
CO2 conversion by reverse water gas shift catalysis: comparison of catalysts, mechanisms and their consequences for CO2 conversion to liquid fuels
Yolanda A. Daza and
John N. Kuhn*
Department of Chemical & Biomedical Engineering, University of South Florida, Tampa, FL 33620, USA. E-mail: jnkuhn@usf.edu; Tel: +1 813 974 6498
First published on 26th April 2016
Abstract
Current society is inherently based on liquid hydrocarbon fuel economies and seems to be so for the foreseeable future. Due to the low rates (photocatalysis) and high capital investments (solar-thermo-chemical cycles) of competing technologies, reverse water gas shift (rWGS) catalysis appears as the prominent technology for converting CO2 to CO, which can then be converted via CO hydrogenation to a liquid fuel of choice (diesel, gasoline, and alcohols). This approach has the advantage of high rates, selectivity, and technological readiness, but requires renewable hydrogen generation from direct (photocatalysis) or indirect (electricity and electrolysis) sources. The goal of this review is to examine the literature on rWGS catalyst types, catalyst mechanisms, and the implications of their use CO2 conversion processes in the future.
1. Introduction
1.1 CO2 availability and current utilization
Recently, the global carbon dioxide atmospheric concentration reached a threshold of 400 ppm, increasing the average world temperature prior to the industrial revolution by 1.5 °C. In 2013, 32.19 gigatonnes (Gt) of CO2 was emitted into the atmosphere,1 and the emissions are expected to increase to 45 Gt per year by 2040. Approximately 22% and 33% of the yearly anthropogenic emissions are absorbed into the oceans and plants, respectively, in the natural photosynthesis cycle, with the remaining 45% contributing to the increasing atmospheric concentrations.2 A drawback with oceanic CO2 absorption is that the gas is not absorbed evenly, but rather 40% of absorption occurs in the Southern Ocean.3 By 2030, the acidification of this Ocean would likely have palpable consequences on its native organisms, which could potentially affect the food web of the area.4 The rapidly increasing atmospheric CO2 concentration and the threat it poses upon the environment has led to increased efforts to reduce or minimize CO2 atmospheric emissions. Among the most widely used approaches is Carbon Capture and Storage (CCS), more commonly called sequestration. Even though the Global CCS Institute estimates that the “large” projects (>0.8 Mt – mega tonnes – for coal-based power plants or >0.4 Mt for other industrial facilities), under evaluation could potentially have a sequestration capacity of 81.5 Mt of CO2/year;5 the actual operational projects only reach a 28.4 Mt sequestration capacity.6 Furthermore, current CO2 utilizations for industrial processes, such as urea and salicylic acid synthesis (Fig. 1), do not exceed 120 Mt per year.7,8 Production of CO2 is more than 150 times higher than its current use and potential sequestration capability (Table 1, current methods). Due to its large-scale, long-term planning of a combination of methods and technologies at all levels of society from industry to individual households, sequestration should be used if we are to significantly reduce CO2 emissions or manufacture it into fuels and chemicals.5,7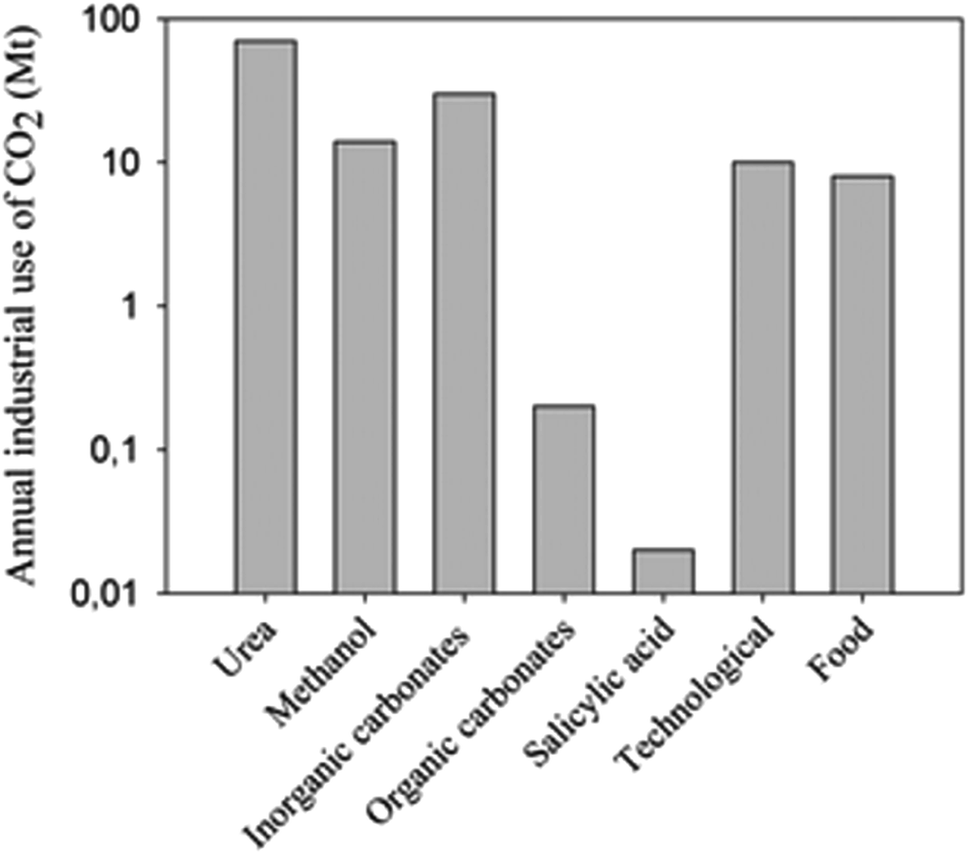 | ||
| Fig. 1 CO2 use in industry. Vertical axis is on logarithmic scale. Reproduced with permission from the Royal Society of Chemistry (from ref. 8). | ||
| Technique | Capacity of CO2 reduction (mega tonnes CO2 per year) | Reduction of total emissionsa | Reduction of atmospheric CO2 influxb | |
|---|---|---|---|---|
| a Calculated using 2013 total emissions as 32.19 gigatonnes per year.1b Calculated using 14.46 gigatonnes CO2 per year absorbed by the atmosphere (45% (ref. 2) of total 2013 emissions).c Estimated from the technology of Job et al.14 and plastics global demand from ref. 15.d In accordance with ref. 16.e Assuming all gasoline as C8H18 with a global demand of 94.83 million barrels per day (ref. 23) and a gallon yield of 45% v/v gasoline.24 | ||||
| Current methods | Sequestration | 81.5 (ref. 5) | 0.25% | 0.56% |
| Fine chemicals synthesis | 120 (ref. 8) | 0.37% | 0.83% | |
| Potential uses | Plastics | 155.5c | 0.48% | 1.07% |
| Methanol | 89.4 (ref. 22) | 0.28%d | 0.62% | |
| Oil derived chemicals | 1200 (ref. 17) | 3.73% | 8.28% | |
| Gasoline | 5364.6e | 16.67% | 37.03% | |
Recently, a variety of technologies for repurposing the vastly abundant carbon dioxide into high value chemicals have emerged. To fulfill the ultimate resolution of environmental remediation, these technologies should be renewable, and the overall process needs to be carbon neutral or negative. Considering the limited sequestration capacity and the costs of CO2 transportation and storage (∼$16.5 per tonne CO2 (ref. 9)), developing technologies for Carbon Capture and Utilization (CCU) may make more sense than simply sequestering CO2. However, the stability of the molecule is another challenge to overcome. CO2 is a very stable form of carbon, making its transformation very energy intensive.
Technologies currently under research to transform CO2 to chemicals of wide use include synthesis of polymers,7 oxalates,10 formates,11 dimethyl ether,12 ethylene and propylene13 and an interesting recently developed technology by Job et al.14 that recycles CO2 into plastics similar to polyurethane (up to 50% CO2 by weight). However, even at the high global demand for plastics (311 Mt in 2014 (ref. 15)), we estimate that less than 0.5% of CO2 emissions would be used even if all the plastic produced in the world was synthesized with this technology (Table 1). Similarly, if all the methanol16 and chemicals (made from oil)17 consumed globally were synthesized from CO2, emissions would not decrease by more than 0.3% and 3.8%, respectively. The comparisons of these values vividly capture the challenge of scale. The key factors of utilization still remain an issue: (i) the need for concentrated CO2 (ref. 18 and 19) and (ii) proven technologies for conversion that can match the scale of CO2 production and produce chemicals of significantly high demand.18–21
1.2 Need for energy-dense transportation fuels
In a worldwide effort to increase environmental friendliness, the use of alternative renewable technologies (such as solar, wind, geothermal, and nuclear) has been steadily increasing and evolved from representing 2.8% of the world energy production in 1973 to 8.4% in 2013.1 The limitation is that these renewable energy sources are mostly used to generate electricity, and in 2013, electricity only represented 18.0% of the global energy consumption.1 Renewables went from representing 32.0% of all the electricity generated in 2011 (ref. 25) to 32.6% in 2013.1 Unfortunately, due to intermittent supply, until new methods for efficiently storing energy generated by alternate renewable sources are developed, energy dense hydrocarbon fuels, currently produced primarily from oil, will still be necessary. Hydrocarbons store substantial chemical energy, which is not possible through various transient processes until batteries or other replacement technologies become viable.Oil represents about 40% of the world energy consumption, and in 2013, 63.8% of all oil products were used to make transportation fuels.1 The amount of oil products that was used to make transportation fuels increased by 44.48 Mtoe (million tonnes of oil equivalent) from 2012 (ref. 26) to 2013.1 The demand for fuels is at least 100 times larger than chemicals.27 Thus, only liquid fuel demand (Table 1, gasoline as example) rivals the scale of CO2 production.19,28,29 In other words, CO2 emissions will continue to outweigh CO2 consumption unless hydrocarbon transportation fuels are produced from CO2 (closed cycle) or they are no longer required. To date, no other type of energy storage vehicle has been able to outrank the practicality of liquid fuels, making energy dense fuels still necessary.30,31 In addition, a world-wide infrastructure for the delivery of liquid hydrocarbon fuels already exists. This avoids a major issue of the H2 economy.
1.3 Cost estimations for CO2 conversion processes
The need for renewable hydrogen poses a crucial problem for using the carbon of CO2 as the backbone of future fuels.32–37 With a minimum levelised cost of renewable electricity (produced by solar towers) of 0.17 USD per kW per h,38 the cost of H2 could be estimated at ∼10 USD per kg H2 (ref. 39) (as opposed to ∼1.6 USD/kg H2 if electricity was not renewable40). This means that if renewable H2 was used to make one GGE of methanol, its selling price would increase by at least 4.43 USD/GGE. More recently, Kim et al. compared the cost of producing methanol with CO2 splitting and different methods for obtaining H2, one from WGS (using water and CO obtained from CO2 splitting)41 and the other through H2O thermochemical splitting to H2.42 They determined that thermochemical splitting of H2O to obtain H2 would allow for a minimal selling point of methanol at 6.73 USD/GGE42 vs. using WGS, which would produce methanol at a minimum selling point of 7.10 USD/GGE.41 Based on these back-of-the-envelope calculations, we estimate that production of renewable H2 would contribute about 65% (4.43 USD/GGE × 100/6.73 USD/GGE) of the total methanol cost. It becomes evident that renewable H2 synthesis is still a technology in development.431.4 Green technologies for CO2 conversion to fuels with large demand
The technologies with the highest readiness level that are focused on converting CO2 to synthetic fuels or their precursors (i.e. CO) are (i) rWGS reaction, (ii) syngas synthesis from methane dry reforming (DR) and (iii) direct hydrogenation of CO2.Approximately 35 megatonnes of CH4 per year are emitted to the atmosphere from landfills.44 If instead, this gas was trapped, it could be reacted with CO2 in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 feed to produce syngas through dry reforming. Even though methane is produced at a much lower scale than CO2 emissions, its use could be advantageous because it is produced naturally. Nonetheless, DR is an endothermic reaction,16 favored at high temperatures (>900 °C), at which catalysts sinter and coke.30 Often, landfill gas contains high levels of sulfur gases that cause catalyst deactivations.16 Low temperature DR has been reported (430–470 °C) with no coking, but using an assembly of noble and transition metal catalysts combined with metal oxides (Pt–Ni–Mg/ceria–zirconia catalysts45) which has not yet been studied for sulfur poisoning.
:1 feed to produce syngas through dry reforming. Even though methane is produced at a much lower scale than CO2 emissions, its use could be advantageous because it is produced naturally. Nonetheless, DR is an endothermic reaction,16 favored at high temperatures (>900 °C), at which catalysts sinter and coke.30 Often, landfill gas contains high levels of sulfur gases that cause catalyst deactivations.16 Low temperature DR has been reported (430–470 °C) with no coking, but using an assembly of noble and transition metal catalysts combined with metal oxides (Pt–Ni–Mg/ceria–zirconia catalysts45) which has not yet been studied for sulfur poisoning.
Direct CO2 hydrogenation is more thermodynamically favored than rWGS. Therefore, it was considered promising for industrialized methanol synthesis46 and has been demonstrated on a pilot scale in Iceland by George Olah and Surya Prakash. However, the CAMERE (carbon dioxide hydrogenation to form methanol via reverse-water-gas-shift reaction) process revealed 20% higher methanol yields when CO2 is converted to CO (through rWGS), and CO to methanol, rather than directly hydrogenating CO2.33
Other methods, such as photo-electro-chemical reduction, are currently not a viable way to convert massive CO2 amounts, because their low rates would highly complicate a process scale-up, which could match CO2 production rates.47,48 Similarly, if using biomass, the atmospheric CO2 concentrations can only be lowered if such biomass is converted to fuels, otherwise it is not a long-term storage of CO2.49,50 Conversion of CO2 to biofuels using biomass that does not compete with food and does not require land would likely involve the use of microalgae. However, the costs of cultivating and maintaining these systems would have to substantially reduce before it becomes feasible.49–51 An upcoming technology, thermochemical CO2 splitting, also referred to as thermochemical cycles (TCs), has the advantage of not requiring an additional reactant (other than CO2). In this technology, CO2 is reduced to CO on the oxygen vacancies of a metal oxide with high oxygen mobility. TCs for CO2 splitting have been demonstrated on several oxides,52–57 but they usually require at least 1000 °C for the formation of oxygen vacancies or several hours to be reduced at lower temperatures. On these oxygen vacant materials, the conversion of carbon dioxide to carbon monoxide has been achieved at ∼900 °C.52,54–56 The high operational temperatures would require specialized gear and an additional equipment (such as solar concentrators) that can generate the required heat input.
The rWGS is an endothermic reaction, favored at high temperatures.36 The most commonly studied catalysts are copper-based58–61 or supported ceria,62–64 potentially less expensive than those used in DR. Its biggest advantage is the formation of CO, which can be used as a building block for a variety of important chemicals such as hydrocarbons in Fischer–Tropsch synthesis, fine chemical synthesis or the purification of nickel. The rWGS is suspected to be a key step in selective methanation of CO2 (ref. 65) and to occur in FT reactors with high CO2 feeds.29,66 It becomes evident that rWGS is a key reaction that should be considered and fully understood.
1.5 Rationale for rWGS catalysis over competing technologies
The rWGS reaction was first observed by Carl Bosch and Wilhelm Wild in 1914, when they attempted (and halfway succeeded) to produce H2 from steam and carbon monoxide on an iron oxide catalyst.67 Currently, it is important in the synthesis of methanol19 and in fixing syngas' H2/CO ratio for various applications.Mallapragada et al.68 compared different routes to transform CO2 into liquid fuels (biomass gasification, rWGS, algae-derived oils and direct photosynthesis) using solar assisted processes and H2 provided by electrolysis. Among the investigated methods, conversion of CO2 to CO by reverse water gas shift reaction followed by CO conversion to fuels with FTS had the highest current and estimated potential efficiency when CO2 is captured from a flue gas or from the atmosphere.68 Furthermore, converting CO2 to CO gives an added versatility in the products that can be obtained from CO transformation.17 The rWGS is also of great interest to be used in space exploration due high (∼95%) atmospheric CO2 concentration on Mars and availability of H2 as a byproduct of oxygen generation.69,70 Therefore, rWGS is a promising reaction, whose products have a wide variety of potential end uses.
The rWGS reaction is advantageous because of its technical feasibility compared to alternative technologies. However, as will be described in Section 1.6, many of the alternative technologies hold much promise if future research advances overcome significant existing challenges. In addition, with the CO2 problem being one of such massive scale and with local resources (e.g., solar insolation, available land and water) varying significantly, a multi-pronged approach is most probable, with the rWGS reaction using renewable hydrogen being one route.
1.6 Goals and limitations of this review
For the arguments already described in this review, conversion of carbon dioxide is an increasingly interesting topic for which many critical advances are needed to make substantial contributions. The readers are directed elsewhere for superb reviews on chemical conversion to a variety of organic products,13,36,71–75 solar-thermal-chemical cycling,76–78 dry reforming79–81 and other reactions with methane82 and photo-electro-catalytic conversion.83–87 Excellent overviews88,89 and reviews on CO2 separation90–92 (including from air93) and the forward water gas shift94 are also already available and may be of interest. Comparatively, there is very little studies summarized for the rWGS reaction even though it is a promising reaction as part of a CO2 conversion system and likely the closest to implementation. Thus, the primary goal of this review is to summarize literature findings for the rWGS reaction, with an emphasis on a discussion of comparing catalyst types, rates, mechanisms, and intensification strategies. Although the forward reaction has been examined in much more depth, this review primarily focuses on literature using CO2 and H2 as the feed, so studies on H2 purification via the forward WGS reaction are not included.In addition, as a secondary goal, the scope of CO2 conversion and the authors' vision for this challenge of scale has been justified in the introduction. The authors envision a society where transportation fuels and chemicals are produced from various CO2 purification and conversion strategies, whereas solar, wind, and geothermal sources are employed for renewable electricity. Since CO2 capture continues to be realized at various degrees, conversion strategies can operate under the assumption that CO2 will be available from flue gas or atmospheric separations (taking a concentration cost but minimizing contaminant issues), which makes the conversion processes a gate-to-grave type comparison. The advantages of the rWGS reaction approach for the conversion are as follows:
• A variety of renewable electricity forms exists with various advantages occurring locally. The rWGS reaction can be implemented with any of them to contribute to a closed carbon loop.
• Hydrogen from electrolysis requires much lower capital costs than using solar-thermal-heating to magnify the low intensity solar flux to practical levels.
• The rWGS reaction produces CO, which is a very flexible chemical intermediate. Alternatively, the hydrocarbon product from photocatalysis is primarily methane, which still requires processing for use.
• Any process that generates CO still requires ∼2 moles H2:1 mol CO to achieve a value-added fuel or chemical. The additional 1 mol H2 for converting CO2 to CO just increases the amount required from H2 generation processes by 50%, not substantiating their existence in the overall process.
• Although not common, the rWGS reaction may be useful in applications where H2 is readily available such as space exploration wherein electrolysis is primarily used for synthetic air production.
For these reasons and the readiness of the rWGS processes, its application in future CO2 conversion strategies seems likely. To reiterate, other strategies such as a closed loop of biomass conversion are also attractive but it is unlikely that one approach would be advantageous globally. With the justification provided above, energy dense liquid hydrocarbon fuels will continue to be a transportation fuel of choice. However, transportation fuels far exceed other chemicals for contributing to the scale of the CO2 problem; therefore, rWGS with methanol synthesis or FTS and biomass conversion to fuels are needed to overcome the challenge of achieving a closed carbon loop. In addition, with either synthetic (chemical) or natural (biological) CO2 separation from air and conversion to plastics as a secondary, albeit smaller scale, route of conversion, it may be possible to decrease atmospheric CO2 concentrations provided that electricity is available primarily from renewable sources.
2. Thermodynamic considerations
The rWGS reaction (eqn (1)) is equilibrium limited and favored at high temperatures due to the endothermic nature of the reaction.| CO2 + H2 ↔ CO + H2O, ΔH0298 = 42.1 kJ mol−1 | (1) |
Additional side reactions include:
Methanation
| CO + 3H2 ↔ CH4 + H2O, ΔH0298 = −206.5 kJ mol−1 | (2) |
| CO2 + 4H2 ↔ CH4 + 2H2O, ΔH0298 = −165.0 kJ mol−1 | (3) |
Thermodynamic evaluations at atmospheric pressure show that CO2 conversion in the rWGS reaction is enhanced when excess H2 is flowing35 and equilibrium conversion increases with temperature35,95 (Fig. 2). Product separation can shift the equilibrium towards the products.27 Whitlow and Parrish from Florida Institute of Technology and NASA, respectively,69 built a rWGS demonstration reactor without a catalyst in the system. They incorporated a membrane reactor to separate the products and achieved close to 100% CO2 conversion (∼5 times the equilibrium conversion). When the H2/CO2 flow is 0.5, CO2 conversion is 1/4 lower than the equilibrium conversion with a 1/1 flow at the same temperature, but when the flow ratio is 2, the conversion is enhanced by 50%. Optimum operating conditions were 310 kPa and 400 °C. Medium pressures were used in the study and it was found that small variations in the pressure (131 to 310 kPa) have no effect on the conversion.69
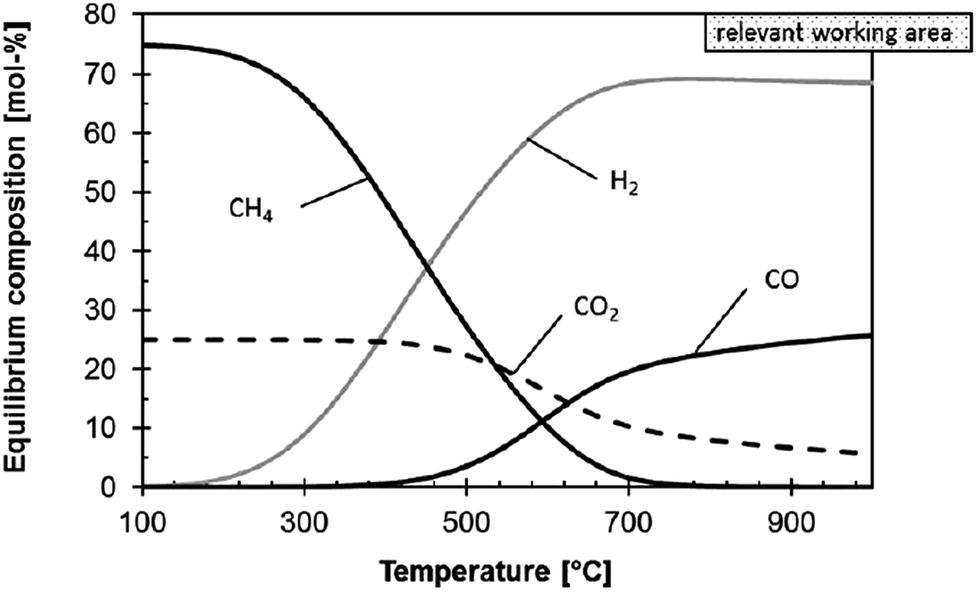 | ||
| Fig. 2 Influence of temperature on the thermodynamic equilibrium of the rWGS reaction at 1 bar and H2/CO2 molar ratio of 3/1. Reproduced with permission from John Wiley and Sons (from ref. 17). | ||
In a PNNL report, VanderWiel et al.70 studied the rWGS and Sabatier reactions for CO2 conversion. rWGS needs to be operated at very low residence times (5 to 64 ms) to achieve the highest CO selectivity (higher than equilibrium) but a methane side product was observed in the rWGS experiments. At residence times of 32 ms, CO selectivity reaches equilibrium at ∼550 °C. No CO2 conversion was observed below 300 °C. Further ways to shift the reaction equilibrium or increase reaction rates involve the use of electricity. Applying an overpotential to the Pd-YSZ electrode increased the rate of the reaction,96 whereas applying 3.0 mA to the 1 wt%Pt:10 mol%La–ZrO2 catalyst was equivalent to increasing the temperature by 100 K.35 In both studies, CO was the only carbonaceous product.
3. Catalyst types
3.1 Supported metal catalysts
The rWGS studies of supported metal catalysts consist primarily of Cu, Pt, and Rh immobilized on a variety supports. Studies on these metals are first highlighted, and then screening studies of a wide variety of metals are discussed. Finally, support effects are reviewed.Chen et al. have several contributions on the rWGS on Cu nanoparticles supported on different metal oxides. In their first study, they determined that supporting Cu NPs on Al2O3 increased the adsorption of formates, which they proposed as the reaction intermediates.100 In their other contributions examining CO2 hydrogenation on Cu nanoparticles105 and Cu nanoparticles supported on SiO2,106 they also concluded that (i) the rWGS mechanism goes through a formate intermediate,105,106 (ii) the CO2 and CO adsorption sites for the forward and reverse mechanisms are independent,105 and (iii) high Cu dispersion on SiO2 enhances CO2 conversion.61 Ginés et al.59 also observed that high Cu dispersion was a characteristic of the catalyst with highest activity in a Cu/ZnO/Al2O3 system.
Chen et al. also studied promoting the reaction with potassium99 and iron60,95 in the Cu/SiO2 system. In general, promoter addition enhanced catalytic activity, but both the metals had slightly different effects. Fe prevented Cu NPs sintering, significantly enhancing the stability and activity of the catalyst,60,95 whereas K increased the surface active sites that can adsorb and decompose formates, enhancing the catalytic activity of the system.99
Meunier's group dominated most of the rWGS studies on Pt supported samples. The group observed different surface reactive compounds in a 2% Pt/CeO2 catalyst depending on the reaction conditions.108 When the reaction intermediates were allowed to accumulate under vacuum, formates were observed as the most reactive, but under steady-state conditions, the most reactive surface compounds were carbonates and carbonyls. These results shed some light on the dispute of carbonates or formates as the main reaction intermediates. High temperature DRIFT and steady-state isotopic transient kinetic analysis (SSITKA) on 2% Pt/CeO2 confirmed that the main reaction intermediates were carbonates and not formates, although CO formation from formates could also occur in minority.109 Observed carbonates could be mono- or bi-dentate.107 On a solid–liquid interface, rWGS was found to occur on a Pt/Al2O3 system by a redox mechanism, where the O adatom (formed from CO2 dissociation) can refill an Al2O3 surface vacancy or recombine with adsorbed H.110
The effect of adsorbed reactants and products has also been investigated in Pt systems. Jacobs and Davis111 studied the effect of H2O and H2 adsorption on 1% Pt/CeO2 during rWGS and observed different spectator species formed under different conditions, suggesting that the forward and backwards WGS mechanisms could be different. Even though Pt/SiO2 systems have achieved higher conversion than Cu/SiO2 at 500 °C,61 poisoning of Pt by CO has been observed in 2% Pt/CeO2 (ref. 112) and on Pt and Ru/Pt alloy electrodes on PEMFCs.113 Bimetallic Co–Pt particles were tested for rWGS but it was found that Pt migrates to the surface, almost inhibiting any Co effect. The selectivity towards CO is highly increased, but there was no mention of CO2 conversion.114
For Rh/SiO2, increasing the surface hydroxyl groups surrounding Rh particles on the catalyst surface increases CO2 conversion and selectivity towards CO because it leads to formation of Rh carbonyl clusters, whereas fewer hydroxyl groups form hydride species on the Rh surface, which can further hydrogenate CO to methane.118 Li was added to an Rh ion-exchanged zeolite (Li/RhY)119 and the selectivity towards CO (vs. CH4) was found to increase with the amount of Li promoter, going from 0.3% at no Li, to 86.6% at 10:1 Li:Rh atomic ratio, but CO2 conversion was decreased to half with Li addition.
Lu et al.121 observed that at low NiO loadings (<3%) on CeO2, the particles were monodispersed on the ceria matrix and lead to 100% selectivity towards CO from 400 to 750 °C, whereas higher loadings lead to aggregation and lower CO selectivity below 650 °C. Sun et al.122 observed similar results on Ni/Ce–ZrO2, increasing Ni loading decreased CO selectivity and CO2 conversion, with the exception of 1% and 3% Ni, which exhibited similar behaviors. In conclusion, Zr appears to lower CO selectivity and CO2 conversion.121,122
Wang et al.64,123,124 demonstrated that different methods for supporting Ni on CeO2 affect CO2 conversion and CO selectivity, where the oxygen vacancies and highly dispersed surface Ni species were found to have the leading role in the reaction activity. The highest rWGS activity was observed on the catalyst synthesized by impregnation because Ni is deposited as NiO, which favors CO formation (as opposed to methane).64 The 1% Ni/CeO2-impregnation catalyst achieved up to 45% conversion and 100% selectivity towards CO in a 1:1H2/CO2 flow at 750 °C.64 Comparing this result to other studies, it appears that increasing Ni loading increases the activity of the catalyst. 2% Ni/CeO2 showed stability for over 9 h and constant CO yield (35% in a 1:1H2/CO2 flow) at 600 °C, and 45% CO selectivity at 750 °C,123 whereas 3% Ni/(Ce–Zr)O2 achieved 50% CO2 conversion and 100% CO selectivity at 750 °C (in a 1:1H2/CO2 flow) for 80 h.122 Supporting nickel on SBA-15 did not have a significant impact on the catalyst activity,125 but incorporation of Cu in a bimetallic Cu–Ni/SBA-15 system improved CO2 conversion and CO selectivity,126 as expected.
Ko et al.127 also performed CO2 dissociation DFT studies on different bimetallic alloy surfaces and determined that Fe alone and Fe-containing bimetallic particles would be the most favored to dissociate CO2 to CO and O. Unsupported Fe-oxide NPs (10 to 20 nm) were tested for 19 h showing high stability and medium CO2 conversion (∼30%). The stability of the sample could have originated from migration of C and O into the catalyst bulk forming iron oxide and iron carbide, which likely prevented the NPs on the surface from agglomerating.128 Kharaji et al.129 determined that the supported bimetallic Mo–Fe/γ-Al2O3 system increased the CO formation rates, CO2 conversion and CO selectivity when compared to the monometallic versions of the catalyst (Fe/γ-Al2O3 or Mo/γ-Al2O3).129 The leading role of the conversion was attributed to Fe, whereas Mo enhanced the stability of iron by increasing the electron deficient state of Fe species, enhancing catalytic activity.129 Addition of Ni to the Mo/Al2O3 system also showed increased activity.130 Incorporation of Fe has also increased CO selectivity in a Rh/TiO2 system, but greatly decreasing CO2 conversion.131 Porosoff et al.132 showed that adding Co into Mo2C enhances CO2 conversion and CO selectivity at 300 °C when compared to Pt–Co and Pd–Ni bimetallic NPs supported on CeO2. However, Ni/Mo2C and Cu/Mo2C have shown higher CO2 conversion and CO selectivity than Co/Mo2C catalysts.133
In2O3 has been found to inhibit CO production,134 but bimetallic In–Pd NPs supported onto SiO2 have achieved 100% CO selectivity on the rWGS,135 although with lower activities than Pd/SiO2. DFT suggested that the bimetallic Pd–In NPs had a weaker CO adsorption than Pd NPs, which suppresses the possibility of further hydrogenating CO to CH4 on the bimetallic system.135
Between Pt/TiO2 and Pt/Al2O3, titania exhibited higher activity and CO selectivity.138 Different lanthanide oxides were tested as Pd supports for the reaction and the activity order was found to be CeO2 > PrO2 > La2O3.139 When ceria has been incorporated into an Fe/Mn/Al2O3 system, CO selectivity was enhanced, but CO2 conversion was slightly decreased.140 Ceria is almost 100% selective towards CO at T ≥ 550 °C,141 most likely because at higher temperatures, the oxygen mobility of the oxide increases. Oxygen vacancies of ceria have been proven to play a leading role on the Pd/CeO2/Al2O3 system, because they can re-oxidize with CO2, whereas the role of Pd is to enhance the reduction of ceria.139 Different shapes of cerium oxide have been tested for the rWGS and it was found that the reaction in ceria is not shape sensitive.141 Moreover, supporting Ni on ceria slightly enhances CO2 conversion but significantly improves CO selectivity,141 as discussed in the previous section.
3.2 Oxide catalysts
The CAMERE process uses a rWGS reaction and a methanol synthesis reactor to convert CO2 to methanol.33 The first catalyst proposed on the CAMERE process consisted of Cu NPs on a ZnO/ZrO2/Ga2O3 support at 250 °C.33 Curiously, ZnO has been shown to be inactive for rWGS at temperatures below 165 °C.97,142 A later CAMERE catalyst consisted of ZnO/Al2O3, which showed enhanced stability (tested for over 100 h) at temperatures above 700 °C.142 The motivation for high temperatures was to favor the reaction thermodynamics. Cu was removed from the catalytic system likely because of low stability due to sample loss from the Cu oxides' reduction.59 ZnO was tested at 600 °C for 60 h and showed high deactivation. The ZnO/Al2O3 catalyst exhibits less CO2 conversion at 600 °C but high stability for over 200 h,143 likely due to the formation of a ZnAl2O4 spinel.142,143Theoretical CO2 adsorption and hydrogenation studies on the In2O3 (110) surface suggested that In2O3 suppressed rWGS due to weak CO2 adsorption144 and has also been found to inhibit CO production.134 Incorporation of CeO2 in In2O3 increased CO2 conversion (at 500 °C in a 1:1H2/CO2 flow) from 2.5% (In2O3) to 20% (In2O3:CeO2, 1:3 w/w ratio) by increasing oxygen mobility, adsorption of CO2 and generation of adsorbed bicarbonate species.62 Similarly, incorporation of ceria into Ga2O3 (Ga:Ce molar ratio of 99:1) increased CO2 conversion by 1.3% when compared to Ga2O3 at the same conditions described above.63 Both studies observed increased amounts of adsorbed bicarbonate species,62,63 which were suspected to be promoted by enhancement of oxygen mobility by ceria,62 but neither study quantified CO selectivity or yield.
Perovskites with La on the A site and Cu145–147 or Co148 on the B site have been studied for CO2 hydrogenation to methane and methanol. CO formation was observed by Kim et al.149 with 97% selectivity and almost 40% CO2 conversion at 600 °C and 1 bar, on a BaZr0.8Y0.16Zn0.04O3 oxide. With a La0.75Sr0.25FeO3 perovskite (for synthesis method see150), we were able to achieve a steady state conversion of 15% at 550 °C (Fig. 3). The sample was reduced for 20 min at 10% H2/He and after 20 min of flushing (100% He), the rWGS reaction (10% CO2/10% H2/He) was performed for 90 min. The obtained rate (1.53 millimol CO per g P per min) was three orders of magnitude larger than those of Goguet et al.112 and Chen et al.100 but at higher temperatures. rWGS on perovskites, BaZr0.8Y0.16Zn0.04O3 (ref. 149) and La0.75Sr0.25FeO3 (this study) exhibited the added advantage of nearly 100% CO selectivity without the use of supported nanoparticles. A comparison of selectivity, conversion and different reaction conditions for multiple catalytic systems can be found in Table 2.
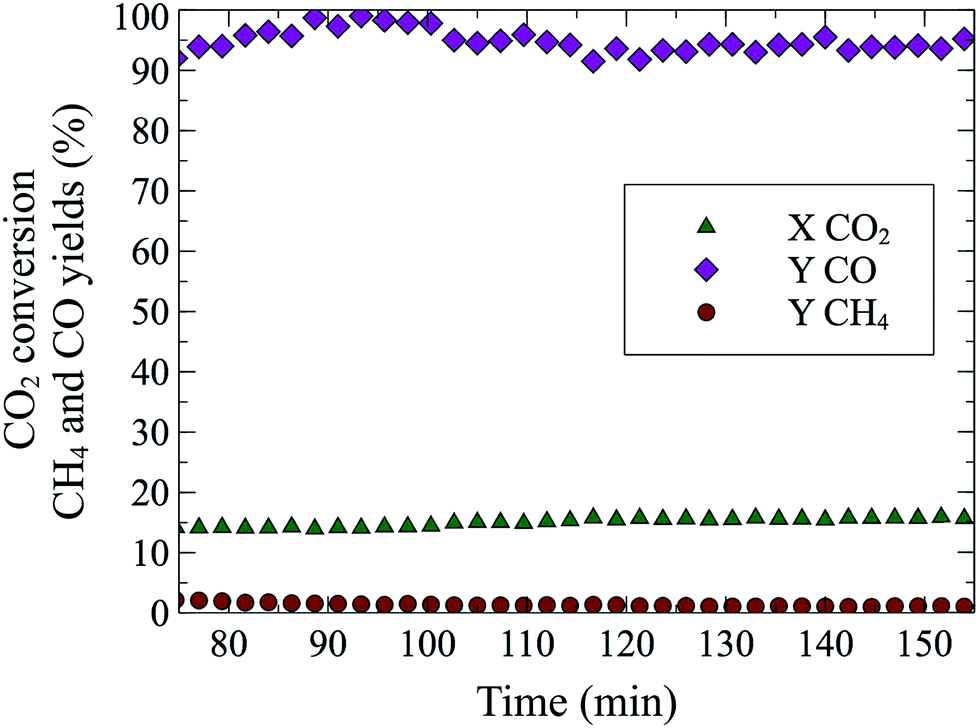 | ||
| Fig. 3 Reverse water gas shift reaction over 78.3 mg of La0.75Sr0.25FeO3 at 550 °C. Total flow 50 sccm (10% H2 10% CO2 v/v, He balance). Previously, catalyst was reduced for 20 min in 10% H2/He at 550 °C. | ||
| Reference | Year | Material | T (°C) | P (bar) | Feed H2/CO2 (v/v) | CO2 conversion (%) | CO selectivity (%) | CO formation (μmol CO per g_cat per s) |
|---|---|---|---|---|---|---|---|---|
| a Calculated as 100-methane selectivity.b For the meaning of G (related to origin of the support) see ref. 137.c Non steady state.d Applying 3.0 mA input current. | ||||||||
| Inoue et al.116 | 1989 | Rh/TiO2 | 300 | 10.13 | 1/1 | 0.82 | ||
| Rh–Na/TiO2 | 260 | 0.43 | ||||||
| Rh/Nb2O5 | 220 | 0.0 | ||||||
| Rh–Na/Nb2O5 | 200 | 0.05 | ||||||
| Rh/MgO | 200 | 0.008 | ||||||
| Rh/Nb2O5 | 200 | 3/1 | 0.078 | |||||
| Rh/ZrO2 | 200 | 0.033 | ||||||
| Rh/TiO2 | 300 | 0.93 | ||||||
| Pettigrew et al.139 | 1994 | Pd/A12O3 | 260 | 1 | 1/1 | 78a | 0.035 (μmol CO2 per g_cat per s) | |
| Pd/La2O3/A12O3 | 70 | 0.027 | ||||||
| Pd/PrO2/A12O3 | 76 | 0.033 | ||||||
| Pd/CeO2 (5)/A12O3 | 87 | 0.045 | ||||||
| Pd/CeO2 (10)/A12O3 | 81 | 0.073 | ||||||
| Ginés et al.59 | 1997 | Commercial CuO/ZnO/A12O3 | 250 | 1 | PH2/PCO2 = 6 | 0.17 | 4.31 | |
| Bando et al.119 | 1998 | Li/RhY (Li:Rh = 0) |
250 | 30 | 3/1 | 24.1 | 0.3 | |
| Li/RhY (Li:Rh = 3) |
12.0 | 3.7 | ||||||
| Li/RhY (Li:Rh = 7) |
11.1 | 27.6 | ||||||
| Li/RhY (Li:Rh = 10) |
13.1 | 86.6 | ||||||
| Chen et al.100 | 2000 | 10 wt% Cu/Al2O3 | 500 | 1 | 1/9 | 60 | 9.0 | |
| Chen et al.95 | 2001 | 10% Cu–0.3% Fe/SiO2 w/w | 600 | 1 | 1/1 | 12 | ||
| Kusama et al.118 | 2001 | 1 wt% Rh/SiO2 | 200 | 50 | 3/1 | 52 | 88.1 | |
| Chen et al.99 | 2003 | 9% Cu/SiO2 w/w | 600 | 1 | 1/1 | 5.3 | ||
| 9% Cu–1.9% K/SiO2 w/w | 12.8 | |||||||
| Chen et al.60 | 2004 | 0.3% Fe/SiO2 | 600 | 1 | 1/1 | 1 | ||
| 10% Cu/SiO2 | 2 | |||||||
| Cu–Fe/SiO2 (Cu/Fe = 10:0.3) |
15 | |||||||
| Cu–Fe/SiO2 (Cu/Fe = 10:0.8) |
16 | |||||||
| Goguet et al.109 | 2004 | 2% Pt/CeO2 by Johnson Matthey | 225 | 4/1 | 13.7 | 2.2 × 10−4 mol CO per g | ||
| Dorner et al.140 | 2010 | Mn 12 wt%/Fe 17 wt%/Al2O3 | 290 | 13.8 | 3/1 | 37.7 | 10.7 (% CO yield) | |
| Ce 2 wt%/Mn 12 wt%/Fe 17 wt%/Al2O3 | 38.6 | 11.5 (% CO yield) | ||||||
| Ce 10 wt%/Mn 12 wt%/Fe 17 wt%/Al2O3 | 35.8 | 17.5 (% CO yield) | ||||||
| Gogate et al.131 | 2010 | 2% Rh/TiO2 | 270 | 20.26 | 1/1 | 7.89 | 14.5 | |
| 2% Rh–2.5% Fe/TiO2 | 9.16 | 28.4 | ||||||
| 2.5% Fe/TiO2 | 2.65 | 73.0 | ||||||
| Kim et al.138 | 2012 | 1% Pt/Al2O3 | 875 | 30/21 | 42 | 0.0104 s−1 (TOF at 300 °C) | ||
| 1% Pt/TiO2 | 48 | 0.0998 s−1 (TOF at 300 °C) | ||||||
| Kim et al.136 | 2012 | Pt/TiO2 (G)b | 300 | 15 | 6480 | |||
| Kharaji et al.129 | 2013 | Fe/Al2O3 | 600 | 10 | 1/1 | 35 (% CO yield) | 96.17 | |
| Mo/Al2O3 | 33 (% CO yield) | 80.14 | ||||||
| Fe–Mo/Al2O3 | 37 (% CO yield) | 128.2 | ||||||
| Lu et al.125 | 2013 | NiO/SBA-15 | 400 | 1 | 1/1 | 5 | 100 | |
| 900 | 55 | 100 | ||||||
| Wang et al.64 | 2013 | Ni–CeO2 | 750 | 1 | 1/1 | 40 | 100 | |
| Lu et al.121 | 2014 | (1 wt% NiO/CeO2)/50% wt SBA-15 | 750c | 1 | 1/1 | 40 | 100 | 10.0 min−1 (TOF at ∼90 °C) |
| (3 wt% NiO/CeO2)/50% wt SBA-15 | 45 | 100 | 4.5 min−1 (TOF at ∼90 °C) | |||||
| Kim et al.149 | 2014 | BaZr0.8Y0.2O3 | 600 | 1/1 | 26.7 | 93 | ||
| BaZr0.8Y0.16Zn0.04O3 | 37.5 | 97 | ||||||
| BaCe0.2Zr0.6Y0.16Zn0.04O3 | 36.3 | 94 | ||||||
| BaCe0.3Zr0.3Y0.16Zn0.04O3 | 22.3 | 92 | ||||||
| BaCe0.7Zr0.1Y0.16Zn0.04O3 | 10.8 | 74 | ||||||
| Oshima et al.35,d | 2014 | 10% mol La–ZrO2 | 150 | 1/1 | 18 | 100 | ||
| 1% wt Pt/10% mol La–ZrO2 | 40 | 99.5 | ||||||
| 1% wt Pd/10% mol La–ZrO2 | 30 | 98.2 | ||||||
| 1% wt Ni/10% mol La–ZrO2 | 28 | 96.5 | ||||||
| 1% wt Fe/10% mol La–ZrO2 | 28 | 100 | ||||||
| 1% wt Cu/10% mol La–ZrO2 | 28 | 100 | ||||||
| Porosoff et al.132 | 2014 | PtCo/CeO2 | 300.85 | 1 | 2/1 | 6.6 | 4.5 (CO:CH4 ratio) |
14.6 min−1 (TOF) |
| PdNi/CeO2 | 2.5 | 0.6 | 5.6 min−1 | |||||
| Mo2C | 8.7 | 14.5 | 25.7 min−1 | |||||
| 7.5 wt% Co/Mo2C | 9.5 | 51.3 | 16.1 min−1 | |||||
| Kim et al.128 | 2015 | Unsupported Fe-oxide NPs | 600 | 1/1 | 38 | >85 | ||
| Xu et al.133 | 2015 | β-Mo2C | 200 | 20 | 5/1 | 6 | 39 | |
| Cu/β-Mo2C | 4 | 44 | ||||||
| Ni/β-Mo2C | 8 | 37 | ||||||
| Co/β-Mo2C | 9 | 31 | ||||||
| Matsubu et al.117 | 2015 | 0.5% w/w Rh/TiO2 | 200 | 1/10 | 3.0 × 10−2 CO molecule per Rh atoms per s (TOF) | |||
| 2% w/w Rh/TiO2 | 0.8 | |||||||
| 4% w/w Rh/TiO2 | 0.4 | |||||||
| 6% w/w Rh/TiO2 | 0.2 | |||||||
| Wang et al.62 | 2016 | In2O3 | 500 | 1/1 | 16 | |||
| In2O3:CeO2 = 3:1 w/w |
17 | |||||||
| In2O3:CeO2 = 1:1 w/w |
20 | |||||||
| In2O3:CeO2 = 1:3 w/w |
11 | |||||||
| In2O3:CeO2 = 1:9 w/w |
9 | |||||||
| CeO2 | 2.5 | |||||||
| This work | 2016 | La0.75Sr0.25FeO3 | 550 | 1 | 1/1 | 15.5 | 95 | 36.4 |
4. Intensified rWGS
The first attempts to achieve an intensified rWGS process emerged from combining chemical looping with DR, but substituting CH4 by H2 due to its higher potential as a reducing agent. In a chemical looping process, the ability of the oxygen carrier to reduce and oxidize under the desired environments is a key factor that can determine the feasibility of the process. In the rWGS process combined with chemical looping, a metal oxide is used as an oxygen carrier (Fig. 4). First, H2 is used to reduce the metal oxide. Subsequently, CO2 serves as an oxidant, returning the metal oxide to an oxidized state while CO is formed. The main advantages of an intensified rWGS-chemical looping process (rWGS-CL) are eliminating the possibility of methanation because the H2/H2O and CO/CO2 flows are kept separate and inherent product separation,150–152 which drives the equilibrium towards the products. In addition, no excess hydrogen is required because the reactions involving the metal oxide are stoichiometric.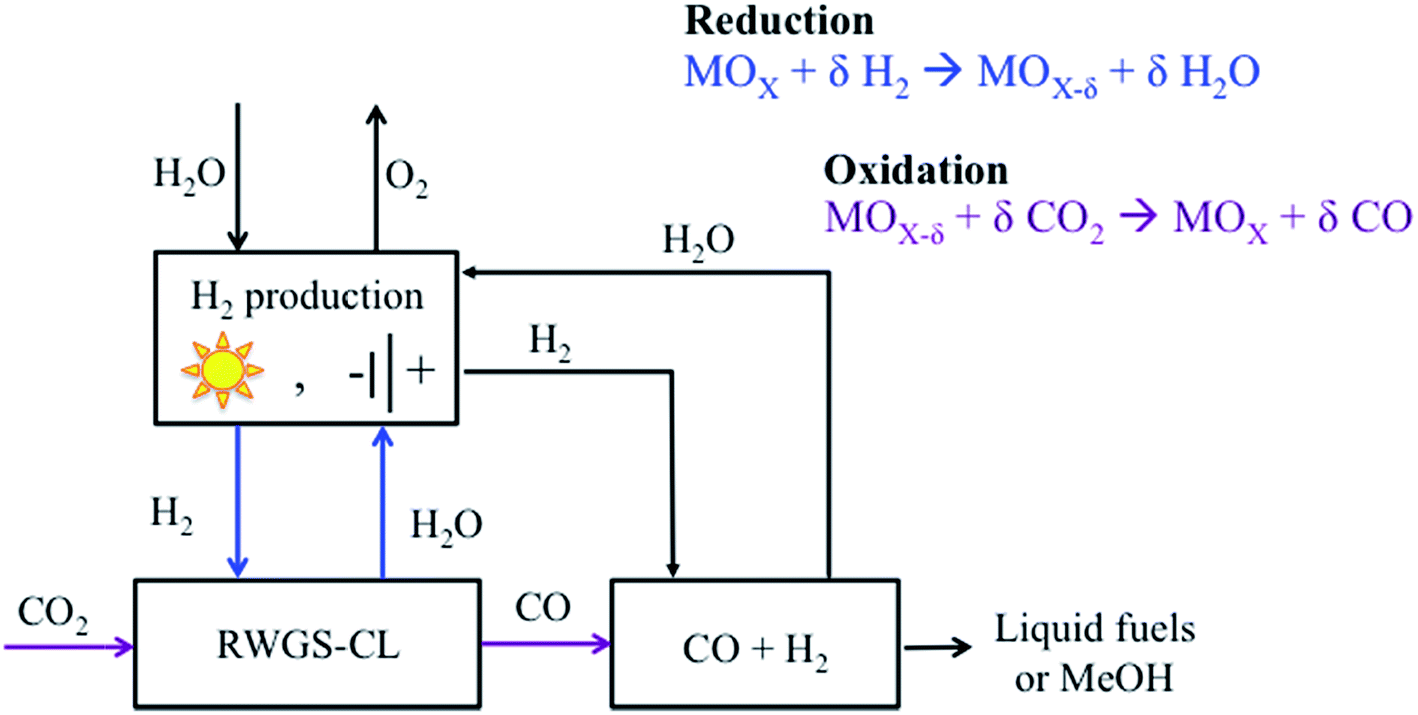 | ||
| Fig. 4 Schematic of the intensified reverse water gas shift-chemical looping process (rWGS-CL). Modified with permission from the American Chemical Society (from ref. 151). | ||
Thermodynamic modeling and experimental screening of transition metal oxides showed that Fe-based materials had one of the best CO2 carrying capacities while having the ability to function in the widest variety of temperatures.153,154 Najera et al.153 observed signs of stability on a 40% w/w Fe-BHA (barium hexaaluminate) porous sample on the intensified rWGS process over 6 reaction cycles and Galvita et al.155 used a Fe2O3–CeO2 composite and found that adding ceria to iron oxide linearly enhanced the stability of the solid solution, but decreased the CO formation capabilities. The same group later studied different weight loadings of Fe2O3 on an Al2O3–MgO system and found that at low loadings of iron oxide (≤30 wt%), the oxygen storage capacity of the samples decreased, but these samples are still preferred for CO2 conversion because of the high stability of the structure that Fe, Mg and Al form during the redox cycles.156
The rWGS-CL process was demonstrated on La(1−X)SrXCoO3 perovskite oxides by Daza et al.,151 but at the studied temperatures, the H2 reduction and CO2 conversion occurred with at least 50 °C difference, so the process was not isothermal. Reduced Fe-based spinels had been used previously for CO2 decomposition to C(s) and O2(g) at 300 °C.157,158 Based on this result, the rWGS-CL process was further examined using La0.75Sr0.25FeO3 and an isothermal process at 550 °C was achieved.150 By substituting cobalt with iron, the reducibility of the material was significantly decreased and it did not decompose under H2 flow. However, the process was not fully stoichiometric, because even though oxygen vacancies were being created, not all the vacancies were re-filled. DFT suggested that the driving force for the CO2 bond cleavage was the increased CO2 adsorption strength at the highest vacancies extent tested. rWGS was tested on La0.75Sr0.25Fe(1−Y)CuYO3, but doping Cu into the B site of the perovskite greatly increased its reducibility and inhibited CO formation.152
CO formation was achieved on both cobalt- and iron-based perovskites at similar reaction conditions, but the different solid state reactions the oxides underwent suggest very different reaction pathways. The high reducibility of the Co-based perovskite151 lead to its reduction to base La2O3 and metallic Co. It is likely that CO2 then adsorbed in the basic lanthanum oxide or lanthanum-based Ruddlesden–Popper phase and dissociated in the metallic cobalt, turning the metal into cobalt oxide (CoO) while yielding CO. On the iron-based material, a surface redox mechanism between oxygen vacancies in the perovskite took place, where CO2 was adsorbed on a lanthanum and oxygen surface termination159 close to an oxygen vacancy, then CO2 could dissociate into CO and an O adatom that re-fills the said oxygen vacancy.150 Introducing Cu into the Fe-based perovskite increases the stability of the perovskite in its reduced state (after forming oxygen vacancies), therefore reducing its oxygen affinity and re-oxidation capabilities; consequently, the observed outcome was a suppression of CO production because CO2 was not able to re-oxidized the reduced copper oxide.152
Throughout the different studies with an intensified version of the conventional rWGS reaction, the highest rates were achieved with Fe-containing solid solutions. A comparison of all studies covered in this section is shown in Fig. 5. Even though it has been shown before that Fe-oxides can decompose CO2 to C(s) and O2,157,158 Fe-based oxides show the highest CO formation, and almost all materials shown in Fig. 5 contain a form of iron. Only one study has tested selectivity towards CO (vs. C(s)) and the process is 30 times more selective towards CO.150 As in conventional rWGS, high temperatures enhance the intensified process for CO2 conversion. The materials with the highest CO formation rates were tested at high temperatures and with high loadings of iron. In addition to being performed at high temperatures and containing a high loading of iron, the Fe2O3–CeO2 mixture exhibited the highest CO formation rates likely due to the high oxygen mobility of ceria.155 Curiously, even though Cu is widely used as a catalyst for the forward and reverse water gas shift reactions, Fe works best for the intensified process.
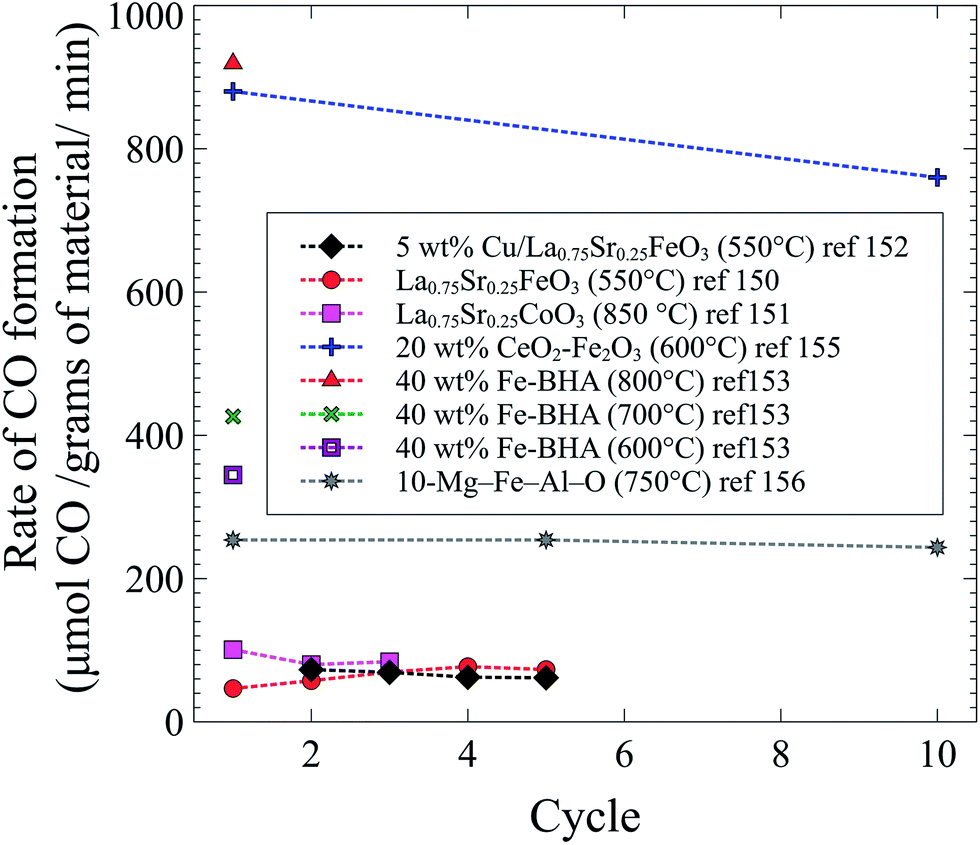 | ||
| Fig. 5 CO formation as a function of cycle in the intensified rWGS-CL process from ref. 150–153, 155 and 156. | ||
5. Mechanistic considerations
5.1 Copper surfaces and supported copper nanoparticles
Studies performed on Cu surfaces58,160 and supported Cu/ZnO systems59 agreed that reaction orders (and therefore the rate limiting step) vary with reaction conditions. Kinetic studies over Cu (100) single crystals58 and commercial Cu/ZnO/Al2O3 (ref. 59) demonstrated that the reaction orders with respect to PH2 and PCO2 change with the partial pressures of the gases.Ernst et al.58 and Ginés et al.59 studied the dependence of the reaction orders for H2 and CO2 in the rWGS reaction. Both studies agreed that at low PCO2/PH2 (below 1/3 for ref. 59 and below 1/10 for ref. 58), the reaction rate is highly dependent on PCO2 (order of ∼1.1 (ref. 59) and 0.6 (ref. 58) for CO2) and independent of H2 (0 order),58,59 likely due to a deconstruction of the surface, which makes it more favorable for CO2 dissociation.58 At intermediate pressures (PCO2/PH2 > 1/3 for ref. 59 and 1/10 < PCO2/PH2 < 1/2 for ref. 58), the studies disagree. Ernst et al. state that within the mentioned pressure interval, the rate depends strongly on PH2 and it is independent of PCO2 (0 order for PCO2), whereas Ginés et al. believe that the reaction rate is dependent on both gases (order 0.3 for PCO2 and 0.8 for H2) (Table 3). At very low PH2, the surface coverage of H2 is lower and cannot form the favored surface;58,59 therefore, the reaction rate is highly dependent on PH2 (2nd order for PH2).58 At higher PCO2/PH2 ratios, the rate is again linearly dependent on CO2 pressure.58,160 High coverage of H atoms adsorbed on Cu surfaces enhance CO2 conversion, regardless of whether hydrogen is provided as molecular hydrogen (H2)58 or electrochemically supplied (H+) in solid oxide fuel cells.161,162
| Ref. | Catalyst | Expression | Assumption |
|---|---|---|---|
| a And other mathematical assumptions.b Redox mechanism and associative mechanism. | |||
| Kaiser et al.17 | 11% Ni/Al12O19 | 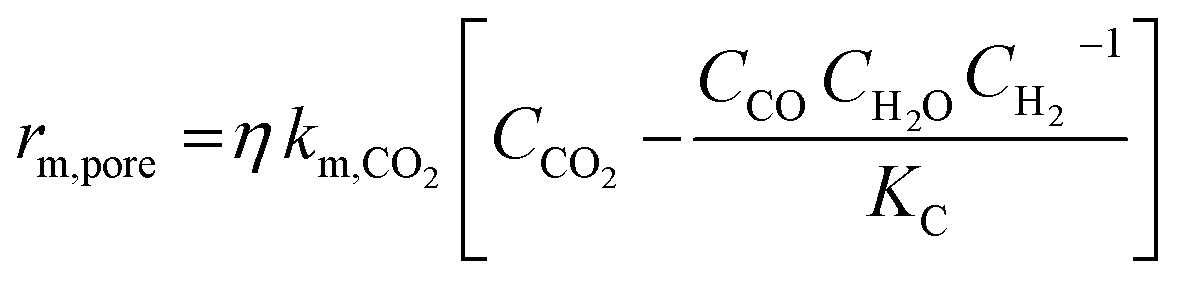 |
Adiabatic. Only accurate if external or internal mass transport occurs, in-between regimes are approximations |
| rm,ext = βAm,ext(CCO2 − CCO,eq) | |||
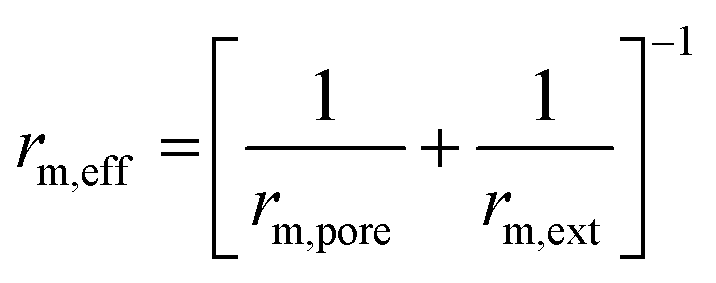 |
|||
| Ginés et al.59 | CuO/ZnO/Al2O3 | 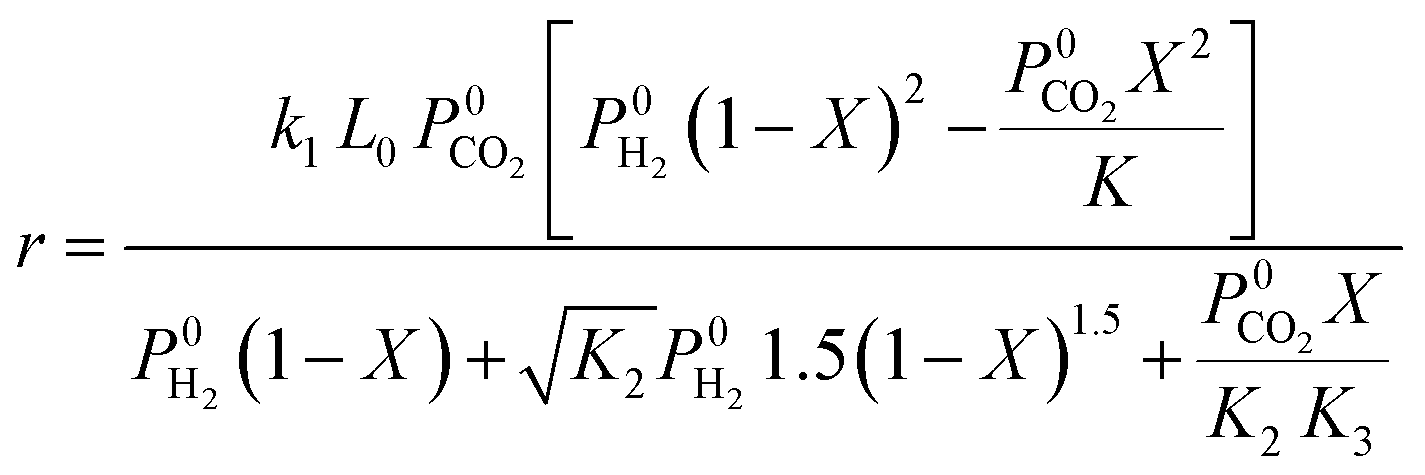 |
CO2 dissociation is the rate-determining step. Rate deduced from Langmuir–Hinshelwood kinetics |
| Chen et al.105 | ALE-Cu/SiO2 | r = 21/2k4K11/2K21/2K3PH21/2PCO21/2 | HCOO–2S → CO–S + OH–S is rate limitinga |
| Kim et al.138,b | Pt/TiO2 and Pt/Al2O3 | 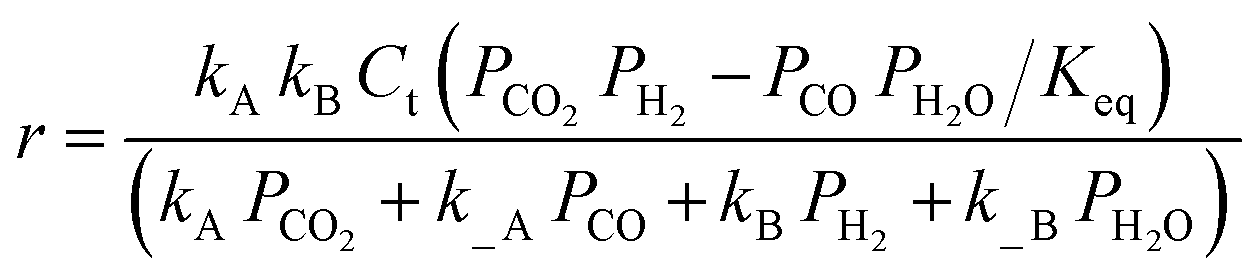 |
The adsorption of CO and H2O was excluded and the dissociation/adsorption step was excluded at low H2 pressure, 1 < 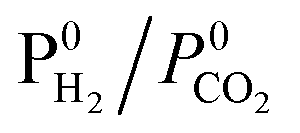 < 4 < 4 |
Reaction rates for the rWGS on Cu(110) and Cu(111) surfaces were comparable to Cu/ZnO except with high H2/CO2 partial pressure ratios. This was consistent with results showing that ZnO is not very active for rWGS97,142 (as mentioned in Section 3.2). In the high H2/CO2 partial pressures case, the CO2 decomposition mechanism seems to be aided by adsorbed H adatoms, which can adsorb in the Cu/ZnO surface but not on Cu(110) and Cu(111)160 (Fig. 6).
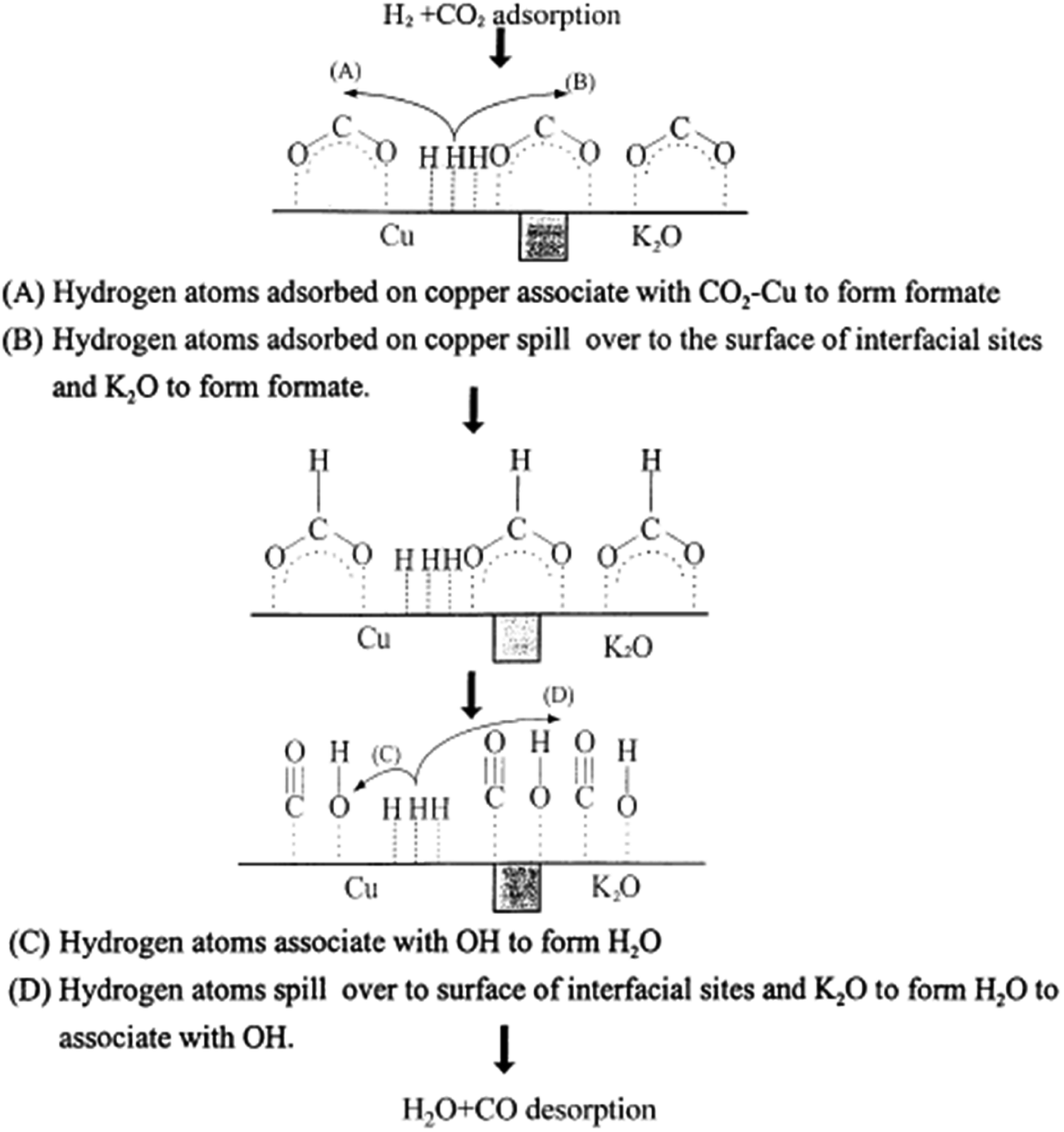 | ||
| Fig. 6 Proposed rWGS mechanism on the Cu/K/SiO2 interface. Reproduced with permission from Elsevier (from ref. 99). | ||
Even though dissociation of CO2 on the Cu atoms is considered as the rate determining step,97 it is worth mentioning that the probability for CO2 dissociation on H-adsorbed Cu surfaces is two orders of magnitude larger than on clean Cu surfaces.160 Therefore, surface modifications by H have been suspected to favor the reaction.160 Rates have increased by one order of magnitude when supplying electrochemical hydrogen (H+) in Cu electrodes in solid oxide fuel cells.162 Furthermore, in UHV conditions, no CO2 dissociation has been observed.101
In general, addition of alkali metals may alter the catalytic system reactivity.163 Adding K as a promoter in a Cu/SiO2 system increases the amount of active sites by increasing the positive charge on the catalyst surface,99 which has been found favorable for the reaction because an increase in surface positive charges is less favorable for CO adsorption and its reduction to methane and other products129 (Fig. 6).
5.2 Interactions of supported platinum nanoparticles with oxygen vacancies of supports
The rWGS mechanism on supported Pt/ceria systems has been highly debated. Jin et al.107 determined that CO2 is converted to CO on the interface between Pt and CeO2 (Fig. 7), but neither on CeO2 nor Pt alone (between 100 and 300 °C). An important observation from this study is that CO (resulting from CO2 decomposition) is adsorbed on Pt in the same way as if CO was flowed directly.107 This suggests that the transport and/or desorption of CO and O species (after CO2 dissociation) is not the rate limiting step, but rather the dissociation of CO2 itself.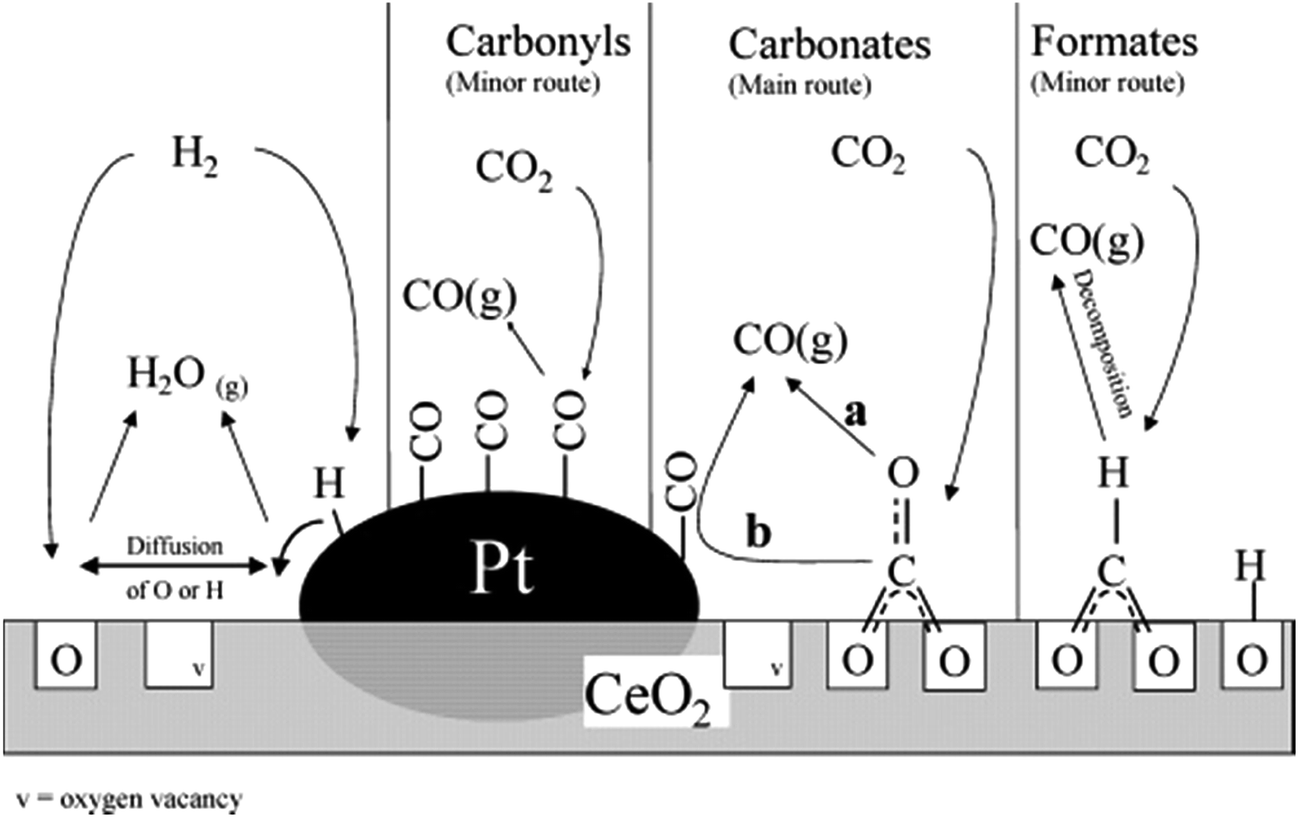 | ||
| Fig. 7 Proposed rWGS mechanism on the Pt/CeO2 interface. Reproduced with permission from the American Chemical Society (from ref. 109). | ||
Formates have been observed as the most reactive intermediate in an inert atmosphere108 and when H2O is included in the rWGS feed.111 Supplying electrochemical hydrogen (H+) in Pt161 electrodes in solid oxide fuel cells has enhanced rWGS rates, likely supporting the claim of the formate route. Nevertheless, steady-state isotopic transient kinetic analysis (SSITKA) combined with diffuse reflectance FT-IR spectroscopy (DRIFTS) revealed that the main intermediate species are carbonates, although the reaction could also take place through minor formates and carbonyl intermediates109 (Fig. 7). CO2 adsorption as carbonates has also been observed on solid–liquid interfaces in the boundaries of a Pt/AlO3 system.110
There is, however, some agreement on the importance of the oxygen vacancies in the support. CO2 is believed to adsorb on a ceria vacancy107,109 near a platinum/ceria boundary109 or a platinum step.164 Goguet et al.109 proposed that after CO2 dissociative chemisorption (to CO and Oa), one Oa re-fills a vacancy and either CO is desorbed or it can migrate to the Pt surface and desorb from there109 where the amount of CO2 decomposition depends on the oxidation state of the local CeO2 interface.107 Even in solid–liquid interfaces on Pt island film deposited on a Al2O3 film, the mechanism for rWGS is suspected to involve an O adatom (formed from CO2 dissociation), which can refill an Al2O3 surface vacancy or recombine with adsorbed H.110
The redox mechanism has been proved by Kim et al. on Pt/TiO2138 and it is suspected to follow mostly a carbonate route, as described by Goguet et al.109 on oxygen-mobile supports. On the contrary, on non-reductive supports (i.e. Al2O3), the carbonyl route is suspected to occur.139
The observation of different spectator species under different reaction conditions suggests that the forward and backwards WGS mechanisms could be different (on Pt/ceria).111
5.3 Role of support
Primarily, the role of support effects on the rWGS mechanism has been focused on oxygen conduction materials such as ceria and perovskite-type oxides. The Au/CeO2 system was proven to be more active than the Au/TiO2 due to the higher oxygen mobility of ceria165 and its ability to be re-oxidized by CO2.139 This oxygen exchange can take place simultaneously (as in rWGS) or subsequently (as in rWGS-CL).165 In2O3 has been shown to be promising for CO2 hydrogenation.144,166 On In2O3–CeO2 catalysts, a volcano-type relationship between oxygen vacancies formation (increasing CeO2) and reactive sites (increasing In2O3) was demonstrated.62 When the ratio of oxides was 1:1, the activity of the rWGS was maximized and no side products were observed.62 CO2 can dissociate on the oxygen vacancies of ceria and on the Ni surface in a Ni/CeO2 catalytic system.141 H2 in the reaction would form more oxygen vacancies on the ceria, but its reduction is suspected to be catalyzed by Ni,141 similar to the mechanism on Pt/CeO2 systems.139
We studied re-oxidation of pre-reduced La0.75Sr0.25CoO3 (Fig. 8) and found that the reactivity of the oxidant was O2 > H2O > CO2. Given the prior results from Wang et al., which suggest that the nature of the oxygen deposited on the reduced ceria surface is similar, whether it came from CO2 or O2 re-oxidation,165 our results suggest that dissociation of CO2 is the rate determining step, and not the Oa migration or H2 dissociation, in agreement with Ernst et al.58
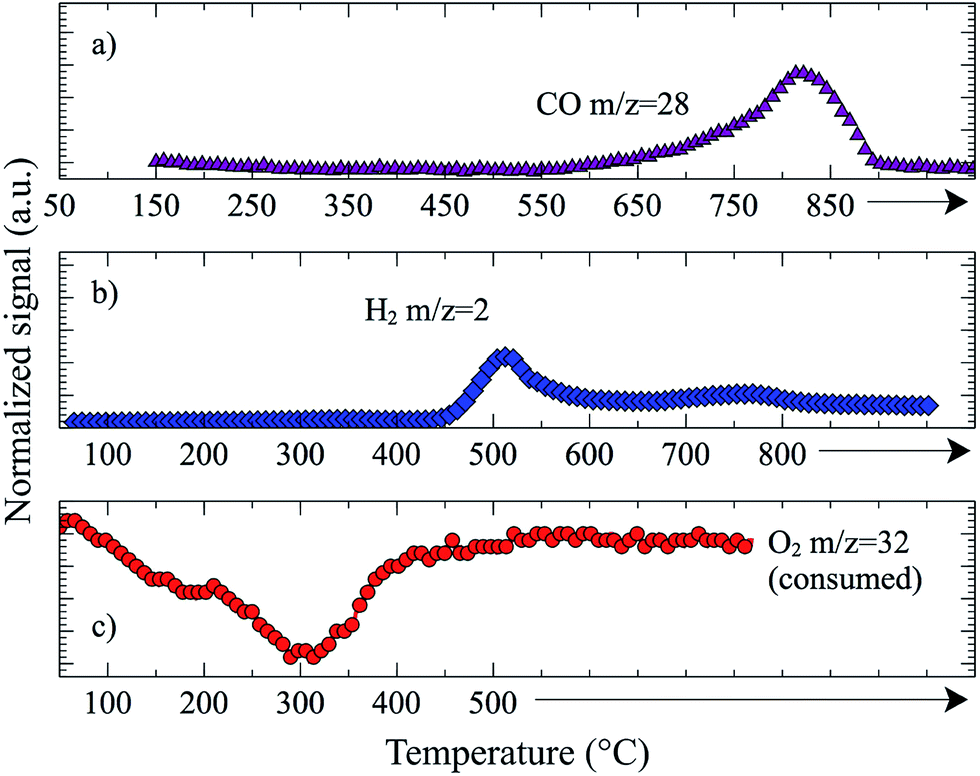 | ||
| Fig. 8 Oxidation of La0.75Sr0.25CoO3 previously reduced with 10% H2/He at 600 °C for 30 min (total flow rate 50 sccm). (a) Oxidation with CO2 forming CO. (b) Oxidation with H2O forming H2. (c) Consumption of O2. | ||
6. Material selection and design principles
A fair and thorough comparison of catalysts is cumbersome because experimental conditions vary widely and in a substantial number of cases, complete information is not reported (i.e. missing rates, conversions or yields). Supported platinum has achieved higher conversion than supported copper at 500 °C.61 However, Cu-based catalysts are generally preferred due to their low cost, high metal abundance and because Pt is highly susceptible to CO poisoning and coke formation.112 The poisoning effect has also been observed on Pt and Ru/Pt alloy electrodes on PEMFCs.113 Among the supports, ceria has been shown to play a key role on the reaction due to its high oxygen mobility.107,109,165 Furthermore, catalytic research is progressing into a material design approach, so that control of metal and support surface faceting, support vacancy amounts and locations for tuning surface properties, is probably on the horizon for rWGS catalysis.In addition, combining Cu and ceria components seems a natural idea. Cu supported on ceria has been previously studied for CO oxidation167,168 but recently, Rodriguez et al. have shown higher selectivity towards rWGS (vs. methanol or methane formation) on ceria supported on Cu surfaces169 and Cu deposited on ceria and titania.170 Therefore, it would likely be advantageous to thoroughly study Cu/ceria systems for the rWGS reaction.
7. Summary and outlook
The rWGS is a promising reaction with high potential use in the near future for the large-scale conversion of CO2 to CO, provided that a technology for production of renewable H2 in large scale is also available. The rWGS reaction also requires lower temperatures (∼200 °C lower) than other conversion technologies that could meet the scale of CO2 emissions. Being only slightly endothermic, the current challenge for rWGS use in fuel synthesis lies in designing materials that can achieve high CO selectivity and high production rates. Intensification strategies have recently been proposed to circumvent thermodynamic and kinetic limitations by using chemical looping to perform stoichiometric reactions rather than catalytic ones. Even though a large number of materials have been studied for the reaction, improvement is still possible. Some reports are often missing key information that allows for an equitable comparison and the effect of non-concentrated CO2 has not been studied. Furthermore, if the rWGS reaction was to play a major role on the reduction of atmospheric CO2 concentration, a catalyst with earth-abundant materials would be preferred.In the interest of adopting earth-abundant metals, iron oxides could be a good substitute for ceria. Fe oxides are also known to have high oxygen mobility and stability, and when added to a Cu system, have increased the rWGS reaction activity.60,95 In a system where Cu particles were to be supported on an iron oxide, Cu would provide high activity for CO formation, whereas Fe oxide would ideally bring high stability and high CO2 adsorption.104 MoC and CoMoC materials are also of interest due to their lack of precious metals and the convenience of employing industrially used metals.
Acknowledgements
The authors would like to acknowledge NSF award 1335817 for financial support. YAD acknowledges the Florida Education Fund for the McKnight Dissertation Fellowship and the NASA Florida Space Grant Consortium for the Dissertation Improvement Fellowship.References
- International Energy Agency, 2015 Key World Energy Statistics, Supply, Consumption and Emissions, 2015, https://www.iea.org/publications/freepublications/publication/key-world-energy-statistics-2015.html.
- U.S. Department of Energy Office of Science, U.S. DOE, Carbon Cycling and Biosequestration: Integrating Biology and Climate Through Systems Science; Report from the March 2008 Workshop, 2008, http://genomicscience.energy.gov/carboncycle/report/index.shtml.
- J.-B. Sallee, R. J. Matear, S. R. Rintoul and A. Lenton, Nat. Geosci., 2012, 5, 579 CrossRef CAS.
- C. Hauri, T. Friedrich and A. Timmermann, Nat. Clim. Change, 2016, 6, 172 CrossRef CAS.
- Global C.C.S. Institute, The Global Status of CCS: 2015, Summary Report, 2015, http://status.globalccsinstitute.com/.
- G.C.C.S. Institute, Last access date February 17, 2016, Last updated 2015, http://www.globalccsinstitute.com/projects/large-scale-ccs-projects.
- M. Aresta and A. Dibenedetto, Dalton Trans., 2007, 2975 RSC.
- M. Mikkelsen, M. Jorgensen and F. C. Krebs, Energy Environ. Sci., 2010, 3, 43–81 CAS.
- J. J. Dooley, R. T. Dahowski and C. L. Davidson, On the Long-Term Average Cost of CO2 Transport and Storage, U.S. Department of Energy under Contract DE-AC05-76RL01830, 2008 Search PubMed.
- R. Angamuthu, P. Byers, M. Lutz, A. L. Spek and E. Bouwman, Science, 2010, 327, 313 CrossRef CAS PubMed.
- S. Sato, T. Arai, T. Morikawa, K. Uemura, T. M. Suzuki, H. Tanaka and T. Kajino, J. Am. Chem. Soc., 2011, 133, 15240 CrossRef CAS PubMed.
- G. A. Olah, A. Goeppert and G. K. S. Prakash, J. Org. Chem., 2009, 74, 487 CrossRef CAS PubMed.
- A. Goeppert, M. Czaun, J.-P. Jones, G. K. Surya Prakash and G. A. Olah, Chem. Soc. Rev., 2014, 43, 7995 RSC.
- G. Job, S. D. Allen, C. Simoneau, R. Valente and J. J. Farmer, Metal complexes, US pat., US20150232496A1, USA, 2015.
- P.E.A.o.P. Manufacturers, Plastics - the Facts 2015, Brussels, Belgium, 2015, p. 1 Search PubMed.
- P. M. Mortensen and I. Dybkjær, Appl. Catal., A, 2015, 495, 141 CrossRef CAS.
- P. Kaiser, R. B. Unde, C. Kern and A. Jess, Chem.-Ing.-Tech., 2013, 85, 489 CrossRef CAS.
- Department for Business, Innovation & Skills (BIS) and Department of Energy & Climate Change (DECC), E.E. Ltd, C.C. Ltd, P.S.E.P. Ltd, I. College, U.o. Sheffield, Demonstrating CO2 capture in the UK cement, chemicals, iron and steel and oil refining sectors by 2025: A Techno-economic Study, Techno-economics of ICCS and CCU in UK, 2014, https://www.gov.uk/government/publications/co2-capture-in-the-uk-cement-chemicals-iron-steel-and-oil-refining-sectors.
- X. Xiaoding and J. A. Moulijn, Energy Fuels, 1996, 10, 305 CrossRef CAS.
- M. B. Ansari and S.-E. Park, Energy Environ. Sci., 2012, 5, 9419 CAS.
- M. Poliakoff, W. Leitner and E. S. Streng, Faraday Discuss., 2015, 183, 9 RSC.
- M. Institute, Last access date Last updated http://www.methanol.org/Methanol-Basics.aspx.
- I.E. Agency, Last access date 02/22/2016, Last updated 02/09/2016, https://www.iea.org/oilmarketreport/omrpublic/.
- U.S.E.I.A., Last access date 02/22/2016, Last updated 11/05/2015, http://www.eia.gov/Energyexplained/index.cfm?page=oil_refining.
- International Energy Agency, 2013 Key World Energy Statistics, Transformation, 2013, http://www.iea.org/publications/freepublications/publication/KeyWorld2013_FINAL_WEB.pdf.
- International Energy Agency, 2014 Key World Energy Statistics, Emissions, 2014, http://www.iea.org/publications/freepublications/publication/key-world-energy-statistics-2014.html.
- G. Centi and S. Perathoner, Catal. Today, 2009, 148, 191 CrossRef CAS.
- S. Chunshan, CO2 Conversion and Utilization: An Overview, CO2 Conversion and Utilization, American Chemical Society, 2002, p. 2 Search PubMed.
- T. Riedel, M. Claeys, H. Schulz, G. Schaub, S.-S. Nam, K.-W. Jun, M.-J. Choi, G. Kishan and K.-W. Lee, Appl. Catal., A, 1999, 186, 201 CrossRef CAS.
- G. Centi, S. Perathoner and G. Iaquaniello, Realizing Resource and Energy Efficiency in Chemical Industry by Using CO2, in CO2: A Valuable Source of Carbon, ed. M. De Falco and G. Iaquaniello and G. Centi, Springer, London, 2013, p. 27 Search PubMed.
- S. Chu and A. Majumdar, Nature, 2012, 488, 294 CrossRef CAS PubMed.
- C. Song, Catal. Today, 2006, 115, 2 CrossRef CAS.
- O.-S. Joo, K.-D. Jung, I. Moon, A. Y. Rozovskii, G. I. Lin, S.-H. Han and S.-J. Uhm, Ind. Eng. Chem. Res., 1999, 38, 1808 CrossRef CAS.
- N. Meiri, Y. Dinburg, M. Amoyal, V. Koukouliev, R. V. Nehemya, M. V. Landau and M. Herskowitz, Faraday Discuss., 2015, 183, 197 RSC.
- K. Oshima, T. Shinagawa, Y. Nogami, R. Manabe, S. Ogo and Y. Sekine, Catal. Today, 2014, 232, 27 CrossRef CAS.
- W. Wang, S. Wang, X. Ma and J. Gong, Chem. Soc. Rev., 2011, 40, 3703 RSC.
- E. V. Kondratenko, G. Mul, J. Baltrusaitis, G. O. Larrazábal and J. Pérez-Ramírez, Energy Environ. Sci., 2013, 6, 3112 CAS.
- International Renewable Energy Agency, IRENA, Renewable Energy Cost Analysis, Concentrating Solar Power, 2012, http://www.irena.org/menu/index.aspx?mnu=Subcat%26PriMenuID=36%26CatID=141%26SubcatID=233.
- J. A. Turner, Science, 2004, 305, 972 CrossRef CAS PubMed.
- A. Hauch, S. D. Ebbesen, S. H. Jensen and M. Mogensen, J. Mater. Chem., 2008, 18, 2331 RSC.
- J. Kim, C. A. Henao, T. A. Johnson, D. E. Dedrick, J. E. Miller, E. B. Stechel and C. T. Maravelias, Energy Environ. Sci., 2011, 4, 3122 CAS.
- J. Kim, T. A. Johnson, J. E. Miller, E. B. Stechel and C. T. Maravelias, Energy Environ. Sci., 2012, 5, 8417 CAS.
- A. Taheri, E. J. Thompson, J. C. Fettinger and L. A. Berben, ACS Catal., 2015, 5, 7140 CrossRef CAS.
- U.S. E.P.A., Global Anthropogenic Non-CO2 Greenhouse Gas Emissions: 1990–2030, O.o.A.P.C.C. Division, U.S. Environmental Protection Agency, Washington, DC, 2012 Search PubMed.
- N. H. Elsayed, N. R. M. Roberts, B. Joseph and J. N. Kuhn, Appl. Catal., B, 2015, 179, 213 CrossRef CAS.
- J. Skrzypek, M. Lachowska and D. Serafin, Chem. Eng. Sci., 1990, 45, 89 CrossRef CAS.
- Q. Zhai, S. Xie, W. Fan, Q. Zhang, Y. Wang, W. Deng and Y. Wang, Angew. Chem., Int. Ed., 2013, 52, 5776 CrossRef CAS PubMed.
- H. Zhou, J. Guo, P. Li, T. Fan, D. Zhang and J. Ye, Sci. Rep., 2013, 3, 1667 Search PubMed.
- F. G. Acién Fernández, C. V. González-López, J. M. Fernández Sevilla and E. Molina Grima, Appl. Microbiol. Biotechnol., 2012, 96, 577 CrossRef PubMed.
- L. Gustavsson, P. Börjesson, B. Johansson and P. Svenningsson, Energy, 1995, 20, 1097 CrossRef CAS.
- K. Gao and K. R. McKinley, J. Appl. Phycol., 1994, 6, 45 CrossRef.
- W. C. Chueh, C. Falter, M. Abbott, D. Scipio, P. Furler, S. M. Haile and A. Steinfeld, Science, 2010, 330, 1797 CrossRef CAS PubMed.
- P. Furler, J. R. Scheffe, M. Gorbar, L. Moes, U. Vogt and A. Steinfeld, Energy Fuels, 2012, 26, 7051 CrossRef CAS.
- J. E. Miller, M. D. Allendorf, R. B. Diver, L. R. Evans, N. P. Siegel and J. N. Stuecker, J. Mater. Sci., 2008, 43, 4714–4728 CrossRef CAS.
- A. Le Gal, S. Abanades and G. Flamant, Energy Fuels, 2011, 25, 4836 CrossRef CAS.
- A. H. McDaniel, E. C. Miller, D. Arifin, A. Ambrosini, E. N. Coker, R. O'Hayre, W. C. Chueh and J. Tong, Energy Environ. Sci., 2013, 6, 2024 Search PubMed.
- A. H. Bork, M. Kubicek, M. Struzik and J. L. M. Rupp, J. Mater. Chem. A, 2015, 3, 15546 CAS.
- K.-H. Ernst, C. T. Campbell and G. Moretti, J. Catal., 1992, 134, 66 CrossRef CAS.
- M. J. L. Ginés, A. J. Marchi and C. R. Apesteguía, Appl. Catal., A, 1997, 154, 155 CrossRef.
- C.-S. Chen, W.-H. Cheng and S.-S. Lin, Appl. Catal., A, 2004, 257, 97 CrossRef CAS.
- C.-S. Chen, J. H. Lin, J. H. You, C. R. Chen and J. Am, J. Am. Chem. Soc., 2006, 128, 15950 CrossRef CAS PubMed.
- W. Wang, Y. Zhang, Z. Wang, J.-M. Yan, Q. Ge and C.-J. Liu, Catal. Today, 2016, 259, 402 CrossRef CAS.
- B. Zhao, Y.-X. Pan and C.-J. Liu, Catal. Today, 2012, 194, 60 CrossRef CAS.
- L. Wang, H. Liu, Y. Liu, Y. Chen and S. Yang, J. Rare Earths, 2013, 31, 969 CrossRef CAS.
- P. Panagiotopoulou, D. I. Kondarides and X. E. Verykios, Catal. Today, 2012, 181, 138–147 CrossRef CAS.
- K.-W. Jun, H.-S. Roh, K.-S. Kim, J.-S. Ryu and K.-W. Lee, Appl. Catal., A, 2004, 259, 221 CrossRef CAS.
- C. Bosch and W. Wild, Producing hydrogen, US pat., US1115776A, 1914.
- D. S. Mallapragada, N. R. Singh, V. Curteanu and R. Agrawal, Ind. Eng. Chem. Res., 2013, 52, 5136 CrossRef CAS.
- J. E. Whitlow and C. F. Parrish, AIP Conf. Proc., 2003, 654, 1116–1123 CrossRef CAS.
- D. P. VanderWiel, J. L. Zilka, Y. Wang, A. Y. Tonkovich and R. S. Wegeng, Carbon Dioxide Conversions in Microreactors, IMRET 4: Proceedings of the 4th International Conference on Microreaction Technology, Topical Conference Proceedings, AIChE Spring National Meeting, American Institute of Chemical Engineers, Atlanta, GA, 2000, p. 187 Search PubMed.
- M. D. Porosoff, B. Yan and J. G. Chen, Energy Environ. Sci., 2016, 9, 62 CAS.
- P. Lanzafame, G. Centi and S. Perathoner, Chem. Soc. Rev., 2014, 43, 7562 RSC.
- G. Centi, E. A. Quadrelli and S. Perathoner, Energy Environ. Sci., 2013, 6, 1711 CAS.
- W. Wang and J. Gong, Front. Chem. Sci. Eng., 2011, 5, 2 CrossRef CAS.
- T. Sakakura, J.-C. Choi and H. Yasuda, Chem. Rev., 2007, 107, 2365 CrossRef CAS PubMed.
- N. P. Siegel, J. E. Miller, I. Ermanoski, R. B. Diver and E. B. Stechel, Ind. Eng. Chem. Res., 2013, 52, 3276 CrossRef CAS.
- G. P. Smestad and A. Steinfeld, Ind. Eng. Chem. Res., 2012, 51, 11828 CrossRef CAS.
- T. Kodama and N. Gokon, Chem. Rev., 2007, 107, 4048 CrossRef CAS PubMed.
- J. R. Rostrup-Nielsen and J. Sehested, Adv. Catal., 2002, 47, 65 CAS.
- M. C. J. Bradford and M. A. Vannice, Catal. Rev.–Sci. Eng., 1999, 41, 1 CAS.
- D. Pakhare and J. J. Spivey, Chem. Soc. Rev., 2014, 43, 7813 RSC.
- V. Havran, M. P. Dudukovi and C. S. Lo, Ind. Eng. Chem. Res., 2011, 50, 7089 CrossRef CAS.
- E. V. Kondratenko, G. Mul, J. Baltrusaitis, G. O. Larrazabal and J. Perez-Ramırez, Energy Environ. Sci., 2013, 6, 3112 CAS.
- S. N. Habisreutinger, L. Schmidt-Mende and J. K. Stolarczyk, Angew. Chem., Int. Ed., 2013, 52, 7372 CrossRef CAS PubMed.
- B. Kumar, M. Llorente, J. Froehlich, T. Dang, A. Sathrum and C. P. Kubiak, Annu. Rev. Phys. Chem., 2012, 63, 541 CrossRef CAS PubMed.
- S. C. Roy, O. K. Varghese, P. Paulose and C. A. Grimes, ACS Nano, 2010, 1259 CrossRef CAS PubMed.
- V. P. Indrakanti, J. D. Kubicki and H. H. Schobert, Energy Environ. Sci., 2009, 2, 745 CAS.
- A. M. Appel, J. E. Bercaw, A. B. Bocarsly, H. Dobbek, D. L. DuBois, M. Dupuis, J. G. Ferry, E. Fujita, R. Hille, P. J. A. Kenis, C. A. Kerfeld, R. H. Morris, C. H. F. Peden, A. R. Portis, S. W. Ragsdale, T. B. Rauchfuss, J. N. H. Reek, L. C. Seefeldt, R. K. Thauer and G. L. Waldrop, Chem. Rev., 2013, 113, 6621 CrossRef CAS PubMed.
- H. Arakawa, M. Aresta, J. N. Armor, M. A. Barteau, E. J. Beckman, A. T. Bell, J. E. Bercaw, C. Creutz, E. Dinjus, D. A. Dixon, K. Domen, D. L. DuBois, J. Eckert, E. Fujita, D. H. Gibson, W. A. Goddard, D. W. Goodman, J. Keller, G. J. Kubas, H. H. Kung, J. E. Lyons, L. E. Manzer, T. J. Marks, K. Morokuma, K. M. Nicholas, R. Periana, L. Que, J. Rostrup-Nielson, W. M. H. Sachtler, L. D. Schmidt, A. Sen, G. A. Somorjai, P. C. Stair, B. R. Stults and W. Tumas, Chem. Rev., 2001, 101, 953 CrossRef CAS PubMed.
- N. von der Assen, P. Voll, M. Peters and A. Bardow, Chem. Soc. Rev., 2014, 43, 7982 RSC.
- L.-S. Fan, L. Zeng, W. Wang and S. Luo, Energy Environ. Sci., 2012, 5, 7254 CAS.
- N. MacDowell, N. Florin, A. Buchard, J. Hallett, A. Galindo, G. Jackson, C. S. Adjiman, C. K. Williams, N. Shah and P. Fennell, Energy Environ. Sci., 2010, 3, 1645 CAS.
- G. Goeppert, M. Czaun, G. K. S. Prakash and G. A. Olah, Energy Environ. Sci., 2012, 5, 7833 Search PubMed.
- C. Ratnasamy and J. P. Wagner, Catal. Rev., 2009, 51, 325 CAS.
- C.-S. Chen, W.-H. Cheng and S.-S. Lin, Chem. Commun., 2001, 1770 RSC.
- S. Bebelis, H. Karasali and C. G. Vayenas, Solid State Ionics, 2008, 179, 1391 CrossRef CAS.
- S.-I. Fujita, M. Usui and N. Takezawa, J. Catal., 1992, 134, 220 CrossRef CAS.
- J. A. Rodriguez, J. Evans, L. Feria, A. B. Vidal, P. Liu, K. Nakamura and F. Illas, J. Catal., 2013, 307, 162 CrossRef CAS.
- C.-S. Chen, W.-H. Cheng and S.-S. Lin, Appl. Catal., A, 2003, 238, 55 CrossRef CAS.
- C.-S. Chen, W.-H. Cheng and S.-S. Lin, Catal. Lett., 2000, 68, 45 CrossRef CAS.
- J. Nakamura, J. A. Rodriguez and C. T. Campbell, J. Phys.: Condens. Matter, 1989, 1, Sb149 CrossRef CAS.
- C. Liu, T. R. Cundari and A. K. Wilson, J. Phys. Chem. C, 2012, 116, 5681 CAS.
- S.-G. Wang, X.-Y. Liao, D.-B. Cao, C.-F. Huo, Y.-W. Li, J. Wang and H. Jiao, J. Phys. Chem. C, 2007, 111, 16934 CAS.
- T. Bligaard, J. K. Nørskov, S. Dahl, J. Matthiesen, C. H. Christensen and J. Sehested, J. Catal., 2004, 224, 206 CrossRef CAS.
- C.-S. Chen, J. H. Wu and T. W. Lai, J. Phys. Chem. C, 2010, 114, 15021 CAS.
- C.-S. Chen and W.-H. Cheng, Catal. Lett., 2002, 83, 121 CrossRef CAS.
- T. Jin, Y. Zhou, G. J. Mains and J. M. White, J. Phys. Chem., 1987, 91, 5931–5937 CrossRef CAS.
- D. Tibiletti, A. Goguet, F. Meunier, J. P. Breen and R. Burch, Chem. Commun., 2004, 1636 RSC.
- A. Goguet, F. C. Meunier, D. Tibiletti, J. P. Breen and R. Burch, J. Phys. Chem. B, 2004, 108, 20240 CrossRef CAS.
- D. Ferri, T. Bürgi and A. Baiker, Phys. Chem. Chem. Phys., 2002, 4, 2667 RSC.
- G. Jacobs and B. H. Davis, Appl. Catal., A, 2005, 284, 31 CrossRef CAS.
- A. Goguet, F. Meunier, J. P. Breen, R. Burch, M. I. Petch and A. F. Ghenciu, J. Catal., 2004, 226, 382 CrossRef CAS.
- T. Gu, W.-K. Lee and J. W. Van Zee, Appl. Catal., B, 2005, 56, 43 CrossRef CAS.
- S. Alayoglu, S. K. Beaumont, F. Zheng, V. V. Pushkarev, H. Zheng, V. Iablokov, Z. Liu, J. Guo, N. Kruse and G. A. Somorjai, Top. Catal., 2011, 54, 778 CrossRef CAS.
- P. G. Jessop, F. Joó and C.-C. Tai, Coord. Chem. Rev., 2004, 248, 2425 CrossRef CAS.
- T. Inoue, T. Iizuka and K. Tanabe, Appl. Catal., 1989, 46, 1 CrossRef CAS.
- J. C. Matsubu, V. N. Yang and P. Christopher, J. Am. Chem. Soc., 2015, 137, 3076 CrossRef CAS PubMed.
- H. Kusama, K. K. Bando, K. Okabe and H. Arakawa, Appl. Catal., A, 2001, 205, 285 CrossRef CAS.
- K. K. Bando, K. Soga, K. Kunimori and H. Arakawa, Appl. Catal., A, 1998, 175, 67 CrossRef.
- Y. Liu and D. Liu, Int. J. Hydrogen Energy, 1999, 24, 351 CrossRef CAS.
- B. Lu and K. Kawamoto, Mater. Res. Bull., 2014, 53, 70 CrossRef CAS.
- F.-M. Sun, C.-F. Yan, Z.-D. Wang, C.-Q. Guo and S.-L. Huang, Int. J. Hydrogen Energy, 2015, 40, 15985 CrossRef CAS.
- L. Wang, S. Zhang and Y. Liu, J. Rare Earths, 2008, 26, 66 CrossRef.
- L. Wang, H. Liu, Y. Liu, Y. Chen and S. Yang, J. Rare Earths, 2013, 31, 559 CrossRef CAS.
- B. Lu, K. Kawamoto and J. Environ, Chem. Eng., 2013, 1, 300 CAS.
- B. Lu, Y. Ju, T. Abe and K. Kawamoto, Inorg. Chem. Front., 2015, 2, 741 RSC.
- J. Ko, B.-K. Kim and J. W. Han, J. Phys. Chem. C, 2016, 120, 3438 CAS.
- D. H. Kim, S. W. Han, H. S. Yoon and Y. D. Kim, J. Ind. Eng. Chem., 2015, 23, 67 CrossRef CAS.
- A. G. Kharaji, A. Shariati and M. A. Takassi, Chin. J. Chem. Eng., 2013, 21, 1007 CrossRef CAS.
- A. G. Kharaji, A. Shariati, M. Ostadi and J. Nanosci, J. Nanotechnol., 2014, 14, 6841 CAS.
- M. R. Gogate and R. J. Davis, Catal. Commun., 2010, 11, 901 CrossRef CAS.
- M. D. Porosoff, X. Yang, A. J. Boscoboinik and J. G. Chen, Angew. Chem., Int. Ed., 2014, 126, 6823 CrossRef.
- W. Xu, P. J. Ramírez, D. Stacchiola, J. L. Brito and J. A. Rodriguez, Catal. Lett., 2015, 145, 1365 CrossRef CAS.
- T. Umegaki, K. Kuratani, Y. Yamada, A. Ueda, N. Kuriyama, T. Kobayashi and Q. Xu, J. Power Sources, 2008, 179, 566 CrossRef CAS.
- J. Ye, Q. Ge and C.-J. Liu, Chem. Eng. Sci., 2015, 135, 193 CrossRef CAS.
- S. S. Kim, H. H. Lee and S. C. Hong, Appl. Catal., B, 2012, 119–120, 100 CAS.
- H. Sakurai, A. Ueda, T. Kobayashi and M. Haruta, Chem. Commun., 1997, 271–272 RSC.
- S. S. Kim, H. H. Lee and S. C. Hong, Appl. Catal., A, 2012, 423–424, 100 CAS.
- D. J. Pettigrew, D. L. Trimm and N. W. Cant, Catal. Lett., 1994, 28, 313 CrossRef CAS.
- R. W. Dorner, D. R. Hardy, F. W. Williams and H. D. Willauer, Catal. Commun., 2010, 11, 816 CrossRef CAS.
- Y. Liu, Z. Li, H. Xu and Y. Han, Catal. Commun., 2016, 76, 1 CrossRef CAS.
- S.-W. Park, O.-S. Joo, K.-D. Jung, H. Kim and S.-H. Han, Appl. Catal., A, 2001, 211, 81 CrossRef CAS.
- O.-S. Joo and K.-D. Jung, Bull. Korean Chem. Soc., 2003, 24, 86 CrossRef CAS.
- J. Ye, C. Liu and Q. Ge, J. Phys. Chem. C, 2012, 116, 7817 CAS.
- L. Jia, J. Gao, W. Fang and Q. Li, J. Rare Earths, 2010, 28, 747 CrossRef CAS.
- H. Zhan, F. Li, P. Gao, N. Zhao, F. Xiao, W. Wei, L. Zhong and Y. Sun, J. Power Sources, 2014, 251, 113 CrossRef CAS.
- L. Jia, J. Gao, W. Fang and Q. Li, Catal. Commun., 2009, 10, 2000 CrossRef CAS.
- M. A. Ulla, R. A. Migone, J. O. Petunchi and E. A. Lombardo, J. Catal., 1987, 105, 107 CrossRef CAS.
- D. H. Kim, J. L. Park, E. J. Park, Y. D. Kim and S. Uhm, ACS Catal., 2014, 4, 3117 CrossRef CAS.
- Y. A. Daza, D. Maiti, R. A. Kent, V. R. Bhethanabotla and J. N. Kuhn, Catal. Today, 2015, 258, 691 CrossRef CAS.
- Y. A. Daza, R. A. Kent, M. M. Yung and J. N. Kuhn, Ind. Eng. Chem. Res., 2014, 53, 5828 CrossRef CAS.
- Y. A. Daza, D. Maiti, B. J. Hare, V. R. Bhethanabotla and J. N. Kuhn, Surf. Sci., 2016, 648, 92 CrossRef CAS.
- M. Najera, R. Solunke, T. Gardner and G. Veser, Chem. Eng. Res. Des., 2011, 89, 1533 CrossRef CAS.
- R. D. Solunke and G. Veser, Ind. Eng. Chem. Res., 2010, 49, 11037 CrossRef CAS.
- V. V. Galvita, H. Poelman, V. Bliznuk, C. Detavernier and G. B. Marin, Ind. Eng. Chem. Res., 2013, 52, 8416 CrossRef CAS.
- N. V. R. A. Dharanipragada, L. C. Buelens, H. Poelman, E. D. Grave, V. V. Galvita and G. B. Marin, J. Mater. Chem. A, 2015, 3, 16251 CAS.
- C. Nordhei, K. Mathisen, I. Bezverkhyy and D. G. Nicholson, J. Phys. Chem., 2008, 112, 6531–6537 CAS.
- C. Nordhei, K. Mathisen, O. Safonova, W. van Beek and D. G. Nicholson, J. Phys. Chem., 2009, 113, 19568 CAS.
- D. Maiti, Y. A. Daza, M. M. Yung, J. N. Kuhn and V. R. Bhethanabotla, J. Mater. Chem. A, 2016, 4, 5137 CAS.
- C. T. Campbell and K.-H. Ernst, Forward and Reverse Water-Gas Shift Reactions on Model Copper Catalysts, Surface Science of Catalysis, American Chemical Society, 1992, p. 130 Search PubMed.
- G. Pekridis, K. Kalimeri, N. Kaklidis, E. Vakouftsi, E. F. Iliopoulou, C. Athanasiou and G. E. Marnellos, Catal. Today, 2007, 127, 337 CrossRef CAS.
- G. Karagiannakis, S. Zisekas and M. Stoukides, Solid State Ionics, 2003, 162–163, 313 CrossRef CAS.
- F. Solymosi, J. Mol. Catal., 1991, 65, 337 CrossRef CAS.
- J. Xu and J. T. Yates Jr, Surf. Sci., 1995, 327, 193–201 CrossRef CAS.
- L. C. Wang, M. Tahvildar Khazaneh, D. Widmann and R. J. Behm, J. Catal., 2013, 302, 20 CrossRef CAS.
- Q. Sun, J. Ye, C.-J. Liu and Q. Ge, Greenhouse Gases: Sci. Technol., 2014, 4, 140–144 CrossRef CAS.
- D. Gamarra, G. Munuera, A. B. Hungría, M. Fernández-García, J. C. Conesa, P. A. Midgley, X. Q. Wang, J. C. Hanson, J. A. Rodríguez and A. Martínez-Arias, J. Phys. Chem. C, 2007, 111, 11026 CAS.
- N. E. Nunez, H. P. Bideberripe, M. L. Casella and G. J. Siri, Curr. Catal., 2014, 3, 187 CrossRef CAS.
- J. A. Rodriguez, P. Liu, D. J. Stacchiola, S. D. Senanayake, M. G. White and J. G. Chen, ACS Catal., 2015, 5, 6696 CrossRef CAS.
- J. Graciani, K. Mudiyanselage, F. Xu, A. E. Baber, J. Evans, S. D. Senanayake, D. J. Stacchiola, P. Liu, J. Hrbek, J. Fernández-Sanz and J. A. Rodriguez, Science, 2014, 345, 546 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |