticipated products generated upon intercalation into acid layered materials, APY1–3 were further N-methylated with iodomethane to afford pyridinium salts MeAPY1–3 in almost quantitative yields.X-ray analysis
Among other spectral characterizations (see the ESI†), the molecular structure of APY3 was also confirmed by single crystal X-ray analysis. Suitable crystals were obtained by slow evaporation of its methanol solution. The ORTEP plot shown in Fig. 2 confirms the proposed molecular structure of APY3 and can also be used to determine the extent of the ICT.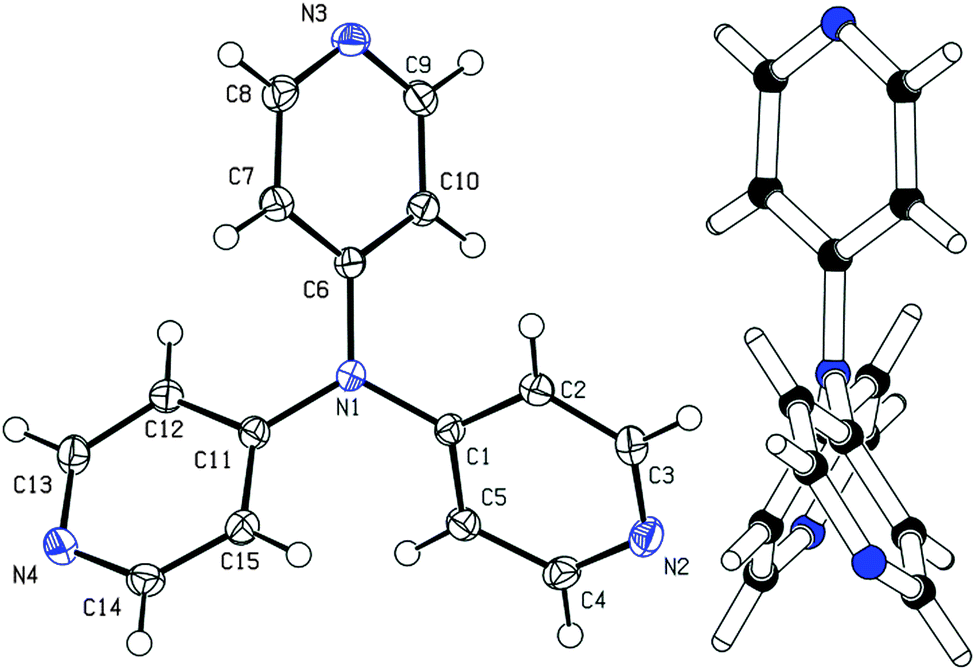 | ||
| Fig. 2 The molecular structure (ORTEP 50% probability level) and the side view of APY3. | ||
In this respect, the involvement of the particular pyridine rings in the ICT can be assessed by their spatial arrangement and bond length alternation (quinoid character δr)19 and aromaticity evaluated by Bird index I6.20 Central nitrogen atom has perfectly planar environment with three nitrogen atoms of pyridin-4-yl substituents, only N4 atom is slightly shifted out of that central plane. The values of interplanar angles of the central plane and pyridyl rings were found in the narrow region of 37.7–39.6°, which is similar to that found for triphenylamine derivatives, e.g. TPPA.21 The quinoid character and aromaticity of pyridin-4-yl rings were calculated to be within the range of δr = 0.009–0.010 and I6 = 94.4–95.9. Unsubstituted benzene has δr and I6 equal to 0 and 100, respectively, whereas unsubstituted pyridine has I6 = 85.7. Both indicators imply that all pyridine rings possess relatively low/high quinoid/aromatic character and, thus are involved in less ICT. However, these findings are in accordance with known tripodal triphenylamine derivatives.1,11
Electrochemistry
Electrochemical behaviour of APY1–3 and MeAPY1–3 were studied by CV, RDV and polarography. The acquired data are summarized in Table 1. The measured electrochemical behavior of APY1 is consistent with the data reported in the literature.22 The oxidations of APY2–3 were not observed in the potential window of the given electrode, electrolyte and solvent (up to +1.7 V vs. SCE). The reduction potentials E1/2(red1) were recorded by polarography. The DME (dropping mercury electrode) allowed a cathodic measurement up to −3 V vs. SCE, while on the Pt-electrode it is only −2 V vs. SCE. Oxidation was observed for pyridinium salts MeAPY1–3 but this is caused by oxidation of the I− anions. The first reductions, which are reversible one-electron processes, were measured only with the Pt-electrode as interactions of MeAPY1–3 with the DME were observed.| Comp. | E 1/2(ox1) (V) | E 1/2(red1) (V) | E HOMO (eV) | E LUMO (eV) | λ max (nm(eV)) | ε (103 M cm−1) | E HOMO (eV) | E LUMO (eV) | ΔEd (eV) | μ (D) |
|---|---|---|---|---|---|---|---|---|---|---|
| a E 1/2(ox1) and E1/2(red1) are half-wave potentials of the first oxidation and reduction, respectively. All potentials were recorded in N,N-dimethylformamide containing 0.1 M Bu4NPF6 and are given vs. SCE. b −EHOMO/LUMO = E1/2(ox1/red1) + 4.35 (ref. 23). c Measured in DMSO (c = 2×10−5 M); sh denotes shoulder. d DFT calculated by (B3LYP/6-311++G(2d,p)//B3LYP/6-311++G(2d,p)) in DMF. | ||||||||||
| APY1 | +1.50 | — | −5.85 | — | 267(4.64)sh | 1.72 | −6.52 | −0.66 | 5.86 | 5.36 |
| APY2 | — | −2.52 | — | −1.83 | 297(4.18) | 29.56 | −6.35 | −1.39 | 4.95 | 3.64 |
| APY3 | — | −2.26 | — | −2.09 | 302(4.11) | 18.43 | −6.30 | −1.58 | 4.72 | 0.02 |
| MeAPY1 | — | −1.36 | — | −2.99 | 273(4.54) | 17.29 | −7.36 | −1.96 | 5.40 | 0.58 |
| MeAPY2 | — | −1.62 | — | −2.73 | 333(3.72)/403(3.08)sh | 23.63/22.97 | −7.75 | −3.21 | 4.54 | 0.16 |
| MeAPY3 | — | −0.90 | — | −3.45 | 334(3.71)/404(3.07) | 30.52/0.62 | −7.96 | −3.55 | 4.41 | 0.06 |
When going from APY2 to APY3, the first reduction potentials decrease by 260 mV, most likely as a result of introducing additional electron withdrawing pyridin-4-yl unit. Quaternization of the pyridine to pyridinium moieties significantly shifted the first reduction potentials to more positive values by 0.9 (APY2vs.MeAPY2) and 1.36 V (APY3vs.MeAPY3). This clearly reflects the improved electron withdrawing ability and thus enhanced ICT from the central amino donor to peripheral pyridinium acceptors. On the contrary, MeAPY2 was reduced at the most negative potential (−1.62 V) among other pyridinium salts – MeAPY1 (−1.36 V) and MeAPY3 (−0.90 V). This is most likely given by a facile loss of HI from MeAPY2 and formation of an imine structure MeAPY2q as shown in the ESI† (Scheme S1). This quinoid cation has also been detected by HR-MALDI-MS as a main peak (Fig. S2, ESI†). In MeAPY2/MeAPY2q, the observed most negative reduction potential reflects enhanced electron density of the central amino donor and subsequent higher saturation of the pyridinium acceptor as expressed by the resonant structures shown in Scheme S1 (ESI†).
UV/Vis absorption spectra of APY1–3 and MeAPY1–3
The optical properties of APY1–3 and MeAPY1–3 in liquid media were investigated by electronic absorption spectroscopy measured in DMSO (Fig. 3). The measured longest-wavelength absorption maxima λmax and the molar absorption coefficients ε are given in Table 1. All of the absorption spectra, except for APY1, are dominated by intense CT-bands with the maxima appearing at 267–302 nm for APY1–3 and 273–404 nm for MeAPY1–3. As expected, these CT-bands are red-shifted with the increasing number of pyridin-4-yl units as well as with their replacement by pyridinium acceptors. MeAPY2 showed two particularly developed CT-bands appearing at 333 and 403 nm. The latter corresponds to facile formation of the quinoid structure MeAPY2q with extended π-conjugated system and stronger electron donor (ICT enhancement). This observation is in accordance with other betaine dyes, especially the Reichardt’s dye used to determine the solvent polarity – ET(30).24 The position of the longest-wavelength absorption maxima and the band-width and shape of betaine dyes are strongly solvent-dependent and methanol seems to be exceptional solvent in this respect.25 Hence, the absorption spectra measured in methanol are shown in the ESI† (Fig. S3). In contrast to DMSO, the spectra of both MeAPY2 and 3 measured in methanol are dominated by intensive low-energy bands appearing at 390 nm. These pronounced bands can be ascribed to a more efficient ICT from the central amino donor to the peripheral pyridinium acceptors and implies higher stabilization of the formed quinoid structure by protic solvents.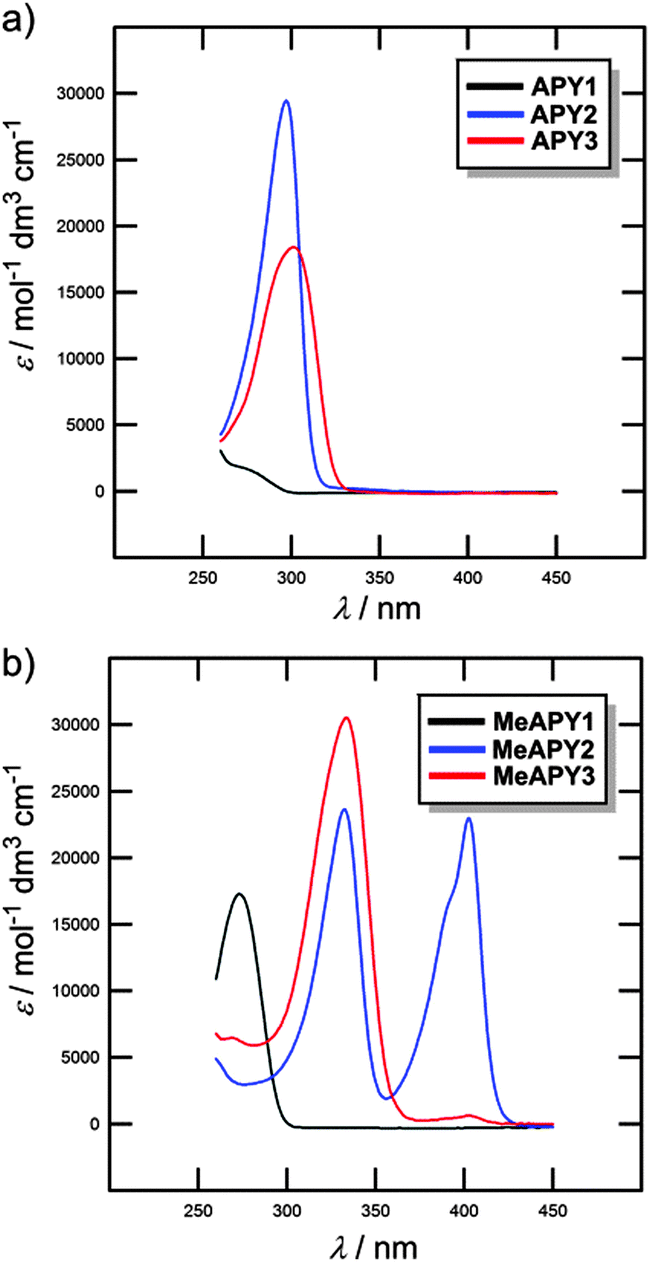 | ||
| Fig. 3 UV/Vis absorption spectra of APY1–3 (a) and MeAPY1–3 (b) measured in DMSO (c = 2 × 10−5 M) | ||
DFT calculations
The spatial and electronic properties of APY1–3 and MeAPY1–3 were also investigated by calculations. Their geometry was optimized by PM3 and subsequently DFT B3LYP methods with B3LYP/6-311++G(2d,p) basis set. The energies of the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO), HOMO–LUMO gap (ΔE), and ground state dipole moment (μ) were calculated by using the B3LYP/6-311++G(2d,p) base, and are summarized in Table 1 (see the ESI† for more details). Only the cationic parts of MeAPY1–3 were considered in DFT calculations. Although the calculated energies of the LUMO differ slightly from those obtained by electrochemical measurements (ΔELUMO = 0.1–1.03 eV), the DFT-derived HOMO–LUMO gap (ΔE) correlate tightly with the energy of the electron transition calculated from the position of the longest-wavelength absorption maxima (1240/λmax) as shown in Fig. S4 (ESI†). Hence, the used DFT calculations are obviously capable of properly describing the trends seen by the optical measurements and can be considered as a reasonable tool for the electronic property description of APY1–3 and MeAPY1–3. The principal changes are seen in the energies of the LUMO, which reflects the increasing number of pyridine and pyridinium acceptors appended to the central amino donor. The calculated HOMO–LUMO gaps decrease from 5.86/5.40 to 4.72/4.41 eV for APY1–3/MeAPY1–3.The visualization of frontier molecular orbitals for APY1–3 and MeAPY1–3 show acceptor-centered LUMO and donor-centred HOMO, and partial charge separation, which confirms their ICT character (Fig. 4). In tripodal molecules APY3 and MeAPY3, the third pyridin-4-yl branch is occupied by the LUMO+1, the HOMO−1 remained on the central amino donor. This is a common feature of tripodal push–pull molecules.26 As expected, the ground state dipole moment μ vanishes with increasing symmetry of the molecule.
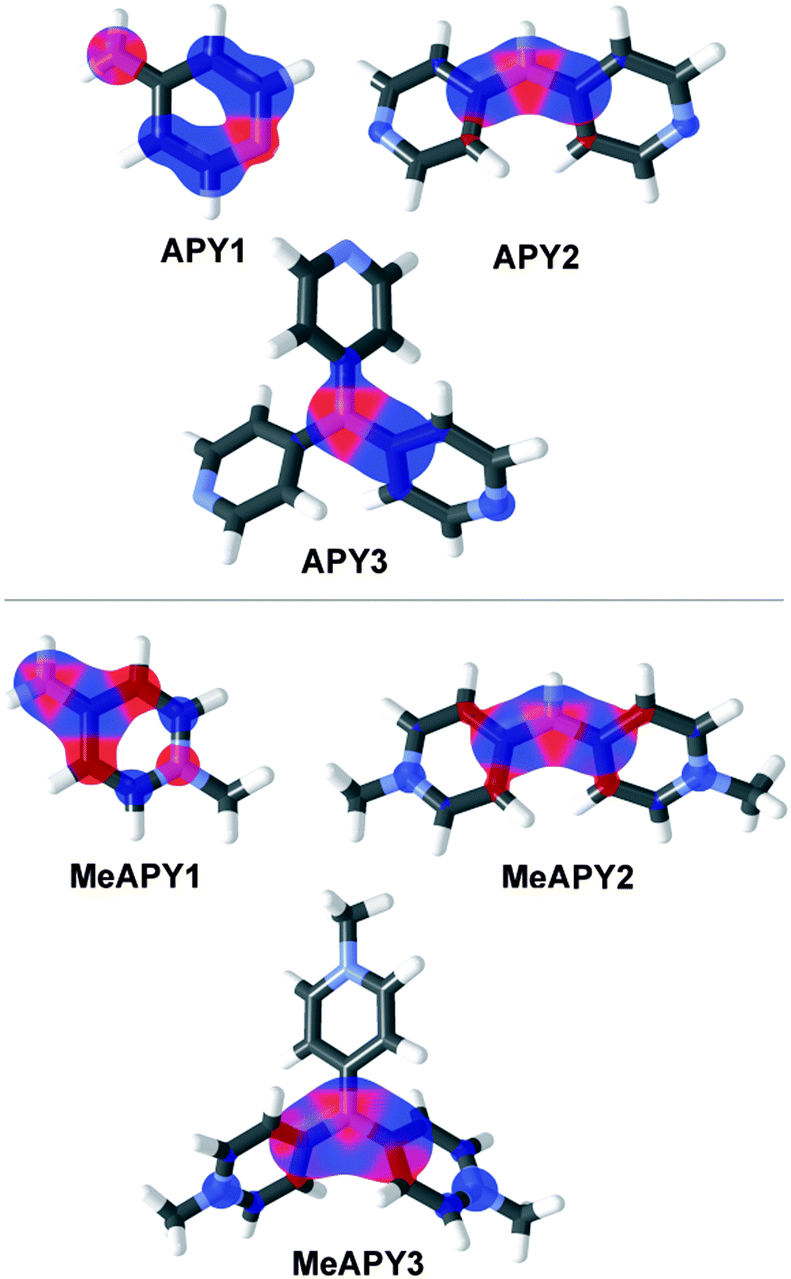 | ||
| Fig. 4 Optimized geometries and HOMO (red) and LUMO (blue) localizations in APY1–3 and MeAPY1–3. | ||
Composition of intercalates
The amount of the intercalate species x in each host is given in Table 2 together with the content of water y. The compositions of the prepared intercalates were calculated on the basis of elemental analysis and thermogravimetric data (thermogravimetry curves are given in Fig. S5–S7 in the ESI†). The general formula can be written as H·xG·yH2O (where H = ZrP, ZrSPP, or TiP and G = APY1, APY2, or APY3). The data in Table 2 shows that the amount of the intercalated guest in the hosts generally decreases in the order ZrSPP > ZrP > TiP. The ratio of the amount the intercalated species in each host is roughly the same and is xAPY1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :xAPY2:xAPY3 = 6:3:2. This ratio is inversely proportional to the charge present at each of the protonated guest species (1:2:3). The higher amount of the guest in the ZrSPP intercalate than in ZrP is in accordance with our previous findings for ZrSPP and ZrP intercalates of tris[4-(pyridin-4-yl)phenyl]amine.11 There is no distinct relation between the amount of the intercalated guest and the amount of water y.
:xAPY2:xAPY3 = 6:3:2. This ratio is inversely proportional to the charge present at each of the protonated guest species (1:2:3). The higher amount of the guest in the ZrSPP intercalate than in ZrP is in accordance with our previous findings for ZrSPP and ZrP intercalates of tris[4-(pyridin-4-yl)phenyl]amine.11 There is no distinct relation between the amount of the intercalated guest and the amount of water y.
| Host H | Guest G | Intercalate | x | y |
|---|---|---|---|---|
| ZrSPP | APY1 | ZrSPP–APY1 | 1.20 | 0.70 |
| ZrSPP | APY2 | ZrSPP–APY2 | 0.50 | 1.50 |
| ZrSPP | APY3 | ZrSPP–APY3 | 0.35 | 1.60 |
| α-ZrP | APY1 | ZrP–APY1 | 0.86 | 1.00 |
| α-ZrP | APY2 | ZrP–APY2 | 0.43 | 2.25 |
| α-ZrP | APY3 | ZrP–APY3 | 0.30 | 1.75 |
| γ-TiP | APY1 | TiP–APY1 | 0.69 | 1.00 |
| γ-TiP | APY2 | TiP–APY2 | 0.35 | 0.75 |
| γ-TiP | APY3 | TiP–APY3 | 0.22 | 2.50 |
On heating, all intercalates release water at relatively low temperature, the weight decrease in all cases starts below 100 °C. For the APY1 intercalates, the second decrease starting at about 300 °C is most probably due to the release of the guest from the interlayer space. Intercalates with APY2 and APY3 release their less volatile guests at higher temperature (500 °C) and the decrease corresponding to this release merges with the weight decrease due to the decomposition of the host.
Structure of the host
The intercalation process is driven by acid–base interactions of acidic active sites present on the surface of the host layers with basic guests APY1–3 (Fig. 1). The intercalation is therefore accompanied by protonation of the peripheral pyridine moieties and generation of pyridinium(s).All three hosts are layered. All contain acidic active sites, formed by oxygen atoms carrying acid protons. In ZrP and TiP these active sites are formed by POH groups whereas in ZrSPP by SOH groups. The surface density of the active sites on one side of the layer can be expressed as the number of sites per area unit of the layer surface.27 A reciprocal value of the surface density then gives an area available for one active site, which is called “free area”. From comparison of the free area with dimension of the intercalated molecule (cross sectional area) we can estimate a maximum amount of the guest which can be intercalated into a given host.
The structure of ZrSPP in its inorganic part is based on the structure of the alpha modification of ZrP.28 From this it follows that ZrP and ZrSPP have the same number of active acidic protons per unit of surface that is the same free area on the layer (24 Å2) per active site, that is 48 Å2 per formula unit of the host.29
The gamma modification of TiP has a different structure.30 Two phosphate groups are present in this gamma modification, one is PO43− in which all oxygen atoms are bonded to titanium, the second is H2PO4− with two oxygen atoms bonded to titanium while other two bear acidic protons. The formula of the gamma modification should therefore be written as Ti(PO4)(H2PO4)·2H2O. The free area surrounding each P(OH)2 group is 32.9 Å2.31
Thickness of the layer defined as the shortest distance between the barycenters of the oxygen atoms situated at the opposite sites of the layer was calculated to be 16.8 Å, 6.3 Å, and 9.2 Å for ZrSPP,28 α-ZrP,32 and γ-TiP,31 respectively. By subtraction of these values from the interlayer distances of the intercalates, we can get the height of the gallery available for the incorporation of the intercalated species. When the gallery height is multiplied by the half of the free area (i.e., 24 Å2 for ZrSPP and ZrP; 16.5 Å2 for TiP) we get the volume which is accessible for the intercalated species in the corresponding host. The corresponding values are given in Table S4 in the ESI.†
Structure of intercalates
The intercalated guest molecules can be placed in the interlayer space in several ways: (i) with the plane of the pyridine rings parallel with the plane of the host layer, either as a one layer of the guest molecules (a “monomolecular” arrangement) or as two layers (a “bimolecular” arrangement); (ii) with the plane of the pyridine ring either perpendicular or tilted with respect to the plane od the host layers. Also in this case the guest molecules can adopt monomolecular or bimolecular arrangement.The basal spacings/interlayer distances for all intercalates are given in Table 3. In all cases the value of the interlayer distance is increased on intercalation, which indicates that the guest molecules are not placed with the pyridine ring planes parallel to the host layers. In that case the height of the gallery should be about 2 or 4 Å only. With the decrease of amount of the guest the interlayer space the guest molecule tends to be more inclined with respect to the plane of the host layer.
| Intercalate | d | d after heating to 210 °Ca | d after rehydrationb | Accessible volume (Å3) |
|---|---|---|---|---|
| a Measured in situ on a heated brass block in the diffractometer. b The samples were rehydrated by standing in a closed vessel over water for one week. | ||||
| ZrSPP–APY1 | 24.282 | 24.14 | 24.22 | 359.136 |
| ZrSPP–APY2 | 23.905 | 23.61 | 23.98 | 341.04 |
| ZrSPP–APY3 | 26.390 | 24.80 | 26.43 | 460.32 |
| ZrP–APY1 | 12.653 | 11.95 | 12.64 | 304.944 |
| ZrP–APY2 | 13.418 | 11.62 | 13.58 + 13.36 | 341.664 |
| ZrP–APY3 | 12.794 | 10.92 | 12.75 | 311.712 |
| TiP–APY1 | 15.704 | 14.52 | 15.02 | 213.9816 |
| TiP–APY2 | 15.156 | 13.77 | 15.16 | 195.9524 |
| TiP–APY3 | 15.480 | 14.29 | 15.45 | 206.612 |
The gallery height in all APY1 intercalates (7.48 Å, 6.35 Å, 6.50 Å for ZrSPP–APY1, ZrP–APY1 and TiP–APY1, respectively) is larger than or equal to the van der Waals length of the APY1 molecule (6.35 Å). It means that this molecule can be arranged in the interlayer space with the plane of the pyridine ring perpendicular to the plane of the host layer most probably in an interdigitated manner.
Roughly the same gallery height (7.1 Å) can be observed for the ZrSPP–APY2 and ZrP–APY2 intercalates while it is much smaller for TiP–APY2 (5.9 Å). In all cases the gallery height is smaller than the dimension of the APY2 molecule along its longest axis (which is about 10.5 Å). Therefore, we can rule out the possibility that the guest molecules are bonded to both layers of the host through their pyridinium groups, forming a kind of pillared structure. The more probable arrangement is that in which each guest molecule is bonded to one layer of the host by both pyridinium nitrogen atoms of APY2 as shown in Fig. 1.
High gallery height found for ZrSPP–APY3 (9.6 Å) indicates that this molecule is in an upright position in ZrSPP, whereas in ZrP and TiP (gallery height 6.5 and 6.3 Å, respectively) it is tilted with respect to the host layer plane under an estimated angle of 45°.
Change on dehydration
To study the influence of dehydration on the interlayer distance the change of the powder X-ray patterns of the intercalates on heating to 200 °C was studied. Almost no change of the interlayer distance was observed for ZrSPP–APY1 intercalate, which contains only 0.7 molecule of water per formula unit. In this case the high degree of intercalation ensures quite rigid arrangement of the guest in the interlayer space which does not allow its change on dehydration. Also small decrease of basal spacing (0.3 Å) was observed for ZrSPP–APY2, even the content of water in this intercalate is higher (y = 1.5). The ZrP–APY1 intercalate exhibits a slight decrease of the basal spacing (0.7 Å), while the decrease for the TiP–APY1 is 1.2 Å. Both these intercalates contain 1 molecule of water per formula unit and show roughly the same increase of the basal spacing on intercalation (4.3 and 4.5 Å). The decrease of the basal spacing on dehydration is given by different amount of the intercalated guest. On the other hand, the intercalates of APY3 are most influenced by dehydration, especially ZrP–APY3 (1.9 Å, y = 1.75) and ZrSPP–APY3 (1.6 Å, y = 1.6). Also high decrease of the basal spacing was observed for the ZrP–APY2 intercalate (1.8 Å) which has the highest amount of water (y = 2.25) among all the intercalates.Infrared spectra
The way how the intercalated guest molecules interact with the host material i.e., the deprotonation of the host and protonation of the guest molecules, was determined on the basis of the IR spectra (see the ESI,† Fig. S8–S16). The peaks in the region from 1150 to 850 cm−1 are narrower in the intercalates compared to those in the pristine hosts. The band at 1231 cm−1 of δP–OH found in the spectrum of the original TiP disappears in the spectra of all three TiP intercalates.12a Analogously this band at 1247 cm−1 in the spectrum of ZrP disappears in the spectra of ZrP–APY1 and ZrP–APY2; this band is retained in the spectrum of ZrP–APY3, nevertheless, with a lower intensity.In pure APY1, δNH scissoring band is observed at 1644 cm−1 and peak of ring stretching appears at 1590, 1558, 1504, 1434 cm−1.33 In this region, a very broad intensive band at 1649 cm−1 and quite intensive band at 1535 cm−1 are observed in methylated APY1 (MeAPY1). The two bands appear also in the TiP–APY1 intercalate. In spectra of ZrSPP–APY1 and ZrP–APY1 a couple of bands at 1650 and 1675 cm−1 and a very broad band at 1540 cm−1 are observed. In addition, in the spectrum of ZrP–APY1 a very weak peak at 1597 cm−1 is observed which is observed also in the pristine APY1. It indicates that in this intercalate a part of the guest remains unprotonated.
In the spectrum of ZrSPP–APY2, the most distinct feature, besides the peaks belonging to the host material, is the existence of peaks at 1624 and 1508 cm−1 (see Fig. S9 in the ESI†). These peaks are not present in the spectrum of APY2 but appear in the spectrum of MeAPY2 at 1654 and 1500 cm−1. In the ZrP–APY2 intercalate, these peaks appear at 1620 and 1506 cm−1 (see Fig. S12 in the ESI†) and in TiP–APY2 at 1626 and 1510 cm−1 (Fig. S15 in the ESI†). In the case of ZrSPP–APY3, the analogous peaks corresponding to methylated (protonated) APY are less distinct (Fig. S10 in the ESI†). While the positions of these peaks are at 1634 and 1512 cm−1 in MeAPY3, only a weak peak at 1628 cm−1 appears in ZrSPP–APY3 and the second peak is shifted to 1498 cm−1. In the ZrP–APY3 intercalate these peaks are more distinct, with the positions at 1628 cm−1 and 1500–1508 cm−1, nevertheless, a peak of the host material (ZrP) observed at 1614 cm−1 might contribute to the intensity of the peak at 1628 cm−1 (Fig. S13 in the ESI†).
Generally, the IR spectra of the intercalates in the region from 1800 to 1150 cm−1 represent a superposition of the peaks found in the host and in the corresponding methylated guest (MeAPY). The IR spectra of the intercalates, when compared with the spectra of all three APY and MeAPY, confirm therefore that these guests are protonated in the intercalates.
Solid state UV/Vis spectra
Beside the measurements in liquid media (see above), the optical properties of APY1–3, MeAPY1–3, and APY1–3 intercalates were also investigated in the solid-state (see the ESI,† Fig. S17–S20).Unprotonated APY1 has an absorption maximum (λmax) at 248 nm, whereas methylated APY1 (MeAPY1) showed a maximum at 272 nm. The maxima of all three APY1 intercalates are roughly at the same position, around 260 nm (Fig. S18, ESI†) The ZrSPP–APY1 intercalate showed slightly bathochromically shifted λmax at 262 nm, which reflects that ZrSPP is a host material with the strongest acidity of the interlayer environment among all three hosts. APY1 protonated by the exposition of this compound to HCl vapours shows a maximum at around 265 nm and its spectrum is similar to those of all three intercalates.
Significant changes in the UV/Vis spectra were observed when going from APY2 to methylated MeAPY2. Whereas unprotonated APY2 showed a single peak at 293 nm, MeAPY2 possesses two maxima at 326 and at 392 nm. This is similar to that observed by measurement in DMSO (see above). Compared to the APY1 intercalates, the difference between the spectra of all three APY2 intercalates is clearly distinguishable (Fig. S19, ESI†). The ZrP–APY2 intercalate has the maximum at the lowest wavelength (297 nm) almost at the same position as the unprotonated APY2 guest, with a shoulder at around 320–330 nm. This means that in ZrP–APY2 we can distinguish between the unprotonated and protonated forms of APY2. This claim is further supported by the fact that APY2 protonated in HCl vapours has a maximum at 321 nm, which is the same region as found for the shoulder in the ZrP–APY2 spectrum. The TiP–APY2 and ZrSPP–APY2 intercalates showed one envelope band with the maxima at 305 and 317 nm, respectively. These bands seem to be superposition of that found for APY2 and its protonated form. From these data we can deduce that APY2 undergoes partial protonation during intercalation into all three hosts and is being most protonated in ZrSPP (most bathochromically shifted maxima). Thus, the acidity of the hosts increases in the order ZrP < TiP < ZrSPP.
In the solid-state, APY3 and MeAPY3 showed the UV/Vis spectra similar to those observed in methanol (see above) with the maxima appearing at 307 and 323/390 nm, respectively. The ZrP–APY3 intercalate possesses almost the same maximum as APY3 with a shoulder at around 325 nm (Fig. S20, ESI†). The spectrum of the TiP–APY3 intercalate shows a very broad unstructured band covering the range of 300–320 nm with the maximum reaching the position observed for APY3 exposed to HCl vapors (λmax = 324 nm). The longest-wavelength absorption maximum of ZrSPP–APY3 (λmax = 323 nm) corresponds tightly to the maximum measured for protonated APY3, but in contrast to MeAPY3, shows no shoulder. This implies that APY3 intercalated into ZrSPP is fully protonated.
NLO properties
To create a long-range ordered non-centrosymmetry the investigated chromophores were embedded into liquid photocomposition of oligoetheracrylate photopolymers,34 which were photo-solidified by nitrogen 337 nm cw laser with power density 55 W cm−2 at an applied electric field successively increasing up to 4 kV cm. The orientation control was done using a polarized absorption. Typical time kinetics of the fundamental and the SHG signals is shown in the ESI† (Fig. S21). The measured principal second-order susceptibility parameters deff jointly with the calculated hyperpolarizabilities β(−2ω;ω,ω) of APY1–3, MeAPY1–3 and intercalates are listed in Table 4.| Comp. | d eff (pm V−1) | β (−2ω;ω,ω) (10−30 esu) |
|---|---|---|
| a Measured in oligoetheracrylate at 1064 (±0.15 pm V−1). b DFT calculated by (B3LYP/6-311++G(2d,p)//B3LYP/6-311++G(2d,p)) in vacuum at 1064 nm. | ||
| APY1 | 1.34 | 1.06 |
| APY2 | 1.56 | 1.44 |
| APY3 | 0.35 | 0.01 |
| MeAPY1 | 1.42 | 1.37 |
| MeAPY2 | 1.67 | 2.61 |
| MeAPY3 | 1.04 | 0.13 |
| ZrSPP–APY1 | 1.78 | — |
| ZrSPP–APY2 | 1.89 | — |
| ZrSPP–APY3 | 1.21 | — |
| ZrP–APY1 | 1.67 | — |
| ZrP–APY2 | 1.72 | — |
| ZrP–APY3 | 1.45 | — |
| TiP–APY1 | 2.01 | — |
| TiP–APY2 | 2.21 | — |
| TiP–APY3 | 1.56 | — |
The NLO data in Table 4 allows evaluation of the structure–property relationships caused by the chromophore arrangement, N-methylation as well as intercalation. The effect of the chromophore arrangement can easily be assessed on the series APY1–3. The linear push–pull aminopyridine APY1 showed a SHG response with deff equal to 1.34 pm V−1. An introduction of the second pyridin-4-yl branch as in APY2 increased the second-order susceptibility to 1.56 pm V−1. On the contrary, tripodal APY3 showed a significantly diminished NLO response with deff = 0.35 pm V−1. This drop reflects the symmetrical arrangement of APY3 (see the X-ray analysis above) and the very low calculated ground-state dipole moment (Table 1). The extent of the ICT and resulting NLO properties of the heteroaromatic D–π–A systems can be improved via alkylation. Thus, the comparison of MeAPY1–3vs.APY1–3 allows evaluation of the effect of the pyridine/pyridinium acceptors. Whereas, the pairs of corresponding chromophores MeAPY1/APY1 and MeAPY2/APY2 showed slightly improved nonlinearities by a factor of 1.06/1.07, the nonlinearity of MeAPY3 is almost three times higher than that found for APY3. This observation is most likely given by a synergistic effect of the ICT enhancement via N-methylation and the symmetry loss caused by iodide counter ions. In general, the SHG response of aminopyridines decreases in the order of APY2 > APY1 > APY3 and MeAPY2 > MeAPY1 > MeAPY3 and MeAPY > APY. The calculated hyperpolarizabilities β(−2ω;ω,ω) showed very similar trends.
The intercalation of APY1–3 into ZrSPP, ZrP, and TiP is accompanied by their protonation as well organization in the bulk. Whereas the effect of protonation can be considered similar to quaternization, the significant SHG improvement seen for all intercalates over MeAPY1–3 must be elucidated as the impact of their organization in the layered host. The measured SHG responses reflect the acidity of the host, where intercalates with the most acidic ZrSPP showed larger deff than ZrP (except APY3). The highest nonlinearities were measured for APY1–3 intercalated into gamma modification of titanium hydrogen phosphate (TiP). In the TiP intercalates, the lowest volume left in the interlayer space of the host after intercalation of APY1–3 were found. This can be expressed as the lowest difference between the accessible volume in the interlayer space of the host VH and the volume of the intercalated molecule Vguest and is documented in Table S4 in the ESI† together with the corresponding calculations. Thus, the arrangement of the guest molecules in TiP turned out to be the most rigid among all three hosts. In agreement with our assumptions given in the Introduction this reduced internal motion of the guests leads to an increased NLO response.
Conclusions
A series of model organic push–pull aminopyridines with various spatial arrangements has been synthesized to examine their intercalation into inorganic layered materials (alpha modification of zirconium hydrogen phosphate, zirconium 4-sulfophenylphosphonate, and gamma modification of titanium hydrogen phosphate). The prepared inorganic–organic hybrids were further investigated as ordered materials for nonlinear optics.A facile synthetic pathway has been developed to novel tripodal tris(pyridin-4-yl)amine (APY3). Its structure was unambiguously confirmed by single crystal X-ray analysis, which revealed almost perfectly planar environment of all three peripheral nitrogen atoms. The fundamental properties of all push–pull aminopyridines were investigated by electrochemistry, UV/Vis absorption spectra and were completed with DFT calculations. It was demonstrated that with increased number of the pyridin-4-yl acceptor units, the HOMO–LUMO gap decreases steadily by more than 1 eV and the longest-wavelength absorption maxima shifts bathochromically (Δλmax ~ 30 nm). N-Quaternization, which takes place exclusively on the peripheral pyridines, caused further reduction of the HOMO–LUMO gap and red-shifts the CT-band. Structural analysis of MeAPY compounds revealed facile formation of the quinoid structures, which significantly affected their electrochemical and optical properties.
Three types of intercalates were prepared with the ratio of the amount of intercalated APY1–3 molecules being 6:3:2. This ratio is inversely proportional to the charge generated at each of the aminopyridine guest (1:2:3). IR and solid-state UV/Vis spectra showed that all aminopyridines underwent protonation during the intercalation process, which extent depends on the number of guest basic centers as well as acidity of the host. Taking spectra of APY1–3 and MeAPY1–3 as limiting, the measured IR spectra of the intercalates are superposition of the peaks found for the host and methylated guest. This observation confirms intercalation process as well as guest protonation. From the UV/Vis spectra, the extent of the protonation can further be assessed. Whereas APY2 underwent only partial protonation upon intercalation, full protonation of APY3 was observed in the most acidic ZrSPP and TiP hosts. Powder X-ray diffraction measurements revealed that the guest molecules are arranged in the interlayer space of the hosts as interdigitated monolayers in the way schematically shown in Fig. 1 for APY2 intercalated in ZrP. In view of a generally low thermal stability of organic push–pull molecules, the prepared inorganic–organic materials resisted thermal decomposition up to 300 (APY1) and 500 °C (APY2–3). This feature makes them thermally very robust.
Second-order nonlinear optical properties of APY1–3, quaternized MeAPY1–3, and APY1–3 intercalates were examined by SHG. The measured as well as calculated optical susceptibilities/hyperpolarizabilities were affected by the following structural features:
• arrangement of the push–pull aminopyridine (a general decrease in the SHG response was observed in the order of APY2 > APY1 > APY3)
• quaternization/protonation (pyridine vs. pyridinium acceptor moieties, ICT enhancement)
• intercalation into layered materials (all intercalates showed significantly higher SHG responses than pure organic push–pull guests)
• acidity of the host material used for intercalation (ZrSPP and TiP proved to be more efficient acids than ZrP)
• structure of the host (compared to ZrSPP and ZrP, the TiP intercalates have more tightly arranged APY molecules and showed higher SHG response).
In this work, we have verified a new strategy on property tuning of organic push–pull molecules. Three aminopyridines with D–π–A, D–(π–A)2, and D–(π–A)3 arrangements were designed, synthesized, and investigated as model guest molecules capable of intercalation into inorganic layered materials providing access to new inorganic–organic hybrid materials. We believe that this multidisciplinary study would serve as useful guide in designing novel NLO-active materials with tailored properties.
Experimental
Aminopyridines
APY1 is commercially available; the synthesis of APY2 and APY3 is outlined in Scheme 1 and described in the ESI.†Intercalates
Three layered host materials were used for the intercalation, namely alpha modification of zirconium hydrogen phosphate with formula Zr(HPO4)2·H2O (denoted further as ZrP), zirconium 4-sulfophenylphosphonate with formula Zr(HO3SC6H4PO3)1.8(C6H5PO3)0.2·2H2O (ZrSPP) and gamma modification of titanium hydrogen phosphate with formula Ti(PO4)(H2PO4)·2H2O (TiP). The syntheses of the host materials (ZrP, ZrSPP and TiP) and the intercalation of APY1, APY2 and APY3 into them are described in the ESI.†Materials and methods
Reagents and solvents were reagent grade and were used as received. 1H and 13C NMR spectra were recorded at 500 and 125 MHz, with a Bruker AVANCE 500 instruments at 25 °C equipped with Prodigy CryoProbe. High resolution MALDI MS spectra were measured on a MALDI mass spectrometer LTQ Orbitrap XL (Thermo Fisher Scientific, Bremen, Germany) equipped with nitrogen UV laser (337 nm, 60 Hz). Elemental analyses were performed on an EA 1108 Fisons instrument. UV/Vis spectra were recorded on a Hewlett–Packard 8453 spectrophotometer. Electrochemical measurements were carried out in N,N-dimethylformamide containing 0.1 M Bu4NPF6 in a three electrode cell by cyclic voltammetry (CV), rotating disk voltammetry (RDV) and polarography. The working electrode was platinum disk (2 mm in diameter) for CV and RDV experiments. As the reference and auxiliary electrodes were used saturated calomel electrode (SCE) separated by a bridge filled with supporting electrolyte and Pt wire, respectively. All potentials are given vs. SCE. Voltammetric measurements were performed using a potentiostat PGSTAT 128N (AUTOLAB, Metrohm Autolab B.V., Utrecht, The Netherlands) operated via NOVA 1.10 software. The X-ray data for the crystals of APY3 were obtained at 150 K using an Oxford Cryostream low-temperature device on a Nonius KappaCCD diffractometer with Mo Kα radiation (λ = 0.71073 Å), a graphite monochromator, and the ϕ and χ scan mode.Powder X-ray diffraction data were obtained with a D8 Advance diffractometer (Bruker AXS, Germany) with Bragg-Brentano θ–θ geometry (40 kV, 30 mA) using CuKα radiation with secondary graphite monochromator. The diffraction angles were measured at room temperature from 2 to 70° (2θ) in 0.02° steps with a counting time of 15 s per step. Powder X-ray diffraction measurements at 210 ± 1 °C were carried out on a heated brass block equipped with a thermocouple in the range from 2 to 35° (2θ) in 0.025° steps with a counting time of 15 s per step. The size of the crystallites of the intercalates was calculated according to the Scherrer formula35 using an EVA software.36
The thermogravimetric measurements (TGA) were done using a home-made apparatus constructed of a computer-controlled oven and a Sartorius BP210 S balance. The measurements were carried in air between 30 and 960 °C at a heating rate of 5 °C min−1.
Infrared spectra in the range of 600–4000 cm−1 were recorded at 64 scans per spectrum at 2 cm−1 resolution using HATR adapter on a Perkin-Elmer FTIR Spectrum BX spectrometer on neat samples. All spectra were corrected for the presence of moisture and carbon dioxide in the optical path.
Second-order non-linear optical susceptibilities were measured by a method similar to that described in the ref. 37 where a polymer supported the crystalline powder. The pulsed Q-switch Nd:YAG laser operating at 1064 nm wavelength with a pulse duration of 18 ns and frequency repetition 10 Hz, power about 1 MW and a pulse repetition about 10 Hz was applied as the fundamental one. The samples were rotated on the travel in order to achieve a maximal SHG which was spectrally separated by a monochromator connected to photomultiplier. The optimal content of chromophore was equal to about 30% in weighting units. LiNbO3 crystals with known parameters of a second-order optical susceptibility were used as reference samples. Using the ratio of the intensities for the reference and the studied samples we have defined the second order susceptibilities with accuracy up to 0.15 pm V−1.
Acknowledgements
This work has been supported by the Czech Science Foundation (13-01061S).Notes and references
- (a) F. Bureš, RSC Adv., 2014, 4, 58826 RSC; (b) M. Kivala and F. Diederich, Acc. Chem. Res., 2009, 42, 235 CrossRef CAS PubMed; (c) H. Meier, Angew. Chem., Int. Ed., 2005, 44, 2482 CrossRef PubMed.
- (a) J. M. Hales, S. Barlow, H. Kim, S. Mukhopadhyay, J.-L. Brédas, J. W. Perry and S. R. Marder, Chem. Mater., 2014, 26, 549 CrossRef CAS; (b) P. N. Prasad and D. J. Williams, Introduction to Nonlinear Optical Effects in Molecules & Polymers, John Wiley & Sons, New York, 1991 Search PubMed; (c) D. R. Kanis, M. A. Ratner and T. J. Marks, Chem. Rev., 1994, 94, 195 CrossRef CAS; (d) J. J. Wolff and R. Wortmann, Adv. Phys. Org. Chem., 1999, 32, 121 CrossRef CAS; (e) G. S. He, L.-S. Tan, Q. Zheng and P. N. Prasad, Chem. Rev., 2008, 108, 1245 CrossRef CAS PubMed.
- M. G. Kuzyk, J. Mater. Chem., 2009, 19, 7444 RSC.
- (a) M. G. Kuzyk, Phys. Rev. Lett., 2000, 85, 1218 CrossRef CAS PubMed; (b) M. G. Kuzyk, J. Chem. Phys., 2006, 125, 154108 CrossRef PubMed; (c) J. Pérez Moreno and M. G. Kuzyk, J. Chem. Phys., 2005, 123, 194101 CrossRef PubMed.
- M. G. Kuzyk, C. W. Dirk and C. W. Characterization Techniques and Tabulations for Organic Nonlinear Optical Materials, Marcel Dekker, New York, 1998 Search PubMed.
- (a) L. R. Dalton, J. Phys.: Condens. Matter, 2003, 15, R897 CrossRef CAS; (b) M. J. Cho, D. H. Choi, P. A. Sullivan, A. J. P. Akelaitis and L. R. Dalton, Prog. Polym. Sci., 2008, 33, 1013 CrossRef CAS; (c) J. Luo, X.-H. Zhou and A. K.-Y. Jen, J. Mater. Chem., 2009, 19, 7410 RSC; (d) L. R. Dalton, P. A. Sullivan and D. H. Bale, Chem. Rev., 2010, 110, 25 CrossRef CAS PubMed.
- (a) N. Epelde-Elezcano, E. Duque-Redondo, V. Martínez-Martínez and H. Manzano, Langmuir, 2014, 30, 10112 CrossRef CAS PubMed; (b) M. Ogawa, M. Takahashi and K. Kuroda, Chem. Mater., 1994, 6, 715 CrossRef CAS; (c) M. Ogawa and A. Ishikawa, J. Mater. Chem., 1998, 8, 463 RSC; (d) G. G. Aloisi, U. Costantino, F. Elisei, L. Latterini, C. Natali and M. Nocchetti, J. Mater. Chem., 2002, 12, 3316 RSC; (e) R. Takenawa, Y. Komori, S. Hayashi, J. Kawamata and K. Kuroda, Chem. Mater., 2001, 13, 3741 CrossRef CAS.
- U. Díaz, Á. Cantín and A. Corma, Chem. Mater., 2007, 19, 3686 CrossRef.
- (a) J. S. O. Evans, S. Bénard, P. Yu and R. Clément, Chem. Mater., 2001, 13, 3813 CrossRef CAS; (b) S. Bénard, A. Léaustic, E. Rivière, P. Yu and R. Clément, Chem. Mater., 2001, 13, 3709 CrossRef; (c) T. Coradin, R. Clément, P. G. Lacroix and K. Nakatani, Chem. Mater., 1996, 8, 2153 CrossRef CAS.
- (a) Y. Suzuki, Y. Tenma, Y. Nishioka and J. Kawamata, Chem. – Asian J., 2012, 7, 1170 CrossRef CAS PubMed; (b) L. Latterini, M. Nocchetti, G. G. Aloisi, U. Costantino and F. Elisei, Inorg. Chim. Acta, 2007, 360, 728 CrossRef CAS; (c) M. Ogawa and K. Kuroda, Chem. Rev., 1995, 95, 399 CrossRef CAS.
- K. Melánová, D. Cvejn, F. Bureš, V. Zima, J. Svoboda, L. Beneš, T. Mikysek, O. Pytela and P. Knotek, Dalton Trans., 2014, 43, 10462 RSC.
- (a) L. M. Nunes and C. Airoldi, Thermochim. Acta, 2005, 435, 118 CrossRef CAS; (b) J. Tarasiewicz, R. Jakubas, I. Majerz, J. Baran, A. Gągor and A. Miniewicz, Vib. Spectrosc., 2013, 66, 93 CrossRef CAS.
- (a) R. L. LaDuca, R. Finn and J. Zubieta, Chem. Commun., 1999, 1669 RSC; (b) S. M. Saylor, R. M. Supkowski and R. L. LaDuca, Inorg. Chim. Acta, 2008, 361, 317 CrossRef CAS; (c) S. H. Qiblawi, L. K. Sposato and R. L. LaDuca, Inorg. Chim. Acta, 2013, 407, 297 CrossRef CAS; (d) M.-L. Han, Y.-P. Wu, J. Zhao, D.-S. Li and Y.-Y. Wang, J. Solid State Chem., 2015, 230, 218 CrossRef CAS; (e) L. E. Weingartz, J. H. Nettleman, G. A. Farnun, R. J. Staples and R. L. LaDuca, Polyhedron, 2015, 89, 168 CrossRef CAS.
- Y. G. Bushuev and G. Sastre, J. Phys. Chem. C, 2010, 114, 19157 CAS.
- (a) E. Koenigs and G. Jung, J. Prakt. Chem., 1933, 137, 141 CrossRef CAS; (b) R. S. Alekseev, A. V. Kurkin and M. A. Yurovskaya, Chem. Heterocycl. Compd., 2012, 48, 1235 CrossRef CAS.
- B. P. Fors, P. Krattiger, E. Strieter and S. L. Buchwald, Org. Lett., 2008, 10, 3505 CrossRef CAS PubMed.
- R. Sarges, H. R. Howard, K. M. Donahue, W. M. Welch, B. W. Dominy, A. Weissman, B. K. Koe and J. Bordner, J. Med. Chem., 1986, 29, 8 CrossRef CAS PubMed.
- (a) E. Cariati, R. Macchi, D. Roberto, R. Ugo, S. Galli, N. Masciocchi and A. Sironi, Chem. Mater., 2007, 19, 3704 CrossRef CAS; (b) T. Cañeque, A. M. Cuadro, J. Alvarez-Builla, J. Pérez-Moreno, K. Clays, G. Marcelo, F. Mendicuti, O. Castaño, J. L. Andrés and J. J. Vaquero, Eur. J. Org. Chem., 2010, 6323 CrossRef; (c) M. A. Ramirez, R. Custodio, A. M. Cuadro, J. Alvarez-Builla, K. Clays, I. Asselberghs, F. Mendicuti, O. Castaño, J. L. Andrés and J. J. Vaquero, Org. Biomol. Chem., 2013, 11, 7145 RSC.
- C. Dehu, F. Meyers and J. L. Brédas, J. Am. Chem. Soc., 1993, 115, 6198 CrossRef CAS.
- (a) C. W. Bird, Tetrahedron, 1986, 42, 89 CrossRef CAS; (b) S. I. Kotelevskii and O. V. Prezhdo, Tetrahedron, 2001, 57, 5715 CrossRef CAS; (c) T. M. Krygowski, H. Szatylowicz, O. A. Stasyuk, J. Dominikowska and M. Palusiak, Chem. Rev., 2014, 114, 6383 CrossRef CAS PubMed.
- (a) C. Hua, P. Turner and D. M. D’Alessandro, Dalton Trans., 2013, 42, 6310 RSC; (b) M.-D. Zhang, C.-M. Di, L. Qin, X.-Q. Yao, Y.-Z. Li, Z.-J. Guo and H.-G. Zheng, Cryst. Growth Des., 2012, 12, 3957 CrossRef CAS.
- (a) I. B. Romanova and L. V. Chervina, Chem. Heterocycl. Compd., 1977, 13, 1321 CrossRef; (b) P. G. Desideri, D. Heimler and L. Lepri, J. Electroanal. Chem., 1978, 87, 275 CrossRef CAS.
- A. A. Isse and A. Gennaro, J. Phys. Chem. B, 2010, 114, 7894 CrossRef CAS PubMed.
- (a) C. Reichardt, Solvents and Solvent Effects in Organic Chemistry, Second, completely revised and enlarged edition, VCH, Weinheim, 1988 Search PubMed; (b) C. Reichardt, Angew. Chem., 1965, 77, 30 CrossRef CAS.
- A. M. Kjaer and J. Ulstrup, J. Am. Chem. Soc., 1987, 109, 1934 CrossRef CAS.
- D. Cvejn, E. Michail, I. Polyzos, N. Almonasy, O. Pytela, M. Klikar, T. Mikysek, V. Giannetas, M. Fakis and F. Bureš, J. Mater. Chem. C, 2015, 3, 7345 RSC.
- G. Alberti and U. Costantino, Layered Solids and their intercalation chemistry, in Comprehensive Supramolecular Chemistry. Two- and Three-dimensional networks, ed. J. L. Atwood, 1996, vol. 7, pp. 1–23 Search PubMed.
- J. Svoboda, V. Zima, K. Melánová, L. Beneš and M. Trchová, J. Solid State Chem., 2013, 208, 58 CrossRef CAS.
- A. Clearfield and U. Costantino, Layered metal phosphates and their intercalation chemistry, in Comprehensive Supramolecular Chemistry, ed. G. Alberti and T. Bein, Pergamon Press, Oxford, 1996, vol. 7, pp. 107–149 Search PubMed.
- A. N. Christensen, E. K. Andersen, I. G. K. Andersen, G. Alberti, M. Nielsen and M. S. Lehmann, Acta Chem. Scand., 1990, 44, 865 CrossRef CAS.
- G. Alberti, Layered metal phosphates and their intercalation chemistry, in Comprehensive Supramolecular Chemistry, ed. G. Alberti and T. Bein, Pergamon Press, Oxford, 1996, vol. 7, p. 142 Search PubMed.
- A. Clearfield and U. Costantino, Layered metal phosphates and their intercalation chemistry, in Comprehensive Supramolecular Chemistry, ed. G. Alberti and T. Bein, Pergamon, New York, 1996, vol. 7, p. 113 Search PubMed.
- Y. Buyukmurat and S. Akyuz, J. Mol. Struct., 2003, 651–653, 533 CrossRef CAS.
- B. Sahraoui, I. V. Kityk, J. Bieleninik, P. Hudhomme and A. Gorgues, Mater. Lett., 1999, 41, 164 CrossRef CAS.
- A. Patterson, Phys. Rev., 1939, 56, 978 CrossRef CAS.
- EVA, ver.19., Diffrac plus Basic Evaluating Package, Bruker AXS GmbH, Germany, 2013.
- S. K. Kurtz and T. T. Perry, J. Appl. Phys., 1968, 39, 3798 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis of APY2–3, MeAPY1–3 and their intercalates, crystallographic data of APY3, structural analysis of MeAPY2, UV/Vis spectra in methanol, further DFT-calculated data, TGA curves, IR spectra, solid-state UV/Vis spectra, further NLO data. CCDC 1405923. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5tc03499j |
| This journal is © The Royal Society of Chemistry 2016 |