
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHydrogenation of CO2 to formic acid with iridiumIII(bisMETAMORPhos)(hydride): the role of a dormant fac-IrIII(trihydride) and an active trans-IrIII(dihydride) species†
S.
Oldenhof
,
J. I.
van der Vlugt
* and
J. N. H.
Reek
*
Homogeneous, Bioinspired & Supramolecular Catalysis, van't Hoff Institute for Molecular Sciences, University of Amsterdam, Science Park 904, 1098 XH Amsterdam, The Netherlands. E-mail: j.i.vandervlugt@uva.nl; j.n.h.reek@uva.nl
First published on 8th October 2015
Abstract
An IrIII-monohydride species bearing a chemoresponsive ligand is active in catalytic CO2 hydrogenation to formic acid with DBU as the exogenous base. Spectroscopic and computational data reveal a trans-IrIII-dihydride as the essential catalytic intermediate and an IrIII(H)3 species as the dormant off-cycle product. This insight will aid future design of improved CO2 reduction catalysts.
Carbon dioxide utilization has attracted much interest in academia and industry. This relates to renewable energy applications and as an alternative C1 carbon building block in synthesis.1 In particular, its reduction to formic acid (HCOOH) has been investigated intensively, given its potential as a reversible hydrogen storage system, alongside other commercial applications in e.g. the rubber, agricultural and textile industries.2 The hydrogenation of CO2 to HCOOH is endergonic by 33 kJ mol−1 mainly because of a large loss in entropy (eqn (1)). Temperature, pressure, solvent and additives can be used to influence the equilibrium of this reaction. CO2 hydrogenation is often performed with addition of an external base such as ammonia or NEt3, as this results in a thermodynamically more stable formate–base ion pair, which drives the equilibrium toward HCOOH formation (eqn (2)).
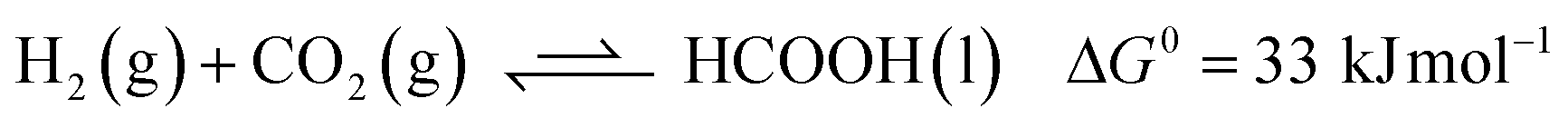 | (1) |
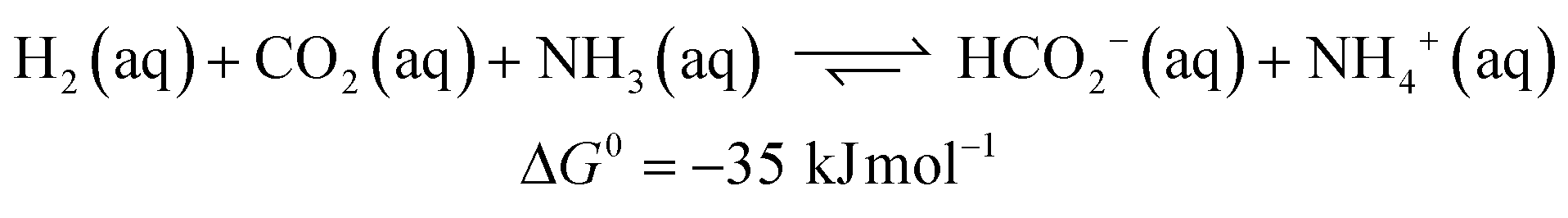 | (2) |
The most active homogeneous catalysts to date for CO2 hydrogenation to HCOOH under basic conditions are based on either Ir or Ru (Fig. 1; A–C).3–5 Outer-sphere interactions such as hydrogen bonding and chemoresponsive ligand reactivity were found to play an essential role in these catalysts to ensure efficient turnover.5–8 The importance of outer-sphere interactions has also been established for various systems specifically reported to catalyze the microscopic reverse process, i.e. formic acid dehydrogenation.9,10 Similar outer-sphere interactions were reported for an iridium-trihydride complex D-CO2 bearing a chemoresponsive PNP ligand that engages in a stabilizing hydrogen bond interaction with CO2.11 DFT calculations have been used to postulate a correlation between the Ir–Haxial bond length and the relative free energy ΔG0 of CO2 insertion: a longer Ir–Haxial bond length (i.e. weaker bond) enhances Ir formate formation (i.e. facilitates CO2 insertion). A related correlation between the hydricity of an Ir–H fragment and the rate of CO2 insertion has recently been formulated, again based on a computational study.12
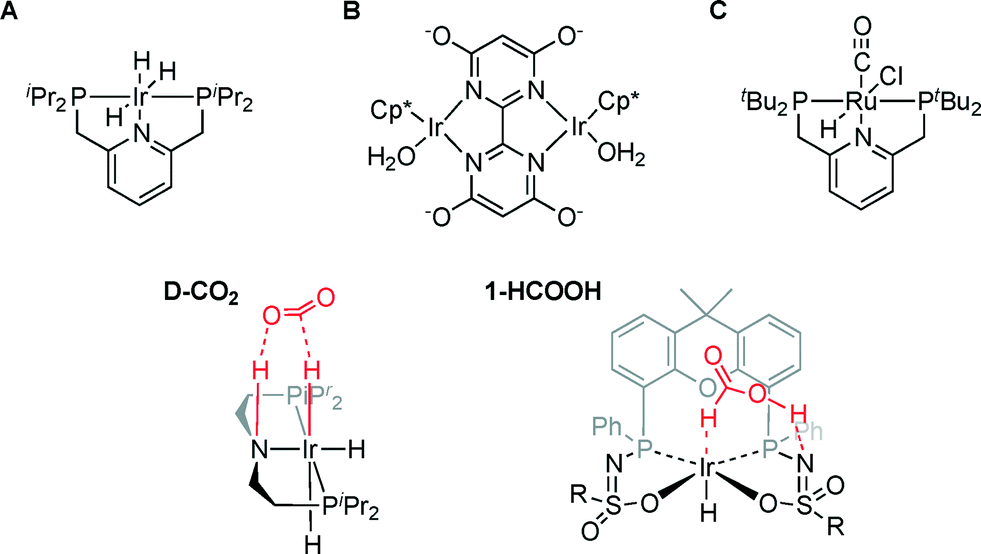 | ||
| Fig. 1 Catalysts A–C and D-CO2 for CO2 hydrogenation to HCOOH and the formic acid adduct of IrIII(H)(bisMETAMORPhos) complex 1 (1-HCOOH; R = 4-butylbenzene). | ||
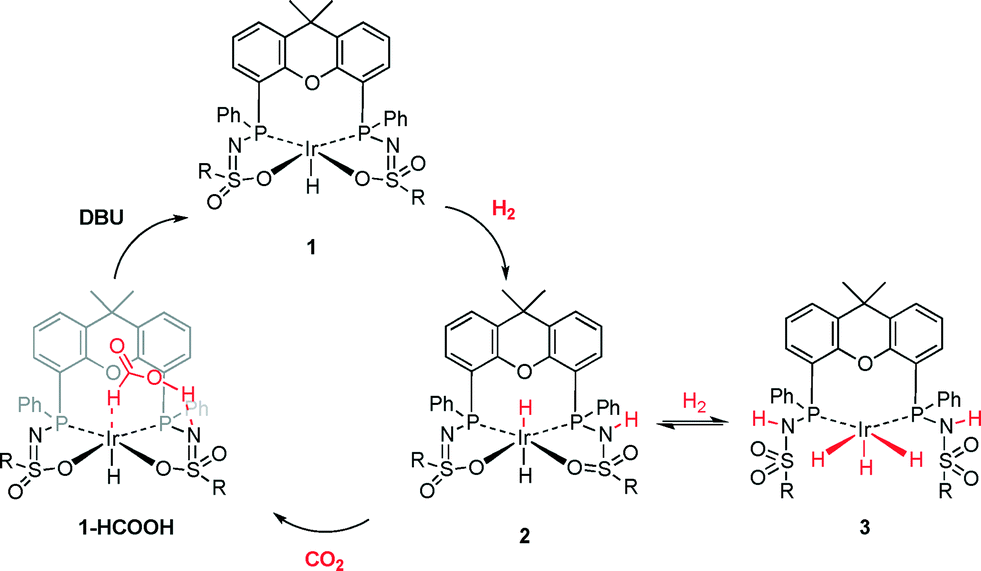
We previously reported the secondary interactions between formic acid and IrIII(H)(bisMETAMORPhos) complex 1 to form 1-HCOOH (Fig. 1) as being relevant for the dehydrogenation of HCOOH.13 The reactive bis(sulfonamidophosphine) ligand in complex 1-HCOOH functions both as an internal base to deprotonate HCOOH and as a hydrogen bond donor/acceptor to pre-assemble HCOOH and stabilize catalytically relevant transition states. Herein, we report initial data for catalytic CO2 hydrogenation with IrIII(H)(bisMETAMORPhos) complex 1 and discuss the role of a relatively unreactive fac-IrIII(H)3 species, which is formed under the applied reaction conditions, based on in situ NMR experiments and DFT calculations. This insight may aid future catalyst design for metal–ligand bifunctional CO2 hydrogenation.
To monitor the catalytic activity of complex 1 in CO2 hydrogenation, high-pressure NMR experiments were performed at 373 K and 50 bar of CO2 and H2 (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio) in DMSO-d6, using DMF (0.5 M) as the internal standard and in the absence of an external base.14 Moderate catalytic activity for CO2 hydrogenation was observed, with a turnover frequency (TOF) of 18 h−1 in the first 30 minutes of the reaction and a turnover number (TON) of 30 after 90 minutes (Fig. 2, green curve). The conversion did not increase significantly between 90 and 180 minutes and a final concentration of 0.015 M HCOOH was obtained.
:1 ratio) in DMSO-d6, using DMF (0.5 M) as the internal standard and in the absence of an external base.14 Moderate catalytic activity for CO2 hydrogenation was observed, with a turnover frequency (TOF) of 18 h−1 in the first 30 minutes of the reaction and a turnover number (TON) of 30 after 90 minutes (Fig. 2, green curve). The conversion did not increase significantly between 90 and 180 minutes and a final concentration of 0.015 M HCOOH was obtained.
 | ||
| Fig. 2 Catalytic CO2 hydrogenation with 1 (0.5 mM) under base-free conditions (green) and with the addition of 1000 equiv. (0.5 M) of NEt3 (red) or DBU (blue). Solvent: DMSO-d6, T = 373 K, total reaction volume = 2 mL. The absolute amount of HCOOH produced in mmol is plotted vs. time in minutes. | ||
When catalysis was performed under the same catalytic conditions but in the presence of 1.0 mmol (0.5 M) of NEt3, only a slight increase in activity was observed (Fig. 2, red curve). In contrast to this negligible effect of NEt3 on the catalytic performance, the addition of 1.0 mmol of DBU (1,8-diazabicyclo[5.4.0]undec-7-ene) led to a significant improvement in the catalytic activity, with a TOF of 636 h−1 between 0–30 minutes and a TON of 685 after 180 minutes (Fig. 2, blue curve), corresponding to a base conversion of 0.685.‡ The remarkable effect of the base on the catalytic activity can be explained by the difference in basicity in DMSO (DBU: pKa 12.0; NEt3: pKa 9.0). Similar differences in the catalytic performance of NEt3 and DBU were observed in system C.5 The formation of HDBU+·HCOO− was monitored over time by the appearance of the HCOO− formate signal at 8.60 ppm in consecutive 1H NMR spectra (see the ESI†). The concentration of H2 increases over time, but is barely detectable in the first 30 minutes of reaction. The determined initial rates are therefore likely limited by mass transfer. Various solvents were used as reaction media but this did not lead to enhanced catalytic activities. In dioxane, a slight decrease in TOF was observed (588 h−1), while in ethylene glycol, the catalytic activity decreased significantly (TOF: 38 h−1). To obtain more insight into the mechanism of CO2 hydrogenation, complex 1 was studied by 1H NMR spectroscopy under combined H2 and CO2 pressure in the absence of a base. When 1 was dissolved in CD2Cl2, a well-defined triplet was observed in the 1H NMR spectrum at δ −28.7 ppm (Fig. 3A) as previously reported.13 However, when 1 was dissolved in DMSO-d6, six different hydride signals were detected in the region from δ −24.0 to −29.0 ppm (Fig. 3B).
 | ||
| Fig. 3 1H NMR spectra of (A) 1 dissolved in CD2Cl2, (B) 1 dissolved in DMSO-d6, and (C) formation of 3 from 1 with H2/CO2 (25/25 bar) at 373 K in DMSO-d6, R = 4-butylbenzene. * indicates a minor impurity.§ | ||
The generation of these species may result from: (1) the coordination of either DMSO, H2O or the oxygen of the xanthene backbone to the vacant axial site of complex 1,¶ (2) the dimer formation to give {(1)2} as previously observed in the solid state13 or (3) the formation of different diastereomers by rotation of the sulfone group. Molecular structures of both a dimer and an axial H2O adduct of complex 1 have been reported.13 Upon pressurizing a DMSO-d6 solution of 1 in a high-pressure sapphire NMR tube with 50 bar CO2/H2 (1:1) at room temperature, no changes were observed in the 1H NMR spectrum after one hour. Heating the sample to 373 K led to the formation of a new species that displayed two broad hydride signals: a doublet-of-doublets at δ −11.9 ppm (2JP–H of 154.3 and 14.9 Hz) and a triplet at δ −15.7 ppm (2JP–H of 17.7 Hz) in a 2:1 ratio (Fig. 3C). The coupling constants observed for the doublet-of-doublets are indicative of trans (154.3 Hz) and cis31P–1H coupling (14.9 Hz), while the triplet originates from coupling of a hydride to two cis-positioned phosphorus nuclei. In the corresponding phosphorus-decoupled 1H NMR spectrum, two singlets were observed. The ratio of the two hydride signals proved to be independent of temperature, suggesting that they belong to a single species. Together, this suggests the formation of five-coordinate trihydride complex 3, fac-IrIII(H)3(bisMETAMORPhos) (see Scheme 1). Related fac-IrIII(H)3 complexes with Xantphos show similar spin systems.15 The 2JH–H couplings, which are typically in the range of 2.6–7.4 Hz, could not be resolved due to broadening of the spectrum at 373 K. The N–H resonances of the protonated ligand arms could not be identified by 1H NMR spectroscopy, as they tend to overlap with aromatic signals.13,16 After releasing the CO2/H2 pressure, 3 remained stable for at least one hour at room temperature. Upon re-heating the depressurized solution to 373 K, the hydride signals corresponding to 3 disappeared and complex 1 was regenerated, concomitant with the formation of H2, showing that the formation of 3 from 1 is reversible (Scheme 1).
 | ||
| Scheme 1 Conversion to 3 from 1 upon addition of two equivalents of H2. | ||
Species 1 is stable under pure CO2, but NMR signals that indicate the slow formation of 3 appear under pure H2 atmosphere. The formation of 3 is suggested to proceed via the formation of intermediate 2 through heterolytic splitting of H2 by 1, as previously described.13,16 Subsequently, another equivalent of H2 is activated, presumably also in a heterolytic fashion, by decoordination of the neutral ligand arm to generate a vacant site and with the anionic ligand arm acting as an internal base, resulting in the square pyramidal fac-IrIII(H)3(bisMETAMORPhos) species 3.
Interestingly, prior to the formation of 3, the generation of 14 equivalents of HCOOH was evidenced by 1H NMR spectroscopy. Upon complete conversion to 3, no further HCOOH generation was observed. This suggests that 3 may be a catalytically dormant species and that 2 is the active species. This hypothesis was further investigated by studying the energetics of the hydride transfer to CO2 for complexes 2 and 3 by DFT calculations (BP86, def2-TZVP), using R = phenyl on the sulfone group for computational simplicity (Fig. 4). Complex 3 is lower in energy than 2 (ΔΔG0298K = −4 kcal mol−1), which is in agreement with the observation of 3 by 1H NMR spectroscopy. For species 2, hydride transfer to CO2via transition state 2-TS has a reasonable activation barrier of 20.1 kcal mol−1, given the applied catalytic conditions. In complex 3, hydride transfer to CO2 could theoretically also occur. However, the transfer of either the axial hydride (3TS-ax: ΔG0298K = 65.6 kcal mol−1) or one of the equatorial hydrides (3TS-eq: ΔG0298K = 44.2 kcal mol−1) is considered too endergonic to be catalytically relevant (see the ESI† for details).
 | ||
| Fig. 4 DFT-calculated potential energy diagram of hydride transfer to CO2 from complexes 2 and 3. ΔG0298K in kcal mol−1, R = phenyl (Turbomole,17 BP86, def2-TZVP). | ||
This observation is in line with the hypothesis that complex 3 is an off-cycle dormant species that is not directly involved in catalytic CO2 hydrogenation (Scheme 2). Upon inspection of the computed structures of 2 and 3, a correlation between the Ir–H bond length and the energy required for CO2 insertion could be deduced (Fig. 5). The Ir–H bonds in species 2 (1.674 and 1.692 Å) are longer than those in 3 (Ir–Heq, 1.631 and 1.632 Å; Ir–Hax, 1.557 Å). The elongation in 2, which results in weaker Ir–H bonds, likely originates from a mutual trans effect of the two hydride ligands. These bond length differences correlate nicely with the lower activation energy found for CO2 insertion in 2 (20.1 kcal mol−1) relative to 3 (44.2 and 65.6 kcal mol−1 for Heq and Hax, respectively). Our results are thus in agreement with the computational findings related to system D, demonstrating that trans-dihydride configurations allow for catalytically accessible energy barriers for CO2 insertion.11,12 Also, all transition states (2-TS, 3TS-ax and 3TS-eq) involve a stabilizing hydrogen bond interaction between the ligand backbone and CO2. Improved catalyst design should focus on favoring the formation of 2 or analogues thereof. Research in this direction is currently ongoing in our laboratories.
 | ||
| Scheme 2 Potential catalytic cycle of CO2 hydrogenation from 1 with the active dihydride intermediate 2 and the dormant species 3 as the proposed off-cycle species. | ||
 | ||
| Fig. 5 Comparison of Ir–H bond lengths in the DFT-calculated optimized structures of complexes 2 and 3 (Turbomole,18 BP86, def2-TZVP). The values are in Å, R = phenyl. | ||
Conclusions
IrIII(H)(METAMORPhos) species 1 is able to catalytically hydrogenate CO2 with a TOF of 18 h−1 in DMSO-d6 at 373 K under 50 bar of CO2/H2 (1:1). A strong effect of the added base on the catalyst activity was observed: triethylamine led to a minor improvement, but DBU gave a significant enhancement of the reaction rate (TOF of 636 h−1). The formation of a tight ion pair between formic acid and DBU (HDBU+·HCOO−) is suggested to provide the thermodynamic driving force. In situ NMR studies reveal that complex 1 is converted to a fac-trihydride complex (3) under CO2/H2 atmosphere (50 bar, 1:1) upon heating to 373 K. DFT calculations suggest that complex 3 is a dormant species in the catalytic cycle and trans-dihydride 2, which is an intermediate in the conversion of 1 to 3, is catalytically relevant. The formation of 3 is reversible, as complex 1 was regenerated upon release of pressure and heating to 373 K. Further studies to tune the reaction conditions for optimal catalytic activity and to design an optimized system should focus on the integration of a trans-dihydride arrangement.
Acknowledgements
This research was funded by a TOP grant from NWO-CW to J.N.H.R. We thank Prof. Dr. Bas de Bruin for helpful suggestions regarding the DFT calculations.Notes and references
- M. Aresta, A. Dibenedetto and A. Angelini, Chem. Rev., 2014, 114, 1709–1742 CrossRef CAS PubMed; C. Maeda, Y. Miyazaki and T. Ema, Catal. Sci. Technol., 2014, 4, 1482–1497 Search PubMed; A. M. Appel, J. E. Bercaw, A. B. Bocarsly, H. Dobbek, D. L. DuBois, M. Dupuis, J. G. Ferry, E. Fujita, R. Hille, P. J. A. Kenis, C. A. Kerfeld, R. H. Morris, C. H. F. Peden, A. R. Portis, S. W. Ragsdale, T. B. Rauchfuss, J. N. H. Reek, L. C. Seefeldt, R. K. Thauer and G. L. Waldrop, Chem. Rev., 2013, 113, 6621–6658 CrossRef PubMed; G. Centi, E. A. Quadrelli and S. Perathoner, Energy Environ. Sci., 2013, 6, 1711–1731 Search PubMed; N. Kielland, C. J. Whiteoak and A. W. Kleij, Adv. Synth. Catal., 2013, 355, 2115–2138 CrossRef; C. Ziebart, C. Federsel, P. Anbarasan, R. Jackstell, W. Baumann, A. Spannenberg and M. Beller, J. Am. Chem. Soc., 2012, 134, 20701–20704 CrossRef PubMed; A. Boddien, D. Mellmann, F. Gärtner, R. Jackstell, H. Junge, P. J. Dyson, G. Laurenczy, R. Ludwig and M. Beller, Science, 2011, 333, 1733–1736 CrossRef PubMed; M. Cokoja, C. Bruckmeier, B. Rieger, W. A. Herrmann and F. E. Kühn, Angew. Chem., Int. Ed., 2011, 50, 8510–8537 CrossRef PubMed; C. Federsel, A. Boddien, R. Jackstell, R. Jennerjahn, P. J. Dyson, R. Scopelliti, G. Laurenczy and M. Beller, Angew. Chem., Int. Ed., 2010, 49, 9777–9780 CrossRef PubMed; C. Federsel, R. Jackstell and M. Beller, Angew. Chem., Int. Ed., 2010, 49, 6254–6257 CrossRef PubMed.
- For a selection of reviews, see: A. K. Singh, S. Singh and A. Kumar, Catal. Sci. Technol., 2015 10.1039/c5cy01276g; W.-H. Wang, Y. Himeda, J. T. Muckerman and E. Fujita, Adv. Inorg. Chem., 2014, 66, 189–222 CrossRef CAS; M. Grasemann and G. Laurenczy, Energy Environ. Sci., 2012, 5, 8171–8181 Search PubMed; T. Schaub and R. A. Paciello, Angew. Chem., Int. Ed., 2011, 50, 7278–7282 CrossRef PubMed; W. Reutemann and H. Kieczka, Formic Acid, Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, 6th edn, 2011 Search PubMed.
- R. Tanaka, M. Yamashita and K. Nozaki, J. Am. Chem. Soc., 2009, 131, 14168–14169 CrossRef CAS PubMed See also: I. Osadchuk, T. Tamm and M. S. G. Ahlquist, Organometallics, 2015 DOI:10.1021/acs.organomet.5b00448.
- J. F. Hull, Y. Himeda, W.-H. Wang, B. Hashiguchi, R. Periana, D. J. Szalda, J. T. Muckerman and E. Fujita, Nat. Chem., 2012, 4, 383–388 CrossRef CAS PubMed.
- G. A. Filonenko, M. P. Conley, C. Copéret, M. Lutz, E. J. M. Hensen and E. A. Pidko, ACS Catal., 2013, 3, 2522–2526 CrossRef CAS; G. A. Filonenko, R. van Putten, E. N. Schulpen, E. J. M. Hensen and E. A. Pidko, ChemCatChem, 2014, 6, 1526–1530 CrossRef See also: G. A. Filonenko, D. Smykowski, B. M. Szyja, G. Li, J. Szczygie, E. J. M. Hensen and E. A. Pidko, ACS Catal., 2015, 5, 1145–1154 CrossRef.
- X. Yang, ACS Catal., 2011, 1, 849–854 CrossRef CAS; M. S. G. Ahlquist, J. Mol. Catal. A: Chem., 2010, 324, 3–8 CrossRef; R. Tanaka, M. Yamashita, L. W. Chung, K. Morokuma and K. Nozaki, Organometallics, 2011, 30, 6742–6750 CrossRef.
- W. Wang, J. T. Muckerman, E. Fujita and Y. Himeda, ACS Catal., 2013, 3, 856–860 CrossRef CAS; W. Wang, J. F. Hull, J. T. Muckerman, E. Fujita and Y. Himeda, Energy Environ. Sci., 2012, 5, 7923–7926 Search PubMed.
- G. A. Filonenko, E. J. M. Hensen and E. A. Pidko, ACS Catal., 2014, 4, 2667–2671 CrossRef CAS; G. A. Filonenko, E. J. M. Hensen and E. A. Pidko, Catal. Sci. Technol., 2014, 4, 3474–3485 Search PubMed.
- E. A. Bielinski, P. O. Lagaditis, Y. Zhang, B. Q. Mercado, C. Würtele, W. H. Bernskoetter, N. Hazari and S. Schneider, J. Am. Chem. Soc., 2014, 136, 10234–10237 CrossRef CAS PubMed.
- C. Yin, Z. Xu, S.-Y. Yang, S. M. Ng, K. Y. Wong, Z. Lin and C. P. Lau, Organometallics, 2001, 20, 1216–1222 CrossRef CAS; C. A. Huff, J. W. Kampf and M. S. Sanford, Organometallics, 2012, 31, 4643–4645 CrossRef; P. Kang, C. Cheng, Z. Chen, C. K. Schauer, T. J. Meyer and M. Brookhart, J. Am. Chem. Soc., 2012, 134, 5500–5503 CrossRef PubMed; C. A. Huff and M. S. Sanford, ACS Catal., 2013, 3, 2412–2416 CrossRef; P. Kang, T. J. Meyer and M. Brookhart, Chem. Sci., 2013, 4, 3497–3502 RSC; L. Cao, C. Sun, N. Sun, L. Meng and D. Chen, Dalton Trans., 2013, 42, 5755–5763 RSC; T. W. Myers and L. A. Berben, Chem. Sci., 2014, 5, 2771–2777 RSC; L. S. Jongbloed, B. de Bruin, J. N. H. Reek, M. Lutz and J. I. van der Vlugt, Chem. – Eur. J., 2015, 21, 7297–7305 CrossRef PubMed; L. S. Jongbloed, B. de Bruin, J. N. H. Reek, M. Lutz and J. I. van der Vlugt, Catal. Sci. Technol. Search PubMed under revision.
- T. J. Schmeier, G. E. Dobereiner, R. H. Crabtree and N. Hazari, J. Am. Chem. Soc., 2011, 133, 9274–9277 CrossRef CAS PubMed.
- B. Mondal, F. Neese and S. Ye, Inorg. Chem., 2015, 54, 7192–7198 CrossRef CAS PubMed; J. T. Muckerman, P. Achord, C. Creutz, D. E. Polyansky and E. Fujita, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15657–15662 CrossRef PubMed.
- S. Oldenhof, B. de Bruin, M. Lutz, M. A. Siegler, F. W. Patureau, J. I. van der Vlugt and J. N. H. Reek, Chem. – Eur. J., 2013, 19, 11507–11511 CrossRef CAS PubMed; S. Oldenhof, M. Lutz, B. de Bruin, J. I. van der Vlugt and J. N. H. Reek, Chem. Sci., 2015, 6, 1027–1034 RSC.
- Related CO2 hydrogenation under acidic conditions: H. Hayashi, S. Ogo and S. Fukuzumi, Chem. Commun., 2004, 2714–2715 RSC; S. Ogo, R. Kabe, H. Hayashi, R. Harada and S. Fukuzumi, Dalton Trans., 2006, 4657–4663 RSC; S. Moret, P. J. Dyson and G. Laurenczy, Nat. Commun., 2014, 5, 4017 Search PubMed.
- D. J. Fox, S. B. Duckett, C. Flaschenriem, W. W. Brennessel, J. Schneider, A. Gunay and R. Eisenberg, Inorg. Chem., 2006, 45, 7197–7209 CrossRef CAS PubMed; B. A. J. Pontiggia, A. B. Chaplin and A. S. Weller, J. Organomet. Chem., 2011, 696, 2870–2876 CrossRef; M. A. Esteruelas, M. Oliván and A. Vélez, Inorg. Chem., 2013, 52, 5339–5349 CrossRef PubMed.
- S. Oldenhof, B. de Bruin, M. Lutz, M. A. Siegler, F. W. Patureau, J. I. van der Vlugt and J. N. H. Reek, Organometallics, 2014, 33, 7293–7298 CrossRef CAS; S. Oldenhof, F. G. Terrade, M. Lutz, J. I. van der Vlugt and J. N. H. Reek, Organometallics, 2015, 34, 3209–3215 CrossRef; S. Oldenhof, J. I. van der Vlugt and J. N. H. Reek, Chem. Commun., 2015, 51, 15200–15203 RSC ; See also: F. F. W. Patureau, S. de Boer, M. Kuil, J. Meeuwissen, P.-A. R. Breuil, M. A. Siegler, A. L. Spek, A. J. Sandee, B. de Bruin and J. N. H. Reek, J. Am. Chem. Soc., 2009, 131, 6683–6685 CrossRef PubMed; F. G. Terrade, M. Lutz, J. I. van der Vlugt and J. N. H. Reek, Eur. J. Inorg. Chem., 2014, 1826–1835 CrossRef.
- R. Ahlrichs, Turbomole Version 5, University of Karlsruhe, Germany, 2002 CrossRef CAS; PQS version 2.4, Parallel Quantum Solutions, Fayettevile, AR (USA), 2001 CrossRef CASThe baker optimizer is available separately from PQS upon request: I. Baker, J. Comput. Chem., 1986, 7, 385–395 CrossRef CAS; P. H. M. Budzelaar, J. Comput. Chem., 2007, 28, 2226–2236 CrossRef PubMed; A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098–3100 CrossRef; J. P. Perdew, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 8822–8824 CrossRef.
- A. Bartoszewicz, N. Ahlsten and B. Martín-Matute, Chem. – Eur. J., 2013, 19, 7274–7302 CrossRef CAS PubMed; R. Crabtree, Acc. Chem. Res., 1979, 12, 331–337 CrossRef; S. P. Smidt, A. Pfaltz, E. Martínez-Viviente, P. S. Pregosin and A. Albinati, Organometallics, 2003, 22, 1000–1009 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental and computational details. See DOI: 10.1039/c5cy01476j |
| ‡ Significant loss of catalytic activity is observed over time, likely due to a pressure drop in the NMR tube during turnover; see the ESI.† |
| § The formation of 3 is accompanied by a species ‘A’ displaying a sharp singlet at −15.0 ppm (*). The ratio of 3 to ‘A’ remains unchanged over time. This complex is thus likely not a derivative of 1, nor does it match previously described deactivation products.18 Stirring Ir(acac)(cod) in DMSO-d6 under 50 bar CO2/H2 (1:1) at 373 K resulted in identical spectral features (Ir(acac)(cod) is added in slight excess (5%) during the synthesis of 1). This unidentified complex is a poor CO2 hydrogenation catalyst (TON of 1.9 after 90 minutes at 373 K). |
| ¶ DMSO is known to have several coordination modes: κ1-O, κ1-S, and κ2-S,O. Species with the xanthene oxygen coordinated to Ir were all found to be close in energy based on DFT calculations [BP86, SV(P)]. |
| This journal is © The Royal Society of Chemistry 2016 |