DOI:
10.1039/C6CS00163G
(Review Article)
Chem. Soc. Rev., 2016,
45, 6327-6344
Applications of N-heterocyclic imines in main group chemistry
Received
29th February 2016
First published on 8th August 2016
Abstract
The imidazolin-2-imino group is an N-heterocyclic imino functionality that derives from the class of compounds known as guanidines. The exocyclic nitrogen atom preferably bonds to electrophiles and its electron-donating character is markedly enhanced by efficient delocalization of cationic charge density into the five-membered imidazoline ring. Thus, this imino group is an excellent choice for thermodynamic stabilization of electron-deficient species. Due to the variety of available imidazoline-based precursors to this ligand, its steric demand can be tailored to meet the requirements for kinetic stabilization of otherwise highly reactive species. Consequently, it does not come as a surprise that the imidazolin-2-iminato ligand has found widespread applications in transition-metal chemistry to furnish pincer complexes or “pogo stick” type compounds. In comparison, the field of main-group metal compounds of this ligand is still in its infancy; however, it has received growing attention in recent years. A considerable number of electron-poor main-group element species have been described today which are stabilized by N-heterocyclic iminato ligands. These include low-valent metal cations and species that are marked by formerly unknown bonding modes. In this article we provide an overview on the present chemistry of main-group element compounds of the imidazolin-2-iminato ligand, as well as selected examples for the related imidazolidin- and benzimidazolin-2-imino system.

Tatsumi Ochiai
| Tatsumi Ochiai was born in Shizuoka, Japan, in 1987. In 2012 he received his MSc degree from the University of Tsukuba under the supervision of Prof. Akira Sekiguchi, for which he received the Dean's Prize. He worked on heteroatom-substituted tetrahedranes. He then moved to Berlin in 2012 starting doctoral work in the research group of Prof. Shigeyoshi Inoue. His research interest mainly focuses on the synthesis of low-coordinate heavier group 14 elements, metallylenes and metallyliumylidene ions. |

Daniel Franz
| Daniel Franz studied chemistry at the Goethe University Frankfurt am Main, Germany, where he received his PhD degree under the supervision of Prof. Matthias Wagner in 2012. Following this he moved to Berlin to carry out postdoctoral studies in the group of Prof. Shigeyoshi Inoue. Today he is pursuing his research interests in coordination chemistry of group 13 and 14 elements at the Technische Universität München under the supervision of Prof. Inoue. |

Shigeyoshi Inoue
| Shigeyoshi Inoue studied chemistry at the University of Tsukuba where he completed his PhD degree under the supervision of Prof. Akira Sekiguchi in 2008. After being a Humboldt Postdoctoral fellow, as well as a JSPS postdoctoral fellow for research abroad with Prof. Matthias Drieß at the Technische Universität Berlin, he began his independent research career as a Sofia Kovalevskaja Professor in 2010 at the same university. Since 2015 he has been a W2-Tenure-Track Professor of Silicon Chemistry at the Technische Universität München. His research interests are focused on the synthesis and reactivity investigation of low-valent main group compounds with the goal of finding novel applications in synthesis and catalysis. |
Introduction
The imidazolin-2-imino group is a potent electron pair donor
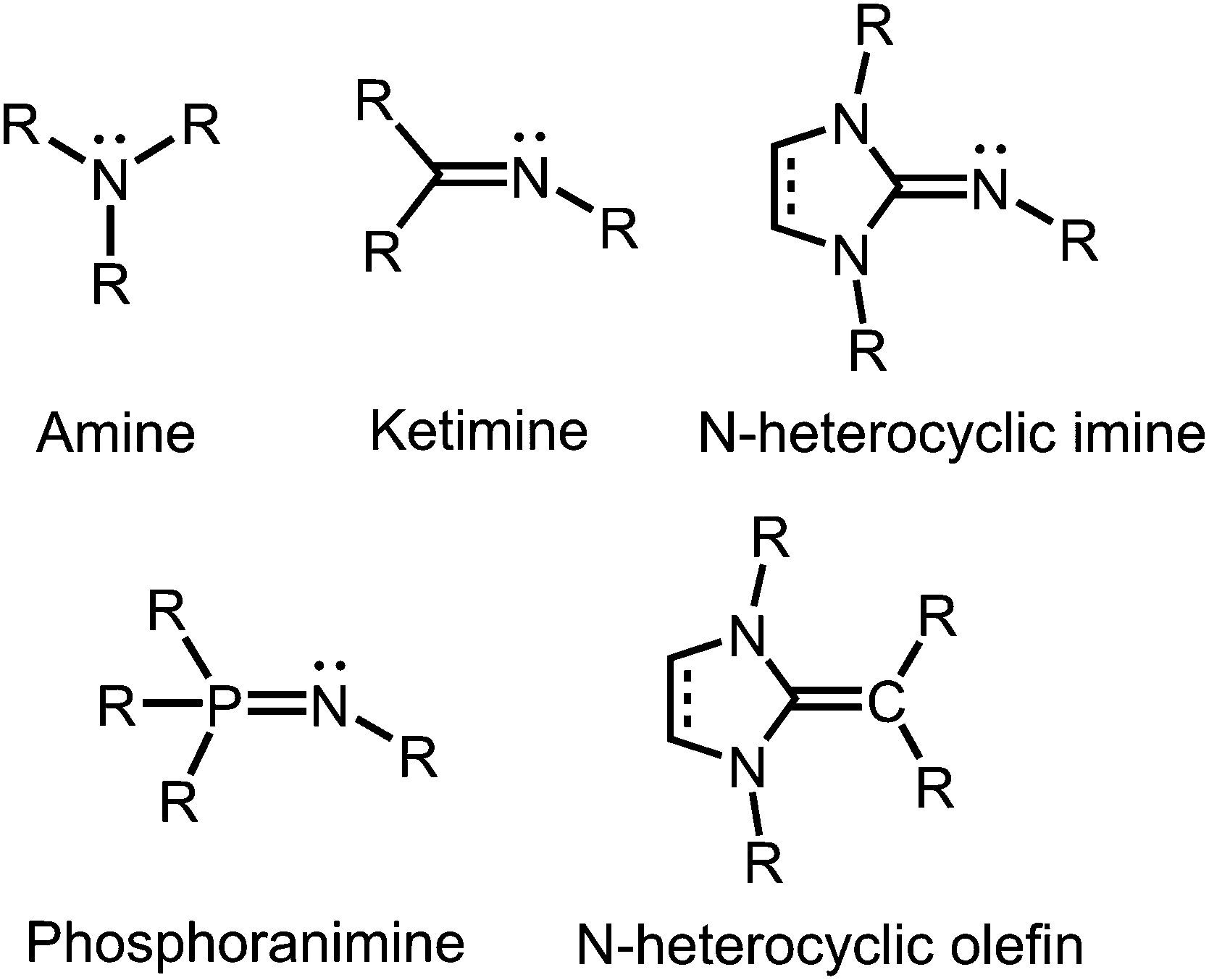
In coordination chemistry nitrogen is particularly recognized for its role as a strong electron-donor atom in ligand systems. Seemingly, this contradicts the fact that this element belongs to the highly electronegative members of the periodic table. However, the trivalent nitrogen atom in amines, as well as in imines has high electron density in the form of a lone pair that it readily shares with various types of hard and soft Lewis acids.
Tertiary amines and secondary ketimines resemble in their nucleophilic properties but, in sharp contrast, the unsaturated carbon atom of the imino functionality is prone to the reaction with nucleophiles or reducing agents whereas the amino carbon atom is inert (Fig. 1). This reactivity results from the π-interaction with the more electronegative nitrogen atom which provides the higher bond order but also drains electron density from the carbon centre in the σ-, as well as the π-scaffold. Due to the orthogonal orientation of the nitrogen lone pair this is not compensated by π back donation. Interestingly, the electronic properties of the imino-nitrogen atom are, vice versa, stronger affected by the characteristics of the carbon atom than it may be the case for the amino-nitrogen centre. In this regard, the electron-rich π-system of an imidazoline ring not only mitigates the electrophilicity of an imino-carbon atom incorporated at the 2-position of the cycle but also pushes electron density to the exocyclic imino-nitrogen atom (Fig. 1). Notably, phosphoranimines (R3P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NR) resemble the imidazolin-2-imines in the electron-donating character of the imino nitrogen atom (Fig. 1). The resemblance of these two ligand classes is reasoned by the similarities in the electronic properties of the parent phosphine and imidazolin-2-ylidene, respectively. Furthermore, one must recognize the isoelectronic relation between imidazolin-2-imines and N-heterocyclic olefins, which function as strong Lewis bases due to the ylide-like nature of the exocyclic alkene bond (Fig. 1).1
NR) resemble the imidazolin-2-imines in the electron-donating character of the imino nitrogen atom (Fig. 1). The resemblance of these two ligand classes is reasoned by the similarities in the electronic properties of the parent phosphine and imidazolin-2-ylidene, respectively. Furthermore, one must recognize the isoelectronic relation between imidazolin-2-imines and N-heterocyclic olefins, which function as strong Lewis bases due to the ylide-like nature of the exocyclic alkene bond (Fig. 1).1
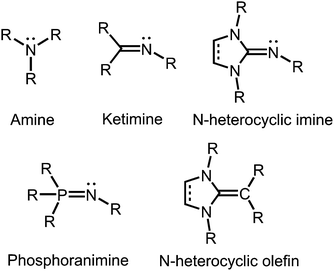 |
| | Fig. 1 Overview of selected N-donor ligands and the related N-heterocyclic olefin (R = organyl or H). | |
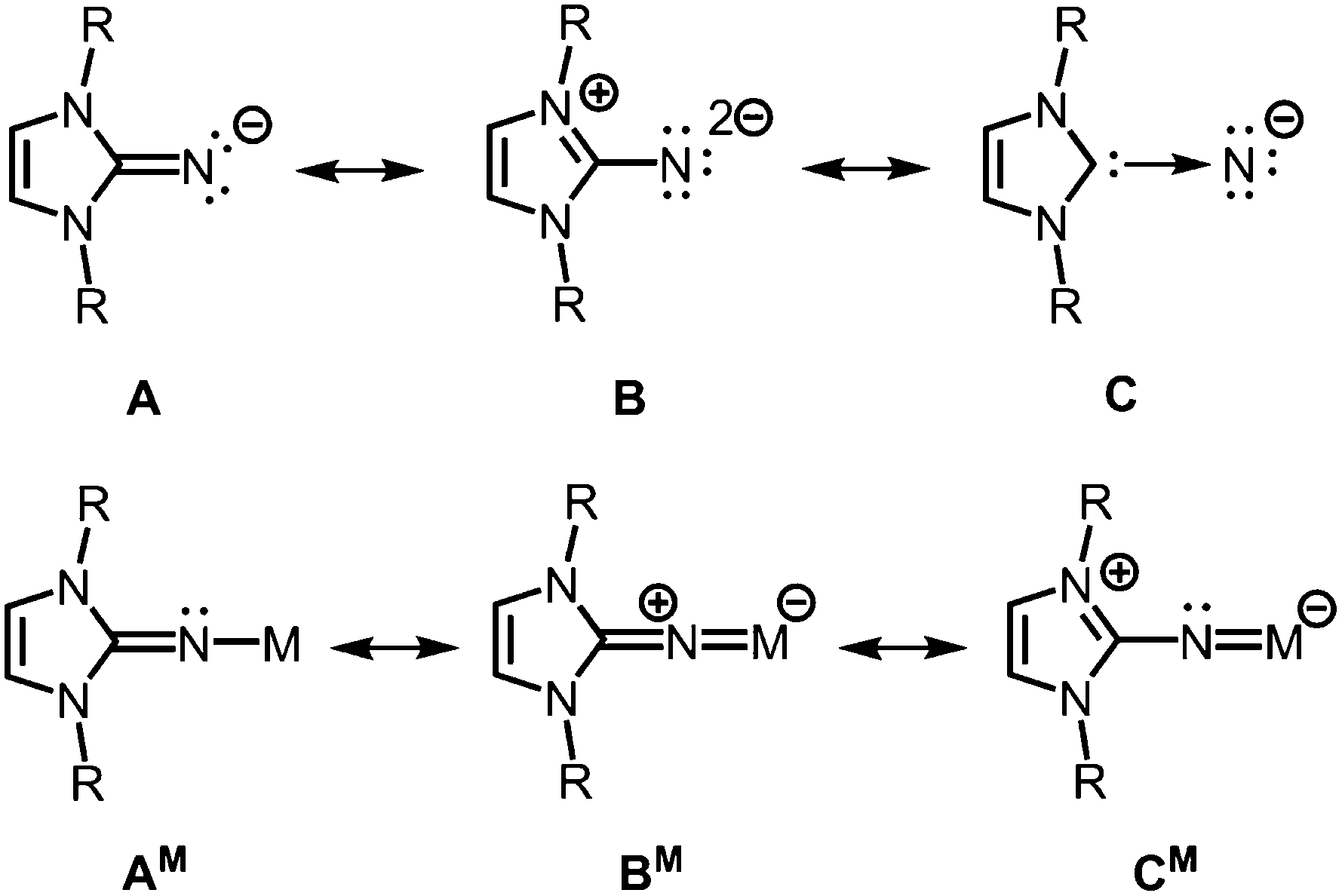
The allocation of electron density from the five-membered imidazoline ring to the exocyclic nitrogen atom is illustrated by conceivable resonance structures of the anionic imidazolin-2-iminato ligand A (Fig. 2).2,3 The canonical form B in which the exocyclic nitrogen atom bears two formal anionic charges suggests a significant boost of its electron-donating properties as compared to ketimines (Fig. 1 and 2). Form C represents the partial N-heterocyclic carbene (NHC) character of the imidazoline moiety (Fig. 2). As apparent from the canonical forms (A–C) the imidazolin-2-iminato ligand represents a 2σ electron donor with potential to contribute an additional two or even four π-electrons. Consequently, its metal complexes (AM) may exhibit significant metalla-2-aza-allene (BM) or metalimide (CM) character (Fig. 2). This manifests in an expansion of the imino group's CN distance and shortening of the N–M bond length. Concomitantly, the C–N–M bond angle is widened to approach the angle of 180° in the ideal CCC allene structure motive. As a result of its electron-donating properties, the imidazolin-2-iminato ligand is an efficient tool for the thermodynamic stabilization of electron-poor species. Moreover, the bulkiness of the imidazoline ring can be conveniently modified to meet individual requirements for kinetic stabilization of otherwise elusive compounds.
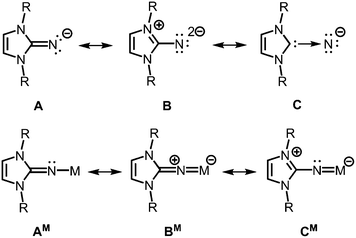 |
| | Fig. 2 Selected resonance structures for the anionic imidazolin-2-iminato ligand, as well as a model complex with M+ (R = organyl). | |
The scope of this review
In this article we focus on the coordination chemistry of the imidazolin-2-iminato ligand, as well as the strongly related imidazolidin-2-imino group and the benzimidazolin-2-imino group with regard to main-group elements. For the latter two only relevant examples will be given. An earlier review of Kuhn, Frenking and coworkers on imidazolin-2-imines includes main-group metal complexes but dates back about 13 years.2 The broad spectrum of transition metal complexes that comprise this ligand class and the methods for the synthesis of the ligand have recently been reviewed by Tamm and coworkers and will be discussed only in part.3
Moreover, only selected examples will be discussed for compounds of this iminato ligand with the non-metals carbon and nitrogen because this belongs to the field of organic chemistry rather than coordination chemistry.
Group 1 and group 2 element complexes
Background
About 20 years ago Kuhn and coworkers started their pioneering studies on the chemistry of imidazolin-2-imines.4 A few alkaline5,6 and alkaline earth7 compounds of the imino group were reported but not investigated thoroughly probably because of the pronounced polar nature of the N–M bond (M = alkaline or alkaline earth metal). This puts them in the role of a reactive intermediate for ligand transfer via salt metathesis rather than a species with its own follow-up reactivity with sustainment of the N–M bond. Accordingly, the chemistry of group 1 and group 2 imidazolin-2-iminato complexes is only explored to a minor degree to date.
Lithium and potassium complexes
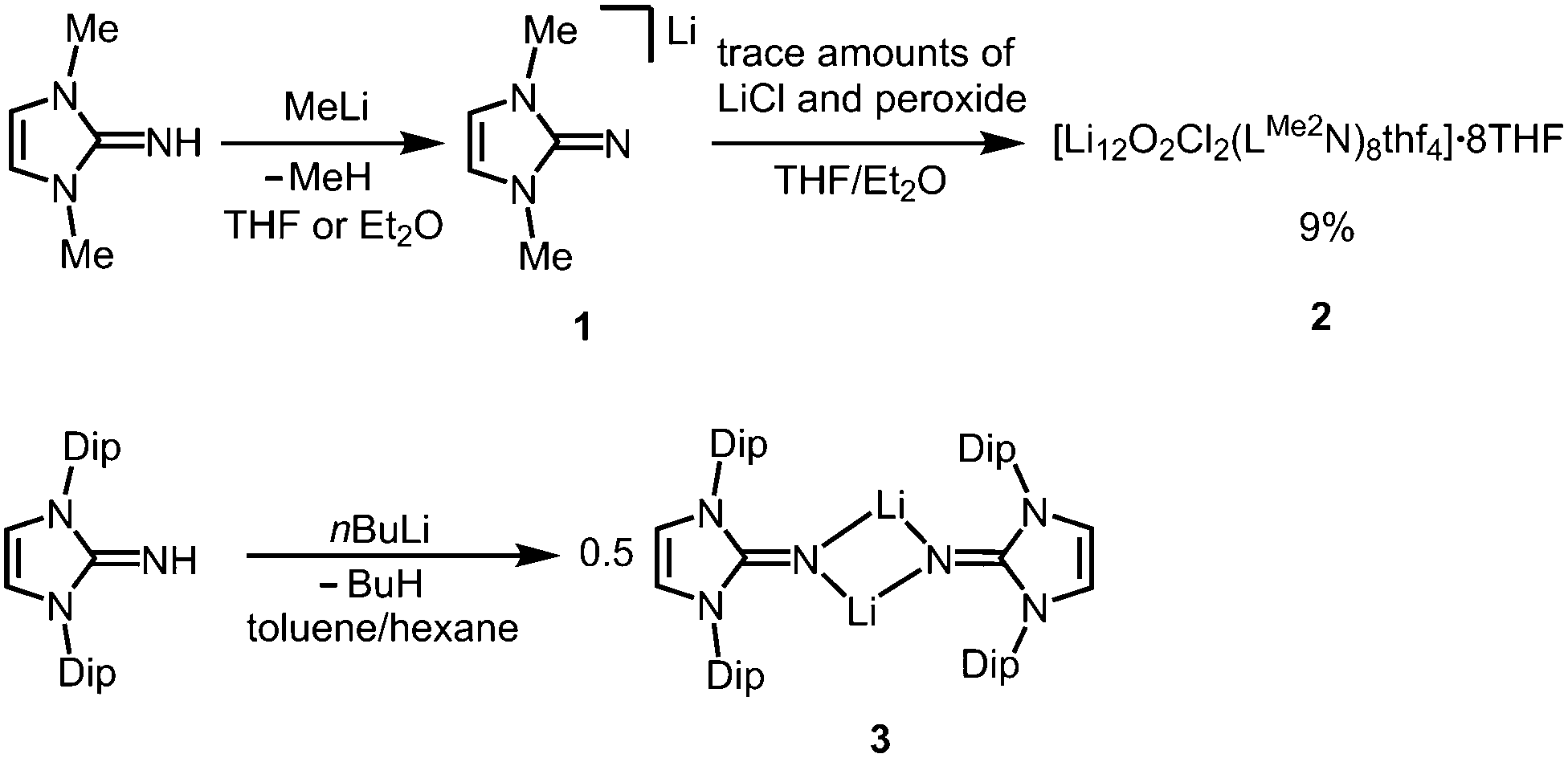
The reaction of LMe2NH (LMe2 = 1,3-dimethyl-imidazolin-2-ylidene) with MeLi in Et2O produces LMe2NLi (1) which is the N-lithiated derivative of the imidazolin-2-imine.5 The species was characterized by 1H NMR analysis and according to the reported CHN elemental analysis no solvent was present in the isolated material. If the conversion was carried out in THF/Et2O with MeLi that was prepared from H3CCl and elemental lithium without prior separation of lithium chloride, crystals of the unexpected composition [Li12O2Cl2(LMe2N)8(thf)4]·8THF (2) were retrieved in low yield (Scheme 1).
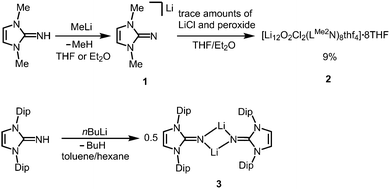 |
| | Scheme 1 Conversion of the imino lithium species 1 into the imino-stabilized LiOCl aggregate 2 (the lithium chloride derives from the methyllithium synthesis and peroxide from contaminated solvent). Formation of the bulky imino lithium dimer 3 (Dip = 2,6-diisopropylphenyl). | |
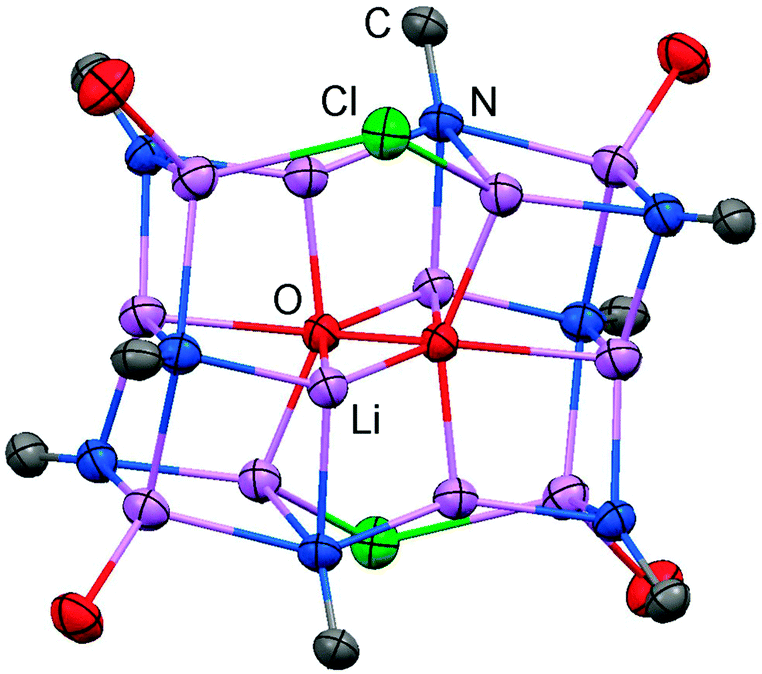
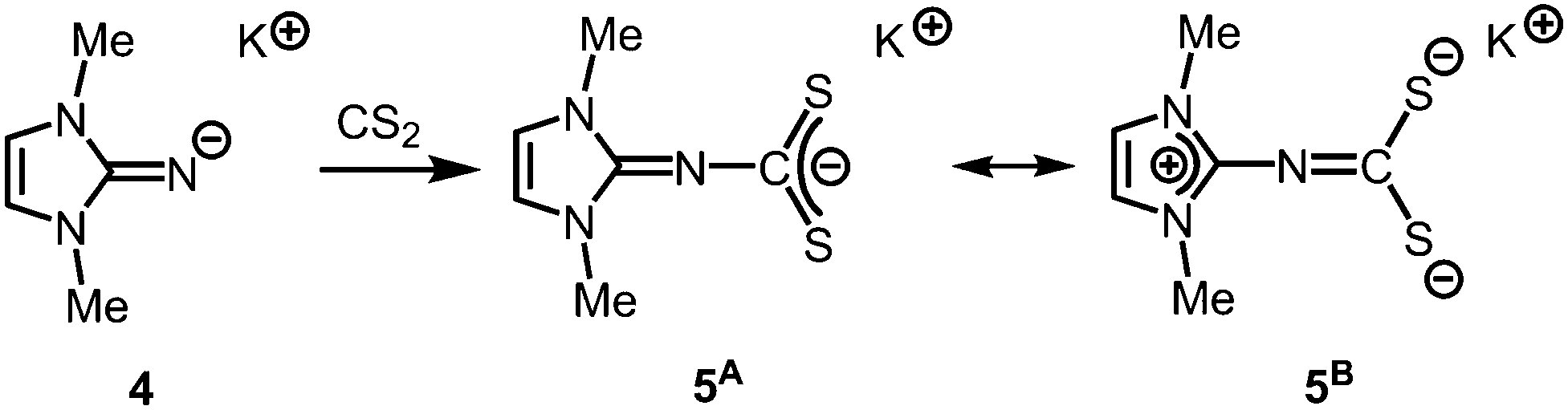
The solid state structure of 2 is marked by a Li12N8O2Cl2 cage that comprises a peroxo moiety in its core (Fig. 3). The authors reasoned that the O22− group resulted from contamination of the solvent with traces of peroxide. Crystals of dimeric [LDipNLi]2·toluene (3·toluene, LDip = 1,3-bis(2,6-diisopropylphenyl)-imidazolin-2-ylidene) were isolated in good yield after the reaction of LDipNH with nBuLi in toluene/hexane.8 Apparently, the formation of higher aggregates is hampered by the bulkier Dip groups (Dip = 2,6-diisopropylphenyl). Bringing into contact LMe2NH and freshly prepared MeK in Et2O afforded the heavier alkaline derivative LMe2NK (4).6 The compound was characterized by elemental analysis and its existence was verified by the synthesis of the dithiocarbiminate LMe2NCS2K (5, Scheme 2). Interestingly, the latter shows structural characteristics that account for a bonding situation as represented by resonance structure 5B (Scheme 2) with the C–Nimino bond length significantly increased (a range from 1.369(16) Å to 1.379(18) Å is observed in the solid state structure; cf.2: C–Nimino = 1.260(4)–1.263(4) Å; 3: C–Nimino = 1.241(3) Å, 1.242(4) Å). Accordingly, the C–S distances in 5 (1.733(13)–1.755(13) Å) resemble typical CS single bond lengths.
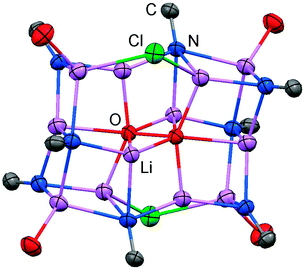 |
| | Fig. 3 Ellipsoid plot (30% level) of the Li12N8O2Cl2 cage in 2 with adjacent imino-carbon atoms and oxygen atoms of coordinated THF. | |
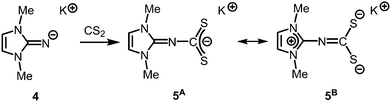 |
| | Scheme 2 Conversion of potassium imide 4 with carbon disulfide to the thiocarbiminate 5 (represented by resonance structures 5A and 5B). | |
Magnesium complexes
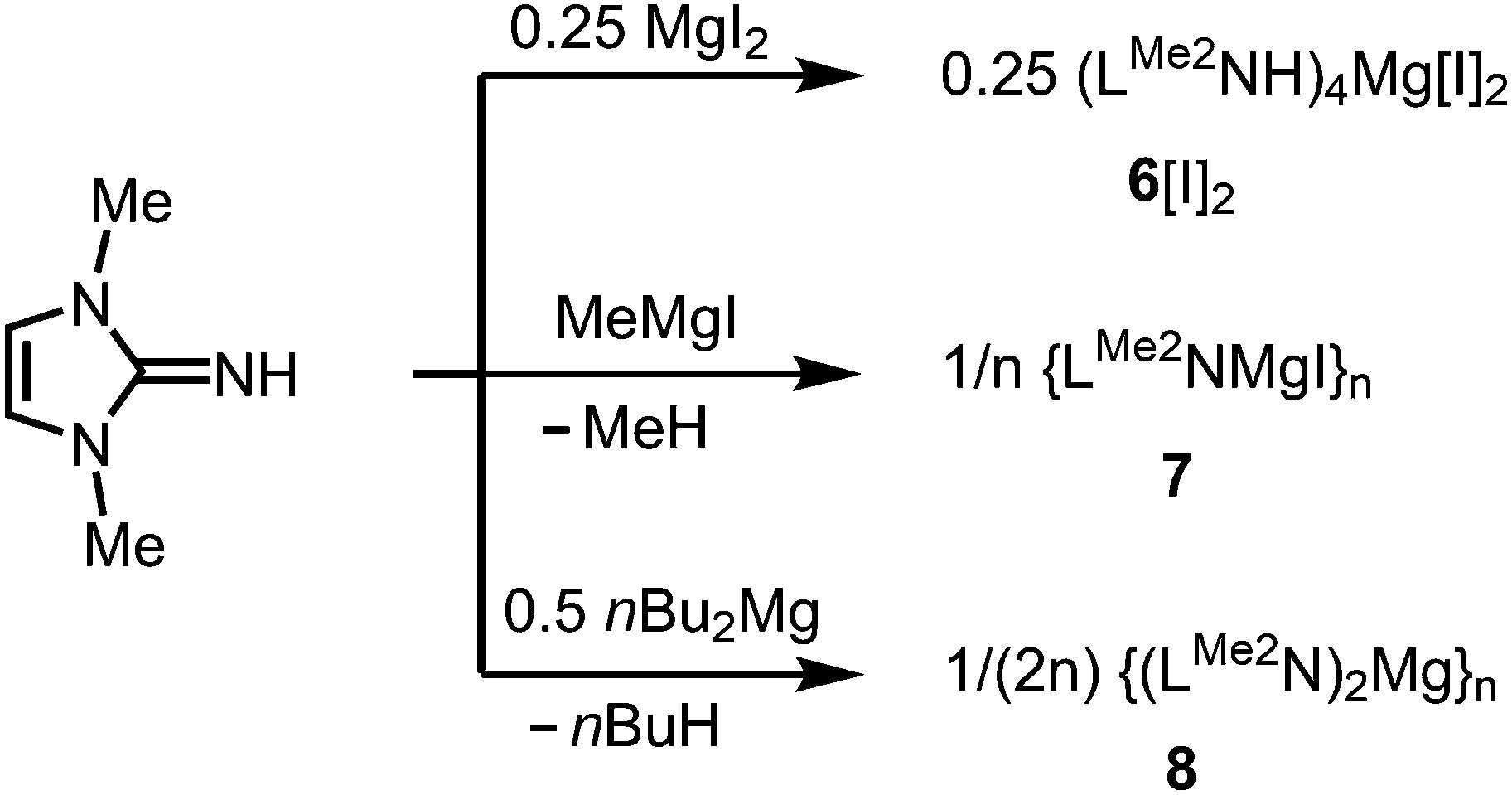
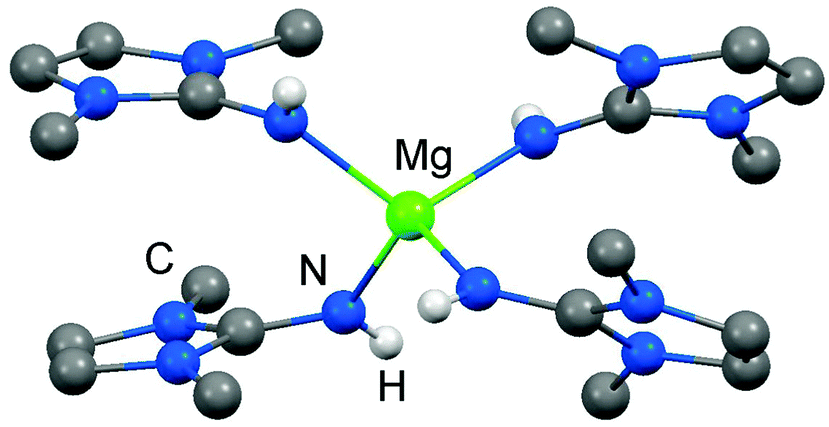
As rare examples for N-heterocyclic iminato complexes of group 2 metals the magnesium compounds (LMe2NH)4MgI2 (6[I]2), as well as {LMe2NMgI}n (7) and {(LMe2N)2Mg}n (8), were reported by Kuhn and coworkers (n ≥ 1).7 They are accessed through LMe2NH via conversion with 0.25 MgI2, MeMgI and (nBu)2Mg, respectively (Scheme 3). Single crystal XRD (X-ray diffraction) data were obtained for 6[I]2 (Fig. 4) while the degree of aggregation (n) of 7 and 8 was not elucidated by structural analysis. Notably, 6[I]2 also formed if less than four equiv. of the imine were reacted with MgI2. The authors attributed this observation to the high basicity of the ligand.
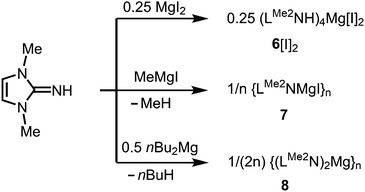 |
| | Scheme 3 Synthesis of the imino-magnesium compounds 6[I]2, 7 and 8. | |
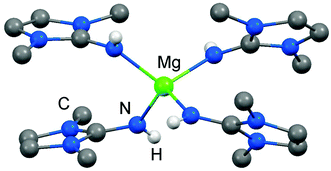 |
| | Fig. 4 Ball and stick representation of (LMe2NH)4Mg2+ (62+) as derived from XRD analysis (non-N-bonded hydrogen atoms have been omitted). | |
Group 13 element complexes
Background
Of group 13 elements only a few aluminium complexes with an N-heterocyclic iminato ligand had been reported until respective research was resumed by our group.6 Reports8–10 in the year 2014 were the first to describe imidazolin-2-imino complexes of boron. In contrast, the coordination chemistry of related phosphoranimines of boron11 and aluminium11a,c,d,12 is thoroughly studied.
Boron complexes
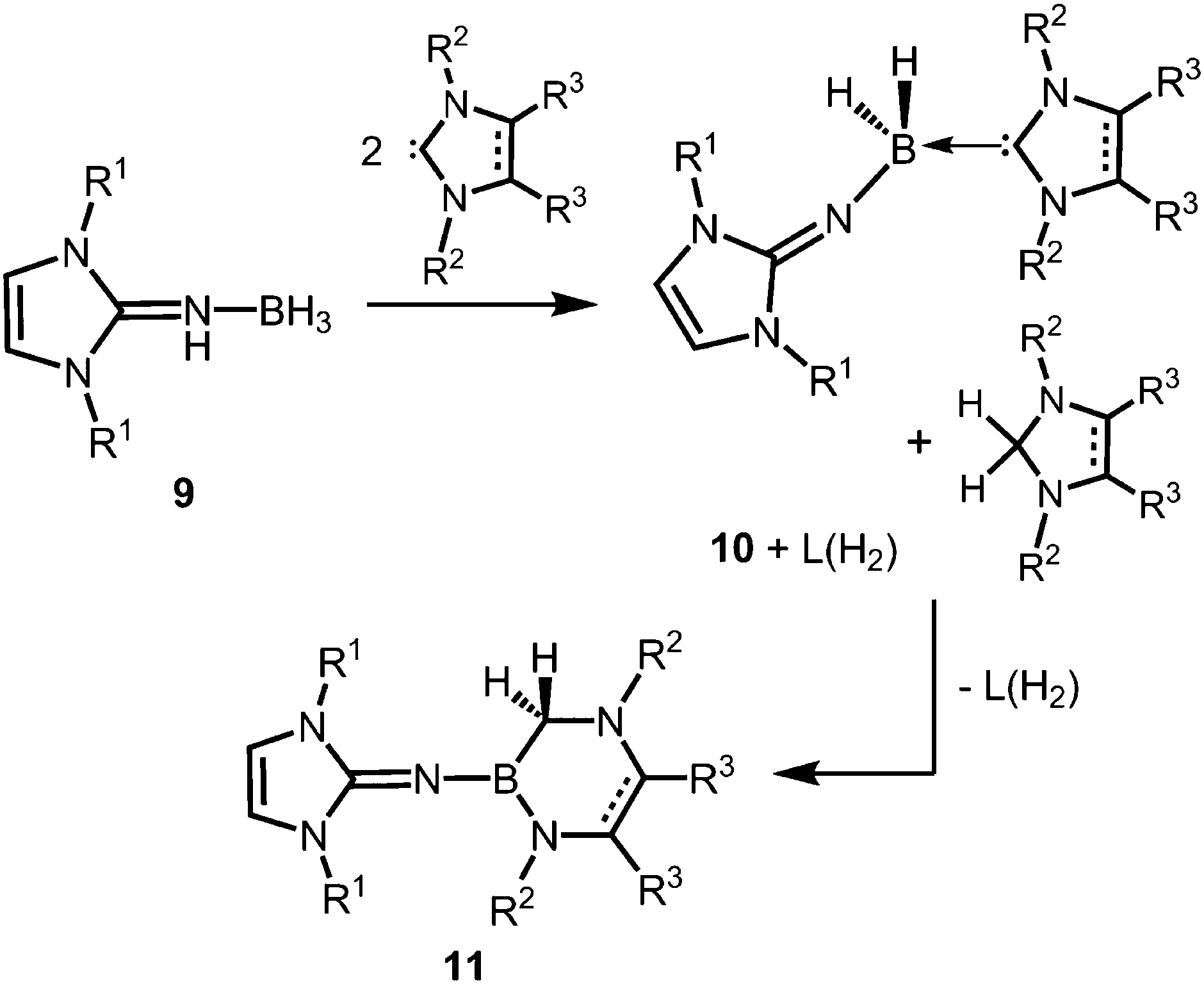
The Lewis acid base adducts LNH(BH3) (9, Scheme 4) between LDipNH, as well as LMesNH (LMes = 1,3-dimesityl-imidazolin-2-ylidene), and the parent borane were isolated after conversion of the imine with Me2S·BH3 in toluene.9 When treated with imidazolin-2-ylidenes (L) dihydrogen is abstracted from the HN–BH moiety of these imine–borane compounds and along with L(H2) (hydrogenated at the formerly carbenic centre) the NHC-adducts of respective imino boron dihydrides (LN(BH2)L) are formed (10, Scheme 4).9 These NHC-adducts undergo hydride-mediated ring-expansion reaction, that is, the boron atom transfers its two hydrides to the adjacent carbon atom and inserts into the CcarbenoidN bond of the NHC (11, Scheme 4).9 Presumably, the interaction between the lone pair at the imino nitrogen atom and the unoccupied p-orbital at the boron centre supports the trigonalization of the metalloid atom. Interestingly, this insertion occurs at higher temperatures with more sterically hindered substituents at the boron atom. Moreover, H2LMes (1,3-dimesityl-imidazolidin-2-ylidene, NHC saturated at the ligand backbone) is subject to ring-expansion reaction at significantly lower temperatures than its congener of very similar sterical encumbrance LMes (NHC unsaturated at the ligand backbone). It was reasoned that the conjugated ring system in LMes is more efficient for the delocalization of positive charge density and, thus, stabilizes the boron dihydride form (LNBH2L).
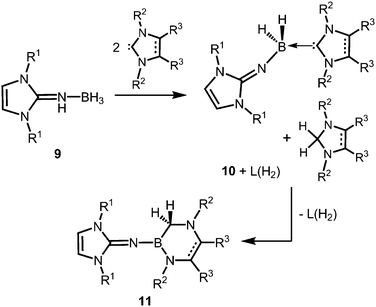 |
| | Scheme 4 Conversion of the imine–borane adduct 9 with NHC to iminoboron dihydride NHC complex 10 and the formation of 11via ring expansion reaction. R1 = Mes or Dip; R2 = Me (for R3 = Me), Mes or Dip (both for R3 = H); R2 = Mes and R3 = H for unsaturated backbone; not all combinations of imine and NHC are viable for 10 and 11.9 | |
Similarly, ring-activation and expansion reaction of LDip took place by heating the amido-substituted hydridoborane LDip(BH2)HNDip reported by Rivard and coworkers.13
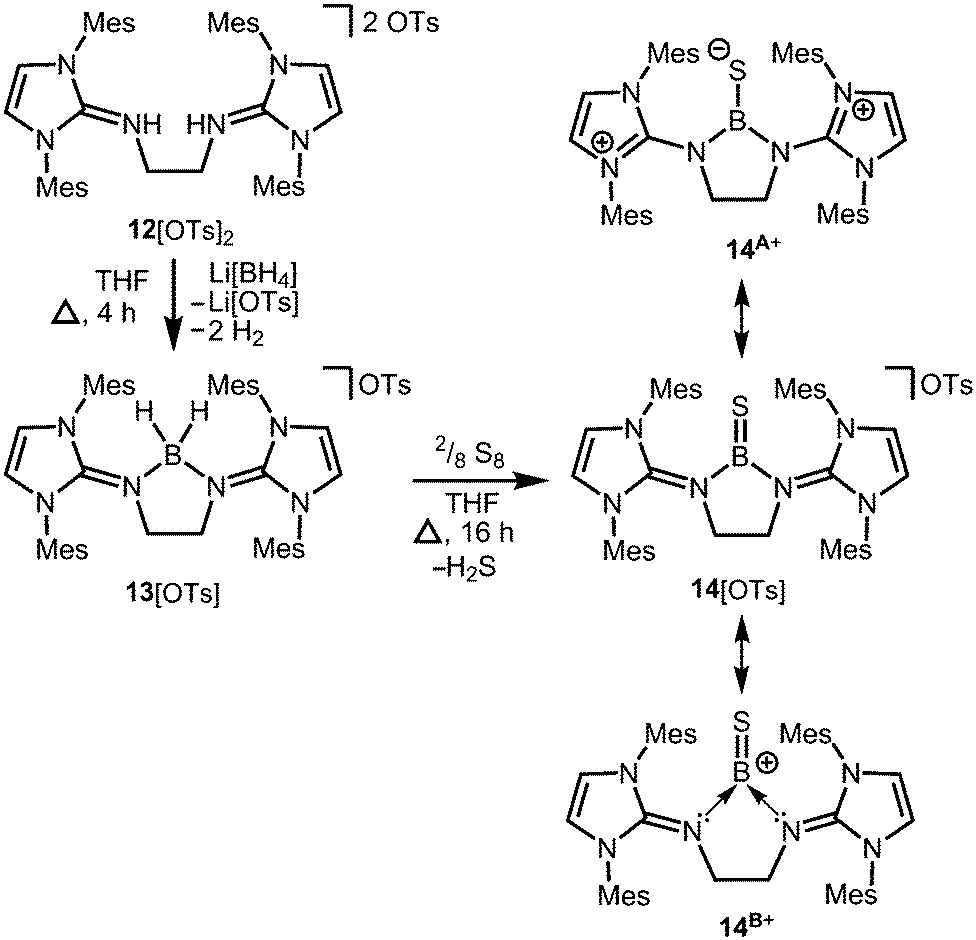
Conversion of the bis(iminiumtosylate) 12[OTs]2 with Li[BH4] furnishes the boronium salt 13[OTs] (Scheme 5).10 The compound reacts with yellow sulfur to give a rare example of a cationic thioxoborane 14[OTs] that was structurally characterized (Scheme 5).10 The B–S bond length (1.710(5) Å) in 14+ is the shortest that has been reported to date for a molecular complex. Notably, the B–N bond lengths significantly decrease upon transformation of tetrahedral 13+ into trigonal-planar 14+ (13+: 1.573(5) Å, 1.577(5) Å; 14+: 1.483(5) Å, 1.493(5) Å) and, consequently, a partial double bond character can be attributed to the boron–nitrogen interactions in 14+. Concomitantly, the C–N distances of the imino groups increase (13+: 1.317(5) Å, 1.318(4) Å; 14+: 1.359(4) Å, 1.363(4) Å) which is in accordance with the formulation of resonance structure 14A+ (Scheme 5) that represents the delocalization of positive charge density into the N-heterocycles and the polarization of the BS bond towards the sulfur atom (NBO charge at S = −0.58; NBO = Natural Bond Orbital). DFT (density functional theory) calculations supported the interpretation of the remarkably short B–S distance in terms of a boron sulfur double bond. For example, the HOMO (highest occupied molecular orbital) shows mainly the sulfur lone pair and the HOMO−1 reveals the BS π-bonding orbital. The NBO charge at the boron centre of 14+ was calculated to be +0.63 which accounts for the boron cation character of the complex as illustrated by the canonical form 14B+ (Scheme 5).
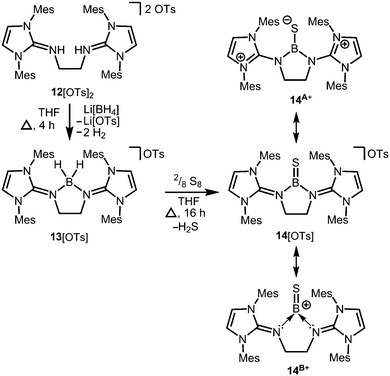 |
| | Scheme 5 Reaction of the bis(iminiumtosylate) 12[OTs]2 with lithium borohydride to the boronium salt 13[OTs] (Ts = tosyl) and its conversion to the thioxoborane salt 14[OTs]. | |
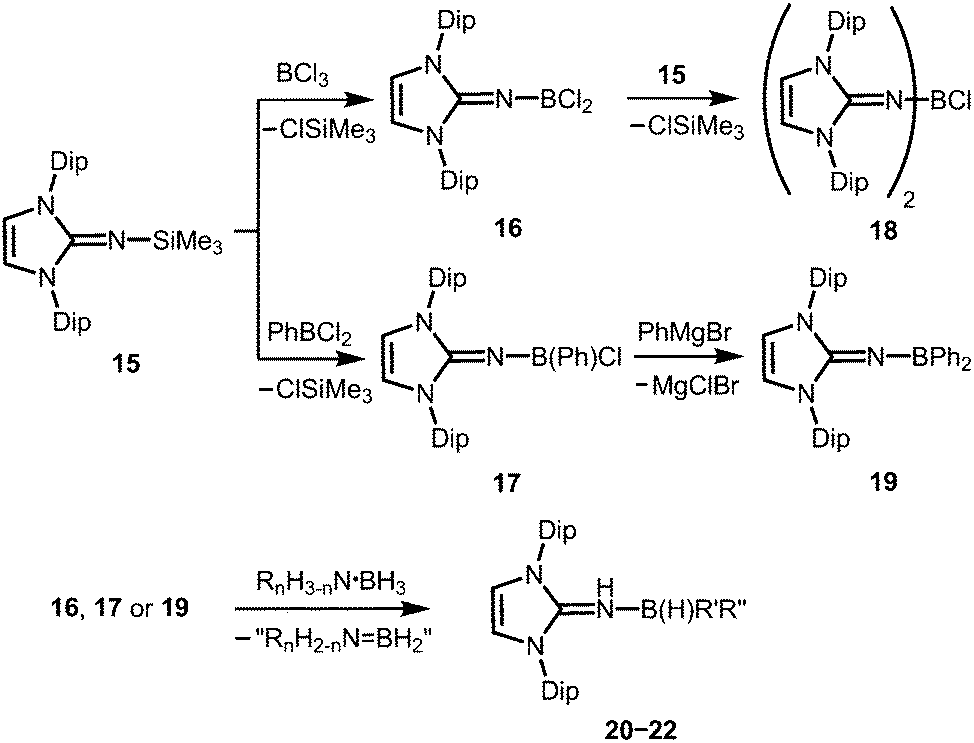
Very recently, Rivard and coworkers described the conversion of the imidazolin-2-imino trimethylsilane 15 to the imino boron dichloride 16 and its organyl derivative 17 by the reaction of 15 with BCl3 and PhBCl2, respectively (Scheme 6).14 The bisimino boron monochloride 18 was furnished in a reaction between 15 and 16 (Scheme 6). Moreover, the synthesis of the diphenyl congener 19 was accomplished by conversion of 17 with phenylmagnesium bromide (Scheme 6). The solid state structure of the dihalide 16 hints toward the significant bora-2-aza-allene properties of the CNB moiety (type BM, Fig. 2) as concluded from the C–N–B bond angle of 180° and the short B–N bond length (1.302(6) Å) which implies high boron–nitrogen double bond character. Remarkably, 16, 17 and 19 react with amine–boranes (RnH3−nN·BH3; R = H, Me; n = 1, 2) to produce the respective dihydrogenated imino boron compounds LDipNH(B(H)R′R′′) 20–22 (R′ = R′′ = Cl for 20; R′ = Cl, R′′ = Ph for 21; R′ = R′′ = Ph for 22) and a mixture of the amine–borane dehydrogenation products (Scheme 6).14 The authors conclude that this imino boron compound acts as an intramolecular frustrated Lewis acid base pair. It should be noted that compound 17 displays catalytic activity in the dehydrocoupling of MeNH2·BH3 to yield [MeNBH]3 along with oligomeric aminoboranes, which shows its great potential with respect to further application in metal-free catalysis for the dehydrocoupling of amine–boranes and related species.
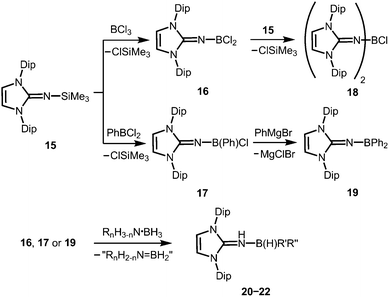 |
| | Scheme 6 Conversion of the imino trimethylsilane 15 to the imino chloroboranes 16–18 and the imino diphenylborane 19. Formation of the imine–borane adducts via abstraction of dihydrogen from amine–borane adducts. | |
Aluminium complexes
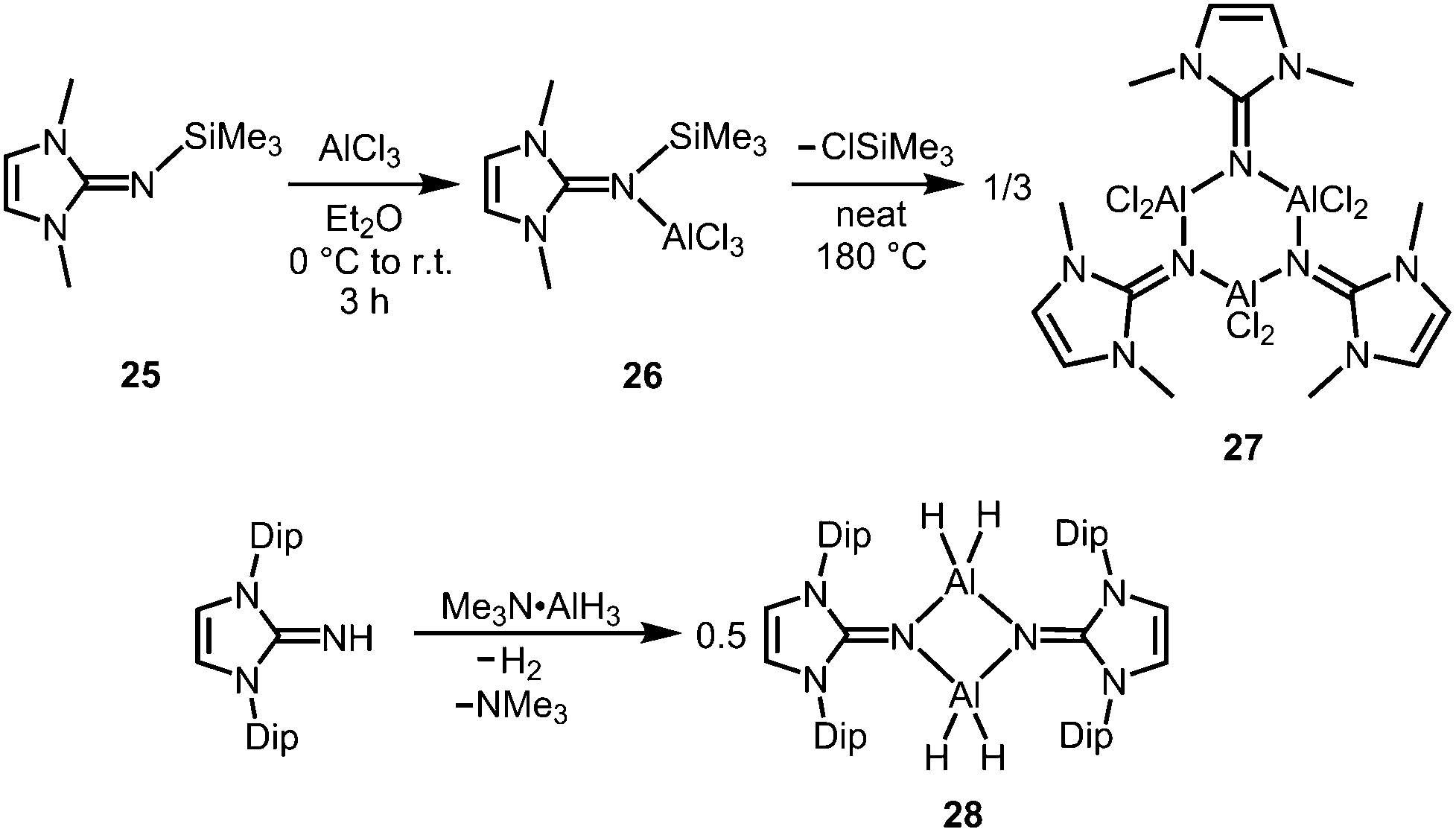
The bisimino aluminium complexes (LMe2NSiMe3)2AlMe2[Cl] (23) and (LMe2NH)2AlMe2[Cl] (24) were synthesized by conversion of LMe2NSiMe3 (25) and LMe2NH, respectively, with 0.5 equiv. of AlMe2Cl.7 The ion-separated forms were postulated on the basis of NMR spectroscopic data. The related Lewis acid base adduct LMe2NSiMe3·AlCl3 (26) releases Me3SiCl upon heating the neat compound to 180 °C and is converted into {LMe2NAlCl2}3 (27, Scheme 7).7 An X-ray crystallographic analysis of compound 27 verified its trimeric structure with a six-membered Al3N3 cycle.
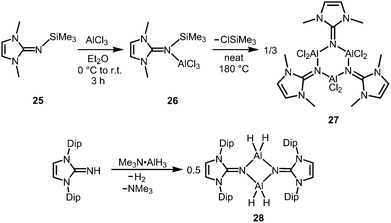 |
| | Scheme 7 Conversion of the trimethylsilylimine 25 to the aluminium trichloride imine complex 26 and its transformation into trimeric 27. | |
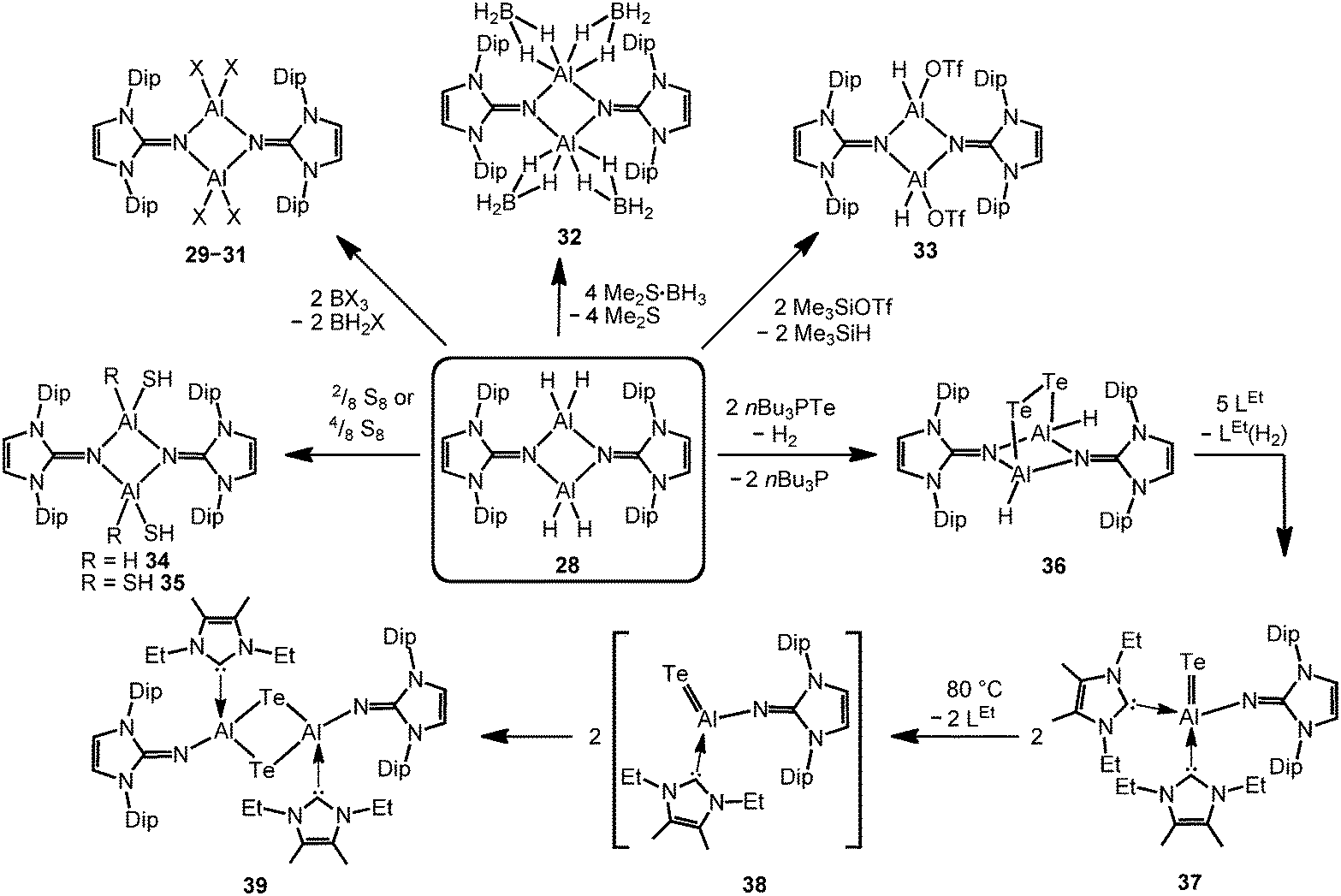
The imino aluminium dihydride {LDipNAlH2}2 (28) results from the reaction of LDipNH with Me3N·AlH3 (Scheme 7).8 From the dihydride one can derive the dihalides {LDipNAlX2}2 (29–31, X = Cl, Br, I) by conversion with BX3 (two equiv.) which were described to form dimers in the solid state, as well as in solution (Scheme 8).7 Obviously, the bulkier iminato ligand in 29 leads to the formation of a four-membered Al2N2 ring with smaller N–Al–N angles (87.8(1)°, 92.3(1)°) in comparison to the six-membered ring in 27 with larger angles (108–110°). Interestingly, the sterically hindered phosphoranimino aluminium dihydride and -dichloride form dimers with four-membered Al2N2 rings ({R3PNAlX2}2; R = iPr, tBu; X = H, Cl), as well.11c,12a Furthermore, the dihydride 28 reacts with the electrophiles Me2S·BH3 (four equiv.) and Me3SiOTf (two equiv., Tf = triflyl) to yield the aluminum borohydride 32 and the aluminium monohydride triflate 33, respectively (Scheme 8).8 Notably, the substitution of both aluminium bonded hydrides in 33 for triflate substituents could not be accomplished by the use of a larger excess of Me3SiOTf, even at elevated temperature. In contrast, the conversion of 28 with only two equiv. of Me2S·BH3 does not afford the expected aluminium monohydride borohydride as a product but yields mixtures of 28 and 32. Obviously, the electron withdrawing triflyl groups in 33 mitigate the hydride-donor strength of the remaining AlH functionality. Accordingly, only aluminium monohydride triflates of the related phosphoraniminato or the 1,3-diketiminato ligand have been reported.11c,15 The conversion of the aluminium dihydride 28 with yellow sulfur affords a rare example of an aluminium hydride hydrogensulfide complex (34) by insertion of a sulfur atom into the AlH bond (Scheme 8).16 Similar to 33 the remaining hydride functionalities at the aluminium centres in 34 are less reactive than in the parent compound. However, the transformation with S8 to form the bis(hydrogensulfide) 35 can be forced onto the system by heating (90 °C for four days, Scheme 8).16 As apparent from the XRD study the Al–S distances in 35 (2.231(1)–2.240(1) Å) are slightly shorter than the respective distances in the monohydrogensulfide 34 (2.250(1) Å and 2.252(1) Å).
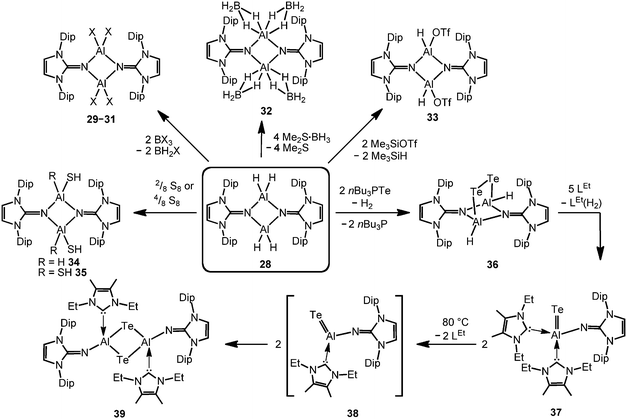 |
| | Scheme 8 Overview on syntheses of imino aluminium compounds derived from the aluminium dihydride 28: reaction of 28 to the dihalides 29–31, the borohydride 32 and the triflate 33 (X = Cl, Br, I). Synthesis of the aluminium mono- and bis(hydrogensulfides) 34, as well as 35. Conversion of 28 to the ditelluride 36 and its monotopic aluminium telluride offspring 37 (LEt = 1,3-diethyl-4,5-dimethyl-imidazolin-2-ylidene). Transformation of 37 to ditopic 39via the presumed intermediate 38. | |
In order to furnish a heavier aluminium chalcogenide of the imidazolin-2-iminato ligand, 28 was converted with the tellurium atom transfer reagent nBu3PTe (two equiv.).17 This conversion yields ditopic aluminium ditelluride 36 as a rare example of an electron-precise aluminium complex with the chalcogen in the oxidation state −1 (Scheme 8). The hydrides left at the aluminium centres in 36 do not react further with excess nBu3PTe. However, the compound converts with NHC (LEt, 5 equiv., LEt = 1,3-diethyl-4,5-dimethyl-imidazolin-2-ylidene) in a dehydrogenative redox process to form the monotopic aluminium telluride 37 (Fig. 5, Scheme 8) with the chalcogen in the oxidation state −2 along with dihydrogenated NHC (LEt(H2)).17
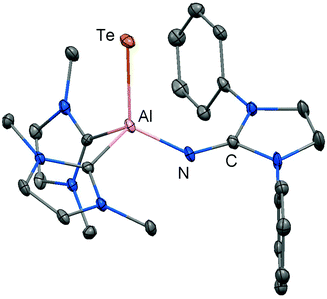 |
| | Fig. 5 Ellipsoid plot (30% level) of the aluminium telluride 37 (hydrogen atoms, isopropyl groups and non-N-bonded methyl groups have been omitted). | |
The structural study of 37 revealed a remarkably short Al–Te distance of 2.5130(14) Å and DFT calculations determined an enhanced aluminium–tellurium interaction (WBIAlTe = 1.20; NPA charges: Al = +1.24, Te = −0.95; WBI = Wiberg bond index, NPA = natural population analysis). It has to be pointed out that the terminal position of the tellurium atom is a very scant structural motif as group 16 atoms commonly assume bridging positions in aluminium chalcogenides. Upon heating a benzene solution of 37 to 80 °C one of the two LEt ligands is released and the putative intermediate LDipN(AlTe)LEt (38) undergoes aggregation to form 39 (Scheme 8).
The reaction pathway via38 was suggested by DFT calculations; however, the isolation of a bulkier congener of this elusive species was not accomplished by the use of more sterically hindered NHC. The structural investigation of 39 revealed significantly increased Al–Te distances (2.6143(14) Å, 2.6211(15) Å) and a decreased bond order for the AlTe interaction (WBIAlTe = 0.75; NPA charges: Al = +1.21, Te = −0.79) with respect to 37.17 It should be noted that in ditopic 39 the aluminium centres are bridged via the tellurium atoms. Notably, 37 and 39 contrast the other given examples for aluminium complexes of the imidazolin-2-iminato ligand in that the aluminium centres are not connected via the nitrogen atoms of the imino groups. Taking into account the marked changes in the Al–Te distances and the values for the WBIAlTe upon transformation of 37 into 39 the nature of the AlTe interaction in 37 was presumed to possess high AlTe double bond character.
Group 14 element complexes
Background
In initial reports on the chemistry of N-heterocyclic iminato ligands Kuhn and coworkers described the imino trimethylsilane 25 (Scheme 7) which was used as an alternative transmetallation reagent to the alkaline metal salts mentioned above.4,7 Presumably, the bulkier LMesNSiMe3 is formed as an intermediate in the synthesis of LMesNH via a Staudinger-type reaction described by Cameron, Jenkins, Clyburne and coworkers in 2001.18 Tamm and coworkers established the general method for the preparation of trimethylsilyl-functionalized bulkier imidazolin-2-iminato ligands such as LMesNSiMe3 and LDipNSiMe3 in 2004.19 This method has tremendous advantages for the convenient and high-yield synthesis of various imidazolin-2-imines. Moreover, a silicon atom was incorporated into the spacer group between the imino- and the arene moiety in oligodentate ligands reported by Tamm and coworkers.20 However, it played a rather passive role in the chemistry of the transition metal complexes derived from this ligand system. As outlined in the following section a considerable time elapsed from Kuhn's initial report until the coordination chemistry of the imidazolin-2-iminato ligand with tetrel atoms was thoroughly investigated.
Silicon complexes
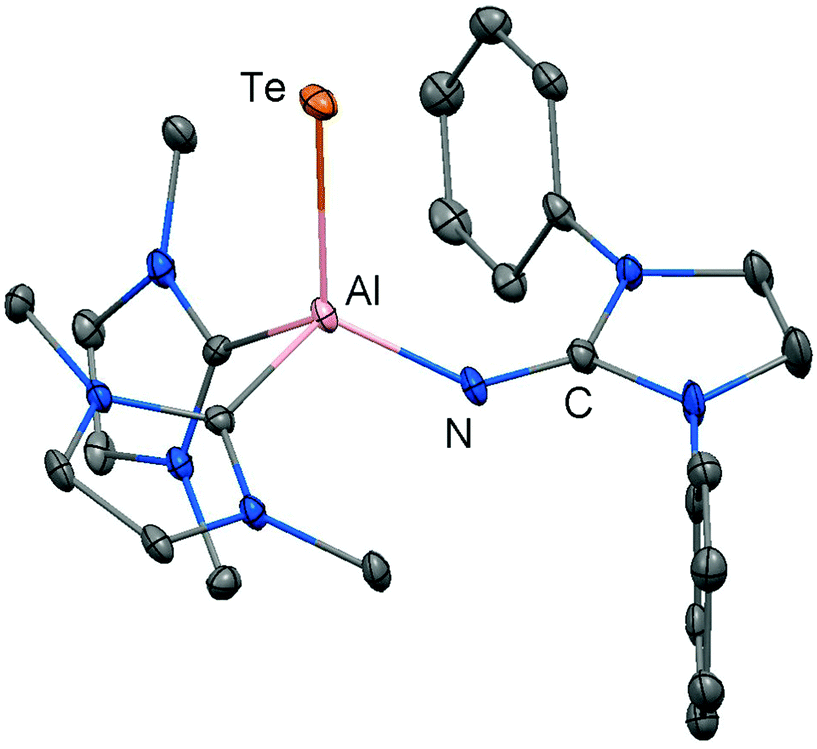
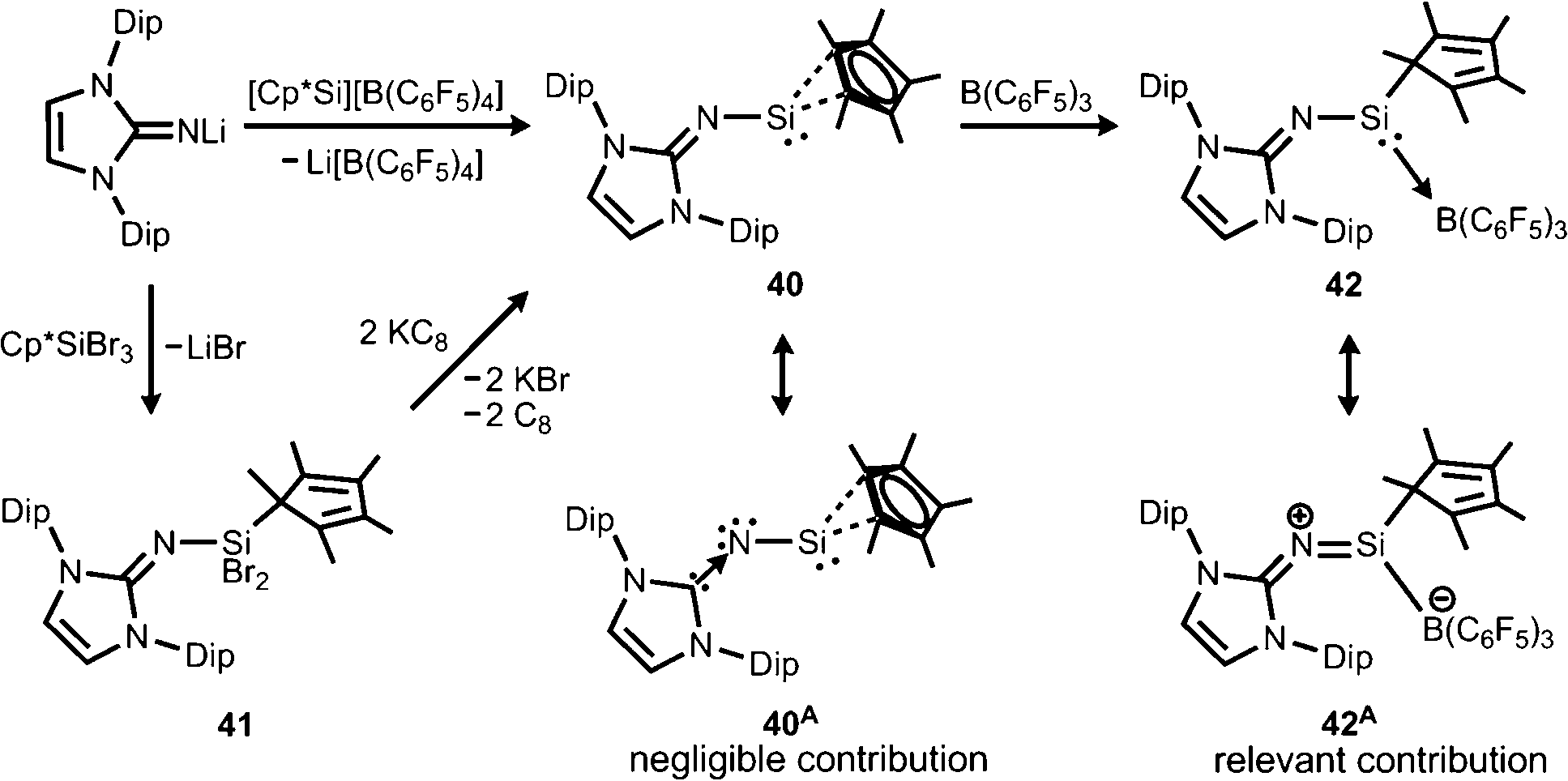
Our group commenced work on main group element complexes of the imidazolin-2-iminato ligand a few years ago and described its complex with a silicon(II) centre in 2012.21 High interest for molecular low-valent silicon compounds originates from their various applications in catalysis and bond activation.22,23 Conversion of LDipNLi with Cp*Si[B(C6F5)4] (Cp* = pentamethylcyclopentadienyl) as a source of silicon(II) afforded the pentamethylcyclopentadienyl imino silylene 40 with η2 coordination of the silicon centre by the organyl ligand (Scheme 9). An alternative synthetic route by which 40 can be accessed is via reaction of LDipNLi with Cp*SiBr3 followed by reductive dehalogenation of LDipNSi(Br2)Cp* (41). Unfortunately, this method affords only very poor yields of the silylene. DFT calculations on 40 show some π bonding interaction between the imino nitrogen lone pair and the unoccupied p-orbital at the silicon centre. The WBISiN of 0.80 and the Si–N bond length (1.691(5) Å) imply single bond character. Thus, multiple bond interaction as illustrated by the general canonical structures BM and CM (Fig. 2) cannot be concluded for 40. A key motivation of the study was to explore potential silylene–nitrene character of complexes between a low-valent silicon atom and the imidazolin-2-iminato ligand as represented by the canonical structure 40A (Scheme 9). However, structural and theoretical investigation verified the imino-substituted silylene formulation 40 with no relevant silylene–nitrene character (40A). Conversion of 40 with tris(pentafluorophenyl)borane furnished the silylene–borane adduct 42. It is interesting to note that the Cp* ligand is coordinated in a η1-mode with one σ bond to the silicon atom in sharp contrast to the precursor 40, in which η2-mode Si–Cp* bonding is observed. As compared to 40 the Si–N bond length is considerably reduced to 1.605(3) Å and its WBISiN is increased to 0.90 which account for partial SiN double bond character. The C–N–Si angle of 158.7(3)° in 42 is wider than in 40 (136.6(4)°). Accordingly, the relevant 1-sila-2-aza-allene nature (cf.BM, Fig. 2) can be attributed to 42 as represented by resonance structure 42A (Scheme 9).
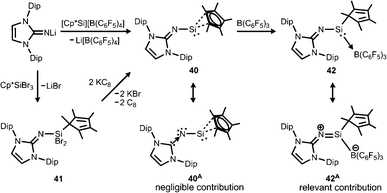 |
| | Scheme 9 Synthesis of Cp*-substituted iminosilylene 40 and its borane adduct 42, as well as the dibromide precursor 41. Silylene–nitrene formulation 40A and sila-2-aza-allene canonical structure 42A. | |
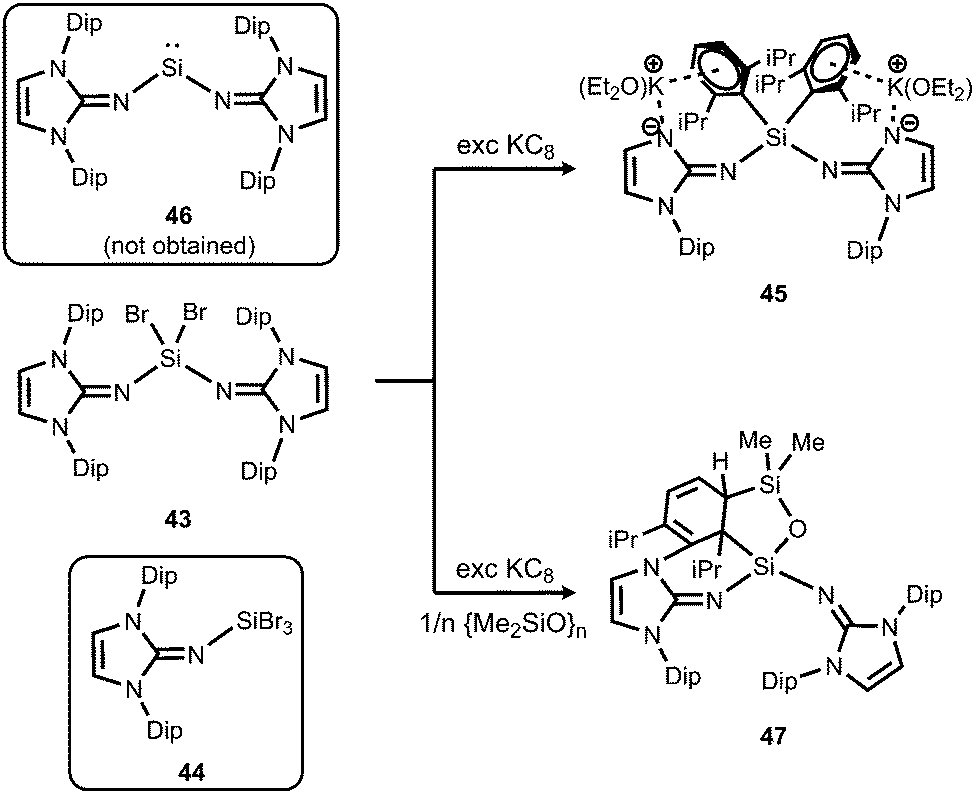
In order to exploit the strongly electron-donating properties of an N-heterocyclic iminato ligand for tuning the reactivity of low-valent silicon species Rivard and coworkers attempted the synthesis of a hypothetical bisiminosilylene. Access to the bisiminodibromosilane precursor 43 is granted by conversion of LDipNSiMe3 (15) with SiBr4 in appropriate stoichiometry (Scheme 10).24 The monoimino derivative LDipNSiBr3 (44) is synthesized in a similar fashion (Scheme 10).24 The reductive dehalogenation of 43 with KC8 (excess) yielded the potassium salt 45 instead of the desired silylene (LDipN)2Si (46, Scheme 10).24 This product (45) was presumed to result from an intermediate potassium silanide via migration of a Dip group. The formation of minor amounts of the siloxane 47 was reasoned by the presence of silicon grease in the reaction mixture (Scheme 10).
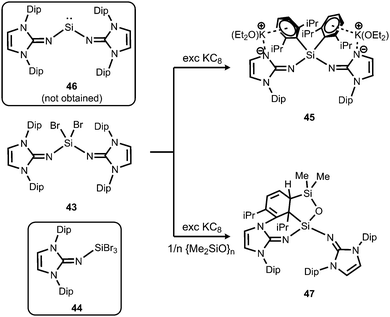 |
| | Scheme 10 Reduction of dibromosilane 43 with KC8 to form the unexpected anionic compound 45 instead of intended 46. The siloxane 47, as well as the tribromide 44. | |
Germanium complexes
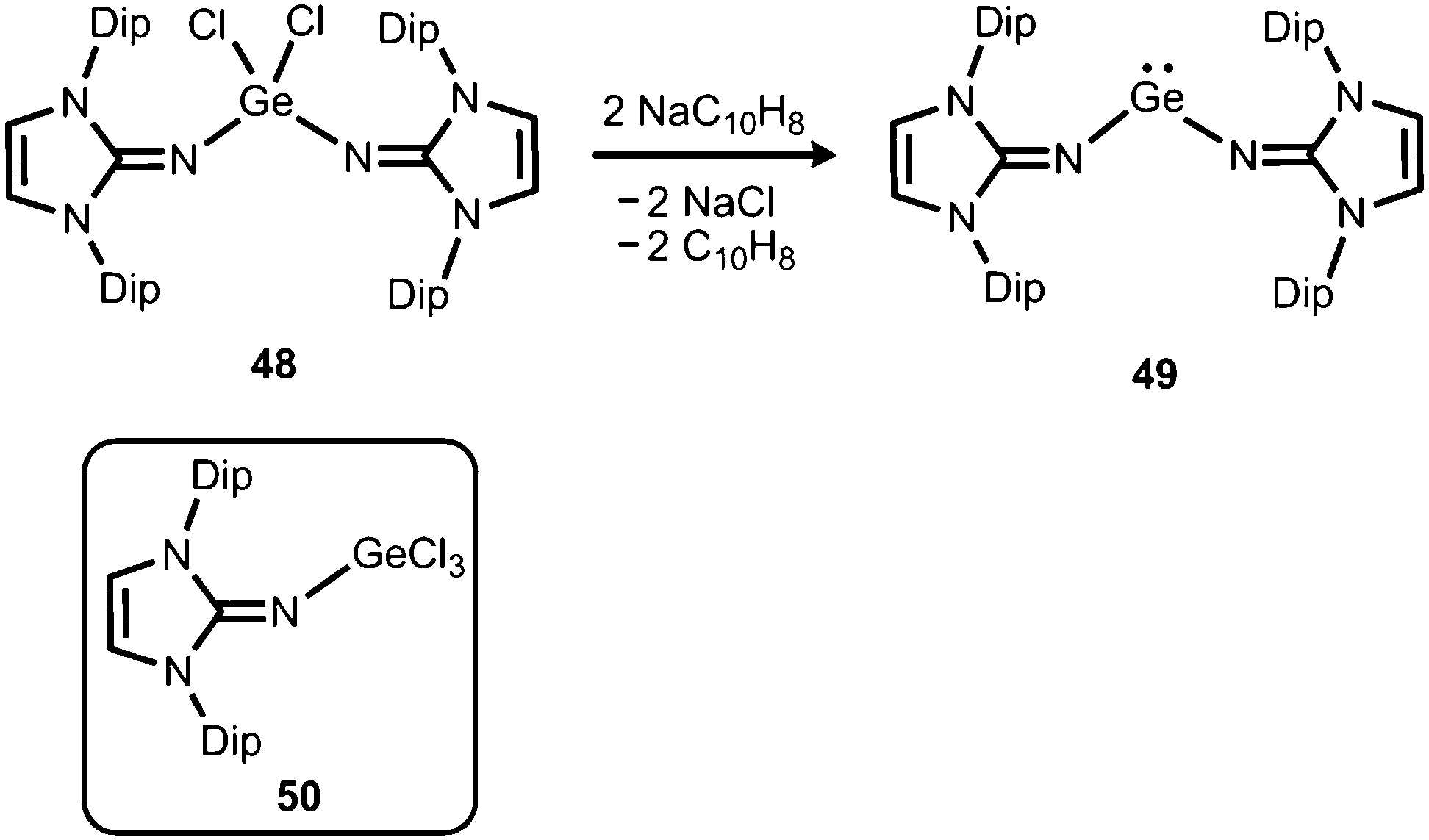
The reductive dehalogenation of the bulky bisiminodichlorogermane 48 with sodium naphthalenide affords the bisiminogermylene 49 as reported by Rivard and coworkers (Scheme 11).24 Notably, the related monoiminotrichlorogermane 50 was also described (Scheme 11). In the solid state the germanium(II) compound (49) exhibits longer Ge–N distances (both: 1.8194(15) Å) and a decreased N–Ge–N bond angle (99.48(10)°) with respect to its halogenated precursor 48 (Ge–N = 1.7528(14) Å, 1.7582(14) Å; N–Ge–N = 106.33(7)°; Fig. 6). These structural features were interpreted by the authors in terms of a higher p-character of the Ge–N bond in 49 as compared to 48. Theoretical calculations indicated a low singlet–triplet gap of 45.8 kcal mol−1 for the bisiminogermylene 49, a value which is similar to that of the elusive bisiminosilylene 46 (44.5 kcal mol−1). This computational study suggests high inclination for the sterically hindered metal centre to insert into element–element bonds of small substrate molecules. However, upon conversion of 49 with dihydrogen Rivard and coworkers observed the formation of LDipNH as the only soluble species instead of the expected (LDipN)2GeH2.24 This may account for the pronounced proton affinity of the imidazolin-2-imino group. Interestingly, the bisiminogermane is also not formed in the reaction of 48 with hydride transfer reagents such as K[BHsBu3] or potassium hydride.24
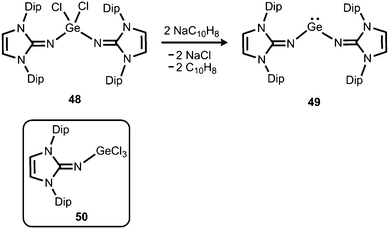 |
| | Scheme 11 Reductive dehalogenation of the dichlorogermane 48 to the bisiminogermylene 49. The trichlorogermane 50. | |
 |
| | Fig. 6 Ellipsoid plot (30% level) of the bisiminogermylene 49 (hydrogen atoms and isopropyl groups have been omitted). | |
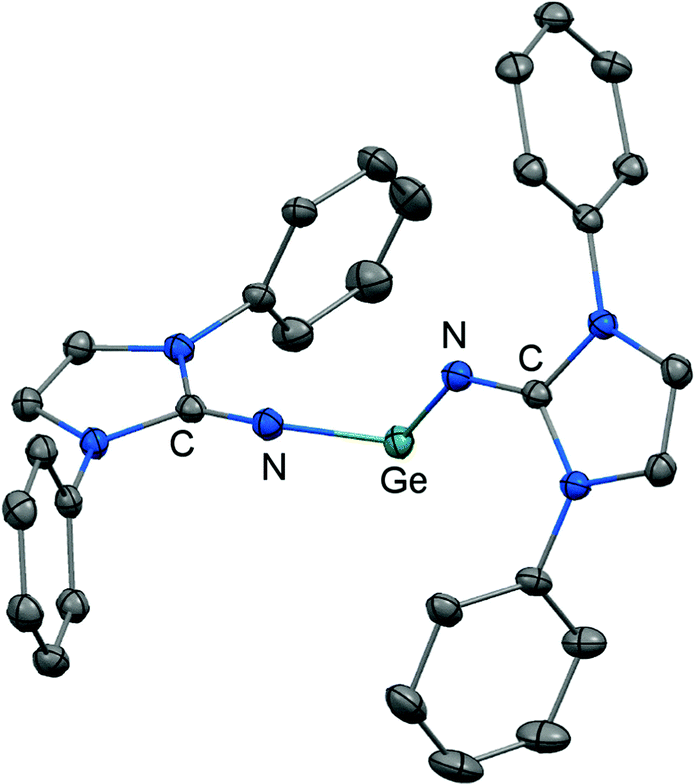
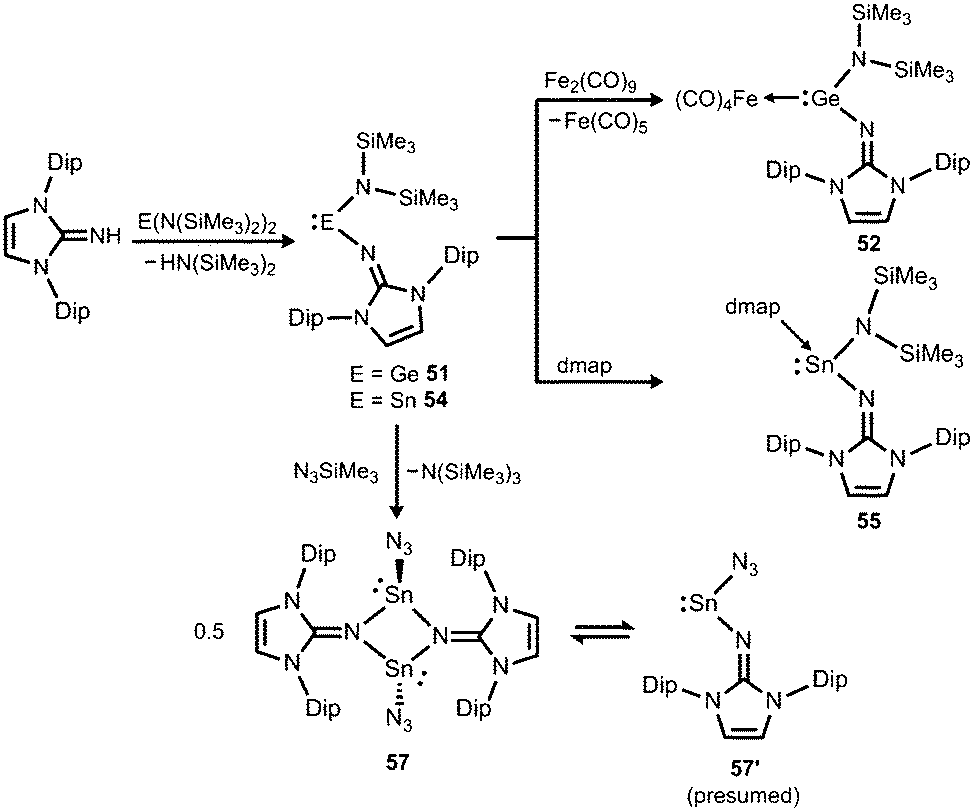
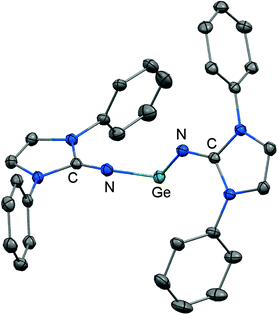
Another synthetic approach to germanium(II) complexes of the imidazolin-2-iminato ligand uses the Lappert's germylene ((Me3Si)2N)2Ge as a low-valent metal source. Its conversion with one equiv. of LDipNH at 50 °C furnishes the amino(imino)germylene 51 in the form of a viscous liquid (Scheme 12).25 It acts as a ligand towards iron carbonyls as demonstrated by the formation of the germylene complex 52 after reaction of 51 with diironnonacarbonyl (Scheme 12).25a The XRD analysis of 52 reveals a Ge–Nimino distance of 1.755(2) Å which is significantly shorter than the Ge–Namino bond length of 1.839(2) Å and also with respect to the free bisiminogermylene 49 (vide supra). Considering the WBIs of the Ge–N bonds in 52 (Ge–Nimino = 0.86, Ge–Namino = 0.60) it is reasonable to assume that the bulky imidazolin-2-iminato ligand bonds stronger to the germanium(II) centre than the bis(trimethylsilyl)amino group, presumably as a result of the iminato ligand's higher electron-donating character.
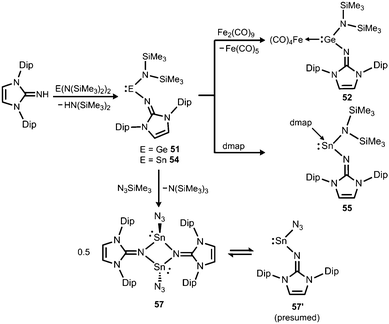 |
| | Scheme 12 Preparation of amino(imino)metallylenes 51 and 54 and the iron carbonyl 52, as well as the dmap adduct 55 (dmap = 4-dimethylamino-pyridine). Conversion of 54 to dimeric stannylene azide 57. | |
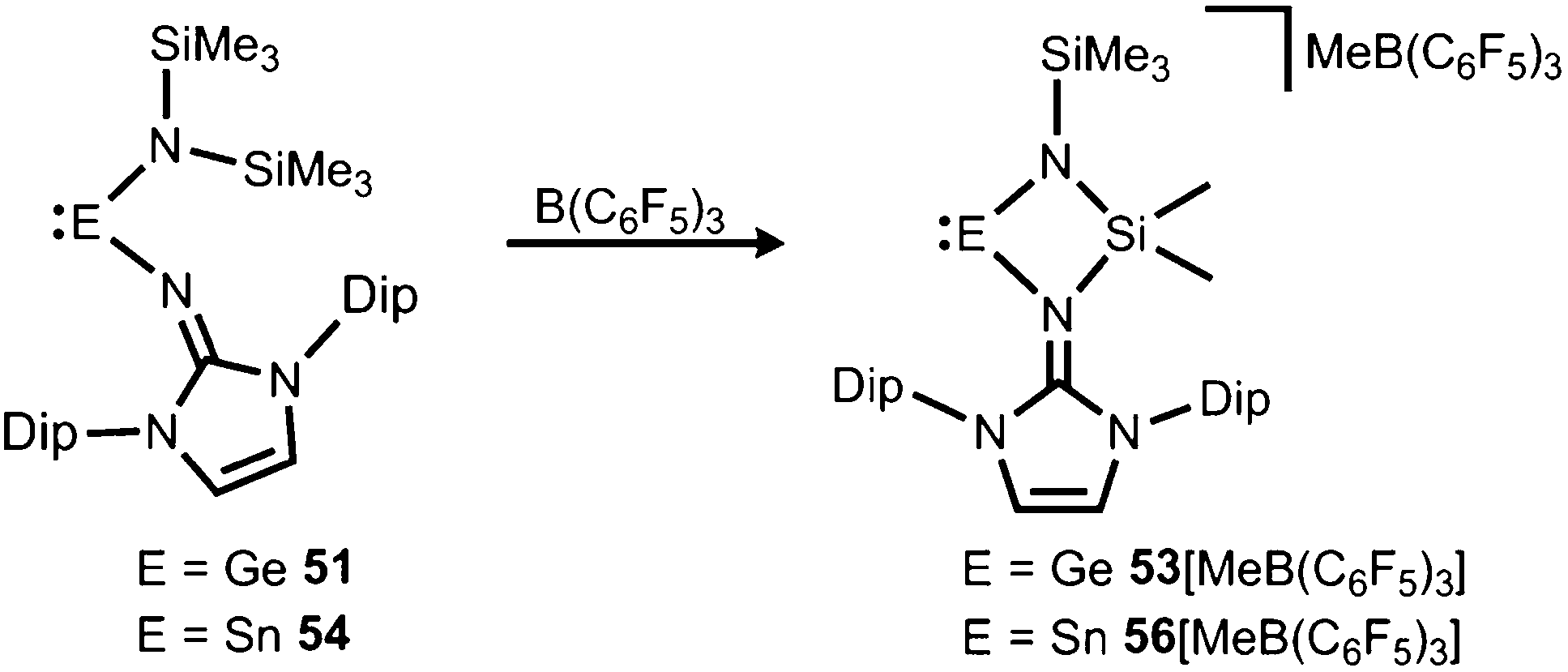
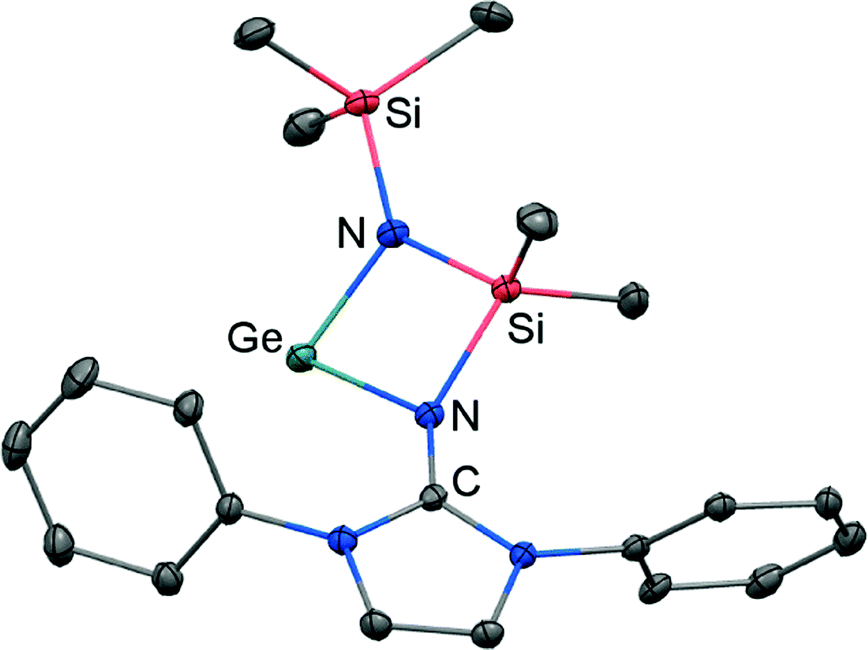
If treated with tris(pentafluorophenyl)borane compound 51 undergoes a methyl-abstraction and ring-closing reaction to form the cyclic germyliumylidene 53[MeB(C6F5)3] as an example for a cationic complex of germanium(II) (Scheme 13, Fig. 7).25a The bonding situation in 53+ is found to be suitably described as an amino-bonded cationic germanium(II) atom that is stabilized via dative bond type interaction with an intramolecularly tethered imino group. This is indicated by a weaker interaction between the Ge(II) centre and the Nimino atom (Ge–Nimino = 1.9694(14) Å, WBIGeN = 0.48) and a stronger bond between the Ge(II) centre and the Namino atom (Ge–Namino = 1.8437(15) Å, WBIGeN = 0.73). This bonding situation between the metal centre and the N atoms appears to be in contrast to the situation in the uncharged congeners 51 and 52 (vide supra). Moreover, the C–Nimino distance of 1.335(2) Å in 53+ is greater in comparison to 52 (C–Nimino = 1.296(3) Å) and hints towards delocalization of cationic charge into the imidazoline ring system similar to the observations reported for 5, as well as 14+. This accounts for the pronounced ability of the imidazolin-2-iminato ligand to stabilize cationic species which was verified yet again by a very recent report on the isolation of bifunctional germylene–germyliumylidenes.25b
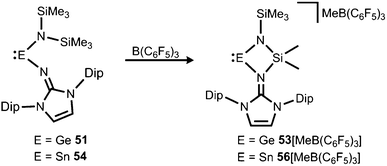 |
| | Scheme 13 Synthesis of four-membered metallyliumylidenes 53+ and 56+ by methyl-abstraction from the amino(imino)metallylenes 51 and 54. | |
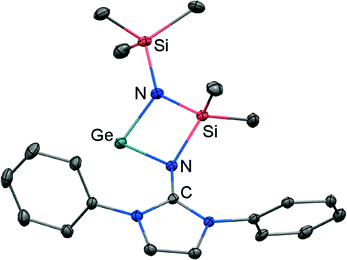 |
| | Fig. 7 Ellipsoid plot (30% level) of the germyliumylidene 53+ (hydrogen atoms and isopropyl groups have been omitted). | |
Tin complexes
In 2015 the formation of the amino(imino)stannylene 54 was reported that proceeds in a similar fashion to the lighter congener 51via reaction of ((Me3Si)2N)2Sn with LDipNH at 60 °C (Scheme 12).26 Notably, the 119Sn NMR chemical shift of −208 ppm for 54 (C6D6) is considerably shifted to higher field with respect to the precursor (767 ppm, C6D6) which was accredited to an aggregated species in solution with a higher coordinate tin(II) centre. The compound (54) was obtained as a pale red powder and reacted with 4-dimethylamino-pyridine (dmap) to give the solid tin(II) adduct 55 that exhibited a resonance at −3 ppm in the 119Sn NMR spectroscopic analysis (Scheme 12).26 The XRD study of 55 shows a shorter Sn–Nimino contact (2.0588(13) Å) and a longer Sn–Namino distance (2.1647(12) Å). This was interpreted in terms of a stronger bond of the metal centre to the iminato ligand and a weaker interaction with the amino group as described for the germanium congener 52, as well (vide supra).25,26 The reaction of 54 with tris(pentafluorophenyl)borane affords the stannyliumylidene salt 56[MeB(C6F5)3] in a methyl abstraction and ring closing reaction similar to the process that afforded the germanium analogue 53[MeB(C6F5)3] (Scheme 13, vide supra). The bonding situations in 56+ and 53+ resemble, that is, an amino bonded metallyliumylidene cation which is stabilized by a dative bond to the imino group. Accordingly, the Sn–Nimino distance of 2.197(2) Å in 56+ is longer than the Sn–Namino bond length of 2.062(2) Å which is an observation that is in contrast to that reported for the uncharged congener 55 that possesses a shorter Sn–Nimino contact. The isolation of 56[MeB(C6F5)3] is another example for the high potential of N-heterocyclic imino systems to stabilize cationic species. Interestingly, the amino(imino)stannylene 54 converts with azido trimethylsilane to the dimeric iminostannylene azide 57 (Scheme 12).26 Apparently, an expected stannaimine of the type (LDipN)((Me3Si)2N)Sn(NSiMe3) is not formed but ligand exchange results in the liberation of (Me3Si)3N from the system. Interestingly, in solution (THF-d8) dimeric 57 (δ(119Sn) = −285 ppm) exists in equilibrium with a monomeric species (δ(119Sn) = 39 ppm; 57′, Scheme 12).
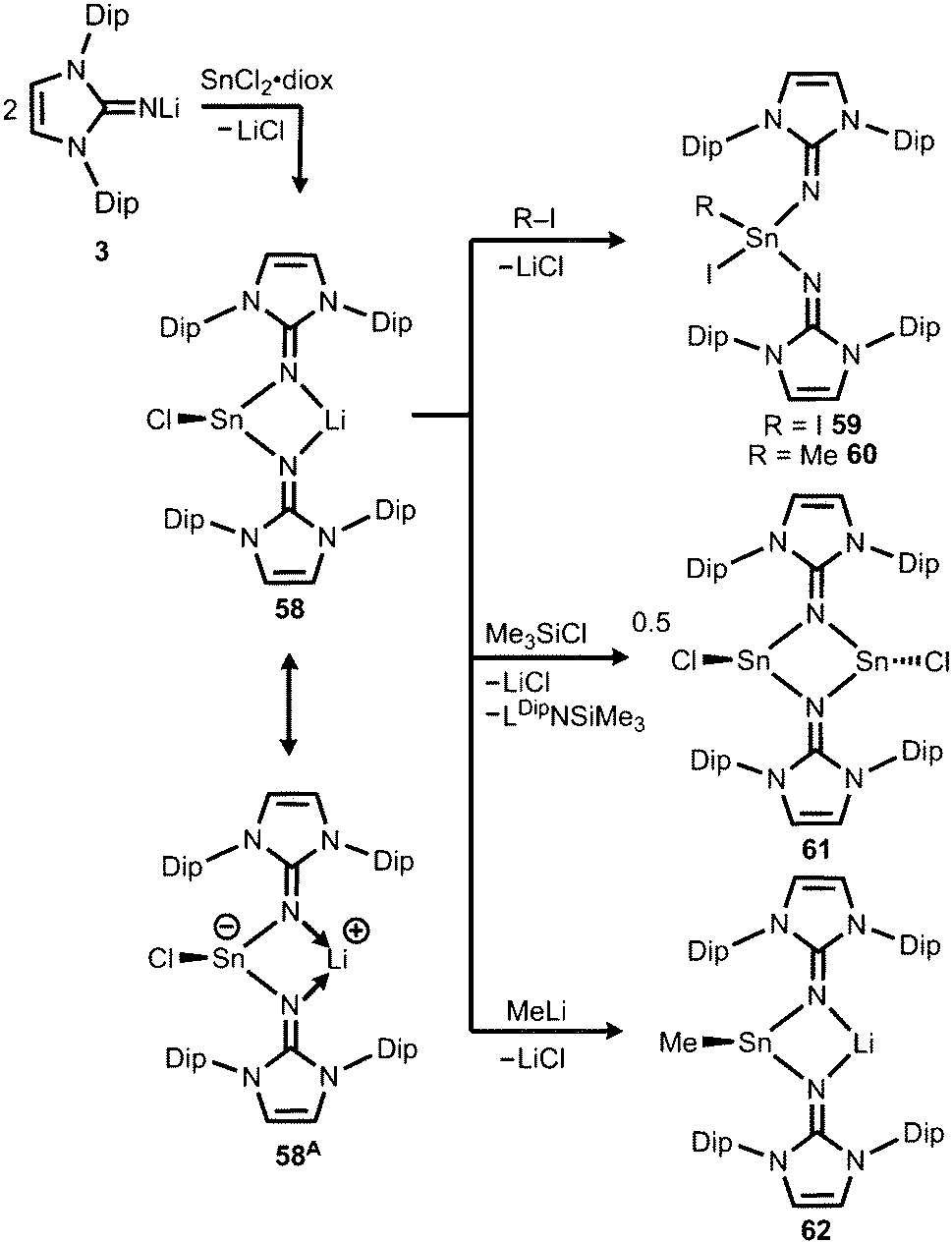
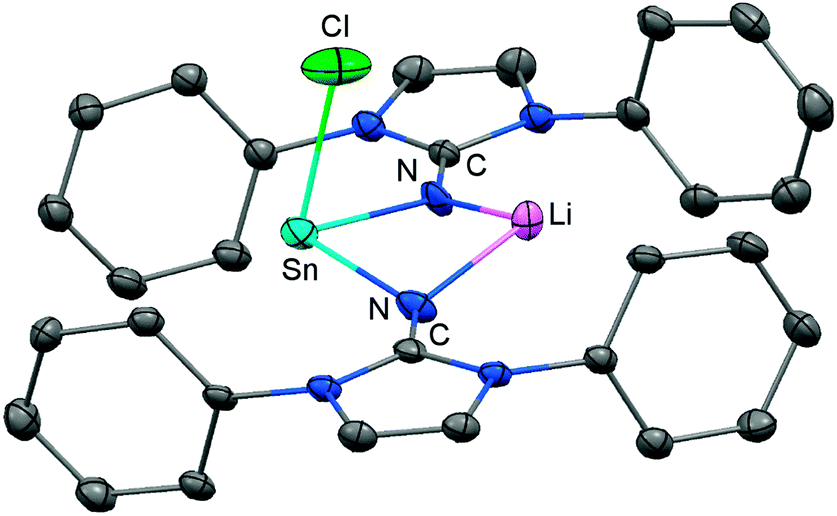
Very recently, our group described the bisiminochlorostannate 58 which forms by the reaction of LDipNLi with half an equivalent of SnCl2·1,4-dioxane (Scheme 14).27 The 119Sn NMR spectrum of 58 (thf-d8) shows a resonance at −18 ppm that is shifted to a lower field in comparison to common monomeric trigonal pyramidal-coordinate 1,3-diketiminato tin(II) chlorides (−118 ppm to −337 ppm).28 The XRD analysis of 58 reveals a butterfly-shaped four-membered SnN2Li stannacycle with no bonding interaction between the Sn atom and the Li atom (Fig. 8). The Sn–Nimino distances of 2.143(5) Å and 2.179(4) Å are longer compared with that of the dmap adduct 55. The Li–Nimino bond lengths of 1.946(9) Å and 2.004(9) Å are comparable to those reported for the imino lithium dimer [LDipNLi]2·toluene (3·toluene).8
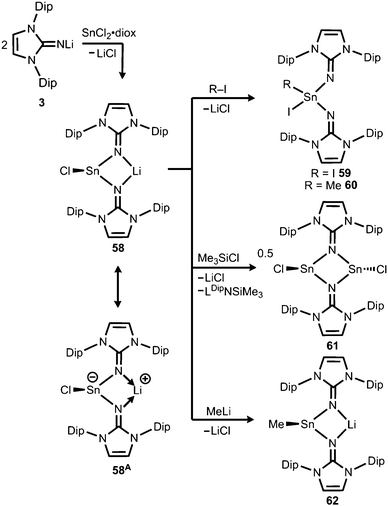 |
| | Scheme 14 Synthesis of bisiminostannylenoid 58 and reactivity with I2, MeI, ClSiMe3 and MeLi to products 59–62 (diox = 1,4-dioxane). | |
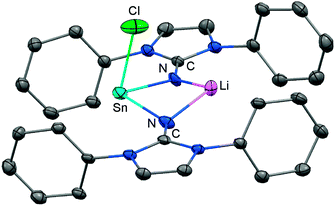 |
| | Fig. 8 Ellipsoid plot (30% level) of the stannylenoid 58 (hydrogen atoms and isopropyl groups have been omitted). | |
Compound 58 reacts with electrophiles such as I2 and MeI to form the oxidative addition products 59 and 60 which demonstrates its stannylene character. For the bulky substrate Me3SiCl the analogous formation of the stannane (Me3Si)ClSn(LDip)2 is suppressed and LDipSiMe3 is formed along with the dimeric chlorostannylene [LDipSnCl]2 (61). The bisiminochlorostannate (58) may also act as an electrophile. Its conversion with MeLi leads to the formation of Me-substituted stannate 62 that exhibited a planar four-membered LiN2Sn ring in the XRD analysis. Theoretical calculations on 58 revealed the high single-bond character of the Sn–Nimino interactions as concluded from the comparison of the WBI values of 58 (0.43 and 0.44) with the ones in 61 (0.24 and 0.24) which mark considerable dative-bond character for the latter. Moreover, the computational study of the natural population analysis (NPA) charge distribution in 58 and 61 shows that the Sn atom in 58 is less positively polarized (+1.22) than that in 61 (+1.42). These theoretical results account for the stannyl anion character of 58 as illustrated by the resonance structure 58A (Scheme 14). However, the ambiphilic reactivity of the tin(II) centre in 58, that is, it functions as a nucleophile in the synthesis of 59 and 60 and as an electrophile in the conversion to 62, has to be pointed out. It allows for the conclusion that the compound (58) possesses high stannylenoid character and thus represents a heavier congener of carbenoids.
Miscellaneous: survey of carbon chemistry
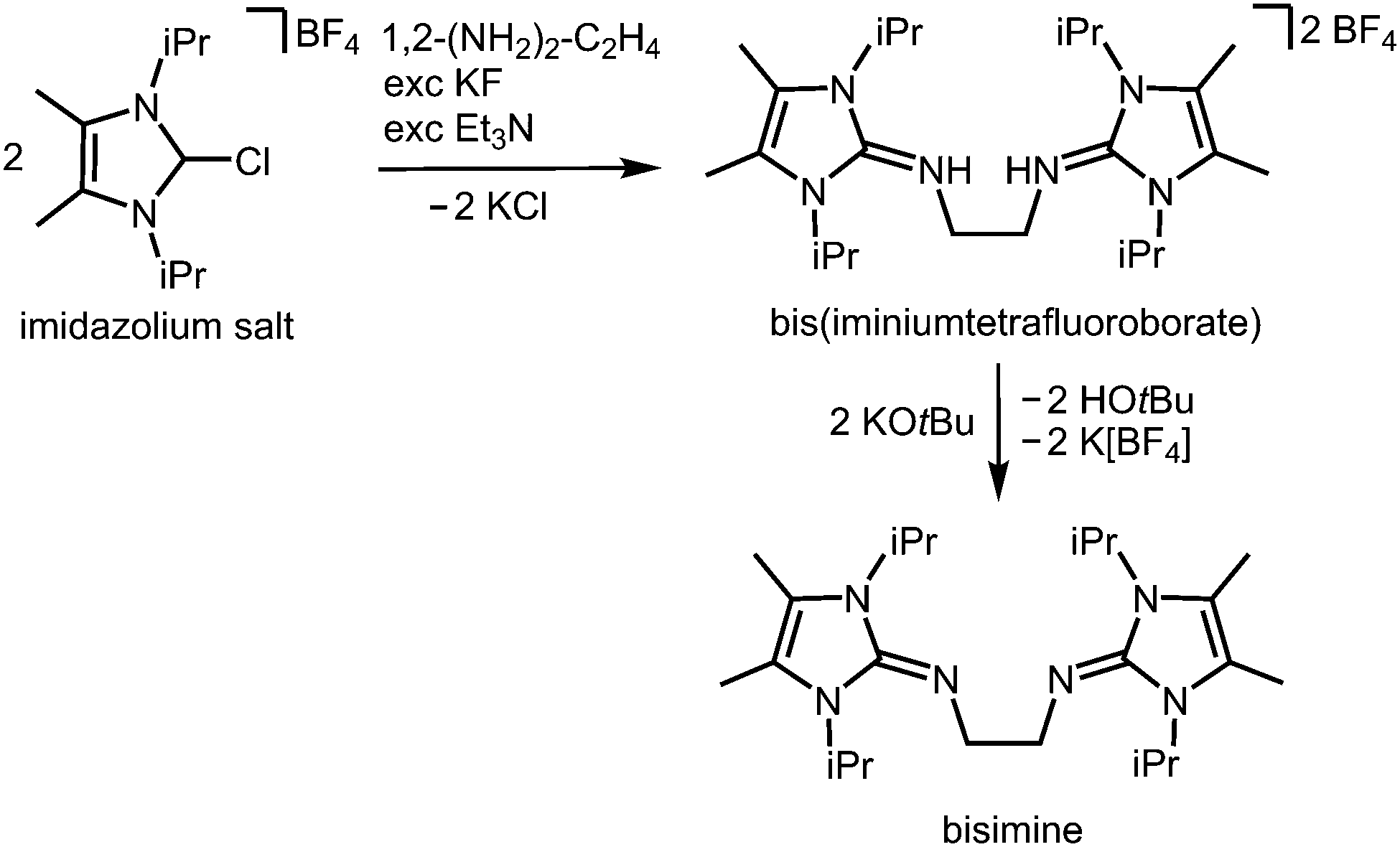
In the field of coordination chemistry the tethering of the exocyclic imino-nitrogen atom of an N-heterocyclic imino group to a carbon atom mostly serves the creation of tailor-made ligand systems. These synthetic methods have been reviewed elsewhere.2,3 They can be complemented by the report of Tamm and coworkers in 2014 on the modified synthesis of the bisimine 1,2-(LiPr2Me2N)2-C2H4 (LiPr2Me2 = 1,3-diisopropyl-4,5-dimethyl-imidazolin-2-ylidene, Scheme 15), a chelate-fashioned ligand system which had been described before in the year 2007.29,30
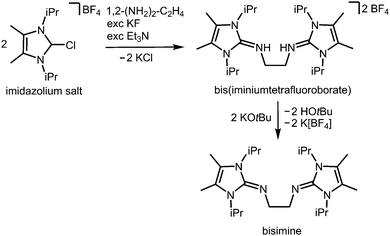 |
| | Scheme 15 Synthesis of the bisimine compound 1,2-(LiPr2Me2N)2-C2H4 (LiPr2Me2 = 1,3-diisopropyl-4,5-dimethyl-imidazolin-2-ylidene) from an imidazolium salt precursor. | |
Group 15 element complexes
Background
For the pnictogen family compounds of phosphorus with the imidazolidin-2-imino group (saturated in the ligand backbone) dominate the field. As outlined in the following section this ligand system is often implemented for the stabilization of phosphorus-centred radicals and suits the requirements for the isolation of cationic species similar to the strongly related imidazolin-2-imino group (unsaturated in the ligand backbone). Most notably, pioneering work on imidazolin-2-imino-substituted phosphanes was reported by Kuhn and coworkers in 1996 and 1998.31 Also, compounds with the imidazolin-2-imino structural motif adjacent to a nitrogen atom are abundant. Examples include common types of organic compounds such as azines of cyclic ureas, cyclic bisguanidines, as well as triazenes and diazotates with the corresponding C3N2 five-membered ring backbone. These will be discussed in the miscellaneous section of this review. Interestingly, the respective chemistry of the heavier pnictogens remains largely unexplored to date.
Phosphorus compounds
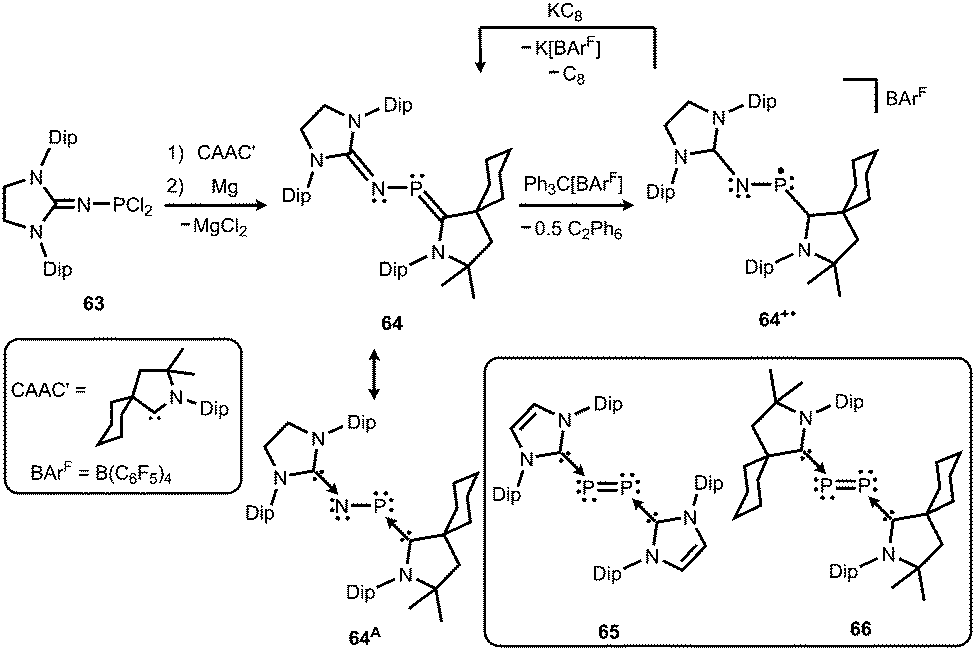
Phosphorus mononitrides and phosphinonitrenes have developed into an established subgenre of the iminato ligand-stabilized phosphorus chemistry and respective research was sparked by Bertrand and coworkers in 2010. They reported the use of the imidazolidin-2-imino lithium reagent (H2)LDipNLi for the synthesis of the phosphorus dichloride 63 which undergoes reductive dehalogenation with magnesium in the presence of a cyclic alkyl(amino) carbene (CAAC) to afford 64 (Scheme 16, Fig. 9).32 The authors demonstrated that this compound (64) can be regarded as a molecular congener of phosphorus mononitride stabilized by a CAAC as a ligand to the phosphorus atom and an NHC at the P-bonded nitrogen atom (64A, Scheme 16). This resonance structure (64A) is reminiscent of the diphosphorus compounds 65 and 66 that bear two NHC ligands or two CAACs, respectively (Scheme 16).33,34
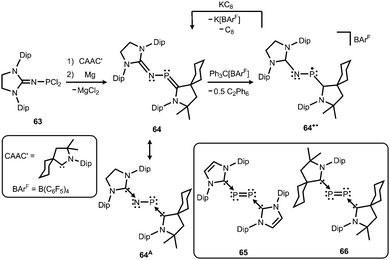 |
| | Scheme 16 Synthesis of phosphazabutadiene 64 and its conversion to the radical cation 64+˙. Phosphorus mononitride formulation 64A. NHC- and CAAC-stabilized diphosphorus complexes 65 and 66. | |
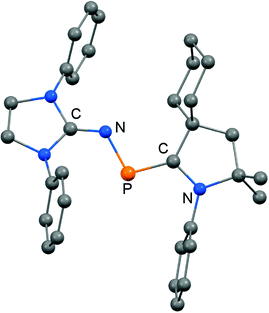 |
| | Fig. 9 Ball and stick representation of the phosphazabutadiene 64 as derived from XRD analysis (hydrogen atoms and isopropyl groups have been omitted). | |
The formulation 64 represents the phosphazabutadiene character of the compound. In the 31P NMR spectroscopic analysis the chemical shift of 64 is observed at 134 ppm which is shifted to a lower field with respect to the heavier congeners 65 and 66 (range: from 59 ppm to −74 ppm). As derived from XRD analysis the geometry of 64 (Fig. 9) was described as trans-bent with a short P–CCAAC bond (1.719(2) Å), as well as an N–CNHC distance (1.282(3) Å) that is in the range of C–N bond lengths of imino groups (vide supra). The P–N distance of 1.7085(16) Å is similar to that of typical P–N single bonds. Oxidation of 64 with Ph3C[B(C6F5)4] (trityl tetrakis(pentafluorophenyl)borate) afforded the radical cation 64+˙ (Scheme 16).32 This process is reversible as was shown by the regeneration of uncharged 64via reduction with potassium graphite (KC8, Scheme 16). In the theoretical analysis of 64 and 64+˙ the shapes of the HOMO and the SOMO (singly occupied molecular orbital), respectively, are very similar. They majorly comprise a π* orbital of the PN group that shows bonding interaction with a p-type orbital at the carbenic atom of the NHC, as well as the CAAC ligand.32 The EPR study of 64+˙ in frozen fluorobenzene at 100 K revealed g-tensors of gx = 2.0052, gy = 2.0087 and gz = 2.0028 which are comparable to the respective values in 65+˙ and 66+˙.32–34
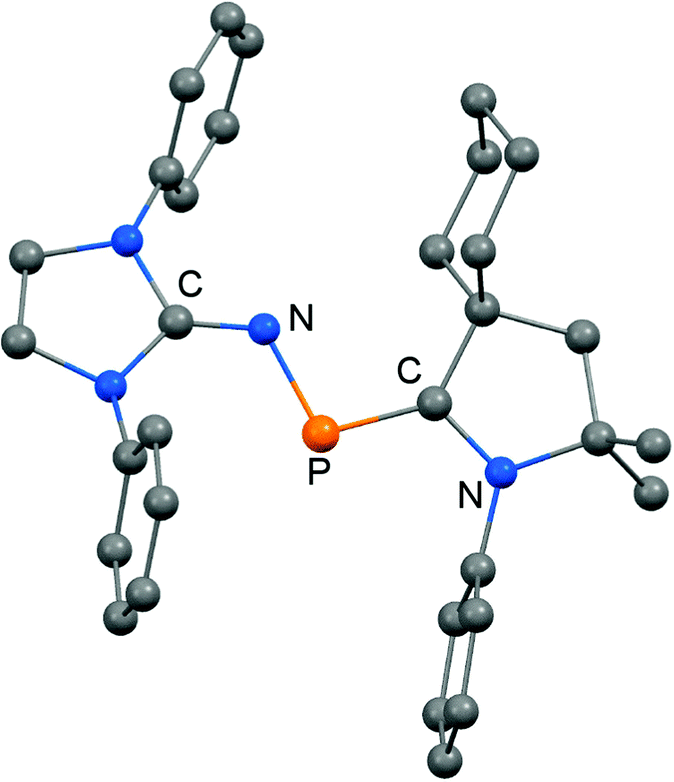
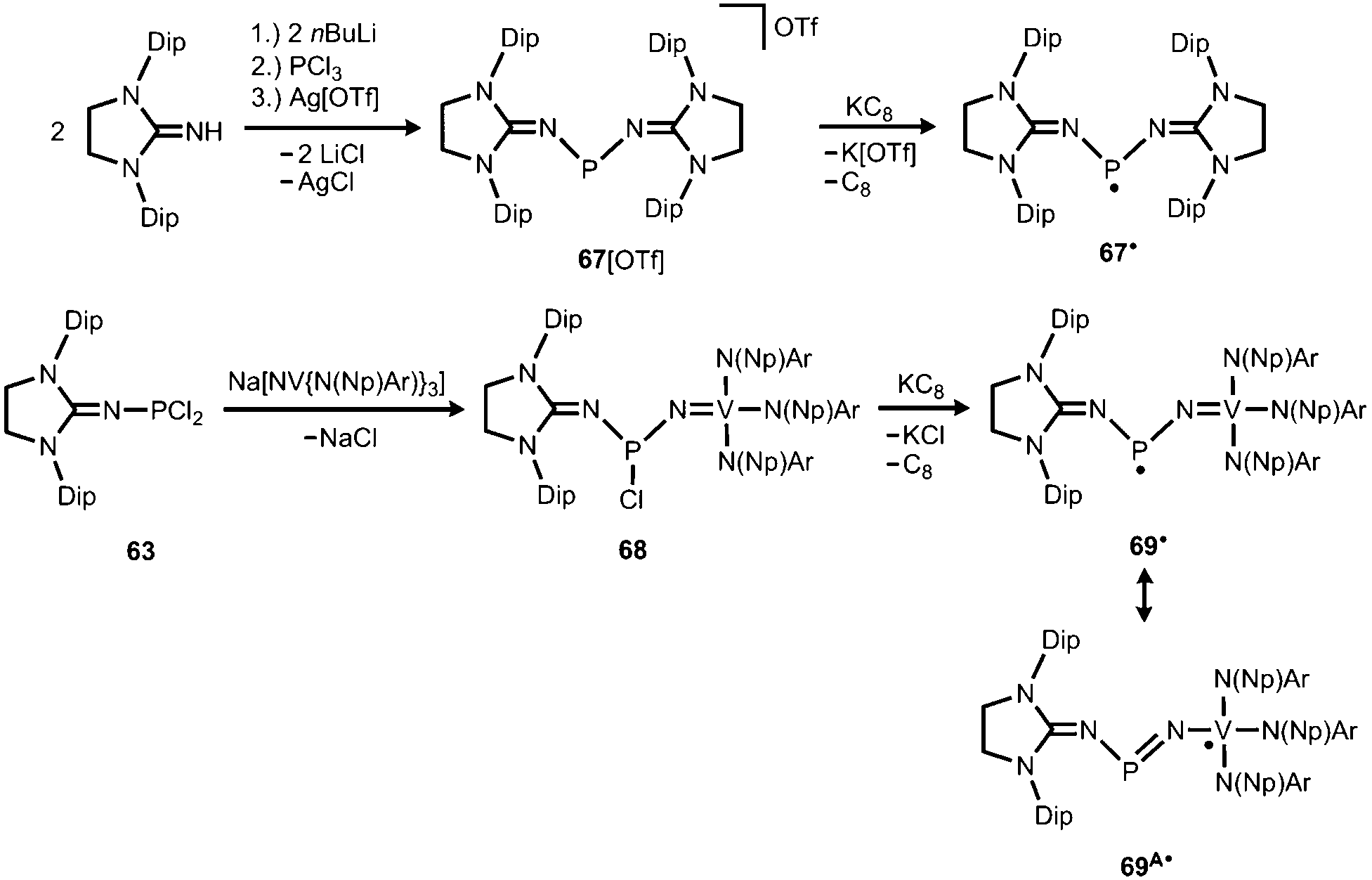
The scope of applications of N-heterocyclic imines in phosphorus chemistry was extended in 2011 when Bertrand and coworkers reported the reduction of the bisimino compound 67[OTf] (Tf = triflyl) with KC8 to the uncharged phosphinyl radical 67˙ (Scheme 17, Fig. 10).35 Notably, the synthesis of 67[OTf] proceeds via the chloride salt 67[Cl] that could not be isolated in analytically pure form at that time but is purified in the course of the anion exchange (chloride vs. triflate, Scheme 17).35 The paramagnetic nature of 67˙ was verified by its EPR study at 100 K in frozen THF from which the g-tensors gx = 2.0074, gy = 2.0062 and gz = 2.0024 were derived. A comparison with hyperfine coupling constants of atomic phosphorus revealed that an unpaired electron is primarily localized on the 3p(P) orbital (62%) with a small contribution of the 3s(P) orbital (2%).
 |
| | Scheme 17 Conversion of the bisiminophosphonium salt 67[OTf] to the phosphinyl radical 67˙. Synthesis of the imino(vanadyl)phosphinyl radical 69˙via chlorophosphine 68. Vanadium-centre formulation 69A˙ (Np = neopentyl). | |
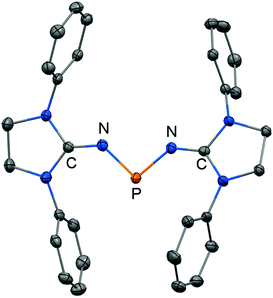 |
| | Fig. 10 Ellipsoid plot (30% level) of the bisiminophosphinyl radical 67˙ (hydrogen atoms and isopropyl groups have been omitted). | |
Bertrand and coworkers extended their investigation of imino-substituted phosphinyl radicals: the phosphorus dichloride 63 served as a precursor to the nitridovanadium-functionalized phosphorus monochloride 68 (Scheme 17).35 In an analogous fashion as for 67˙ the reduction of 68 with KC8 furnished the phosphinyl radical 69˙ (Scheme 17).35 From the EPR study of 69˙g-tensors of gx = 1.9726, gy = 2.0048 and gz = 1.9583 were determined. Taking into account the hyperfine coupling constants to 51V, as well as 31P it was concluded that the spin density in 69˙ mainly resides on the vanadium centre (67%) and is only localized to a minor degree on the phosphorus atom and the NHC moiety. In contrast, the spin density of the bisimino derivative 67˙ was found to reside with 62% in the 3p(P) orbital and with 2% in the 3s(P) orbital. In line with the structural and theoretical analysis the authors concluded that the bisimino radical 67˙ is a phosphorus centred radical with little spin delocalization over the iminato ligands. The nitridovanadium congener 69˙, however, is best represented by the canonical structure 69A˙, that is a vanadium(IV) complex with a phosphinimide ligand.
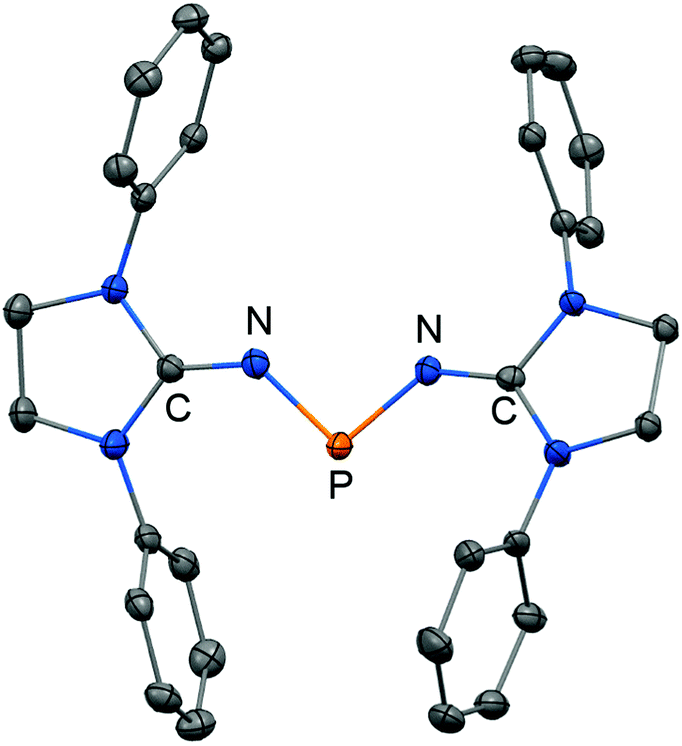
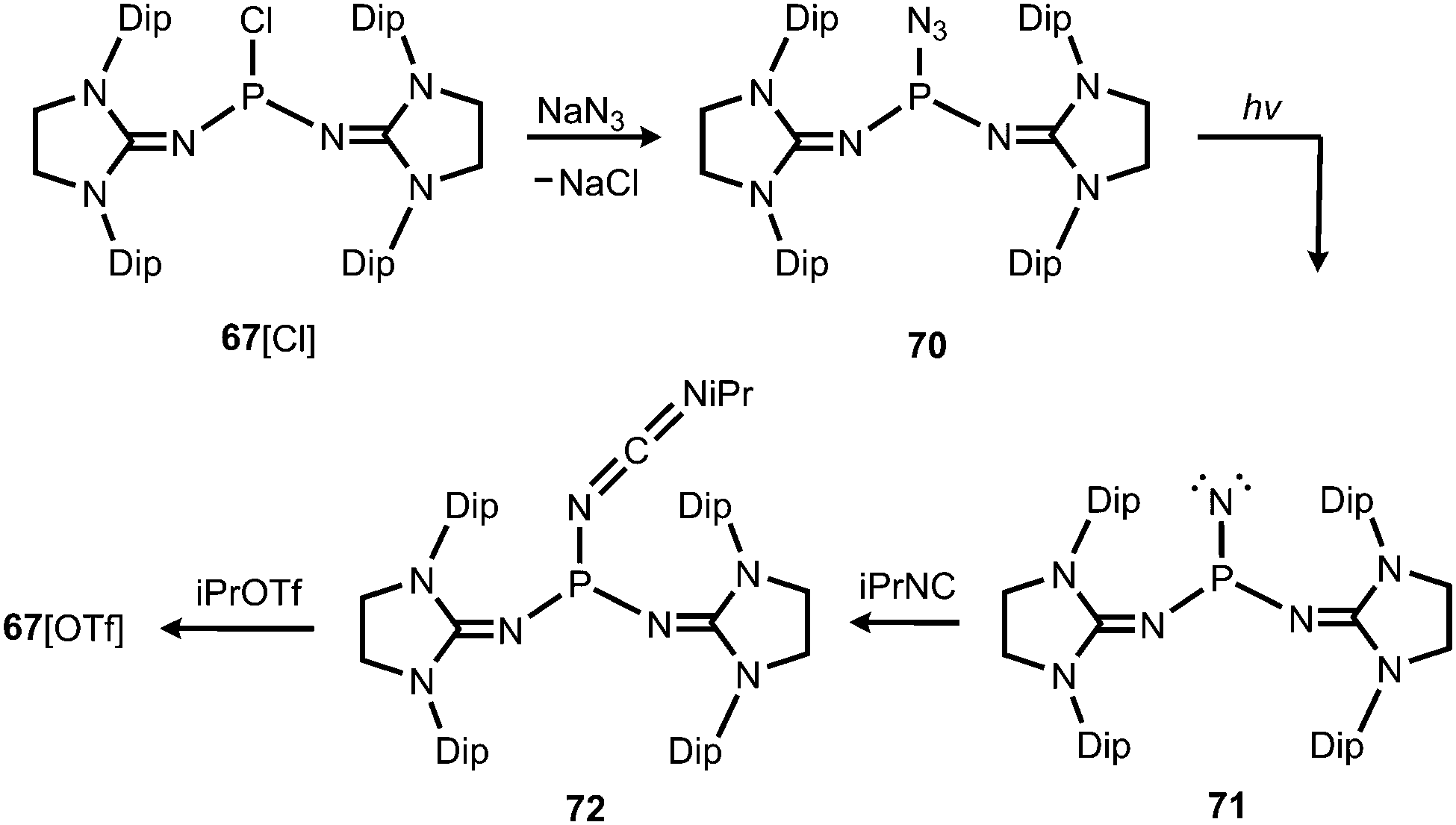
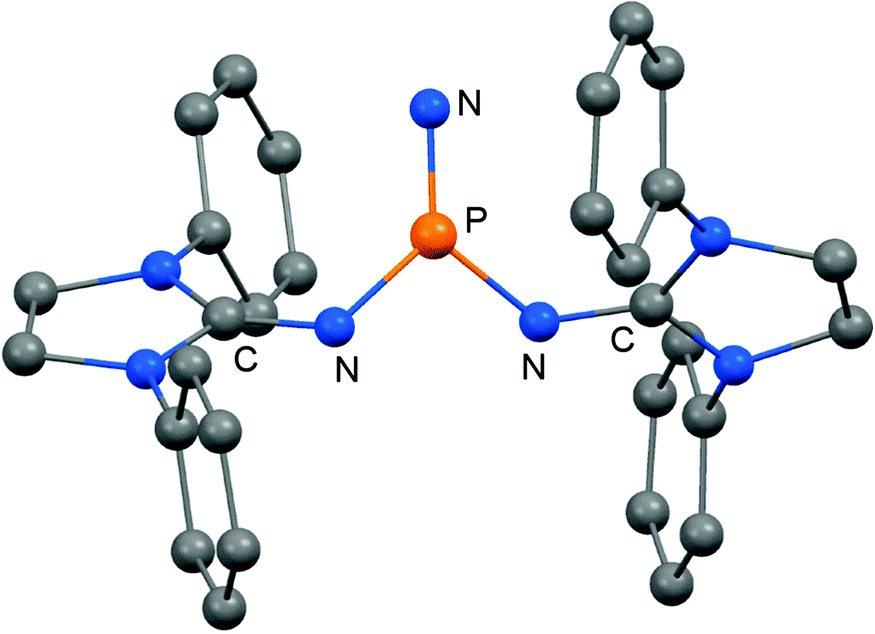
In 2012 Betrand and coworkers reported the remarkable transformation of the azido bisiminophosphane 70 to the phosphinonitrene species 71via irradiation at 254 nm (Scheme 18).36 As a starting material the bisiminophosphenium salt 67[Cl] was used that had also been implemented in the synthesis of the phosphinyl radical 67˙ (Scheme 17).35,36 The theoretical analysis of the nitrene (71) suggested that the back donation of a nitrogen lone-pair into accessible σ* orbitals at the phosphorus atom significantly contributes to the thermodynamic stability of the compound. The phosphorus atom in 71 is the centre of a trigonal-plane and the sum of the angles around the P atom amounts to 360° (Fig. 11). Notably, the P–Nnitrene bond length of 1.457(8) Å in 71 is significantly shorter than the P–Nimino distances (1.618(8) Å, 1.629(8) Å), as well as the P–Nazido distance of 1.895(11) Å in the precursor (70). This is in good agreement with the upfield shift in the 31P NMR spectrum of 71 (8 ppm) in comparison to 70 (111 ppm) which indicates the multiple-bond character of the PN functionality. The phosphinonitrene (71) reacts with isopropyl isonitrile (iPrNC) to yield the carbodiimide 72 that was not structurally characterized (Scheme 18).36 In consequence, the created NCNiPr group can be abstracted from the phosphorus atom by implementing isopropyltriflate as an alkylating agent. In the outcome the starting material 67+ is generated in the form of the triflate salt (67[OTf], Scheme 18).
 |
| | Scheme 18 Synthesis of phosphinonitrene 71 by photoirradiation of azidophosphane 70 and conversion to carbodiimide 72. | |
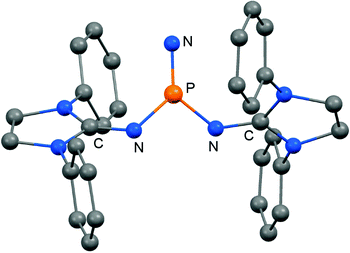 |
| | Fig. 11 Ball and stick representation of the phosphinonitrene 71 as derived from XRD analysis (hydrogen atoms and isopropyl groups have been omitted). | |
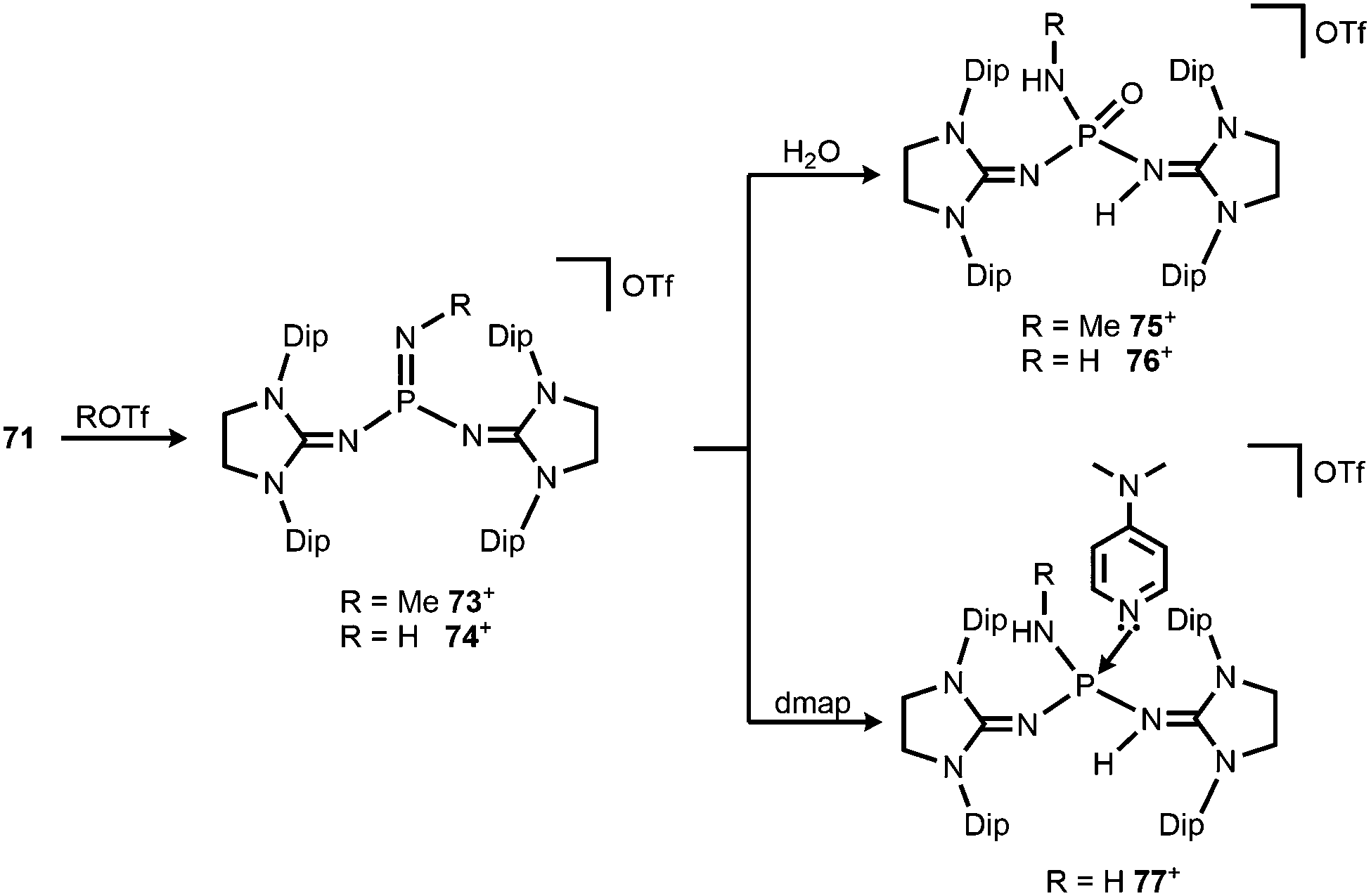
Bertrand and coworkers described the transformation of the phosphinonitrene 71 to iminophosphonium triflates in 2013.37 The methylation or protonation of 71 using methyltriflate or triflic acid, respectively, furnished 73[OTf] or 74[OTf] (Scheme 19). The PN bond length of the phosphoranimine functionality in 74+ amounts to 1.526(2) Å which is longer than the distance of these atoms in the precursor 71 (vide supra, note that the structural parameters of 73+ are not discussed due to poor data quality). On the other hand, the P–Nimino distances in 74+ are decreased to 1.553(2) Å and 1.559(2) Å in comparison to 71 (vide supra). This suggests a stronger interaction between the P-centre and the imino nitrogen atoms and accounts for the potential of N-heterocyclic imino systems in stabilizing cationic species. The addition of water to 73[OTf] or 74[OTf] yielded the cationic phosphine oxides 75[OTf] or 76[OTf], respectively (Scheme 19).37 The expected electrophilic properties of 74+ were verified by its conversion with dmap that generated the Lewis acid base adduct 77[OTf] (Scheme 19).37 Notably, the 31P NMR chemical shift of 77+ was observed at a significantly higher field (−1 ppm) compared to the precursor 74+ (73 ppm).
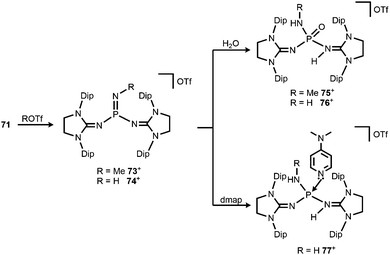 |
| | Scheme 19 Methylation and protonation of 71 to 73[OTf] and 74[OTf] and their reactivity towards H2O and dmap to produce (75–77)[OTf]. | |
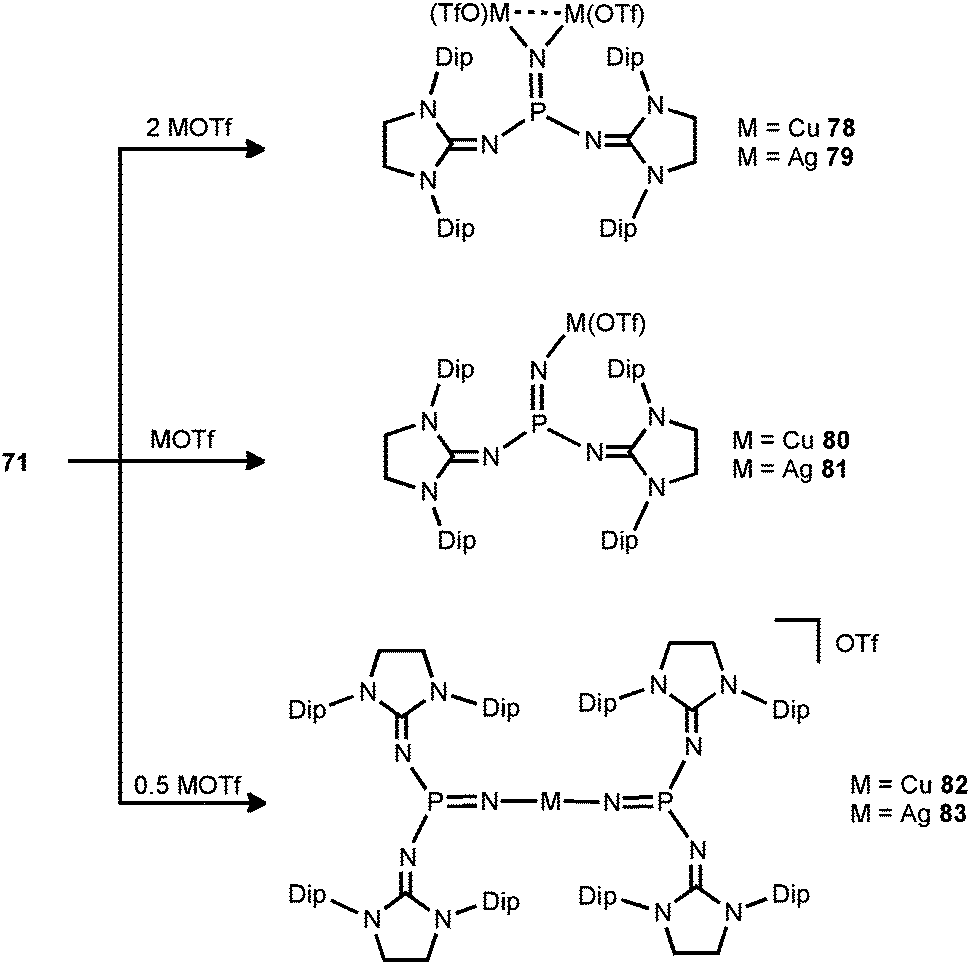
In 2014 the literature on the phosphinonitrene species (71) was enriched by Bertrand and coworkers with their investigation of coinage metal–nitrene compounds.38 By conversion of 71 with a corresponding equivalent of copper- or silver triflate (MOTf) the respective complexes with bridging or terminal phosphinonitrene ligands are generated (78–83, Scheme 20). The reaction of 71 with two equiv. of MOTf furnished the bimetallic complexes 78 or 79 with a bridging nitrenic atom. These showed similar structural features in the solid state, that is, a planar coordination environment of the phosphorus atom and significantly increased P–Nnitrenic bond lengths (1.510(5) Å for 78 and 1.528(3) Å for 79) with respect to 71. Furthermore, the P–Nimino bond lengths are shortened (1.573(3) Å for 78 and 1.561(3) Å for 79) and, vice versa, the C–Nimino distances are lengthened (range of 1.31–1.36 Å for 78 and 79) which indicates the stronger allocation of electron density from the imidazolidin-2-imino system to the phosphorus atom than in the precursor. After conversion of two equiv. of 71 with MOTf the linear complexes 82 and 83 with terminal bis(phosphinonitrene) ligands were obtained.38 Notably, the M–Nnitrenic bond lengths (1.801(2) Å, 1.807(3) Å for 82 and 2.017(3) Å, 2.029(4) Å for 83) in the linear complexes are decreased in comparison to the bimetallic systems (1.817(3) Å for 78 and 2.080(3) Å, 2.086(3) Å for 79). Interestingly, the conversion of the phosphinonitrene and MOTf in a one to one ratio afforded 80 and 81, respectively, as confirmed by NMR spectroscopic analysis. However, these compounds were found in a dynamic equilibrium with their bridging and terminal congeners (78, 82 for 80 and 79, 83 for 81).38
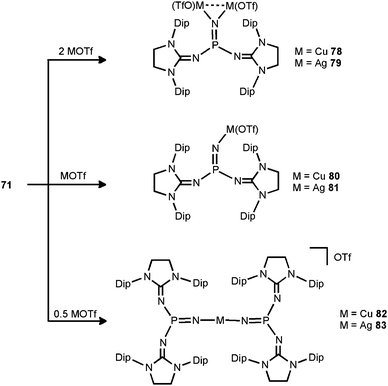 |
| | Scheme 20 The formations of metal–nitrene complexes 78–83 from phosphinonitrene 71. | |
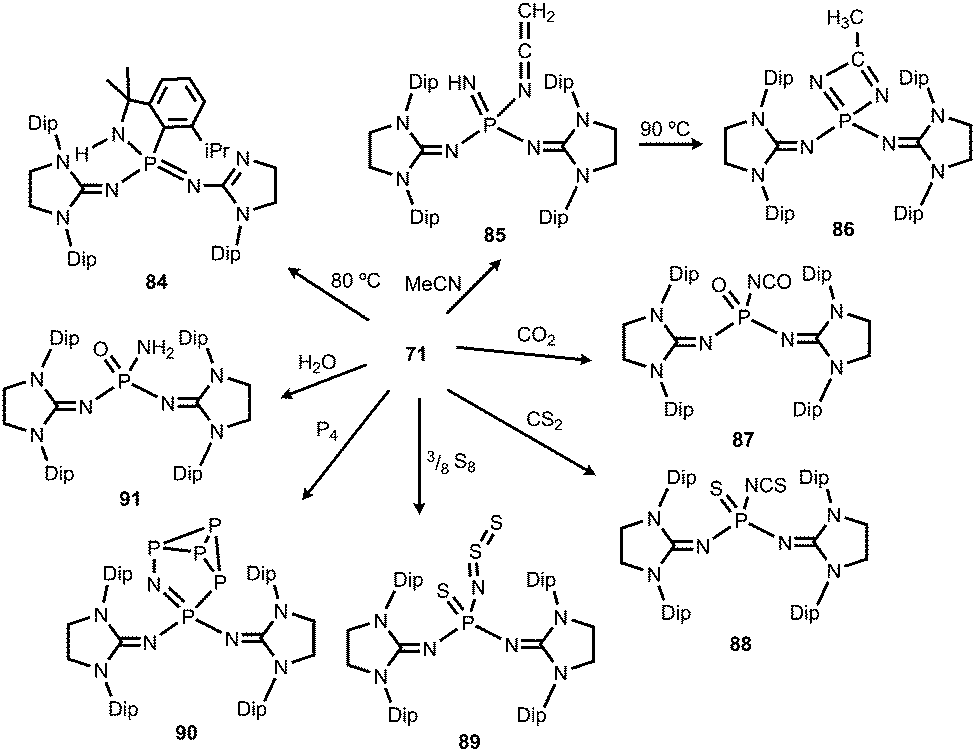
A thorough study on the reactivity of the phosphinonitrene 71 was published in 2015.39 The authors described its thermal transformation to the iminophosphorane 84, as well as several conversions with typical small molecule substrates (Scheme 21). At elevated temperature quantitative rearrangement of 71 was observed. The nitrenic atom inserts into a tertiary carbon CH bond of an isopropyl side chain followed by migration of the Dip moiety to the phosphorus centre to create the five membered PNC3 ring in 84. The addition of an excess amount of acetonitrile to the phosphinonitrene (71) afforded a mixture (16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) of the ketenimine 85 and the diazaphosphete 86 (Scheme 21).39 Notably, the ketenimine is transformed into the diazaphosphete at elevated temperature (90 °C). This process was reasoned by the initial deprotonation of acetonitrile by the nitrenic centre and nucleophilic attack of the generated cyanomethylanion at the phosphorus atom to afford 85. Subsequent cyclization and proton migration leads to the formation of 86. Reaction of the phosphinonitrene (71) with carbon dioxide or carbon disulfide yields the isocyanate 87 or the isothiocyanate 88, respectively (Scheme 21).39 One should point out the cleavage of the thermodynamically stable CE double bond (E = O, S) in this process. Compound 71 activates elemental sulfur (S8), as well as white phosphorus (P4).39 The reaction with S8 furnishes the phosphine sulfide 89 that bears a thiosulfinylamino group at the phosphorus atom (Scheme 21). The conversion of 71 with P4 affords phosphorus enriched 90 with a unique P5N moiety via insertion of the PNnitrene fragment into a P–P single bond of the P4 cluster (Scheme 21). With a slight excess of water the phosphinonitrene (71) reacted to yield the aminophosphine oxide 91 as the product of the addition of H2O to the PNnitrene bond (Scheme 21).39
:1) of the ketenimine 85 and the diazaphosphete 86 (Scheme 21).39 Notably, the ketenimine is transformed into the diazaphosphete at elevated temperature (90 °C). This process was reasoned by the initial deprotonation of acetonitrile by the nitrenic centre and nucleophilic attack of the generated cyanomethylanion at the phosphorus atom to afford 85. Subsequent cyclization and proton migration leads to the formation of 86. Reaction of the phosphinonitrene (71) with carbon dioxide or carbon disulfide yields the isocyanate 87 or the isothiocyanate 88, respectively (Scheme 21).39 One should point out the cleavage of the thermodynamically stable CE double bond (E = O, S) in this process. Compound 71 activates elemental sulfur (S8), as well as white phosphorus (P4).39 The reaction with S8 furnishes the phosphine sulfide 89 that bears a thiosulfinylamino group at the phosphorus atom (Scheme 21). The conversion of 71 with P4 affords phosphorus enriched 90 with a unique P5N moiety via insertion of the PNnitrene fragment into a P–P single bond of the P4 cluster (Scheme 21). With a slight excess of water the phosphinonitrene (71) reacted to yield the aminophosphine oxide 91 as the product of the addition of H2O to the PNnitrene bond (Scheme 21).39
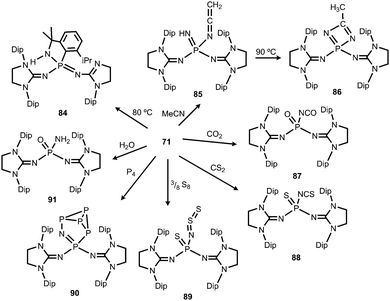 |
| | Scheme 21 Thermal conversion of phosphinonitrene 71 to 84 and reactivity with small molecules to imino complexes 85–91. | |
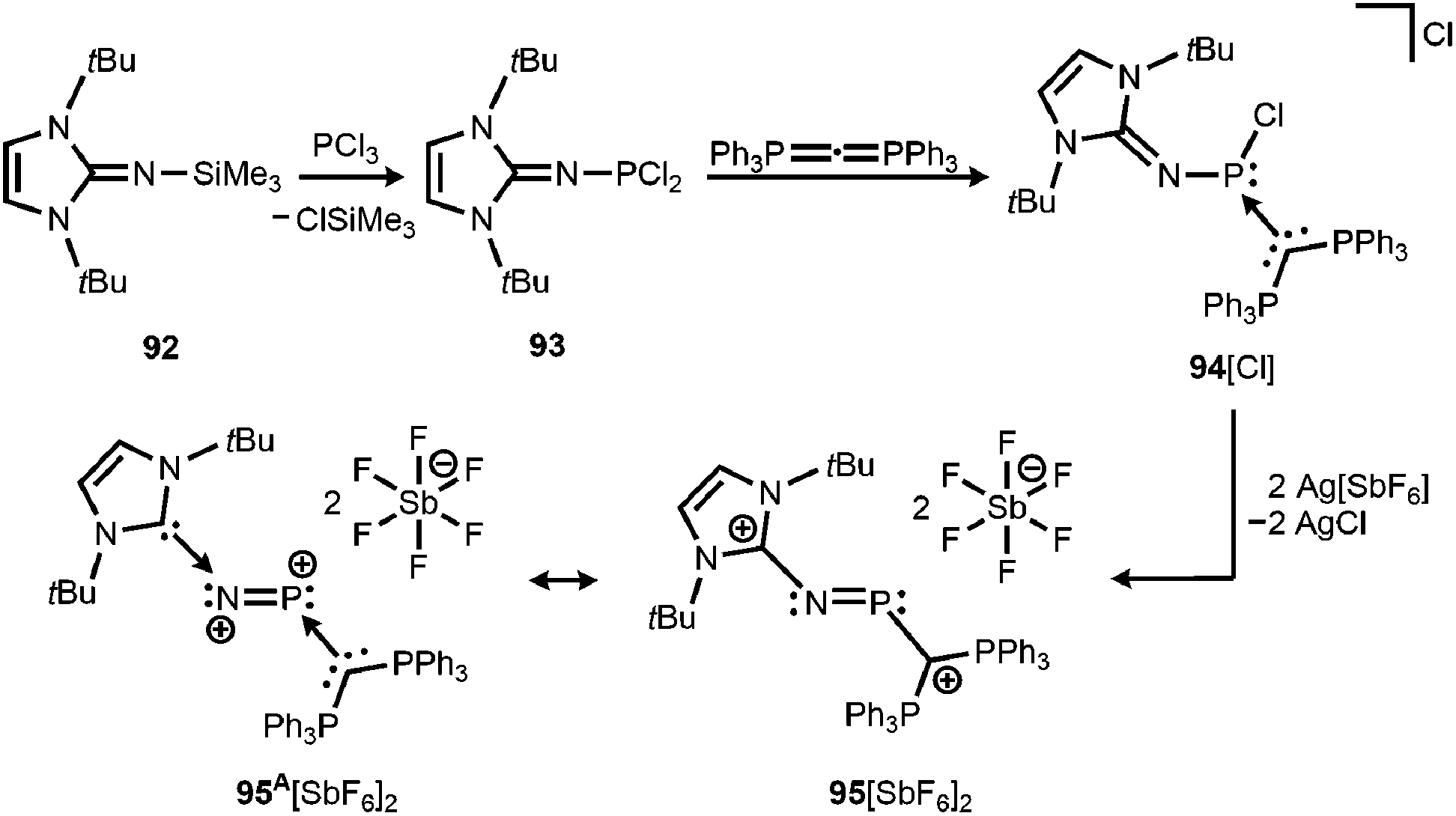
By implementing the imidazolin-2-imino trimethylsilane 92 Vidović and coworkers synthesized the imino phosphorus dichloride 93 that was reacted with carbodiphosphorane to yield the phosphenium salt 94[Cl] (Scheme 22).40 The latter was subjected to chloride abstraction with two equiv. of silver hexafluoroantimonate and in the outcome the dicationic phosphinimine 95[SbF6]2 was formed (Scheme 22).40 The dication assumes a trans-bent structure motif and the P–N distance of 1.594(6) Å is significantly shorter than the respective bond lengths in the CAAC congener 64 (1.7085(16) Å) and its radical cation 64+˙ (1.645(4) Å). This suggests relevant double bond character for the PN fragment in 952+. As concluded from the theoretical analysis of the dication the authors attributed the increased PN interaction to the removal of electrons from the HOMO which majorly comprises the PN π* antibonding orbital. Taking into account structural parameters such as the comparably long C–Nimino bond (1.367(8) Å) and bond polarizations derived from the NBO analysis it was presumed that 952+ possesses dicationic phosphorus mononitride character to a minor degree (95A2+, Scheme 22). Regardless of the dominant resonance structure of 952+ its isolation confirms the potential of the imidazolin-2-imino ligand for stabilizing cationic species.
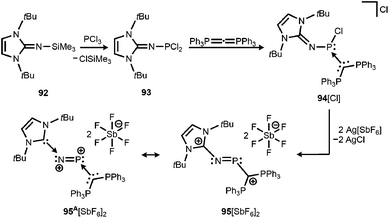 |
| | Scheme 22 Reaction of imino phosphorus dichloride to the phosphenium salt 94[Cl] and its conversion to the dicationic phosphinimine 952+. Phosphorus mononitride formulation 95A2+. | |
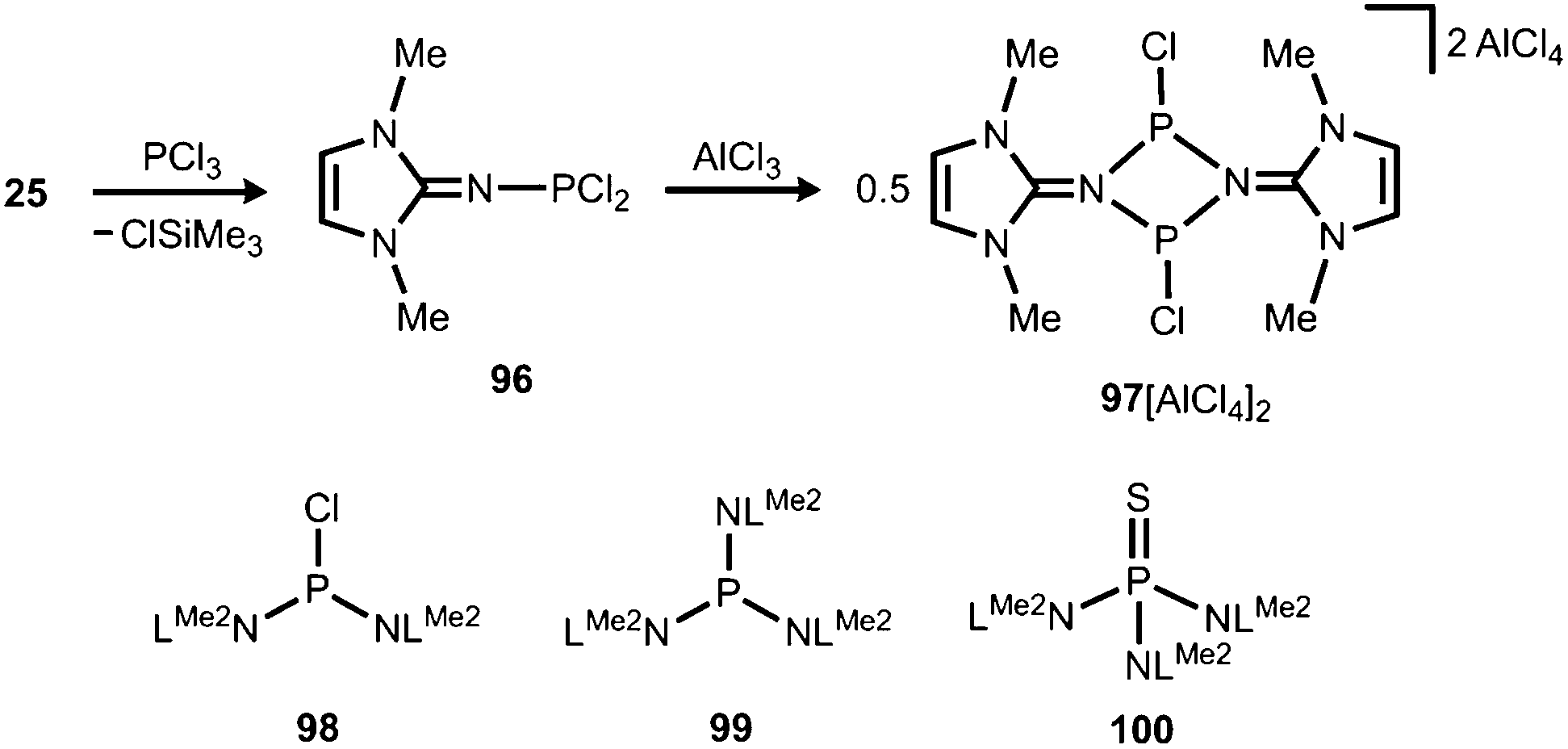
The application of (benz)imidazolin-2-imino substituents as supporting groups for P-based ligands has recently emerged as a subgenre of the field of phosphorus compounds of the iminato ligand. The chemistry relies on the pioneering work of Kuhn and coworkers, who converted LMe2SiMe3 (25) to the imino dichlorophosphane 96 (Schemes 7 and 23).31a If treated with AlCl3 this compound reacts to yield the ditopic phosphenium salt 97[AlCl4]2, the structural formulation of which was based on NMR spectroscopic characteristics (Scheme 23).31a The authors described that in solution 972+ is in equilibrium with 96 depending on the nucleophilic properties of the solvent. Moreover, Kuhn and coworkers described the iminophosphanes 98 and 99, as well as the conversion of 99 to the iminophosphorane 100 (Scheme 23).31b
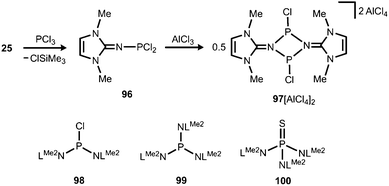 |
| | Scheme 23 Preparation of the imino phosphorus dichloride 96 from the imino trimethylsilane 25. Reaction to the dimeric phosphenium cation 972+. The iminophosphanes 98 and 99, as well as the iminophosphorane 100. | |
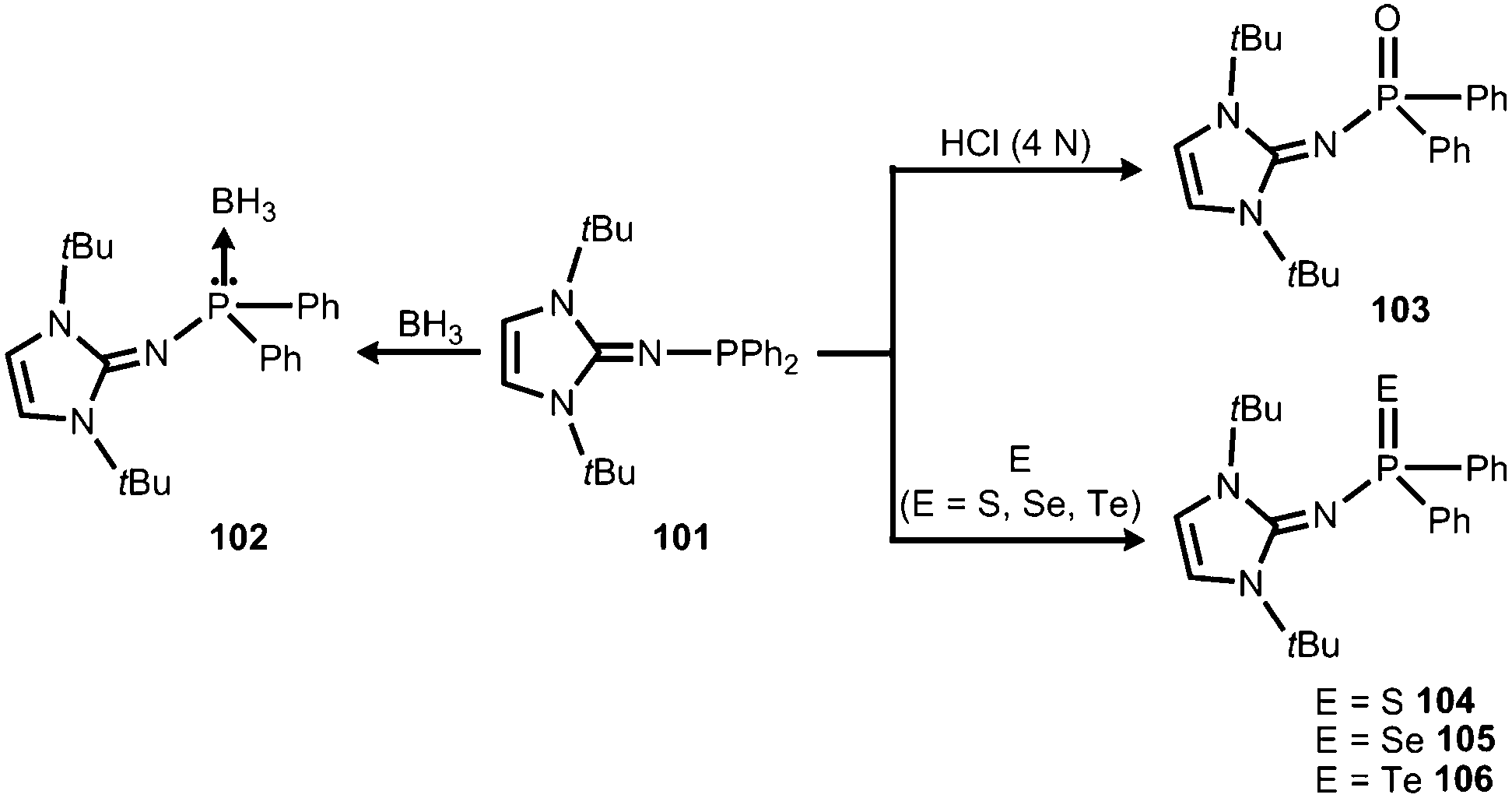
In 2015 Mallik, Panda and coworkers reported the imino diphenylphosphine 101 that was converted to the borane adduct 102, as well as the phosphorus chalcogenides 103–106 (Scheme 24).41
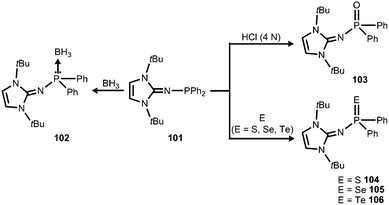 |
| | Scheme 24 Reaction of the imino diphenylphosphine 101 to the borane-adduct 102, as well as the phosphorus chalcogenides 103–106. | |
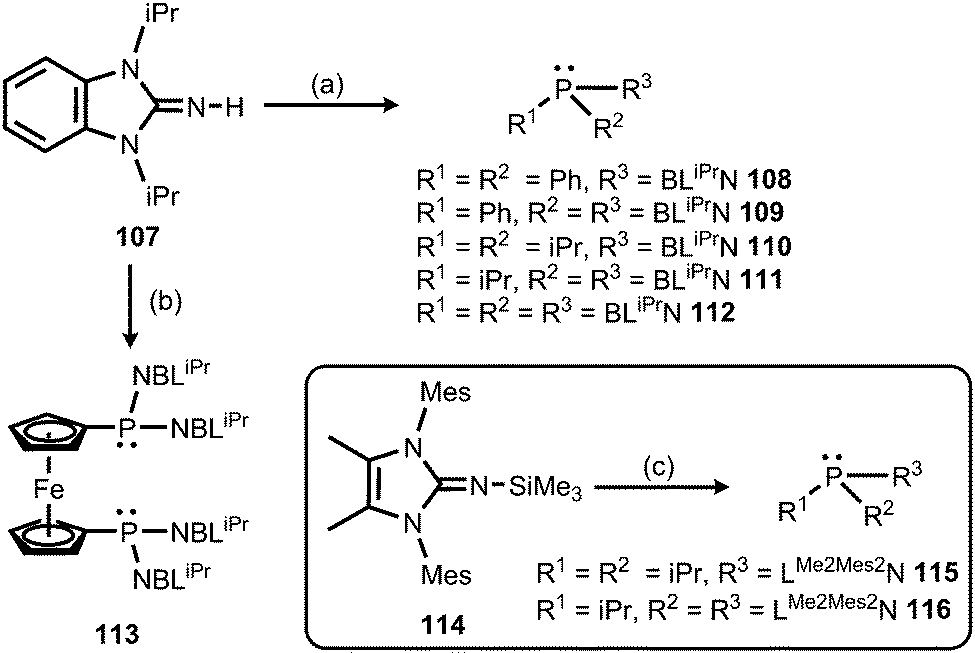
Also in 2015 Dielmann and coworkers established the use of imidazolin-2-imino-substituted phosphines as electron-rich ligands to transition metals.42 By lithiation of the benzimidazolin-2-imine BLiPrNH (107, BLiPr = 1,3-diisopropylbenzimidazolin-2-ylidene) and reaction with the corresponding chlorophosphines the synthesis of the iminophosphines 108–113 was accomplished (Scheme 25).42 In addition, the conversion of the bulkier LMe2Mes2NSiMe3 (114, LMe2Mes2 = 1,3-dimesityl-4,5-dimethyl-imidazolin-2-ylidene) with the respective chlorophosphines led to the iminophosphines 115 and 116 in the outcome (Scheme 25).42 To assess the electron-donor strength of these phosphorus-based ligands the Tolman electronic parameters (TEP) of their nickel tricarbonyl complexes were determined.43 Moreover, they were evaluated according to the Huynh's method. The 13C NMR-spectroscopic shift of the carbene carbon of the BLiPr group in the trans-{PdBr2(BLiPr)ligand} is sensitive to the donor strength of the ligand, in which the carbene resonance of the BLiPr is downfield shifted with increasing donor strength of the ligand trans to the BLiPr group.44 The Huynh's parameters of the iminophosphines show the same qualitative trend as the TEP analysis. As a result, many of the iminophosphines were found to be more potent electron-pair donors than most electron-rich trialkylphosphines. Remarkably, the authors concluded that the iminophosphines 111 and 112, as well as 115 and 116, are stronger donor ligands than classical NHCs. In addition, the bisimine 116 was presumed to be a more potent donor ligand than the very strongly electron donating abnormal NHCs exceeding the capability of monoimine 115, as well as the bis- and the trisimine 111 and 112. Consequently, it was reasoned that the imidazolin-2-iminato ligand is a stronger π-electron donor than the related benzimidazolin-2-iminato ligand.
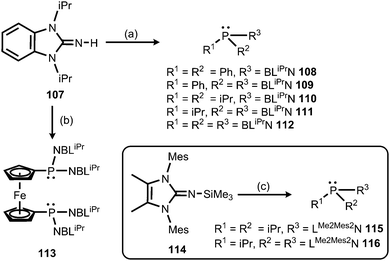 |
| | Scheme 25 Synthesis of benzimidazolin-2-imino-substituted phosphines 108–113, as well as the imidazolin-2-imino-substituted phosphines 115 and 116. (a) (1) nBuLi, THF, −78 °C; (2) chlorophosphine, room temperature. (b) (1) nBuLi, THF, −78 °C; (2) [Fe(C5H4PCl2)2]. (c) 115: iPr2PCl, THF; 116: (1) PCl3, THF, −78 °C; (2) iPrMgCl, THF, −78 °C. | |
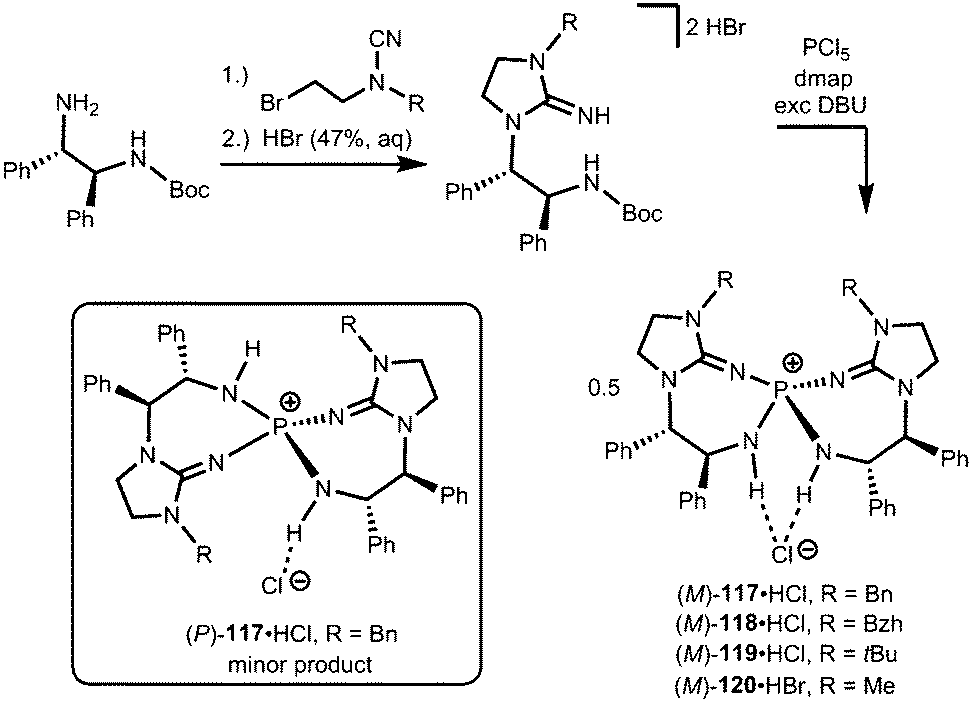
Uncharged organosuperbases that comprise the imidazolidin-2-imino fragment as a chiral bis(guanidine)iminophosphorane were described by Takeda and Terada in 2013.45 The respective iminophosphonium salts 117·HCl, 118·HCl, 119·HCl and 120·HBr were synthesized by conversion of respective aminoguanidinium halides with phosphorus pentachloride in the presence of base followed by acidic work-up (Scheme 26).45 The stability of the iminophosphonium hydrohalide salt, and thus the high Brønsted basicity of the uncharged compounds, relies on the properties of the iminophosphorane as an electron-rich oligodentate ligand. The free base was not characterized but generated by reaction with potassium tertbutoxide and used in situ for the assessment of catalytic activity in the electrophilic amination of tetralones with azodicarboxylate.45 Notably, no particular reason for the use of the imidazolidin-2-imino group instead of acyclic guanidino functionalities was pointed out by the authors. We assume that its implementation rather follows synthetic applicability for furnishing the chiral bis(guanidine)iminophosphorane species. Notably, the scope of catalytic applications of this compound as a chiral uncharged organosuperbase was expanded in recent years.46
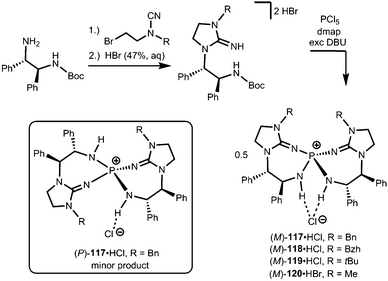 |
| | Scheme 26 Preparation of the chiral imino phosphonium halides (M)-117·HCl–(M)-120·HBr and the stereoisomer (P)-117·HCl. | |
Miscellaneous: survey of related nitrogen compounds
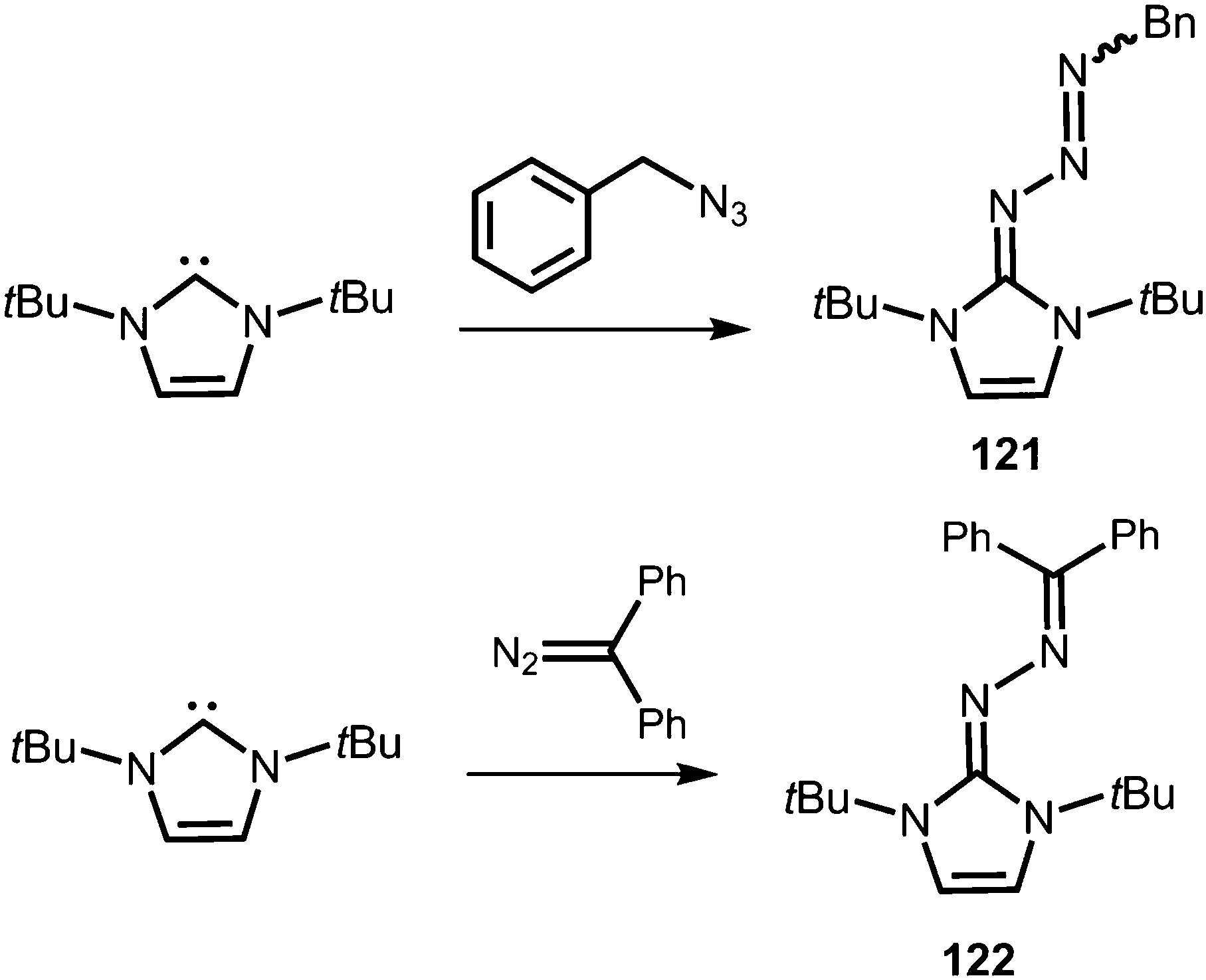
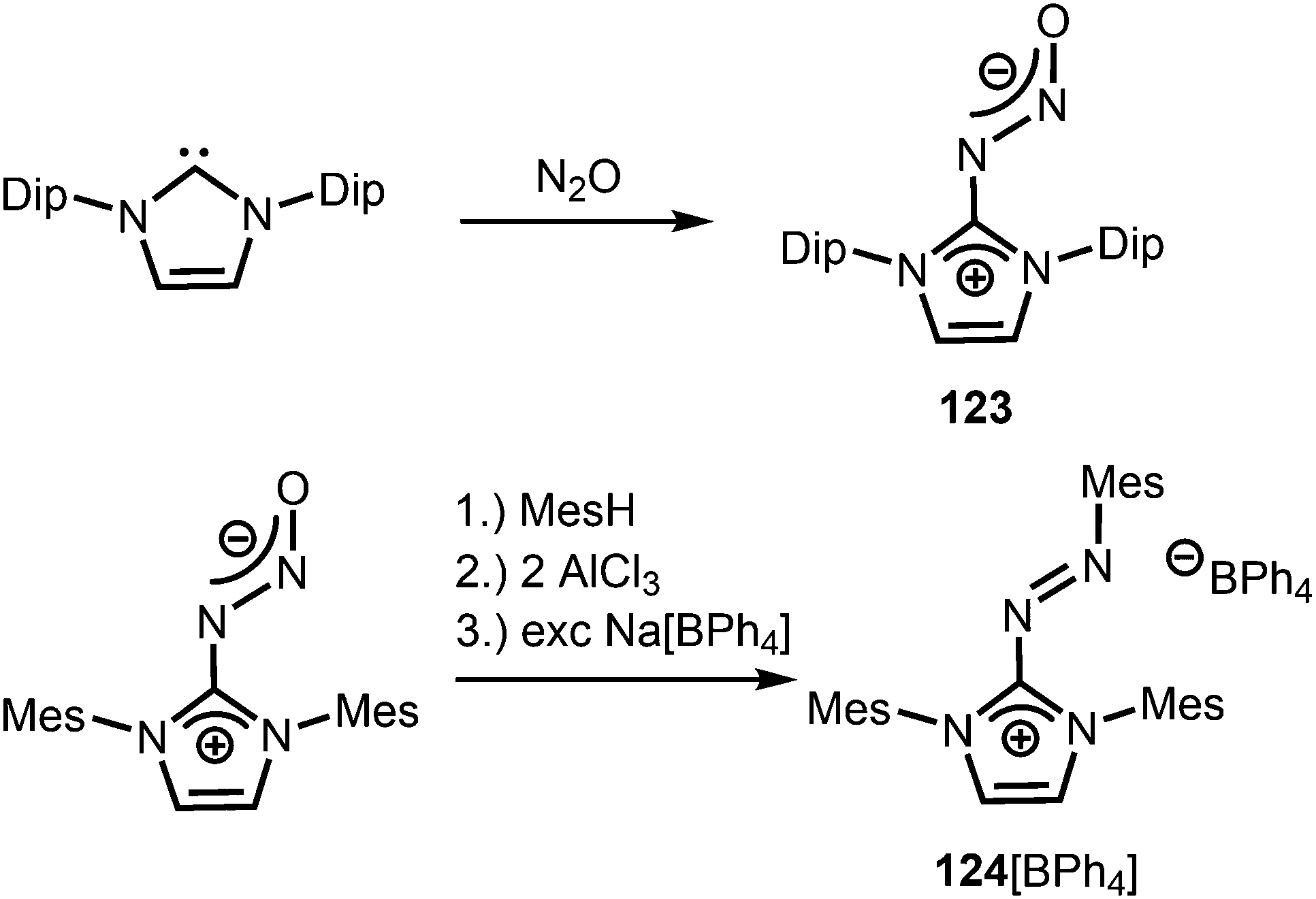
Compounds in which the exocyclic nitrogen atom of an N-heterocyclic imino fragment bonds to another nitrogen atom are abundant in the literature but may be accounted for in the field of classical organic chemistry rather than inorganic or organometallic coordination chemistry which is the focus of this review. They can be categorized into triazenes47 (representative example: 121, Scheme 27), azines18,48 (122, subcategory: bisguanidines, Scheme 27), as well as diazotates49 (123, Scheme 28) and their azoimidazolium50 derivatives (124[BPh4], Scheme 28).
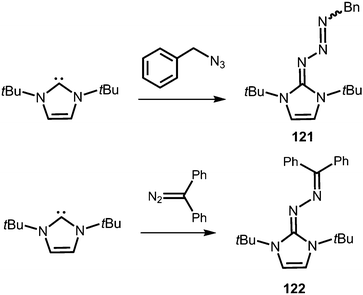 |
| | Scheme 27 Synthesis of representative examples for the compound classes: triazenes (121) and azines (122). | |
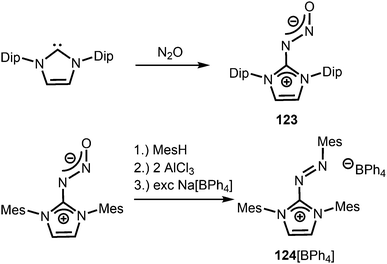 |
| | Scheme 28 Synthesis of representative examples for the compound classes: diazotates (123) and azoimidazolium salts (124[BPh4]). | |
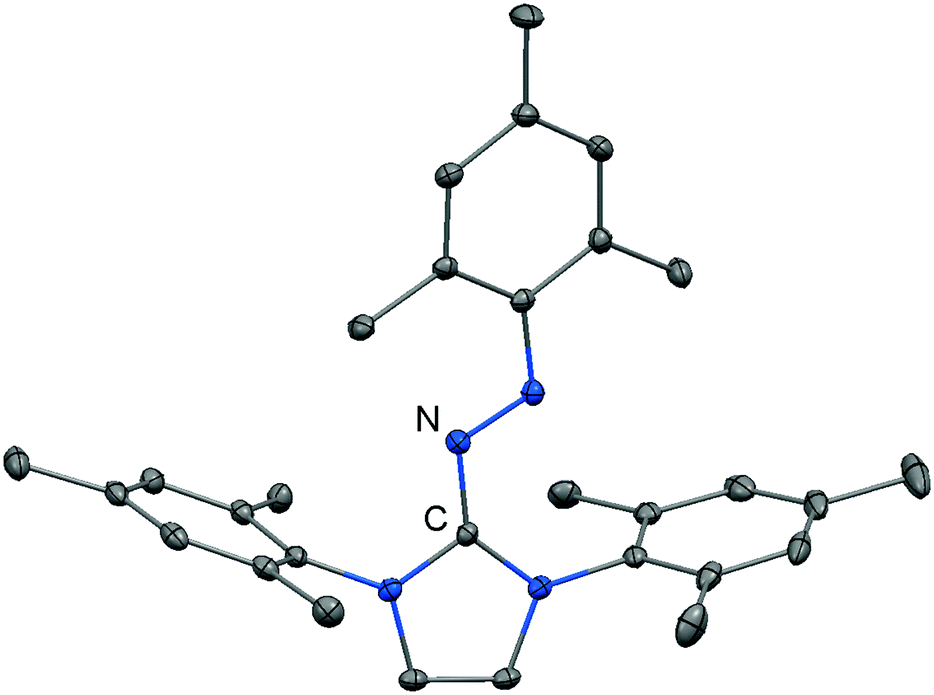
Imidazolyl triazenes (121) release dinitrogen under thermal conditions which leads to the formation of imino organyls whereas their exposure to acidic conditions generates a diazonium species along with the imine. The latter accounts for the pronounced proton affinity of the imidazolin-2-imino group. Apart from applications in organic chemistry azines (122) and diazotates (123), as well as their azoimidazolium spin-offs (124[BPh4]), are found to be employed as ligands to main group elements or transition metals in rare instances. The mechanism for the formation of 124[BPh4] is proposed to involve AlCl3-mediated oxygen abstraction to afford the dicationic diazonium compound, followed by its azo coupling with mesitylene. As an interesting difference in their bonding modes the C–Nimino distance in azines is generally shorter than in the reported diazotates. This suggests high CN double bond character for the former and considerable single bond character along with delocalization of positive charge into the imidazoline ring for the latter. Similarly, the azoimidazolium cation 124+ exhibits a comparably long C–Nimino bond length of 1.386(2) Å which indicates that the positive charge is majorly distributed among the atoms of the five-membered ring (Scheme 28, Fig. 12).
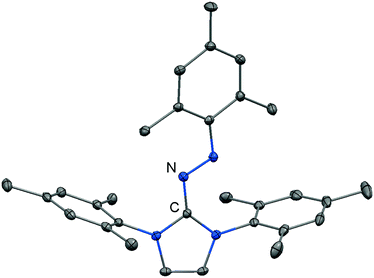 |
| | Fig. 12 Ellipsoid plot (30% level) of the azoimidazolium cation 124+ (hydrogen atoms have been omitted). | |
Conclusions
This survey of the coordination chemistry of main group element complexes with N-heterocyclic imines shows that these ligands are suitable for the isolation of otherwise elusive species (e.g. AlTe double bond, stannylenoid, phosphinonitrene). In particular, they have proven valuable for the thermodynamic stabilization of electron-deficient central atoms, and thus enabled the isolation of rare types of low-coordinate cationic metal complexes (e.g. cationic thioxoborane, germyliumylidene). The strongly electron-donating character of the imidazolin-2-iminato ligand derives from the efficient delocalization of cationic charge density into the five-membered ring system. The lengthening of the C–Nimino distance is an indicator for the allocation of electron density by the ligand as it is often observed upon transformation of an uncharged species into a cationic offspring. The exploration of the phosphorus chemistry of this imino ligand demonstrates its applicability for the stabilization of charged, as well as uncharged phosphorus-centred radicals. Moreover, the electron-rich nature of the imidazolin-2-imino group has resulted in a new class of phosphines that bear supporting imino groups and act as highly electron donating phosphorus-centred ligands.
The chemistry of N-heterocyclic iminato complexes of main group elements is still in its infancy as compared to the widespread field of metal amides.51 However, the growing interest in N-heterocyclic imines in recent years underlines their usefulness as ancillary ligands and distinguishes them from other classes of nitrogen-based ligand systems. Future work should study yet unexplored complexes of the imidazoli(di)n-2-imino group with heavier main group metals and focus on catalytic applications of the respective systems.
Acknowledgements
Financial support of the WACKER Chemie AG, as well as the European Research Council (SILION 637394) is gratefully acknowledged.
Notes and references
-
(a) N. Kuhn, H. Bohnen, J. Kreutzberg, D. Bläser and R. Boese, J. Chem. Soc., Chem. Commun., 1993, 1136 RSC
 ;
(b) S. M. Ibrahim Al-Rafia, A. C. Malcolm, S. K. Liew, M. J. Ferguson, R. McDonald and E. Rivard, Chem. Commun., 2011, 47, 6987 RSC ;
(c) S. M. Ibrahim Al-Rafia, M. J. Ferguson and E. Rivard, Inorg. Chem., 2011, 50, 10543 CrossRef PubMed ;
(d) S. Kronig, P. G. Jones and M. Tamm, Eur. J. Inorg. Chem., 2013, 2301 CrossRef CAS ;
(e) Y. Wang, M. Y. Abraham, R. J. Gilliard, Jr., D. R. Sexton, P. Wei and G. H. Robinson, Organometallics, 2013, 32, 6639 CrossRef CAS .
;
(b) S. M. Ibrahim Al-Rafia, A. C. Malcolm, S. K. Liew, M. J. Ferguson, R. McDonald and E. Rivard, Chem. Commun., 2011, 47, 6987 RSC ;
(c) S. M. Ibrahim Al-Rafia, M. J. Ferguson and E. Rivard, Inorg. Chem., 2011, 50, 10543 CrossRef PubMed ;
(d) S. Kronig, P. G. Jones and M. Tamm, Eur. J. Inorg. Chem., 2013, 2301 CrossRef CAS ;
(e) Y. Wang, M. Y. Abraham, R. J. Gilliard, Jr., D. R. Sexton, P. Wei and G. H. Robinson, Organometallics, 2013, 32, 6639 CrossRef CAS .
- N. Kuhn, M. Göhner, M. Grathwohl, J. Wiethoff, G. Frenking and Y. Chen, Z. Anorg. Allg. Chem., 2003, 629, 793 CrossRef CAS .
-
(a) A. G. Trambitas, T. K. Panda and M. Tamm, Z. Anorg. Allg. Chem., 2010, 636, 2156 CrossRef CAS ;
(b) Y. Wu and M. Tamm, Coord. Chem. Rev., 2014, 260, 116 CrossRef .
- N. Kuhn, R. Fawzi, M. Steimann, J. Wiethoff, D. Bläser and R. Boese, Z. Naturforsch., 1995, 50b, 1779 Search PubMed .
- N. Kuhn, U. Abram, C. Maichle-Mößmer and J. Wiethoff, Z. Anorg. Allg. Chem., 1997, 623, 1121 CrossRef CAS .
- N. Kuhn, R. Fawzi, M. Steimann, J. Wiethoff and G. Henkel, Z. Anorg. Allg. Chem., 1997, 623, 1577 CrossRef CAS .
- N. Kuhn, R. Fawzi, M. Steimann and J. Wiethoff, Z. Anorg. Allg. Chem., 1997, 623, 554 CrossRef CAS .
- D. Franz, E. Irran and S. Inoue, Dalton Trans., 2014, 43, 4451 RSC .
- D. Franz and S. Inoue, Chem. – Asian J., 2014, 9, 2083 CrossRef CAS PubMed .
- D. Franz, E. Irran and S. Inoue, Angew. Chem., Int. Ed., 2014, 53, 14264 CrossRef CAS PubMed .
-
(a) K. Dehnicke and F. Weller, Coord. Chem. Rev., 1997, 158, 103 CrossRef CAS ;
(b) S. Courtenay, J. Y. Mutus, R. W. Schurko and D. W. Stephan, Angew. Chem., Int. Ed., 2002, 41, 498 CrossRef CAS ;
(c) S. Courtenay, D. Walsh, S. Hawkeswood, P. Wei, A. K. Das and D. W. Stephan, Inorg. Chem., 2007, 46, 3623 CrossRef CAS PubMed ;
(d) O. Alhomaidan, E. Hollink and D. W. Stephan, Organometallics, 2007, 26, 3041 CrossRef CAS ;
(e) M. H. Holthausen, I. Mallov and D. W. Stephan, Dalton Trans., 2014, 43, 15201 RSC ;
(f) K. Spannhoff, R. Rojas, R. Fröhlich, G. Kehr and E. Erker, Organometallics, 2011, 30, 2377 CrossRef CAS ;
(g) K. Jaiswal, B. Prashanth, S. Ravi, K. R. Shamasundar and S. Singh, Dalton Trans., 2015, 44, 15779 RSC .
-
(a) C. M. Ong, P. McKarns and D. W. Stephan, Organometallics, 1999, 18, 4197 CrossRef CAS ;
(b) K. Aparna, R. McDonald, M. Ferguson and R. G. Cavell, Organometallics, 1999, 18, 4241 CrossRef CAS ;
(c) Z.-X. Wang and Y.-X. Li, Organometallics, 2003, 22, 4900 CrossRef CAS ;
(d) J. Guo, J.-S. Lee, M.-C. Foo, K.-C. Lau, H.-W. Xi, K. H. Lim and C.-W. So, Organometallics, 2010, 29, 939 CrossRef CAS ;
(e) C. V. Cárdenas, M. Á. M. Hernández and J.-M. Grévy, Dalton Trans., 2010, 39, 6441 RSC .
- S. M. I. Al-Rafia, R. McDonald, M. J. Ferguson and E. Rivard, Chem. – Eur. J., 2012, 18, 13810 CrossRef CAS PubMed .
- M. W. Lui, N. R. Paisley, R. McDonald, M. J. Ferguson and E. Rivard, Chem. – Eur. J., 2016, 22, 2134 CrossRef CAS PubMed .
- Z. Yang, M. Zhong, X. Ma, S. De, C. Anusha, P. Parameswaran and H. W. Roesky, Angew. Chem., Int. Ed., 2015, 54, 10225 CrossRef CAS PubMed .
- D. Franz and S. Inoue, Chem. – Eur. J., 2014, 20, 10645 CrossRef CAS PubMed .
-
(a) D. Franz, T. Szilvási, E. Irran and S. Inoue, Nat. Commun., 2015, 6, 10037 CrossRef CAS PubMed ;
(b) D. Franz and S. Inoue, Dalton Trans., 2016, 45, 9385 RSC .
- J. M. Hopkins, M. Bowdridge, K. N. Robertson, T. S. Cameron, H. A. Jenkins and J. A. C. Clyburne, J. Org. Chem., 2001, 66, 5713 CrossRef CAS PubMed .
-
(a) M. Tamm, S. Randoll, T. Bannenberg and E. Herdtweck, Chem. Commun., 2004, 876 RSC ;
(b) M. Tamm, D. Petrovic, S. Randoll, S. Beer, T. Bannenberg, P. G. Jones and J. Grunenberg, Org. Biomol. Chem., 2007, 5, 523 RSC .
- S. Randoll, P. G. Jones and M. Tamm, Organometallics, 2008, 27, 3232 CrossRef CAS .
- S. Inoue and K. Leszczyńska, Angew. Chem., Int. Ed., 2012, 51, 8589 CrossRef CAS PubMed .
- M. Asay, C. Jones and M. Driess, Chem. Rev., 2011, 111, 354 CrossRef CAS PubMed .
- G.-H. Lee, R. West and T. Müller, J. Am. Chem. Soc., 2003, 125, 8114 CrossRef CAS PubMed .
- M. W. Lui, C. Merten, M. J. Ferguson, R. McDonald, Y. Xu and E. Rivard, Inorg. Chem., 2015, 54, 2040 CrossRef CAS PubMed .
-
(a) T. Ochiai, D. Franz, X.-N. Wu and S. Inoue, Dalton Trans., 2015, 44, 10952 RSC ;
(b) T. Ochiai, T. Szilvási, D. Franz, E. Irran and S. Inoue, Angew. Chem., Int. Ed., 2016 DOI:10.1002/anie.201605636 .
-
(a) T. Ochiai, D. Franz, E. Irran and S. Inoue, Chem. – Eur. J., 2015, 21, 6704 CrossRef CAS PubMed ;
(b) T. Ochiai and S. Inoue, Phosphorus, Sulfur Silicon Relat. Elem., 2016, 191, 624 CrossRef CAS .
- T. Ochiai, D. Franz, X.-N. Wu, E. Irran and S. Inoue, Angew. Chem., Int. Ed., 2016, 55, 6983 CrossRef CAS PubMed .
-
(a) A. Akkari, J. J. Byrne, I. Saur, G. Rima, H. Gornitzka and J. Barrau, J. Organomet. Chem., 2001, 622, 190 CrossRef CAS ;
(b) Y. Ding, H. W. Roesky, M. Noltemeyer, H.-G. Schmidt and P. P. Power, Organometallics, 2001, 20, 1190 CrossRef CAS ;
(c) P. B. Hitchcock, J. Hu, M. F. Lappert and J. R. Severn, Dalton Trans., 2004, 4193 RSC ;
(d) A. P. Dove, V. C. Gibson, E. L. Marshall, H. S. Rzepa, A. J. P. White and D. J. Williams, J. Am. Chem. Soc., 2006, 128, 9834 CrossRef CAS PubMed ;
(e) S. L. Choong, C. Schenk, A. Stasch, D. Dange and C. Jones, Chem. Commun., 2012, 48, 2504 RSC ;
(f) R. Olejník, Z. Padělková, R. Mundil, J. Merna and A. Růžička, Appl. Organomet. Chem., 2014, 28, 405 CrossRef .
- J. Volbeda, P. G. Jones and M. Tamm, Inorg. Chim. Acta, 2014, 422, 158 CrossRef CAS .
- D. Petrovic, T. Glöge, T. Bannenberg, C. G. Hrib, S. Randoll, P. G. Jones and M. Tamm, Eur. J. Inorg. Chem., 2007, 3472 CrossRef CAS .
-
(a) N. Kuhn, R. Fawzi, M. Steimann and J. Wiethoff, Chem. Ber., 1996, 129, 479 CrossRef CAS ;
(b) N. Kuhn, H. Kotowski and J. Wiethoff, Phosphorus, Sulfur Silicon Relat. Elem., 1998, 133, 237 CrossRef CAS .
- R. Kinjo, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2010, 49, 5930 CrossRef CAS PubMed .
- Y. Wang, Y. Xie, P. Wie, R. B. King, H. F. Schaefer, III, P. v. R. Schleyer and G. H. Robinson, J. Am. Chem. Soc., 2008, 130, 14970 CrossRef CAS PubMed .
- O. Back, B. Donnadieu, P. Parameswaran, G. Frenking and G. Bertrand, Nat. Chem., 2010, 2, 369 CrossRef CAS PubMed .
- O. Back, B. Donnadieu, M. v. Hopffgarten, S. Klein, R. Tonner, G. Frenking and G. Bertrand, Chem. Sci., 2011, 2, 858 RSC .
- F. Dielmann, O. Back, M. Henry-Ellinger, P. Jerabek, G. Frenking and G. Bertrand, Science, 2012, 337, 1526 CrossRef CAS PubMed .
- F. Dielmann, C. E. Moore, A. Rheingold and G. Bertrand, J. Am. Chem. Soc., 2013, 135, 14071 CrossRef CAS PubMed .
- F. Dielmann, D. M. Andrada, G. Frenking and G. Bertrand, J. Am. Chem. Soc., 2014, 136, 3800 CrossRef CAS PubMed .
- F. Dielmann and G. Bertrand, Chem. – Eur. J., 2015, 21, 191 CrossRef CAS PubMed .
- Y. K. Loh, C. Gurnani, R. Ganguly and D. Vidović, Inorg. Chem., 2015, 54, 3087 CrossRef CAS PubMed .
- K. Naktode, S. D. Gupta, A. Kundu, S. K. Jana, H. P. Nayek, B. S. Mallik and T. K. Panda, Aust. J. Chem., 2015, 68, 127 CrossRef CAS .
- M. A. Wünsche, P. Mehlmann, T. Witteler, F. Buß, P. Rathmann and F. Dielmann, Angew. Chem., Int. Ed., 2015, 54, 11857 CrossRef PubMed .
- C. A. Tolman, Chem. Rev., 1977, 77, 313 CrossRef CAS .
- H. V. Huynh, Y. Han, R. Jothibasu and J. A. Yang, Organometallics, 2009, 28, 5395 CrossRef CAS .
- T. Takeda and M. Terada, J. Am. Chem. Soc., 2013, 135, 15306 CrossRef CAS PubMed .
-
(a) T. Takeda and M. Terada, Aust. J. Chem., 2014, 67, 1124 CrossRef CAS ;
(b) A. Kondoh, M. Oishi, T. Takeda and M. Terada, Angew. Chem., Int. Ed., 2015, 54, 15836 CrossRef CAS PubMed ;
(c) T. Takeda, A. Kondoh and M. Terada, Angew. Chem., Int. Ed., 2016, 55, 4734 CrossRef CAS PubMed .
-
(a) D. M. Khramov and C. W. Bielawski, Chem. Commun., 2005, 4958 RSC ;
(b) D. M. Khramov and C. W. Bielawski, J. Org. Chem., 2007, 72, 9407 CrossRef CAS PubMed ;
(c) D. J. Coady, D. M. Khramov, B. C. Norris, A. G. Tennyson and C. W. Bielawsky, Angew. Chem., Int. Ed., 2009, 48, 5187 CrossRef CAS PubMed ;
(d) A. G. Tennyson, D. M. Khramov, C. D. Varnado Jr., P. T. Creswell, J. W. Kamplain, V. M. Lynch and C. W. Bielawski, Organometallics, 2009, 28, 5142 CrossRef CAS ;
(e) A. G. Tennyson, E. J. Moorhead, B. L. Madison, J. A. V. Er, V. M. Lynch and C. W. Bielawski, Eur. J. Org. Chem., 2010, 6277 CrossRef CAS ;
(f) R. J. Ono, Y. Suzuki, D. M. Khramov, M. Ueda, J. L. Sessler and C. W. Bielawski, J. Org. Chem., 2011, 76, 3239 CrossRef CAS PubMed ;
(g) D. Jishkariani, C. D. Hall, A. Demircan, B. J. Tomlin, P. J. Steel and A. R. Katritzky, J. Org. Chem., 2013, 78, 3349 CrossRef CAS PubMed ;
(h) S. Patil, K. White and A. Bugarin, Tetrahedron Lett., 2014, 55, 4826 CrossRef CAS ;
(i) F. W. Kimani and J. C. Jewett, Angew. Chem., Int. Ed., 2015, 54, 4051 CrossRef CAS PubMed .
-
(a) B. Bildstein, M. Malaun, H. Kopacka, K.-H. Ongania and K. Wurst, J. Organomet. Chem., 1999, 572, 177 CrossRef CAS ;
(b) M. Reinmuth, C. Neuhäuser, P. Walter, M. Enders, E. Kaifer and H.-J. Himmel, Eur. J. Inorg. Chem., 2011, 83 CrossRef CAS ;
(c) J. Tauchman, K. Hladíková, F. Uhlík, I. Císařová and P. Štěpnička, New J. Chem., 2013, 37, 2019 RSC ;
(d) H. Herrmann, M. Reinmuth, S. Wiesner, O. Hübner, E. Kaifer, H. Wadepohl and H.-J. Himmel, Eur. J. Inorg. Chem., 2015, 2345 CrossRef CAS .
-
(a) A. G. Tskhovrebov, E. Solari, M. D. Wodrich, R. Scopelliti and K. Severin, Angew. Chem., Int. Ed., 2012, 51, 232 CrossRef CAS PubMed ;
(b) A. G. Tskhovrebov, E. Solari, M. D. Wodrich, R. Scopelliti and K. Severin, J. Am. Chem. Soc., 2012, 134, 1471 CrossRef CAS PubMed ;
(c) A. G. Tskhovrebov, B. Vuichoud, E. Solari, R. Scopelliti and K. Severin, J. Am. Chem. Soc., 2013, 135, 9486 CrossRef CAS PubMed .
- A. G. Tskhovrebov, L. C. E. Naested, E. Solari, R. Scopelliti and K. Severin, Angew. Chem., Int. Ed., 2015, 54, 1289 CrossRef CAS PubMed .
-
(a)
M. F. Lappert, P. P. Power, A. V. Protchenko and A. L. Seeber, Metal Amide Chemistry, Wiley-VCH, Weinheim, 2009 Search PubMed ;
(b) D. L. Kays, Chem. Soc. Rev., 2016, 45, 1004 RSC .
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a