 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Carbon Capture and Storage: introductory lecture
Berend
Smit
ab
aLaboratory of Molecular Simulation, Institut des Sciences et Ingénierie Chimiques, Valais, Ecole Polytechnique Fédérale de Lausanne (EPFL), Rue de l'Industrie 17, CH-1951 Sion, Switzerland. E-mail: berend.smit@epfl.ch
bDepartment of Chemical and Biomolecular Engineering, University of California, Berkeley, CA 94720-1462, USA
First published on 29th June 2016
Abstract
Carbon Capture and Storage (CCS) is the only available technology that allows us to significantly reduce our CO2 emissions while keeping up with the ever-increasing global energy demand. Research in CCS focuses on reducing the costs of carbon capture and increasing our knowledge of geological storage to ensure the safe and permanent storage of CO2. This brief review will discuss progress in different capture and storage technologies.
Introduction
Why do we need a Faraday Discussion on carbon capture and storage? If we simply leave all carbon in the ground there is no need to develop new technologies to capture CO2 from flue gasses and/or to refine our knowledge on how to permanently store CO2 in geological formations. Indeed, there is little doubt that in the long term we will have developed technologies to completely decarbonize our energy production. There is also little doubt that 80% of all of tomorrow’s energy production will still be based on fossil fuels. In fact, most energy production scenarios assume that the relative importance of fossil fuels will decrease in the coming years. However, as our total energy consumption is expected to increase faster than renewable energies can grow, these scenarios also predict that in absolute terms the total fossil fuel production will continue to increase.1Carbon Capture and Storage (CCS) is aimed at making this transition towards decarbonized energy production as smooth as possible. As we will see in the rest of these Discussions, we do have the technology to capture and store significant amounts of emitted CO2. However, the costs associated with such a reduction of CO2 emissions are large. These Discussions show that we are generating many options to reduce these costs. These costs, however, will never be zero and society will have to pay more for energy. At present one can emit CO2 without any costs. It is only if we, as a society, put a price on emitting carbon or regulate the maximum allowed global CO2 emission, that the technologies described here will be of any use.2
The ideas underlying carbon capture and storage are simple (see Fig. 1).3–6 The first step is to separate CO2 from a stationary source, which can, for example, be a coal or gas fired power plant or a cement factory. The next step is to transport CO2 through a pipeline, and subsequently store CO2 in a geological formation. There are no fundamental technological barriers for each of these three steps, and hence to implement large-scale CCS.
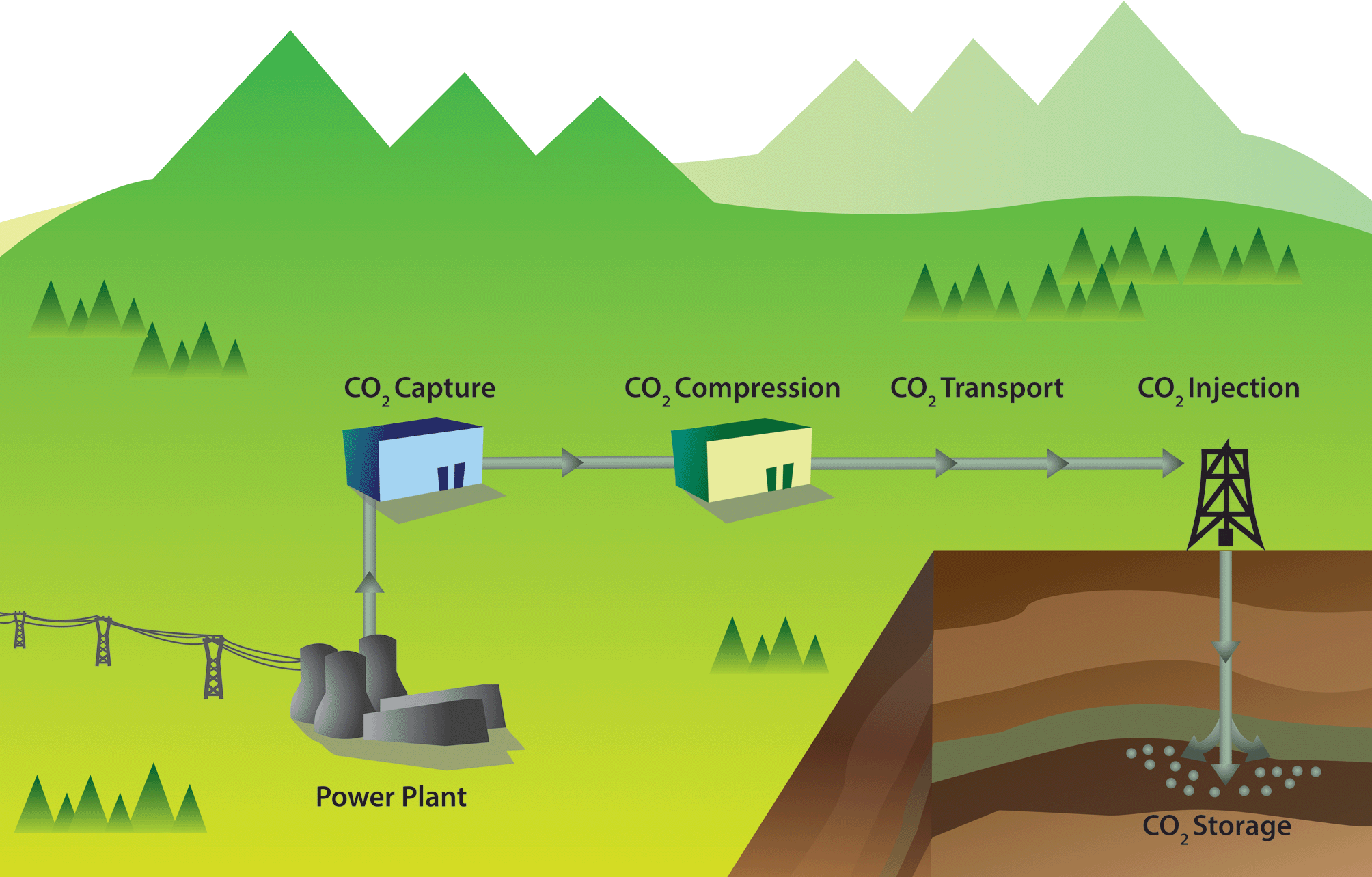 | ||
| Fig. 1 Schematic drawing of a carbon capture and storage project (figure adopted from Smit et al.3). | ||
A carbon-capture unit can be based on the many years of experience we have with sweetening natural gas. Most natural gas contains significant amounts of CO2, which needs to be removed before commercial use. The current technology of gas sweetening is based on scrubbing with amine solutions, which was developed in 1930 by Bottoms.7 Amine scrubbing can also be used to capture carbon from flue gasses.8 For many years, oil companies have used CO2 to enhance the recovery of oil. Enhanced oil recovery involves the transport and injection of CO2 in those geological formations where oil is produced. This technology can be adopted to inject CO2 into geological formations for permanent storage.
A question one often poses is whether we should not recycle the CO2 that is produced from a power plant.9 CO2 is a waste product, so why does the title of this article not include recycling or re-use of CO2? First of all, CO2 is used in several applications. For example, most CO2 for enhanced oil recovery is obtained from geological formations; using CO2 from anthropogenic sources is simply more expensive. Of course, one would like that all the CO2 that is at present used commercially should come from anthropogenic sources. However, as our CO2 production from power plants alone is orders of magnitude larger than what we can use, using anthropogenic CO2 for these applications will be important to develop the technologies, but will have a minor effect on emissions. We simply produce so much CO2 that we will saturate any conceivable market if we convert CO2 into useful chemicals.10
There are only two products that are produced on the scale of CO2: water and fuels. One could therefore envision closing the combustion cycle and converting CO2 back into a fuel. This scheme will only reduce CO2 emissions if we have a source of renewable, non-CO2 emitting, energy. However, given such a source of renewable energy there might be little need to use fossil fuels in the first place. One of the issues with renewable energy is its intermittency, which requires additional technologies to store energy. If we produce electricity from solar energy during the day, we rely on storage for the night, or wind energy might be available in excess during the winter, but not enough during the summer. Fossil fuels have excellent energy storage capacity and one can envision using an excess of renewable energy to convert CO2 into a synthetic fuel for storage or transportation fuels. But, in the case of transportation fuels, only if the CO2 is coming directly from the air can one truly close the carbon loop.
It is also important to realize that a fraction of the fossil fuels are used by the chemical industry as a feedstock for chemicals, and ideally one would also like to replace these fossil fuels with CO2 as the source of carbon. Therefore utilization of CO2 in the context of decarbonizing the chemical industry is an important topic of research.11
Given the enormous volumes of CO2 that fossil fuel power plants generate, the only viable option to significantly reduce CO2 emissions is to carry out large-scale CCS. At present CCS is only possible for stationary sources, which amounts to approximately 50% of all CO2 emissions (see Fig. 2). That the technologies to carry out large-scale CCS exist does not imply that no further research is needed. On the capture side one has to realize that the separation of CO2 from flue gasses of a power plant costs energy and requires additional investments, which increase the price of electricity significantly. In addition, large-scale CO2 storage raises questions on how we can guarantee the long-term safety of the injected CO2. These two issues, the concern of the public about the cost of capture and the safety of geologically stored CO2, are also the driving force of most of the research underlying this Faraday Discussions.
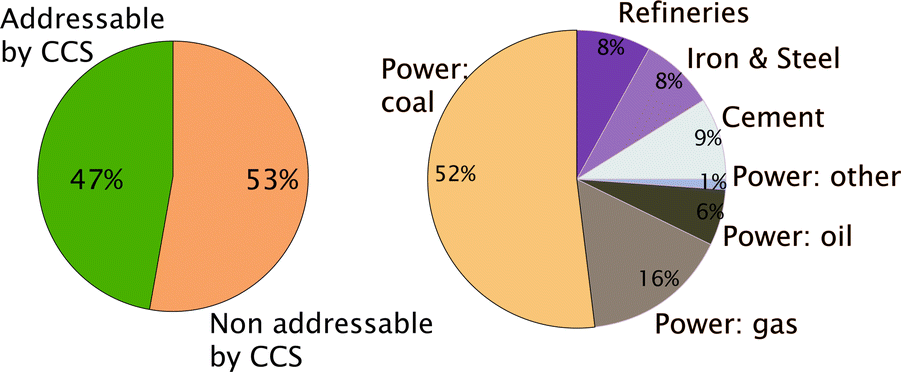 | ||
| Fig. 2 (Left) Fraction of the total CO2 emission from stationary sources and (right) CO2 from the different stationary sources (figure adopted from Smit et al.3). | ||
This Faraday Discussions brings together researchers from several different fields. In this Introductory Lecture the context is given on how these different contributions will help us in addressing the main challenges in CCS. These contributions range from pilot-plant studies and cost calculations, to obtaining a detailed fundamental understanding of the materials that are involved.
Carbon capture
There are different technologies to capture CO2.12,13 If we assume existing power plants as the emission sources, the simplest option is post-combustion carbon capture. If there is enough space at the power plant, a post combustion power plant can simply be added without major changes to the existing plant (Fig. 3). However, one has to realize that not only are the capital investments associated with a capture unit significant, but also that the separation of CO2 from flue gasses does require significant amounts of energy.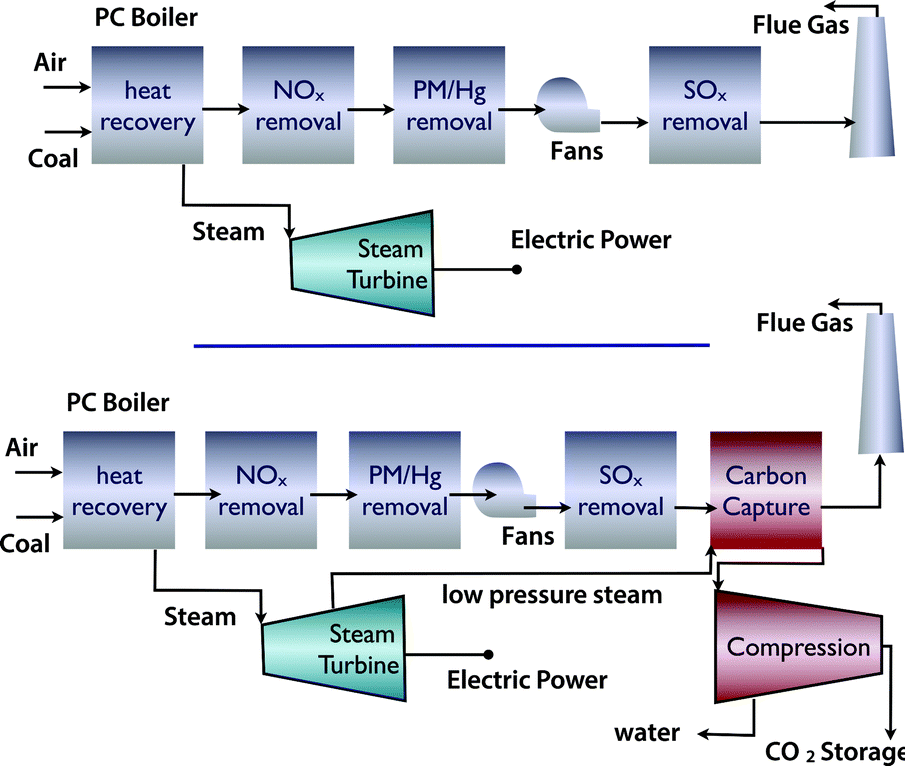 | ||
| Fig. 3 (a) Coal fired power plant and (b) coal fired power plant with carbon capture and compression for geological storage (figure adopted from Smit et al.3). | ||
The currently available technologies are mostly based on amine scrubbing. An often-used reference is monoethanolamine (MEA). MEA is relatively cheap and has high CO2 absorption capacity. However, MEA has also some important disadvantages: it is toxic and corrosive at elevated temperatures. In addition, as it binds CO2 strongly the regeneration is energy intensive. Therefore finding alternative absorbents is an active area of research. In addition, research exists to find other post-combustion capture technologies such as solid adsorption or membrane separations. These technologies are in an earlier stage of development, but have the potential of decreasing the costs associated with CCS.
Post-combustion CCS has as its main advantage that it has the smallest impact on existing power plants. If one wanted to construct new power plants or further modify existing power plants, there are alternative technologies.
The problem of carbon capture from flue gasses is the nitrogen in the air. If one burns fossil fuels with pure oxygen, one would produce only CO2 and H2O, which can be separated by simply condensing the water. For such a process the bottleneck is the separation of oxygen from air. At present this is mainly done by cryogenic distillation (see Fig. 4) or through chemical looping (see Fig. 5). In chemical looping one uses a solid oxygen carrier that is looping between two fluidized beds. In one bed the solid adsorbs oxygen from the air and the oxygen-loaded solid particles are then transported to the combustion part where it releases the oxygen and the fossil fuel can be burned without the nitrogen.14
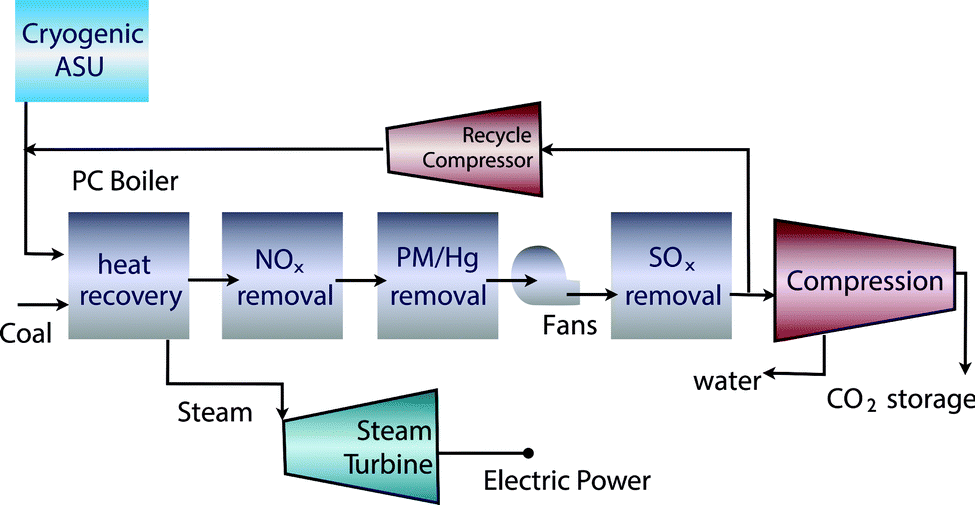 | ||
| Fig. 4 Oxy-combustion coal fired power plant with carbon capture and storage (figure adopted from Smit et al.3). | ||
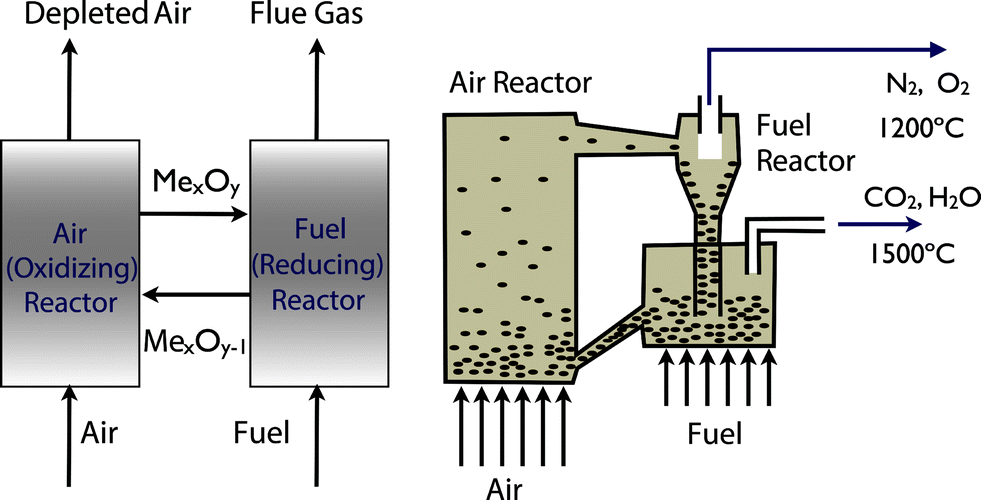 | ||
| Fig. 5 Chemical looping: the left figure shows the schematics of metal oxide (MeO) used to transport oxygen form the air into the reactor. The right figure illustrates how this solid is looping form the air reactor to the fuel reactor and back (figure adopted from Smit et al.3). | ||
Modern power plants often use a combined cycle. The idea of a combined cycle is that one can increase the efficiency of a heat engine by increasing the operational temperature. Conventional power generation uses steam as a working fluid. However, using steam as a working fluid at high temperatures requires relatively large amounts of (prohibitively) expensive steel. Therefore the use of inexpensive steel imposes a practical limit on the maximum operational temperature, and hence the efficiency of steam turbines. For gas turbines, however, the amount of required expensive steel is far less. In a combined cycle power plant, one uses the flue gas to drive such gas turbines and the heat of the gas turbines’ exhaust is used to generate steam as in conventional power plants. In Natural Gas Combined Cycle (NGCC) the fuel is natural gas while in an Integrated Gasification Combined Cycle (IGCC) one uses syngas obtained from coal gasification. From a carbon capture point of view the important difference is that in NGCC one has to separate CO2 from flue gas at much higher temperatures, while in IGCC the CO2 separation is from syngas, which involves a high temperature H2/CO2 separation.
Separation technologies
One of the main driving forces for the development of novel carbon capture technologies is reducing costs. For CCS the costs include separation and compression, and include both equipment and operational costs. One way to express the operational costs is to use the parasitic energy, which is defined as the loss of electricity production caused CCS: we redirect the energy from a power plant for the separation and the compression.15,16 Huck et al.16 showed that for flue gas from a coal-fired power plant (14% CO2–86% N2) the minimum energy for separation is 168 kJ kgCO2−1 and the compression to 150 bar, which is required for transport and geological storage, is 358 kJ kgCO2−1. A typical MEA plant has a parasitic energy of 1060 kJ kgCO2−1.15,17 Comparing these numbers shows that the parasitic energy for only the separation in the MEA process is 702 kJ kgCO2−1, which is 4 times the thermodynamic minimum. Comparisons between typical separations in the chemical process industry shows that a factor 2 should be achievable, which shows that one should be able to reduce the parasitic energy to 700 kJ kgCO2−1. In these parasitic energy calculations one only considers operational costs. Capital costs are an equally important factor, but these are significantly more difficult to estimate for novel materials.18Liquid-adsorption
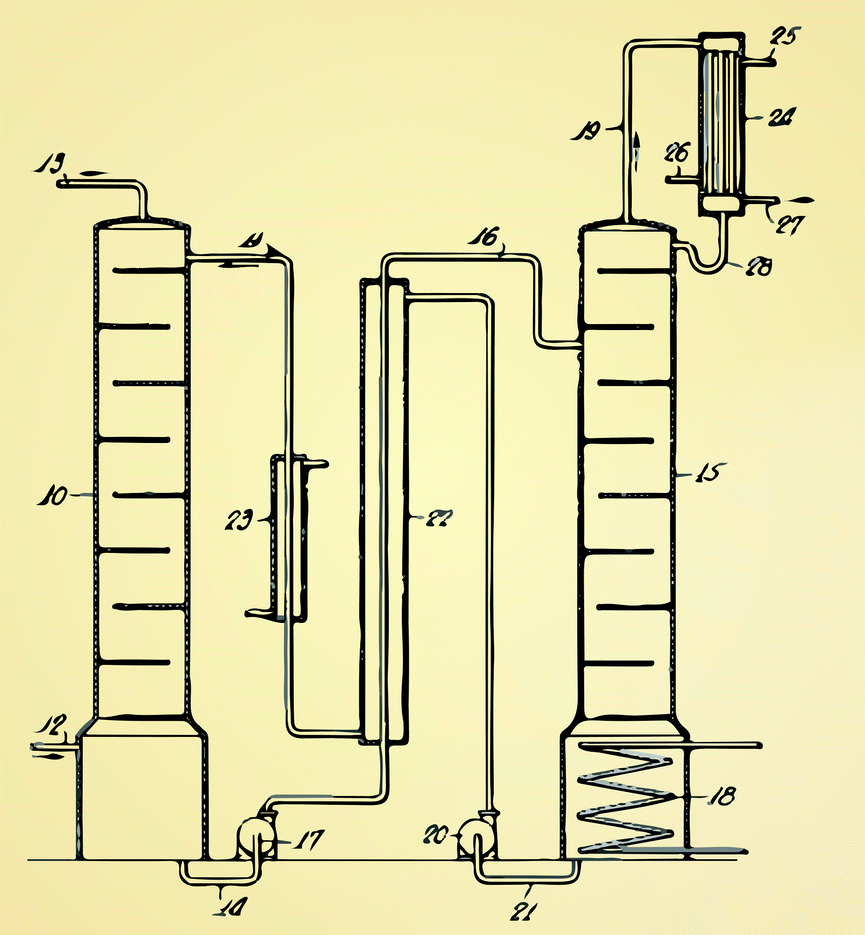 | ||
| Fig. 6 The original design by Bottoms7 of an amine scrubbing using the removed CO2 from natural gas. The current technology for liquid absorption looks very similar: flue gas is injected at (12) and the CO2 is captured by an amine solution is the absorber (10). In the stripper (15) the amine solution is regenerated using steam (18) from the power plant. Heat losses are minimized by using a heat exchanger (27). | ||
Lin et al. show from Aspen simulation studies that one of the main bottlenecks for improving the efficiency of the capture process is the solvent's absorption rate and capacity.
To speed up the screening process for new solvents Brand et al. (DOI: 10.1039/C6FD00041J) introduce in this volume a thermodynamic model based on the statistical mechanical SAFT approach to predict the basic thermodynamic and transport data to predict the performance of a new solvent.
An important requirement for any novel materials, and certainly for materials that are aimed at mitigating green house gas emissions, is that their production does not create another environmental problem. For this it is important to carry out a life cycle assessment. The problem is that for novel materials essential data are lacking to carry out a complete assessment. Cuéllar-Franca et al. (DOI: 10.1039/C6FD00054A) developed an alternative method in which the aim is to identify hotspots in the synthesis process. Cuéllar-Franca et al. show that for the current synthesis of one of the most promising ionic liquids, [P66614][124Triz], the environmental impacts are worse than for MEA. However, as Cuéllar-Franca et al. point out this calculation is done per kg of material and we lack information to carry out the same calculation per kg of removed CO2. Nevertheless it is important to have such information available at an early stage of the research as ionic liquids are tunable and it would be desirable to add LCA aspects to the selection criteria.
Mercy et al. (DOI: 10.1039/C6FD00081A) discuss a computational study on the binding mechanism in two types of ionic liquids: superbase and amino acid ionic liquids. Mercy studied the mechanism of CO2 adsorption in these ionic liquids to obtain insight on how to tune the strength of the interactions with CO2. Ideally for carbon capture one would like to have a strength sufficiently strong to selectively adsorb CO2 at a sufficiently high capacity, but not too strong as this may make regeneration more expensive.
A practical issue that is often observed in ionic liquids is that they tend to be viscous. An increased viscosity reduces mass transfer and makes these ionic liquids less efficient, as one would expect on purely thermodynamic grounds. To mitigate these negative effects of ionic liquids Stolaroff et al. (DOI: 10.1039/C6FD00049E) discuss a methodology to encapsulate ionic liquids with a semi-permeable membrane. This membrane is permeable to CO2 but not to the solvent. Stolaroff et al. showed that for these Micro-Encapsulated CO2 Sorbents (MECS) the rate of CO2 absorption is enhanced by a factor 3.5 compared to a film of ionic liquids.
A different solution for the mass-transfer limitations of ionic liquids is proposed in the Discussions by Dowson et al. (DOI: 10.1039/C6FD00035E). Dowson et al. use an ionic liquid that is in the solid phase at the condition of interest. To increase the surface area of this solid ionic liquid (SoIL), the solids were finely ground. In a pressure swing operation, Dowson et al. illustrated the feasibility of the process: compared to similar ILs the capacity was decreased but the uptake rate was significantly faster.
Solid adsorption
An alternative to liquid absorption is to use solid adsorbents. The focus of most of this scientific research is to find novel materials that can capture CO2 more efficiently.24 However, from a process engineering point of view solids are more difficult to handle compared to liquids. For example, liquids can be pumped, which makes it simple to develop a continuous process with full heat integration. Solid adsorption is usually envisioned as a batch process with two beds of adsorbents: one bed is in adsorption mode while the other is in regeneration mode. It is possible to use solids in continuous operation by using a fluidized bed or by using a simulated moving bed.3 Alternatively one can make a slurry of solid particles in a solvent, which can be pumped.25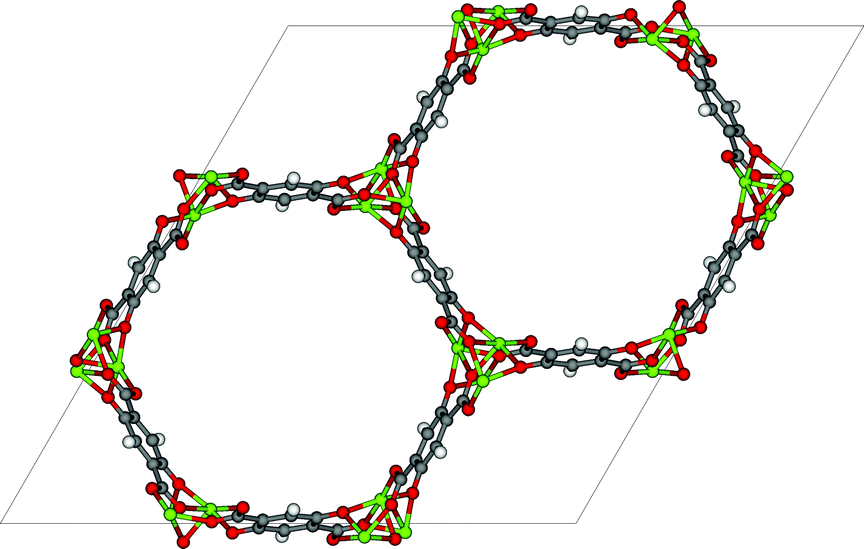 | ||
| Fig. 7 Example of a metal–organic framework (MOF-74 or CPO-27) (figure by Matthew Witman). | ||
To compare different materials one needs to have a metric. Huck et al.16 used the parasitic energy (i.e. the loss of produced electricity), to evaluate the performance of MOFs for carbon capture and sequestration. Most of these calculations rely on accurate thermodynamic data. If accurate experimental data are missing, one can rely on molecular simulations to predict the adsorption behavior in these materials. Or, as discussed in the article by Cresswell et al. (DOI: 10.1039/C6FD00026F), to predict the effects of impurities on the thermodynamic properties of CO2.
For some practical applications it may not be necessary to compress CO2 or have CO2 at such a high purity. Joos et al. (DOI: 10.1039/C6FD00031B) discuss how changing the purity requirement influences the selection of an adsorption material.
The thermodynamic variables are not the only important criteria discussed by Mangano et al. (DOI: 10.1039/C6FD00045B), who report the stability of Mg- and Ni-CPO-27 (also referred to as MOF-74, see Fig. 7) upon adsorption of wet flue gasses. Mangano et al. show that Mg-CPO-27 rapidly degenerates, while Ni-CPO-27 remains relatively stable. In this context it is important to realize that MOFs represent a class of materials; some of MOFs are unstable in water but some others can be boiled in acids. In addition to chemical stability it is also important to study the mechanical stability of these materials. In practical applications the pressure exerted on these materials can be very high.
Most MOFs that can selectively adsorb CO2 from flue gasses require dry flue gasses, as those materials that are selective for CO2 often have an even greater affinity for water. To mitigate the negative interference from water, amine-appended MOFs have been developed.28 Of particular interest are those amine appended MOFs that show a phase transition. This phase transition is associated with the collective behavior of the amines, which form chains upon the adsorption of CO2.29–31 In addition by changing the metal the CO2 pressure at which the transition takes place can be tuned.29–31 Hefti et al. (DOI: 10.1039/C6FD00040A) discuss the potential of these new materials for carbon capture in a temperature swing adsorption process. Hefti et al. showed that the specific energy requirement of this process operated with these materials is lower than for a commercial 13X zeolite. In particular, the fact that these isotherms show a step allows for a smaller temperature swing compared to conventional materials.
Membranes separations
An alternative for either solid adsorption or liquid absorption is to use membranes to separate CO2. For flue gas separation one of the key issues is the low CO2 partial pressure. As the difference in partial pressure of CO2 is the main driving force for diffusion across the membrane, there will only be a flux if (see Fig. 8):where p is the pressure, x the composition, and sub- and superscripts P and F refer to the permeate and feed, respectively. If we take a typical flue gas from a gas-fired power plant,
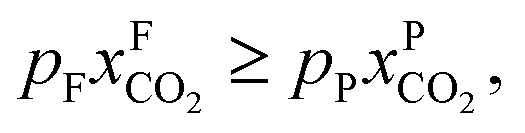
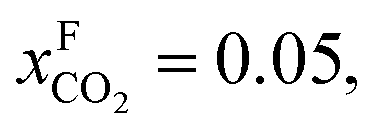 and assume 90% capture,
and assume 90% capture, 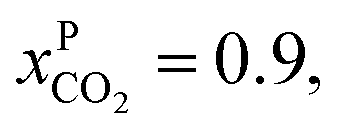 we see that the ratio between the pressure of the feed (flue gas) and the permeate on the other side of the membrane has to be:
we see that the ratio between the pressure of the feed (flue gas) and the permeate on the other side of the membrane has to be: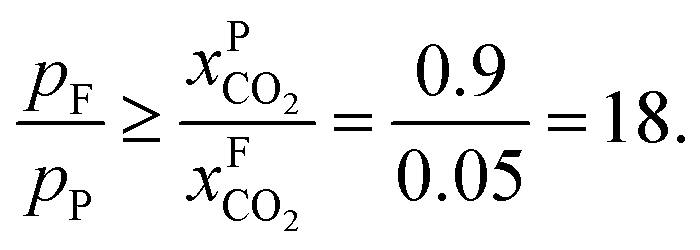
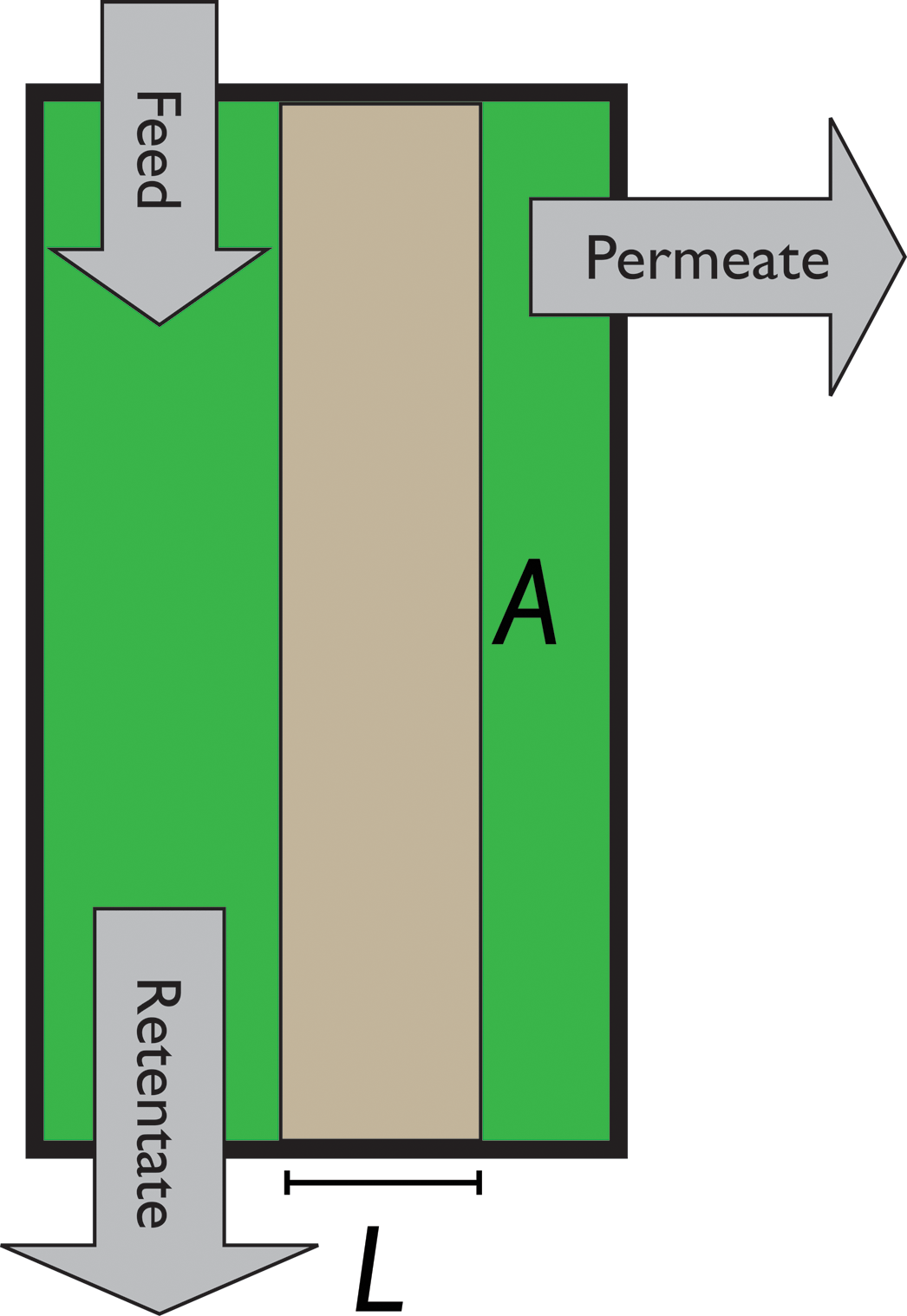 | ||
Fig. 8 Schematics of a membrane where feed is the flue gas that enters the membrane with a pressure pF and composition 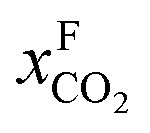 . Most membranes have a higher permeability for CO2 and therefore the permeate is rich in CO2 . Most membranes have a higher permeability for CO2 and therefore the permeate is rich in CO2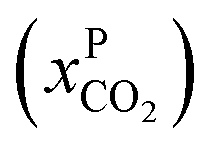 while the retentate is CO2 poor (figure adopted from Smit et al.3). while the retentate is CO2 poor (figure adopted from Smit et al.3). | ||
One can create this ratio in pressures by compressing flue gas or pulling vacuum on the permeate side. However, given the large volume of flue gas, such a large ratio would require more power than is produced by the power plant, and in practice the maximum ratio that can be achieved is 5. This simple argument shows that a single stage membrane separation cannot achieve the requirements for carbon capture. Of course, one can achieve the required purity with a two-stage process (i.e. two membranes) but at the expense of twice the compression costs, which makes such a two-stage membrane separation impractical. Merkel et al.32 developed an alternative design using two membranes where in the first membrane an air sweep is used to increase the concentration of CO2 in the air, which is subsequently used for the combustion. As the partial pressure of CO2 in the air is very small even at atmospheric pressure on the feed side, there is still a very large driving force. As we now burn fossil fuels with air, which has much higher CO2 concentrations, we are “artificially” increasing 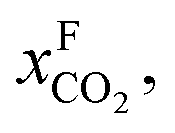 hence we now have a pressure ratio that is much smaller.
hence we now have a pressure ratio that is much smaller.
From a material design point of view the issue of designing a membrane is a balance between selectivity and permeability. For most polymer membranes one is confronted with the Robeson upper bound.33,34 Robeson observed that for most polymers if one would like to enhance the flux through the membrane, one needs to have a material with “bigger” cavities to increase the permeability, but creating bigger cavities reduces the selectivity. Hence, for a given permeability there is an upper boundary of the selectivity for a given permeability. Interestingly such an upper boundary does not exist for membranes composed of nanoporous materials.35
Membrane separations are based on differences in the permeability of the gasses when they pass through the material. The permeability is the product of the solubility and diffusion coefficient of these gasses. For most materials CO2 has the highest permeability. One can enhance the permeability if in the material CO2 gets converted to H2CO3. This so-called facilitated transport is used by systems in nature to selectively transport CO2.
In these Faraday Discussions we have contributions from Anantharaman et al. (DOI: 10.1039/C6FD00038J) and Zhang et al. (DOI: 10.1039/C6FD00030D).
Anantharaman et al. (DOI: 10.1039/C6FD00038J) discuss the potential of highly CO2-selective dual-phase membranes. These membranes consist of a porous solid phase matrix filled with a molten carbonate phase. Unlike conventional polymer membranes these materials operate at high temperatures (400 °C). At these high temperatures CO2 can be ionized to form CO32−, which is highly mobile in the molten carbonate phase. In such a system the driving force is the gradient of the electrochemical potential. Because this membrane operates at high temperatures, it is less useful for the conventional low-temperature post-combustion carbon capture. However, alternative processes such as the Natural Gas Combined Cycle (NGCC) or Integrated Gasification Combined Cycle (IGCC) operate at exactly these high temperatures.
The issue with NGCC is that, like in conventional post-combustion, carbon capture occurs at a low concentration of CO2. Anantharaman et al. (DOI: 10.1039/C6FD00038J) show that this low concentration makes a high temperature membrane less favorable compared to amine scrubbing. For IGCC, however, Anantharaman et al. show that this separation with these high temperatures is cheaper compared to MEA scrubbing. Zhang et al. (DOI: 10.1039/C6FD00030D) discuss addressing the low concentration of CO2 in the IGCC process using a membrane-amine hybrid solution. Zhang et al. propose to use a membrane with air sweep to obtain a CO2 enriched flue gas, in which the CO2 is subsequently captured using amine (piperazine) scrubbing.
Chemical looping
Whereas the main focus of chemical looping is to provide pure oxygen for combustion, it is also possible to use a solid to remove CO2 from flue gasses.36 In this application a metal oxide is used to capture CO2 from the flue gas:| Capture: MxO + CO2 ⇄ MxCO3 |
| Regeneration: MxCO3 ⇄ MxO + CO2 |
This Faraday Discussions contains contributions on chemical looping from Dunstan et al. (DOI: 10.1039/C6FD00047A), Li et al. (10.1039/C6FD00019C), Naeem et al. (DOI: 10.1039/C6FD00042H), and Erans et al. (DOI: 10.1039/C6FD00027D).
Dunstan et al. (DOI: 10.1039/C6FD00047A) discuss a range of experimental techniques to study the degeneration and the effect of steam on the CaO–CaCO3 system upon repeated cycles of CO2 adsorption and desorption. For CaO to react it is important that there are no mass transfer limitations and also one needs space for the CaCO3 to form.37 Therefore it is important to understand the pore-size distribution during the conversion of CaCO3 to CaO. Li et al. (DOI: 10.1039/C6FD00019C) discuss a model for describing the evolution of the pore-size distribution during the calcination of the CaCO3 particles.
Naeem et al. (DOI: 10.1039/C6FD00042H) discuss a synthesis strategy to tune the pore-size of CaO using a carbon-based sacrificial templating agent. These synthetic materials Naeem et al. developed showed an increased performance compared to limestone.
In practice these materials are not used as pure crystals but as pellets. This granulation process allows us to add binders (e.g. bentonites, kaolin, etc.) to improve the mechanical strength or add other components to improve the performance. Erans et al. (DOI: 10.1039/C6FD00027D) discuss the performance of the pure materials with those in biomass-templated calcium aluminate pellets.
Costs of CCS
Compared to emitting CO2 directly in the atmosphere, the additional costs for capturing CO2, transporting CO2, and geological sequestration are significant. For example, conventional technologies to capture CO2 from flue gasses of a coal-fired power plant would add 30–40% to the coal consumption of a plant.38 Therefore, for any new carbon-capture technology that will be implemented in a power plant, a key question the general public will ask is by how much will this increase the price of electricity. In this Faraday Discussions costs are addressed by Porrazzo et al. (DOI: 10.1039/C6FD00033A) and Adderley et al. (DOI: 10.1039/C6FD00046K).Porrazzo et al. (DOI: 10.1039/C6FD00033A) discuss the electricity costs of carbon capture using MEA and chemical looping, and show that chemical looping can result in lower electricity costs. Porrazzo et al. show that a key variable in these costs is the lifetime of the solid particles.
Adderley et al. (DOI: 10.1039/C6FD00046K) discuss the potential cost savings if carbon capture is implemented in future low carbon power generation from the new natural gas combined cycle (NGCC). In their study they focused on plants that are expected to be built in the UK in the 2020s and 2030s and that will continue to be operated into the 2050s.
Cement production
Most of the focus of CCS is on electricity production as this is the biggest stationary source of CO2 production. The production of cement is another important stationary source of CO2 emission (see Fig. 2). The total cement production is responsible for 7–13% of the global anthropogenic CO2 emissions.39,40 Cement production, actually, represents a double burden. It is not only an energy intensive process, but it also uses CaCO3 as an ingredient and during cement production limestone is converted into CaO and CO2. The CO2 is emitted with the flue gasses. CCS is therefore an important technology for the cement industry to reduce their CO2 emissions. The flue gas composition from cement production is slightly different from the flue gas from power plants; therefore one can use the post-combustion carbon capture technologies used in power plants.41 One can also opt for pre-combustion carbon capture and here we can also use CO2 diluted oxygen for the combustion.Even if one uses air or oxygen to burn fossil fuels this has little impact on electricity production as it does not influence the quality of the steam that is produced. Cement that is produced in an oxy-combustion environment has different properties from cement that is produced traditionally by combustion in air. In this Faraday Discussions Zheng et al. (DOI: 10.1039/C6FD00032K) report on the quality of cement that is produced by oxy-combustion and show that high-quality cement can be successfully produced in an oxy-fuel atmosphere.
Negative CO2 emissions
If one looks at the fate of CO2 emitted in the atmosphere, one can see that about 30% ends up as increased terrestrial biomass and 25% in the surface of the ocean either as biomass or dissolved in the water. The remainder stays in the atmosphere. If we were to stop emitting any CO2 tomorrow it would take over 200 years before these CO2 levels decreased to preindustrial levels.3 The reason for these long times is the slow mixing of the surface of the ocean with the deep ocean. For all practical purposes we can therefore assume that 45% of all the CO2 we emit now will stay “forever” in the atmosphere. This implies that if we overshoot our target atmospheric CO2 concentration we cannot wait until nature takes care of this overshoot; the only solution is to actively reduce the CO2 concentration in the atmosphere. Direct air capture is a technology that aims to achieve this.42–46 As air has a much lower concentration of CO2, the costs of capturing a CO2 molecule once it has been emitted in the atmosphere are significantly larger than capturing it from a more concentrated source.47 A cheaper alternative for direct air capture is to co-fire biomass in a coal-fired plant and subsequently sequester the CO2. Bioenergy with Carbon Capture and Storage (BECCS) is seen as a promising technology to achieve negative emissions using current infrastructures. A key question is the source of the biomass. Initially the focus will be on waste biomass, however, there is a limit to the total available waste biomass, and if one needs to grow biomass for BECCS there is the question of competition with food production and changes in land use.In this Faraday Discussions Mac Dowell and Fajardy (DOI: 10.1039/C6FD00051G) compare BECCS in combination with post-combustion and oxy-combustion carbon capture. They showed that going to negative CO2 emission does not come for free. The efficiency of power plants with BECCS will depend on the moisture content of the biomass and co-firing rate, but in general the efficiency will be lower than the efficiency of conventional power plants with CCS.
Geological storage
Given the extremely large volumes of CO2 we are producing, converting CO2 into useful products or fuels can only mitigate a small fraction of the CO2 we produce. Therefore, if we want to have an impact on total emissions, the bulk of the CO2 that is being produced will have to be permanently stored. Geological formations are an option for permanent CO2 storage. Geological analysis shows that we have a sufficient number of geological formations to store all the CO2 that we will be producing.An important factor in geological storage is the perception of the public. In the context of enhanced oil recovery we have quite some experience in transporting CO2 and injecting CO2 in geological formations. However, we do not have experience with injecting CO2 on such a large scales as is required to make a significant impact on emissions. Therefore research on geological storage is focused on increasing our knowledge of how to ensure the safe and permanent sequestration of CO2.
Of particular importance for geological storage are aquifers, which are sandstone formations in which the pores are filled with brine. If one injects CO2 in these formations one has to understand the chemical interactions between the CO2 and the reservoir fluids and rocks. For example, the dissolved CO2 makes the brine solution more acidic and this acidic solution may interact with the rocks and change the properties of the formation. In this Faraday Discussions Peng et al. (DOI: 10.1039/C6FD00048G) study the dissolution rate of several typical carbonate minerals, which one can find in these formations, in CO2-saturated water or brine at typical sequestration conditions. Quantifying these rates is important as these are the input for the computational models that are used to predict the fate of CO2 injected in these aquifers. These reservoir simulators are important as they are used to guide the CO2 injection process.
Outlook
The first question to ask is whether any of the technologies that are discussed in this article will be implemented. The most important factor in answering this question will be whether there will be a sufficiently large price on emitting CO2. None of the technologies that we have discussed will make the burning of fossil fuels any cheaper; hence with such a price on carbon it is essential to recover the costs. Whether we like it or not reducing CO2 emissions will cost money. In this context it is important to realize that, according to the IPCC report, not employing CCS will make the reduction of CO2 emissions significantly more expensive.1 Hence, if we are serious in reducing CO2 emissions, CCS will be part of the mix of technologies that will be employed.A second question is which of the carbon capture technologies that we have discussed will be employed. It is clear that the first generation of carbon-capture technologies will be solvent based; several processes have already been developed. Whether the second or third generation carbon-capture plants will still use this technology is an open question. From a research point of view it is important to generate as many options as possible. Novel solvents, solid adsorption, membranes, or hybrid-amine adsorptions are interesting alternatives. Cost calculations are normally used to select the most promising technologies for further development. It would be interesting to use these cost calculations at an early stage of research. This would give the researchers some feedback on which aspects of a process with a novel material could become a potential bottleneck for large-scale employment.
From a scientific point of view it is interesting to see research focused on tailor-made materials for a given separation. Ionic liquids can be tuned to have optimal properties for carbon capture, as is the case for metal–organic frameworks (MOFs). These studies will give us important insights in what would be the ideal molecular structure to achieve such a separation. As these studies involve screening of thousands of materials, many of these studies are integrated with computational approaches (i.e. materials genomics).15,23 As a first step these studies are very successful in identifying the best materials for highly idealized separations (e.g. flue gasses without water),16 with materials that may not be ideal for such large-scale separations. Therefore it will be important to see how this knowledge can be translated to materials that do meet all the requirements for practical applications.
New materials may also require different processes. For example, solid adsorption or membrane separations are, from a process-engineering point of view, more difficult than liquid adsorption. This raises the question as to whether, if the most ideal solid adsorbent or membrane is found, the process technology is sufficiently developed to fully take advantage of these novel materials. In these Discussions we see some nice examples related to chemical looping on the type of progress that can be made in process engineering.
There is an important difference between mitigating CO2 emissions and mitigating acid rains. SOx and NOx emissions cause acid rain, but as soon as we stop emitting SOx and NOx these gasses will be removed from the atmosphere with the acid rain, and the problem is solved. However, of all the emitted CO2, about 45% will stay for at least 200 years in the atmosphere. Even if we stop investing in fossil fuels tomorrow, it is unlikely that existing investments will be demolished. Hence, we are already committed to CO2 emissions by our past investments.48 This makes it likely that we will overshoot our targets and will therefore need a much more aggressive search for technologies that reduce CO2 concentration in the atmosphere.
Acknowledgements
I would like to thank Matthew Witman for providing Fig. 7 and a careful proofreading of the manuscript. The research leading to these results has received funding from the European Research Council under the European Union's Seventh Framework Programme (FP/2007-2013)/ERC Grant Agreement n.666983 – MaGic.Notes and references
- R. K. Pachauri and L. A. Meyer, Climate Change 2014: Synthesis Report. Contribution of Working Groups I, II and III to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change, ed. IPCC, IPCC, Geneva, Switzerland, 2014 Search PubMed.
- W. D. Nordhaus, The Climate Casino: Risk, Uncertainty, and Economics for a Warming World, Yale University Press, New Haven, 2013 Search PubMed.
- B. Smit, J. R. Reimer, C. M. Oldenburg and I. C. Bourg, Introduction to Carbon Capture and Sequestration, Imperial College Press, London, 2014 Search PubMed.
- J. Wilcox, Carbon Capture, Springer, New York, 2012 Search PubMed.
- M. E. Boot-Handford, J. C. Abanades, E. J. Anthony, M. J. Blunt, S. Brandani, N. Mac Dowell, J. R. Fernandez, M. C. Ferrari, R. Gross, J. P. Hallett, R. S. Haszeldine, P. Heptonstall, A. Lyngfelt, Z. Makuch, E. Mangano, R. T. J. Porter, M. Pourkashanian, G. T. Rochelle, N. Shah, J. G. Yao and P. S. Fennell, Energy Environ. Sci., 2014, 7, 130–189 CAS.
- N. MacDowell, N. Florin, A. Buchard, J. Hallett, A. Galindo, G. Jackson, C. S. Adjiman, C. K. Williams, N. Shah and P. Fennell, Energy Environ. Sci., 2010, 3, 1645–1669 CAS.
- R. Bottoms, Separating acid gases, US Pat., 1783901, 1930 Search PubMed.
- G. T. Rochelle, Science, 2009, 325, 1652–1654 CrossRef CAS PubMed.
- B. Smit, Greenhouse Gases: Sci. Technol., 2013, 3, 159–160 CrossRef.
- A. S. Bhown and B. C. Freeman, Environ. Sci. Technol., 2011, 45, 8624–8632 CrossRef CAS PubMed.
- B. Smit, A.-H. A. Park and G. Gadikota, Front. Energ., 2014, 2, 55 Search PubMed.
- J. D. Figueroa, T. Fout, S. Plasynski, H. McIlvried and R. D. Srivastava, Int. J. Greenhouse Gas Control, 2008, 2, 9–20 CrossRef CAS.
- J. P. Ciferno, J. J. Marano and R. K. Munson, Chem. Eng. Prog., 2011, 107, 34–44 CAS.
- L. S. Fan, L. Zeng and S. W. Luo, Chem. Eng. Prog., 2015, 111, 30–38 CAS.
- L.-C. Lin, A. H. Berger, R. L. Martin, J. Kim, J. A. Swisher, K. Jariwala, C. H. Rycroft, A. S. Bhown, M. W. Deem, M. Haranczyk and B. Smit, Nat. Mater., 2012, 11, 633–641 CrossRef CAS PubMed.
- J. M. Huck, L.-C. Lin, A. Berger, M. N. Shahrak, R. L. Martin, A. Bhown, M. Haranczyk, K. Reuter and B. Smit, Energy Environ. Sci., 2014, 7, 4136–4146 Search PubMed.
- M. Ramezan, T. J. Skone, N. ya Nsakala and G. N. Liljedahl, Carbon Dioxide Capture from Existing Coal-fired Power Plants, Report DOE/NETL-401/110907, National Energy Technology Laboratory, US Department of Energy, 2007 Search PubMed.
- R. Sathre and E. Masanet, RSC Adv., 2013, 3(15), 4964–4975 RSC.
- G. Rochelle, E. Chen, S. Freeman, D. Van Wagener, Q. Xu and A. Voice, Chem. Eng. J., 2011, 171, 725–733 CrossRef CAS.
- J. L. Anthony, S. N. V. K. Aki, E. J. Maginn and J. F. Brennecke, Int. J. Environ. Technol. Manage., 2004, 4, 105–115 CrossRef CAS.
- J. E. Brennecke and B. E. Gurkan, J. Phys. Chem. Lett., 2010, 1, 3459–3464 CrossRef CAS.
- B. Gurkan, B. F. Goodrich, E. M. Mindrup, L. E. Ficke, M. Massel, S. Seo, T. P. Senftle, H. Wu, M. F. Glaser, J. K. Shah, E. J. Maginn, J. F. Brennecke and W. F. Schneider, J. Phys. Chem. Lett., 2010, 1, 3494–3499 CrossRef CAS.
- F. Y. Yan, M. Lartey, K. Jariwala, S. Bowser, K. Damodaran, E. Albenze, D. R. Luebke, H. B. Nulwala, B. Smit and M. Haranczyk, J. Phys. Chem. B, 2014, 118, 13609–13620 CrossRef CAS PubMed.
- D. M. D'Alessandro, B. Smit and J. R. Long, Angew. Chem., Int. Ed., 2010, 49, 6058–6082 CrossRef PubMed.
- H. Liu, B. Liu, L. C. Lin, G. J. Chen, Y. Q. Wu, J. Wang, X. T. Gao, Y. N. Lv, Y. Pan, X. X. Zhang, X. R. Zhang, L. Y. Yang, C. Y. Sun, B. Smit and W. C. Wang, Nat. Commun., 2014, 5, 5147 CrossRef CAS PubMed.
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 974 CrossRef CAS PubMed.
- K. Sumida, D. L. Rogow, J. A. Mason, T. M. McDonald, E. D. Bloch, Z. R. Herm, T.-H. Bae and J. R. Long, Chem. Rev., 2012, 112, 724–781 CrossRef CAS PubMed.
- T. M. McDonald, W. R. Lee, J. A. Mason, B. M. Wiers, C. S. Hong and J. R. Long, J. Am. Chem. Soc., 2012, 134, 7056–7065 CrossRef CAS PubMed.
- T. M. McDonald, J. A. Mason, X. Q. Kong, E. D. Bloch, D. Gygi, A. Dani, V. Crocella, F. Giordanino, S. O. Odoh, W. S. Drisdell, B. Vlaisavljevich, A. L. Dzubak, R. Poloni, S. K. Schnell, N. Planas, K. Lee, T. Pascal, L. W. F. Wan, D. Prendergast, J. B. Neaton, B. Smit, J. B. Kortright, L. Gagliardi, S. Bordiga, J. A. Reimer and J. R. Long, Nature, 2015, 519, 303–308 CrossRef CAS PubMed.
- B. Vlaisavljevich, S. O. Odoh, S. K. Schnell, A. L. Dzubak, K. Lee, N. Planas, J. B. Neaton, L. Gagliardi and B. Smit, Chem. Sci., 2015, 6, 5177–5185 RSC.
- N. Planas, A. L. Dzubak, R. Poloni, L.-C. Lin, A. McManus, T. M. McDonald, J. B. Neaton, J. R. Long, B. Smit and L. Gagliardi, J. Am. Chem. Soc., 2013, 135, 7402 CrossRef CAS PubMed.
- T. C. Merkel, H. Q. Lin, X. T. Wei and R. Baker, J. Membr. Sci., 2010, 359, 126–139 CrossRef CAS.
- L. M. Robeson, J. Membr. Sci., 1991, 62, 165–185 CrossRef CAS.
- L. M. Robeson, J. Membr. Sci., 2008, 320, 390–400 CrossRef CAS.
- J. Kim, M. Abouelnasr, L.-C. Lin and B. Smit, J. Am. Chem. Soc., 2013, 135, 7545–7552 CrossRef CAS PubMed.
- E. J. Anthony, Ind. Eng. Chem. Res., 2008, 47, 1747–1754 CrossRef CAS.
- P. S. Fennell, R. Pacciani, J. S. Dennis, J. F. Davidson and A. N. Hayhurst, Energy Fuels, 2007, 21, 2072–2081 CrossRef CAS.
- R. S. Haszeldine, Science, 2009, 325, 1647–1652 CrossRef CAS PubMed.
- Climate Change 2014: Mitigation of Climate Change, Contribution of Working Group III to the Fifth Assessment, ed. O. Edenhofer, R. Pichs-Madruga, Y. Sokona, E. Farahani, S. Kadner, K. A. A. Seyboth, I. Baum, S. Brunner, P. Eickemeier, B. Kriemann, J. Savolainen, S. Schlömer, C. von Stechow, T. Zwickel and J. C. Minx, IPCC, 2014 Search PubMed.
- Report of the Intergovernmental Panel on Climate Change, Cambridge University Press, Cambridge, United Kingdom and New York, NY, USA, 2014 Search PubMed.
- T. Hills, D. Leeson, N. Florin and P. Fennell, Environ. Sci. Technol., 2016, 50, 368–377 CrossRef CAS PubMed.
- K. S. Lackner, Eur. Phys. J.: Spec. Top., 2009, 176, 93–106 CrossRef.
- K. S. Lackner, S. Brennan, J. M. Matter, A. H. A. Park, A. Wright and B. van der Zwaan, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 13156–13162 CrossRef CAS PubMed.
- D. W. Keith, Science, 2009, 325, 1654–1655 CrossRef CAS PubMed.
- D. W. Keith, M. Ha-Duong and J. K. Stolaroff, Clim. Change, 2006, 74, 17–45 CrossRef CAS.
- D. W. Keith, K. Heidel and R. Cherry, in Geo-engineering Climate Change : Environmental Necessity or Pandora's Box?, ed. B. E. Launder and M. T. Thompson, Cambridge University Press, Cambridge, UK; New York, 2010, pp. 107–126 Search PubMed.
- R. Socolow, M. Desmond, R. Aines, J. Blackstock, O. Bolland, T. Kaarsberg, N. Lewis, M. Mazzotti, A. Pfeffer, K. Sawyer, J. Siirola, B. Smit and J. Wilcox, Direct Air Capture of CO2 with Chemicals: A Technology Assessment for the APS Panel on Public Affairs, American Physical Society, 2011 Search PubMed.
- S. J. Davis, K. Caldeira and H. D. Matthews, Science, 2010, 329, 1330–1333 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |