
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The evolution of bicontinuous polymeric nanospheres in aqueous solution†
B. E.
McKenzie
a,
J. F.
de Visser
a,
G.
Portale
b,
D.
Hermida-Merino
b,
H.
Friedrich
a,
P. H. H.
Bomans
ac,
W.
Bras
b,
O. R.
Monaghan
d,
S. J.
Holder
*d and
N. A. J. M.
Sommerdijk
*ac
aLaboratory of Materials and Interface Chemistry and Soft Matter Cryo-TEM Research Unit, Eindhoven University of Technology, P.O. Box 513, 5600 MB Eindhoven, The Netherlands. E-mail: N.Sommerdijk@tue.nl
bNetherlands Organization for Scientific Research (NWO), European Synchrotron Radiation Facility (ESRF), DUBBLE-CRG, Grenoble, F-38043, France
cInstitute for Complex Molecular Systems, Eindhoven University of Technology, PO Box 513, 5600 MB Eindhoven, The Netherlands
dFunctional Materials Group, School of Physical Sciences, University of Kent, Canterbury, Kent CT2 7NH, UK. E-mail: S.J.Holder@kent.ac.uk
First published on 1st April 2016
Abstract
Complex polymeric nanospheres in aqueous solution are desirable for their promising potential in encapsulation and templating applications. Understanding how they evolve in solution enables better control of the final structures. By unifying insights from cryoTEM and small angle X-ray scattering (SAXS), we present a mechanism for the development of bicontinuous polymeric nanospheres (BPNs) in aqueous solution from a semi-crystalline comb-like block copolymer that possesses temperature-responsive functionality. During the initial stages of water addition to THF solutions of the copolymer the aggregates are predominantly vesicles; but above a water content of 53% irregular aggregates of phase separated material appear, often microns in diameter and of indeterminate shape. We also observe a cononsolvency regime for the copolymer in THF–water mixtures from 22 to 36%. The structured large aggregates gradually decrease in size throughout dialysis, and the BPNs only appear upon cooling the fully aqueous dispersions from 35 °C to 5 °C. Thus, the final BPNs are ultimately the result of a reversible temperature-induced morphological transition.
Introduction
Amphiphilic block copolymers self-assemble in aqueous solution into morphologies analogous to those observed from low molecular weight lipid amphiphiles. Most commonly reported are spherical micelles, cylindrical micelles and vesicles.1,2 Extensive studies into the fundamental contributions to block copolymer self-assembly have enabled the formation and specific tailoring of more complex morphologies in solution. Examples include multi-compartmental micelles,3–5 Janus particles,6,7 helical structures,8–10 and disk-like micelles.11–13 Aggregates with complex internal phase separation are being targeted for the simultaneous solubilization of chemically different components and for the compartmentalization of reactions.4,14,15A relatively new complex morphology is represented by bicontinuous polymeric nanospheres (BPNs) – discrete nanoparticles comprising intertwined but non-intersecting hydrophilic and hydrophobic phases. Much like their lipid counterparts (lipid cubosomes),16–18 the high internal surface area and concurrent hydrophobic and hydrophilic domains make BPNs promising candidates for use as catalytic nanoreactors,19 delivery vehicles20 and as frameworks for the organization of inorganic material.21 Furthermore, their formation from block copolymers with different compositions and chemistries presents possibilities for the formation of BPNs with additional functionality.22,23 For example, we have previously reported the formation of BPNs from a semi-crystalline block copolymer of poly(ethylene oxide)-block-poly(octadecyl methacrylate) with temperature responsive behavior.24,25
Comparatively few examples of this morphology have been reported and so far the criteria for its formation are somewhat ambiguous. To date BPNs have been observed from triblock copolymers,26–28 single24 and double29 comb-like block copolymers, branched copolymers,30,31 Janus dendrimers32 and block copolymers with ionic composition.28,33,34 From the available reported examples, it would appear that structural complexity of the copolymer is a necessity for the formation of BPNs; however, we have recently reported their formation from simple amorphous block copolymers of poly(ethylene oxide)-block-poly(butyl methacrylate) (PEO-b-PBMA).35 In the same work, we reported that choosing a cosolvent with higher affinity for the hydrophobic block than for the hydrophilic block aids the formation of the bicontinuous phase, shedding further light on what were previously elusive ideas on the solvent contribution to the phase.33,36
The challenge in studying such structures has always been to be able to discern the morphology whilst preserving the native state of the aggregates so as not to alter the self-assembly behavior or introduce artifacts, as is the concern when using conventional dry TEM techniques. To that end, the more wide-spread application of 2- and 3D cryoTEM to the assessment of block copolymer self-assembly has made a significant contribution to the characterization of new morphologies such as those aforementioned, and allows the assessment of the effects of external factors such as choice of solvent, temperature and pH on the aggregate morphology in the native solvated state.32,37–39 Moreover, SAXS is applicable to different length scales and so is able to probe molecular contributions to morphological changes. Although much emphasis has rested upon observing the morphology post-processing, insight into the intermediary stages yields additional key information regarding the polymer properties and behavior that may have implications for the intended applications of the resulting complex nanospheres, and may significantly aid in the tailored design of such complex morphologies.
Herein, we present the evolution of complex poly(ethylene oxide)-b-poly(octadecyl methacrylate) (PEO-b-PODMA) BPNs from THF–water mixtures through 2- and 3D cryoTEM and SAXS analysis; and we reveal their development from a series of preliminary lamellar phases. We show that the process is accompanied by the appearance of structure or demixing within the THF–water mixtures over a certain composition range. Ultimately the removal of THF leads to the formation of dispersed fluid microphase separated large ‘fields’ and spheroids. Furthermore, we report that the bicontinuous morphology is ultimately the result of a morphological change driven by traversing the thermal transitions of the block copolymer accompanied by a change in packing of the hydrophobic PODMA side chains. Moreover, the temperature-responsive behavior of the system is reversible.
Experimental
Block copolymer synthesis
A PEO macroinitiator was synthesized using poly(ethylene glycol) methyl ether (Mn = ca. 2 kDa) and 2-bromoisobutyryl bromide. The block copolymer PEO52-b-PODMA26 was then synthesized by copper(I) mediated atom transfer radical polymerization of the PODMA block from the PEO macroinitiator. The polymer was isolated by precipitation from cold methanol and fully characterized by NMR and GPC. Details are in the ESI.†Polymer dispersions
Briefly, the dispersions were formed by the nanoprecipitation method (water addition process): 50 mg of the block copolymer PEO52-b-PODMA26 was dissolved in 4 mL of 450 nm filtered tetrahydrofuran (THF), and a predetermined volume of MilliQ water (0.2–6 mL; 5–63 wt%) was added to the stirring polymer–THF solutions at a rate of 0.07 mL min−1. The addition process was conducted at 35 °C – above the PEO-b-PODMA melting temperature – and aliquots taken from the resulting dispersions were vitrified at 35 °C for cryoTEM analysis. The solution containing THF![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :water 4:6 mL (5 g L−1 polymer) was placed in a dialysis membrane and dialyzed against 5 L of water at 35 °C for 24 h, during which time samples were collected and vitrified for cryoTEM analysis. The aqueous dispersion was then cooled stepwise to 5 °C, equilibrated at each predetermined temperature for 15 min and vitrified at the corresponding temperatures for cryoTEM analysis.
:water 4:6 mL (5 g L−1 polymer) was placed in a dialysis membrane and dialyzed against 5 L of water at 35 °C for 24 h, during which time samples were collected and vitrified for cryoTEM analysis. The aqueous dispersion was then cooled stepwise to 5 °C, equilibrated at each predetermined temperature for 15 min and vitrified at the corresponding temperatures for cryoTEM analysis.
CryoTEM analysis
Visualization was conducted using the TU/e CryoTitan (FEI Company). Grids were prepared using an automated robot (Vitrobot™ Mark III, FEI Company), which was kept at appropriate temperatures between 5 and 35 °C. 20 nm gold nanoparticles were added to an aliquot of the dispersion taken for cryoTEM analysis and are used as fiducial markers for tomography. The excess of liquid on the grid was blotted away with filter paper to form a thin film of the dispersion; and to vitrify, the grid was plunged rapidly into liquid ethane. It was not possible to saturate the vitrobot with a THF-containing atmosphere due to the sensitivity of internal components to THF and the feasibility of maintaining the necessary THF:water ratio within the humidifier. Consequently all samples were prepared in a 100% water atmosphere.
Small angle X-ray scattering (SAXS) analysis
Measurements were performed at the Dutch–Belgian BM26B beamline at the ESRF in Grenoble (France). An X-ray photon energy of 10 keV (X-ray wavelength of 1.24 Å) and two different sample-to-detector distances (7 m and 5 m) were used, in order to explore a wide q range (0.35 nm−1 < q < 2.9 nm−1, where q is the modulus of the scattering vector = q = 4πsin(θ/2)/λ, with λ the X-rays wavelength and θ the scattering angle). The SAXS images were recorded using a 2D Pilatus1M photon counting detector. The samples were contained in 2 mm borosilicate capillaries and left for 2 min to equilibrate at the appropriate temperature before the measurement was conducted. Measurements of pure THF and water, and the solvent mixtures without polymer, were measured and were used for background subtraction of the polymer-containing samples. Further details can be found in the ESI.†
Dynamic light scattering (DLS) analysis
Measurements on samples taken during the dialysis procedure were carried out using a Zeta-sizer Nano series (Nano-ZS) machine, supplied by Malvern Instruments. The z-average, number average and dispersity were measured at 35 °C for the dialysis samples and at 5 °C and 35 °C for the post-dialysis samples. Measurements were taken over a period of 90 seconds with an automatically generated number of scans, ranging from 12–19.Results and discussion
There are three major steps involved in the formation of the BPN dispersions: the water addition process, dialysis, and temperature decrease from 35 to 4 °C (Fig. 1A). Therefore, the following results will be addressed in the corresponding sections.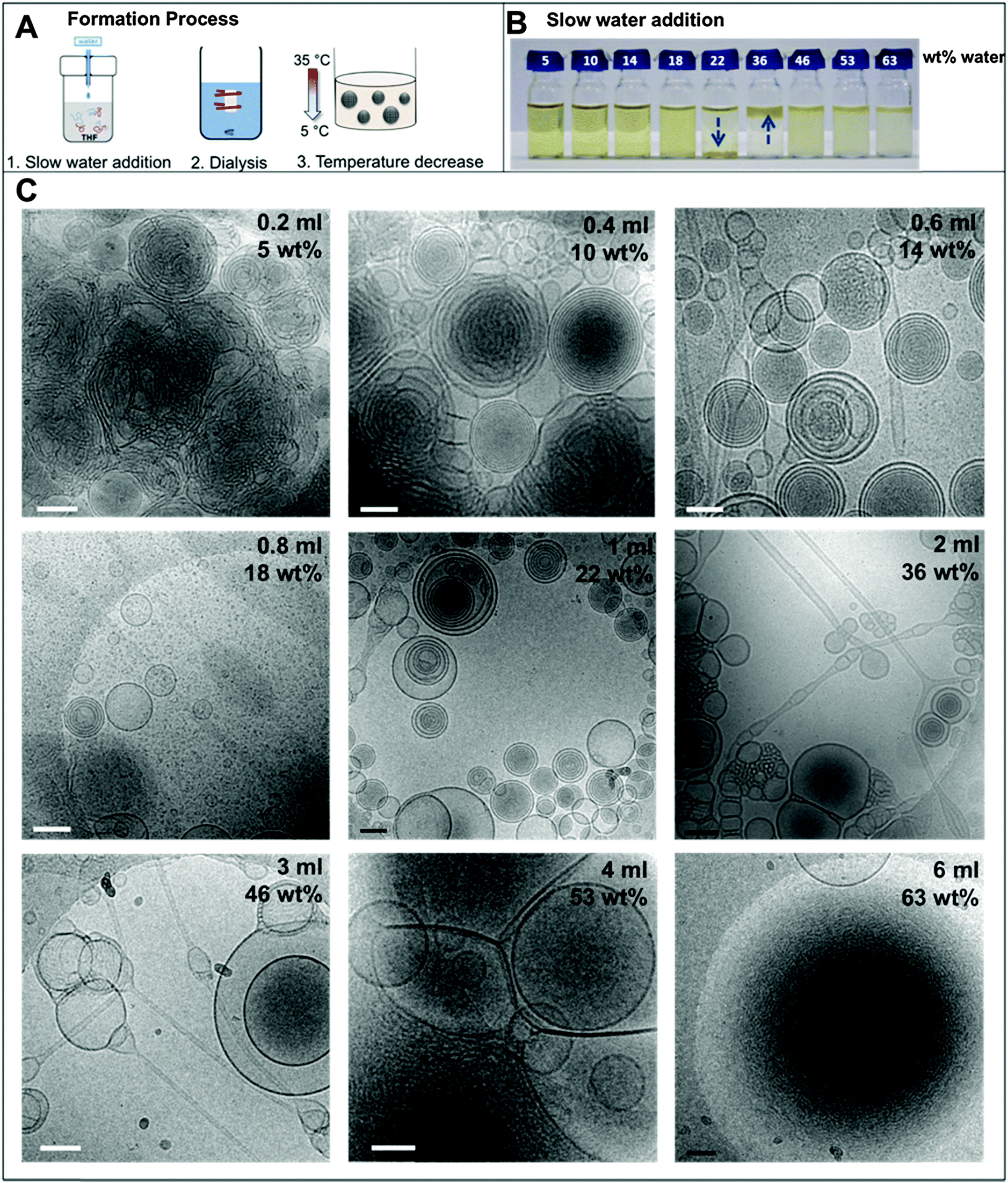 | ||
| Fig. 1 (A) Schematic of the complete formation process; (B) photograph of the individual THF–polymer–water solutions. The arrows highlight the position of the floc; (C) selected cryoTEM micrographs of the dispersions at different water contents vitrified at 35 °C, showing representative morphologies. Scale bars represent 200 nm. | ||
1. Water addition
In the nanoprecipitation method, the polymer is initially completely dissolved in the cosolvent and the aggregate formation is induced by the slow addition of water. The optimised addition rate was kept at 0.07 mL min−1; faster addition rates had previously been observed to lead to precipitation rather than dispersion for high water contents. The hydrophobic components aggregate to form a core segregated from the increasingly hydrophilic environment. To form the dispersions, PEO52-b-PODMA26 was dissolved in 4 mL THF and a predetermined volume of water was added slowly (0.067 mL min−1). The subsequent dispersions were analyzed by cryoTEM (Fig. 1C). A general decrease in sample densities with lower THF content was observed (Fig. 3C, left to right and down) that most likely results from the dilution of the sample with increased volumes of water. The effect of the concentration in THF–water mixtures on morphology was not investigated but previous work has demonstrated that the changing sample concentration has no effect on the final aggregate morphology only on aggregate size.47We observed that below 10 wt% water content, dense regions of ill-defined multi- and unilamellar vesicular structures formed. Increasing the water content (14 wt%) caused segregation of the aggregates into better-defined spherical multi- and unilamellar vesicles, and nanospheres with internal phase separation. Elongated tubular structures were also observed. At 18 wt%, a very polydisperse population of aggregates formed with sizes ranging from 10–400 nm, consisting of micelles and unilamellar vesicles. At 22 wt% water, the major morphology observed was multi-lamellar vesicles with membrane thicknesses of 17 ± 3 nm. Between 22 and 50 wt% water, the tubular structures reformed and complex structured connections between vesicles were observed. Some nanospheres exhibited internal phase separation, the morphology of which could not be ascertained. Between 53 and 63 wt% water, far fewer unilamellar vesicles were observed and the formation of large dense polymer aggregates of more than a micron in size were obtained. SAXS analysis of the multi-walled vesicles formed at 22 wt% water yielded spacings of 17 nm (Fig. S9, ESI†), which are in good agreement with values for membrane thickness obtained from the cryoTEM micrographs.
The appearance of the dispersions changed with water content from first being clear solutions (<18 wt% water) to the presence of floc phase separated from the bulk solution (22–36 wt% water) to becoming a milky dispersion (>36 wt%) (Fig. 1B). At 22 wt% water the floc remains at the bottom of the solution and then resides at the top when the water content reaches 36 wt%. The 22 and 36 wt% samples were homogenised before analysis. Unsuccessful attempts were made to perform SAXS upon the floc separately (if this was successful it would not have been verifiable by TEM because of the high polymer concentration). TEM/SAXS on the solutions revealed very few aggregates and insufficient scattering against the background, suggesting that the bulk of the copolymer resided in the floc. To investigate this variation in solvent quality the corresponding mixtures of THF/water in the absence of copolymer were separately analyzed by SAXS. These measurements revealed that the solvent mixtures contained features of 3 nm over the range 18 to 54% water with the scattering intensity for this feature increasing up to 36 wt% water and decreasing with higher water contents (Fig. 2). The presence of the features could not be confirmed by cryoTEM due to insufficient electron contrast between THF and water.
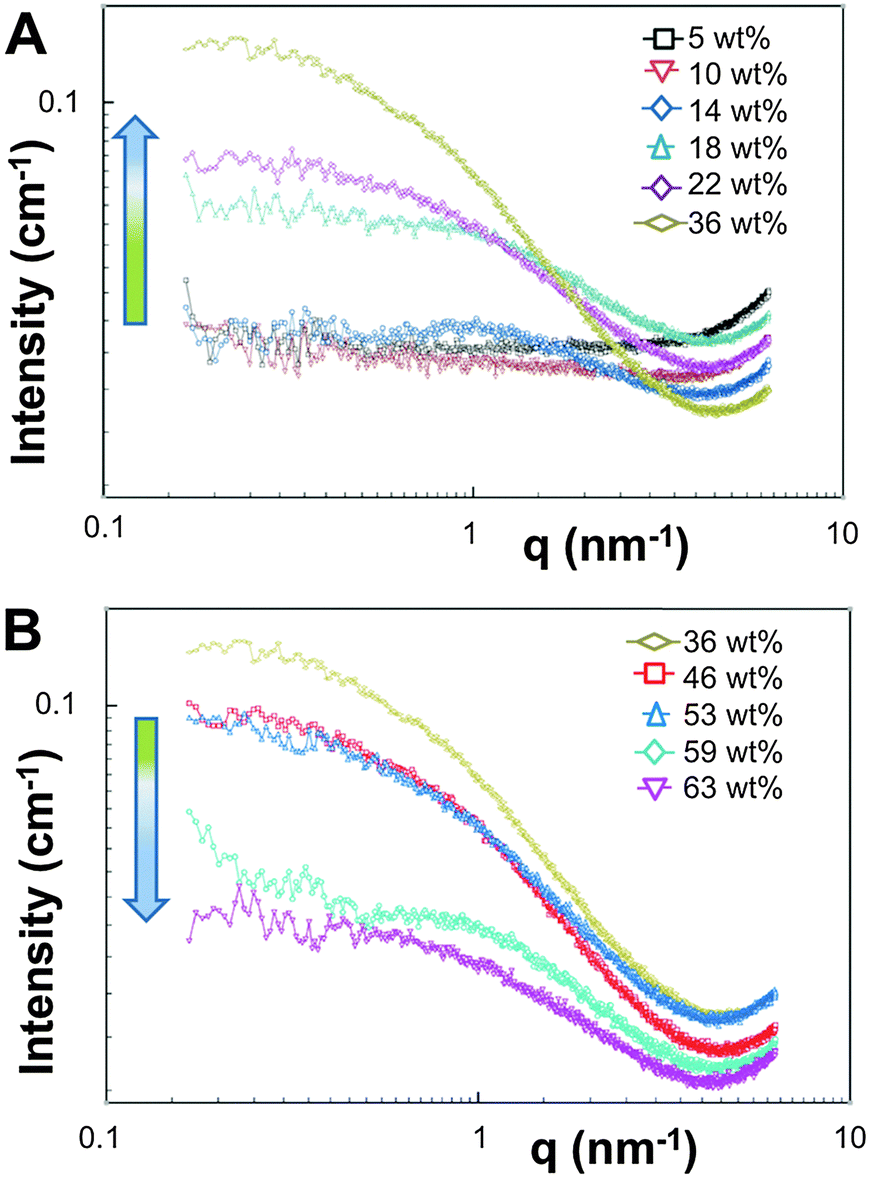 | ||
| Fig. 2 SAXS data recorded at 35 °C for THF–water mixtures without polymer from (A) 5–36 wt% water and (B) 36–63 wt% water. | ||
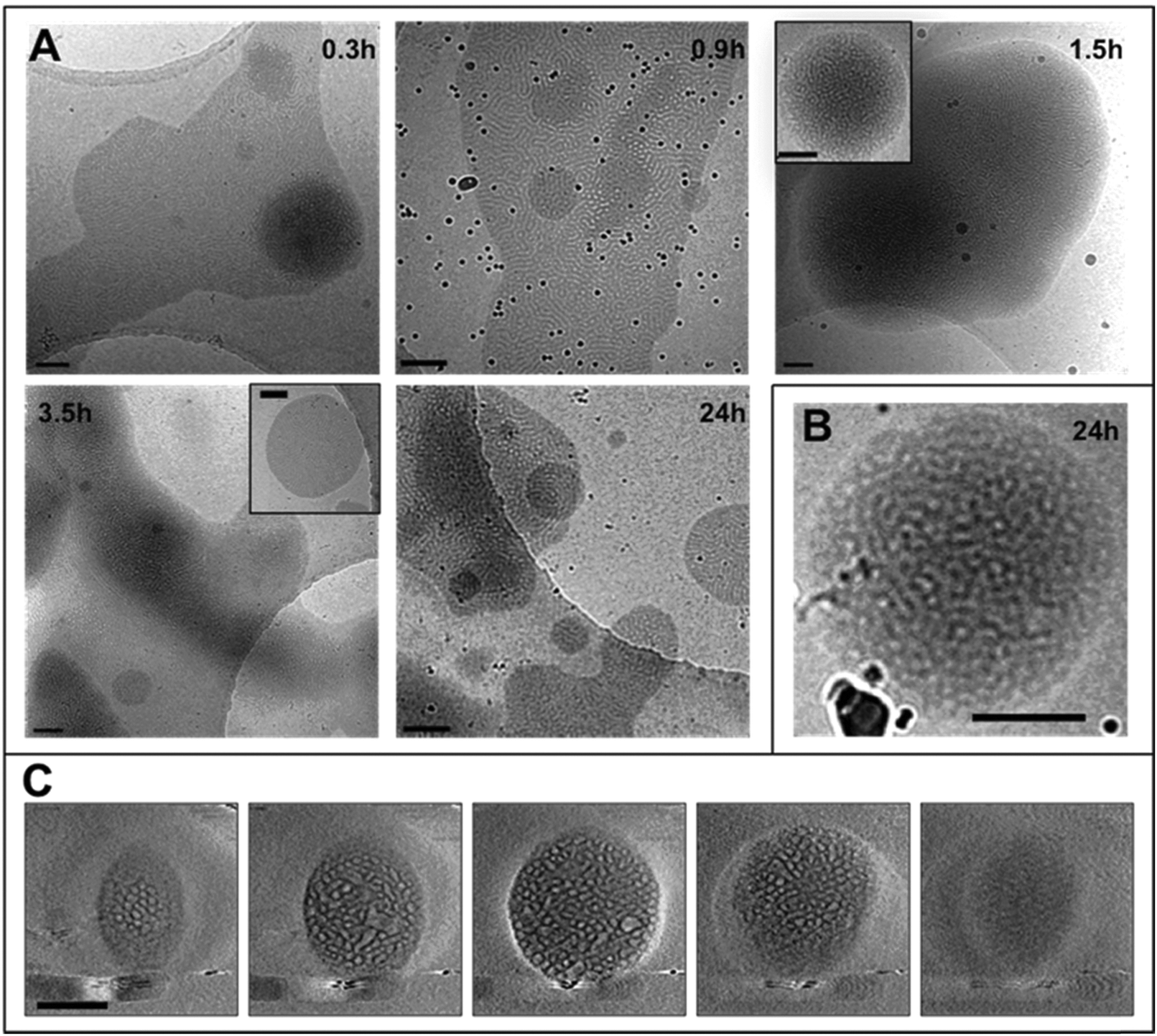 | ||
| Fig. 3 (A) CryoTEM micrographs of the dispersions at different times during the dialysis period, black spheres are gold nanoparticles used as fiducial markers; (B) cryoTEM image of a structured oblate spheroid from the dispersion at 24 h; (C) gallery of z-slices showing different cross sections of a 3D SIRT (simultaneous iterative reconstruction technique) reconstruction of a tomographic series recorded from the object shown in B. Scale bars represent 200 nm. | ||
The reason for this phenomenon, known as cononsolvency, is that at certain volume fractions small dynamic clusters of THF and water exist, forming a “new solvent” which is a non-solvent for the polymer.40,41 By increasing the content of one of the solvents the clusters disintegrate, and the polymer is again solvated, as the system again resembles the properties of a single solvent. The cononsolvency phenomenon has been well documented for poly(N-isopropylacrylamide) (PNIPAM) and its derivatives in water–alcohol/cycloether mixtures.42–44 In the present case, the floc that forms between 18 and 22 wt% water settles at the bottom of the vial and resides at the top of the vial at 36 wt% water (Fig. 1B). The change in position of the floc is in response to the changing density of the solution relative to the density of the block copolymer in its non-solvated state. The estimated density of the THF:water solvent mix changes from 0.910 g cm−3 at 22 wt% to 0.939 g cm−3 at 36 wt% whereas the estimated density of the PEO-b-PODMA is 0.936 g cm−3 (full details on density calculations and estimates are presented in the ESI†).45,46 Whilst these calculations have large degrees of error (due to uncertainties with regard to crystalline content, solvent content, and estimates from the original literature data), it is apparent that the change in the position of the floc is simply a reflection of the THF–water density change with composition. Nevertheless, the polymorphism exhibited reveals the beauty and complexity of the self-assembly of PEO-b-PODMA. Although all of the morphological intricacies are difficult to individually characterize, the observation can be made that the general morphology is lamellar. Surprisingly, the familiar BPNs were not observed at this stage but much later in the formation process.
2. Dialysis
The THF–polymer–water dispersion was placed in a dialysis membrane and dialysed against large volumes of water to remove the THF. Samples of the solution were taken between 0.3 and 24 h and vitrified at 35 °C for cryoTEM analysis (Fig. 3A). Already in the early stages of dialysis (0.3 h) the micrographs revealed that the unilamellar vesicular structures were no longer present. Instead, vast networks of microphase separated structures (‘fields’) with solvated PEO regions with diameters of 16 ± 4 nm were observed. Whilst these ‘fields’ of microphase separated PEO–PODA appear to be sheet-like in the cryoTEM images, variable contrast across the structures suggested they were of varying thickness (e.g.Fig. 1C at 6 mL and Fig. 1A at 3 h). Given that the temperature at which dialysis was conducted was above the Tm of the PODA block these ‘fields’ are likely to be fluid and therefore confinement in the liquid layer after blotting in the cryoTEM preparation may be artificially flattening these aggregates and giving them the appearance of sheets. DLS studies (Fig. 4a) later confirmed that these aggregates were very large in the bulk water dispersions, with apparent hydrodynamic diameters in excess of 10 microns. Thus the term ‘field’ will be used to describe these large aggregates. After 0.9 h of dialysis, the structured ‘fields’ begin to bud into rounded objects with diameter of 100–300 nm that possess the same internal structure as the fields. CryoET confirmed that the rounded aggregates were indeed flattened and not spherical (Fig. 3B and C), reminiscent of the oblate spheroids previously reported for PEO39-b-PODMA17 at 45 °C.24 The structured oblate spheroids are smaller isolations of the large structured fields. The thickness of the PODMA domains as measured by FFT of the cryoTEM micrographs from an average of 50 measurements is 21 ± 4 nm (Fig. S6), which is in good agreement with the SAXS data (22 ± 2 nm) (Fig. S9, ESI.†). To confirm this observation, dynamic light scattering (DLS) analysis was conducted on the solutions. This revealed the sample to be highly polydisperse with at least two distinct size ranges of aggregate after 0.3 h of dialysis; namely, very large structures with sizes often larger than the upper analysis limits of the DLS instrument, alongside a population of smaller aggregates. The z-average was 10400 (±8400) nm, and for the smaller aggregates CONTIN analysis gave a size distribution of 86 (±60) nm. Analysis of the larger distribution was not possible for the aforementioned reason. The resulting correlation functions for the dialysis samples are shown in Fig. 4A. During the course of the dialysis at 35 °C it can be seen that the distribution of the sample gradually decreases with the larger aggregates disappearing, until after 24 hours the z-average distribution has decreased to 610 (±45) nm. CONTIN analysis indicates that two species exist with the predominant one having a number average size of 66 (±6) nm and a significantly smaller fraction with a number average size of 410 (±37) nm (Fig. 4B).
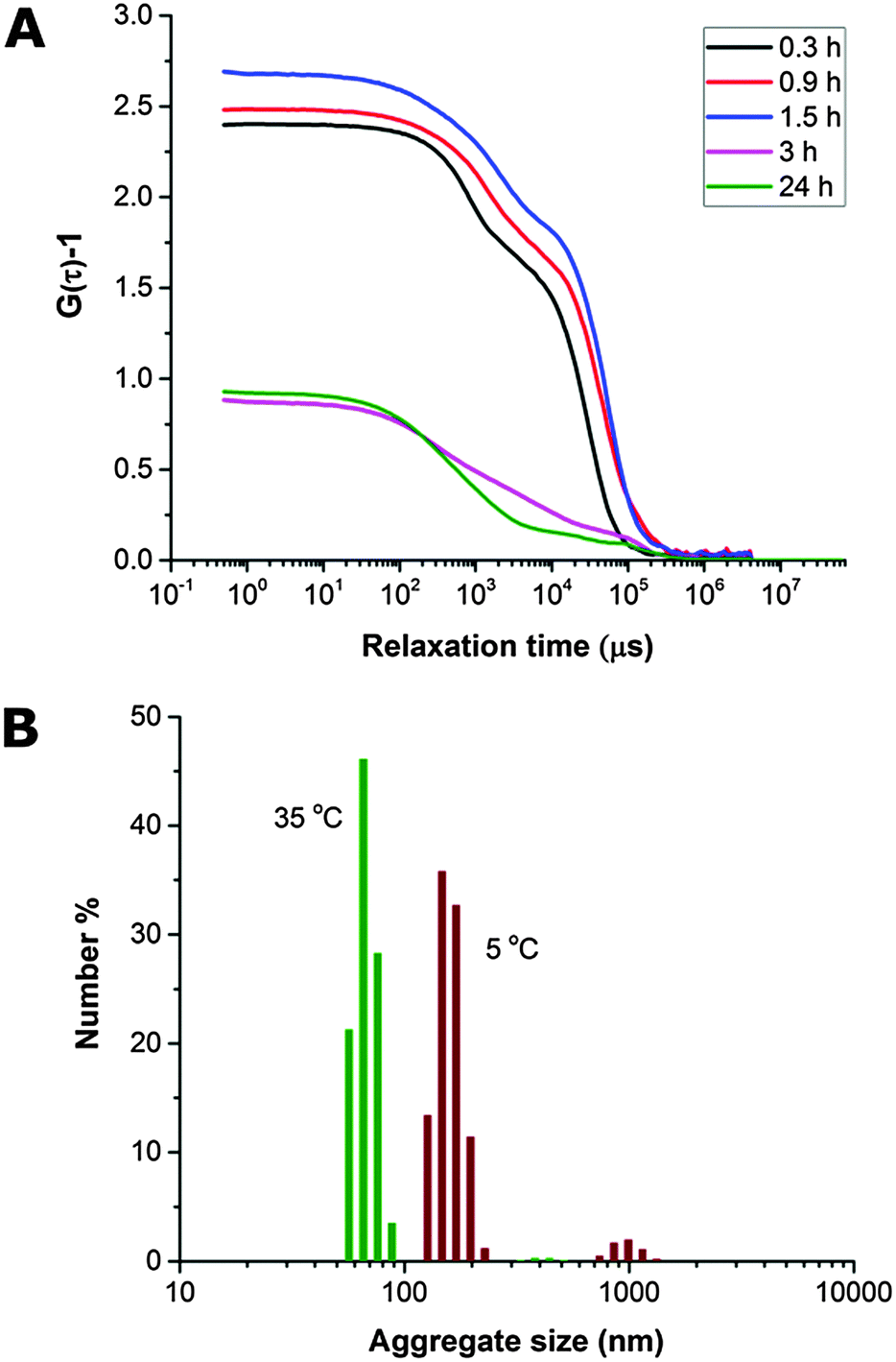 | ||
| Fig. 4 (A) Correlograms illustrating change in correlation function with time during dialysis of PEO-b-PODMA THF/water dispersions, suggesting a decrease in particle size. (B) Number average particle size distributions (CONTIN analysis) for PEO-b-PODMA dispersion after 24 h of dialysis (THF removed) at 35 °C and after cooling to 5 °C. | ||
3. Temperature decrease
The dialyzed solution was cooled stepwise from 35 °C to 5 °C (Fig. 5). CryoTEM analysis of the effect of decreasing the temperature revealed the following: above 25 °C the structured films formed during dialysis persist (Fig. 5A). At 25 °C, the films begin to break into strands and the structure of the films becomes less well defined. The familiar BPNs are first observed at 20 °C and coexist with the strands. At and below 10 °C, the strands are no longer visible and only the structured nanospheres remain. Previously reported multi-lamellar “onion” vesicles47 with structured cores and nanospheres with “pearl” membranes were also present, but the major morphology was bicontinuous (Fig. 5B). DLS analysis demonstrated that this transition was accompanied by an increase in the overall aggregate size (Fig. 4B). Whether or not this increase in size is a consequence of crystallization of the PODMA or is the result of Ostwald ripening is at the moment unknown. When the temperature was again increased the nanospheres were lost and the structured fields and oblate spheroids were reformed, demonstrating the reversibility of the system (Fig. 5C).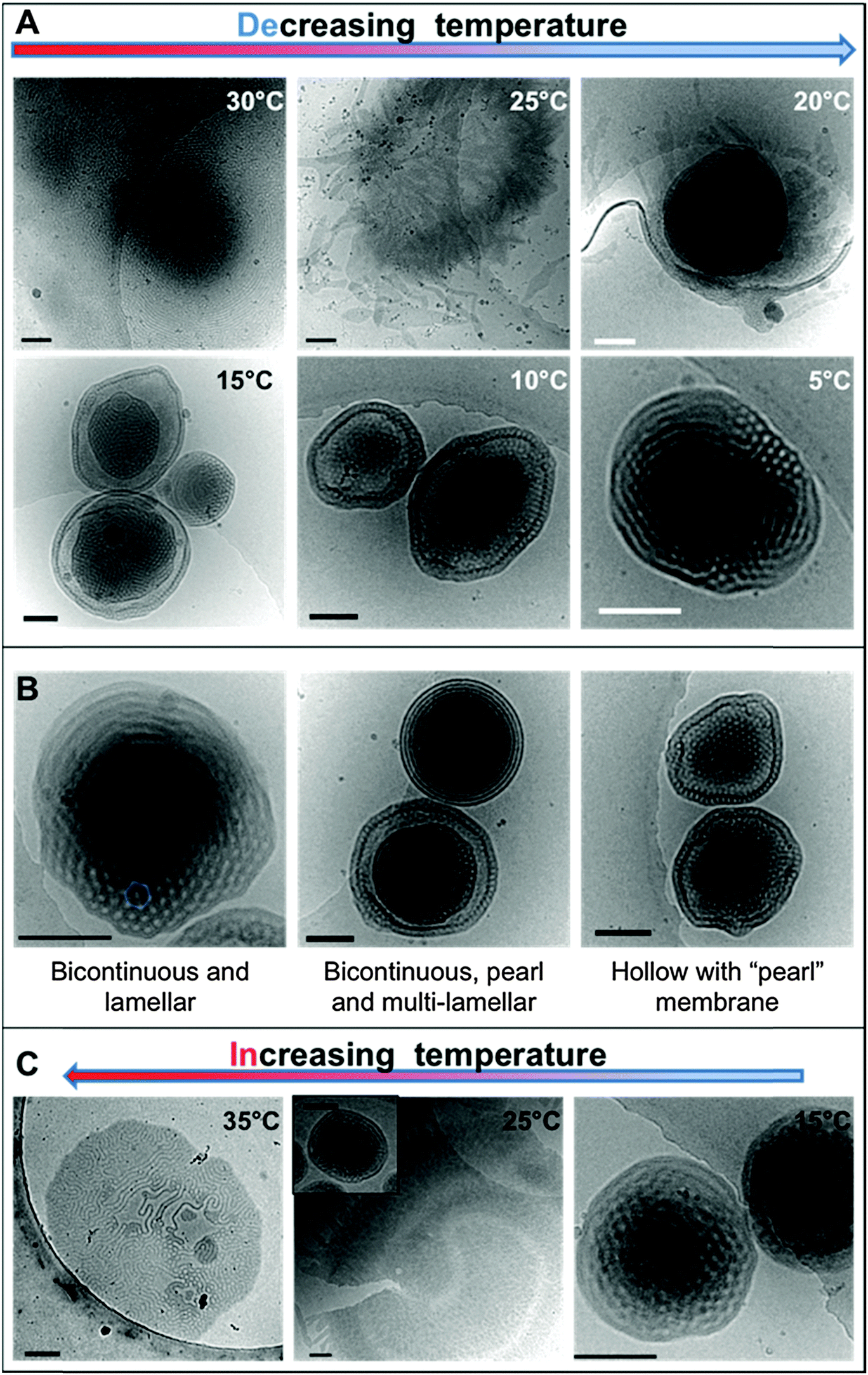 | ||
| Fig. 5 CryoTEM micrographs of the dispersions with (A) decreasing temperature and (C) increasing temperature. Images in (B) are taken from vitrified films of the dialyzed sample at 5 °C, exemplifying the different morphologies present in the dispersion. Scale bars represent 200 nm. | ||
Temperature-resolved SAXS measurements revealed that the morphological transition is accompanied by changes in the ordering and packing of the polymer chains from a disordered state to a more ordered arrangement (Fig. 6). Measurements conducted on the bulk material revealed the bicontinuous cubic phase lm3m with a lattice spacing of 15.1 nm (Fig. 6A). In temperature-resolved measurements, the reflections corresponding to this morphology (observed between 0.6–1.7 nm−1) were most prominent above 25 °C (above the Tc of the PODMA block). Below 25 °C, the intensity of the lm3m reflections becomes weaker; however, the broad peak at q ∼ 2.7 nm−1 sharpens and a secondary peak at 4.4 nm−1 can be seen. This confirmed the crystallization of the PODMA side chains with a spacing of 3 nm between polymer molecule back-bones. This proved significant, as the same peak was observed when temperature-resolved SAXS measurements were conducted on the nanospheres in aqueous solution (Fig. 6D). Assuming a fully extended side chain, the spacing between the main chain methacrylate backbones of adjacent polymer molecules would be 7 nm. This therefore suggests that in solution, the side chain of adjacent molecules interdigitate as the transition from large fields and spheroids to BPNs is made (at 25 °C and below, Fig. 6C).
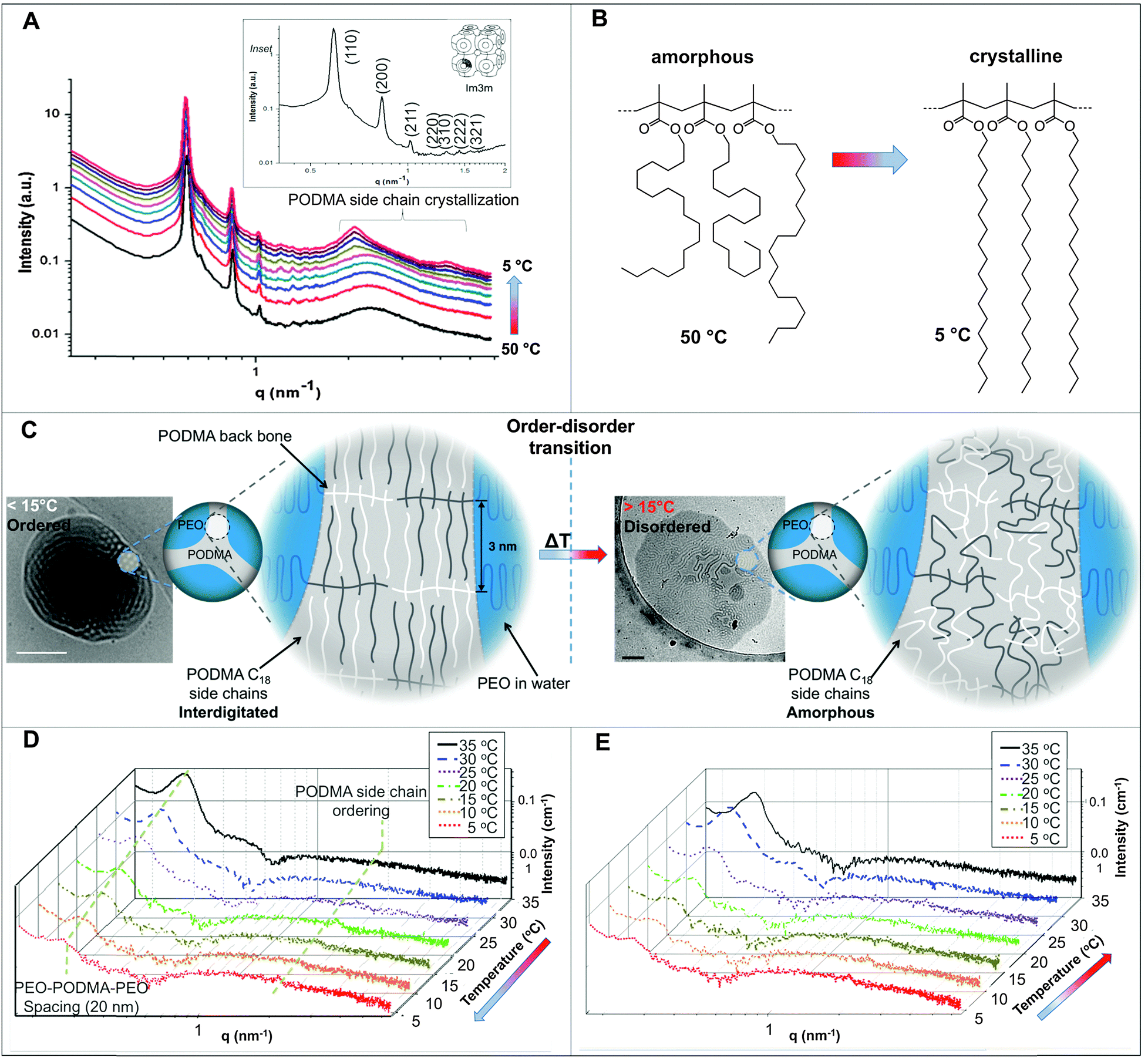 | ||
| Fig. 6 (A) Temperature-resolved SAXS of the bulk material revealing the evolution of peaks corresponding to PODMA side chain crystallization between 2–5 nm−1; inset is an expansion of the q range 0–1.5 nm−1, showing reflections of the lm3m phase in the bulk PEO-b-PODMA at 30 °C. (B) representation of the change in the PODMA side chains at 50 and 5 °C. Temperature-resolved SAXS for the aqueous dispersion with (C) decreasing temperature and (D) increasing the temperature again. (D) Schematic representation of the change in polymer molecule arrangement with temperature and the corresponding cryoTEM micrographs. Scale bars represent 200 nm. | ||
Discussion
To our knowledge, this is the first demonstration of the (experimental) formation mechanism of complex nanospheres in solution. Following the process in this manner makes it evident that the change in temperature contributes significantly to the changes in morphology. The coexistence of vesicular structures and vesicular nodules is often reported in the formation of lipid cubosomes,17,48,49 which suggests that the phase forms through their coalescence. Evidence for a similar mechanism can be seen in the cryoTEM micrographs of dispersions with 36–46 wt% content, where several vesicles are conjoined by “bicontinuous” connections (Fig. 1C).Most significantly, the water addition procedure does not trap the morphologies, as reported previously for vesicle systems.2 In fact the BPNs are not even observed after the water addition process but only after the complete removal of THF by dialysis followed by a decrease in temperature. Thus, it appears that the gradual addition of water to the THF solution plays no significant part in the actual evolution of the BPNs themselves. A possible role may be to prevent the formation of lamellar structures (such as vesicles and/or onion-like multi-lamellar aggregates) and it would be interesting to study the effect of different solvents on this aspect of the process. For instance, the use of dioxane in place of THF results in the formation of such structures for PEO-b-PBMA block copolymers.35 Furthermore, although the cononsolvency of the polymer occurs concomitantly it does not appear to have an effect upon the BPN formation process itself. PEO (the stabilizing block) is soluble in both THF and water, but the block copolymer precipitates in the form of a floc in the “new solvent” between 22 and 36 wt% water, which suggests the possible cononsolvency of PEO in the THF–water mixtures. This is under further investigation. Nevertheless, it is ultimately the removal of THF during dialysis that appears to lead to the initial formation of discrete aggregates. Whether discrete aggregates would develop in the absence of THF is uncertain. It is important to note that whilst dispersions of this polymer cannot be formed by direct dissolution into water, even at elevated temperatures they can be dispersed directly into water at 40 °C by the use of a homogenizer. This is akin to the formation of bicontinuous lipid cubosomes from cubic liquid crystalline precursors.50,51 It therefore appears that the principle role of the THF is to allow for dispersion of the copolymer into an aqueous solution facilitating the formation of microphase separated fields that upon cooling break down and form discrete aggregates. Overall, the bicontinuous morphology is locally thermodynamically stable and certainly kinetically stable with respect to the disordered lamellar structure of the fields, as is exemplified in the reproducible reversibility of the morphological transition.
In the bulk material, the bicontinuous structure is well defined at elevated temperatures (>20 °C) as was resolved by SAXS (Fig. 6A). When the temperature is decreased below the polymer Tc (10 °C), the lm3m peaks become less defined and the broad diffuse peak at 2.5 nm−1 sharpens, with an additional peak appearing at 4 nm−1. These correspond to the 1st and 2nd order crystallization of the PODMA side chains respectively, where the initial 1st order process involves crystallization of the chain ends parallel to the backbone. The 2nd order crystallization occurs by coarsening of the crystallites perpendicular to the backbone. The side chain crystallization disturbs the bicontinuous organization of the bulk material, and so the corresponding peaks are broader and less well-defined. The peak at 2.5 nm−1 corresponds to a spacing (from one PODMA backbone to another) of 3 nm, and shows that the side chains are interdigitated, confirming previous reports on the crystallization behavior of the comb-like polymer.52 Measurement of the bicontinuous dispersion in a similar temperature-resolved manner does not reveal the same lm3m reflections (Fig. 6D and E), although the internal connectivity of the nanospheres is established by cryoET. The bicontinuity of the nanospheres in solution is therefore more disordered than is found in the bulk structure and more disordered than the structure often reported in lipid cubosomes27,50 and other bicontinuous nanospheres.30 However, what does persist is the ordering within the PODMA regions below 25 °C, which is ascertained by the (re)appearance of the peak at 2.7 nm−1 (although with lower intensity than in the bulk). The geometric transformation from fluid fields to spheres is accompanied by alterations in the PODMA packing that are induced by traversing the order–disorder transition at the melting temperature of the PODMA block (Fig. 6C).24 It is probable that extensive crystallization is inhibited by the geometric constraints imposed on the chains in the sphere.
Conclusions
Combining cryoTEM and SAXS analysis reveals that bicontinuous polymeric nanospheres of PEO-b-PODMA form in initial THF–water mixtures from the coalescence of preliminary lamellar phases. The BPNs are not formed by the addition of water to the THF–polymer solution but are the result of a transition from irregular dispersed fields of microphase separated fluid copolymer into discrete aggregates during dialysis of the THF solution. The process is accompanied by precipitation and re-dispersion of the block copolymer, which although does not affect the BPN formation, it may be a result of the cononsolvency of PEO in THF–water mixtures, and is under further investigation. The bicontinuous morphology of the BPNs arises from a molecular rearrangement driven by the temperature-responsive nature of the hydrophobic PODMA block upon cooling. Thus, the system presents significant possibilities for application as a temperature-responsive release mechanism. It is evident that careful selection of the solvent is still necessary for the formation of porous morphologies, and applying this concept presents exciting prospects for the design of BPNs from different block copolymers. The work presented here illustrates how combining the morphological evidence provided by cryoTEM with the nanoscale structural information provided by SAXS reveals the interim processes involved in self-assembly. In a broader sense, the analysis of the system in its native solvated state as presented here reveals that more complex transitions occur during the formation of the block copolymer aggregates than first thought. It is likely that similar complex transition sequences are playing a role in other amphiphilic polymer systems, and that the mechanisms of formation of some of such systems may need to be revisited.Competing financial interest
The authors declare no competing financial interest.Acknowledgements
Portions of this work were performed at the Dutch Belgian Beamline (DUBBLE) BM6 at the European Synchrotron Radiation Facility (ESRF) in Grenoble, France. The authors would also like to thank Dr Christopher Silles from the School of Computing at the University of Kent for assistance with computer artwork.Notes and references
- A. Blanazs, S. Armes and A. Ryan, Macromol. Rapid Commun., 2009, 30, 267 CrossRef CAS PubMed.
- D. E. Discher and A. Eisenberg, Science, 2002, 297, 967 CrossRef CAS PubMed.
- A. O. Moughton, M. A. Hillmyer and T. P. Lodge, Macromolecules, 2011, 45, 2 CrossRef.
- J.-N. l. Marsat, M. Heydenreich, E. Kleinpeter, H. v. Berlepsch, C. Böttcher and A. Laschewsky, Macromolecules, 2011, 44, 2092 CrossRef CAS.
- N. Saito, C. Liu, T. P. Lodge and M. A. Hillmyer, Macromolecules, 2008, 41, 8815 CrossRef CAS.
- R. Erhardt, M. Zhang, A. Böker, H. Zettl, C. Abetz, P. Frederik, G. Krausch, V. Abetz and A. H. E. Müller, J. Am. Chem. Soc., 2003, 125, 3260 CrossRef CAS PubMed.
- A. Walther and A. H. E. Muller, Soft Matter, 2008, 4, 663 RSC.
- N. A. J. M. Sommerdijk, S. J. Holder, R. C. Hiorns, R. G. Jones and R. J. M. Nolte, Macromolecules, 2000, 33, 8289 CrossRef CAS.
- J. J. L. M. Cornelissen, M. Fischer, N. A. J. M. Sommerdijk and R. J. M. Nolte, Science, 1998, 280, 1427 CrossRef CAS.
- J. Dupont, G. Liu, K.-i. Niihara, R. Kimoto and H. Jinnai, Angew. Chem., Int. Ed., 2009, 48, 6144 CrossRef CAS PubMed.
- J. Zhu, S. Zhang, K. Zhang, X. Wang, J. W. Mays, K. L. Wooley and D. J. Pochan, Nat. Commun., 2013, 4, 2297 Search PubMed.
- Z. Li, Z. Chen, H. Cui, K. Hales, K. Qi, K. Wooley and D. Pochan, Langmuir, 2005, 21, 7533 CrossRef CAS PubMed.
- S. J. Holder and N. A. J. M. Sommerdijk, Polym. Chem., 2011, 2, 1018 RSC.
- T. P. Lodge, A. Rasdal, Z. Li and M. A. Hillmyer, J. Am. Chem. Soc., 2005, 127, 17608 CrossRef CAS PubMed.
- M. Marguet, C. Bonduelle and S. Lecommandoux, Chem. Soc. Rev., 2013, 42, 512 RSC.
- G. Garg and S. Saraf, Biol. Pharm. Bull., 2007, 30, 350 CAS.
- J. Barauskas, M. Johnsson and F. Tiberg, Nano Lett., 2005, 5, 1615 CrossRef CAS PubMed.
- J. Gustafsson, H. Ljusberg-Wahren, M. Almgren and K. Larsson, Langmuir, 1997, 13, 6964 CrossRef CAS.
- A. Angelova, B. Angelov, B. Papahadjopoulos-Sternberg, M. Ollivon and C. Bourgaux, Langmuir, 2005, 21, 4138 CrossRef CAS PubMed.
- Y.-D. Dong, I. Larson, T. Hanley and B. J. Boyd, Langmuir, 2006, 22, 9512 CrossRef CAS PubMed.
- B. Smarsly and M. Antonietti, Eur. J. Inorg. Chem., 2006, 1111 CrossRef CAS.
- Z. Li, K. Hur, H. Sai, T. Higuchi, A. Takahara, H. Jinnai, S. M. Gruner and U. Wiesner, Nat. Commun., 2014, 5, 3247 Search PubMed.
- Y. La, C. Park, T. J. Shin, S. H. Joo, S. Kang and K. T. Kim, Nat. Chem., 2014, 6, 534 CrossRef CAS PubMed.
- B. E. McKenzie, F. Nudelman, P. H. H. Bomans, S. J. Holder and N. A. J. M. Sommerdijk, J. Am. Chem. Soc., 2010, 132, 10256 CrossRef CAS PubMed.
- S. J. Holder, G. Woodward, B. McKenzie and N. A. J. M. Sommerdijk, RSC Adv., 2014, 4, 26354 RSC.
- A. G. Denkova, P. H. H. Bomans, M. O. Coppens, N. A. J. M. Sommerdijk and E. Mendes, Soft Matter, 2011, 7, 6622 RSC.
- U. Mansfeld, S. Hoeppener, K. Kempe, J.-M. Schumers, J.-F. Gohy and U. S. Schubert, Soft Matter, 2013, 9, 5966 RSC.
- K. Hales, Z. Chen, K. Wooley and D. Pochan, Nano Lett., 2008, 8, 2023 CrossRef CAS PubMed.
- A. L. Parry, P. H. H. Bomans, S. J. Holder, N. A. J. M. Sommerdijk and S. C. G. Biagini, Angew. Chem., Int. Ed., 2008, 47, 8751 CrossRef.
- T. H. An, Y. La, A. Cho, M. G. Jeong, T. J. Shin, C. Park and K. T. Kim, ACS Nano, 2015, 9, 3084 CrossRef CAS PubMed.
- Y. La, T. H. An, T. J. Shin, C. Park and K. T. Kim, Angew. Chem., Int. Ed., 2015, 54, 10483 CrossRef CAS PubMed.
- V. Percec, D. A. Wilson, P. Leowanawat, C. J. Wilson, A. D. Hughes, M. S. Kaucher, D. A. Hammer, D. H. Levine, A. J. Kim, F. S. Bates, K. P. Davis, T. P. Lodge, M. L. Klein, R. H. DeVane, E. Aqad, B. M. Rosen, A. O. Argintaru, M. J. Sienkowska, K. Rissanen, S. Nummelin and J. Ropponen, Science, 2010, 328, 1009 CrossRef CAS PubMed.
- K. Yu, L. Zhang and A. Eisenberg, Langmuir, 1996, 12, 5980 CrossRef CAS.
- E. Karjalainen, N. Chenna, P. Laurinmaki, S. J. Butcher and H. Tenhu, Polym. Chem., 2013, 4, 1014 RSC.
- B. E. McKenzie, J. F. de Visser, H. Friedrich, M. J. M. Wirix, P. H. H. Bomans, G. de With, S. J. Holder and N. A. J. M. Sommerdijk, Macromolecules, 2013, 46, 9845 CrossRef CAS.
- I. C. Riegel, A. Eisenberg, C. L. Petzhold and D. Samios, Langmuir, 2002, 18, 3358 CrossRef CAS.
- B. E. McKenzie, S. J. Holder and N. A. J. M. Sommerdijk, Curr. Opin. Colloid Interface Sci., 2012, 17, 343 CrossRef.
- F. Nudelman, G. de With and N. A. J. M. Sommerdijk, Soft Matter, 2011, 7, 17 RSC.
- H. Friedrich, P. M. Frederik, G. de With and N. A. J. M. Sommerdijk, Angew. Chem., Int. Ed., 2010, 49, 7850 CrossRef CAS PubMed.
- J. Hao, H. Cheng, P. Butler, L. Zhang and C. C. Han, J. Chem. Phys., 2010, 132, 154902 CrossRef PubMed.
- C. Yang, W. Li and C. Wu, J. Phys. Chem. B, 2004, 108, 11866 CrossRef CAS.
- F. M. Winnik, M. F. Ottaviani, S. H. Bossmann, W. Pan, M. Garcia-Garibay and N. J. Turro, Macromolecules, 1993, 26, 4577 CrossRef CAS.
- H. G. Schild, M. Muthukumar and D. A. Tirrell, Macromolecules, 1991, 24, 948 CrossRef CAS.
- K. Pagonis and G. Bokias, Polymer, 2004, 45, 2149 CrossRef CAS.
- A. Eliassi, H. Modarress and G. A. Mansoori, J. Chem. Eng. Data, 1998, 43, 719 CrossRef CAS.
- J. N. Nayak, M. I. Aralaguppi, B. V. Kumar Naidu and T. M. Aminabhavi, J. Chem. Eng. Data, 2004, 49, 468 CrossRef CAS.
- B. E. McKenzie, H. Friedrich, M. J. M. Wirix, J. F. de Visser, O. R. Monaghan, P. H. H. Bomans, F. Nudelman, S. J. Holder and N. A. J. M. Sommerdijk, Angew. Chem., Int. Ed., 2015, 54, 2457 CrossRef CAS PubMed.
- B. J. Boyd, S. B. Rizwan, Y.-D. Dong, S. Hook and T. Rades, Langmuir, 2007, 23, 12461 CrossRef CAS PubMed.
- L. E. Scriven, Nature, 1976, 263, 123 CrossRef CAS.
- M. Nakano, A. Sugita, H. Matsuoka and T. Handa, Langmuir, 2001, 17, 3917 CrossRef CAS.
- B. Siekmann, H. Bunjes, M. H. J. Koch and K. Westesen, Int. J. Pharm., 2002, 244, 33 CrossRef CAS PubMed.
- E. Hempel, H. Budde, S. Horing and M. Beiner, J. Non-Cryst. Solids, 2006, 352, 5013 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Materials and methods; details of block copolymer synthesis and characterization by 1H NMR and size exclusion chromatography; cryoTEM micrographs of PEO-b-PODMA THF–water dispersions; graph of PODMA domain thickness with increasing water content; graph of density with increasing alkyl side chain length; graph of changing solution density with increasing water content; SAXS data. See DOI: 10.1039/c6sm00053c |
| This journal is © The Royal Society of Chemistry 2016 |