
Exploiting developments in nanotechnology for the preferential delivery of platinum-based anti-cancer agents to tumours: targeting some of the hallmarks of cancer
James P.
Parker
,
Ziga
Ude
and
Celine J.
Marmion
*
Centre for Synthesis and Chemical Biology, Department of Pharmaceutical & Medicinal Chemistry, Royal College of Surgeons in Ireland, 123 St. Stephen's Green, Dublin 2, Ireland. E-mail: cmarmion@rcsi.ie; Tel: +353 1 4022161
First published on 16th November 2015
Abstract
Platinum drugs as anti-cancer therapeutics are held in extremely high regard. Despite their success, there are drawbacks associated with their use; their dose-limiting toxicity, their limited activity against an array of common cancers and patient resistance to Pt-based therapeutic regimes. Current investigations in medicinal inorganic chemistry strive to offset these shortcomings through selective targeting of Pt drugs and/or the development of Pt drugs with new or multiple modes of action. A comprehensive overview showcasing how liposomes, nanocapsules, polymers, dendrimers, nanoparticles and nanotubes may be employed as vehicles to selectively deliver cytotoxic Pt payloads to tumour cells is provided.
1. Cancer and its treatment
Cancer is not one disease, but an umbrella term for over 100 different types of distinctive diseases. All forms of cancer are characterised by abnormal cell growth resulting from spontaneous, inherited, or environmentally induced genetic mutations. These cells can then invade adjoining parts of the body and spread to other organs, a process referred to as metastasis which is the major cause of death from cancer.1 Identifying differences between cancer cells, cancerous tumours, normal cells and normal tissues is of paramount importance if one is to rationally design more efficacious cancer therapies to overcome drawbacks associated with those currently in use.Numerous strategies have been investigated to combat cancer. These include but are not limited to hormonal therapies and monoclonal antibodies to antibody drug candidates and oncolytic viruses, to molecular therapies such as angiogenesis inhibitors or agents that interfere with the immune system (e.g. immune check point inhibitors and adoptive cell therapy) or the use of multi-targeted or drug combination regimes.2 An alternative approach is to employ small-interfering RNA or siRNA as a therapeutic platform to modulate the expression of disease-related genes.3 The purpose of this review however is to showcase how different types of nanotechnologies may be exploited to selectively deliver Pt drugs to tumour cells. While the earliest reports on the therapeutic use of transition metal complexes in cancer date from the sixteenth century it was, however, the serendipitous discovery of the anti-cancer properties of cisplatin, cis-[PtCl2(NH3)2], by Barnett Rosenberg in 1965 and its subsequent clinical introduction in 1978 that propelled the scientific community into conducting expansive studies on the development of metal-based drugs for therapeutic gain. Now half a century since the anti-cancer properties of cisplatin were first discovered and despite the large number of metals available, the exploitation of Pt for cancer treatment dominates other metal complexes with nearly 50% of all anti-cancer therapies being Pt based.4 While clinically very successful, it is surprising to note that only three Pt drugs boast worldwide clinical approval for the treatment of cancer, namely cisplatin, 1, carboplatin, 2, and oxaliplatin, 3, while three others have gained regional limited approval, namely nedaplatin, 4, heptaplatin, 5, and lobaplatin, 6, in Japan, South Korea and China respectively, Fig. 1.4–6
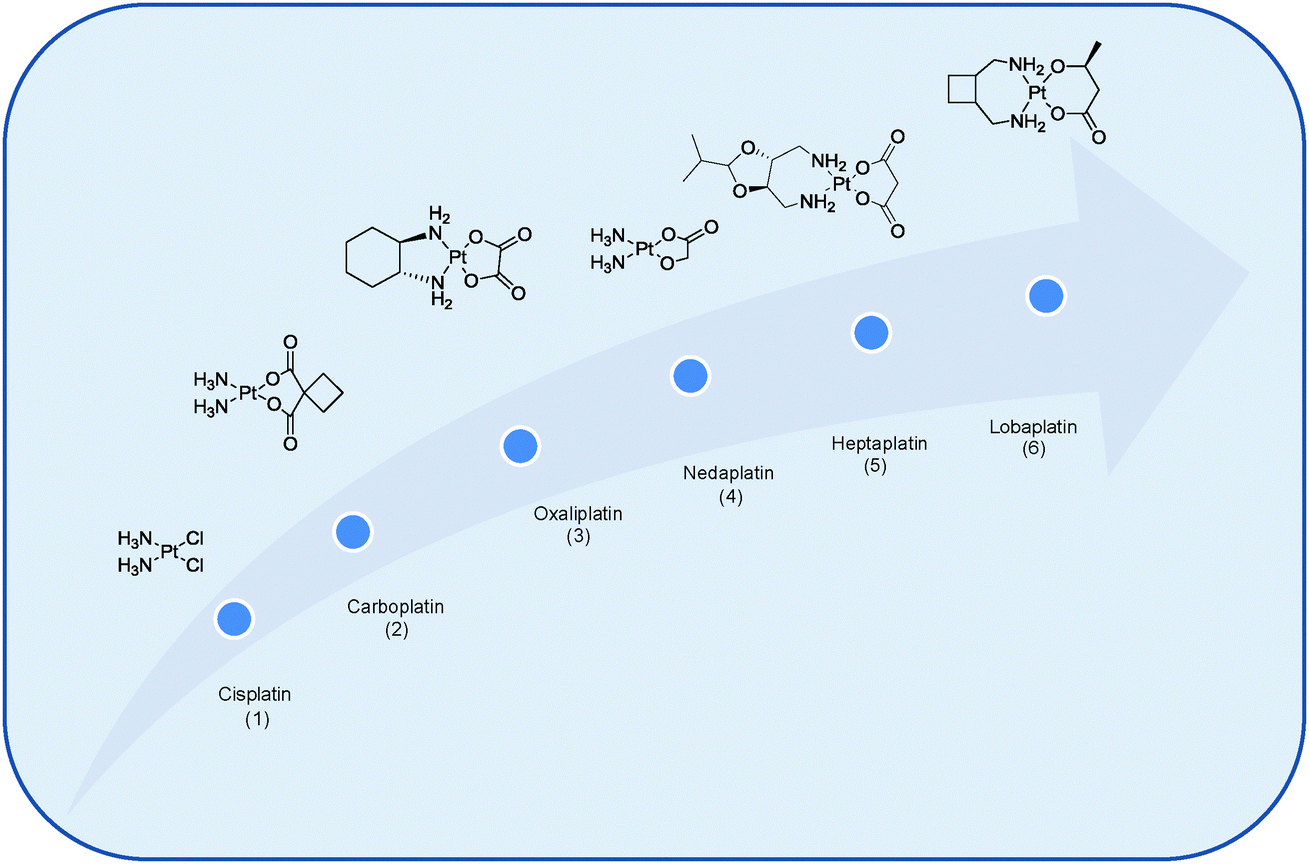 | ||
| Fig. 1 Chemical structures of Pt-based anti-cancer agents that have received worldwide clinical approval, namely cisplatin (1), carboplatin (2) and oxaliplatin (3) and regionally approved drugs nedaplatin (4), heptaplatin (5) and lobaplatin (6).6 | ||
In total, 22 Pt drug candidates have reached clinical trials.7 This is surprising given the number of anti-cancer agents that have been investigated. The clinical evaluation of 14 of these drugs was discontinued, Fig. 2, because of severe and/or unpredictable side effects, because of a lack of activity in phase II/III trials, or for economic reasons.7
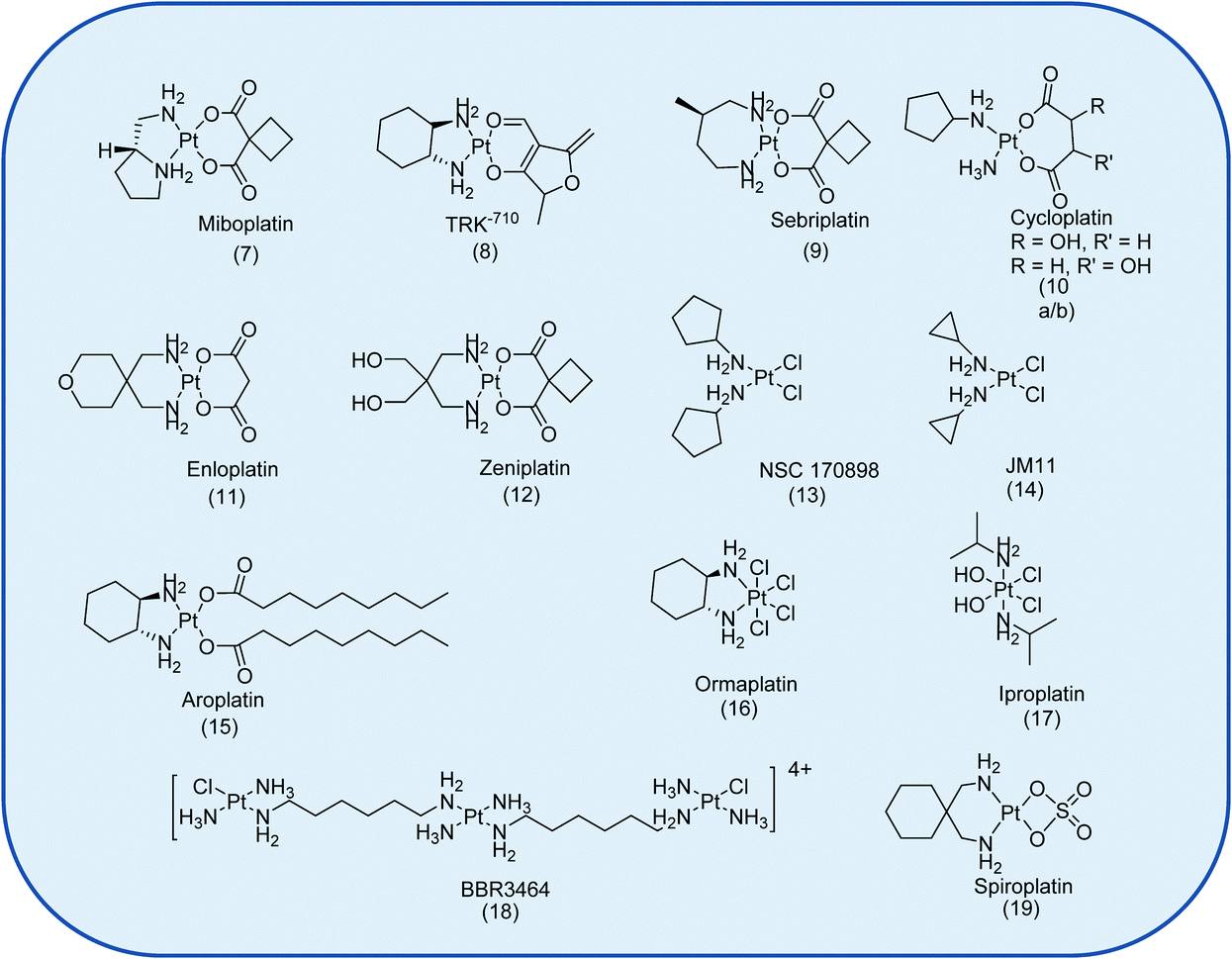 | ||
| Fig. 2 The Pt-based anti-cancer drugs discontinued from clinical trials.7 Not shown is the liposomal formulation of SP1-77 with Aroplatin™. | ||
Currently there are 4 drugs in various phases of clinical trials, namely the Pt(IV) satraplatin, 20 and the liposomal formulation Lipoplatin™, the polymeric delivery system ProLindac™, 21, and picoplatin, 22, Fig. 3.6,7 No new small molecule Pt drug has entered clinical trials since 1999.7 That said, Pt drug development still remains the subject of many current investigations with a clear shift in focus from drug discovery towards targeted drug delivery in the quest to add to the existing armamentarium of chemotherapeutic agents.
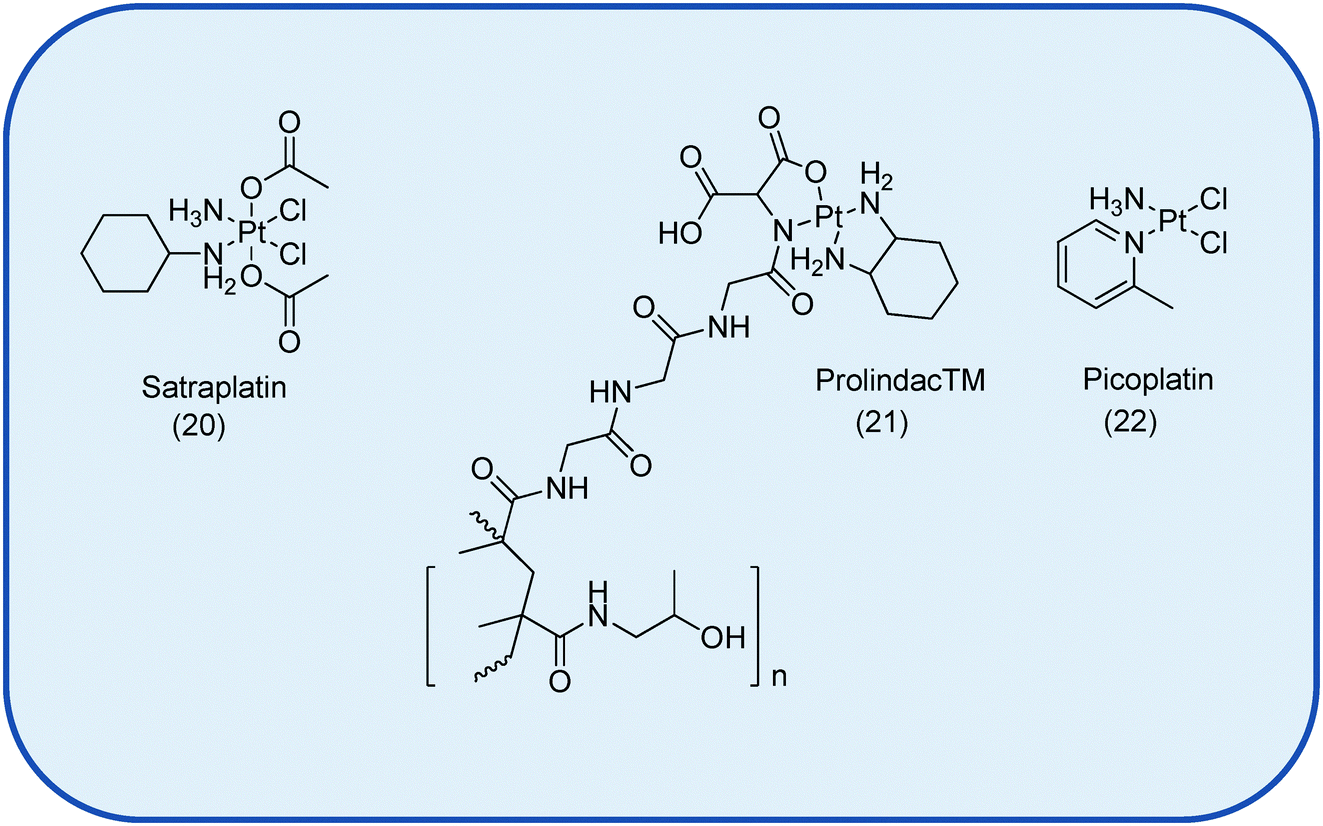 | ||
| Fig. 3 The Pt-based drugs currently undergoing clinical trials as anti-cancer therapeutics. Not shown is the liposomal formulation Lipoplatin™.7 | ||
2. Recent advances in platinum-based cancer chemotherapy
The search for Pt drugs which (i) target cancer cells and/or (ii) have a different mode of action to classical Pt drugs remains the subject of intense investigation. By targeting characteristics unique to cancer we can hope to reduce unwanted side-effects brought about by non-selective targeting while new modes of action may circumvent the intrinsic and/or acquired resistance associated with Pt drugs.2.1 Platinum drug delivery systems
Extensive studies into tumour vasculature revealed abnormal molecular and fluid transport dynamics. Certain molecules of particular sizes (typically liposomes, nanoparticles, and macromolecular drugs) were identified as having the ability to accumulate in solid tumour tissues much more so than in normal tissues.8–10 This is not possible for low molecular-weight molecules because of rapid washout by capillary blood flow. This seemingly selective accumulation, coined as the enhanced permeability and retention (EPR) effect, subsequently stimulated efforts into tumour targeting using macromolecular delivery systems such as liposomes, polymers and dendrimers. Such molecular nanostructures with well-defined particle size and shape capable of targeting tumours may overcome some of the shortcomings of existing therapies, including but not limited to poor drug bioavailability, non-specific systemic drug distribution and inadequate drug concentrations reaching the tumour. While this targeting and delivery strategy clearly offers many advantages, there have as yet been no mainstream drug delivery/targeting technologies approved to date for Pt drugs although some are currently undergoing clinical evaluation. | ||
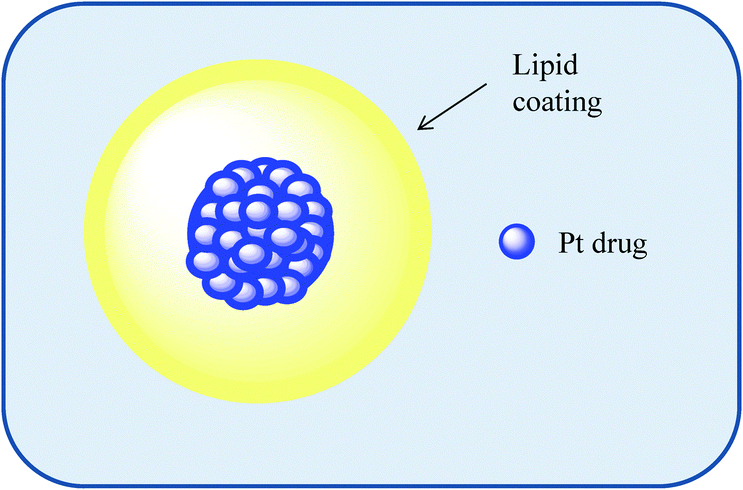
| Fig. 4 An illustration of a liposome as a macromolecular carrier for the selective delivery of drug molecules. | ||
Liposomes offer the advantage of being able to carry hydrophilic as well as hydrophobic drugs; water soluble drugs can be trapped in the interior of the liposome-enclosed aqueous core while the encapsulating bilayer can be used to deliver hydrophobic drugs. Liposomes have several advantages such as biocompatibility, versatility allowing encapsulation (and thus protection from metabolic degradation of biologically active drugs) and the capacity to deliver drugs to a desired location reducing side effects. Their surface properties, size and charge are easily modified during formulation. Such systems have been clinically approved for the anti-cancer therapeutics doxorubicin and paclitaxel.11,12 The liposomal formulation of doxorubicin and albumin-based delivery systems for paclitaxel utilise the EPR effect to allow better permeation in cancer tissue, as well as improved retention.12
Exploitation of liposomes as delivery vehicles to selectively deliver Pt drugs to tumours is not a new phenomenon. For example, Asefa et al. demonstrated time-dependent enhanced cytotoxicity of Pt drugs when loaded into mesoporous silica nanoparticles, MCM-41 and SBA-15.13,14 There are now several liposomal formulations of Pt drugs under evaluation with some currently in clinical trials and one already receiving orphan drug status.15–21 Lipoplatin™, which is a promising cisplatin–liposome formulation, is currently in advanced stages of clinical trials for the treatment of non-small cell lung cancer (NSCLC),19 HER2/neu negative metastatic breast cancer18 and advanced gastric cancer.17 The macromolecular targeting entity, comprising ∼9% cisplatin to ∼91% lipids (w/w) and having a particle size of approximately 110 nm, displays preferential uptake and retention in cancerous tissue compared to surrounding non-cancerous tissue. The formulation also shows reduced toxicity lacking the serious side effects associated with cisplatin treatment while retaining the same efficacy of cisplatin.15–21 Lipoplatin™ was granted orphan drug status by the European Medicines Agency for the treatment of pancreatic adenocarcinoma.20 The molecular mechanisms of Lipoplatin, together with pre-clinical and clinical data, are comprehensively reviewed by Staphopoulos and Boulikas.16
LiPlaCis, another cisplatin–liposomal system, reached phase I clinical trials but the trials were terminated as results indicated no additional benefit over standard cisplatin treatment.22 SPI-77, another liposomal formulation encapsulating cisplatin, advanced to several phase II studies of patients with inoperable head and neck cancer,23 advanced NSCLC24 or Pt-sensitive recurrence of ovarian cancer, but failed to progress despite being less toxic compared to cisplatin, most likely due to slow and inefficient release of its Pt payload.
More recently, the cationic modification of liposomes using the transfection agent polycation polyethylenimine (PEI), rarely used to generate liposomes for anti-tumour drugs, was employed to successfully deliver cisplatin to A549 lung cells.25 A follow up in vivo study on a H22 hepatoma-bearing mouse model indicated the liposomal system retained the efficacy of cisplatin and demonstrated reduced nephrotoxicity.26
Replacement of the chlorido ligands of cisplatin with either one or two caprylate ligands generated caprylate–cisplatin analogues which were successfully loaded into liposomes with an encapsulation efficiency in the region of 96% demonstrating unprecedented drug loading (0.21 mg cisplatin/mg of lipids) and comparable efficacy to cisplatin against A549 lung tumour cells.27
Liposomes, with their capacity to deliver water insoluble drugs, have also been investigated in vitro as potential delivery vehicles for the water insoluble Pt(II) complex, 2-(4-(tetrahydro-2H-pyran-2-yloxy)-undecyl)-propane-1,3-diamminedichloroplatinum(II). Of the seven tumour cell lines evaluated, the liposomal formulation was found to be more effective than cisplatin against cisplatin resistant testicular TGCT 1411HP and anaplastic thyroid carcinoma SW1736 cell lines.28
Several phase I and phase II clinical studies of a liposomal formulation of an oxaliplatin analogue, cis-bis-neodecanoato-trans-R,R-1,2-diaminocyclohexane platinum(II) or Aroplatin™ have been undertaken; for example phase I studies in patients with pleural mesothelioma, ovarian cancer and peritoneal carcinomatosis and sarcomatosis and in phase II for mesothelioma and advanced colorectal cancer.15 In the latter case, in patients with advanced colorectal cancer resistant to 5-fluorouracil/leucovorin, capecitabine or irinotecan, the study indicated good tolerability but limited tumour response.21 Interestingly, the liposomal carrier itself is thought to play an instrumental role in the cytotoxicity and anti-tumour activity of Aroplatin™.
Lipoxal, another liposomal oxaliplatin formulation, in contrast, was found to be well tolerated by patients with advanced disease of the gastrointestinal system with peripheral neuropathy being the most common toxic side effect.29
The transferrin (Tf)-conjugated glutaryl phosphatidylethanolamine liposomal formulation of oxaliplatin, MBP-426, has also demonstrated promise. It binds to transferrin receptors which appear to facilitate preferential tumour targeting. It is currently in phase II clinical trials.30–32
Pre-clinical studies of polyethylene glycol (PEG)ylated carboplatin liposomes on SGC-7901 gastric cell bearing mice indicated promising anti-tumour and anti-metastatic effects.33
Oxaliplatin is currently used to treat certain (wild-type KRAS) metastatic colorectal cancers expressing epidermal growth factor receptor (EGFR) in combination with the monoclonal antibody Cetuximab. Garrido et al. successfully linked Cetuximab to oxaliplatin-loaded EGFR-targeted liposomes in a pioneering study. This facilitated the selective delivery of both drug entities in a single therapeutic approach. The liposomes demonstrated enhanced tumour drug accumulation as compared to free oxaliplatin or a non-targeted liposome as well as having improved efficacy in mice inoculated with a colorectal cancer cell line which over expresses this receptor. This approach was also shown to overcome resistance associated with classical Pt drugs in that targeted delivery was also demonstrated in oxaliplatin resistance cell lines.34
The reader is directed to a series of reviews on the use of liposomes to selectively deliver Pt drugs to cancer cells. Liu et al. provide a comprehensive review of the lipid compositions, physical properties, loading methods and drug-to-lipid ratios of Aroplatin, SPI-77, Lipoplatin, Lipoxal and LiPlCis together with their pharmacokinetic, biodistribution and toxicity profiles and therapeutic efficacies both in pre-clinical animal studies and in patients.35 Wang and Guo review different drug targeting and delivery (DTD) strategies for enhancing efficacy of Pt drugs whilst reducing side effects.36 Despite the advancement of some liposomal formulations of Pt drugs in clinical trials, challenges remain as outlined in reviews by Kieler-Ferguson et al.37 and Zalba and Garrido.38 The use of liposomes, polymeric nanocarriers and carbon nanotubes to selectively deliver oxaliplatin to tumours are reviewed by Lila et al.39
Despite the aforementioned success in exploiting liposomes as Pt-drug nanocarriers, there remains a need to overcome the existing drawbacks with liposomal formulation. These include poor storage stability, rapid clearance from the bloodstream, and non-specific uptake by the mononuclear phagocytic system. Furthermore, the limited volume of the lipid bilayer makes the delivery of hydrophobic drugs highly inefficient as well as the current lack of functionalisation for targeted delivery to non-tumour based cancers. In contrast, other nanostructures offer a higher degree of functionalisation accommodating the possibility of creating specific cell targeted structures.
 | ||
| Fig. 5 An illustration of a nanocapsule as a macromolecular carrier for the selective delivery of Pt drug molecules. | ||
In 2002, Burger et al. utilised this technology to encapsulate cisplatin with high encapsulation efficiency resulting in the formation of small aggregates of cisplatin enveloped by a single lipid bilayer. The resulting Pt-loaded nanocapsules had unprecedented drug to lipid ratio and up to 1000-fold greater in vitro cytotoxicity as compared to the free drug.40 Building on this work, the same group examined the molecular architecture of these Pt-loaded nanocapsules using solid state NMR spectroscopy and demonstrated that the nanocapsule core consisted of solid cisplatin (with ∼90% present as the dichloro species) and was devoid of water.41 A further study demonstrated that while Pt loading in nanocapsules was highly efficient, stability was a potential issue. Adjusting the formulation to include a poly(ethylene glycol 2000) (PEG)-derivatised phosphatidylethanolamine and cholesterol in the bilayer coat was shown to extend the lifetime of the cisplatin nanocapsules in mouse serum.42 The high cytotoxicity associated with the cisplatin nanocapsules in ovarian cancer cells was later shown to require caveolin-1-dependent endocytosis.43 An in vivo study was conducted in nude mice bearing human ovarian carcinoma OVCAR-3 xenografts to investigate the anti-cancer efficacy and biodistribution of PEGylated cisplatin nanocapsules as compared with those of the free drug. The Pt-nanocapsules and cisplatin were found to inhibit the growth of the OVCAR-3 xenografts in nude mice to a similar extent and the concentration of Pt in both plasma and tumour were found to be similar for both formulations. The Pt derived from the nanocapsules was however shown to rapidly accumulate in the liver (4.5-fold higher accumulation as compared to cisplatin alone), and was also shown, albeit at a slower rate, to accumulate to a high concentration in the spleen. Rapid clearance from circulation appeared to be a factor contributing to the efficacy of these Pt-nanocapsules.44
Nanocapsules incorporating carboplatin have also been developed and have been found to exhibit 1000-fold greater cytotoxicity against a range of tumour cell lines as compared to carboplatin alone. This significant increase in cytotoxicity is thought to be associated with greater Pt accumulation in tumour cells resulting from uptake of the formulation by endocytosis.45
Bryde and de Kroon provide a comprehensive review of Pt nanocapsules and their potential as chemotherapeutic agents.46
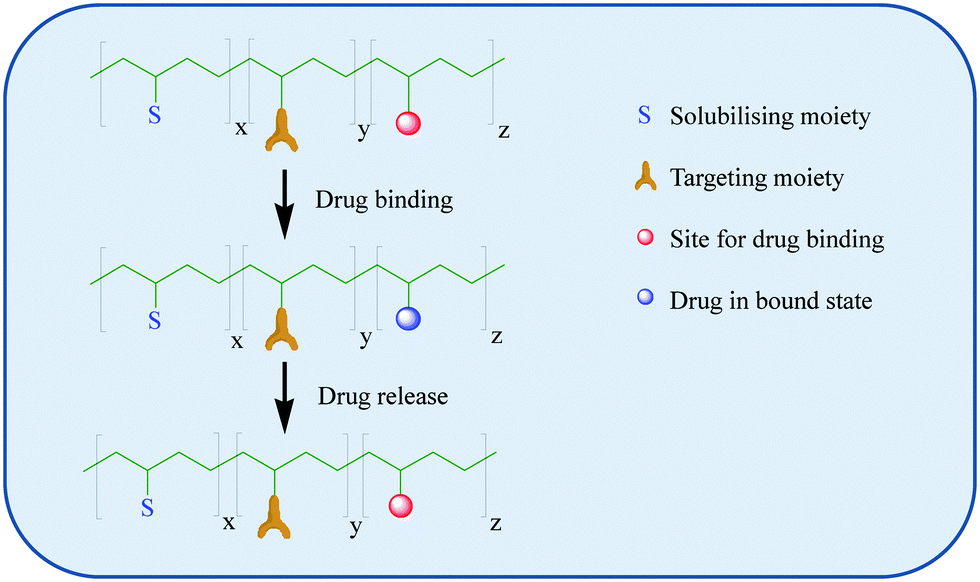 | ||
| Scheme 1 A schematic representation of the binding of a drug to a polymer and its release at its intended target. Adapted from Neuse et al.49 | ||
Early studies focussed on conjugating carboplatin to HMPA via a malonate end group of the polymer (AP5280).50 Although progressing to clinical trials, it is the nanoparticulate polymer pro-drug bound to the Pt anti-cancer agent, ProLindac™ (AP5346), 21, Fig. 3, which is dominant in this domain. It utilises a 25 kDa delivery vehicle based on hydrophilic HMPA to target the active form of oxaliplatin to tumour cells.51,52 The oxaliplatin analogue was covalently bound to the polymer via a pH sensitive amidomalonate linker that releases the active form of oxaliplatin in the more acidic environment associated with many solid tumours, compared to surrounding normal tissues. This macromolecular system was designed to be relatively non-toxic while in general circulation. Pre-clinical data in 10 tumour models showed that the polymeric delivery system was never inferior to oxaliplatin and often markedly superior as evident in the B16 melanoma tumour model of the study. Studies extended into phase I and phase II clinical trials for patients with reoccurring ovarian cancer and these indicated that ProLindac™ exhibited equal or greater efficacy than oxaliplatin.51 The patients also demonstrated a higher tolerability attributed to the ability of ProLindac™ to deliver the oxaliplatin directly to the tumour. However, its clinical evaluation was subsequently discontinued due to inconsistencies in its formulation. Several phase II combination studies in which ProLindac™ is administered in combination with other chemotherapeutics such as paclitaxel and gemcitabine in patients with solid tumours including colorectal and ovarian cancer are currently underway.15
Exploitation of the lower critical solution temperature (LCST) i.e. the critical temperature below which the components of a mixture are miscible for all compositions is another avenue receiving attention of late. This property has been exploited in the development of thermosensitive Pt(II)-cyclotriphosphazenes in which (diammine)Pt(II)-cyclotriphosphazenes were conjugated to alkoxy polyethylene glycol. These were found to exhibit variable LCST in the wide range of 12 to 92 °C and were subsequently assessed for their anti-cancer activity both in vitro and in vivo.53,54 These conjugates offer several advantages over unconjugated cancer drugs or those physically loaded in polymeric matrices such as liposomes. For example, the conjugates with LCST below body temperature offer the possibility of direct application of the nanoparticulate to the tumour via intra-tumoural injection where degradation of the phosphazene ring results in the release of the (diammine)Pt(II) cytotoxic agent in vivo. Another advantage is the reproducible formulation of the Pt–polymer conjugates as well as their high degree of functionality enabling modification of their LCST and solubility. They are also capable of carrying and delivering high quantities of the therapeutic agent. Each investigated conjugate showed a markedly higher accumulation within the chosen cells when compared to cisplatin demonstrating potent anti-cancer activity both in vitro and in vivo against murine L1210 with the effective dose (ED50) comparable to cisplatin and a mean survival time for the mice much greater than those treated with cisplatin or carboplatin.
Other polyphosphazene–Pt derivatives have also been synthesised and their in vivo potential evaluated.53,55 One of the more promising results involved the modification of an amphiphilic polyphosphazene using pegylation and its conjugation to the anti-tumour diaminocyclohexane Pt(II) moiety. The resulting Pt–polymer conjugate was shown to accumulate in tumour tissue to a much greater extent as compared to normal tissue (tumour/tissue ratio > 4) and exhibited high in vitro cytotoxicity against human cancer cell lines.56 Amphiphilic polyphosphazene–Pt conjugates capable of assembling into stable nanoparticles with a mean diameter of approximately 90–200 nm have also more been reported.57 These conjugates exhibited good activity against a selection of human tumour cell lines but had lower in vitro cytotoxicity as compared to cisplatin.
Jain et al. developed orally active hyaluronic acid coupled chitosan nanoparticles bearing oxaliplatin encapsulated in Eudragit S100 for colonic delivery of oxaliplatin. Superior efficacy was demonstrated in a HT-29 colorectal murine tumour model with high local accumulation of the nanoparticles in colonic tumours over a prolonged period of time.58
Lippard et al. loaded the Pt(IV) prodrug of cisplatin into poly(D,L-lactic-co-glycolic acid) (PLGA)-PEG-functionalized polymers decorated with prostate-specific membrane antigen (PSMA) targeting aptamers for enhanced targeted delivery to and in vitro cytotoxicity against prostate cancer cells. Release of the prodrug was facilited upon cell entry whereupon reduction to cisplatin occurred with subsequent formation of cisplatin 1,2-intrastrand d(GpG) cross-links on nuclear DNA. The efficacy of the Pt-loaded nanoparticles against the PSMA(+) LNCaP cells was found to be an order of magnitude greater than cisplatin alone.59 A later in vivo study confirmed the therapeutic efficacy of these Pt-PLGA-b-PEG-Apt-nanoparticle with improved pharmacokinetics, biodistribution, tolerability and efficacy when compared to cisplatin.60
The expression of integrins, transmembrane proteins involved in cell adhesion and cell signalling, is commonly upregulated in inflammatory diseases and in cancers. The α(v)β(3) integrin in particular is differentially upregulated on angiogenic endothelial cells in addition to many tumour cells. As such, integrins have also been the subject of investigation for targeted drug delivery. Lippard et al. rationally designed cisplatin prodrug loaded PLGA-PEG nanoparticles with differential targeting to the α(v)β(3) integrin on cancer cells using the cyclic pentapeptide c(RGDfK). These nanoparticles, as compared to cisplatin, had enhanced efficacy against prostate and breast cancer epithelial cells as well as being more efficacious and better tolerated in an orthotopic human breast cancer xenograft model in vivo.61
Sadhukha and Probha likewise successfully exploited PLGA nanoparticles but, in their case, for the targeted delivery of carboplatin. The greater cytotoxicity of the carboplatin-loaded nanoparticles against several cell lines as compared to carboplatin alone was attributed to enhanced and more rapid intracellular accumulation and greater distribution within the cell nucleus.62
While much research has focused on exploiting pH or the reduction environment of tumour cells to mediate drug release from nanocarriers, there are only a few examples of systems specifically responsive to physiological levels of H2O2 (50–100 mM).63–65 Guo, He et al. have recently developed an innovative H2O2-responsive PLGA-based nanocarrier incorporating cisplatin and catalase, the latter acting as an O2-generating agent. Upon intracellular H2O2 penetration, catalysed by catalase, O2 is released which results in an increase in pressure and generation of high levels of ROS species. The increase in pressure causes rupturing of the nanoparticle and release of the drug payload. An enhancement in cytotoxicity was observed for the carrier system as compared to cisplatin. Moreover, release of O2 was found to overcome hypoxia-induced multi-drug resistance in cancer cells. This system is the first of its kind to integrate chemotherapy and oxygen therapy in a synergistic manner.66
A rationally designed biodegradable beta-casein–chitosan nanocarrier system loaded with the bipyridine morpholine dithio-carbamate Pt(II) nitrate developed by Razmi et al. has also shown promise for targeted oral delivery applications. This study investigated the influence of pH on the formation of these stable colloidal systems with a pH of 5.7 being optimal for particle formation. This carrier system was found to have enhanced cellular uptake into and greater efficacy against colorectal carcinoma HCT116 cells as compared to the free Pt complex.67
Condensation polymerisation was employed to generate backbone Pt(IV)-coordination polymers using either diamminedichlorodihydroxyplatinum or its dicarboxyl derivative diamminedichlorodisuccinatoplatinum as comonomers.68,69In vitro studies confirmed that these polymers were cytotoxic against a range of tumour cell lines and an in vivo study demonstrated that, in comparison to the monomer diamminedichlorodisuccinatoplatinum, the polymers had slower blood clearance, enhanced tumour accumulation and lower levels of Pt were found in most normal organs.69
Self-assembly, under aqueous conditions, of linear-brush grafted copolymers forming inter-polymer complexes by H-bonding via pH control afforded robust cross linked polymers capable of conjugating cisplatin with high loading efficiency and steady release rate.70
Combining drug delivery with biomedical imaging can be highly advantageous as you can not only selectively deliver your drug payload to its intended target but you can track its journey along the way. This is precisely what Lin et al. set out to achieve. They rationally designed and developed multifunctional upconversion nanocrystals/polymer nanocomposites for delivery of the Pt(IV) prodrug of cisplatin to tumours by linking the Pt(IV) prodrug to the amphiphilic tri-block copolymer, methoxyl-poly(ethylene glycol)-block-poly(3 caprolactone)-block-poly(L-lysine) or mPEG-b-PCL-b-PLL. Rhodamine B was linked to the same polymer to form conjugates which could co-assemble into fluorescent micelles, facilitating both in vitro and in vivo imaging. Entry into HeLa cells and tumour tissue was shown to be via endocytosis whereupon release of the cytotoxic Pt load took place. This study is an excellent example of how theranostics may be exploited to not only deliver cytotoxic payloads but to gain a deeper insight into the delivery route and mechanism of action of the drug molecules.71 Guo, Wang et al. have likewise developed fluorescent theranostic maghemite nanoparticles incorporating cisplatin that display high cytotoxicity towards cisplatin-resistant cell lines. They too were able to track drug distribution both in vitro and in vivo using confocal fluorescence imaging.72
Jing, Zhang et al. developed camplatin, a Pt(IV) hybrid prodrug derived from cisplatin and the medicinal plant camphor. Conjugation of camplatin to the amine groups of the amphiphilic biodegradable polymer MPEG-b-PCL-b-PLL afforded a macromolecular prodrug which was found to have superior efficacy as compared to cisplatin to both cisplatin sensitive and cisplatin resistant cell lines. This enhanced cytotoxicity was attributed to the ability of the macromolecular prodrug to efficiently and effectively enter the tumour cells via endocytosis and, upon release, down-regulate the anti-apoptotic gene Bcl-2. There was little effect on the pro-apoptotic gene Bax.73
Kim et al. have reviewed progress over the past five years in relation to the application of polymeric nanoparticles for the delivery of Pt-based chemotherapeutics.74
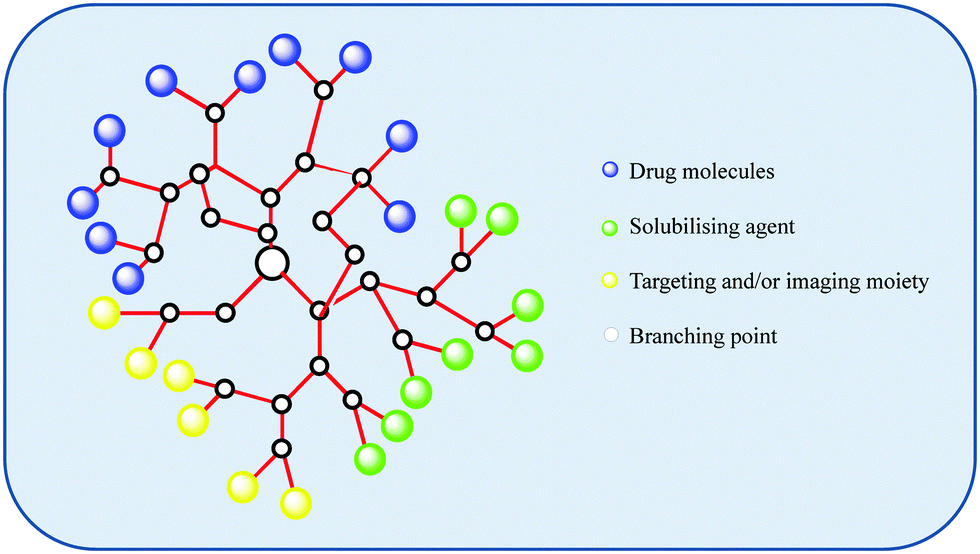 | ||
| Fig. 6 An illustration of a possible design for a dendritic drug carrier which displays the flexibility and versatility that dendritic systems provide. | ||
The most frequently reported dendrimers are polyamidoamines (PAMAM).75 These have been conjugated to cisplatin via a functionalised sodium carboxylate surface.76 The conjugates demonstrated increased solubility, high loading capacity, decreased systemic toxicity, selective accumulation in solid tumours and the ability to slowly release cisplatin in vitro. Of note was the ability of this Pt–dendrimer to retard the growth of the subcutaneous B16F10 murine melanoma in contrast to cisplatin alone which failed to demonstrate any anti-tumour activity. An investigation by Wheate et al. into the use of anionic PAMAM dendrimers as delivery vehicles for the passive targeting of Pt drugs to solid tumours exploiting the EPR effect demonstrated that only a fraction of the Pt drug is released, most likely a function of non-reversible coordinate bond formation between the Pt moiety and the amine and amide groups within the dendrimer branches. Whilst the dendrimer exhibited no cytotoxicity in A2780 ovarian tumour cell lines, moderate cytotoxicity was observed for the Pt–dendrimer conjugate. In vivo studies using an A2780 tumour xenograft showed more promising results.77
The encapsulation efficiency, in vitro cytotoxicity and cellular accumulation of cisplatin-loaded biotinylated PAMAM dendrimers have also been reported. Encapsulation efficiency ranged from 5–21%. The cisplatin–dendrimers exhibited enhanced cytotoxicity as compared to cisplatin alone against a range of ovarian cancer cell lines and improved cellular accumulation.78
Oxaliplatin–pegylated dendrimer conjugates as pH responsive drug delivery vehicles have also been recently investigated.79 These pH-sensitive conjugates demonstrated greater efficacy as compared to oxaliplatin alone in a SKOV-3 human ovarian xenograft without inducing toxicity.
Optimisation of a carboxylate-terminated Pt-PAMAM dendrimer formulation has very recently been reported with respect to varying dendrimer core, generation, drug entrapment, purification, yield, reproducibility, stability, storage and in vitro release.80
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000000:1.81
000000:1.81
 | ||
| Fig. 7 An illustration of a SWCNT as a Pt drug carrier. | ||
CNTs provide very high surface area per unit weight facilitating high drug loading. Plenty of inner spaces facilitate incorporation of drugs while the tube walls allow for drugs and other functional molecules (imaging/targeting agents) to be physically adsorbed. Also, the ends and side holes can be oxidised to afford functional groups where covalent attachment of useful moieties is possible. The surface holes can be also plugged with drugs.
Pioneering work by Ajima et al. demonstrated that cisplatin could be incorporated into and subsequently released from SWCNTs.82 They later found that incorporation and release of cisplatin from these SWCNTs could be enhanced through chemical modification of the SWCNTs structural holes83 or by changing the solvent system during the preparation of these ‘nanohorns’84 with promising in vitro and in vivo results.
A separate molecular modelling study conducted by Hilder and Hill demonstrated that in order for a nanotube to host cisplatin, its radius must be a minimum of 4.8 Å while the maximum uptake of cisplatin was observed when the radius of the nanotube was approximately 5.3 Å.85
Cisplatin encapsulation into ultra-short SWNTs wrapped with Pluronic-F108 surfactant (used to control cisplatin release) resulted in nanotubes which exhibited enhanced cytotoxicity over free cisplatin against two different breast cancer cells lines, MCF-7 and MDA-MB-231 after 24 hours.86
Lippard et al. utilised an amine-functionalised SWCNT as an effective tool to deliver a Pt(IV) prodrug, cis,cis,trans-[Pt(NH3)2Cl2(OEt)(O2CCH2CH2CO2H)] to tumour cells. cis,cis,trans-[Pt(NH3)2Cl2(OEt)(O2CCH2CH2CO2H)] was tethered to the nanotube via a peptide linkage. The resulting water soluble nanotube was shown, by atomic absorption spectroscopy, to carry an average of 65 Pt(IV) centres per nanotube. They anticipated that once inside the reducing environment of tumour cells, the Pt(IV) prodrug would be reduced to the cytotoxic cis-Pt(NH3)2Cl2 or cisplatin. The Pt(IV) precursor and the amine-functionalised SWCNT alone were shown to be relatively non-toxic as compared to cisplatin against the testicular carcinoma cell line NTera-2. The SWCNT–Pt(IV) conjugate, in contrast, was significantly more cytotoxic as compared to the free Pt(IV) prodrug and was shown to surpass that of cisplatin when compared on a per Pt basis.87 They also exploited the use of folic acid (FA) as a means of drug targeting given that many cancer cells overexpress the folate receptor. The Pt(IV) complex cis,cis,trans-[Pt(NH3)2Cl2(O2CCH2CH2CO2H)(O2CCH2CH2CONH-PEG-FA)], bearing the folate derivative in the axial position, was tethered to the surface of an amine-functionalised SWCNT through multiple amide linkages. Release of the Pt payload was facilitated upon reduction of the Pt(IV) to Pt(II) inside tumour cells whereupon the Pt(II) adduct was shown to form 1,2-intrastrand cross links with nuclear DNA. This is the first example of a construct containing both the targeting and delivery moieties in one molecule.88
Lippard et al. subsequently exploited gold nanoparticles as an alternative delivery system in which the Au-NPs were functionalised with thiolated 28 mer oligonucleotides containing a terminal dodecyl amine for conjugation. cis,cis,trans-[Pt(NH3)2Cl2(OH)(O2CCH2CH2CO2H)], an inactive Pt(IV) cisplatin prodrug, was tethered to an amine functionalised DNA–Au nanoparticle surface via amide linkages which could then be activated by reduction in the acidic environment in cancer cells.89In vitro studies confirmed the parent Pt(IV) complex to be relatively inactive but subsequently made active against several cancer cell lines when attached to Au–DNA nanoparticles. Fluorescence spectroscopy revealed HeLa cells incubated with the conjugate displayed its localisation within cell vesicles after 6 hours and within the cytosol after 12 hours. The cytotoxicity profiles of Pt–DNA–Au nanoparticles in human lung carcinoma A549, human prostate cancer PC3, cervical cancer HeLa, and human osteosarcoma U2OS cells showed the conjugate to be more toxic relative to cisplatin.
Liu et al. employed pegylated Au nanorods conjugated to the Pt(IV) prodrug of cisplatin whereupon cell entry, the Pt(IV) is reduced to Pt(II) with subsequent release of cisplatin from its carrier, Fig. 8.90 The rationale for choosing pegylated Au nanotubes was based on the premise that they have been proven to be highly stable and relatively non-toxic in vivo. In addition, such carriers can mask the pegylated agent from the host's immune system thus reducing immunogenicity and antigenicity.91 These Pt-loaded nanotubes were found to be stable under physiological conditions, demonstrated enhanced cellular accumulation of the Pt prodrug and had a significantly higher cytotoxicity profile as compared to cisplatin against a range of cancer cell lines.90 Building on this work, the same group demonstrated that this same Pt(IV)-carrier system avoided the types of drug resistance associated with cisplatin use. For example, endocytosis was found to be the route of entry for these carriers in contrast to the resistance-associated uptake mediated by the copper transport protein Ctr1. Utilising the more inert Pt(IV) prodrug of cisplatin overcame issues around deactivation by glutathione-S-transferase and metallothioneins, found in high concentrations in the resistant A549R lung cell lines tested. These pegylated Au nanorods conjugated to the Pt(IV) prodrug of cisplatin were found to be highly cytotoxic to these resistant cells.92
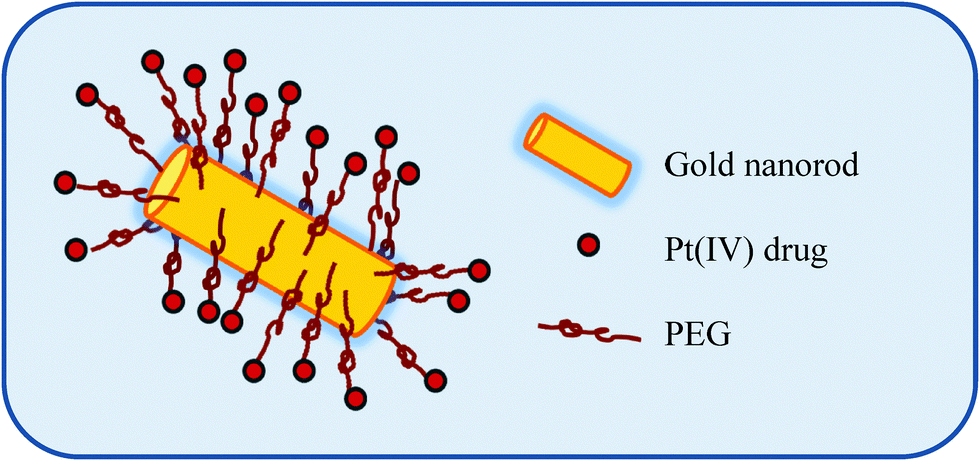 | ||
| Fig. 8 An illustration of a Au nanoparticle as a delivery vehicle for Pt(IV) prodrugs of cisplatin. | ||
A nanotube consisting of a modified SWNT attached to cisplatin and epidermal growth factor (EGF) was reported by Rusling, Gutkind, Patel et al. This bioconjugate capitalises on the specific affinity for the EGF for its cognate cell surface receptor, expressed on most squamous cancer cells. In vitro and in vivo imaging and cytotoxicity studies indicated that the targeted drug delivery system selectively and effectively targeted squamous cancer cells that over express EGF receptors with cell entry occurring via a receptor-mediated endocytosis pathway, accompanied by a less specific and less efficient secondary cell-internalisation.93
In another approach to selectively target cancer cells, Liang, Wang, Zhang et al. developed neuropilin-1-targeted Au nanoparticles to enhance tumour penetration of Pt(IV) drugs, thereby increasing their therapeutic efficacy. Neurophil-1 (Nrp-1) is a transmembrane glycoprotein which is expressed by a large variety of tumours. They constructed glutathione-stabilised Au nanoparticles together with a Pt(IV) prodrug of cisplatin and functionalised with the targeting neuropilin-1 targeting peptide CRGDK in a single platform. Glutathione was chosen because of its well established anti-oxidant properties which can lead to tumour regression. These Au nanoparticles were found to be more cytotoxic towards prostate cancer cells that overexpress Nrp-1 receptors due to greater cell penetration and internalization efficiency as compared to the nanoparticles without the targeting peptide.94
Nanotubes have also been exploited as carriers of carboplatin with activity against urological tumour cell lines enhanced following treatment with the carboplatin-loaded nanotubes as compared to the free drug.95
The use of PEGylated MWCNTs for encapsulation and sustained release of oxaliplatin has also been investigated and compared with non-PEGylated CNTs by Wu et al. After 20 hours, 80% of oxaliplatin had been released from the non-PEGylated CNT in contrast to 50% of the PEGylated derivative. These findings were consistent with relative cytotoxicities measured over this timeframe. Cytotoxicities were greatly enhanced after 48 and 96 hours most likely due to increased cellular accumulation of oxaliplatin.96
In another study, the influence of functionalised CNTs encapsulating either cisplatin or an inert Pt(IV) complex, cis,cis,trans-[Pt(NH3)2Cl2(O2CC6H5)2], on the biodistribution of the Pt complexes was investigated. Interestingly, Pt accumulation in vital organs suggested that the functionalised CNTs did not affect cisplatin distribution but significantly enhanced that of the Pt(IV) derivative in certain tissues. Enhanced accumulation was observed for example in lung tissue with a reduction in accumulation in both kidney and liver tissues thus demonstrating their potential to safely and efficiently deliver Pt drugs to target organs.97
Whilst nuclear DNA is thought to be the major target of classical Pt drugs, Yoong et al. investigated the use of CNTs functionalised with the mitochondrial targeting lipophilic cation rhodium-110 (Rho-110), to selectively deliver a Pt(IV) prodrug of cisplatin to the mitochondria. The CNT-Rho110 alone was neither cytotoxic to cells nor detrimental to mitochondrial function. In contrast, the Pt-encapsulated CNT-Rho110 augmented cytotoxicity relative to cisplatin. Co-encapsulation of the Pt(IV) prodrug with 3-bromopyruvate, a chemo-sensitiser, resulted in a synergistic effect in the cell lines tested.98
More recently, tetrameric nanotubes, formed through self-assembly from α-helical right handed coiled coils (RHCC), encapsulating a Pt(IV) derivative have shown promise in that they exhibit superior in vitro and in vivo chemotherapeutic efficacy and an improved selectivity towards human malignant glioblastoma cells when compared to the free Pt(IV) compound.99
The use of nanotubes for targeted and ‘remote control’ dual drug delivery of doxorubicin and a cisplatin prodrug has also been explored. Shanmugam et al. developed Au nanorods in which the 5′ thiol ends of single stranded DNA were conjugated to nanorods, following which the complimentary DNA strands were hybridised. In so doing, this new entity was able to bind doxorubicin through intercalation into the CG base pairs of a DNA duplex. The complimentary DNA was designed such that it incorporated a 5′ amine functional moiety free to tether the Pt(IV) prodrug of cisplatin through the formation of an amide bond upon reaction of this amine with a carboxylate functionality in one of the axial ligands. The other axial ligand housed folic acid and served as a targeting agent in its own right for folate receptors overexpressed on cancer cells. Upon cell entry, exposure of these gold nanorods to near infrared (NIR) radiation resulted in doxorubicin and Pt prodrug release. The Pt prodrugs were subsequently reduced, under the reducing environment of tumour cells, yielding the cytotoxic Pt(II) species. This novel ‘external stimulus combination drug delivery system’ was found to be highly effective both in vitro and in vivo.100
Tuning the diameter of nanotubes to selectively deliver Pt drugs has also been investigated. The incorporation of a hydrophobic Pt(IV) complex within the inner chamber of two different diameter functionalised nanotubes resulted in nanotubes with differing cytotoxicity profiles as compared to the free drug. While both induced cell death after 72 hours, it was the larger diameter nanotubes which proved more efficacious due to more prolonged release of the drug cargo. Interestingly, these nanotubes, regardless of diameter, were only poorly cytotoxic on macrophages. No pro-inflammatory cytokine production nor cell activation was observed. This study demonstrated that fine-tuning the diameter of nanotubes can lead to effective drug delivery systems without inducing an inflammatory response.101
Wong et al. provide a comprehensive review of the use of CNTs for the delivery of small molecule drugs including Pt-based drugs.102
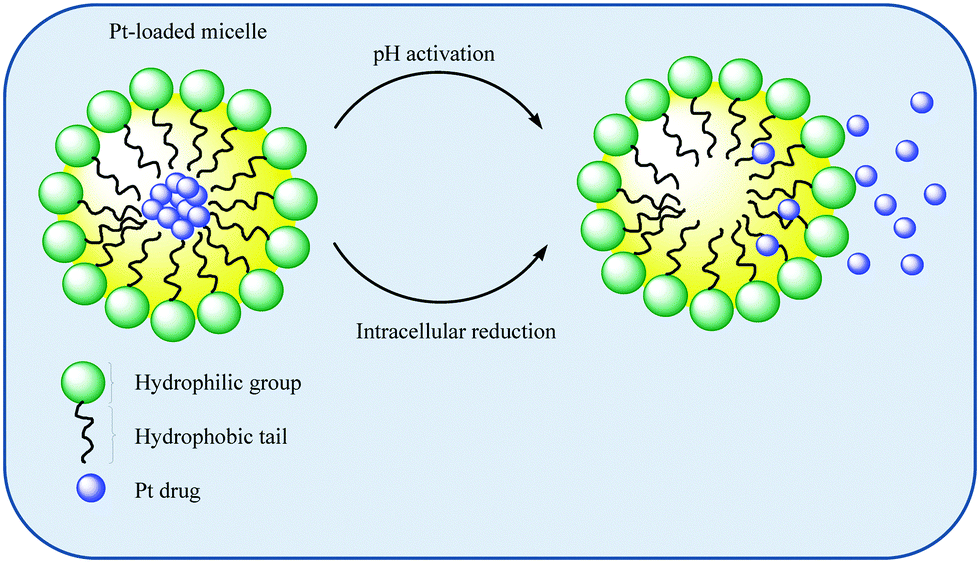 | ||
| Scheme 2 An illustration of Pt-loaded micelles and their intracellular mode of activation. | ||
Hydrophobic drugs tend to distribute into the micelle core while more polar drugs tend to occupy peripheral positions. Those which are peripherally located tend to be predisposed to release. The use of block ionomers which have the ability to form polymer–metal complexes have been exploited for Pt drug delivery. Pt drugs form coordinate bonds with the polyion block of the copolymer which also facilitates micelle formation.112 The use of polycarboxylates as ionic blocks has been particularly exploited given the propensity of polycarboxylates to substitute anionic ligands such as chlorido ligands in, for example, cisplatin.113 Pt drug release can be affected by external conditions such as salt concentrations, pH, reductants, and overall micelle stability, Scheme 2. In addition, biologically abundant counter ions can further enhance the promotion of drug release by ligand exchange.112
The most widely used copolymers for Pt drug delivery are poly(amino acid) based, such as poly(aspartic acid), PAsp and poly(glutamic acid), PGlu.114 Pioneering studies by Kataoka et al.113,115,116 in which cisplatin was complexed to PEG PAsp copolymers resulted in the spontaneous formation of stable polymer micelles with sizes in the range of 20–100 nm and high drug loading. They also demonstrated that release of the Pt drug from the micelles was dependent on the PAsp block length and occurred via chlorido ion exchange.113 Joining the PEG-PAsp block ionomers with PAsp homopolymer was shown to alter micelle size, micelle decay and cisplatin release.116 Moreover, studies in mice demonstrated that incorporation of cisplatin into such polymer micelles prevented kidney toxicity in contrast to cisplatin treatment alone which is highly nephrotoxic, and enhanced exposure of the drug in tumours.115
Incorporation of cisplatin into PGlu-based block copolymers resulted in micelles with enhanced stability as compared to PAsp derivatives. Preclinical studies demonstrated prolonged blood circulation and a 20-fold higher accumulation in solid tumours (Lewis lung carcinoma cells) as compared to cisplatin alone. While both cisplatin and the cisplatin-loaded micelles had significant in vivo cytotoxicity in C26 bearing mice, the micelles demonstrated complete regression of tumours with no significant body loss in contrast to cisplatin alone which did result in tumour survivals and body weight loss.117 These micelles were also found to have a safer toxicity profile as compared to cisplatin in a guinea pig model.118 Phase I clinical studies demonstrated that these micelles, under the development name NC-6004, were well tolerated by patients with a range of advanced solid tumour types. However, hypersensitivity reactions induced by NC-6004 were more frequent than those caused by cisplatin.119 This formulation is currently undergoing phase III clinical trials in Asia (Nanoplatin; Nanocarrier Co., Ltd; Japan).15 A phase II study in which patients with locally advanced or metastatic pancreatic cancer were treated with NC-6004 in combination with gemcitabine demonstrated that Pt hypersensitivity was not an issue if prophylactic treatment was used with no need for pre-hydration.15,120 Survival rates were better (12.3 months) using this combination compared to the overall median survival (7.5 months) reported for cisplatin/gemcitabine combination.121 An FDA application for an investigational new drug (IND) based on this combination was submitted in 2013 in the US.15
Another approach incorporating cisplatin using polymer micelles was based on the biodegradable polyester block polymer PEG-b-polycaprolactone (PEG-b-PCL). Anti-tumour activity of such micelles was demonstrated both in vitro and in vivo with high encapsulation efficiency.122 pH sensitive micelles incorporating cisplatin have also been developed with rapid endosomal cisplatin release.123In vivo studies of cisplatin-loaded core cross-linked micelles of poly(ethylene glycol)-b-poly(methacrylic acid) demonstrated prolonged blood circulation, enhanced tumour accumulation, a decrease in renal exposure as well as enhanced efficacy as compared to cisplatin alone.124
The use of graft copolymers for Pt drug delivery has also been exploited. Carboxylic acid-functionalized poly(beta-aminoester)graft-poly(ethylene glycol) copolymers were complexed to cisplatin resulting in 100–200 nm negatively charged nanogels with a PEG outer layer. Whilst these demonstrated significantly lower in vitro cytotoxicity against SKOV-3 ovarian cancer cells as compared to cisplatin alone, they exhibited similar anti-cancer activity toward SKOV-3 tumours xenografted to immunocompromised mice.125 Micelles incorporating mPEG-g-alpha,beta-poly[(N-amino acidyl)-DL-aspartamide] (mPEG-g-PAAsp) complexed to cisplatin were not found to be as cytotoxic as compared to cisplatin against Bel-7402 hepatoma cells.126 The development of carboxylic acid conjugated, hydrophobically derivatized, hyperbranched polyglycerols as nanoparticulate drug carriers for cisplatin has also been described. These biocompatible carriers were found to inhibit proliferation of KU-7-luc bladder cancer cells.127 Folate-decorated nanogels incorporating cisplatin have also been developed as targeted therapeutic agents for the treatment of ovarian cancer where folate receptors are overexpressed. These cisplatin-containing nanogels were found to possess superior anti-tumour properties in vivo as compared to the free Pt drug.128
Whilst the use of micelles as drug delivery vehicles has been extensively investigated, their dynamic nature can often lead to disassembly in vivo which can in turn negatively impact on their biodistribution and cellular uptake. Cross-linking can overcome this in many ways by locking the micelle into its desired spherical form.129
An interesting in vitro and in vivo study by Zhang, Chen et al. describes cisplatin cross-linked pH-sensitive dextran-nanoparticles as efficient vehicles for the selective delivery of doxorubicin to cancer cells. Here the cisplatin is employed as a cross linker and this cross linking appears to significantly enhance the surface charge and stability of the nanoparticles leading to improved tolerability, in vivo pharmacokinetics, biodistribution and anti-cancer efficacy. In the A549 lung xenograft model investigated, a reduction in tumour size as well as drug-related multi-organ toxicity was observed.130
In another study, complexation of cisplatin to the pendant carboxyl groups on the poly(e-caprolactone) core of methoxy poly(ethylene oxide)-block-poly-(a-carboxylate-e-caprolactone) or PEO-b-PCCL generated pH-responsive micelles in which cisplatin release was triggered upon exposure to electrolytes and/or pH change mimicking that of the extracellular tumour microenvironment or intracellular organelles. These demonstrated promising in vitro activity against breast cancer cell lines.131
The potential of methoxy poly(ethylene glycol)-block-poly(glutamic acid) nanoparticles as carriers of cisplatin for the treatment of solid tumours has already been highlighted in numerous studies including but not limited to those by Nishiya et al.119 and Yamasoda et al.118 and Chen et al.132 Rapid release of cisplatin facilitated by the higher chloride ion concentration in blood plasma compared to chloride ion concentration inside tumour cells has been a challenge. To overcome this, Tang, Shah, Chen et al. reported the first example of a miscellar methoxy poly(ethylene glycol)-block-poly(glutamic acid) carrier incorporating a hydrophobic moiety poly(L-phenylalanine), [mPEG-b-P(Glu-co-Phe)], for targeted delivery of cisplatin. Cisplatin loading was facilitated through metal conjugation with the carboxyl groups of the poly(L-glutamic acid) block while the hydrophilicity of the poly(ethylene glycol) shell protected the carrier from phagocytosis. The presence of the poly(L-phenylalanine) afforded the system hydrophobicity to control cisplatin drug release. Two cisplatin-loaded [mPEG-b-P(Glu10-co-Phe10) (PGlu10) and mPEG-b-P(Glu20-co-Phe10) (PGlu20)] were developed and their drug release, cell viability, plasma clearance, and pharmacokinetic profile compared. Both nanoparticles demonstrated controlled and sustained release at physiological and lysosomal pH. Both showed dose- and time-dependent cytotoxicity against the human breast cancer cell line ZR-75-30. The in vivo pharmacokinetic profile for both demonstrated prolonged blood circulation times in contrast to cisplatin alone.133
Wang et al. rationally designed core shell corona polyion complex nanoparticles for Pt drug delivery utilising positively charged and Pt(IV)–prodrug-conjugating micellar nanoparticles and the pH responsive negatively charged pegylated diblock copolymer PPC-DA. pH activation led to release of the positively charged micellar nanoparticles which further facilitated nanoparticle internalisation and subsequent release of cisplatin upon exposure to the intracellular reducing environment. Prolonged circulation times and tumour growth inhibition in an A549 lung tumour xenograft model were observed.134
Antibody fragment-installed polymeric micelles have also recently been investigated as a potential means to selectively deliver Pt drugs to pancreatic tumours. The Pt-loaded micelles demonstrated more than 15-fold increased cellular binding within the first hour and rapid cellular internalisation compared to non-targeted micelles which ultimately led to enhanced in vitro cytotoxicity. These Pt-loaded micelles were also found to significantly suppress the growth of pancreatic tumour xenografts.135
Polymer–albumin micelles loaded with Pt drugs have also been developed. These conjugates self-assemble in water with a nanoparticulate size of approximately 80 nm attributed to the hydrophobic nature of the Pt drugs. These albumin coated polymers were taken up readily by ovarian cancer cell lines and were found to have superior efficacy over a control sample without albumin coating.136
Micelles incorporating oxaliplatin have also demonstrated promise.137,138 Their relatively small size results in deep tumour penetration even in poorly permeable tumours such as intractable pancreatic139 and scirrhous gastric cancers.140 Their ability to selectively dissociate within late endosomes and thus enhance delivery of the Pt drug to DNA relative to oxaliplatin alone, results in these micelles generally exhibiting greater efficacy as compared to oxaliplatin alone even against oxaliplatin-resistant tumours.141 For example, chitosan-based polymer micelles encapsulating oxaliplatin, formed by stearic acid-grafted chitosan oligosaccharide, resulted in in vitro anti-tumour activity against drug sensitive gastric SGC-7901, ovarian SKOV3, hepatoma BEL-7402, leukemia K562 and breast MCF-7 and the multidrug resistant cells MCF-7/Adr. Significantly enhanced cytotoxicity was observed for the oxaliplatin-loaded micelles over oxaliplatin alone. They were also active against the multi-drug resistant cells tested.142
Cross-linked micelles carrying oxaliplatin, generated by using block ionomer complexes of poly(ethylene oxide)-b-polymethacrylate (PEO-b-PMA) copolymer and divalent metal cations as templates, have also been investigated and were found to be not only stable but also exhibited pH-dependent sustained release of the Pt drug. Up to 25% w/w loading was achieved as a result of the core's ionic character. The drug loaded micelles demonstrated significantly higher in vitro cytotoxicity as compared to oxaliplatin alone which increased with exposure time.143 The core cross-linked block ionomer micelles were utilised by the same group as pH-responsive carriers for cisplatin but with more efficient loading (up to 42% w/w). This core cross-linking helped to stabilise the micelles against structural disintegration while also preventing premature drug release.144
Amphiphilic biodegradable polymers bearing pendant carboxyl groups, mPEG-b-P(LA-co-MCC), have been utilised to bind the Pt of dichloro(1,2-diaminocyclohexane)platinum(II) resulting in the assembly of water soluble micelles incorporating the oxaliplatin analogues. They demonstrated dose-dependent cytotoxicities against breast cancer EMT6 cells which were comparable to free oxaliplatin.145 Another amphiphilic biodegradable copolymer, mPEG-b-P(LAco-MAC/TMA), bearing 1,2-dicarboxyl groups capable of chelating the (DACH)Pt of oxaliplatin has also been developed. This system too self-assembles into micelles with desirable acid-responsive drug release kinetics and in vitro cytotoxicity against ovarian SKOV-3 and breast MCF-7 cancer cells. They were also found however to display reduced toxicity to HeLa cells as compared with oxaliplatin.146
Biodegradable polymers have also been exploited to co-deliver oxaliplatin and daunomycin. Polymers with a similar backbone were conjugated to oxaliplatin analogues bearing axial carboxyl groups and to daunomycin and these were able to co-assemble into composite micelles. Release of the oxaliplatin derivative was facilitated upon reduction in tumour cells and daunomycin release was facilitated via acid hydrolysis. In vitro and in vivo results demonstrated that these composites exhibited reduced systemic toxicity and enhanced synergy over the combination of both drugs.147
More recently, an in vivo study using a transgenic model of spontaneous pancreatic cancer indicated that these micelles loaded with oxaliplatin prolonged mice survival for more than 100 days preventing the onset of intraperitoneal metastasis in contrast to those treated with oxaliplatin alone where 50% of the mice were dead after 50 days.148 The anti-tumour efficacy of these micelles and their association with peripheral neuropathy has also been evaluated given that the latter is a primary dose-limiting factor in oxaliplatin therapy. Their efficacy was found to be superior to that of oxaliplatin in an in vivo rat model bearing the human carcinoma KB. The animals did not experience acute cold hypersensitivity, often felt by patients following oxaliplatin administration.149 This micelle formulation is being developed under the name NC-4016 (NanoCarrier Co., Ltd; Japan) and is advancing to phase I/II clinical trials in the US against a range of solid tumours.15 Incorporation of the oxaliplatin motif into cross-linked block copolymer micelles has also shown potential both in vitro and in vivo with improved efficacy as compared to oxaliplatin alone against A2780 ovarian cancer cells and in an ovarian tumor xenograft model respectively.150
Yong et al. utilised mPEG-Ad@β-CD-7PLGA/CDDP nano-sized supramolecular micelles fabricated from β-CD-7PLGA/CDDP and mPEG-Ad as vehicles for cisplatin but compared to cisplatin alone, the micelles demonstrated decreased cytotoxicity against KB cells.151
Whilst many Pt-loaded micelles developed to date target solid tumours, the ability of micellar nanoparticles as oxaliplatin transporters for the treatment of liver metastases of bioluminescent murine colon adenocarcinoma C-26, during overt and pre-angiogenic metastatic stages has also recently been investigated. These oxaliplatin-loaded micelles effectively inhibited tumour growth in the metastases models investigated. Anti-tumour activity in the overt model was associated with selective accumulation of the micelles in cancerous tissues having neovasculature. In contrast, a correlation was found between the ability of the micelles to target preangiogenic metastases and the inflammatory microenvironment of the niche.152
Whilst most studies to date have focussed on micelles encapsulating cisplatin and to a lesser extent, oxaliplatin, Duong et al. reported novel micelles incorporating a Pt(IV) derivative of cisplatin. Isocyanate groups in the poly(oligo(ethylene glycol) methyl ether methacrylate)-block-poly(styrene-co-3-isopropenyl-R,R-dimethylbenzyl isocyanate) (POEGMA-block-P(STY-co-TMI)) micelle core reacted with amine groups in the Pt(IV) derivative to generate stable micelles with promising in vitro activity. The rationale behind the approach was that the Pt(IV) would be reduced in the reducing environment of tumour cells releasing the active cisplatin and the resulting non-toxic micelle subsequently excreted.153
The incorporation of bioactive ligands in the axial positions of the Pt(IV) analogue of cisplatin is not a new phenomenon. For example, Lippard et al. developed mitaplatin, a dual-functioning Pt(IV) prodrug incorporating the DNA-binding drug cisplatin and the mitochondria-targeting drug dichloroacetate (DCA) which demonstrated cytotoxicity comparable to cisplatin.154 Zhang et al. developed paclitaxel–cisplatin(IV) conjugates but these, in contrast, were found to be non-toxic and inactive against tumour cells, most likely due to their inability to enter cancer cells.155 Xiao et al. sought to overcome drawbacks such as instability, poor solubility and bioavailability, short blood circulation time, etc., often associated with small molecular drugs such as these by designing micelles capable of encapsulating mitaplatin156 and the paclitaxel–cisplatin(IV) conjugates157 with a view to enhancing both efficacy and tolerance. They employed succinic acid as a linker between the Pt(IV)–DCA complex and the carrier biodegradable and amphiphilic copolymer MPEG-b-PCL-b-PLL, forming a polymer–Pt(IV) conjugate and its micelles. Under simulated intracellular conditions, the micelles rapidly released the drug and demonstrated higher cytotoxicity towards SKOV-3 human ovarian cancer cells than its precursors alone.156 The same group co-assembled the Pt(IV) cisplatin prodrug and paclitaxel into single carrier composite micelles. Release of the cisplatin prodrug was facilitated upon cellular reduction and paclitaxel via acid hydrolysis following entry into tumour cells. Moreover, a synergistic effect was observed in vitro. In vivo studies provided evidence that the micelles carrying the two drug cargos displayed safer and more efficacious inhibition towards cervical U14 tumour growth as compared to the drugs administered in combination.157 Scarano et al. likewise employed a dual-drug delivery approach to transport curcumin and Pt(IV) prodrugs in polymeric micelles. When tested against A2780 ovarian cancer cells, co-administration of curcumin and the Pt drug without the carrier demonstrated synergy with a combination index from 0.4–0.8. This synergy was enhanced when the two were co-delivered in these micelles resulting in a combination index of 0.2–0.35.158
Site-specific activation of photosensitive Pt(IV) prodrugs is now possible through the use of lasers and fibre optics capable of reaching any tissue in the body, thus minimising the severe toxic side effects associated with Pt(II) drugs. Sadler et al. were the first to develop photo-sensitive Pt(IV) prodrugs whereby the prodrug would only be activated upon exposure to visible light releasing the highly reactive Pt(II) species capable of binding rapidly and stereospecifically to nucleotides forming established cisplatin-nucleotide cross-links. These Pt(IV)–azide complexes offer a distinct advantage over photodynamic therapy in that they do not require photosensitizing catalysts or oxygen-rich environments.159–161 Recognising the potential of these Pt(IV)–photosensitiser systems, Bilgicer, Jing et al. recently developed a novel series of cisplatin and oxaliplatin Pt(IV)-photosensitiser prodrugs in micellar nanoparticle formulations with a miscellar diameter of 100–200 nm, sizes which are known to induce EPR effects in vivo. These formulations, which were stable in the dark, exhibited high sensitivity towards UVA irradiation, releasing their cytotoxic Pt(II) agents which were subsequently found to form DNA cross links, Scheme 3. These photosensitiser prodrugs were found to be up to 8-fold and 13-fold more effective in vitro as compared to cisplatin and oxaliplatin respectively. They were also found to be highly effective in an in vivo H22 murine hepatocarcinoma model upon UVA activation with enhanced blood circulation times, greater tumour growth inhibition and reduced systemic toxicity.162
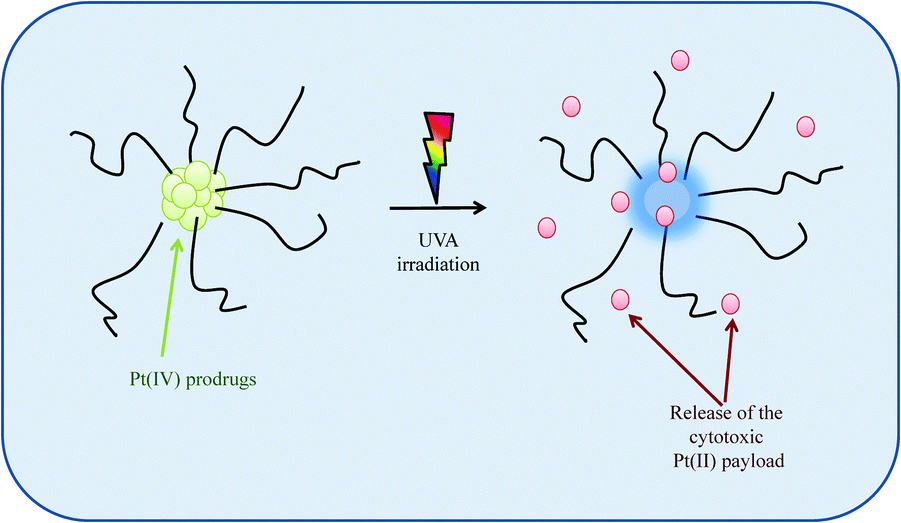 | ||
| Scheme 3 A schematic to illustrate how photosensitive Pt(IV) prodrugs, upon irradiation with UVA light, release their cytotoxic Pt(II) payloads. | ||
Stenzel et al. conducted a comprehensive in vivo investigation into the effectiveness of folate decorated cross-linked micelles for the delivery of the Pt(IV) prodrug of cisplatin. They used a selection of fluorophore-labelled micelles (with and without folate) cross-linked with 1,8-diaminooctane with sizes ranging from 75 to 200 nm and with both spherical and worm-like conformations. Using optical imaging, they established that accumulation in organs, especially in the liver and kidneys, was enhanced when the micelles were cross-linked and decorated with folic acid while micelles that were not conjugated with folate and not cross-linked were rapidly clearly from the mice. The micelles with worm-like conformations had a slower clearance rate as compared to spherical micelles.129
A Pt(IV)–thermo gel polymer, capable of self-assembling into micelles in water has recently been investigated in which a Pt(IV) prodrug was covalently linked to the hydrophobic end of two methoxyl poly(ethylene glycol)-b-poly(D,L-lactide) (mPEG-PLA) copolymer chains, resulting in the formation of Bi(mPEG-PLA)–Pt(IV). This novel conjugate was shown to accumulate in cells via endocytosis with sustained release of its Pt cargo up to 2 months. Bi(mPEG-PLA)–Pt(IV) was also found to have enhanced in vitro cytotoxicity as compared to cisplatin against breast MDA-MB-231 cancer cells.163
In vitro activity of micelles (NP-UVA-Pt2) incorporating a photosensitive platinum(IV) prodrug (UVA-Pt2) conjugated to a biodegradable polymer (PE, methoxyl-poly(ethylene glycol)-block-poly(lactide-co-2-methyl-2-carboxyl-propylene carbonate-ethanol amine)) demonstrated improved cytotoxicity against ovarian SKOV-3 cells as compared to cisplatin. As anticipated, cellular accumulation appeared to be facilitated via endocytosis rather than passive diffusion, and did not involve the use of copper transporter protein (Ctr1). NP-UVA-Pt2 was found to be highly responsive to photo-irradiation while the micelles were stable at physiological pH in the dark.164
Cyclotriphosphazene micelles incorporating a hydrophobic and water insoluble Pt(II) complex cis-bis(cyclohexylamine) dinitratoPt(II) demonstrated both potent in vitro and in vivo cytotoxicity as well as a favourable pharmacokinetic profile in vivo as compared to carboplatin. The micelles were shown to be particularly cytotoxic to stomach tumour cells (SNU638), which is one of the least responsive cancers to chemotherapeutics currently in clinical use.165 An amphiphilic polyphosphazene–Pt conjugate designed to selectively deliver oxaliplatin to tumours has also been developed. The (dach)Pt[HEDM] where dach is trans-(±)-1,2-diaminocyclohexane and HEDM is 2-hydroxyethoxydiethylmalate was designed such that the HEDM could serve as a linker between the Pt and the polyphazene backbone generating a novel amphiphilic polyphosphazene–Pt conjugate, [NP(MPEG550)(dach)Pt(EM)]n [MPEG550]: methoxy poly(ethylene glycol) which could self-assemble into stable polymeric micelles of a mean diameter of 130 nm, suitable for passive tumour targeting by the EPR effect. This novel polyphosphazene–Pt conjugate was found to have a superior pharmacokinetic and cytotoxicity profile as compared to oxaliplatin.166
A strategy employed to overcome some of the drawbacks associated with classical Pt drugs was the development of non-classical Pt drugs capable of binding DNA in a different manner to cisplatin and its analogues. BBR-3464 (18), Fig. 2, is an example of the first and only ‘non-classical’ Pt drug to undergo clinical evaluation.167 This is a tri-nuclear positively charged Pt(II) drug which can form flexible and non-directional and long range DNA adducts resulting in conformational changes to both A- and Z-type DNA.168–170
Inspired by the success of cisplatin and oxaliplatin and more recently that of multi-nuclear Pt drugs, polymer-di–Pt(IV) conjugates derived from cisplatin and oxaliplatin were developed which were assembled into micelles, the rationale being that these new micelles would be internalised by tumour cells via endocytosis increasing drug concentrations and reducing dose-limiting systemic toxicity. Once inside the reducing environment of tumour cells, the prodrugs would be reduced releasing the cytotoxic dinuclear Pt(II) adducts free to bind DNA. These micelles did indeed demonstrate reduced systemic toxicity, relatively long blood circulation and enhanced tumour efficacy as anticipated171
The synthetic, biodegradable, and water soluble polypeptide methoxy-polyethylene glycol-block-poly(glutamic acid) (MPEG-PGA) bearing pendant negatively charged carboxyl moieties has been exploited as a drug carrier due to its biodegradability and biocompatibility. The presence of the negatively charged carboxyl groups are ideally suited to interact with positively charged drug molecules via electrostatic interactions. Xiao et al. used this strategy to develop novel micellar nanoparticle incorporating positively charged multi-nuclear Pt drugs. Drug loading can be adjusted by varying the stoichiometric ratios of the multi-nuclear Pt drugs to the negatively charged carboxyl groups present on the polypeptide. These multi-nuclear Pt-loaded micelles exhibited not only efficient Pt loading but improved cellular accumulation and in vitro as well as in vivo activity.172
Malzert-Fréon et al. specifically review nanocarriers including polymeric conjugates, dendrimers, inclusion molecules: cyclodextrines, polymeric micelles, nanogels, nanoparticles and liposomes for the targeted treatment of ovarian cancers with a particular emphasis on their use in preclinical development.173 Lippard et al. describe recent advances in the development of Pt(IV) prodrugs and Pt(IV) drug nanoconstructs for their selective in vivo delivery.174 Liang, Gottesman et al. focus on abnormal membrane protein trafficking in their review of nanoscale drug delivery platforms to overcome Pt-based resistance in cancer cells.175 Cabral and Kataoka provide a comprehensive review of polymeric micelles and their performance in human studies.176 Guo, Wang and Wang review the functionalization of Pt complexes for both targeted drug delivery and as theranostic agents.177 Mumper et al. draw from the expertise of leaders in biology, chemistry, materials science, pharmaceutics, toxicology, chemical engineering, imaging, physiology, oncology and regulatory affairs and provide an insightful commentary into the ‘six tennets’ of biotargeted cancer nanomedicines required to translate basic science into clinical applications.178 Other, more general reviews are provided by Oberoi et al.,15 Sadler et al.,179 and Kieler-Ferguson et al.37
3. Conclusion
The rational design and development of innovative anti-cancer Pt drug candidates to overcome drawbacks associated with those currently in the clinic has produced an inspiring armamentarium of possible chemotherapeutics. However, in the 50 years since the discovery of the anti-cancer properties of cisplatin, none to date has been as successful as cisplatin, carboplatin or oxaliplatin. Recent advances in this field have included the search for nanotechnologies to essentially protect the Pt from deactivation until such time as the drug reaches the tumour site whereupon the technology is capable of releasing its Pt payload. An alternative approach is to incorporate vectors onto existing nanotechnologies to serve as homing devices with a view to enhancing drug targeting. Exploiting the use of nanotechnology to preferentially deliver and deposit Pt drug payloads to tumours has resulted in the employment of liposomes, nanocapsules, polymers, dendrimers, nanoparticles, nanotubes and others as vehicles for this purpose. We have attempted to provide an overview of progress in this exciting domain. There has undoubtedly been much success in this field with some technologies already undergoing clinical evaluation. Future work should focus on more fully understanding the complexity of the mechanisms underlying nanoparticle targeting. The advent of nanotechnologies with theranostic applications will certainly help to improve our understanding in this regard and we look forward to further developments in this field. There remains a need however, through multi-disciplinary research, to further optimise the synthesis (homogeneity), characterisation and scale-up, physicochemical profiles, stability in systemic circulation, and delivery and therapeutic efficacy of these systems as well as reflecting on data generated to date to inform the future design of these systems if such technologies are to successfully cross the interface between pre-clinical and clinical application for cancer treatment.Acknowledgements
This material is based upon works supported by the Science Foundation Ireland under Grant No. [11/RFP.1/CHS/3095] and [12/TIDA/B2384]. This work has also been funded under the Programme for Research in Third-Level Institutions and co-funded under the European Regional Development fund (BioAT programme). The authors would also like to acknowledge COST CM1105 for being a platform to progress fruitful collaborations.References
- D. Hanahan and R. A. Weinberg, Cell, 2000, 100, 57–70 CrossRef CAS PubMed
.
- G. Awada, H. R. Kourie and A. H. Awada, Discov. Med., 2015, 20, 33–41 Search PubMed
- Y. Wen and W. S. Meng, J. Pharm. Sci. Innovation, 2014, 9, 158–173 CrossRef PubMed
- M. G. Apps, E. H. Choi and N. J. Wheate, Endocr.-Relat. Cancer, 2015, 22, R219–R233 Search PubMed
- L. Kelland, Nat. Rev. Cancer, 2007, 7, 573–584 CrossRef CAS PubMed
- C. Monneret, Ann. Pharm. Fr., 2011, 69, 286–295 CrossRef CAS PubMed
- N. J. Wheate, S. Walker, G. E. Craig and R. Oun, Dalton Trans., 2010, 39, 8113–8127 RSC
- Y. Matsumura and H. Maeda, Cancer Res., 1986, 46, 6387–6392 CAS
- H. Maeda, G. Y. Bharate and J. Daruwalla, Eur. J. Pharm. Biopharm., 2009, 71, 409–419 CrossRef CAS PubMed
- H. Maeda, Adv. Enzyme Regul., 2001, 41, 189–207 CrossRef CAS PubMed
- G. Bozzuto and A. Molinari, Int. J. Nanomed., 2015, 10, 975–999 CrossRef CAS PubMed
- B. W. Harper, A. M. Krause-Heuer, M. P. Grant, M. Manohar, K. B. Garbutcheon-Singh and J. R. Aldrich-Wright, Chemistry, 2010, 16, 7064–7077 CrossRef CAS PubMed
- Z. Tao, B. Toms, J. Goodisman and T. Asefa, ACS Nano, 2010, 4, 789–794 CrossRef CAS PubMed
- Z. Tao, Y. Xie, J. Goodisman and T. Asefa, Langmuir, 2010, 26, 8914–8924 CrossRef CAS PubMed
- H. S. Oberoi, N. V. Nukolova, A. V. Kabanov and T. K. Bronich, Adv. Drug Delivery Rev., 2013, 65, 1667–1685 CrossRef CAS PubMed
- G. P. Stathopoulos and T. Boulikas, J. Drug Delivery, 2012, 2012, 581363 CAS
- M. I. Koukourakis, A. Giatromanolaki, M. Pitiakoudis, G. Kouklakis, P. Tsoutsou, I. Abatzoglou, M. Panteliadou, K. Sismanidou, E. Sivridis and T. Boulikas, Int. J. Radiat. Oncol., Biol., Phys., 2010, 78, 150–155 CrossRef CAS PubMed
- F. Farhat, J. Kattan, K. Ibrahim, N. Bitar, N. Haddad, S. Tamraz, H. Hatoum and A. Shamseddine, EJC Suppl., 2010, 8, 192 CrossRef
- C. Kosmas, J. Angel, A. Athanasiou, A. Rapti, C. Karanikas, S. Lambaki, N. Politis and N. Mylonakis, EJC Suppl., 2009, 7, 531 CrossRef
- T. Boulikas, Expert Opin. Invest. Drugs, 2009, 18, 1197–1218 CrossRef CAS PubMed
- T. Dragovich, D. Mendelson, S. Kurtin, K. Richardson, D. Von Hoff and A. Hoos, Cancer Chemother. Pharmacol., 2006, 58, 759–764 CrossRef CAS PubMed
- M. J. de Jonge, M. Slingerland, W. J. Loos, E. A. Wiemer, H. Burger, R. H. Mathijssen, J. R. Kroep, M. A. den Hollander, D. van der Biessen, M. H. Lam, J. Verweij and H. Gelderblom, Eur. J. Cancer, 2010, 46, 3016–3021 CrossRef CAS PubMed
- K. J. Harrington, C. R. Lewanski, A. D. Northcote, J. Whittaker, H. Wellbank, R. G. Vile, A. M. Peters and J. S. Stewart, Ann. Oncol., 2001, 12, 493–496 CrossRef CAS PubMed
- S. C. White, P. Lorigan, G. P. Margison, J. M. Margison, F. Martin, N. Thatcher, H. Anderson and M. Ranson, Br. J. Cancer, 2006, 95, 822–828 CrossRef CAS PubMed
- X. Sun, J. Chen, H. Chen and W. Liang, Die Pharmazie, 2012, 67, 426–431 CAS
- X. Sun, J. Chen, X. Gu, W. Liang and J. Wang, Die Pharmazie, 2014, 69, 281–286 CrossRef CAS PubMed
- I. Vhora, N. Khatri, J. Desai and H. P. Thakkar, AAPS PharmSciTech, 2014, 15, 845–857 CrossRef CAS PubMed
- G. N. Kaluderovic, A. Dietrich, H. Kommera, J. Kuntsche, K. Mader, T. Mueller and R. Paschke, Eur. J. Med. Chem., 2012, 54, 567–572 CrossRef CAS PubMed
- G. P. Stathopoulos, T. Boulikas, A. Kourvetaris and J. Stathopoulos, Anticancer Res., 2006, 26, 1489–1493 CAS
- N. N. Senzer, K. Matsuno, N. Yamagata, T. Fujisawa, E. Wasserman, W. Sutherland, S. Sharma and A. Phan, Mol. Cancer Ther., 2009, 8, C36 Search PubMed
- K. K. Sankhala, A. C. Mita, R. Adinin, L. Wood, M. Beeram, S. Bullock, N. Yamagata, K. Matsuno, T. Fujisawa and A. Phan, J. Clin. Oncol., 2009, 27, 2535 Search PubMed
- A. Phan, C. Takimoto, R. Adinin, L. Wood, H. Xiong, K. Matsuno, S. Konno, T. Fujisawa and M. Beeram, Mol. Cancer Ther., 2007, 6, 3563s–3564s Search PubMed
- J. Zhang, C. Huang and H. Huang, Oncol. Lett., 2014, 8, 2209–2214 Search PubMed
- S. Zalba, A. M. Contreras, A. Haeri, T. L. Ten Hagen, I. Navarro, G. Koning and M. J. Garrido, J. Controlled Release, 2015, 210, 26–38 CrossRef CAS PubMed
- D. Liu, C. He, A. Z. Wang and W. Lin, Int. J. Nanomed., 2013, 8, 3309–3319 Search PubMed
- X. Wang and Z. Guo, Chem. Soc. Rev., 2013, 42, 202–224 RSC
- H. M. Kieler-Ferguson, J. M. Frechet and F. C. Szoka, Jr., Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2013, 5, 130–138 CrossRef CAS PubMed
- S. Zalba and M. J. Garrido, Expert Opin. Drug Delivery, 2013, 10, 829–844 CrossRef CAS PubMed
- A. S. Lila, H. Kiwada and T. Ishida, Biol. Pharm. Bull., 2014, 37, 206–211 Search PubMed
- K. N. Burger, R. W. Staffhorst, H. C. de Vijlder, M. J. Velinova, P. H. Bomans, P. M. Frederik and B. de Kruijff, Nat. Med., 2002, 8, 81–84 CrossRef CAS PubMed
- V. Chupin, A. I. de Kroon and B. de Kruijff, J. Am. Chem. Soc., 2004, 126, 13816–13821 CrossRef CAS PubMed
- M. J. Velinova, R. W. Staffhorst, W. J. Mulder, A. S. Dries, B. A. Jansen, B. de Kruijff and A. I. de Kroon, Biochim. Biophys. Acta, 2004, 1663, 135–142 CrossRef CAS PubMed
- I. H. Hamelers, R. W. Staffhorst, J. Voortman, B. de Kruijff, J. Reedijk, P. M. van Bergen en Henegouwen and A. I. de Kroon, Clin. Cancer Res., 2009, 15, 1259–1268 CrossRef CAS PubMed
- R. W. Staffhorst, K. van der Born, C. A. Erkelens, I. H. Hamelers, G. J. Peters, E. Boven and A. I. de Kroon, Anti-Cancer Drugs, 2008, 19, 721–727 CrossRef CAS PubMed
- I. H. Hamelers, E. van Loenen, R. W. Staffhorst, B. de Kruijff and A. I. de Kroon, Mol. Cancer Ther., 2006, 5, 2007–2012 CrossRef CAS PubMed
- S. Bryde and A. I. de Kroon, Future Med. Chem., 2009, 1, 1467–1480 CrossRef CAS PubMed
- J. Kopecek and H. Bazilova, Eur. Polym. J., 1973, 9, 7–14 CrossRef CAS
- P. A. Vasey, S. B. Kaye, R. Morrison, C. Twelves, P. Wilson, R. Duncan, A. H. Thomson, L. S. Murray, T. E. Hilditch, T. Murray, S. Burtles, D. Fraier, E. Frigerio, J. Cassidy and C. R. C. P. I. I. Comm, Clin. Cancer Res., 1999, 5, 83–94 CAS
- E. W. Neuse, Met.-Based Drugs, 2008, 2008, 469531 Search PubMed
- E. Gianasi, R. G. Buckley, J. Latigo, M. Wasil and R. Duncan, J. Drug Targeting, 2002, 10, 549–556 CrossRef CAS PubMed
- D. P. Nowotnik and E. Cvitkovic, Adv. Drug Delivery Rev., 2009, 61, 1214–1219 CrossRef CAS PubMed
- R. Duncan and M. J. Vicent, Adv. Drug Delivery Rev., 2010, 62, 272–282 CrossRef CAS PubMed
- S. C. Song, S. B. Lee, B. H. Lee, H. W. Ha, K. T. Lee and Y. S. Sohn, J. Controlled Release, 2003, 90, 303–311 CrossRef CAS PubMed
- B. Klajnert and M. Bryszewska, Acta Biochim. Pol., 2001, 48, 199–208 CAS
- H. Baek, Y. Cho, C. O. Lee and Y. S. Sohn, Anti-Cancer Drugs, 2000, 11, 715–725 CrossRef CAS PubMed
- R. Song, Y. Joo Jun, J. Ik Kim, C. Jin and Y. S. Sohn, J. Controlled Release, 2005, 105, 142–150 CrossRef CAS PubMed
- J. Y. Yu, Y. J. Jun, S. H. Jang, H. J. Lee and Y. S. Sohn, J. Inorg. Biochem., 2007, 101, 1931–1936 CrossRef CAS PubMed
- A. Jain, S. K. Jain, N. Ganesh, J. Barve and A. M. Beg, Nanomedicine, 2010, 6, 179–190 CrossRef CAS PubMed
- S. Dhar, F. X. Gu, R. Langer, O. C. Farokhzad and S. J. Lippard, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 17356–17361 CrossRef CAS PubMed
- S. Dhar, N. Kolishetti, S. J. Lippard and O. C. Farokhzad, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 1850–1855 CrossRef CAS PubMed
- N. Graf, D. R. Bielenberg, N. Kolishetti, C. Muus, J. Banyard, O. C. Farokhzad and S. J. Lippard, ACS Nano, 2012, 6, 4530–4539 CrossRef CAS PubMed
- T. Sadhukha and S. Prabha, AAPS PharmSciTech, 2014, 15, 1029–1038 CrossRef CAS PubMed
- M. S. Shim and Y. Xia, Angew. Chem., Int. Ed., 2013, 52, 6926–6929 CrossRef CAS PubMed
- K. E. Broaders, S. Grandhe and J. M. Frechet, J. Am. Chem. Soc., 2011, 133, 756–758 CrossRef CAS PubMed
- C. de Gracia Lux, S. Joshi-Barr, T. Nguyen, E. Mahmoud, E. Schopf, N. Fomina and A. Almutairi, J. Am. Chem. Soc., 2012, 134, 15758–15764 CrossRef CAS PubMed
- H. Chen, W. He and Z. Guo, Chem. Commun., 2014, 50, 9714–9717 RSC
- M. Razmi, A. Divsalar, A. A. Saboury, Z. Izadi, T. Haertle and H. Mansuri-Torshizi, Colloids Surf., B, 2013, 112, 362–367 CrossRef CAS PubMed
- J. Yang, W. Mao, M. Sui, J. Tang and Y. Shen, J. Controlled Release, 2011, 152(suppl 1), e108–e109 CrossRef CAS PubMed
- J. Yang, W. Liu, M. Sui, J. Tang and Y. Shen, Biomaterials, 2011, 32, 9136–9143 CrossRef CAS PubMed
- J. Xu, Q. Fu, J. M. Ren, G. Bryant and G. G. Qiao, Chem. Commun., 2013, 49, 33–35 RSC
- P. Ma, H. Xiao, X. Li, C. Li, Y. Dai, Z. Cheng, X. Jing and J. Lin, Adv. Mater., 2013, 25, 4898–4905 CrossRef CAS PubMed
- J. Wang, X. Wang, Y. Song, C. Zhu, K. Wang and Z. Guo, Chem. Commun., 2013, 49, 2786–2788 RSC
- R. G. Qi, H. H. Xiao, S. H. Wu, Y. X. Li, Y. Zhang and X. B. Jing, J. Mater. Chem. B, 2015, 3, 176–179 RSC
- J. Kim, S. Pramanick, D. Lee, H. Park and W. J. Kim, Biomater. Sci., 2015, 3, 1002–1017 RSC
- R. Esfand and D. A. Tomalia, Drug. Discovery Today, 2001, 6, 427–436 CrossRef CAS PubMed
- N. Malik, E. G. Evagorou and R. Duncan, Anti-Cancer Drugs, 1999, 10, 767–776 CrossRef CAS PubMed
- G. J. Kirkpatrick, J. A. Plumb, O. B. Sutcliffe, D. J. Flint and N. J. Wheate, J. Inorg. Biochem., 2011, 105, 1115–1122 CrossRef CAS PubMed
- V. K. Yellepeddi, A. Kumar, D. M. Maher, S. C. Chauhan, K. K. Vangara and S. Palakurthi, Anticancer Res., 2011, 31, 897–906 CAS
- D. Pan, W. She, C. Guo, K. Luo, Q. Yi and Z. Gu, Biomaterials, 2014, 35, 10080–10092 CrossRef CAS PubMed
- H. Kulhari, D. Pooja, M. K. Singh and A. S. Chauhan, Drug Dev. Ind. Pharm., 2015, 41, 232–238 CrossRef CAS PubMed
- X. Wang, Q. Li, J. Xie, Z. Jin, J. Wang, Y. Li, K. Jiang and S. Fan, Nano Lett., 2009, 9, 3137–3141 CrossRef CAS PubMed
- K. Ajima, M. Yudasaka, T. Murakami, A. Maigne, K. Shiba and S. Iijima, Mol. Pharmaceutics, 2005, 2, 475–480 CrossRef CAS PubMed
- K. Ajima, M. Yudasaka, A. Maigne, J. Miyawaki and S. Iijima, J. Phys. Chem. B, 2006, 110, 5773–5778 CrossRef CAS PubMed
- K. Ajima, T. Murakami, Y. Mizoguchi, K. Tsuchida, T. Ichihashi, S. Iijima and M. Yudasaka, ACS Nano, 2008, 2, 2057–2064 CrossRef CAS PubMed
- T. A. Hilder and J. M. Hill, Nanotechnology, 2007, 18, 275704 CrossRef
- A. Guven, I. A. Rusakova, M. T. Lewis and L. J. Wilson, Biomaterials, 2012, 33, 1455–1461 CrossRef CAS PubMed
- R. P. Feazell, N. Nakayama-Ratchford, H. Dai and S. J. Lippard, J. Am. Chem. Soc., 2007, 129, 8438–8439 CrossRef CAS PubMed
- S. Dhar, Z. Liu, J. Thomale, H. Dai and S. J. Lippard, J. Am. Chem. Soc., 2008, 130, 11467–11476 CrossRef CAS PubMed
- S. Dhar, W. L. Daniel, D. A. Giljohann, C. A. Mirkin and S. J. Lippard, J. Am. Chem. Soc., 2009, 131, 14652–14653 CrossRef CAS PubMed
- Y. Min, C. Mao, D. Xu, J. Wang and Y. Liu, Chem. Commun., 2010, 46, 8424–8426 RSC
- F. M. Veronese and G. Pasut, Drug Discovery Today, 2005, 10, 1451–1458 CrossRef CAS PubMed
- Y. Min, C. Q. Mao, S. Chen, G. Ma, J. Wang and Y. Liu, Angew. Chem., Int. Ed., 2012, 51, 6742–6747 CrossRef CAS PubMed
- A. A. Bhirde, V. Patel, J. Gavard, G. Zhang, A. A. Sousa, A. Masedunskas, R. D. Leapman, R. Weigert, J. S. Gutkind and J. F. Rusling, ACS Nano, 2009, 3, 307–316 CrossRef CAS PubMed
- A. Kumar, S. Huo, X. Zhang, J. Liu, A. Tan, S. Li, S. Jin, X. Xue, Y. Zhao, T. Ji, L. Han, H. Liu, J. Zhang, G. Zou, T. Wang, S. Tang and X. J. Liang, ACS Nano, 2014, 8, 4205–4220 CrossRef CAS PubMed
- M. Arlt, D. Haase, S. Hampel, S. Oswald, A. Bachmatiuk, R. Klingeler, R. Schulze, M. Ritschel, A. Leonhardt, S. Fuessel, B. Buchner, K. Kraemer and M. P. Wirth, Nanotechnology, 2010, 21, 335101 CrossRef CAS PubMed
- L. Wu, C. Man, H. Wang, X. Lu, Q. Ma, Y. Cai and W. Ma, Pharm. Res., 2013, 30, 412–423 CrossRef CAS PubMed
- J. Li, A. Pant, C. F. Chin, W. H. Ang, C. Menard-Moyon, T. R. Nayak, D. Gibson, S. Ramaprabhu, T. Panczyk, A. Bianco and G. Pastorin, Nanomedicine, 2014, 10, 1465–1475 CrossRef CAS PubMed
- S. L. Yoong, B. S. Wong, Q. L. Zhou, C. F. Chin, J. Li, T. Venkatesan, H. K. Ho, V. Yu, W. H. Ang and G. Pastorin, Biomaterials, 2014, 35, 748–759 CrossRef CAS PubMed
- T. Thanasupawat, H. Bergen, S. Hombach-Klonisch, J. Krcek, S. Ghavami, M. R. Del Bigio, S. Krawitz, G. Stelmack, A. Halayko, M. McDougall, M. Meier, J. Stetefeld and T. Klonisch, Nanomedicine, 2015, 11, 913–925 CrossRef CAS PubMed
- V. Shanmugam, Y. H. Chien, Y. S. Cheng, T. Y. Liu, C. C. Huang, C. H. Su, Y. S. Chen, U. Kumar, H. F. Hsu and C. S. Yeh, ACS Appl. Mater. Interfaces, 2014, 6, 4382–4393 CAS
- L. Muzi, C. Menard-Moyon, J. Russier, J. Li, C. F. Chin, W. H. Ang, G. Pastorin, G. Risuleo and A. Bianco, Nanoscale, 2015, 7, 5383–5394 RSC
- B. S. Wong, S. L. Yoong, A. Jagusiak, T. Panczyk, H. K. Ho, W. H. Ang and G. Pastorin, Adv. Drug Delivery Rev., 2013, 65, 1964–2015 CrossRef CAS PubMed
- C. Allen, D. Maysinger and A. Eisenberg, Colloids Surf., B, 1999, 16, 3–27 CrossRef CAS
- C. Wang, J. Mallela and S. Mohapatra, Curr. Drug Metab., 2013, 14, 900–909 CrossRef CAS PubMed
- L. Bromberg, J. Controlled Release, 2008, 128, 99–112 CrossRef CAS PubMed
- M. F. Francis, M. Cristea and F. M. Winnik, Pure Appl. Chem., 2004, 76, 1321–1335 CrossRef CAS
- F. Mathot, L. van Beijsterveldt, V. Preat, M. Brewster and A. Arien, J. Controlled Release, 2006, 111, 47–55 CrossRef CAS PubMed
- H. Gao, Y. W. Yang, Y. G. Fan and J. B. Ma, J. Controlled Release, 2006, 112, 301–311 CrossRef CAS PubMed
- P. Tengamnuay and A. K. Mitra, Pharm. Res., 1990, 7, 370–375 CrossRef CAS
- A. K. Gupta, S. Madan, D. K. Majumdar and A. Maitra, Int. J. Pharm., 2000, 209, 1–14 CrossRef CAS PubMed
- J. Liaw, S. F. Chang and F. C. Hsiao, Gene Ther., 2001, 8, 999–1004 CrossRef CAS PubMed
- K. J. Haxton and H. M. Burt, J. Pharm. Sci., 2009, 98, 2299–2316 CrossRef CAS PubMed
- N. Nishiyama, M. Yokoyama, T. Aoyagi, T. Okano, Y. Sakurai and K. Kataoka, Langmuir, 1998, 15, 377–383 CrossRef
- A. Lavasanifar, J. Samuel and G. S. Kwon, Adv. Drug Delivery Rev., 2002, 54, 169–190 CrossRef CAS PubMed
- N. Nishiyama, Y. Kato, Y. Sugiyama and K. Kataoka, Pharm. Res., 2001, 18, 1035–1041 CrossRef CAS
- N. Nishiyama and K. Kataoka, J. Controlled Release, 2001, 74, 83–94 CrossRef CAS PubMed
- N. Nishiyama, S. Okazaki, H. Cabral, M. Miyamoto, Y. Kato, Y. Sugiyama, K. Nishio, Y. Matsumura and K. Kataoka, Cancer Res., 2003, 63, 8977–8983 CAS
- M. Baba, Y. Matsumoto, A. Kashio, H. Cabral, N. Nishiyama, K. Kataoka and T. Yamasoba, J. Controlled Release, 2012, 157, 112–117 CrossRef CAS PubMed
- R. Plummer, R. H. Wilson, H. Calvert, A. V. Boddy, M. Griffin, J. Sludden, M. J. Tilby, M. Eatock, D. G. Pearson, C. J. Ottley, Y. Matsumura, K. Kataoka and T. Nishiya, Br. J. Cancer, 2011, 104, 593–598 CrossRef CAS PubMed
- Y. Matsumura, Jpn. J. Clin. Oncol., 2014, 44, 515–525 CrossRef PubMed
- V. Heinemann, D. Quietzsch, F. Gieseler, M. Gonnermann, H. Schonekas, A. Rost, H. Neuhaus, C. Haag, M. Clemens, B. Heinrich, U. Vehling-Kaiser, M. Fuchs, D. Fleckenstein, W. Gesierich, D. Uthgenannt, H. Einsele, A. Holstege, A. Hinke, A. Schalhorn and R. Wilkowski, J. Clin. Oncol., 2006, 24, 3946–3952 CrossRef CAS PubMed
- X. Li, R. Li, X. Qian, Y. Ding, Y. Tu, R. Guo, Y. Hu, X. Jiang, W. Guo and B. Liu, Eur. J. Pharm. Biopharm., 2008, 70, 726–734 CrossRef CAS PubMed
- P. Xu, E. A. Van Kirk, W. J. Murdoch, Y. Zhan, D. D. Isaak, M. Radosz and Y. Shen, Biomacromolecules, 2006, 7, 829–835 CrossRef CAS PubMed
- H. S. Oberoi, N. V. Nukolova, F. C. Laquer, L. Y. Poluektova, J. Huang, Y. Alnouti, M. Yokohira, L. L. Arnold, A. V. Kabanov, S. M. Cohen and T. K. Bronich, Int. J. Nanomed., 2012, 7, 2557–2571 CrossRef CAS PubMed
- W. Jin, P. Xu, Y. Zhan, Y. Shen, E. A. Van Kirk, B. Alexander, W. J. Murdoch, L. Liu and D. D. Isaak, Drug Delivery, 2007, 14, 279–286 CrossRef CAS PubMed
- C. Wang, Y. Gong, N. Fan, S. Liu, S. Luo, J. Yu and J. Huang, Colloids Surf., B, 2009, 70, 84–90 CrossRef CAS PubMed
- L. Ye, K. Letchford, M. Heller, R. Liggins, D. Guan, J. N. Kizhakkedathu, D. E. Brooks, J. K. Jackson and H. M. Burt, Biomacromolecules, 2011, 12, 145–155 CrossRef CAS PubMed
- N. V. Nukolova, H. S. Oberoi, S. M. Cohen, A. V. Kabanov and T. K. Bronich, Biomaterials, 2011, 32, 5417–5426 CrossRef CAS PubMed
- J. Eliezar, W. Scarano, N. R. Boase, K. J. Thurecht and M. H. Stenzel, Biomacromolecules, 2015, 16, 515–523 CrossRef CAS PubMed
- M. Li, Z. Tang, S. Lv, W. Song, H. Hong, X. Jing, Y. Zhang and X. Chen, Biomaterials, 2014, 35, 3851–3864 CrossRef CAS PubMed
- M. Shahin, N. Safaei-Nikouei and A. Lavasanifar, J. Drug Targeting, 2014, 22, 629–637 CrossRef CAS PubMed
- W. Song, M. Li, Z. Tang, Q. Li, Y. Yang, H. Liu, T. Duan, H. Hong and X. Chen, Macromol. Biosci., 2012, 12, 1514–1523 CrossRef CAS PubMed
- Z. Ahmad, Z. Tang, A. Shah, S. Lv, D. Zhang, Y. Zhang and X. Chen, Macromol. Biosci., 2014, 14, 1337–1345 CrossRef CAS PubMed
- X. Z. Yang, X. J. Du, Y. Liu, Y. H. Zhu, Y. Z. Liu, Y. P. Li and J. Wang, Adv. Mater., 2014, 26, 931–936 CrossRef CAS PubMed
- J. Ahn, Y. Miura, N. Yamada, T. Chida, X. Liu, A. Kim, R. Sato, R. Tsumura, Y. Koga, M. Yasunaga, N. Nishiyama, Y. Matsumura, H. Cabral and K. Kataoka, Biomaterials, 2015, 39, 23–30 CrossRef CAS PubMed
- A. Dag, Y. Jiang, K. J. Karim, G. Hart-Smith, W. Scarano and M. H. Stenzel, Macromol. Rapid Commun., 2015, 36, 890–897 CrossRef CAS PubMed
- H. Cabral, N. Nishiyama and K. Kataoka, J. Controlled Release, 2007, 121, 146–155 CrossRef CAS PubMed
- H. Cabral, N. Nishiyama, S. Okazaki, H. Koyama and K. Kataoka, J. Controlled Release, 2005, 101, 223–232 CrossRef CAS PubMed
- H. Cabral, Y. Matsumoto, K. Mizuno, Q. Chen, M. Murakami, M. Kimura, Y. Terada, M. R. Kano, K. Miyazono, M. Uesaka, N. Nishiyama and K. Kataoka, Nat. Nanotechnol., 2011, 6, 815–823 CrossRef CAS PubMed
- M. Rafi, H. Cabral, M. R. Kano, P. Mi, C. Iwata, M. Yashiro, K. Hirakawa, K. Miyazono, N. Nishiyama and K. Kataoka, J. Controlled Release, 2012, 159, 189–196 CrossRef CAS PubMed
- M. Murakami, H. Cabral, Y. Matsumoto, S. Wu, M. R. Kano, T. Yamori, N. Nishiyama and K. Kataoka, Sci. Transl. Med., 2011, 3, 64ra62 Search PubMed
- Y. Y. Xu, Y. Z. Du, H. Yuan, L. N. Liu, Y. P. Niu and F. Q. Hu, J. Drug Targeting, 2011, 19, 344–353 CrossRef CAS PubMed
- H. S. Oberoi, N. V. Nukolova and T. K. Bronich, PMSE preprints American Chemical Society. Division of Polymeric Materials: Science and Engineering. Meeting, 2011, 104, 630–631 Search PubMed
- H. S. Oberoi, F. C. Laquer, L. A. Marky, A. V. Kabanov and T. K. Bronich, J. Controlled Release, 2011, 153, 64–72 CrossRef CAS PubMed
- H. Xiao, Y. Fan, S. Liu, X. Chen, Y. Huang and X. Jing, J. Controlled Release, 2011, 152(suppl 1), e103–e104 CrossRef CAS PubMed
- H. Xiao, D. Zhou, S. Liu, R. Qi, Y. Zheng, Y. Huang and X. Jing, Macromol. Biosci., 2012, 12, 367–373 CrossRef CAS PubMed
- H. Xiao, W. Li, R. Qi, L. Yan, R. Wang, S. Liu, Y. Zheng, Z. Xie, Y. Huang and X. Jing, J. Controlled Release, 2012, 163, 304–314 CrossRef CAS PubMed
- H. Cabral, M. Murakami, H. Hojo, Y. Terada, M. R. Kano, U. I. Chung, N. Nishiyama and K. Kataoka, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 11397–11402 CrossRef CAS PubMed
- T. Ueno, K. Endo, K. Hori, N. Ozaki, A. Tsuji, S. Kondo, N. Wakisaka, S. Murono, K. Kataoka, Y. Kato and T. Yoshizaki, Int. J. Nanomed., 2014, 9, 3005–3012 CrossRef CAS PubMed
- H. S. Oberoi, N. V. Nukolova, Y. Zhao, S. M. Cohen, A. V. Kabanov and T. K. Bronich, Chemother. Res. Pract., 2012, 2012, 905796 Search PubMed
- D. Yong, Y. Luo, F. Du, J. Huang, W. Lu, Z. Dai, J. Yu and S. Liu, Colloids Surf., B, 2013, 105, 31–36 CrossRef CAS PubMed
- H. Wu, H. Cabral, K. Toh, P. Mi, Y. C. Chen, Y. Matsumoto, N. Yamada, X. Liu, H. Kinoh, Y. Miura, M. R. Kano, H. Nishihara, N. Nishiyama and K. Kataoka, J. Controlled Release, 2014, 189, 1–10 CrossRef CAS PubMed
- H. T. Duong, V. T. Huynh, P. de Souza and M. H. Stenzel, Biomacromolecules, 2010, 11, 2290–2299 CrossRef CAS PubMed
- S. Dhar and S. J. Lippard, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 22199–22204 CrossRef CAS PubMed
- S. Aryal, C. M. Jack Hu, V. Fu and L. F. Zhang, J. Mater. Sci., 2012, 22, 994–999 CAS
- H. Xiao, L. Yan, Y. Zhang, R. Qi, W. Li, R. Wang, S. Liu, Y. Huang, Y. Li and X. Jing, Chem. Commun., 2012, 48, 10730–10732 RSC
- H. Xiao, H. Song, Q. Yang, H. Cai, R. Qi, L. Yan, S. Liu, Y. Zheng, Y. Huang, T. Liu and X. Jing, Biomaterials, 2012, 33, 6507–6519 CrossRef CAS PubMed
- W. Scarano, P. de Souza and M. H. Stenzel, Biomater. Sci., 2015, 3, 163–174 RSC
- P. Muller, B. Schroder, J. A. Parkinson, N. A. Kratochwil, R. A. Coxall, A. Parkin, S. Parsons and P. J. Sadler, Angew. Chem., Int. Ed., 2003, 42, 335–339 CrossRef CAS PubMed
- F. S. Mackay, J. A. Woods, P. Heringova, J. Kasparkova, A. M. Pizarro, S. A. Moggach, S. Parsons, V. Brabec and P. J. Sadler, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 20743–20748 CrossRef CAS PubMed
- P. J. Bednarski, F. S. Mackay and P. J. Sadler, Anti-Cancer Agents Med. Chem., 2007, 7, 75–93 CrossRef CAS PubMed
- H. Xiao, G. T. Noble, J. F. Stefanick, R. Qi, T. Kiziltepe, X. Jing and B. Bilgicer, J. Controlled Release, 2014, 173, 11–17 CrossRef CAS PubMed
- W. Shen, J. Luan, L. Cao, J. Sun, L. Yu and J. Ding, Biomacromolecules, 2015, 16, 105–115 CrossRef CAS PubMed
- R. Du, H. Xiao, G. Guo, B. Jiang, X. Yan, W. Li, X. Yang, Y. Zhang, Y. Li and X. Jing, Colloids Surf., B, 2014, 123, 734–741 CrossRef CAS PubMed
- V. B. Jadhav, Y. J. Jun, J. H. Song, M. K. Park, J. H. Oh, S. W. Chae, I. S. Kim, S. J. Choi, H. J. Lee and Y. S. Sohn, J. Controlled Release, 2010, 147, 144–150 CrossRef CAS PubMed
- P. G. Avaji, H. I. Joo, J. H. Park, K. S. Park, Y. J. Jun, H. J. Lee and Y. S. Sohn, J. Inorg. Biochem., 2014, 140, 45–52 CrossRef CAS PubMed
- D. I. Jodrell, T. R. Evans, W. Steward, D. Cameron, J. Prendiville, C. Aschele, C. Noberasco, M. Lind, J. Carmichael, N. Dobbs, G. Camboni, B. Gatti and F. De Braud, Eur. J. Cancer, 2004, 40, 1872–1877 CrossRef CAS PubMed
- V. Brabec, J. Kasparkova, O. Vrana, O. Novakova, J. W. Cox, Y. Qu and N. Farrell, Biochemistry, 1999, 38, 6781–6790 CrossRef CAS PubMed
- A. Johnson, Y. Qu, B. Van Houten and N. Farrell, Nucleic Acids Res., 1992, 20, 1697–1703 CrossRef CAS PubMed
- T. D. McGregor, Z. Balcarova, Y. Qu, M. C. Tran, R. Zaludova, V. Brabec and N. Farrell, J. Inorg. Biochem., 1999, 77, 43–46 CrossRef CAS PubMed
- H. Xiao, H. Song, Y. Zhang, R. Qi, R. Wang, Z. Xie, Y. Huang, Y. Li, Y. Wu and X. Jing, Biomaterials, 2012, 33, 8657–8669 CrossRef CAS PubMed
- H. Xiao, J. F. Stefanick, X. Jia, X. Jing, T. Kiziltepe, Y. Zhang and B. Bilgicer, Chem. Commun., 2013, 49, 4809–4811 RSC
- J. Tomasina, S. Lheureux, P. Gauduchon, S. Rault and A. Malzert-Freon, Biomaterials, 2013, 34, 1073–1101 CrossRef CAS PubMed
- T. C. Johnstone, J. J. Wilson and S. J. Lippard, Inorg. Chem., 2013, 52, 12234–12249 CrossRef CAS PubMed
- X. Xue, M. D. Hall, Q. Zhang, P. C. Wang, M. M. Gottesman and X. J. Liang, ACS Nano, 2013, 7, 10452–10464 CrossRef CAS PubMed
- H. Cabral and K. Kataoka, J. Controlled Release, 2014, 190, 465–476 CrossRef CAS PubMed
- X. Wang, X. Wang and Z. Guo, Acc. Chem. Res., 2015, 48, 2622–2631 CrossRef CAS PubMed
- M. S. Goldberg, S. S. Hook, A. Z. Wang, J. W. Bulte, A. K. Patri, F. M. Uckun, V. L. Cryns, J. Hanes, D. Akin, J. B. Hall, N. Gharkholo and R. J. Mumper, Nanomedicine, 2013, 8, 299–308 CrossRef CAS PubMed
- J. S. Butler and P. J. Sadler, Curr. Opin. Chem. Biol., 2013, 17, 175–188 CrossRef CAS PubMed
| This journal is © The Royal Society of Chemistry 2016 |