
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Cellular uptake of metallated cobalamins
Mai Thanh Quynh
Tran†
a,
Stefan
Stürup
*b,
Ian Henry
Lambert
c,
Bente
Gammelgaard
b,
Evelyne
Furger‡
d and
Roger
Alberto
*a
aInstitute of Inorganic Chemistry, University of Zürich, Winterthurerstr. 190, 8057 Zürich, Switzerland. E-mail: ariel@aci.uzh.ch
bDepartment of Pharmacy, Faculty of Health Sciences, University of Copenhagen, Universitetsparken 2, 2100 Copenhagen, Denmark. E-mail: stefan.sturup@sund.ku.dk
cDepartment of Biology, University of Copenhagen, Universitetsparken 13, 2100 Copenhagen, Denmark
dCenter for Radiopharmaceutical Science, Paul Scherrer Institute, CH-5232 Villigen PSI, Switzerland
First published on 22nd December 2015
Abstract
Cellular uptake of vitamin B12-cisplatin conjugates was estimated via detection of their metal constituents (Co, Pt, and Re) by inductively coupled plasma mass spectrometry (ICP-MS). Vitamin B12 (cyano-cob(III)alamin) and aquo-cob(III)alamin [Cbl-OH2]+, which differ in the β-axial ligands (CN− and H2O, respectively), were included as control samples. The results indicated that B12 derivatives delivered cisplatin to both cellular cytosol and nuclei with an efficiency of one third compared to the uptake of free cisplatin cis-[PtIICl2(NH3)2]. In addition, uptake of charged B12 derivatives including [Cbl-OH2]+, [{Co}-CN-{cis-PtCl(NH3)2}]+, [{Re}-{Co}-CN-{cis-PtCl(NH3)2}]+, and [{Co}-CN-{trans-Pt(Cyt)(NH3)2}]2+ (Cyt = cytarabin) was high compared to neutral B12, which implied the existence of an additional internalization pathway for charged B12 vitamin analogs. The affinities of the charged B12 derivatives to the B12 transporters HC, IF and TC were similar to that of native vitamin B12.
Introduction
Cyano-cob(III)alamin, also designated vitamin B12, is an essential, water soluble, and non-toxic vitamin even at high doses. Vitamin B12 is involved in the methionine synthesis and the tricarboxylic acid cycle. B12 comprises an octahedral CoIII complex with a tetradentate corrin ring, an α-nucleotide loop and a CN− ligand in the β-axial position. The cyanide ligand can be exchanged for other monodentate functions (Fig. 1).1 Intracellular reduction of the CoIII center subsequently forms square pyramidal cob(II)alamin and square planar cob(I)alamin, respectively.2,3 Enzymatic alkylation of cob(I)alamin in the cytosol and mitochondria leads to the formation of two B12-coenzymes with methyl and adenosyl as β-ligands.4,5 B12 is protected from degradation inside living systems through its binding to the proteins haptocorrin (HC, present in saliva/gastric fluids), intrinsic factor (IF, secreted by parietal cells), and transcobalamin (TC, plasma protein).6,7 Binding of B12 to TC is required for facilitating the uptake of the B12–TC complex via the TC-receptor or CD320 (across most membranes) and megalin (reabsorbed from the urine in kidney cells).7 The B12 specificity follows the order: IF > TC ≫ HC.8–11 Numerous functional groups on the corrin side chains, β-ligand, and α-nucleotide loop (Fig. 1) are available to coordinate or conjugate drugs and analytical modalities.12 The β-axial ligand is a preferred derivatization site owing to its ease-of-release in cellular environment and its little involvement in interaction with proteins. Nitrogen mustard chlorambucil,13 colchicine,14 cisplatin,15 and insulinomimetic vanadate (VV) compound16 are drugs which have been bound to B12 at this site. In addition, fluorophores17 and radionuclides18,19 have also been attached to B12 at this position. The ribose-hydroxyl group is another convenient derivatization site as it is not involved in protein interaction.20 Fluorophores,21,22 cytotoxic drugs,23 insulin,24 and appetite suppressing peptide hPYY25 have been coupled to B12 via this position.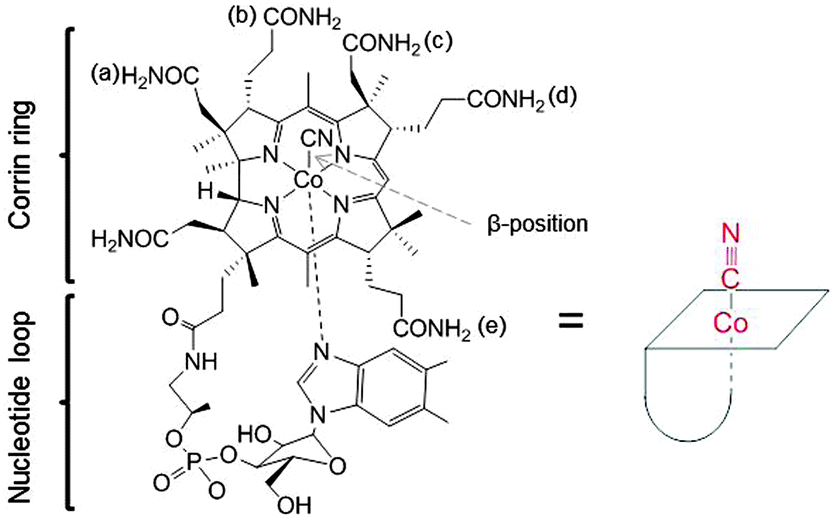 | ||
| Fig. 1 Structure of Cyano-cob(III)alamin, vitamin B12 (left) and sketch of B12 for the following figures. | ||
Derivatization of B12, however, is not limited to the β- and ribose-positions. Radioactive 111In-DTPA and 99mTc-DTPA were coupled to B12 via the b-side chain.26,27 The {99mTc(CO)3} moiety was coupled to B12 via ligands coupled to the b- and d-side chains28 and biotin via either the b-, c-, d-, or e-side chains.12 Increasing structural knowledge of B12 and of its transport proteins is the base for fine-tuning the structures of biologically active B12 derivatives and mimetics. Several B12 antagonists were reported, including B12-[c-lactam],29 and B12 with de novo peptide backbones.30,31 In addition, tissue distribution was interfered by modifying the B12 structure at a critical site (b-side chain) for protein interaction.32
Research programs have been dedicated to explore the correlation of B12 requirements and DNA synthesis. This, however, is not the only advantage of using B12 as a carrier for anti-cancer drugs. Its water solubility allows convenient administration of the attached insoluble drugs.15 B12 protects protein–peptide drugs24,25,33 from hydrolysis in the acidic condition of the gastrointestinal track.34 Though the utilization of B12 as drug carrier appeared to be restrained by its cellular loading capacity,35 a recent study showed that tissue accumulation of B12 increased significantly (up to 350%) upon overdosing of the vitamin.36
In this paper, B12 was applied as a drug carrier for cisplatin and as a carrier of the {fac-Re(CO)3}+ moiety, which is a model moiety for Tc and Re radiopharmaceuticals. Cisplatin was chosen as it is the most often used platinum based drug in the clinic, its mode of action is binding to DNA in the nucleus, which triggers apoptosis and cell death. Cisplatin is taken up by the cells via passive diffusion or via active transport through the copper transporters (CTR1 and CTR2) and the organic cation transporters (OCTs) and possible also other pathways.37 Once in the cell cisplatin undergoes aquation, chloride exchanged by water, forming positive ions which are more reactive towards the main biological target, DNA.38 We determined the cytosolic and nucleic uptake of cisplatin, vitamin B12, [{Co}-CN-{cis-PtCl(NH3)2}]+ (1), [{Re}-{Co}-CN-{cis-PtCl(NH3)2}]+ (2), and [{Co}-CN-{trans-Pt(Cyt)(NH3)2}]2+ (3) (Fig. 2) in human leukemia cells (K562 cells) and non-adherent (EATC)/adherent (ELA) Ehrlich ascites tumor cells. The uptake was determined by ICP-MS, where the contents of Co, Pt, and Re represent the uptake of B12 and the platinum/rhenium drug, respectively.
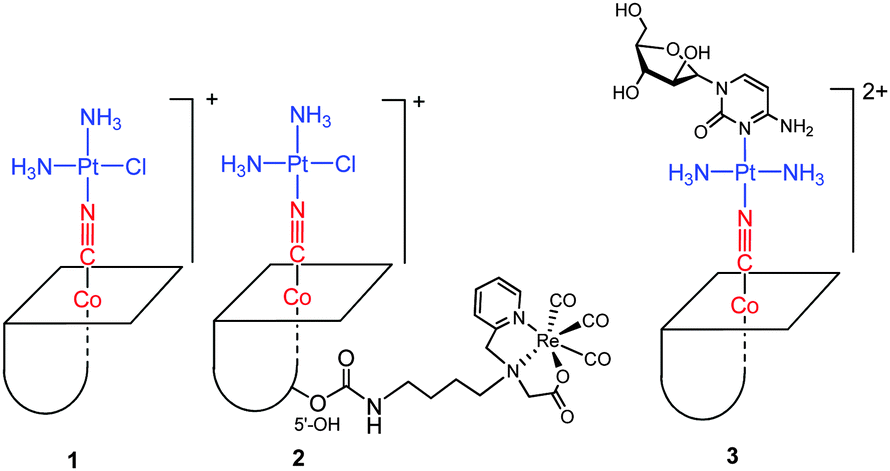 | ||
| Fig. 2 Structures of B12 derivatives including [{Co}-CN-{cis-PtCl(NH3)2}]+ (1), [{Re}-{Co}-CN-{cis-PtCl(NH3)2}]+ (2), and [{Co}-CN-{trans-Pt(Cyt)(NH3)2}]2+ (3). | ||
Results and discussion
Synthesis of [{Re}-{Co}-CN-{cis-PtCl(NH3)2}]+
The tridentate NNO ligand (6) was chelated to the {fac-Re(CO)3}+ moiety prior to conjugation to the B12 structure (step iv and v,), see Scheme 1. In the next step, the β-CN− was coordinated to [cis-PtCl(OH2)(NH3)2]+ by substituting the aquo-ligand bound to PtII (step vi) to yield 2. This synthesis scheme produced a high yield (80%) of [{Re}-{Co}-CN-{cis-PtCl(NH3)2}]+ (2, Scheme 1).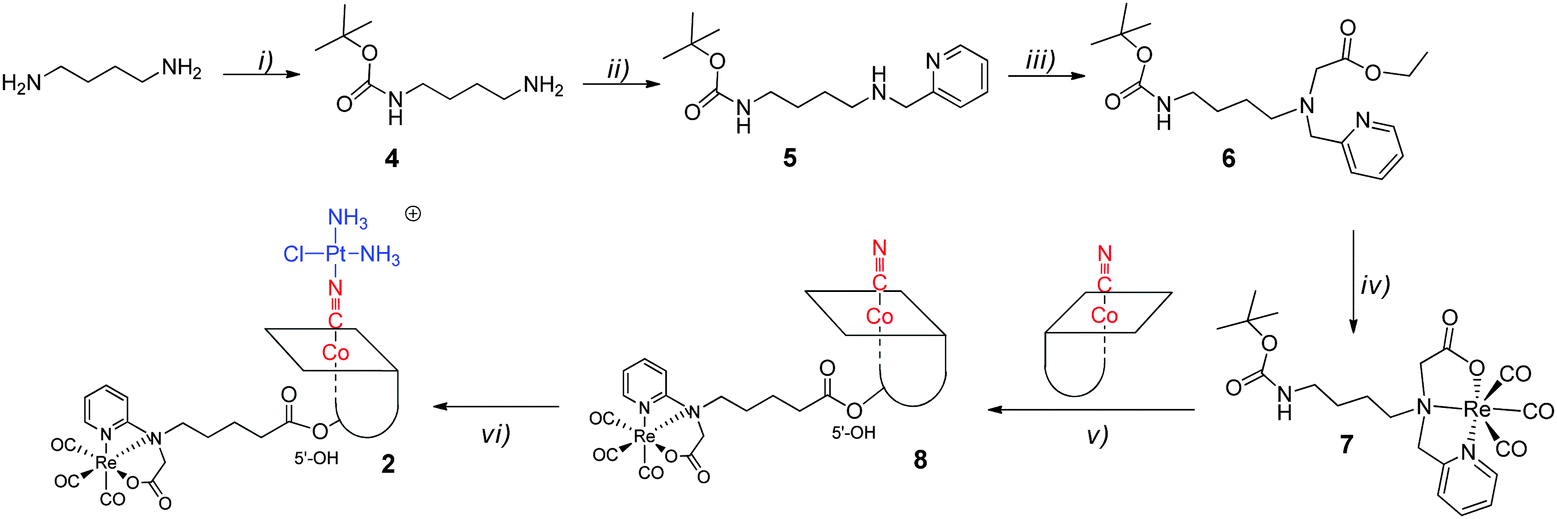 | ||
| Scheme 1 Synthesis scheme towards [{Re}-{Co}-CN-{cis-PtCl(NH3)2}]+ (2): (i) Boc2O; (ii) py-2-CHO, NaBH4; (iii) BrCH2CO2Et; (iv) [NEt4]2[ReBr3(CO)3]; (v) TFA and then CDT, TEA; (vi) cisplatin, AgNO3. | ||
The IR stretching frequency of CN− was higher in 2 (2200 cm−1) than in B12 (2134 cm−1). It confirmed the formation of {Co}}–CN{–{Pt} coordinative bond in 2, as it was previously reported that CN− stretching frequency, bridging two metal centers ({M}}–CN{–{M′}), appears at higher frequency than terminal CN− ({M}}–CN).15,39 The doubly charged species (m/z 1076.6) on ESI-MS confirmed the formation of 2 and the NMR spectrum is in agreement with its authenticity. The single signal in the 195Pt NMR spectrum at −2354 ppm is in the same range as previously reported B12 derivatives containing {Co}–CN{–{Pt} bonds.15,40 Compared to the 1H NMR spectrum of [{Co}-CN-{PtCl(NH3)2}]+ previously published40 the spectrum of [{Re}-{Co}-CN-{cis-PtCl(NH3)2}]+ shows four additional 1H signals (from the Re-[chelate]-ligand) in the fingerprint aromatic region >5.5 ppm (unmarked peaks in Fig. 3), which further confirmed the presence of the rhenium complex.
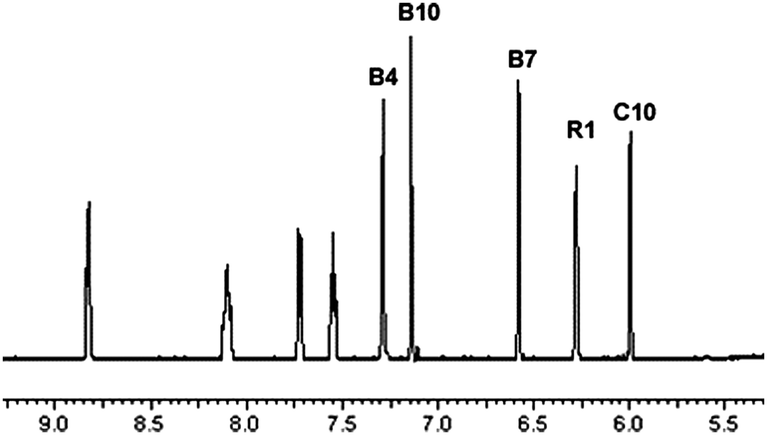 | ||
| Fig. 3 1H NMR spectrum of [{Re}-{Co}-CN-{cis-PtCl(NH3)2}]+ (2). | ||
Affinity of B12 derivatives towards transport proteins
Affinity to transport proteins of B12 derivatives was measured by Differential Scanning Fluorometry (DSF), where a fluorescent dye interacts with the hydrophobic sites of unfolded proteins and the melting temperature (Tm) of proteins can be inferred from the recorded fluorescence intensity.41,42 Following ligand binding, the protein stability is enhanced and the ligand-bound proteins unfold at higher temperature with respect to the free proteins. B12 derivatives which bind to B12 transport proteins would produce comparable ΔTm values to those of B12 while ΔTm values would be much lower if these derivatives do not bind to B12 transport proteins. In Fig. 4, B12 was used as a positive control for the three transport proteins and cobinamide (Cbi) was used as a negative control.42 Despite the chemical modification of the B12 structure, compound 2 retained good affinity to all B12 transport proteins as it has comparable ΔTm values to those of B12.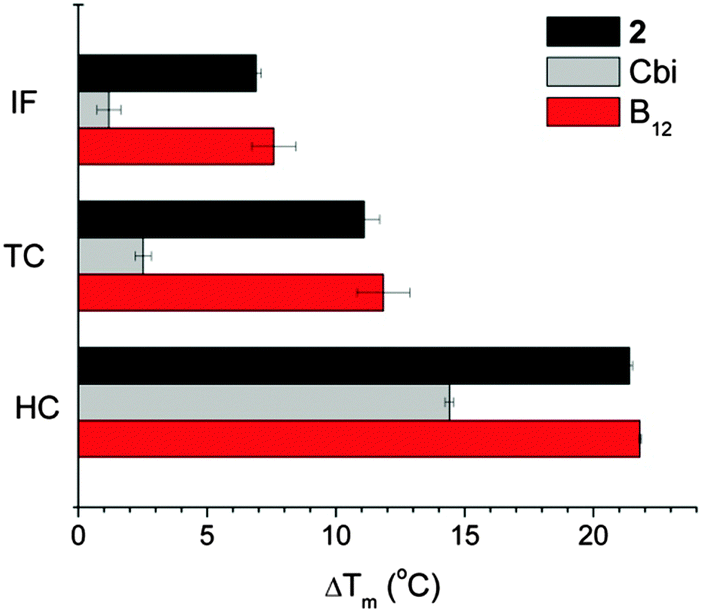 | ||
| Fig. 4 Binding of B12 and [{Re}-{Co}-CN-{cis-PtCl(NH3)2}]+ (2) to haptocorrin (HC), intrinsic factor (IF), and transcobalamin (TC) determined by differential scanning fluorometry. High ΔTm values indicate strong binding. | ||
Cellular uptake of [{Co}-CN-{cis-PtCl(NH3)2}]+, [{Re}-{Co}-CN-{cis-PtCl(NH3)2}]+ and [{Co}-CN-{trans-Pt(Cyt)(NH3)2}]2+
To obtain information on the action of these compounds, knowledge on the amount of drug taken up by cells and accumulated in the nucleus with and without B12 is required and is the base for evaluating the efficiency of B12 as a tumor-targeting drug carrier molecule. The metals (Co, Re, Pt) in B12–drugs conjugates can be measured by ICP-MS, i.e., cytosolic and nucleic determination of 59Co, 195Pt, and 187Re reveal the intracellular distribution of these compounds. Fig. 5A shows the cellular cisplatin uptake in K562 cells, determined as the content in the cytosol. The uptake of platinum containing B12 derivatives (1, 2, and 3) is approximately one third compared to the uptake of cisplatin. Similarly it is seen from Fig. 5B that the platinum uptake into cellular nuclei of 1, 2, and 3 was low compared to cisplatin. The low nucleic platinum uptake of 3 was expected as it has a transplatin-based structure.43,44 More interestingly, substantial extracellular Pt-release from the B12 structures, which would have resulted in comparative cellular uptake to cisplatin, was not observed. These data indicate that Pt is not released in the extracellular media but taken up together with B12. The complexes are protected when conjugated to the B12 structure. Unexpectedly, the cytosolic uptake of B12 was considerably lower than of [Cbl-OH2]+, 1, 2, and 3 (Fig. 5). This indicated that the observed uptake was not mediated by TC as this protein has similar affinities to B12, 2 (Fig. 4), and 3.42 Therefore TC would have allowed similar accumulation of these compounds in cellular components.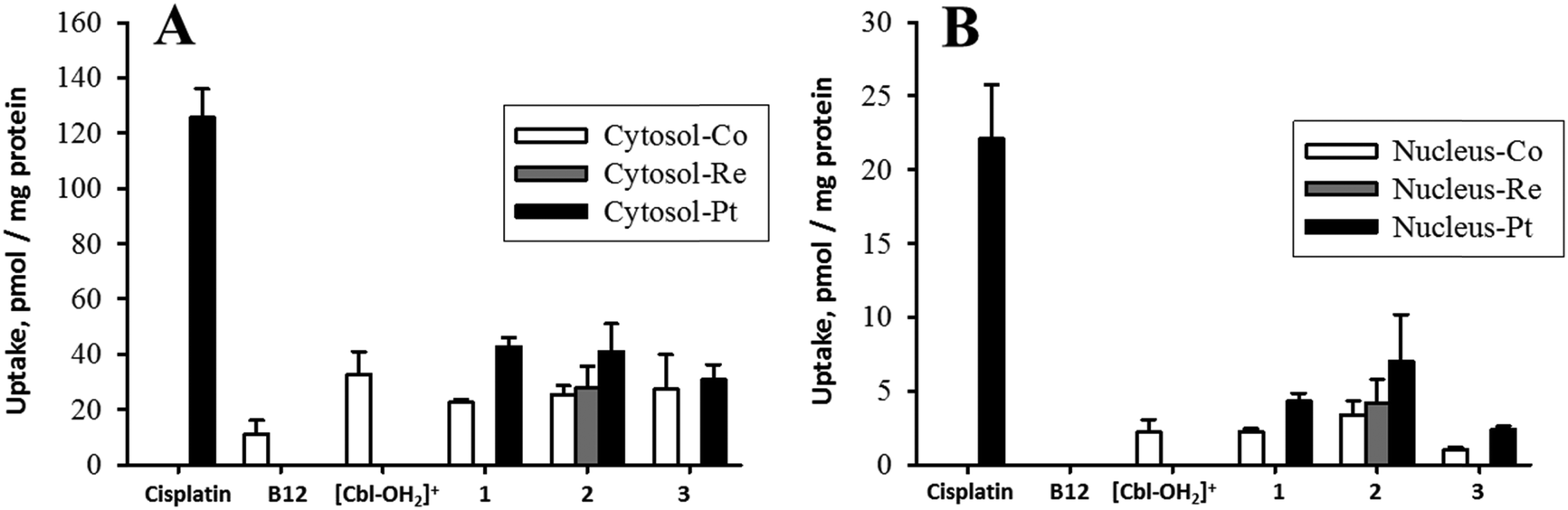 | ||
| Fig. 5 Uptake in K562 cellular cytosol (A) and nucleus (B) of [{Co}-CN-{cis-PtCl(NH3)2}]+ (1), [{Re}-{Co}-CN-{cis-PtCl(NH3)2}]+ (2), and [{Co}-CN-{trans-Pt(Cyt)(NH3)2}]2+ (3). Cisplatin, B12, [Cbl-OH2]+ were included as control samples. | ||
It was previously reported that B12 derivatives are able to enter cells without the support of TC. This uptake was, nevertheless, less efficient.45–48 HeLa cells internalized only 1–2% of free B12 with respect to holo-TC.45 It was later concluded that free B12 supported cell growth ca. 100 to 1000-fold less efficiently than holo-TC.49 However, human skin fibroblasts could internalize free B12 better than holo-TC, up to more than 20% were reported.46 Berliner et al. proposed a free B12-specific receptor on the cell membrane46 while Hitzig et al. suggested an α2-globulin-like protein in serum as the alternative pathway for cell to internalize free B12 derivatives.50
In this study, we observed higher accumulation of [Cbl-OH2]+ as compared to B12. In a previous report, it was found that [Cbl-OH2]+ was internalized and retained better than B12 in human skin fibroblasts culture51 but the root of this difference was not fully understood. As we also found comparable cellular uptake among compounds 1, 2, 3, and [Cbl-OH2]+, we conclude that the positive charge carried by these compounds allows cells to internalize them better than the neutral B12 derivatives (Fig. 5). Behavioral differences between charged and neutral B12 derivatives were also previously observed.52 It is possible that charged B12 compounds were either distinctly recognized by a B12-specific receptor or they simply passed through the cationic channels on cell membranes. This, however, requires further clarification. In any case, the experimental results highlighted once more the complex multichannel uptake pathways of B12 vitamin analogues including internalization of holo-TC, free B12 derivatives, and charged B12 derivatives. The preferable cellular import of charged B12 compounds should be kept in perspective for future design of pharmaceutical B12 derivatives.
Drugs like cisplatin and cytarabine attached to the β-CN− of B12 are released by reducing enzymes in the cytosol.15,40,42 Furthermore, subsequent cytotoxic activities of the released drugs from [{Co}-CN-{cis-PtCl(NH3)2}]+ (1) and [{Co}-CN-{trans-Pt(Cyt)(NH3)2}]2+ (3) were also demonstrated.42,52 Cisplatin interferes with cell division by binding to DNA. Previous studies demonstrated accumulation of cisplatin in the cytosol and the nucleus in non-adherent EATC and adherent ELA cells.53 It is therefore especially important for compound 1 and 2 to deliver platinum to nucleus for DNA interaction. As evident from Fig. 6, platinum accumulated in the nucleus in EATC and in ELA cells similar to K562 cells. Furthermore, we found that the absolute amount of platinum bound to DNA in cells exposed for 18 hours to 10 μM and 50 μM [{Re}-{Co}-CN-{cis-PtCl(NH3)2}]+ (2) was 19 and 83 pmol μg−1 DNA (EATC) and 5 and 20 pmol μg−1 DNA (ELA). Hence, cisplatin coupled to B12 derivative did accumulate in the nucleus and was bound to DNA.
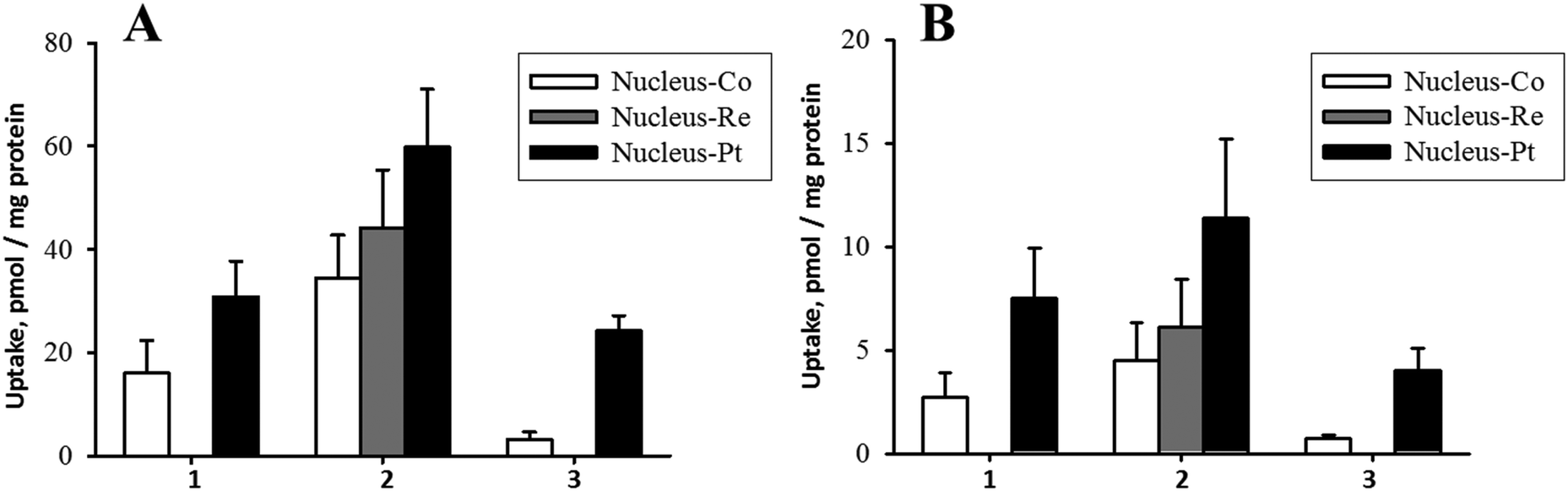 | ||
| Fig. 6 Nuclear uptake of [{Co}-CN-{cis-PtCl(NH3)2}]+ (1), [{Re}-{Co}-CN-{cis-PtCl(NH3)2}]+ (2), and [{Co}-CN-{trans-Pt(Cyt)(NH3)2}]2+ (3) in non-adherent Ehrlich cells (EATC) and adherent Ehrlich Lettre cells (ELA). Uptake was determined as Co, Re, and Pt content in EATC (A) and ELA (B) measured by ICPMS. | ||
The uptake of Pt into cellular components was always higher than Co (Fig. 5 and 6). A correlation between cobalt and platinum uptake for the three compounds 1, 2 and 3 is shown in Fig. 7. The equation for the trend line is y = 1.60X + 4.9. The one outlier point for EATC, cytosol was omitted. The correlation shows that platinum is taken up by a factor of 1.6 higher than cobalt as estimated from the cytosolic and nucleic content. This observation could reflect a more efficient efflux of Co as compared to platinum.
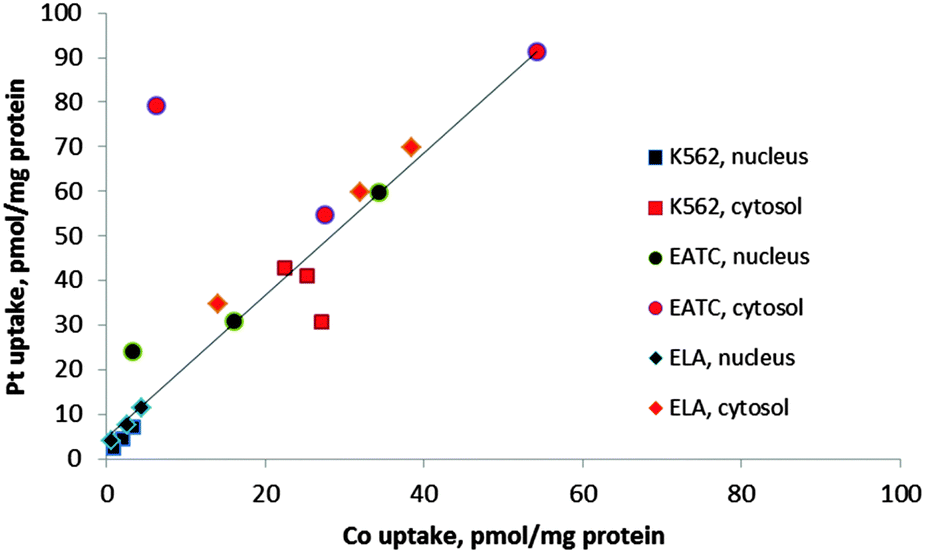 | ||
| Fig. 7 Correlation between drug (Pt) and vitamin B12 (Co) in K562, EATC, and ELA cells. Uptake was determined as the nuclear (black symbols) and cytosolic (red symbols) Pt and Co content measured by ICPMS. | ||
Conclusions
We reported here comparable uptake in cellular nuclei and cytosol of different B12–drug conjugates, including [{Co}-CN-{cis-PtCl(NH3)2}]+, [{Re}-{Co}-CN-{cis-PtCl(NH3)2}]+, and [{Co}-CN-{trans-Pt(Cyt)(NH3)2}]2+. The results underline that these B12 derivatives deliver cispaltin and cisplatin-related drugs to both, cytosol and nuclei, in two different cancer cell lines. It was found that charged B12 derivatives were preferentially taken up into cellular compartments as compared to neutral and native B12. We therefore propose the existence of an additional uptake pathway for charged B12 derivatives in addition to the previously known TC-mediated B12 uptake pathway. Accumulation of released platinum in the nucleus was clearly observed and explains the cytotoxic action of these compounds as previously described.50Experimental
Materials and methods
Analytical high performance liquid chromatography (HPLC) analyses were performed using a Merck-Hitachi L-7000 system equipped with a diode-array UV/Vis spectrometer. Reaction progresses to 2, 3, 7 and 8 were monitored by HPLC. Preparative HPLC purification was performed on Varian Prostar system with two Prostar 215 pumps and a Prostar 320 UV/Vis detector. Reaction progresses to 4, 5, and 6 were assessed by TLC. After purification the B12 derivatives were liophilized. Purity was assessed by analytical HPLC, IR, ESI-MS (Bruker Daltonic) and NMR. NMR spectra were recorded by Bruker DRX 500 MHz spectrometer. Chemical shifts are reported relative to residual solvent protons and carbons. The chemical shifts of 195Pt NMR spectra were relative to Na2PtCl6 in D2O.Phosphate-buffered saline (PBS) contained 137 mM NaCl, 2.6 mM KCl, 6.5 mM Na2HPO4, and 1.5 mM KH2PO4. Lysis buffer was prepared from150 mM NaCl, 20 mM HEPES buffer, 1 mM EDTA, 0.5% Triton X-100 (Pharmacia Biotech AB), 1% protease inhibitor cocktail (freshly added), 1 mM NaVO3 (freshly added). All aqueous solutions for ICPMS measurement were prepared in purified water (>18.2 MΩ) from Milli-Q deionization unit. Standards and samples were prepared in a mixture of HNO3 0.65%![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :HCl 0.1%.
:HCl 0.1%.
Syntheses
N-tert-Butyloxycarbonyldiamines (4),54 [NEt4]2[ReBr3(CO)3],55 and [{Co}-CN-{trans-Pt(Cyt)(NH3)2}]2+ (3)42 were prepared according to literature procedures. Compounds (5) and (6) were prepared as described in literature56 but with the following modifications: 2 g (4) (11 mmol) was dissolved in 100 ml MeOH. 1.3 g pyridine-2-carbaldehyde (12 mmol) was added dropwise. After stirring for 3 hours at room temperature, the solvent was removed and the imine dissolved in 50 ml ethanol (EtOH). Reduction with 1.5 g NaBH4 in 2 h, removal of solvent, re-dissolution in diluted NaHCO3 and extraction into CH2Cl2 gave the raw product. The purification of 5 was performed with silicagel column (ethylacetate, methanol 1:3) (2.8 g, 91%). 1H-NMR (200 MHz, MeOD, δ in ppm): δ = 8.63(m, 1H), 7.87(m, 1H), 7.49(m, 1H), 7.41 (m, 1H), 4.37 (s, 2H), 3.32 (m, 2H), 3.29 (m, 2H), 1.94-1.42 (m, 13H). Rf = 0.28 (NH4OH/methanol/ethylacetate, 1:10:89). ESI-MS: m/z = 302.3 [M + H + Na]2+. 1.5 g (5) was mixed with 1 ml triethylamine in 50 ml EtOH. The mixture was cooled to 0 °C. 600 μl bromo-acetic acid ethylester was added dropwise within 5 min. After stirring for 30 minutes at 0 °C and 16 hours at room temperature, the solvent was removed in vacuo. The residue was dissolved in CH2Cl2. Extraction was performed with H2O. The organic phase was dried over MgSO4, filtered and the solvent was removed. The product (6) (1.2 g, 60%) was collected as oil. 1H-NMR (200 MHz, MeOD, δ in ppm): δ = 8.51 (m, 1H), 7.63 (m, 1H), 7.47 (d, J = 7.8 Hz, 1H), 7.13 (m, 1H), 4.73 (m, 1H), 4.13 (q, J = 7.2, 7, 7.2 Hz, 2H), 3,89 (s, 2H), 3.36 (s, 2H), 3.06 (d, J = 6 Hz, 2H), 2.65 (t, J = 6.6, 6.8 Hz, 2H), 2.61–1.20 (m, 15H). ESI-MS: m/z = 366.3 [M + H]+.
Affinity to transport proteins
Binding to HC/IF and TC was measured by differential scanning fluorimetry (DSF). Details of the method, protein expression and purification was previously described.42DNA was isolated according to the following procedure. Cells were centrifuged two times for 5 s at 13000–16000 g. The precipitate was subsequently lysed with 300 μl lysis solution. 100 μl of Protein Precipitation Solution was added and mixed well. After the proteins were precipitated, the supernatant was transferred into isopropanol (300 μl). This mix is centrifuged for 1 minute at 13000–16000 g to collect DNA as precipitate. The DNA fractions were washed with 70% ethanol. DNA hydration solution (50 μl) was then added to the dry DNA. The mixture was then incubated in a water bath at 65 °C for 1 hour to dissolve DNA, followed by incubating at room temperature overnight. Both subcellular fractions were diluted with 0.65% HNO3/0.1% HCl for ICPMS measurement.
Acknowledgements
Financial support by the University of Zürich and the Chemical and Molecular Science Graduate School CMSZH is gratefully acknowledged. COST action CM1105 is acknowledged for their support.References
- L. Randaccio, S. Geremia, G. Nardin and J. Wuerges, Coord. Chem. Rev., 2006, 250, 1332–1350 CrossRef CAS.
- M. D. Wirt, I. Sagi, E. Chen, S. M. Frisbie, R. Lee and M. R. Chance, J. Am. Chem. Soc., 1991, 113, 5299–5304 CrossRef CAS.
- M. D. Wirt, I. Sagi and M. R. Chance, Biophys. J., 1992, 63, 412–417 CrossRef CAS PubMed.
- G. A. Walker, S. Murphy and F. M. Huennekens, Arch. Biochem. Biophys., 1969, 134, 95–102 CrossRef CAS PubMed.
- M. V. Fonseca and J. C. Escalante-Semerena, J. Biol. Chem., 2001, 276, 32101–32108 CrossRef CAS PubMed.
- S. N. Fedosov, Subcell. Biochem., 2012, 56, 347–367 CAS.
- M. J. Nielsen, M. R. Rasmussen, C. B. Andersen, E. Nexø and S. K. Moestrup, Nat. Rev. Gastroenterol. Hepatol., 2012, 9, 345–354 CrossRef CAS PubMed.
- J. F. Kolhouse and R. H. Allen, J. Clin. Invest., 1977, 60, 1381–1392 CrossRef CAS PubMed.
- E. Stupperich and E. Nexø, Eur. J. Biochem., 1991, 199, 299–303 CrossRef CAS PubMed.
- S. N. Fedosov, N. U. Fedosova, B. Kräutler, E. Nexø and T. E. Petersen, Biochemistry, 2007, 46, 6446–6458 CrossRef CAS PubMed.
- J. Wuerges, S. Geremia and L. Randaccio, Biochem. J., 2007, 403, 431–440 CrossRef CAS PubMed.
- P. M. Pathare, D. S. Wilbur, S. Heusser, E. V. Quadros, P. McLoughlin and A. C. Morgan, Bioconjugate Chem., 1996, 7, 217–232 CrossRef CAS PubMed.
- W. A. Howard, A. Bayomi, E. Natarajan, M. A. Aziza, O. El-Ahmady, C. B. Grissom and F. G. West, Bioconjugate Chem., 1997, 8, 498–502 CrossRef CAS PubMed.
- J. D. Bagnato, A. L. Eilers, R. A. Horton and C. B. Grissom, J. Org. Chem., 2004, 69, 8987–8996 CrossRef CAS PubMed.
- S. Mundwiler, B. Spingler, P. Kurz, S. Kunze and R. Alberto, Chem. – Eur. J., 2005, 11, 4089–4095 CrossRef CAS PubMed.
- R. Mukherjee, E. G. Donnay, M. A. Radomski, C. Miller, D. A. Redfern, A. Gericke, D. S. Damron and N. E. Brasch, Chem. Commun., 2008, 3783–3785 RSC.
- C. C. Smeltzer, M. J. Cannon, P. R. Pinson, J. D. Munger, F. G. West and C. B. Grissom, Org. Lett., 2001, 3, 799–801 CrossRef CAS PubMed.
- S. Kunze, F. Zobi, P. Kurz, B. Spingler and R. Alberto, Angew. Chem. Int. Edit., 2004, 43, 5025–5029 CrossRef CAS PubMed.
- P. Ruiz-Sanchez, S. Mundwiler, A. Medina-Molner, B. Spingler and R. Alberto, J. Organomet. Chem., 2007, 692, 1358–1362 CrossRef CAS.
- J. Wuerges, G. Garau, S. Geremia, S. N. Fedosov, T. E. Petersen and L. Randaccio, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 4386–4391 CrossRef CAS PubMed.
- M. Lee and C. B. Grissom, Org. Lett., 2009, 11, 2499–2502 CrossRef CAS PubMed.
- N. Viola-Villegas, A. E. Rabideau, M. Bartholoma, J. Zubieta and R. P. Doyle, J. Med. Chem., 2009, 52, 5253–5261 CrossRef CAS PubMed.
- P. Siega, J. Wuerges, F. Arena, E. Gianolio, S. N. Fedosov, R. Dreos, S. Geremia, S. Aime and L. Randaccio, Chem. – Eur. J., 2009, 15, 7980–7989 CrossRef CAS PubMed.
- A. K. Petrus, A. R. Vortherms, T. J. Fairchild and R. P. Doyle, ChemMedChem, 2007, 2, 1717–1721 CrossRef CAS PubMed.
- C. H. Fazen, D. Valentin, T. J. Fairchild and R. P. Doyle, J. Med. Chem., 2011, 54, 8707–8711 CrossRef CAS PubMed.
- D. A. Collins and H. P. Hogenkamp, J. Nucl. Med., 1997, 38, 717–723 CAS.
- J. Q. Yang, Y. Li, J. Lu and X. B. Wang, J. Radioanal. Nucl. Chem., 2005, 265, 467–472 CrossRef CAS.
- B. Spingler, S. Mundwiler, P. Ruiz-Sánchez, D. R. van Staveren and R. Alberto, Eur. J. Inorg. Chem., 2007, 2641–2647 CrossRef CAS.
- J. H. Matthews, Blood, 1997, 89, 4600–4607 CAS.
- K. Zhou and F. Zelder, Angew. Chem., Int. Ed., 2010, 49, 5178–5180 CrossRef CAS PubMed.
- K. Zhou, R. M. Oetterli, H. Brandl, F. E. Lyatuu, W. Buckel and F. Zelder, ChemBioChem, 2012, 13, 2052–2055 CrossRef CAS PubMed.
- R. Waibel, H. Treichler, N. G. Schaefer, D. R. van Staveren, S. Mundwiler, S. Kunze, M. Kuenzi, R. Alberto, J. Nuesch, A. Knuth, H. Moch, R. Schibli and P. A. Schubiger, Cancer Res., 2008, 68, 2904–2911 CrossRef CAS PubMed.
- A. K. Petrus, D. G. Allis, R. P. Smith, T. J. Fairchild and R. P. Doyle, ChemMedChem, 2009, 4, 421–426 CrossRef CAS PubMed.
- A. K. Petrus, T. J. Fairchild and R. P. Doyle, Angew. Chem., Int. Ed., 2009, 48, 1022–1028 CrossRef CAS PubMed.
- S. M. Clardy, D. G. Allis, T. J. Fairchild and R. P. Doyle, Expert Opin. Drug Delivery, 2010, 8, 127–140 CrossRef PubMed.
- D. L. Lildballe, E. Mutti, H. Birn and E. Nexø, PLoS One, 2012, 7, e46657 CAS.
- T. C. Johnstone, G. Y. Park and S. J. Lippard, Anticancer Res., 2014, 34, 471–476 CAS.
- D. Wang and S. J. Lippard, Nat. Rev. Drug Discovery, 2005, 4, 307–320 CrossRef CAS PubMed.
- D. A. Dows, A. Haim and W. K. Wilmarth, J. Inorg. Nucl. Chem., 1961, 21, 33–37 CrossRef CAS.
- P. Ruiz-Sanchez, S. Mundwiler, B. Spingler, N. R. Buan, J. C. Escalante-Semerena and R. Alberto, JBIC, J. Biol. Inorg. Chem., 2008, 13, 335–347 CrossRef CAS PubMed.
- F. H. Niesen, H. Berglund and M. Vedadi, Nat. Protoc., 2007, 2, 2212–2221 CrossRef CAS PubMed.
- M. T. Q. Tran, E. Furger and R. Alberto, Org. Biomol. Chem., 2013, 11, 3247–3254 CAS.
- N. P. Johnson, J. D. Hoeschele, N. B. Kuemmerle, W. E. Masker and R. O. Rahn, Chem. – Biol. Interact., 1978, 23, 267–271 CrossRef CAS PubMed.
- N. P. Johnson, J. D. Hoeschele and R. O. Rahn, Chem. – Biol. Interact., 1980, 30, 151–169 CrossRef CAS PubMed.
- C. A. Hall, W. H. Hitzig, P. D. Green and J. A. Begley, Blood, 1979, 53, 251–263 CAS.
- N. Berliner and L. E. Rosenberg, Metabolism, 1981, 30, 230–236 CrossRef CAS PubMed.
- M. Ramasamy, D. H. Alpers, C. Tiruppathi and B. Seetharam, Am. J. Physiol., 1989, 257, G791–797 CAS.
- K. S. Ramanujam, S. Seetharam, M. Ramasamy and B. Seetharam, Am. J. Physiol., 1991, 260, G416–422 CAS.
- G. R. McLean, E. V. Quadros, S. P. Rothenberg, A. C. Morgan, J. W. Schrader and H. J. Ziltener, Blood, 1997, 89, 235–242 CAS.
- W. H. Hitzig, U. Dohmann, H. J. Pluss and D. Vischer, J. Pediatr., 1974, 85, 622–628 CrossRef CAS PubMed.
- I. Mellman, H. F. Willard, P. Youngdahl-Turner and L. E. Rosenberg, J. Biol. Chem., 1979, 254, 11847–11853 CAS.
- P. Ruiz-Sanchez, C. Konig, S. Ferrari and R. Alberto, JBIC, J. Biol. Inorg. Chem., 2011, 16, 33–44 CrossRef CAS PubMed.
- H. S. Tastesen, J. B. Holm, J. Moller, K. A. Poulsen, C. Moller, S. Sturup, E. K. Hoffmann and I. H. Lambert, Cell. Physiol. Biochem., 2010, 26, 809–820 CrossRef CAS PubMed.
- J.-F. Pons, J.-L. Fauch”re, F. Lamaty, A. Molla and R. Lazaro, Eur. J. Org. Chem., 1998, 853–859 CrossRef CAS.
- R. Alberto, R. Schibli, A. Egli, P. August Schubiger, W. A. Herrmann, G. Artus, U. Abram and T. A. Kaden, J. Organomet. Chem., 1995, 493, 119–127 CrossRef CAS.
- A. Stichelberger, Nucl. Med. Biol., 2003, 30, 465–470 CrossRef CAS PubMed.
- M. Q. Tran, Y. Nygren, C. Lundin, P. Naredi and E. Bjorn, Anal. Biochem., 2010, 396, 76–82 CrossRef CAS PubMed.
Footnotes |
| † 13 Nguyen Tuan Street, Ward 3, Go Vap District, Ho Chi Minh City, Vietnam. |
| ‡ Present address: Therwilerstrasse 47, 4153 Reinach, Switzerland. |
| This journal is © The Royal Society of Chemistry 2016 |