
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photodimerisation of the α′-polymorph of ortho-ethoxy-trans-cinnamic acid occurs via a two-stage mechanism at 343 K yielding 100% α-truxillic acid†
M. A.
Fernandes
* and
D. C.
Levendis
*
Molecular Sciences Institute, School of Chemistry, University of the Witwatersrand, PO Wits 2050, Johannesburg, South Africa. E-mail: Manuel.Fernandes@wits.ac.za; Fax: +27 11 7176749; Tel: +27 11 7176723
First published on 6th July 2016
Abstract
Three different polymorphs of o-ethoxy-trans-cinnamic acid (OETCA) are known to behave quite differently when irradiated with ultraviolet light. While the α and β polymorphs photodimerise to yield ethoxy-truxillic acid and ethoxy-truxinic acid respectively, the γ polymorph is unreactive. A fourth polymorph, α′, can be obtained by heating the α polymorph to 333 K and stabilising it by partially reacting it. This polymorph contains two distinctly different reaction sites in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio. Irradiation of the stabilised α′ polymorph at 293 K yielded an ordered single crystal product that went to 67% completion (corresponding to the ratio of the different reaction sites). Here we report on the reaction of the α′-polymorph at a higher temperature, 343 K, which now occurs in two separate stages. In stage 1, only molecules in one of the sites react (in a single-crystal-to-single-crystal manner in a similar way to that of the 293 K reaction). In stage 2 the crystal undergoes a phase transition to a polycrystalline phase in which molecules in the second site react, driving the reaction to 100% completion. The powder X-ray diffraction pattern of the final product corresponds to the calculated pattern of the recrystallized ethoxy-truxillic acid photodimer. Examination of close contacts in the crystal structure after stage 1 of the reaction, and using lattice energy calculations to evaluate different arrangements of molecules in the crystal after stage 1, reveals that the reaction occurs in an ordered and cooperative manner even though the product solves as a disordered structure.
:1 ratio. Irradiation of the stabilised α′ polymorph at 293 K yielded an ordered single crystal product that went to 67% completion (corresponding to the ratio of the different reaction sites). Here we report on the reaction of the α′-polymorph at a higher temperature, 343 K, which now occurs in two separate stages. In stage 1, only molecules in one of the sites react (in a single-crystal-to-single-crystal manner in a similar way to that of the 293 K reaction). In stage 2 the crystal undergoes a phase transition to a polycrystalline phase in which molecules in the second site react, driving the reaction to 100% completion. The powder X-ray diffraction pattern of the final product corresponds to the calculated pattern of the recrystallized ethoxy-truxillic acid photodimer. Examination of close contacts in the crystal structure after stage 1 of the reaction, and using lattice energy calculations to evaluate different arrangements of molecules in the crystal after stage 1, reveals that the reaction occurs in an ordered and cooperative manner even though the product solves as a disordered structure.
Introduction
Organic solid-state reactions, including the first observation of the photodimerisation of cinnamic acid into α-truxillic acid in 1895, were investigated by a number of eminent chemists in the second half of the 19th century, as elegantly documented by Roth in his review of the history of organic photochemistry.1 The [2 + 2] photocycloaddition is recognized as being one of the most important and frequently used photochemical reactions, as detailed in an extensive recent review that includes a more general survey of the dimerisation of cinnamic acids.2 Solid-state dimerisation, including the ‘crystal engineering’ of topochemical reactions involving different alkenes and also involving metal organic frameworks, has been discussed in other reviews.3,4In the 1960s Schmidt and Cohen published a series of articles on the [2 + 2] photodimerisations of a range of trans-cinnamic acids5 which led to the formulation of Schmidt's celebrated topochemical principles in 1971 i.e. solid-state reactions occur with minimal atomic motion if the reacting groups are within 4.2 Å from each other.6 In a recent commentary by Williams7 on the question of mechanical properties as a relatively new way to understand solid-state reactions, he notes that although topochemical, the reactions hardly ever occur in a single-crystal-to-single-crystal (SCSC) fashion. Decades after Schmidt's pioneering work, Enkelmann et al. (1993)8 discovered that SCSC reactions were indeed possible using ‘tail absorption’, and demonstrated the progressive SCSC structural change of α-trans-cinnamic acid to the head-to-tail α-truxillic acid dimer. They later confirmed their discovery9 after criticisms of errors in their structure of polymorph II, the product of the one-phase solid-state reaction.
However, many examples have been found to contravene these principles, where molecular crystals were unreactive despite the reactive groups being closer than 4.2 Å, whilst others reacted even though the reactive groups were beyond the 4.5 Å limit. Kaupp, using primarily atomic force microscopic (AFM) data, postulated that many solid-state organic reactions were in fact accompanied by large movements of the molecules.10 He proposed a three-step solid-state reaction theory10 involving phase rebuilding, phase transformation and crystal disintegration, disagreeing with Schmidt's topochemical idea. Therefore some phototransformations involve large structural changes, seemingly impossible to take place in a SCSC fashion. However, such molecular transformations can occur through the motion of a pair of molecular fragments in a way that is analogous to the pedal motion of a bicycle.11 The contemporary use of the term SCSC, its shortcomings and other possible interpretations of results were also recently highlighted by Halasz.12
Another important concept introduced in the early studies of organic photochemistry was that of a ‘reaction cavity’, a flexible space facilitating the reaction.13,14 However, as noted in our previous work,15 the movement of bulkier groups, such as the o-ethoxy group, and larger separations between the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds can actually expedite the required rearrangement to the truxillic acid. It has been noted in other cases that photodimerisation is accompanied by a large shift of one of the substituents on the reacting molecules, such as the 180° flip of the light sensitive host in the [4 + 4] dimerisation of pyridinones in a host–guest complex.16 Turowska-Tyrk's research group has described17 how structural transformations in crystals are induced by both radiation and pressure, where in some cases UV radiation causes structural changes in crystals even though no photochemical reaction occurs, including degradation of crystallinity, a feature not often emphasized sufficiently in these studies. The importance of the decrease of crystal diffracting power after molecular crystals have been heated or exposed to UV radiation has also been noted by them.18 Other important mechanisms in SCSC reactions, such as cooperativity and feedback resulting from changes in intermolecular interactions during Diels Alder reactions were described by one of us previously.19 This point was also noted by us in a study of the thermal Diels Alder dimerisation of bis-(N-allylimino)-1,4-dithiin and 9-bromoanthracene, which shows both topochemical and topotactic behavior.20
C bonds can actually expedite the required rearrangement to the truxillic acid. It has been noted in other cases that photodimerisation is accompanied by a large shift of one of the substituents on the reacting molecules, such as the 180° flip of the light sensitive host in the [4 + 4] dimerisation of pyridinones in a host–guest complex.16 Turowska-Tyrk's research group has described17 how structural transformations in crystals are induced by both radiation and pressure, where in some cases UV radiation causes structural changes in crystals even though no photochemical reaction occurs, including degradation of crystallinity, a feature not often emphasized sufficiently in these studies. The importance of the decrease of crystal diffracting power after molecular crystals have been heated or exposed to UV radiation has also been noted by them.18 Other important mechanisms in SCSC reactions, such as cooperativity and feedback resulting from changes in intermolecular interactions during Diels Alder reactions were described by one of us previously.19 This point was also noted by us in a study of the thermal Diels Alder dimerisation of bis-(N-allylimino)-1,4-dithiin and 9-bromoanthracene, which shows both topochemical and topotactic behavior.20
Numerous other studies of the photodimerisation of cinnamic acids or their derivatives have been undertaken in the past few years.21–26 A recent study by Mishra and co-workers27 examined the crystal chemistry and photomechanical behavior of two polymorphs of 3,4-dimethoxycinnamic acid using nanoindentation. Their results indicate that where molecular movement is facile (in the triclinic form I), gross changes in mutual orientations lead to slower reaction and lower final yields. Conversely, the photoreaction in the monoclinic form II is slower with higher yields, and probably governed more by ‘Schmidt-type’ topochemistry.
In this work we investigate the role of increased temperature on the solid-state reactivity (photodimersation) of the fourth polymorph15 of o-ethoxy-trans-cinnamic acid (OETCA), α′, in terms of rate, yield and reaction mechanism. The α′ polymorph is obtained by a reversible phase change from the α polymorph at 333 K. Partially reacting this crystal stabilises the α′-polymorph preventing the reverse phase change to the α polymorph. The relationship between all polymorphs is shown in Scheme 1. While the α polymorph contains a single reaction site28 the α′ polymorph has two different reaction environments (referred to as the AB and CC sites which are present in a 2:1 ratio),29 allowing us an opportunity to study the effect of these on the material's solid-state reactivity as well as any possible cooperative effects. Molecules in the AB are pseudo-centrosymmetrically related with the distance between the reacting CC bonds being less than 3.6 Å. By contrast, molecules in the CC sites are centrosymmetricallly related to each other, but the distance between the potentially reacting CC bonds is around 4.8 Å. Consequently, according to Schmidt's criterion6 molecules in the AB site should react while those in the CC site should not. In a previous paper we reported on the photodimerisation of the α′ polymorph at 293 K (Scheme 1b).15 Here we report on the effect of irradiating the α′ polymorph with UV light at 343 K (Scheme 1c).
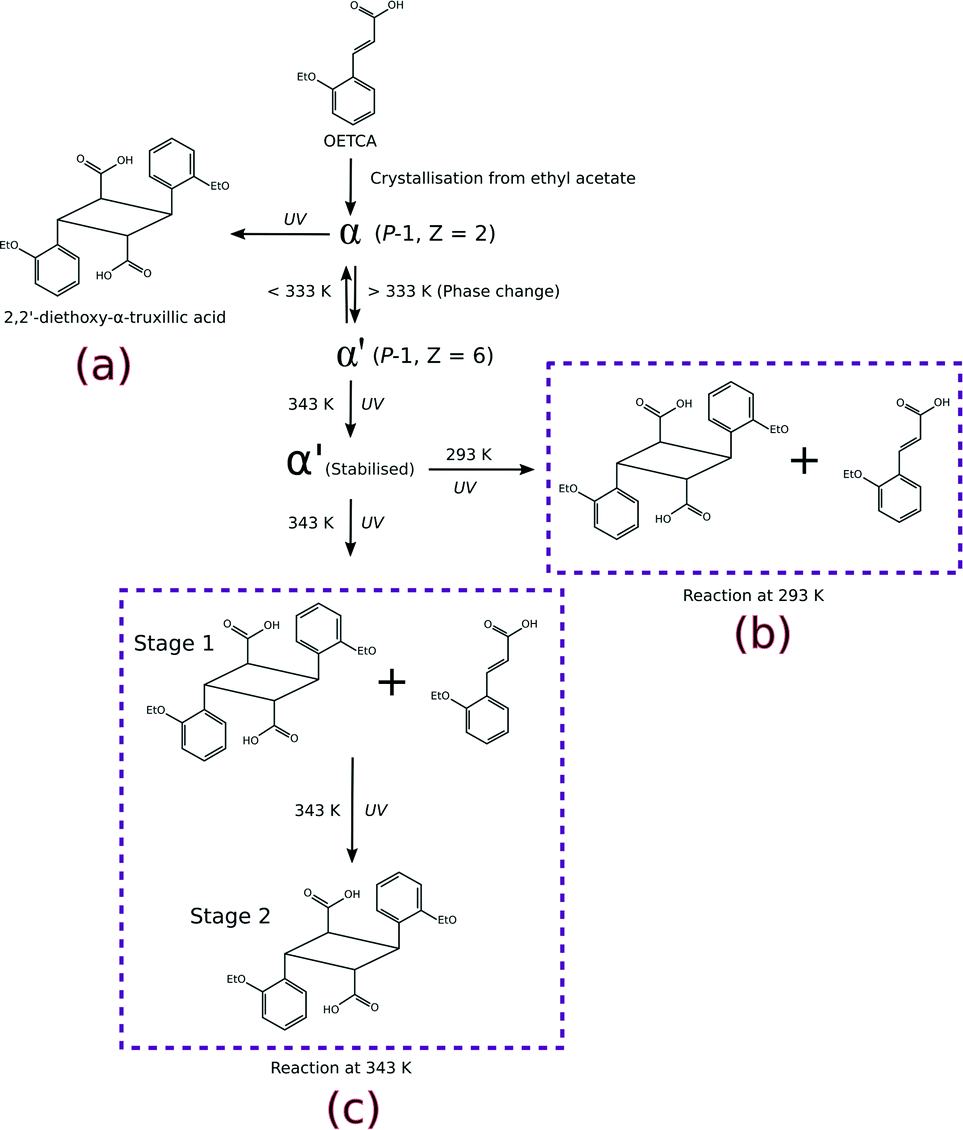 | ||
| Scheme 1 Different reaction pathways which can be explored starting from the α polymorph of OETCA; a) reaction product from the α polymorph;5 b) 293 K reaction product of the α′ polymorph (obtained by phase transition of the α polymorph at 333 K);15,29 c) reaction of the α′ polymorph at 343 K (this work). | ||
Experimental
An extended experimental description can be found in the ESI.†Crystallisation and solid-state reaction
Crystals of the α-polymorph were grown by slow evaporation from a saturated solution of OETCA in ethyl acetate to yield prism-like crystals. A sample of these was then placed on a Kofler hot stage at 343(3) K leading to a SCSC phase transformation to the α′-polymorph which was then exposed to UV light. Crystals were then extracted from the hot stage at 6 hour intervals and analyzed by single crystal X-ray diffraction (SCXRD). Data sets were collected after 6, 12, 18, 24, 36, 48, 60, 72 and 120 hours of UV exposure (all on different crystals), and are referred to as structures 6 h to 120 h in the remaining text.Crystal structure solution and refinement
Where possible, crystals were chosen according to their ability to uniformly polarize light under a microscope, and then examined using SCXRD. This approach was successful for crystals which had been exposed for 24 hours or less. The surfaces of crystals which had been exposed for longer periods of time became white with longer reaction times. This was presumably due to reaction and phase transition (stage 2) at the surface in competition with SCSC reaction in the body of the crystals. These were also examined under a polarizing microscope but had to be cut in order to extract fragments suitable for a SCXRD study. Intensity data for crystals were collected at 173 K on a Bruker SMART 1K CCD area detector diffractometer with graphite monochromated Mo Kα radiation. Software used in this work were as follows: data collection: SMART;30 cell refinement and data reduction: SAINT;31 structure solution and refinement: SHELX-2014;32 molecular graphics: ORTEP-3,33SCHAKAL-99,34VMD-1.91,35 and CrystalExplorer-3.1;36 software used to prepare material for publication: WinGX-2014.1 (ref. 37) and PLATON.38The labelling system used in our two previous papers involving the α′ polymorph – namely on the formation of the α′ polymorph by phase transition from the α polymorph,29 and the photodimerisation of the α′ polymorph at 293 K (ref. 15) – has been retained (Fig. 1a). In crystals where the AB predimer pair (made up of molecules A and B) has photodimerised, the photodimer has been labelled as the DE product where D and E were the original A and B molecules respectively (Fig. 1b and 3). Structure solutions of the α′-polymorph exposed for more than 24 h (67% conversion of AB predimer) indicate that the DE product molecule changes conformation during the course of the reaction to an alternative conformation that we have labelled as the FG conformer. Here the F and G labelled atoms were the original A and B molecules, respectively (Fig. 1c and 3). Crystallographic data for crystals at various reaction times are given in Table S1 in the ESI.†
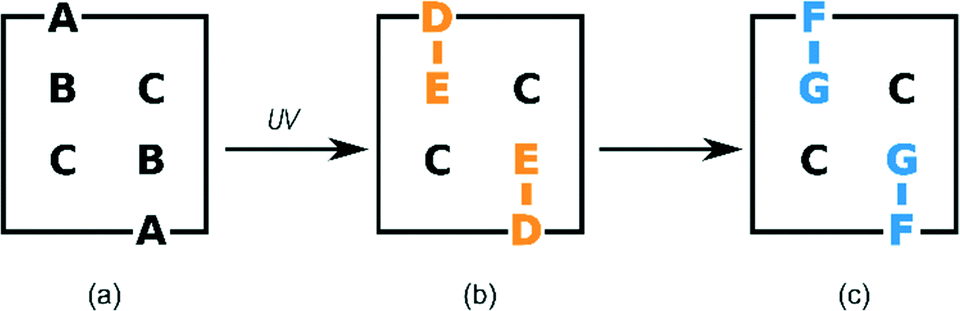 | ||
| Fig. 1 Schematic representation of the labelling system used to describe the different molecules during the photodimerisation; (a) molecules in the monomer structure; (b) photoproduct (DE) derived directly from the A and B monomers; (c) photoproduct FG after conformation change from DE in the solid-state. Molecule C remains unreacted during stage 1. | ||
Calculations
Intramolecular energies for DE, FG and C molecules were calculated with M06-2X39/6-311G(d,p) using Gaussian-09.40 Lattice and molecule⋯molecule interaction energies were calculated using the August 2014 version of AA-CLP.41Results and discussion
Reaction by UV radiation
The overall reaction scheme for the α′-polymorph is shown in Fig. 2. The photodimerisation of molecules in the α′-polymorph at 343 K can be thought of as taking place in two stages: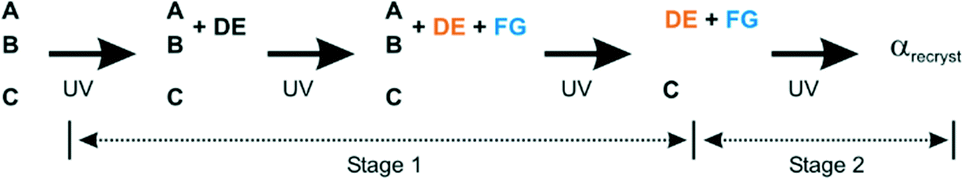 | ||
| Fig. 2 Overall reaction scheme for the α′ polymorph at 343 K. | ||
i. In stage 1 molecules A and B photodimerise to form the DE product (see also Fig. 3b). As the reaction proceeds beyond 67% conversion, some of the DE product undergoes a conformational rearrangement to the FG conformation (see also Fig. 3c). At the end of stage 1, when all the molecules in the AB reaction site have reacted, the photodimerisation product (DE) and its conformer (FG) occupy the former AB reaction site in an almost 1:1 ratio. Molecule C does not react during this stage. This process was monitored by SCXRD.
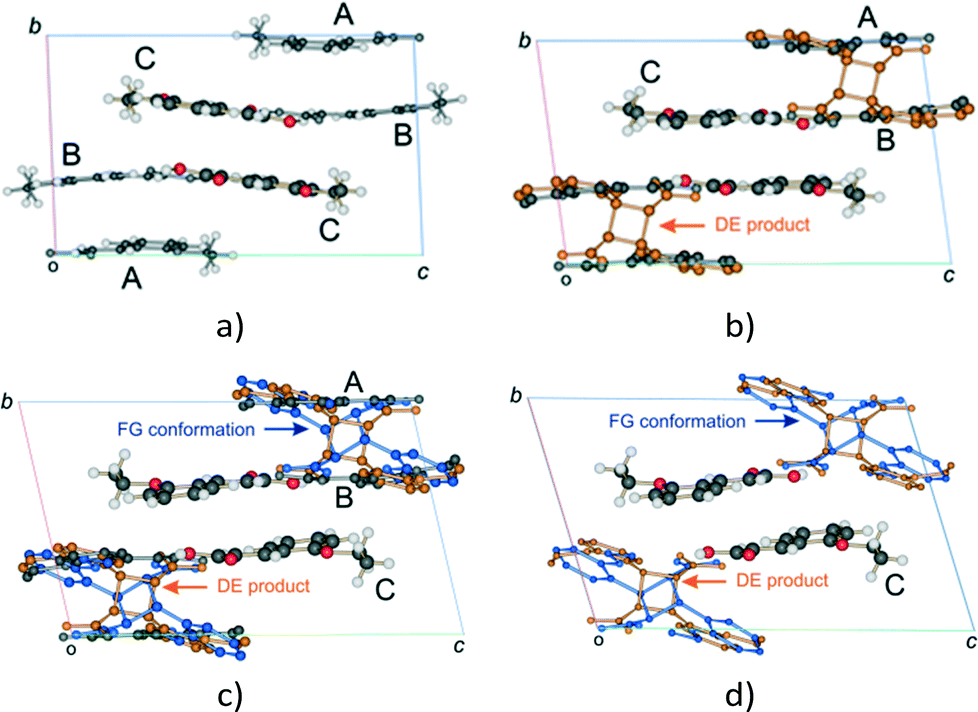 | ||
Fig. 3 Changes in the unit cell contents (viewed down the a axis) of the α′ polymorph (P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , Z = 6) with UV exposure time; (a) the α′ polymorph at 343 K showing the AB and CC reaction sites; (b) the unit cell after 6 to 24 hours (12–67% conversion) of UV exposure; (c) after 30 to 48 hours of UV exposure the AB site is occupied by the A and B molecules, the DE product and an alternate conformer of the product designated FG; (d) the final stage 1 product crystal (see Fig. 4) containing the DE and FG conformations of the product as well as the unreacted C molecules. At this point the crystal starts undergoing a phase change (stage 2), forming a phase that allows the C molecule to react. , Z = 6) with UV exposure time; (a) the α′ polymorph at 343 K showing the AB and CC reaction sites; (b) the unit cell after 6 to 24 hours (12–67% conversion) of UV exposure; (c) after 30 to 48 hours of UV exposure the AB site is occupied by the A and B molecules, the DE product and an alternate conformer of the product designated FG; (d) the final stage 1 product crystal (see Fig. 4) containing the DE and FG conformations of the product as well as the unreacted C molecules. At this point the crystal starts undergoing a phase change (stage 2), forming a phase that allows the C molecule to react. | ||
ii. The second stage involves a combination of conformational and phase transformations in the crystal enabling molecules in the CC (unreacted) site to dimerise yielding 100% of the photodimer (100% photoreaction). Due to the degradation of the crystals during the transformation processes (crystals become completely white and brittle), this stage cannot be monitored by SCXRD, and so other techniques such as powder diffraction must be used.
Details on the solid state reaction will therefore be presented in two parts – each concentrating on one of the two reaction stages described.
Stage 1
C bonds in molecules making up the CC site is around 4.8 Å (see ref. 29 for a detailed description) and hence too far for a photodimerisation reaction to take place. The distance between the CC bonds in the AB site is within 3.6 Å. Consequently, only the progress of the photodimerisation during stage 1 and not the reaction as a whole is shown in Fig. 5 and discussed in the text in terms of site occupancy factor (SOF) of the molecules in the AB site.
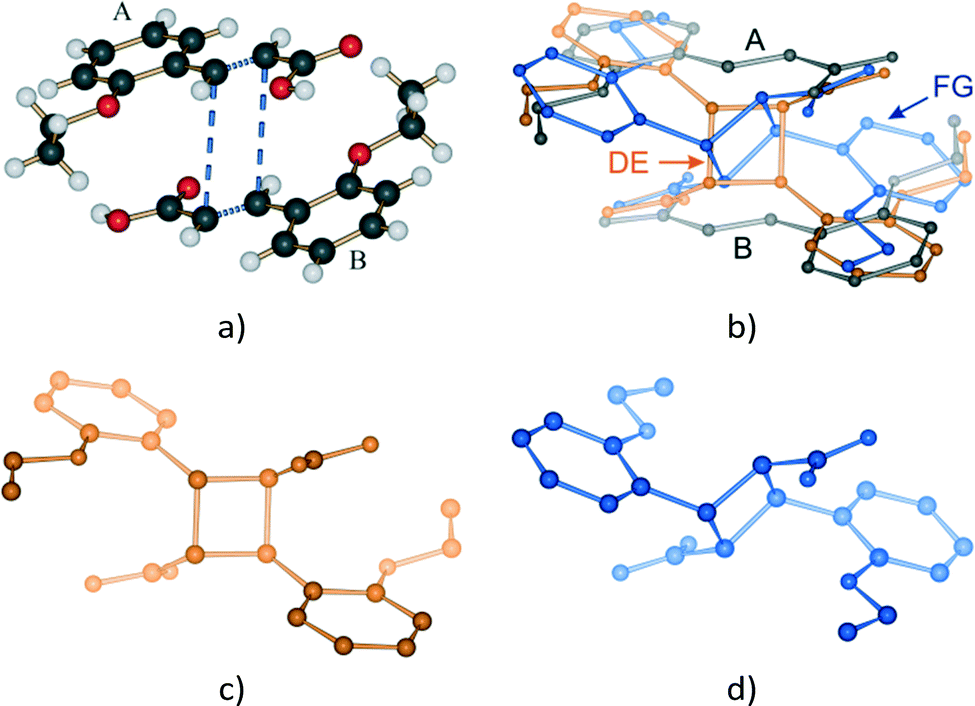 | ||
| Fig. 4 The AB reaction site before reaction and after 48 hours of UV exposure; (a) the AB predimer; (b) after 48 hours of UV exposure showing the remaining reactant molecules, the DE (or direct) photodimer and FG conformer; (c) the DE photodimer; (d) the FG conformer. The relative orientation of the ethoxy groups is maintained in the DE photodimer while in the FG conformer the ethoxy groups point in the opposite direction to those in the parent AB predimer. | ||
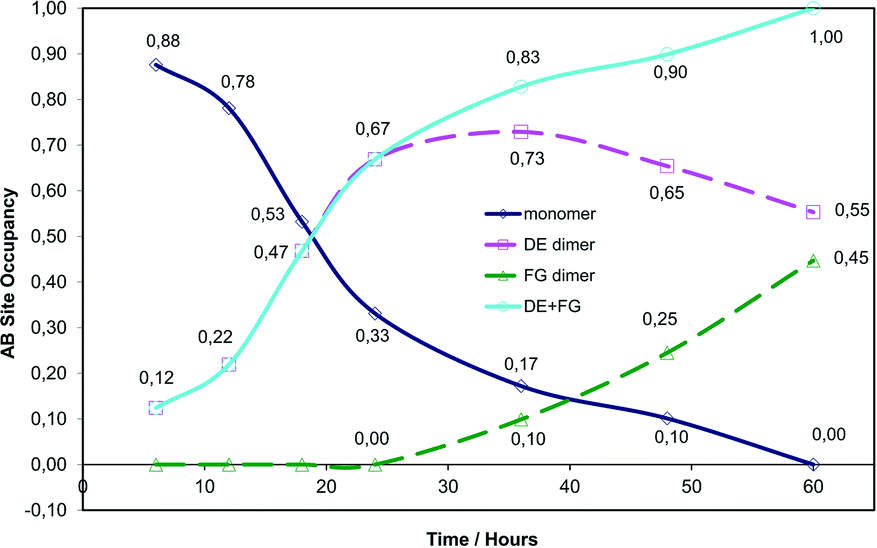 | ||
| Fig. 5 Graph of site occupancy (SOF) versus time for each component in the AB reaction site. | ||
We have previously reported on the reaction of the α′ polymorph at 293 K (ref. 15) and will make comparisons with the reaction at 343 K as there are many similarities and differences. The photodimerisation of the α′ polymorph at 343 K initially (6 to 24 hours of irradiation, 12 to 67% monomer conversion in the AB site) follows a similar path as at 293 K in that the photodimerisation occurs only at the AB site and only the DE (or direct) photoproduct is obtained at this point (reaction at 293 K effectively only involves the processes shown in Fig. 3a and b with the product crystal being composed of an ordered arrangement of DE and C molecules; the reaction occurs in one stage and there are no obvious side reactions). After 24 hours of irradiation (67% conversion in the AB site) the two reaction paths start to diverge. After 24 hours of irradiation at 343 K, the DE molecules start undergoing a conformational change in the solid state, forming the FG product (or conformation; Fig. 3c and d and 4) with monomer molecules still present at this point. The occupancy of DE molecules in the AB reaction site increases with reaction time reaching a maximum after 36 hours (83% conversion) and then decreases to a final site occupancy of 0.55 (55% conversion; Fig. 5). By contrast the FG conformer of the photodimer starts forming after 24 hours of irradiation to reach 45% conversion after all of the monomer molecules in the AB site have reacted (Fig. 3d).
The occupancy ratio of DE to FG molecules in the final stage 1 product is 0.55:0.45. Structure solutions of crystals that had been irradiated for 72 hours (72 h) and 120 hours (120 h) indicate that this ratio does not change. Upon closer examination this almost 1:1 ratio between the DE and FG component has structural implications which will be discussed later. A plot of ln(SOF) versus time for the monomer is linear indicating that the reaction is probably first order, suggesting that changes in the crystal environment during the reaction have minimal effect on the rate of reaction (Fig. S1†).
Plots showing changes in unit cell parameters with respect to dimer formation (sum of DE and FG occupancies) in the AB site are shown as solid lines in Fig. 6. Corresponding data from the photodimerisation study at 293 K are superimposed as dotted lines. In contrast to the reaction at 293 K, the unit cell parameters change dramatically as the reaction proceeds at 343 K. Initially, the reaction at 343 K is structurally equivalent to that at 293 K but deviates significantly after 67% conversion has been reached. This is a consequence of the conformational changes occurring in the crystal during the reaction. The plots indicate that the a axis contracts by 0.27 Å (3.1% compared with 3.9% at 293 K) in an apparently linear manner as the reaction progresses. By contrast the b axis expands, slowly at first, then abruptly after about 80% conversion. The total change is 0.38 Å (3.5%) with most of the change (0.34 Å; 3.1%) occurring after 67% conversion. The c axis initially expands by 0.11 Å (0.64%) until the dimer occupancy reaches 67% then contracts from this point by 0.31 Å (1.8%). The total change in the c axis is 0.20 Å (1.1%). Interplanar unit cell angles (α and γ; the α′ polymorph being a layered structure) change significantly during the course of the reaction. Initially they follow very similar paths to those at 293 K but deviate significantly once 67% conversion has been achieved. The α angle increases by about 13.59° (14.7%) or 6.30° more than at 293 K. The γ angle decreases by 8.061° (7.16%) or 5.89° more than at 293 K. Both the α and β angles end up at around 105° once the molecules in the AB site have fully reacted. By contrast the β angle, which lies in the relatively incompressible molecular plane containing the phenyl groups, changes by only 0.52° (0.56%). As at 293 K, cell volumes decrease as the reaction proceeds but do not reach an equivalent minimum. The largest difference between the starting volume and final volume is −13.9 Å3 (−0.93%) for reaction at 343 K while it is −24.3 Å3 (−1.6%) at 293 K. The larger cell volume for crystals irradiated at 343 K (Fig. 6) is probably a consequence of the molecules being disordered leading to less efficient packing.
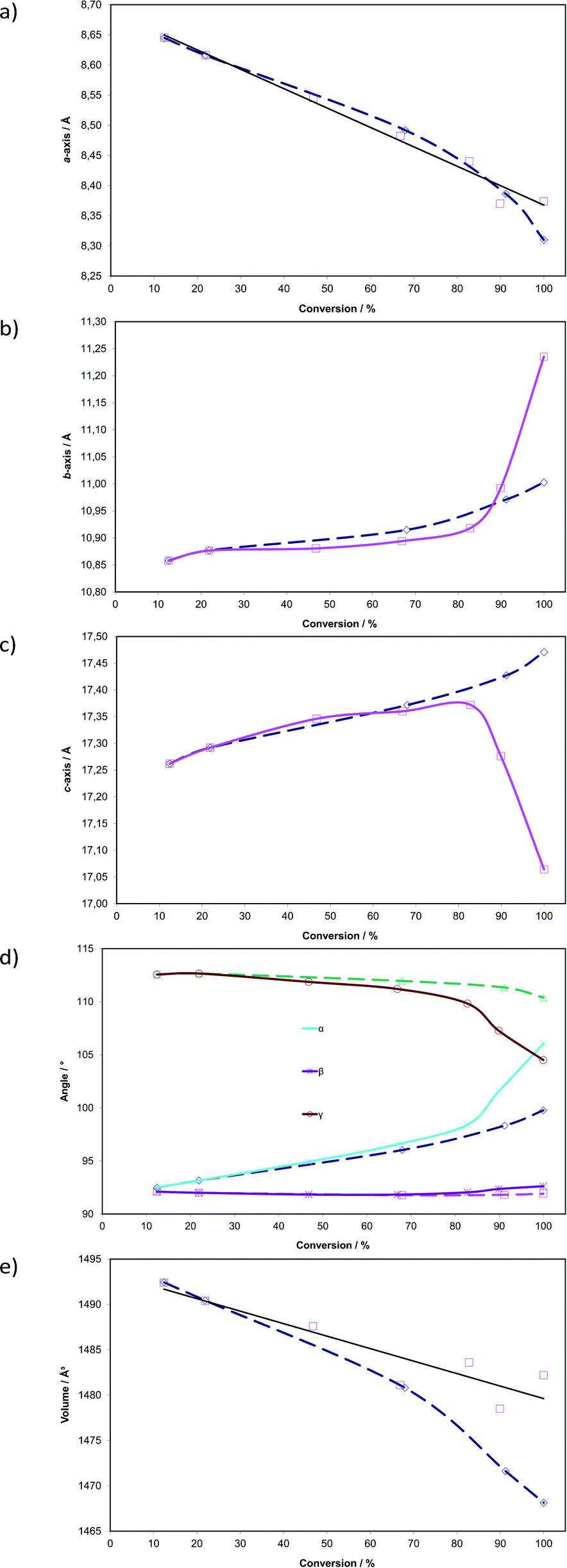 | ||
| Fig. 6 Changes in unit cell parameters with respect to photodimer formation (sum of the site occupancies of DE and FG conformers; see Fig. 5) in the AB reaction site. Corresponding data from the photodimerisation study at 293 K (ref. 15) are superimposed as dashed lines (see text for details). | ||
These large changes in the unit cell parameters are a consequence of changes in the AB reaction site, as the molecules in the site have to adapt to the shape of the photoproduct and its conformer as the reaction proceeds in order to maintain crystal integrity. In addition the geometry of the CC site will also be affected by changes in the AB site. However, detailed analysis of the crystal structures indicates that the relative distances between the reactant molecules in the AB and CC sites change by less than 0.2 Å during the course of the reaction until 80% conversion is achieved, while at the same time the relative orientation of the molecules changes by less than 5°. These changes are similar to those occurring at 293 K. Larger changes occur after 80% conversion as the crystal anneals towards the final stage 1 product, but none of these provide any direct indication for how the conformation from the DE to FG product might occur even though the conformational change predominantly occurs after 80% conversion. A detailed description of the structural changes occurring in the AB and CC sites can be found in the ESI.†
Molecular structure
ORTEP diagrams of the stage 1 product crystal at 343 K are shown in Fig. S2.† The ORTEP diagrams for molecules DE and FG (Fig. S2b and c†) reveal that while most ellipsoids are spherical and well behaved those of the D ethoxy group (O21D, C21D and C22D) in the DE conformer are quite large. This feature is present in the structure solutions after 48, 60 and 72 h of UV exposure, and may provide a clue to the origin of the conformation change as well as the phase change in stage 2 of the reaction process. Due to the α′ polymorph having two reaction sites, one photo-reactive and one photo-stable (in stage 1) at 343 K, it is possible to monitor the change in conformation of molecule C as a result of stresses caused by the reaction process. Measuring the r.m.s. (root-mean square) deviation from planarity for C molecules at various reaction times indicates that it increases steadily from 0.065 Å after 6 hours (structure 6 h; 12% conversion) of UV exposure and reaches a maximum of 0.107 Å after 48 hours (structure 48 h; 92% conversion). The r.m.s. deviation from planarity then drops to 0.103 Å after 60 hours (structure 60 h; 100% conversion) of reaction. The deviation from planarity is due to changes in the conformation of the carboxylic and ethoxy groups in molecule C (Fig. 7). The superimposition shown in Fig. 7 indicates that the conformation of the ethoxy group (C21C and C22C) changes in a regular manner in molecule C as the reaction proceeds. The position of C22C undergoes a total change of about 0.81 Å during the reaction. The conformation of the carboxylic acid group also changes in a regular manner up to a conversion of 92% (48 hours of irradiation). The total change in the C13C position is about 0.37 Å at this point. The carboxylic acid group then moves about 0.20 Å (more than half way) back towards the starting position when 100% conversion has been achieved (60 h of irradiation). Since the carboxylic acid is hydrogen bonded to dimer molecules in the AB reaction site, this indicates that large changes occur in the structure of the AB site (and crystal) as the reaction nears completion. Calculating the intramolecular energy of the C molecule in all the reacted crystals using M06-2X39/6-311G(d,p) indicates that the difference between the lowest energy conformation (molecule C in 18 h) and the highest energy conformation (molecule C in 48 h) is only about 2.74 kJ mol−1, with the energies of the C molecules in the other structures being between these values (Fig. 7b). The reaction therefore has a very small effect on the energy of the C molecule. However, the distribution of energies in Fig. 7b allows one to use molecule C as a barometer to gain speculative insight into the pressure experienced by molecules within the crystal at different levels of conversion. At low conversion (points associated with 12.4 and 21.9% conversion), the photodimer has to fit within the structure of the monomer and as a result creates stress within the crystal. Consequently molecule C adopts a higher energy conformation. DE is also unable to convert to FG at this point. During the intermediate stages of the reaction (points associated with 46.8, 66.9, and 82.8% conversion) molecule C adopts the lowest energy conformations suggesting that the crystal is now flexible (presumably due to space created by the reaction) enough to allow the molecule to relax. Coinciding with this is the DE to FG conformational change discussed earlier. As the reaction nears completion (89.9% conversion and above) the energy of the C molecule suddenly reaches a maximum (coinciding with the large changes in the cell parameters shown in Fig. 6), but decreases in energy as the reaction nears completion and the crystal adapts (anneals) towards the final stage 1 product structure.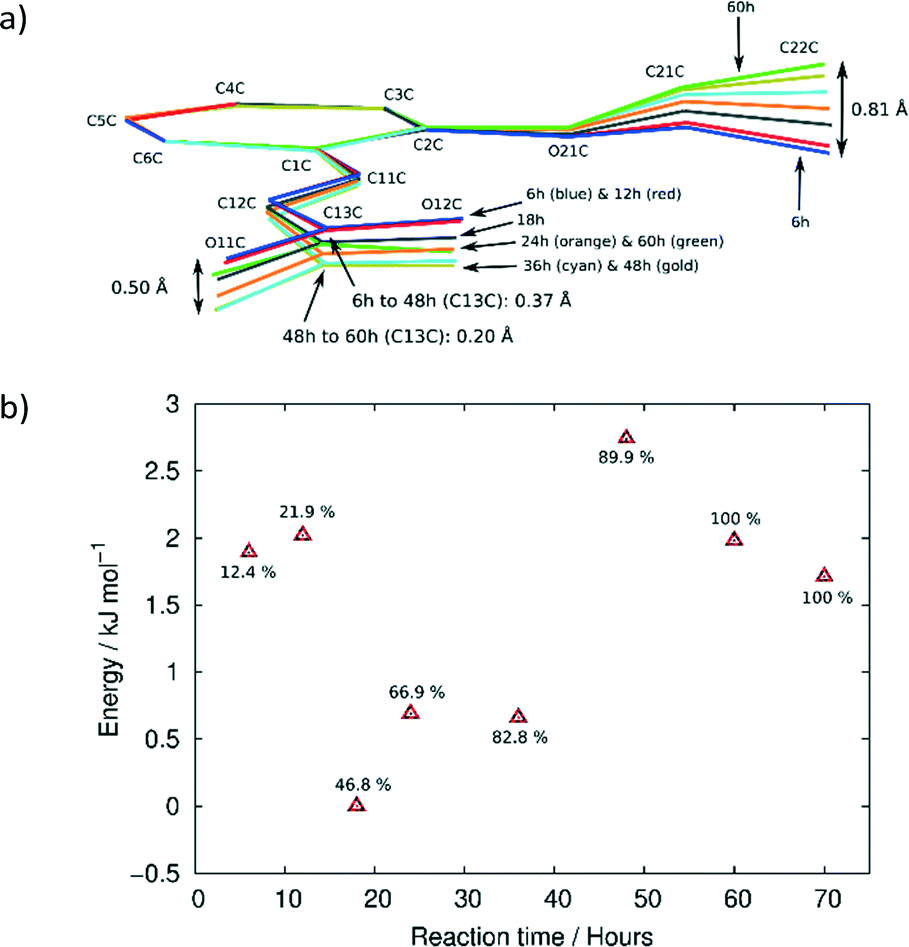 | ||
| Fig. 7 (a) Superimposition of C molecules from the 6, 12, 18, 24, 36, 48 and 60 h structures by least squares fit of corresponding phenyl (C1C–C6C) atoms. (b) Relative energies of the C molecules at different reaction times. Indicated next to each point is the degree of conversion in the AB site. | ||
Upon undergoing a photodimerisation reaction in the AB site, the DE photodimer retains a very similar conformation to the AB predimer (Fig. 4). Most torsion angles for molecules A, B, C, and the DE photodimer differ by less than 20° (Table 1). This similarity is understandable as the dimer product has to fit within the reaction cavity containing the substrate A and B molecules and is therefore derived from the geometric relationship between the A and B molecules. The FG conformation of the product differs mainly by a rotation around the C1–C11 bond leading to the ethoxy group pointing in a direction opposite to that in the DE conformer (Fig. 4). This results in the ethoxy group lying below and above the cyclobutane ring in the FG conformer instead of pointing away from the cyclobutane ring as in the DE conformer. The difference in the C2–C1–C11–C12 torsion angle between the DE and FG conformer is about 140° (Table 1). In addition there is also some rotation around C12–C13 causing the relative orientations in the carboxylic acids to differ by about 38° between the DE and FG conformers.
| Moleculea | C2–C1–C11–C12 | O11–C13–C12–C11 | O12–C13–C12–C11 |
|---|---|---|---|
| a Data for molecules A, B and C were obtained from structure 6 h. Data for molecules DE and FG were obtained from structure 60 h. | |||
| A | −177.8 (2) | 179.4 (2) | −1.2 (4) |
| B | −172.8 (2) | −174.2 (2) | 6.0 (4) |
| C | −176.9 (2) | −175.92 (19) | 4.9 (3) |
| D | −164.5 (4) | −168.3 (6) | 16.6 (11) |
| E | 167.9 (8) | 173.4 (13) | −9.7 (18) |
| F | 51.8 (6) | 159.0 (12) | −24 (3) |
| G | −55.8 (12) | −157.0 (18) | 22 (2) |
The conformation of the DE and FG molecules from structures 48, 60 and 72 h were compared by least squares superimposition to the DE product obtained by reaction at room temperature (293 K; labelled r48 h in the remaining text)15 as well as the photodimer obtained after recrystallisation from solution (2,2′-diethoxy-α-truxillic acid structure published by Gopalan and Kulkarni in 2001;42 CSD43 refcode XOSKEV). The result of a superimposition of DE molecules from the 48, 60, 72 h and r48 h structures is shown in Fig. 8. The superimposition indicates that the conformation of the DE molecule is identical in structures at 60 and 72 h. The conformation of the DE molecule from 48 h lies in between that of 60 h and r48 h. The largest difference between r48 h and 60 h is in the orientation of the phenyl ethoxy group, with the difference in the position of C22D in the two structures being about 0.77 Å. There are also differences in the orientation of the carboxylic acid with the largest difference being the position of O11E at about 0.31 Å. The conformation of the rigid cyclobutane containing core of the molecule made up of carbon atoms 1D, 11D, 12D, 13D, 1E, 11E, 12E and 13E does not vary much among the four structures.
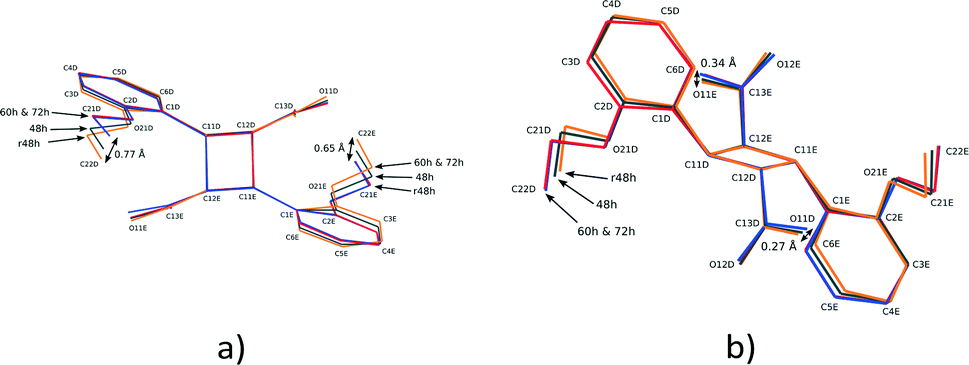 | ||
| Fig. 8 Two different views of the superimposition of the DE photodimer molecules from the r48 h (final DE product crystal structure after 48 h reaction at 293 K), 48, 60 and 72 h structures. | ||
The result of a superimposition of FG molecules from the 48, 60 and 72 h structures, as well as the photodimer obtained after recrystallization from solution (XOSKEV), is shown in Fig. 9. The superimposition indicates that the conformation of the FG molecule is identical in structures 48, 60 and 72 h, and very similar to XOSKEV. The largest difference in conformation between these structures and XOSKEV is again the orientation of the phenyl ethoxy group. The maximum differences in atom positions in the XOSKEV and other structures is about 0.41 Å for the ethoxy groups and 0.29 Å for the carboxylic acid groups. The differences in the molecules are an indication of the effect of packing forces on molecular conformation, with molecules obtained under the most different conditions showing the largest structural differences.
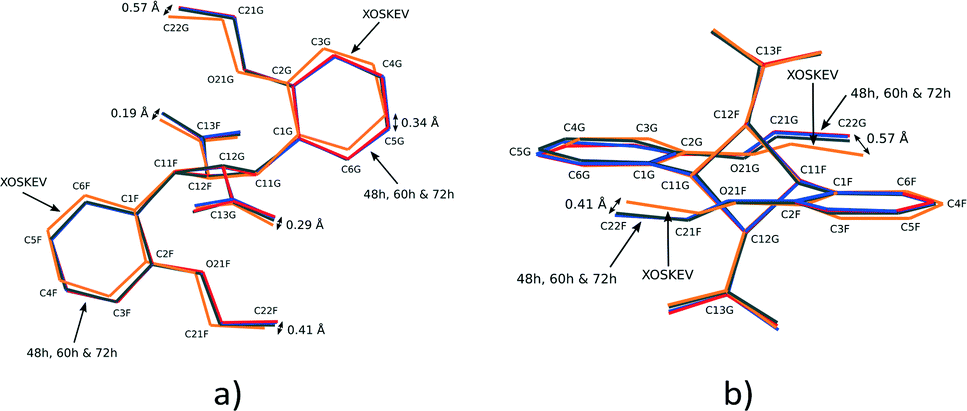 | ||
| Fig. 9 Two different views of the superimposition of FG photodimer molecules from XOSKEV (recrystallized photodimer), 48, 60 and 72 h. | ||
Hydrogen bonding
The asymmetric unit of 60 and 72 h (the final stage 1 product crystals) contains three molecules capable of hydrogen bonding, potentially forming the following hydrogen bonded string of molecules: C⋯DE⋯DE⋯C or C⋯FG⋯FG⋯C or C⋯DE⋯FG⋯C. Graphical comparison of hydrogen bonding involving these different combinations of molecules is shown in Fig. 10. Though distorted, the hydrogen bonding geometry between molecule C and both conformers of the photoproduct, DE and FG, is typical of carboxylic acids, with an average D⋯A distance of 2.63 Å (Table 2). By contrast, DE molecules do not form reasonable hydrogen patterns with each other for two reasons: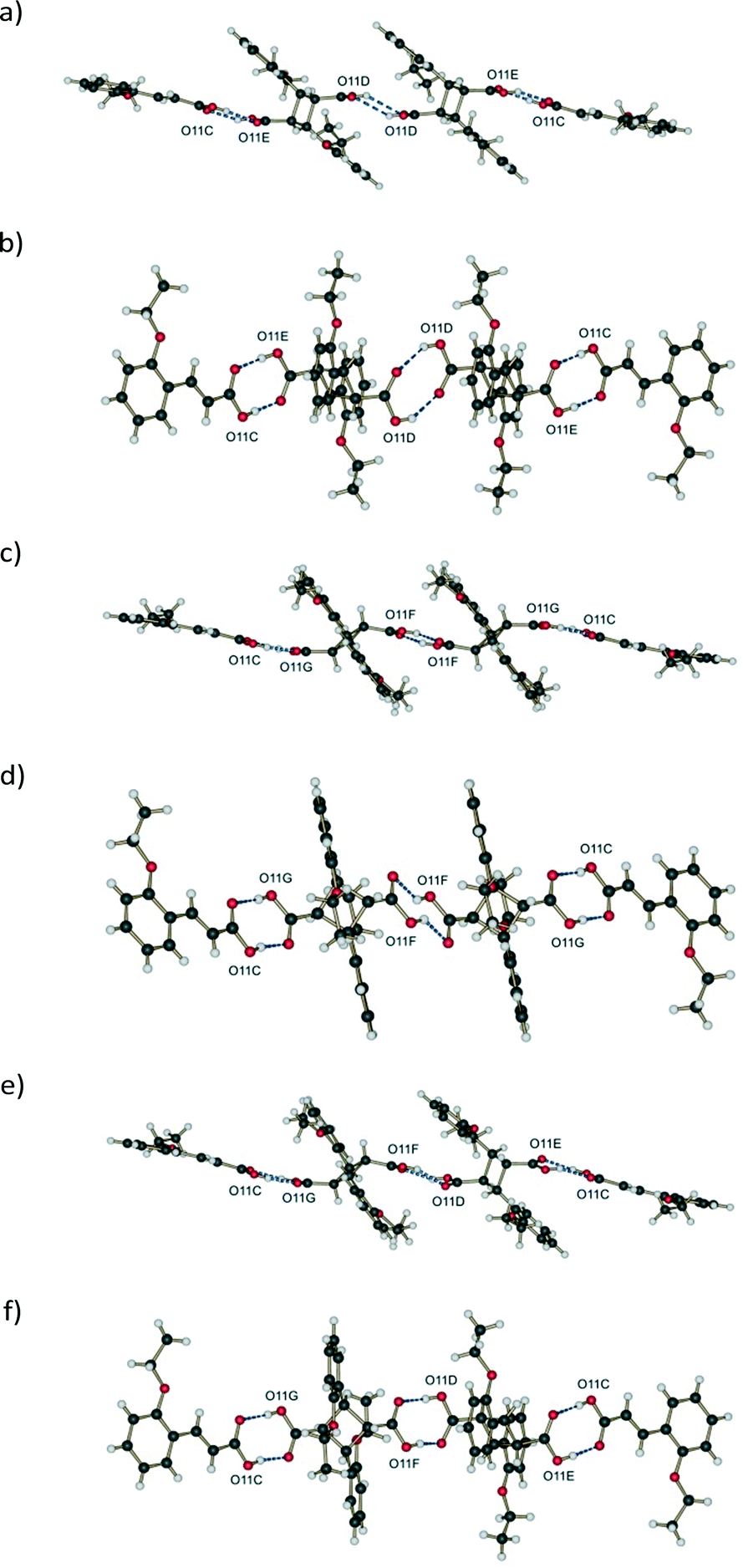 | ||
| Fig. 10 Graphical comparison of hydrogen bonding involving different combinations of molecules in the asymmetric unit of d60 h showing two different views for each; (a) and (b) DE:DE; (c) and (d) FG:FG; (e) and (f) DE:FG. | ||
| Structure | D–H⋯A | D–H | H⋯A | D⋯A | <D–H⋯A |
|---|---|---|---|---|---|
| 60 h | O11D–H1D⋯O12Fi | 0.84 | 1.82 | 2.610(6) | 156 |
| 60 h | O11F–H1F⋯O12Di | 0.84 | 1.85 | 2.659(9) | 160 |
| 60 h | O11C–H1C⋯O12Eii | 0.84 | 1.87 | 2.712(13) | 176 |
| 60 h | O11E–H1E⋯O12Cii | 0.84 | 1.84 | 2.645(16) | 160 |
| 60 h | O11C–H1C⋯O12Gii | 0.84 | 1.68 | 2.504(18) | 166 |
| 60 h | O11G–H1G⋯O12Cii | 0.84 | 1.85 | 2.670(16) | 165 |
| 60 h (unlikely) | O11D–H1D⋯O12Di | 0.84 | 2.60 | 3.314(8) | 144 |
| 60 h (unlikely) | O11F–H1F⋯O12Fi | 0.84 | 1.65 | 2.313(6) | 134 |
| r48 h | O11D–H1D⋯O12Di | 0.92(2) | 1.80(2) | 2.7138(18) | 169(2) |
| r48 h | O11C–H1C⋯O12Eii | 0.87(2) | 1.80(2) | 2.6619(18) | 176(2) |
| r48 h | O11E–H1E⋯O12Cii | 1.00(2) | 1.64(3) | 2.6350(18) | 169(2) |
| 6 h | O12A–H1A⋯O11Ai | 0.84 | 1.73 | 2.566(3) | 170 |
| 6 h | O11C–H1C⋯O12Bii | 0.84 | 1.80 | 2.622(3) | 166 |
| 6 h | O11B–H1B⋯O12Cii | 0.84 | 1.76 | 2.591(2) | 168 |
| α | O11–H11a⋯O12iv | 0.84 | 1.80 | 2.637(2) | 172 |
| β | O12–H110⋯O11v | 0.84 | 1.78 | 2.618(2) | 177 |
| γ | O11–H110⋯O12vi | 0.84 | 1.80 | 2.6249(17) | 165 |
| α-Dimer (XOSKEV) | O2–H2a⋯O1vii | 0.96(2) | 1.67(2) | 2.627(3) | 174(5) |
i. The donor and acceptor oxygen atoms are too far from each other. The D⋯A distance is 3.31 Å which is very unusual for OETCA (Table 2),
ii. The carboxylic acids are vertically and horizontally separated (Fig. 10a and b) by a large distance.
The FG molecules also display unusual hydrogen bond geometry in that the carboxylic acid groups are horizontally displaced from each other (Fig. 10c and d).
The last combination, FG hydrogen bonded to DE (Fig. 10e and f), shows a reasonable (if distorted) hydrogen bond geometry with reasonable D⋯A distances. The distorted geometry is probably an artefact of the disorder model used to solve the structures as well as using calculated carboxylic hydrogen positions during structure refinement which further exaggerate the distortion. The geometry is, however, not very different from the DE to DE hydrogen bond geometry present in the 293 K product crystal (see 48 h in ref. 15). Based on the above considerations the most likely hydrogen bonded combination is C⋯DE⋯FG⋯C. This string of molecules is also consistent with the arrangement of molecules in the crystal (see crystal packing discussion below). However, since from the structure refinements the DE:FG ratio is 55:45, the C⋯DE⋯DE⋯C hydrogen bonded combination has to be partially present in 60 h. Hydrogen bond distances for the different geometries are given in Table 2.
In addition to the classical hydrogen bonding, 60 h is also kept together by weak C–H⋯O interactions. As for the reaction at room temperature (r48 h; final reacted crystal for the reaction at 293 K),15 these are derived from the parent α′ polymorph crystal and stabilize the final stage 1 product crystal. However, since the predominant hydrogen bonded string is C⋯DE⋯FG⋯C, where the conformation of the FG molecules disrupts the C–H⋯O hydrogen bond pattern, C–H⋯O interactions do not play as large a role in stabilizing the 343 K structure. The C–H⋯O interactions between C molecules do, however, still exist. These act to stabilize the assembly of C⋯DE⋯FG⋯C molecules forming a ribbon down the a axis similar to r48 h.
Crystal packing
Like the room temperature product crystal (r48 h), the stage 1 product crystal 60 h has features in common with its parent crystal (Fig. 4a and d). The corrugated sheet structure and the boundaries of the original ribbons in the α′ polymorph can still be seen in the structure of 60 h (Fig. 11). Consequently the structure and crystal packing of the 60 h can be viewed as derived from its parent crystal. Whereas the hydrogen bonded OETCA dimer is the basic building block of the α′ polymorph, the hydrogen bonded string of molecules made up of two C molecules and the two dimer molecules (C⋯DE⋯FG⋯C; Fig. 10e and f; see hydrogen bonding section) can be viewed as the basic building block of 60 h. These hydrogen bonded units are then further assembled via C–H⋯O interactions (derived from the monomer crystal) to form a new ribbon running down the a axis. These ribbons are then held together by other weak interactions to form a layer of molecules. The upper and lower parts of the hydrogen bonded units can be thought of as defining the limits of a layer (marked with dotted lines in Fig. 11), which stack on each other along the b axis.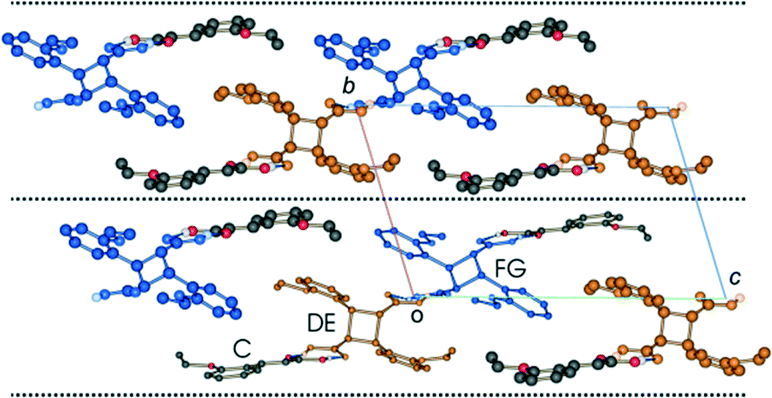 | ||
| Fig. 11 Crystal packing of the structure determined after 60 h of UV exposure (67% conversion) at 343 K as viewed down the a axis. | ||
Examining close contacts between the DE and FG molecules (Table 3) in structures 48, 60 and 72 h provides good evidence for an ordered arrangement of DE and FG molecules in the crystals. Fig. 12 graphically represents a close contact between DE molecules related by a centre of inversion on the ac face of the unit cell in 60 and 72 h. Here the C22D⋯C22D contact distance is about 3.13 Å. This is about 0.5 Å closer than in 48 h (about 90% conversion; DE:FG ratio 67:25) and r48 h (final 293 K product crystal; Table 3). No close contact exists between DE and FG molecules when a FG is substituted into one of the DE positions which implies that an alternating relationship exists between DE and FG molecules along the a axis. Examining the FG molecules for close contacts indicates that there is an unreasonably close contact between O12F and C22F (about 2.3 Å; Fig. 13) as well as a very close contact between O12G and C22G (2.85 Å). Here the FG molecules involved are related by translational symmetry along the a axis. Testing an alternating arrangement of FG and DE molecules along the a axis produces no close contacts, which implies that an alternating arrangement exists between DE and FG molecules down the a axis. The arrangement of molecules that results from taking these considerations into account is shown in Fig. 14. This is the most likely local arrangement between molecules within a layer in the 60 and 72 h structures. This arrangement also produces the C⋯DE⋯FG⋯C string of molecules mentioned in the hydrogen bond section. It also indicates that there is a very high level of cooperativity in the DE to FG conformation change. Hirshfeld surfaces and Divs. De plots36 for molecules in the various arrangements are given in Fig. S3 in the ESI.† Included for comparison are Hirshfeld surfaces for the DE molecule from the α′-polymorph reacted at 298 K (r48 h) where the final product is ordered, as well as the FG molecule after recrystallization. Very close contacts can be clearly seen around the DE ethoxy groups in a hypothetical crystal composed of C⋯DE⋯DE⋯C only (Fig. S3a†). Very close contacts are also present around the FG carboxylic acid and ethoxy groups in a hypothetical crystal composed of C⋯FG⋯FG⋯C only (Fig. S3d†). These close contacts mostly disappear when the packing arrangement in Fig. 14 is considered (Fig. S3b and e†).
| 72 h | 60 h | 48 h | r48 h | |
|---|---|---|---|---|
| FG⋯FG | ||||
| O12F⋯C22F | 2.305 | 2.295 | 2.408 | — |
| O12G⋯C22G | 2.831 | 2.854 | 2.878 | — |
| C13F⋯C13F | 3.482 | 3.483 | 3.478 | |
| DE⋯DE | ||||
| C22D⋯C22D | 3.159 | 3.134 | 3.600 | 3.648 |
| C13D⋯C13D | 4.355 | 4.360 | 4.121 | 3.874 |
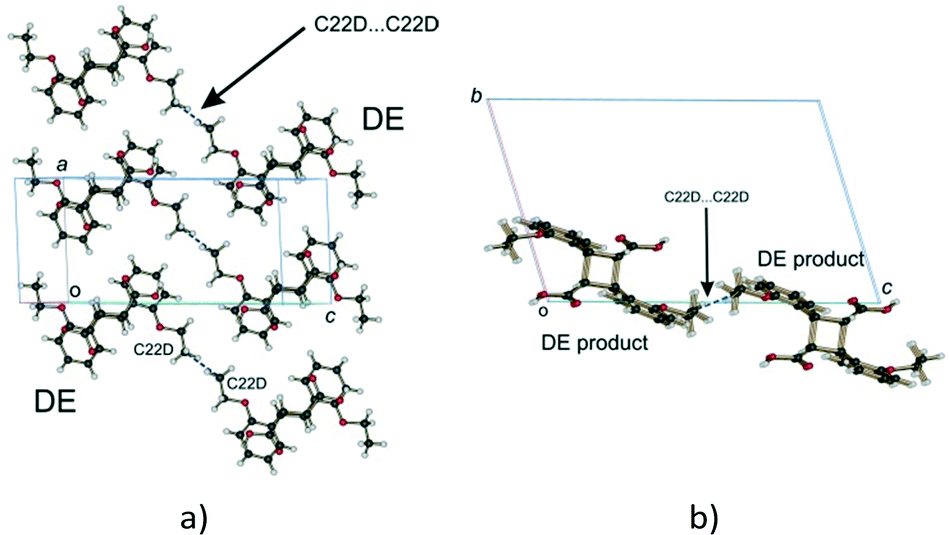 | ||
| Fig. 12 Close contacts between DE molecules; (a) view perpendicular to the ac plane; (b) view down the a axis. | ||
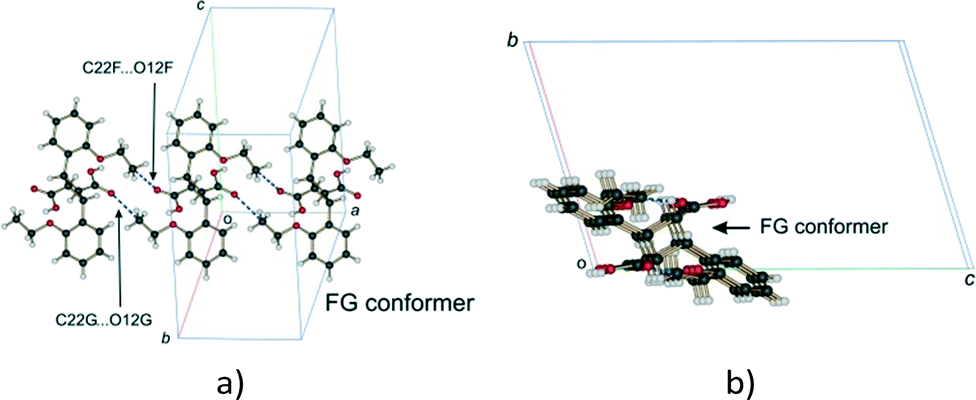 | ||
| Fig. 13 Close contacts between FG molecules; (a) view perpendicular to the molecular plane; (b) view down the a axis. | ||
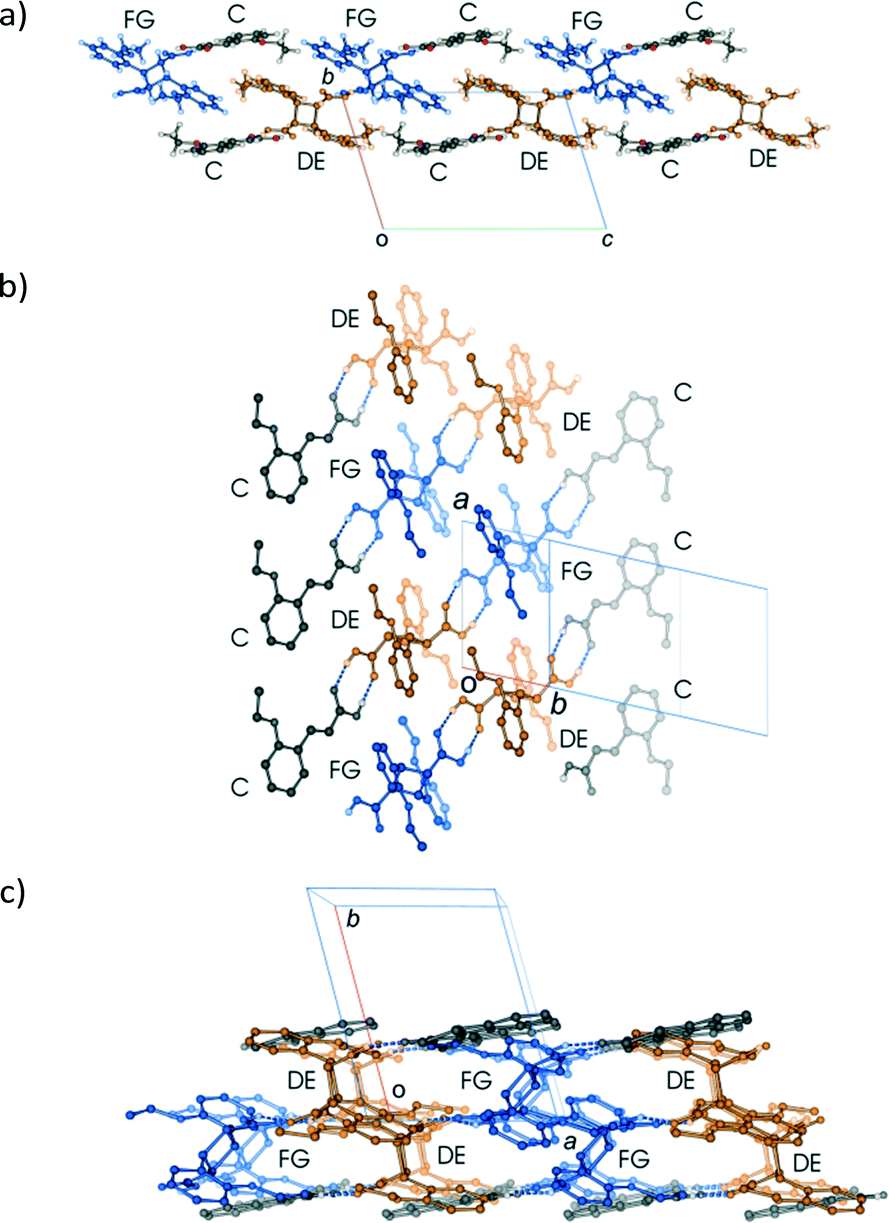 | ||
| Fig. 14 Different views of the alternating arrangement of DE and FG molecules within a layer; (a) view down the a axis; (b) view approximately perpendicular to the ribbon of molecules; (c) view down the c axis. The diagram in (c) indicates that the alternating arrangement of DE and FG molecules form alternating stacks of DE and FG molecules, each stack aligned along the c axis providing clues for possible conformational change mechanisms. | ||
To confirm that this packing arrangement is energetically feasible, lattice energy calculations were carried using AA-CLP41 on a hypothetical crystal consisting of the packing arrangement shown in Fig. 14 and compared with the lattice energy of the previously room temperature product crystal (r48 h). For the arrangement shown in Fig. 14 the lattice energy was found to be −167.5 kJ mol−1, while the lattice energy for r48 h was found to be −174.9 kJ mol−1 – a difference of only 7.4 kJ mol−1. In fact the Coulombic, polarisation, dispersion and repulsion energy contributions to the lattice energy are all about the same for the two structures: −41.8, −48.3, −177.2, and 99.7 kJ mol−1 respectively for the arrangement in shown in Fig. 16, versus −43.9, −47.8, −177.8, and 94.5 kJ mol−1 respectively for r48 h. Part of the energy difference between the two structures comes from the higher repulsion energy in the Fig. 14 arrangement (about 5.2 kJ mol−1). Another reason for the difference is that the molecule⋯molecule interaction energy (including the hydrogen bond) for the DE⋯FG arrangement (−57.7 kJ mol−1) in Fig. 14 is much less than the DE⋯DE arrangement (−70.7 kJ mol−1) in r48 h – a difference of about 13 kJ mol−1. One reason for the higher energy of the DE⋯FG arrangement is that the hydrogen bond geometry between these molecules is distorted due to approximations introduced in the disorder model, even though it is better than the alternatives (Fig. 10). In addition, the hydrogen positions were calculated rather than placed. By contrast there is no disorder in the r48 h structure so all atoms were placed directly and the carboxylic hydrogen atoms refined. In addition, the molecule⋯molecule energy (including hydrogen bond) for the C⋯DE arrangement in r48 h is −52.9 kJ mol−1, while the C⋯DE and C⋯FG arrangements contribute −50.8 and −28.5 kJ mol−1 respectively to the arrangement in Fig. 14. In the latter case the O⋯O distances for the hydrogen bond between molecules C and FG are quite short (average 2.58 Å) with the hydrogen bond angle and hydrogen bond position being non-ideal. It is therefore likely that the energy difference between the two arrangements is very small, and that in fact the arrangement in Fig. 14 is more stable than r48 h which favours the conformation change. In addition, intramolecular energy calculations using Gaussian-09 (ref. 40) and M06-2X39/6-311G(d,p) (bond and torsion angles fixed, bond lengths and hydrogen atom positions allowed to optimize – see experimental in ESI†) indicate that the FG conformation is significantly more stable than the DE conformation. The relative intramolecular energies are as follows: photodimer obtained after recrystallization from solution (XOSKEV; same conformation as FG) and therefore expected to be in the most stable conformation: 0.0 kJ mol−1, FG conformer present in 60 h: +15.9 kJ mol−1, DE molecule present in r48 h: +20.5 kJ mol−1, and DE molecule present in 60 h: +36.4 kJ mol−1. The conversion from DE to FG is therefore energetically favoured. The arrangement shown in Fig. 14 is therefore probably more stable than r48 h for both intra- and intermolecular reasons.
Lattice energy calculations were carried out on several alternative arrangements of C, DE and FG molecules but all were found to be less stable. For example, an hypothetical crystal composed of the C⋯DE⋯FG⋯C arrangement in a P1 unit cell but ignoring the alternating relationship of DE and FG molecules along the a axis (effectively the packing shown in Fig. 11), leads to a lattice energy of −97.0 kJ mol−1. The lattice energy for a crystal containing the C⋯DE⋯DE⋯C arrangement only is −144.5 kJ mol−1, while for a crystal containing the C⋯FG⋯FG⋯C arrangement it is +1163.5 kJ per mole. As mentioned before, the DE:FG ratio at the end of the stage 1 is 55:45. The excess DE molecules will therefore form an excess amount of C⋯DE⋯DE⋯C arrangements in the crystal which, while not optimal, is still energetically favourable. An excess of FG molecules and hence an excess of the C⋯FG⋯FG⋯C arrangement would be very energetically unfavourable.
Since the photodimerisation occurs randomly in the solid state, the ordered arrangement of DE and FG molecules is probably restricted to only part of a layer, forming a domain which is surrounded by domains where the positions of the DE and FG molecules are swapped around. There seems to be no restriction on the relationship between the layers along the b axis (Fig. 11). An ordered arrangement of layers is therefore unlikely. The average of this random arrangement of layers and domains within a layer, would appear in an X-ray diffraction experiment as a centrosymmetric crystal, with the FG and DE molecules disordered in the asymmetric unit as experimentally observed in this work.
Stage 2
In the second stage of the photodimerisation process, the stage 1 crystal product undergoes a phase change in which the C molecules dimerise and a final 100% photodimer product is obtained. Though the final product crystal has little in common with the stage 1 product crystal it is the result of molecules in the AB site (stage 1) and CC site (stage 2) photodimerising. Due to the degradation of the crystals during the phase transformation process, this stage cannot be monitored by SCXRD, so other techniques such as powder diffraction have to be used. Two powder patterns of the final stage 2 product are compared with the powder pattern of the recrystallized photodimer in Fig. 15.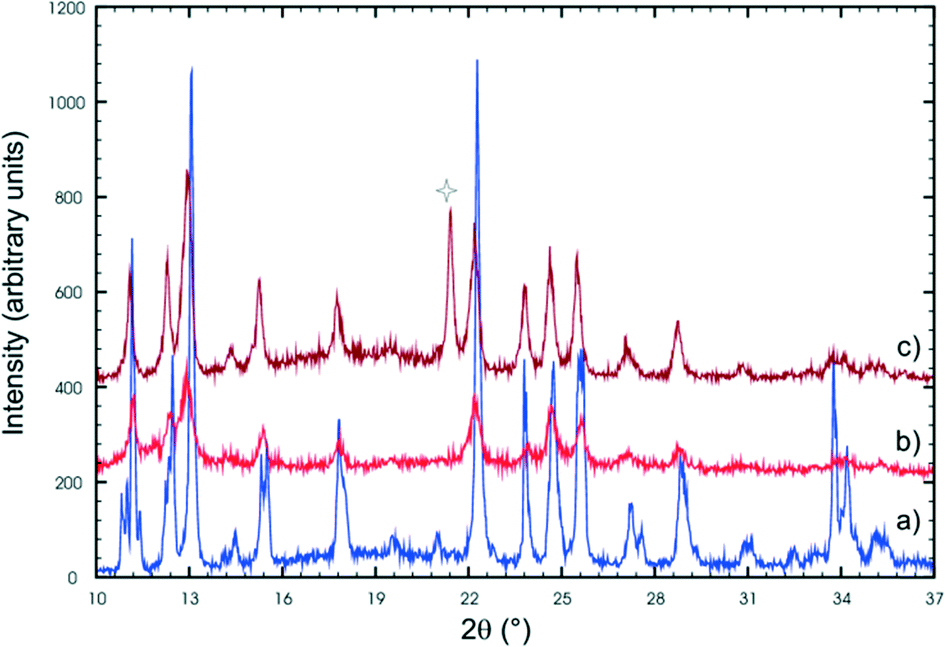 | ||
| Fig. 15 X-ray powder patterns of (a) the recrystallized photodimer, and the final stage 2 photoproduct (b and c). The peak indicated by a star is due to a contaminant (probably dust as the powders were reacted in an open laboratory environment). | ||
Powder patterns of the final stage 2 photoproduct are identical to that of the recrystallized photodimer. The structure of the recrystallized photodimer has already been reported by Gopalan & Kulkarni in 2001 [space group P21/n; a = 8.3547(6) Å, b = 8.4793(2) Å, c = 14.196(1) Å, β = 106.810(2)°, V = 962.7(1) Å3, Z = 2; CSD recode XOSKEV]42 and only two details will be presented here. The most obvious is that the recrystallized photodimer has a conformation nearly identical to that of the FG conformer (Fig. 9). This means that the orientation of the ethoxy groups is completely different in the final product (ethoxy groups sitting above the cyclobutane ring) when compared to the conformation in the predimers (ethoxy groups pointing away from the cyclobutane ring). The stage 2 phase change and final photodimerisation reaction therefore involves a rearrangement of the ethoxy groups. The photodimerisation in stage 2 eventually joins neighbouring layers from stage 1 together to form an infinite chain of hydrogen bonded photodimer molecules (Fig. 16).
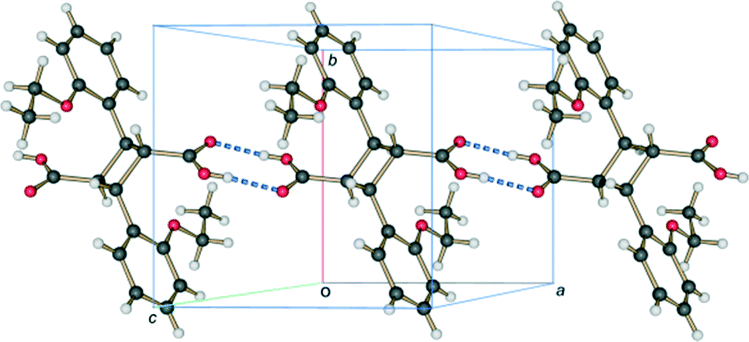 | ||
| Fig. 16 Hydrogen bonded carboxylic acid dimer chain, running parallel to the a axis, present in the crystal structure of 2,2′-diethoxy-α-truxillic acid (CSD refcode XOSKEV) – the final photoproduct at the end of stage 2. | ||
Though not much is known about the stage 2 mechanism, the phenomenon starts from the surface of the crystal and works its way inwards towards the core of the crystal. Evidence for this comes from the fact that the surface of unreacted crystals which are initially colourless turn white as the reaction proceeds. This was found to be especially noticeable if a crystal had been reacted for 36 hours or more. In addition, cutting such a crystal revealed a relatively transparent inner core. Also, as the reaction proceeds the contribution of powder rings (from the final product) to the diffraction pattern increases (Fig. 17a). These were minimal or not visible in samples that had been reacted for less than 60 hours but started becoming more visible (but not very significant) in the 60 and 72 h samples. Keeping a sample under the reaction conditions for 120 hours and taking a rotation photograph of a crystal yielded a powder pattern corresponding to that of the recrystallised photodimer (Fig. 17b). However, very long exposure times on the diffractometer (120 seconds) revealed peaks that when indexed corresponded with the unit cell parameters of the stage 1 product. This indicated that a small single crystal core still existed and allowed a SCXRD experiment to be carried out. The data was very weak but using the atom coordinates from the 72 h structure solution and refining the position of each molecule as a rigid body yielded an R factor of about 15% – acceptable under the circumstances. Crystal data for 72 h and 120 h is given in Table S1.† The r.m.s. (root-mean square) deviation of respective C and O atoms in the asymmetric unit of 120 h from those in 60 h was found to be 0.074 Å.
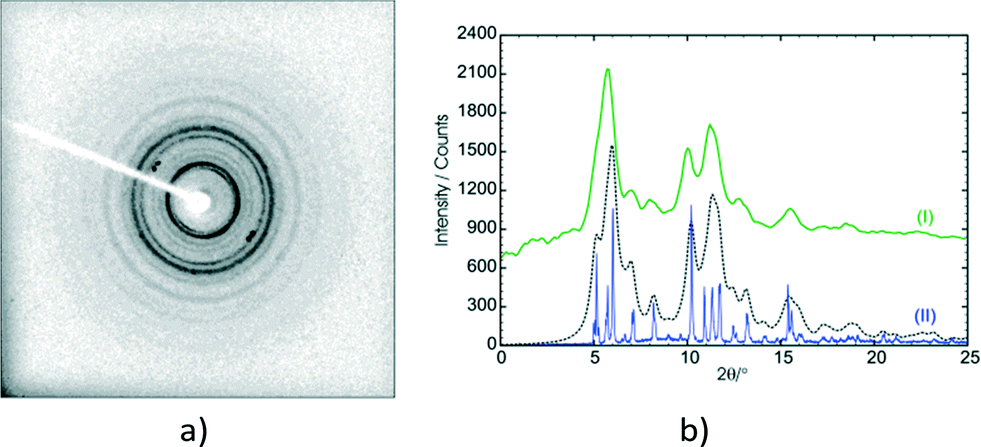 | ||
| Fig. 17 Diffractograms of the α′ polymorph after 120 h of UV irradiation. (a) Rotation photograph taken on a 1K CCD SMART diffractometer showing the powder rings of the final stage 2 photoproduct and some single crystal diffraction peaks of the final stage 1 photoproduct. (b) A comparison of the integrated powder rings from the rotation photograph (I; green) with a powder scan of the recrystallised photodimer (II; blue). To aid the comparison a calculated powder pattern, using the coordinates of the recrystallised α photodimer (XOSKEV) and wide Gaussian peak profiles (to increase the broadness of the peaks), is also shown (black dotted line). | ||
In summary, the stage 2 transformation is most likely a combination of molecular rearrangement at a local level on the surface (hence the powder pattern), followed by photodimerisation of the C molecules, followed by further rearrangements to produce the final product crystal/material. This transformation takes a relatively long time to occur (hours) because it has to work its way from the crystal surface into the crystal core and needs UV light (so that the C molecules photodimerise) as well as heat to occur.
Final comments on the DE to FG conformation change
Though the conformational change in going from the DE to the FG molecule in the solid state is unusual, similar phenomena have been found in numerous organic compounds.16,20,44–48 Such conformational changes are linked to phase changes or associated with solid state reactions. In addition, many studies have revealed a dynamic relationship between the populations of conformers with temperature in crystals. The DE to FG conformational change is especially unusual as the change involves the whole molecule (examples in literature tend to involve only part of a molecule or involve relatively small molecules). In addition, the rotation involved in changing the orientation of the ethoxy groups is huge – at least 140° (Fig. 18). In order to check whether the DE to FG conversion has a dependence on temperature alone or on a combination of UV irradiation and temperature, an α′ crystal which had been reacted for 48 h at room temperature (all molecules in the AB site reacted, molecules in CC site unreacted) was kept for 24 hours sequentially at 323, 333 and 343 K and examined by SCXRD after each temperature. It was found that the overall data intensity would decrease after each stage of heating. After the last heating stage at 343 K powder rings were visible in the diffraction pattern of the sample. In all three cases it was possible to index the individual diffraction peaks and integrate the SCXRD data as usual. All three data sets yielded structure solutions corresponding to the 293 K (r48 h) final product, i.e. no DE to FG conversion had occurred. Though not studied further, the loss of data intensity with temperature is most probably due to part of the stage 2 transformation mentioned above occurring. The fact that no DE to FG conversion occurred indicates that the conversion is dependent not only on temperature (no conversion occurs under identical irradiation conditions at 293 K), but also on the photodimerisation process. The photodimer has a smaller molecular volume (about 481 Å3) compared to two OETCA monomer molecules (about 497 Å3) and it is possible that the DE to FG conversion depends on space (or small voids) created during the photodimerisation process. The r48 h crystal is the final product of the reaction at 293 K and voids caused by the photodimerisation reaction have most probably already been minimized by molecular adjustments within the crystal. The plot of cell volume with reaction time shown in Fig. 6e seems to confirm this, as the final 293 K product crystal has a lower cell volume than the final 343 K stage 1 product crystal.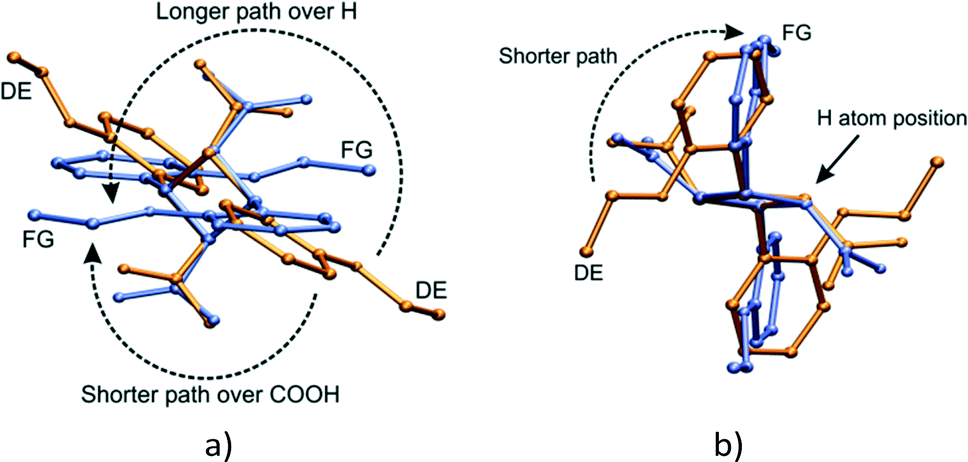 | ||
| Fig. 18 Superimposition of the DE and FG molecules (by least-squares fit of respective C1, C11, C12 and C13 cyclobutane atoms) after 60 h of UV irradiation; (a) approximate top view of the cyclobutane ring showing the two possible paths taken by the phenyl-ethoxy group of the FG molecule undergoing a conformational change from the DE molecule; (b) side view of the cyclobutane ring giving an indication of the size of the carboxylic acid group. | ||
A superimposition by least squares of the DE and FG molecules after 60 h of UV irradiation is shown in Fig. 18. Two routes exist for the movement of the phenyl-ethoxy group to convert the DE to the FG conformation. The shorter path requires a rotation around the C1–C11 bonds of about 140° over the bulky carboxylic acid groups. The carboxylic acid group most probably interferes with this rotation, hence energy in the form of heat is required to drive the conformation change. This would occur within the layers discussed earlier (see Fig. 19a). The alternative path involves a rotation of about 220° over a cyclobutane ring hydrogen. Though the less sterically hindered path in solution, this route would most probably cause the maximum amount of disturbance to the surrounding environment, and is therefore less likely.
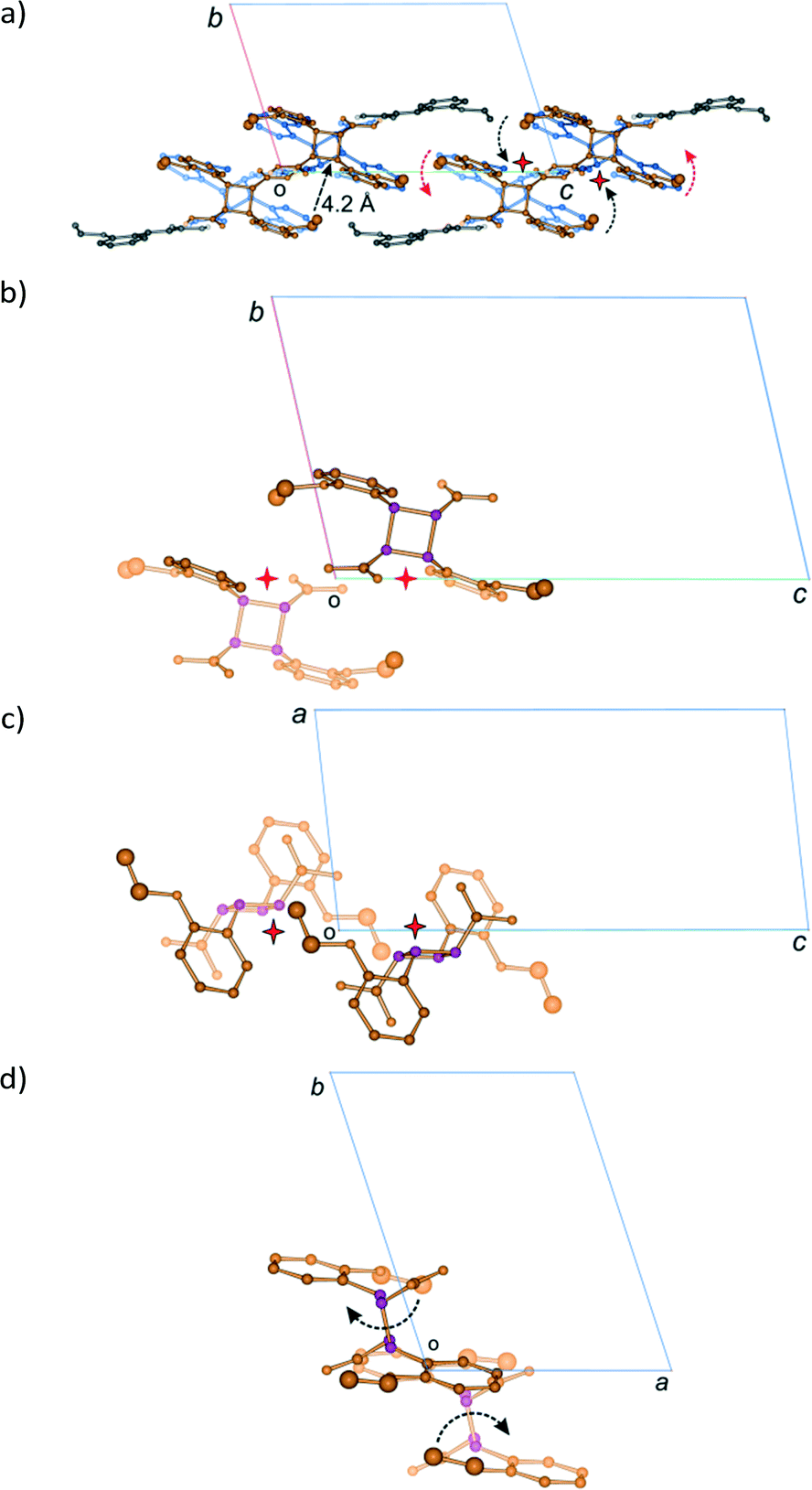 | ||
| Fig. 19 Proposed mechanism for the DE to FG conformation change. The radii of the ethoxy group carbon atoms (C21D, C22D, C21E and C22E) have been increased so as to indicate their position relative to the cyclobutane ring atoms [drawn in purple in (b)–(d)]. The distance from C22 (the terminal ethoxy carbon) to C11 and C12 (the cyclobutane atoms) is about 4.2 Å. (a) View down the a axis showing the likely movement of ethoxy groups during the conformation change; (b)–(d) views down the a, b and c axis, respectively, of the pair of DE molecules, showing the relative orientation of the ethoxy and cyclobutane groups. | ||
A possible mechanism for the DE to FG conformation change is shown in Fig. 19. The approximate position of the voids created by the dimerisation of the A and B monomer molecules to form the DE molecule are indicated with stars. Ethoxy groups on DE molecules related by a centre of inversion would probably move cooperatively through the space created by the photodimerisation reaction (shown with black arrows in Fig. 19a). The other ethoxy group in each DE molecule might then move over the face of the C molecule to complete the conformation change (shown with red arrows in Fig. 19a). Both movements would probably take the shortest path over the carboxylic acid groups. This conformation change process would produce the alternating arrangement of stacks of DE and FG molecules shown in Fig. 14c.
Conclusions
The α polymorph of o-ethoxy-trans-cinnamic acid (OETCA) undergoes a reversible phase transition to the related α′-polymorph at 333 K which contains two reaction sites, AB and CC, present in a 2:1 ratio. Partially reacting this crystal stabilises the α′-polymorph preventing a reverse phase change to the α polymorph. We previously reported a study of the reaction of the α′-polymorph at 293 K (after stablisation at 343 K) which led to a single crystal product composed of an ordered product molecule in the AB site (the DE product) as well as a the unreacted C molecule in the CC site.15,29 Here we presented the reaction of the α′-polymorph at 343 K which occurs in two stages: stage 1 where only molecules in the AB site react in a single-crystal-to-single-crystal manner which can be studied by single crystal X-ray diffraction, and stage 2 where the crystal undergoes a phase transition to a polycrystalline phase in which molecules in the CC site react. In stage 1 the reaction occurs in a similar way to the reaction at 293 K resulting in the exclusive formation of the DE product in the AB site until 67% conversion has occurred. At this point some of the DE molecules undergo a conformation change to the FG conformation by rotation of a phenyl-ethoxy group by at least 140° so that it points in the opposite direction to that in the DE product. This leads to significant changes in the unit cell parameters (especially cell angles) which diverge greatly from those from the reaction at 293 K. At the end of stage 1 the crystal is composed of an apparently disordered combination of DE and FG molecules present in a 55:45 ratio in the AB site, and the unreacted C molecule in the CC site. Examining close contacts in the crystal structure after stage 1 of the reaction, and using lattice energy calculations (AA-CLP) to evaluate different arrangements of DE and FG molecules in the crystal after stage 1, reveals that in the reaction the DE to FG conformation change occurs in an ordered and cooperative manner.
Acknowledgements
The authors wish to thank the University of the Witwatersrand and the South African National Research Foundation (GUN 76914 and 77122) for financial support.References
- H. D. Roth, Angew. Chem., Int. Ed. Engl., 1989, 28, 1193–1207 CrossRef.
- S. Poplata, A. Tröster, Y.-Q. Zou and T. Bach, Chem. Rev., 2016 DOI:10.1021/acs.chemrev.5b00723.
- I. G. Georgiev and L. R. MacGillivray, Chem. Soc. Rev., 2007, 36, 1239–1248 RSC.
- K. Biradha and R. Santra, Chem. Soc. Rev., 2013, 42, 950–967 RSC.
- (a) M. Cohen and G. Schmidt, J. Chem. Soc., 1964, 1996–2000 RSC; (b) M. Cohen, G. Schmidt and F. Sonntag, J. Chem. Soc., 1964, 2000–2013 RSC; (c) G. Schmidt, J. Chem. Soc., 1964, 2014–2021 RSC.
- G. M. J. Schmidt, Pure Appl. Chem., 1971, 27, 647–678 CrossRef CAS.
- I. D. Williams, IUCrJ, 2015, 2, 607–608 CrossRef CAS PubMed.
- V. Enkelmann, G. Wegner, K. Novak and K. B. Wagener, J. Am. Chem. Soc., 1993, 115, 10390–10391 CrossRef CAS.
- I. Abdelmoty, V. Buchholz, L. Di, C. Guo, K. Kowitz, V. Enkelmann, G. Wegner and B. M. Foxman, Cryst. Growth Des., 2005, 5, 2210–2217 CAS.
- G. Kaupp, CrystEngComm, 2003, 5, 117–133 RSC.
- (a) J. Harada, H. Uekusa and Y. Ohashi, J. Am. Chem. Soc., 1999, 121, 5809–5810 CrossRef CAS; (b) Y. Ohashi, Metastable or Unstable Intermediates in Reversible Processes in Crystalline State Photoreactions, Springer, 2014, pp. 151–184 Search PubMed; (c) J. Harada and K. Ogawa, Cryst. Growth Des., 2014, 14, 5182–5188 CrossRef CAS.
- I. Halasz, Cryst. Growth Des., 2010, 10, 2817–2823 CAS.
- M. D. Cohen, Angew. Chem., Int. Ed. Engl., 1975, 14, 386–393 CrossRef.
- Y. Ohashi, in Crystalline State Photoreactions, Springer, 2014, pp. 5–18 Search PubMed.
- M. A. Fernandes and D. C. Levendis, Acta Crystallogr., Sect. B: Struct. Sci., 2004, 60, 315–324 CAS.
- T. V. Sreevidya, D.-K. Cao, T. Lavy, M. Botoshansky and M. Kaftory, Cryst. Growth Des., 2013, 13, 936–941 CAS.
- (a) J. Bąkowicz, R. Siedlecka and I. Turowska-Tyrk, J. Chem. Crystallogr., 2012, 42, 593–599 CrossRef; (b) J. Bąkowicz and I. Turowska-Tyrk, CrystEngComm, 2014, 16, 6039–6048 RSC; (c) K. Konieczny, J. Bąkowicz and I. Turowska-Tyrk, CrystEngComm, 2015, 17, 7693–7701 RSC.
- K. Konieczny, J. Bąkowicz and I. Turowska-Tyrk, J. Chem. Crystallogr., 2016, 46, 77–83 CrossRef CAS.
- S. Khorasani and M. A. Fernandes, Cryst. Growth Des., 2013, 13, 5499–5505 CAS.
- S. Khorasani, D. Botes, M. A. Fernandes and D. C. Levendis, CrystEngComm, 2015, 17, 8933–8945 RSC.
- (a) S. D. Atkinson, M. J. Almond, G. A. Bowmaker, M. G. Drew, E. J. Feltham, P. Hollins, S. L. Jenkins and K. S. Wiltshire, J. Chem. Soc., Perkin Trans. 2, 2002, 1533–1537 RSC; (b) S. L. Jenkins, M. J. Almond, S. D. Atkinson, M. G. Drew, P. Hollins, J. L. Mortimore and M. J. Tobin, J. Mol. Struct., 2006, 786, 220–226 CrossRef CAS.
- M. A. Khoj, C. E. Hughes, K. D. Harris and B. M. Kariuki, Cryst. Growth Des., 2013, 13, 4110–4117 CAS.
- J. Inkinen, J. Niskanen, T. Talka, C. J. Sahle, H. Müller, L. Khriachtchev, J. Hashemi, A. Akbari, M. Hakala and S. Huotari, Sci. Rep., 2015, 5, 1–8 Search PubMed.
- V. Arjunan, R. Anitha, S. Thenmozhi, M. Marchewka and S. Mohan, J. Mol. Struct., 2016, 1113, 42–54 CrossRef CAS.
- (a) M. Bertmer, R. C. Nieuwendaal, A. B. Barnes and S. E. Hayes, J. Phys. Chem. B, 2006, 110, 6270–6273 CrossRef CAS PubMed; (b) I. Fonseca, M. Baias, S. E. Hayes, C. J. Pickard and M. Bertmer, J. Phys. Chem. C, 2012, 116, 12212–12218 CrossRef CAS.
- J. B. Benedict and P. Coppens, J. Phys. Chem. A, 2009, 113, 3116–3120 CrossRef CAS PubMed.
- M. K. Mishra, A. Mukherjee, U. Ramamurty and G. R. Desiraju, IUCrJ, 2015, 2, 653–660 CAS.
- M. A. Fernandes, D. C. Levendis and C. De Koning, Cryst. Eng., 2001, 4, 215–231 CrossRef CAS.
- M. A. Fernandes, D. C. Levendis and F. R. L. Schoening, Acta Crystallogr., Sect. B: Struct. Sci., 2004, 60, 300–314 Search PubMed.
- Bruker, SMART-NT, Version 5.050, Bruker AXS Inc., Madison, Wisconsin, USA, 1998 Search PubMed.
- Bruker, SAINT+, Version 6.02 (includes XPREP and SADABS), Bruker AXS Inc., Madison, Wisconsin, USA, 1999 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 1997, 30, 565 CrossRef CAS.
- E. Keller, SCHAKAL-99, University of Freiberg, Germany, 1999 Search PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14(1), 33–38 CrossRef CAS PubMed.
- (a) S. K. Wolff, D. J. Grimwood, J. J. McKinnon, M. J. Turner, D. Jayatilaka and M. A. Spackman, CrystalExplorer, Version 3.1, University of Western Australia, Crawley WA, Australia, 2012 Search PubMed; (b) M. A. Spackman, J. J. McKinnon and D. Jayatilaka, CrystEngComm, 2008, 10, 377–388 CAS.
- L. J. Farrugia, J. Appl. Crystallogr., 1999, 32, 837–838 CrossRef CAS.
- A. L. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148–155 CrossRef CAS PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision A.02, Gaussian Inc., Wallingford, CT, 2009 Search PubMed.
- A. Gavezzotti, New J. Chem., 2011, 35, 1360–1368 RSC.
- R. S. Gopalan and G. U. Kulkarni, Proc. - Indian Acad. Sci., Chem. Sci., 2001, 113, 307–324 CrossRef CAS.
- F. H. Allen, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 380–388 CrossRef.
- T. B. Brill and R. J. Karpowicz, J. Phys. Chem., 1982, 86, 4260–4265 CrossRef CAS.
- G. A. Sim, Acta Crystallogr., Sect. B: Struct. Sci., 1990, 46, 676–686 CrossRef.
- Y. Ohashi, Acta Crystallogr., Sect. A: Found. Crystallogr., 1998, 54, 842–849 CrossRef.
- A. Katrusiak, Acta Crystallogr., Sect. B: Struct. Sci., 2000, 56, 872–881 Search PubMed.
- R. S. Bogadi, D. C. Levendis and N. J. Coville, J. Am. Chem. Soc., 2002, 124(6), 1104–1110 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Crystal coordinates as CIF files for all the structures reported in this work have been deposited as a compressed file, as well as a PDF file containing supplementary information discussed in the text. The CIF files can also be obtained from the Cambridge Crystallographic Data Centre (CCDC). CCDC 1472988–1472996. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ce00809g |
| This journal is © The Royal Society of Chemistry 2016 |