
Chemical and electrochemical hydrogenation of CO2 to hydrocarbons on Cu single crystal surfaces: insights into the mechanism and selectivity from DFT calculations†
Lihui Ou
College of Chemistry and Chemical Engineering, Hunan University of Arts and Science, Changde 415000, China. E-mail: oulihui666@126.com; Tel: +86-736-7186115
First published on 17th June 2015
Abstract
Atomic level mechanistic insights into the chemical and electrochemical reduction of CO2 on the Cu(111) and Cu(100) surfaces are presented based on DFT-based thermodynamic and kinetic calculations. On Cu(111), COads is firstly formed by the dissociative hydrogenation of CO2, and CHOads and CH2Oads are the key intermediates towards the chemical and electrochemical reduction of CO2 into methanol and CH4. Despite being thermodynamically or kinetically favoured, it is likely that CH2OHads instead of CH3Oads is the intermediate for methanol and CH4 formation. Based on the activation barriers, CH2OHads intermediate either forms CH3OH by direct hydrogenation or forms CH2ads by hydrogenative dissociation, which may be a parallel path in the CO2 reduction mechanism on the Cu(111) surface. Finally, the CH2 intermediate leads to formation of the hydrocarbons. On Cu(100), CO2 reduction takes a different pathway in the early stages; CO is formed through the direct dissociation of CO2 rather than hydrogenative dissociation as on the Cu(111) surface due to stronger bonding of CO. Further reduction of CO also undergoes a different pathway, in which CO dimerization is more easy to achieve, whereas CO hydrogenation is difficult on the Cu(100) surface. This explains why C2H4 is formed more favorably on the Cu(100) surface and CH4 is predominantly produced on the Cu(111) surface under both chemical and electrochemical conditions. In addition, DFT results showed for the first time that the electrochemical reduction would be expected to be highly favored at potentials of interest by CO2 reduction compared with chemical reduction and that the carbon dioxide anion radical (˙CO2−) is involved in the initial stage of CO2 electroreduction. Simultaneously, the results also explain partly why CH3OH is formed in gas phase chemistry and only CH4 is observed in electrochemistry on copper surfaces. By analyzing the chemical and electrochemical reduction paths, important mechanistic information is deduced on the Cu single crystal surfaces. The study of CO2 reduction mechanisms on copper will lead to a deeper understanding of the reaction chemistry and could eventually lead to the design of more efficient and selective catalysts.
1. Introduction
Converting CO2 into organic fuels and/or chemicals would positively impact the global carbon balance and offer a novel solution to the dilemma of growing energy demands and global warming we face nowadays.1,2 Among various CO2 fixation methods, reduction with hydrogen gas3,4 or electricity1,5–9 may represent the most promising pathways since either of them can be generated on large scales by sustainable energies such as solar, hydro and wind; moreover, they provide ways of storing these intermittently available sustainable energies. The two reduction methods similarly involve the successive hydrogenation of CO2 molecules, leading to a variety of products such as formic acid, methanol and hydrocarbons. In comparison, the electrochemical reduction of CO2 offers several advantages such as room temperature and ambient pressure operation, tunable reaction rate and selectivity by electrode potential, and the capability of producing hydrocarbons such as methane (CH4) and ethylene (C2H4),1,5–13 which are particularly desired due to their high energy densities and widespread use in the current energy and industrial infrastructures. Among electrocatalyst materials explored to date, metallic Cu is the most capable of converting CO2 into hydrocarbons with high Faraday efficiency.5,7,8,10–19 Therefore, the electrochemical reduction of CO2 on Cu-based materials has received growing interest in recent years.Although CO2 can be converted into CH4 and C2H4 with high Faraday efficiency on Cu electrodes, considerable overpotentials are required for the reaction to occur, with onset potentials being around −0.7 V and −0.8 V (vs. RHE), respectively, for the formation of C2H4 and CH4.5–7,20 At less negative potentials, formate and CO are the main products. Studies on single crystal surfaces showed that more C2H4 was formed on the Cu(100) surface, whereas the Cu(111) surface produced more CH4.7,21,22 Understanding the atomic-level origin of the product distribution and the surface structure effect in CO2 electroreduction are crucial for electrocatalyst optimization. This requires a detailed microscopic view on the reaction pathways and kinetics. It has been found that the electroreduction of CO exhibits very similar potential and surface structure dependence of the CH4 and C2H4 formation, while experiments starting with HCOOH showed no detectable products,20,23 which indicates that CO is one of the intermediates involved in CO2 electroreduction to these hydrocarbons and CO is formed in a separate pathway from formate formation.8,13,21,24,25 However, the reaction pathways and intermediates for CO formation remain unclear.
Based on the reaction free energies calculated using density functional theory (DFT) for various possible elementary steps, Nørskov and coworkers26,27 recently proposed that the formation of CH4 and C2H4 from CO2 on various fcc Cu facets similarly involves the hydrogenative reduction of CO to an adsorbed formyl (CHO*) as the rate-determining step (rds), which is followed by successive hydrogenative reduction steps to form adsorbed formaldehyde (CH2O*) and methoxy species (CH3O).26 However, experimental results obtained recently by Koper and coworkers28 and earlier by Hori et al.25 seem to suggest that the reaction pathways of CH4 and C2H4 formation are separated at an early stage of CO reduction. Results from a more recent experimental study of CO electroreduction on single-crystal copper electrodes by Koper and coworkers29 further implied that there are two separate pathways for C2H4 formation: one that shares an intermediate with the pathway to CH4 and one that occurs mainly on Cu(100) and probably involves the formation of a CO dimer as the key intermediate.8 The latest DFT calculation study by Nie et al.30 suggested an alternative reaction path for both CH4 and C2H4 production through a hydroxymethylidyne (COH) intermediate from CO electroreduction. These controversial results and propositions have left both the rate-determining and selectivity-determining steps for the formation of hydrocarbon products from CO2 and CO electroreduction on Cu still uncertain. In addition, several other questions in the reaction mechanism remain, such as why methanol, i.e. the dominant product on copper catalysts in chemical reduction, is nearly absent in the electrochemical reduction; whether methylene (:CH2) proposed in earlier studies8 is involved in the hydrocarbon formation; and why C2H4 formation is favored over CH4 at lower overpotentials.
In an attempt to ascertain the dominant reaction pathways for the formation of various products in CO2 reduction, we present a systematic DFT calculation on the reaction free energies and activation barriers of various possible elementary steps on the two most common types of Cu surface, namely, the closed-packed Cu(111) surface and the open Cu(100) surface. Both chemical reduction by gaseous hydrogen and electrochemical reduction through proton-electron pairing are considered and their similarities and differences are discussed. We calculated the free energies of the reaction and the activation barriers for the chemical reduction, according to which the optimized pathways for chemical reduction are deduced. With the theoretical hydrogen electrode model, the effect of electrode potentials on the steps in these pathways is discussed. Based on the calculated reaction free energy, the pathways with water in the double layer as the hydrogenation agent in the electrochemical environment are discussed. A mechanism for the electrochemical reduction is proposed. In chemical reduction, the adsorbed hydrogen atoms (H*) should be the main reducing and hydrogenation agent, while in electrochemical reduction, the hydrogenative reduction can be either accomplished by H* or by protons from the double layer plus electrons from electrode. Thermodynamically, the two ways are not distinguished, but they could be different kinetically as the transition states in the two cases could be totally different.
2. Computational method and modeling
Calculations were performed in the framework of DFT using the generalized gradient approximation (GGA) of Perdew–Burke–Ernzerhof (PBE)31 and employing ultrasoft pseudopotentials32 for nuclei and core electrons. Calculations of reaction free energies were performed using periodic super-cells with the Cu electrodes modeled by four-layer slabs with a 3 × 3 surface. A vacuum space of 16 Å was placed above the slabs and adsorption is allowed on only one of the two surfaces exposed. The Kohn–Sham orbitals were expanded in a plane-wave basis set with a kinetic energy cutoff of 26 Ry and a charge density cutoff of 260 Ry. The Fermi-surface effects were treated by the smearing technique of Methfessel and Paxton using a smearing parameter33 of 0.02 Ry. Calculations were carried out with spin-polarization, which is essential to properly represent the electronic structure of adsorbed CO2. The PWSCF codes contained in the Quantum ESPRESSO distribution34 were used to implement all calculations, while figures of the chemical structures were produced with the XCRYSDEN35–37 graphics package. BZ integrations were performed using a (3 × 3 × 1) uniformly shifted k-mesh for a (3 × 3) supercell.The calculated equilibrium lattice constant for Cu was 3.66 Å, which agreed well with theoretical and experimental values (3.66 and 3.62 Å, respectively).38,39 During the calculations, the structure of the bottom two layers were fixed at the theoretical bulk positions, whereas the top two layers and the adsorbates were allowed to relax and all the other structural parameters were optimized so as to minimize the total energy of the system. Structural optimization was performed until the Cartesian force components acting on each atom were brought below 10−3 Ry Bohr−1 and the total energy converged to within 10−5 Ry.
The climbing-image nudged elastic band (CI-NEB) method was used to determine the minimum energy paths (MEPs) for all the elementary steps.40,41 The transition state of the optimized reaction coordinate was approximated by the image of highest energy. The transition state images from the CI-NEB calculations were optimized using the quasi-Newton method, which minimizes the forces to find the saddle point. Geometry optimization was performed for each intermediate point in MEPs, in which a three-layer Cu slab with a 2 × 3 surface unit cell was used. Considering the high cost of the CI-NEB calculation, the bottom two layers of metal atoms were fixed, while the top layer of metal atoms and all other non-metal atoms were allowed to relax.
The adsorption energy (Ead) of adsorbate “A” was calculated according to Ead(A) = E(slab-A) − E(slab) − E(A), where E(slab-A), E(slab) and E(A) refer to the total energy of a slab with an adsorbed “A”, the total energy of a slab, and the total energy of the free “A”, respectively. The co-adsorption energy between adsorbates “A” and “B” was calculated according to Ead(A, B) = E(slab-A, B) − E(slab) − E(A) − E(B), in which E(slab-A, B) refers to the calculated total energy of the slab with co-adsorbed “A” and “B”. The difference value of “Ead(A, B) − (Ead(A) + Ead(B))” should represent the interaction between “A” and “B” on the surface.
3. Results and discussion
3.1 Chemical reduction of CO2
Table 1 lists the possible reaction steps in the chemical reduction of CO2 by H2 and the DFT-calculated reaction free energies for these steps on the Cu(111) and Cu(100) surfaces. The preferred adsorption configurations for all the reactants, intermediates and products listed in Table 1 on the two surfaces are given in Tables S1–S6 and Fig. S1–S3 in the ESI.† For the sake of simplicity, we directly use an H2 molecule as the reactant in the reaction free energy calculations for all the hydrogenation steps. In reality, the hydrogenation agent in the elementary reaction steps of chemical reduction should be the adsorbed hydrogen atoms (H*) formed through H2 dissociation. Therefore, the hydrogenation steps listed in Table 1 are not the elementary reaction steps. Although they are not the elementary steps, the calculated reaction free energies in Table 1 should still represent the general thermodynamic preference of various hydrogenation steps.| Possible reaction steps | Reaction free energies, ΔGreacb (kJ mol−1) | |
|---|---|---|
| Cu(111) | Cu(100) | |
| a The asterisk (*) indicates that the species is adsorbed on the surface.b In the calculations, the entropies obtained from the literature of Nørskov and coworkers26,27 are considered for gaseous molecules, including CO2, H2, CH3OH, CH4 and C2H4, whereas the entropies of the adsorbed species are ignored. The zero point energies (ZPE) for all species, which are taken from the literature of Nørskov and coworkers,26,27 are included in the calculations. For steps involving the co-adsorption of two species, the co-adsorption structures were optimized and used in calculations. For instance, for a reaction step of (A + B)* → (C + D)*, the reaction free energy is calculated according to “E(slab-C, D) − E(slab-A, B) + ZPE(C*) + ZPE(D*) − ZPE(A*) + ZPE(B*)”. | ||
| 1/2H2(g) → H* | −45.67 | −41.34 |
| (1a) CO2(g) + H* → HCOO* | −28.47 | −50.66 |
| (1b) CO2(g) + H* → COOH* | 32.91 | 36.35 |
| (1c) CO2(g) → CO* + O* | 91.47 | 22.57 |
| (1d) COOH* → CO* + OH* | −36.22 | −49.87 |
| (2a) CO* + 1/2H2(g) → COH* | 96.92 | 63.59 |
| (2b) CO* + 1/2H2(g) → CHO* | 60.04 | 41.41 |
| (2c) CO* → OCCO* | 132.55 | 75.59 |
| (2d) CO* → C* + O* | 263.66 | 154.73 |
| (2e) COH* → CHO* | −36.88 | −22.18 |
| (3a) CHO* + 1/2H2(g) → CH2O* | −40.49 | −63.45 |
| (3b) COH* + 1/2H2(g) → CHOH* | −25.13 | 2.30 |
| (3c) COH* → C* + OH* | 78.26 | 5.38 |
| (3d) CHO* → CH* + O* | 67.59 | 27.82 |
| (4a) CH2O* + 1/2H2(g) → CH2OH* | −29.99 | −1.64 |
| (4b) CH2O* + 1/2H2(g) → CH3O* | −127.37 | −99.55 |
| (4c) CH2O* + 1/2H2(g) → CH2* + OH* | −42.45 | −47.05 |
| (4d) CH2O* → CH2* + O* | 41.87 | 10.11 |
| (4e) CHOH* + 1/2H2(g) → CH2OH* | −82.22 | −89.57 |
| (4f) CHOH* → CH2O* | −52.23 | −87.93 |
| (4g) CHOH* → CH* + OH* | −22.44 | −110.11 |
| (5a) CH2OH* + 1/2H2(g) → CH3OH(l) | −112.67 | −96.53 |
| (5b) CH2OH* → CH2* + OH* | −12.46 | −45.41 |
| (5c) CH3O* + 1/2H2(g) → CH3OH(l) | −15.29 | 1.38 |
| (5d) CH2OH* → CH3O* | −97.38 | −97.91 |
| (5e) CH2OH* + 1/2H2(g) → CH3* + OH* | −118.31 | −126.97 |
| (5f) CH3O* + 1/2H2(g) → CH3* + OH* | −20.93 | −29.07 |
| (6a) CH2* + 1/2H2(g) → CH3* | −105.85 | −61.37 |
| (6b) CH3* + 1/2H2(g) → CH4(g) | −99.41 | −78.61 |
| (6c) CH2* → 1/2C2H4* | −42.70 | −92.59 |
According to calculated adsorption free energies (Table S1†), CO2 cannot form stable chemical adsorption states on the Cu(111) and Cu(100) surfaces. Therefore, gaseous CO2 is used in the free energy calculations. The physisorption state of CH4 and chemisorption state of C2H4 are obtained on the Cu(111) surface. The corresponding adsorbed states of CH4 and C2H4 are also obtained on the Cu(100) surface. Moreover, the physisorption state of CH3OH is also obtained on the Cu(111) surface. Although the chemical reduction in industry should be operated practically at elevated temperatures, the reaction free energies in Table 1 were calculated for room temperature (298 K) so that the potential effect can be easily determined.
Although the chemical hydrogenation reactions of CO2 to CH3OH, CH4 and C2H4, namely, CO2(g) + 3H2(g) → CH3OH + H2O, CO2(g) + 4H2(g) → CH4 + 2H2O and CO2(g) + 3H2(g) → 1/2C2H4 + 2H2O, have rather negative overall reaction free energies of −109.45 kJ mol−1, −232.43 kJ mol−1 and −180.46 kJ mol−1, respectively, some of the elementary reaction steps, especially the reaction steps of CO, have rather positive reaction free energies. This might be the reason why these reactions are practically difficult.
The DFT-calculated activation energies for H2 dissociation are 0.52 eV and 0.45 eV on the Cu(111) and Cu(100) surfaces, respectively, which are relatively small compared to activation barriers for other steps in CO2 reduction as shown later. Therefore, H2 dissociation should also play a relatively minor role in the kinetics of the chemical reduction of CO2 by H2.
In the following, we will first deduce the most possible pathways for the chemical reduction of CO2 on the two types of Cu surfaces according to the calculated reaction free energies shown in Table 1 and the activation barriers determined from the CI-NEB method. The mechanisms of electrochemical reduction will then be discussed by analyzing the effect of electrode potentials on the steps in these pathways on the basis of the theoretical hydrogen electrode model and by considering water in the double layer as the hydrogenation agent in the electrochemical environment.
Fig. 1 displays the calculated MEPs for these possible initial steps on the Cu(111) surface. In these calculations, the initial state contains a physisorbed CO2 molecule and a chemisorbed H* at the fcc site. The calculated activation energies for these steps follow the same trend as the reaction free energies, i.e., the direct dissociation of CO2 exhibits the highest activation barrier (∼1.54 eV), while the formation of formate by the direct hydrogenation of CO2 requires the lowest activation energy (∼0.86 eV). The activation barrier for the formation of carboxyl (COOH) is ca. 1.18 eV. The dissociative hydrogenation of CO2 to adsorbed CO and OH is not an elementary step. It involves COOH as an intermediate, whose dissociation requires activation energy of only ca. 0.2 eV. Thus, both the reaction free energies and activation energies from DFT calculations suggest that formate is the most preferred initial product in the chemical reduction of CO2 on the Cu(111) surface. The formation of CO from CO2 is most probably through the COOH intermediate, which is formed with an activation barrier slightly higher than the formate formation. Experiments starting with formate showed no detectable products, and CO product was suppressed at a less negative potential when starting with CO2.20,23
 | ||
| Fig. 1 Minimum energy paths for the three possible initial steps in the chemical reduction of CO2 on the Cu(111) surface. Oxygen atoms are red, hydrogen atoms are white, carbon atoms are gray, and copper atoms are blue. | ||
Similar to the Cu(111) surface, the formation of formate is a relatively strong exergonic process (∼92.00 kJ mol−1). Compared to that on the Cu(111) surface, however, the formation of COOH becomes much less exergonic, while the direct dissociation of the CO2 becomes much less endergonic on the Cu(100) surface. If considering that hydrogenation may occur through adsorbed H atoms, the reaction free energy for the formation of COOH could be more endergonic than the dissociation of CO2. As will be shown in later MEP calculations, the direct dissociation of CO2 on the Cu(100) surface requires a lower activation energy than its hydrogenation to COOH.
As shown in Fig. 2, CO formation through the direct dissociation of CO2 requires considerably lower activation energy (1.29 eV) than that through the hydrogenative dissociation (1.79 eV) on the Cu(100) surface and is different from that on the Cu(111) surface. Therefore, the direct dissociation of CO2 would be the preferred pathway for CO formation on the Cu(100) surface.
 | ||
| Fig. 2 Minimum energy path of CO formation on the Cu(100) surface by the direct dissociation and hydrogenation of CO2. Oxygen atoms are red, hydrogen atoms are white, carbon atoms are gray, and copper atoms are blue. | ||
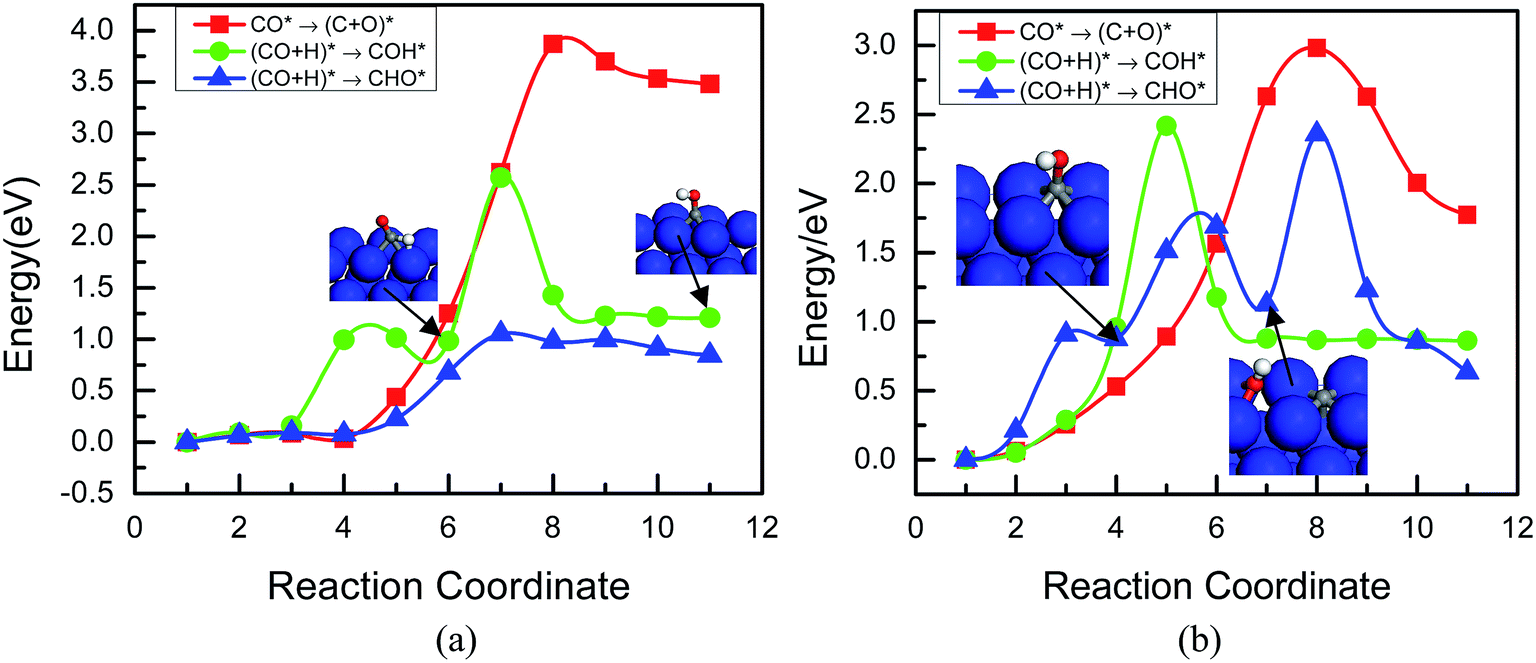 | ||
| Fig. 3 Minimum energy path of CO dissociation and hydrogenation to form C, COH and CHO (a) on the Cu(111) surface and (b) on the Cu(100) surface. Oxygen atoms are in red, hydrogen atoms are in white, carbon atoms are in gray, and copper atoms are in blue. | ||
The calculated MEPs for the steps on the Cu(100) surface following CO formation suggest they are very similar to that on the Cu(111) surface. The activation energy barriers for the direct dissociation of CO and hydrogenation to form COH* and CHO* are 3.01, 2.43, and 2.36 eV, respectively, as shown in Fig. 3b. These results indicate that CO hydrogenation requires higher activation energy barriers on the Cu(100) surface, which is more easy to achieve on the Cu(111) surface. By carefully inspecting the intermediate images in the calculated MEPs for the two hydrogenation reaction paths on the Cu(100) surface, one finds that CHO* formation involves COH* and the dissociative adsorption state C* + OH* as the intermediates.
In fact, the formation of a formyl intermediate has been recognized in other reactions involving CO hydrogenation, for examples, alcohol production from carbon monoxide and molecular hydrogen on the Rh(111) surface,42 Rh/TiO2 (ref. 43) and Rh/SiO2 catalysts,44,45 and formaldehyde and methanol synthesis from CO and H2 on the Ni(111) surface.46
 | ||
| Fig. 4 Minimum energy paths on the Cu(111) surface of CHO* dissociation and hydrogenation into CH, C, CHOH and CH2O. Oxygen atoms are red, hydrogen atoms are white, carbon atoms are gray, and copper atoms are blue. | ||
In a previous study on the mechanism of ethanol synthesis from syngas on the Rh(111) surface, Liu et al.42 showed that the CHO* intermediate formed through CO hydrogenation can be more easily hydrogenated to form CH2O* than the dissociation. Earlier studies13,23 have also shown that CH4 can be synthesized on Cu with formaldehyde (CH2O) as the starting material.
CH2O may undergo direct dissociation to methylene (:CH2) and O*, direct hydrogenation to form CH3O* and/or CH2OH, and/or hydrogenated dissociation to methylene (:CH2) and OH. Calculated reaction free energies for the formation of CH3O* and/or CH2OH* are −127.37 kJ mol−1 and −29.99 kJ mol−1, respectively, while it is −49.08 kJ mol−1 for :CH2 formation. This seems to suggest that the CH3O* pathway is preferable on the Cu(111) surface. As shown by the MEP calculation results in Fig. 5, the formation of CH3O* and the hydrogenated dissociation of CH2O* to :CH2 actually proceed via a CH2OH* intermediate, which has a lower activation energy than the CH2O* direct dissociation. Therefore, although the reaction free energy of CH2OH* formation is less exergonic than CH3O*, the preferred reaction pathway of CH2O* should be hydrogenation to form CH2OH* rather than CH3O*.
 | ||
| Fig. 5 Minimum energy paths of the direct hydrogenation, the hydrogenative dissociation, and the direct dissociation of CH2O on the Cu(111) surface. Oxygen atoms are in red, hydrogen atoms are in white, carbon atoms are in gray, and copper atoms are in blue. | ||
It can be seen (Fig. 5) that the formation of CH2 and OH via CH2O* hydrogenative dissociation is not an elemental step. The hydrogenation of CH2O* first produces a CH2OH* intermediate with an activation barrier of about 0.48 eV; the transformation of CH2OH* to CH2 and OH has an activation barrier of about 1.04 eV (Fig. 5). As shown by the calculated MEPs in Fig. 6, the activation barriers for the direct hydrogenation of CH2OH* to form CH3OH and the hydrogenative dissociation of CH2OH* to form CH2 are ∼0.68 eV and 0.66 eV, respectively. Thus, the CH2OH* intermediate either forms CH3OH by direct hydrogenation or forms CH2 by hydrogenative dissociation. Based on the activation barriers, the simultaneous occurrence of both the paths can be concluded, which may be a parallel path in CO2 reduction mechanism on the Cu(111) surface. In addition, the hydrogenative reduction of CH2OH* to CH3OH and the hydrogenative dissociation to form CH2 and H2O are also easier than the abovementioned CH2OH direct dissociation. The presented results explain the experimental results in which CH3OH was observed in CO2 chemical reduction on copper surfaces.
 | ||
| Fig. 6 Minimum energy paths of the hydrogenative dissociation of CH2OH to CH2 and H2O and the direct hydrogenation to CH3OH, respectively, on the Cu(111) surface. Oxygen atoms are in red, hydrogen atoms are in white, carbon atoms are in gray, and copper atoms are in blue. | ||
The formation of CH4 through CH2 hydrogenation is drastically exergonic on the Cu(111) surface. In comparison, C2H4 formation is less exergonic. This explains the experimental result that CH4 is more likely formed on the Cu(111) surface. Fig. 7a shows the MEPs for the formation of CH4 and C2H4 from CH2. The activation barriers for the two hydrogenation steps in CH4 formation are 0.63 eV and 1.03 eV. For the association of two CH2 to form C2H4, the activation barrier is 0.21 eV, which is less than that of CH2 hydrogenation to CH3. This seemingly indicates that CH2 dimerization is kinetically more favorable. However, as shown by the calculated MEPs in Fig. 6, CH3 and OH intermediates are observed during the course of CH2OH hydrogenative dissociation to CH2 and H2O, and the reaction free energy of forming CH3 and OH intermediates is more negative than that of forming CH2 and H2O; an activation barrier of ∼0.53 eV is required from CH3 and OH to CH2 and H2O, namely, the CH3 intermediate may be easier to form on the Cu(111) surface. Considering that the surface coverage of the CH2 should be very low, the dimerization reaction would thus still have a much lower reaction rate than the hydrogenation reaction on the Cu(111) surface.
 | ||
| Fig. 7 Minimum energy paths of CH2 successive hydrogenation into hydrocarbons CH3 and CH4 and dimerization into C2H4 (a) on the Cu(111) surface and (b) on the Cu(100) surface. | ||
On the Cu(100) surface, however, we find that the CH2 dimerization into C2H4 is a non-activated process, as shown in Fig. 7b. The paths for CH3 and CH4 formation have activation energy barriers of 0.34 and 0.55 eV, respectively. The dimerization of CH2 to C2H4 has a more negative reaction free energy than that of CH2 hydrogenation to CH3. Thus, C2H4 formation should be more easy on the Cu(100) surface than CH4 formation.
The results from Koper and coworkers29 suggested that there are two separate pathways for the formation of C2H4, one that shares an intermediate with the pathway to CH4, as they observed on the Cu(111) surface and below −0.8 V on the Cu(100) surface at pH 7, and a second pathway that occurs only on the Cu(100) surface. For this second pathway, they suggest that the formation of a CO dimer is the key intermediate in the formation of C2H4. As shown in Fig. 8, the activation energy barrier for the formation of CO dimer on the Cu(111) and Cu(100) surfaces is 1.59 and 1.26 eV, respectively. The results indicate that CO dimerization has a higher activation energy barrier than CO hydrogenation to form CHO* on the Cu(111) surface, whereas on the Cu(100) surface, CO dimerization requires a lower activation energy barrier than CO hydrogenation to form CHO*. Therefore, CO hydrogenation is more easy on the Cu(111) surface, whereas CO dimerization into (CO)2 is more easy on the Cu(100) surface. Such a surface dimer could explain the unique selectivity for C2H4 (for detailed arguments, see ref. 7) and is in agreement with the suggestion of Gattrell et al.11 who proposed that this CO dimer would be more stable on the Cu(100) surface. In addition, our results may somewhat agree with Koper and coworkers.
 | ||
| Fig. 8 Minimum energy paths of CO dimerization into OCCO* on the Cu(111) and Cu(100) surfaces. | ||
Based on the abovementioned reaction free energy and MEP calculations, the optimal reaction paths for CO2 reduction to hydrocarbons on the Cu(111) surface can be summarized in Table 2. CO is first formed by the dissociative hydrogenation of CO2, which is the most feasible reaction path for CO is its hydrogenation to a CHO* intermediate. The key intermediate CH2O can be more easily formed through further CHO hydrogenation, and the preferred reaction pathway of CH2O should be the hydrogenation to form CH2OH, which is based on the activation energy barriers. The CH2OH intermediate either forms CH3OH by direct hydrogenation or forms CH2 by hydrogenative dissociation, which may be a parallel path in the CO2 reduction mechanism on the Cu(111) surface; finally, the CH2 intermediate leads to formation of the hydrocarbons. From a kinetic viewpoint, the relatively slow steps on the Cu(111) surface include CO2(g) + H* → (CO + OH)* and (CO + H)* → CHO*. The reason may be that the weak Cu–CO2 interaction is an obstacle to CO2 dissociative hydrogenation and therefore slows down the overall conversion. The differences in the reaction mechanism on the Cu(111) and Cu(100) surfaces are that the CO formation and further reduction undergoes a different pathway. Simultaneously, the preference for the formation of CH4 and C2H4 is also different; the formation of C2H4 is easier on the Cu(100) surface, and the experimental results are explained to some degree on the selectivity of the Cu single crystal surface. The C2H4 formation mechanisms via CO dimerization will be studied systematically in our future work.
| Reaction paths | Eact (eV) |
|---|---|
| a The values in parentheses are the activation energy barriers of the corresponding processes on the Cu(100) surface. | |
| CO2(g) + * → (CO + O)* | 1.54 (1.29)a |
| CO2(g) + H* → (CO + OH)* | 1.18 (1.79) |
| (CO + H)* → CHO* | 1.06 |
| (CHO + H)* → CH2O* | 0.72 |
| (CH2O + H)* → (CH2 + OH)* | 1.12 |
| (CH2O + H)* → CH2OH* | 0.95 |
| (CH2O + H)* → CH3O* | 1.86 |
| (CH2OH + H)* → CH3OH(l) | 0.68 |
| (CH2OH + H)* → CH2* + H2O(l) | 0.66 |
| (CH2 + H)* → CH3* | 0.63 (0.34) |
| (CH3 + H)* → CH4(g) | 1.03 (0.56) |
| 2CH2* → C2H4* | 0.21 (∼0) |
3.2 Electrochemical reduction of CO2
One of the major differences between the electrochemical reduction and the chemical reduction is that the H2 molecules will be replaced by the (H+ + e) pair. The overall reaction for the electrochemical reduction of CO2 to methanol and hydrocarbons can be expressed as follows,| CO2(g) + 8(H+ + e−) → CH4 + 2H2O | (1) |
| CO2(g) + 6(H+ + e−) → 1/2C2H4 + 2H2O | (2) |
| CO2(g) + 6(H+ + e−) → CH3OH + H2O | (3) |
Since the chemical potential (μ) of the electron changes with the electrode potential (E), the reaction free energies will vary with E. If the potential of the reversible hydrogen electrode (RHE) is used as the zero potential, the relationship would be μ(H+ + e−) = 1/2μ(H2) − eE.47 This relationship provides an elegant way to calculate the potential-dependent free energies of the reaction steps in the electrochemical reduction by avoiding the explicit treatment of solvated protons.
In electrochemical reduction, the reaction steps involving hydrogenation will be enhanced thermodynamically as the potential becomes negative. In addition, for reaction steps which produce O* or OH*, the reaction may be promoted since these oxygenated species can be easily removed from the surface at negative potentials. Experimental results9,16,20 have shown that when the electrode potential is −0.50 V (vs. RHE), formate is the main product; when the electrode potential is −0.67 V (vs. RHE), CO is the main product; when the electrode potential is more negative, i.e. −0.90 V (vs. RHE), hydrocarbons begin to form. We chose these three electrode potentials to calculate the reaction free energies for various steps and the results are given in Table 3.
| Possible reduction steps | Reaction free energies, ΔGreac (kJ mol−1) | |||||
|---|---|---|---|---|---|---|
| Cu(111) | Cu(100) | |||||
| −0.50 V | −0.67 | −0.90 | −0.50 V | −0.67 | −0.90 | |
| (a) CO2(g) + H+ + e− → HCOO* | −98.06 | −114.47 | −136.66 | −119.84 | −136.25 | −158.44 |
| (b) CO2(g) + H+ + e− → COOH* | −16.29 | −32.70 | −54.89 | −32.83 | −49.24 | −71.43 |
| (c) COOH* + H+ + e− → CO* + H2O(l) | −84.07 | −101.48 | −123.67 | −73.12 | −89.53 | −111.72 |
| (d) HCOO* + H+ + e− → CO* + H2O(l) | −3.30 | −19.71 | −41.90 | 13.89 | −2.52 | −24.71 |
| (e) CO* + H+ + e− → CHO* | 11.79 | −4.62 | −26.81 | −6.84 | −23.25 | −45.44 |
| (f) CO* + H+ + e− → COH* | 48.67 | 32.26 | 10.07 | 15.34 | −1.07 | −23.26 |
| (g) CHO* + H+ + e− → C* + H2O(l) | 84.63 | 68.22 | 46.03 | −25.48 | −41.89 | −64.08 |
| (h) CHO* + H+ + e− → CH* + OH* | −58.95 | −75.36 | −97.55 | −133.88 | −150.29 | −172.48 |
| (i) CH* + OH* + H+ + e− → CH* + H2O(l) | −48.84 | −65.25 | −87.44 | −23.25 | −39.66 | −61.85 |
| (j) CHO* + H+ + e− → CH2O* | −88.74 | −105.15 | −127.34 | −111.70 | −128.11 | −150.30 |
| (k) CHO* + H+ + e− → CHOH* | −36.50 | −52.91 | −75.10 | −23.77 | −40.18 | −62.37 |
| (l) CH2O* + H+ + e− → CH3O* | −175.62 | −192.03 | −214.22 | −147.80 | −164.21 | −186.40 |
| (m) CH2O* + H+ + e− → CH2OH* | −78.24 | −94.65 | −116.84 | −49.89 | −66.30 | −88.49 |
| (n) CH2OH* + H+ + e− → CH3OH(l) | −160.92 | −177.33 | −199.52 | −144.78 | −161.19 | −183.38 |
| (o) CH3O* + H+ + e− → CH3OH(l) | −63.54 | −79.95 | −102.14 | −46.87 | −63.28 | −85.47 |
| (p) CH2O* + H+ + e− → CH2* + OH* | −90.71 | −107.12 | −129.31 | −95.30 | −111.71 | −133.90 |
| (q) CH2* + OH* + H+ + e− → CH2* + H2O(l) | −48.84 | −65.25 | −87.44 | −23.25 | −39.66 | −61.85 |
| (r) CH2* + H+ + e− → CH3* | −154.10 | −170.51 | −192.70 | −129.82 | −146.23 | −168.42 |
| (s) CH3* + H+ + e− → CH4(g) | −147.66 | −164.07 | −186.26 | −146.09 | −162.50 | −184.69 |
| (t) CH2* → 1/2C2H4* | −85.40 | −85.40 | −85.40 | −185.18 | −185.18 | −185.18 |
The free energies, ΔGreac, on the Cu(111) surface for the overall reactions (1) are −618.43 kJ mol−1, −749.67 kJ mol−1, and −927.23 kJ mol−1; the ΔGreac for the overall reaction (2) are −469.96 kJ mol−1, −568.39 kJ mol−1, and −701.56 kJ mol−1; and the ΔGreac for the overall reaction (3) are −389.95 kJ mol−1, −497.38 kJ mol−1, and −630.55 kJ mol−1 when the electrode potential is −0.50 V, −0.67 V, and −0.90 V (vs. RHE), respectively, which are all highly exergonic. The ΔGreac in the electrochemical environment is more negative than that in chemical reduction environment; therefore, these reactions may be more favorable in an electrochemical environment.
According to the calculated electrochemical reaction free energies, the formation of formate and COOH will both be promoted at negative electrode potentials. In the electrochemical condition, the hydrogenated dissociation of COOH to CO would become a favorable step as the potential goes more negative than −0.67 V. The subsequent protonation of CO also gradually becomes highly exergonic. Therefore, the hydrocarbons would become the favored products in the electrochemical reduction, especially at more negative potentials. These results are in accordance with the experimental results.9,16,20
Under the electrochemical condition, CHO* could undergo a hydrogenated dissociation to CH as the OH and O can be easily removed from the surface. As the potential is more negative than −0.67 V, the formation of CH could become more favored than the formation of CH2O*. However, as shown in Fig. 4, the activation energy for CHO hydrogenation to form CH2O* is ca. 0.7 eV and the dissociation to CH* + O* requires activation energies above 2.0 eV. CH2O* could undergo a hydrogenated dissociation to CH2* + H2O or be hydrogenated directly to CH3O* or CH2OH*. As the potential is more negative than −0.90 V, the formation of CH2* + H2O and CH3O* are strong exergonic processes, and the free energies, ΔGreac, are approximately equal, indicating that CH2* + H2O and CH3O* may be formed simultaneously by further the reduction of CH2O*. However, as shown in Fig. 6, CH3O* formation requires a higher activation energy. Thus, the formation of CH2* + H2O could become more favored. It is worth considering why CH3OH is formed in gas phase chemistry48 and CH4 is formed in electrochemistry16,17 on copper surfaces. In the current study, we considered that adsorbed CH3O* or CH2OH* may form CH3OH via a proton transfer reaction. However, as our calculations showed that the free energies, ΔGreac, of CH2OH* formation are more positive than the CH2O* hydrogenated dissociation to CH2* + H2O, and CH3O* formation requires a higher activation energy, this would be in favor of CH4 formation, in agreement with the electrochemical experimental results in which only CH4, and not CH3OH, was observed. Accordingly, we speculate that the optimum electrochemical reduction paths of CO2 on the Cu(111) surfaces are CO2(g) + 2(H+ + e−) → CO* + H2O(l), CO* + (H+ + e−) → CHO*, CHO* + (H+ + e−) → CH2O*, CH2O* + 2(H+ + e−) → CH2* + H2O(l), CH2* + (H+ + e−) → CH3*, and CH3* + (H+ + e−) → CH4(g), in which CO* + (H+ + e−) → CHO* may be the potential-limiting step. A similar conclusion has been obtained by Nørskov and coworkers.26
In the calculations conducted by Nørskov et al.,26,27 direct hydrogenation was considered as the major reaction of CH2O*. The key intermediate for the formation of hydrocarbons in their reaction mechanism is therefore CH3O*. By comparing the calculated reaction free energies and activation energies for the two pathways, we find that the hydrogenated dissociation pathway is actually more favorable. Koper et al.28,29 reported their studies of the reduction of CO2 on two basal planes, Cu(111) and Cu(100), using online electrochemical mass spectrometry (OLEMS) to investigate the path to CH4 and C2H4. This tip-based sampling technique allows the formation of volatile reaction intermediates and products to be followed while the potential at the electrode is changed. One can measure the reduction of the various species online while changing the potential and therefore follow the formation and consumption of intermediates during the reaction. Their experimental results show that it is very likely that CHOads is the key intermediate toward the breaking of the C–O bond and therefore the formation of CH2 and CH3 species and CH4. CH3O cannot be reduced to CH4 on the copper electrodes. The experimental observations appear to be in conflict with the DFT results from Nørskov et al., which suggest that CHOads is the intermediate in the formation of methane, but the energetically favored route to CH4 is through adsorbed CH3O species.
In addition, it has been proposed in earlier studies49 that the electrocatalytic reduction of CO2 on Cu electrodes involves an initial stage forming the carbon dioxide anion radical, CO2˙−, which is not observed on the Cu(111) surface in our previous calculations since the chemisorption state of the CO2 molecule is not obtained on the Cu(111) surface during the geometry optimization and minimum energy path analysis. Although the chemisorption state of CO2 molecule is obtained on the Cu(100) surface, the adsorption energies of the chemisorption states of the CO2 molecule are positive, which indicates that the chemisorption state is a metastable state. Indeed, in the electrochemical reduction of CO2, formate and COOH species are usually formed through anion radical ˙CO2− protonation. Due to a large reorganizational energy between the linear molecule and bent radical anion, the outer-sphere single electron reduction of CO2 to carbon dioxide anion radical should be difficult from both kinetics and thermodynamics.1 However, when the electrode potential is more negative, the electron level of the electrode could be aligned with the LOMO of CO2 and electron transfer to the CO2 molecule is possible. If a proton transfer occurs simultaneously, the products could be more stable. This phenomenon can be verified by the geometry optimization of the CO2 molecule in the presence of a hydronium ion. Accordingly, in this section, we used the hydronium ion H3O+⋯(H2O)3 as a model of solvation in an acid solution in order to model the first proton-coupled electron transfer step during CO2 reduction at the Cu/electrolyte interface and evaluate the role of the proton, for which the rationalization of the model has been validated in our previous work.50 Fig. 9 gives the geometry structural optimization plot of the protonation of CO2 in the presence of hydronium ion H3O+⋯(H2O)3. Optimized results indicate that the anion radical ˙CO2− is formed initially and then the hydrated proton, H9O4+, preferentially combined with O atom in the chemisorbed CO2 to form a COOH species (Fig. 9a and b), rather than a formate species, when the CO2 molecule adsorbed on the Cu(111) and Cu(100) surfaces for the protonation process, even if the hydrated proton H9O4+ was closer to the C atom in the initial structure. In other words, formate species formation may require a higher activation energy than COOH under the electrochemical environment. Based on the geometry optimization, it can be concluded that the anion radical ˙CO2− is formed primarily under electrochemical conditions and then successive proton and electron transfer occurs, eventually leading to the formation of hydrocarbons. Moreover, the experimental result51 also indicated that the adsorption of alkali metal on the Cu surfaces promoted the formation of the carbon dioxide anion radical ˙CO2−. Thus, the reaction in experiment conditions may be also promoted by alkali metal halides.
 | ||
| Fig. 9 Geometry optimization plot of the protonation of CO2 in the presence of hydrated proton H9O4+: (a) COOH adsorbed on the Cu(111) surface and (b) COOH adsorbed on the Cu(100) surface. | ||
Simultaneously, the abovementioned DFT calculations have also shown that there are two possibilities for hydrocarbon formation: one is chemical reduction through direct hydrogenation via adsorbed hydrogen, which occurs by a surface chemistry reaction, for example:
| (CH2 + H)* → CH3*, (CH3 + H)* → CH4(g) |
The other possibility to produce hydrocarbons is by direct electrochemical reaction:
| CH2* + H+ + e− → CH3*, CH3* + H+ + e− → CH4(g) |
The two routes are compared in Fig. 10 for the Cu(111) surface at −0.90 V (vs. RHE). As can be seen, in the direct electrochemical route, the intermediate CH2 formation is exergonic. CH2 formation can be seen to be ‘uphill’ in energy, and the free energies of CH2 hydrogenation to form CH4 are more positive in the hydrogenation route than in the direct electrochemical route. Thus, the direct electrochemical route would be expected to be highly favored at potentials of interest to CO2 reduction. Simultaneously, under electrochemical conditions, the free energy of CH4 formation via two protonation steps of CH2 is more negative on the Cu(111) surface than on the Cu(100) surface (as shown in Fig. 10), and the free energy of C2H4 formation is more negative than that of CH2 protonation to form CH3 on the Cu(100) surface (as shown in Table 3). Thus, DFT study indicated that CH4 is favorably formed on the Cu(111) surface and C2H4 is preferably formed on the Cu(100) surface under electrochemical conditions, explaining the experimental results.
 | ||
| Fig. 10 Comparison of the production of CH4(g) from CH2* via the direct electrochemical route and the combination of adsorbed CH2* and adsorbed H* on the Cu(111) surface, and effect of crystal face on selectivity of product. | ||
The possible optimized reaction paths of CO2 reduction to form hydrocarbons on Cu single crystal electrode surfaces and the degree of difficulty of the reaction have been revealed, and a systematic understanding of the CO2 reduction mechanism has also been achieved using density functional theory (DFT) method in the first principle on the geometry structural optimization, the reaction energy calculations and the minimum energy paths analysis. On this basis, the effect of combining the activation barriers of rate-determining steps and reaction pathways in C2H4 production via CO dimerization on the Cu(100) surface will be studied in the future. The research results have great significance for understanding in depth the selectivity of CO2 electrochemical reduction processes, the sensitivity of surface structure and the size effect of catalysts. Simultaneously, the research results can provide a scientific basis for designing the catalysts for CO2 electrochemical reduction.
4. Conclusions
In this study, a complete thermodynamic and kinetic description of CO2 reduction on two Cu single crystal surfaces is carried out for the first time based on a systematic DFT exploration. New insight has been provided on the chemical and electrochemical reduction of CO2 into CH4 at the atomic level on the Cu surfaces.In agreement with the present experiment and theoretical studies, our DFT calculated results indicated that CHOads and CH2OHads are the key intermediates toward the chemical and electrochemical reduction of CO2 into hydrocarbons. CO dimerization is more easy to achieve; CO hydrogenation is difficult to occur on the Cu(100) surface, explaining the unique selectivity for C2H4. However, our DFT calculated results indicated CH3O cannot be reduced to CH4 on copper electrodes; the main reaction of the CHOads is the hydrogenated dissociation to :CH2, eventually leading to the formation of CH3 species and CH4, which are consistent with the present experimental results but conflict the present theoretical results. In addition, DFT calculated results showed for the first time that the electrochemical reduction would be expected to be highly favored at potentials of interest to CO2 reduction compared with the chemical reduction, and the carbon dioxide anion radical (˙CO2−) is involved in the initial stage of CO2 electroreduction.
By analyzing the chemical reduction pathways, we deduced important mechanistic information. The possible optimal pathway on the Cu(111) surface is CO2(g) + H* → COOH* → CO* + OH*, CO* + H* → CHO*, CHO* + H* → CH2O*, CH2O* + H* → CH2OH* → CH2* + OH*, CH2* + H* → CH3*, CH3* + H* → CH4(g). CO is first formed by the dissociative hydrogenation of CO2. The most possible reaction path for CO is its hydrogenation to a CHO* intermediate. The key intermediate CH2O can be more easily formed through further CHO hydrogenation; the preferred reaction pathway of the CH2O should be hydrogenation to form CH2OH. The CH2OH intermediate either forms CH3OH by direct hydrogenation or CH2 by hydrogenative dissociation based on the activation barriers, which may be a parallel path in the CO2 reduction mechanism on the Cu(111) surface; finally, the CH2 intermediate leads to the formation of the hydrocarbons. From a kinetic viewpoint, the relatively slow steps on the Cu(111) surface include CO2(g) + H* → (CO + OH)* and (CO + H)* → CHO*. The reason may be that the weak Cu–CO2 interaction is an obstacle to CO2 dissociative hydrogenation and therefore slows down the overall conversion. The differences in the reaction mechanism on the Cu(111) and Cu(100) surfaces are that the CO formation and further reduction undergoes a different pathway. Simultaneously, the preference in the formation of CH4 and C2H4 is also different, the formation of C2H4 is easier on the Cu(100) surface, and the experimental results are explained to some degree based on the selectivity of the Cu single crystal surface.
The free energies for various steps in the electrochemical reduction of CO2 were calculated under different electrode potentials. The results indicated that formate and CO are mainly formed when the potential is more positive than −0.50 V (vs. RHE). The protonated dissociation of CO2 to form CO and the subsequent protonation of CO become increasingly exergonic as the potential goes negative, so that hydrocarbon gradually becomes the favored product during the electrochemical reduction. Under electrochemical conditions, the possible optimum reaction path on the Cu(111) surface is CO2(g) + 2(H+ + e−) → CO* + H2O(l), CO* + (H+ + e−) → CHO*, CHO* + (H+ + e−) → CH2O*, CH2O* + 2(H+ + e−) → CH2* + H2O(l), CH2* + (H+ + e−) → CH3*, CH3* + (H+ + e−) → CH4(g), in which CO* + (H+ + e−) → CHO* may be the potential-limiting step. Simultaneously, the calculated results also partly explained why CH3OH is formed in gas phase chemistry and only CH4 is observed in electrochemistry on copper surfaces.
Acknowledgements
This study is financially supported by the National Natural Science Foundation of China (Grant No. 21303048), the Hunan Provincial Natural Science Foundation of China (Grant No. 13JJ4101), the Construct Program of the Key Discipline in Hunan Province (Applied Chemistry) and the Doctoral Start-up Fund of Hunan University of Arts and Science.References
- F. Hasegawa, S. H. Yokoyama and K. Imou, Bioresour. Technol., 2010, 101, S109–S111 CrossRef CAS PubMed
.
- M. Abu-zahra, L. H. Schneiders, J. P. M. Niederer, P. H. Feron and G. F. Versteeg, Int. J. Greenhouse Gas Control, 2007, 1, 37–46 CrossRef CAS
- B. P. Sullivan, Electrochemical and Electrocatalytic Reduction of Carbon Dioxide, Elsevier, Amsterdam, 1993 Search PubMed
- C. M. Sanchez-Sanchez, V. Montiel, D. A. Tryk, A. Aldaz and A. Fujishima, Pure Appl. Chem., 2001, 73, 1917–1927 CrossRef CAS
- M. M. Halmann, Greenhouse Gas Carbon Dioxide Mitigation, Lewis Publishers, Washington, DC, 1999 Search PubMed
- T. Inui, M. Anp, K. Izui, S. Yanagida and T. Yamaguchi, Advances in Chemical Conversions for Mitigating Carbon Dioxide, Elsevier, Amsterdam, 1998 Search PubMed
- Y. Hori, K. Kikuchi, A. Murata and S. Suzuki, Chem. Lett., 1986, 15, 897–898 CrossRef
- Y. Hori, A. Murata, R. Takahashi and S. Suzuki, J. Am. Chem. Soc., 1987, 109, 5022–5023 CrossRef CAS
- J. J. Kim, D. P. Summers and K. W. Frese Jr, J. Electroanal. Chem., 1988, 245, 223–244 CrossRef CAS
- Y. Hori, Electrochemical CO2 reduction on metal electrodes, in Modern Aspects of Electrochemistry, Springer, New York, 2008 Search PubMed
- M. Gattrell, N. Gupta and A. Co, J. Electroanal. Chem., 2006, 594, 1–19 CrossRef CAS PubMed
- W. M. Ayers, Carbon Dioxide Chemistry: Environmental Issues, The Royal Society of Chemistry, U. K., 1994 Search PubMed
- K. Ito, S. H. Ikeda and H. Noda, Solar World Congress Proceedings Biennial Congress International Solar Energy Society, Pergamon, Oxford, 1992 Search PubMed
- R. L. Cook, R. C. MacDuff and A. F. Sammells, J. Electrochem. Soc., 1989, 136, 1982–1984 CrossRef CAS PubMed
- D. W. DeWulf, T. Jin and A. J. Bard, J. Electrochem. Soc., 1989, 136, 1686–1691 CrossRef CAS PubMed
- Y. Hori, A. Murata and R. Takahashi, J. Chem. Soc., Faraday Trans. 1, 1989, 2309–2326 RSC
- Y. Hori, H. Wakebe, T. Tsukamoto and O. Koga, Electrochim. Acta, 1994, 39, 1833–1839 CrossRef CAS
- H. De Jesús-Cardona, C. del Moral and C. R. Cabrera, J. Electroanal. Chem., 2001, 513, 45–51 CrossRef
- C. Delacourt, P. L. Ridgway, J. B. Kerr and J. Newman, J. Electrochem. Soc., 2008, 155, B42–B49 CrossRef CAS PubMed
- H. Noda, S. Ikeda, Y. Oda and K. Ito, Chem. Lett., 1989, 18, 289–292 CrossRef
- J. Krause, D. Borgmann and G. Wedler, Surf. Sci., 1996, 347, 1–10 CrossRef CAS
- I. A. Bonicke, W. Kirstein and F. Thieme, Surf. Sci., 1994, 307−309, 177–181 CrossRef
- Y. Hori, A. Murata, T. Tsukamoto, H. Wakebe, O. Koga and H. Yamazaki, Electrochim. Acta, 1994, 39, 2495–2500 CrossRef CAS
- J. Pritchard, Surf. Sci., 1979, 79, 231–244 CrossRef CAS
- Y. Hori, O. Koga, H. Yamazaki and T. Matsuo, Electrochim. Acta, 1995, 40, 2617–2622 CrossRef CAS
- A. A. Peterson, F. Abild-Pedersen, F. Studt, J. Rossmeisl and J. K. Nørskov, Energy Environ. Sci., 2010, 3, 1311–1315 CAS
- W. J. Durand, A. A. Peterson, F. Studt, A. A. Peterson and J. K. Nørskov, Surf. Sci., 2011, 605, 1354–1359 CrossRef CAS PubMed
- K. J. P. Schouten, Y. Kwon, C. J. M. van der Ham, Z. Qin and M. T. M. Koper, Chem. Sci., 2011, 2, 1902–1909 RSC
- K. J. P. Schouten, Z. Qin, E. P. Gallent and M. T. M. Koper, J. Am. Chem. Soc., 2012, 134, 9864–9867 CrossRef CAS PubMed
- X. Nie, M. R. Esopi, M. J. Janik and A. Asthagiri, Angew. Chem. Int. Ed., 2013, 52, 2459–2462 CrossRef CAS PubMed
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS
- D. Vanderbilt, Phys. Rev. B: Condens. Matter Mater. Phys., 1990, 41, 7892–7895 CrossRef
- M. Methfessel and A. T. Paxton, Phys. Rev. B: Condens. Matter Mater. Phys., 1989, 40, 3616–3621 CrossRef CAS
- S. Baroni, A. Dal Corso, S. de Gironcoli and P. Giannozzi, PWSCF and PHONON: Plane-Wave Pseudo-Potential Codes, 2001, http://www.pwscf.org Search PubMed
- A. Kokalj, J. Mol. Graphics Modell., 1999, 17, 176–179 CrossRef CAS
- A. Kokalj and M. Causà, Scientific Visualization in Computational Quantum Chemistry, In Proceedings of High Performance Graphics Systems and Applications European Workshop, CINECA-Interuniversity Consortium, Bologna, Italy, 2000 Search PubMed
- A. Kokalj and M. Causà, XCrySDen: X-Window CRYstalline Structures and DENsities, 2001, http://www-k3.ijs.si/kokalj/xc/XCrySDen.html Search PubMed
- J. Greeley, A. A. Gokhale, J. Kreuser, J. A. Dumesic, H. Topsøe, N. Y. Topsøe and M. Mavrikakis, J. Catal., 2003, 213, 63–72 CrossRef CAS
- CRC Handbook of Chemistry and Physics, CRC Press/Taylor and Francis, Boca Raton, FL, 91st edn, (Internet Version 2011), 2011 Search PubMed
- G. Henkelman and H. Jonsson, J. Chem. Phys., 2000, 113, 9978–9985 CrossRef CAS PubMed
- G. Henkelman, B. P. Uberuaga and H. Jonsson, J. Chem. Phys., 2000, 113, 9901–9904 CrossRef CAS PubMed
- Y. M. Choi and P. Liu, J. Am. Chem. Soc., 2009, 131, 13054–13061 CrossRef CAS PubMed
- A. Deluzarche, J. P. Hindermann, R. Kieffer, R. Breault and A. Kiennemann, J. Phys. Chem., 1984, 88, 4993–4995 CrossRef CAS
- A. Kiennemann, R. Breault, J. P. Hindermann and M. Laurin, J. Chem. Soc., Faraday Trans. 1, 1987, 83, 2119–2128 RSC
- C. Diagne, H. Idriss, J. P. Hindermann and A. Kiennemann, Appl. Catal., 1989, 51, 165–180 CrossRef CAS
- I. N. Remediakis, F. Abild-Pedersen and J. K. Nørskov, J. Phys. Chem. B, 2004, 108, 14535–14540 CrossRef CAS
- J. K. Nørskov, J. Rossmeisl, A. Logadottir and L. Lindqvist, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef
- J. B. Hansen and P. E. Højlund Nielsen, Methanol synthesis, in Handbook of Heterogeneous Catalysis, ed. G. Ertl, H. Knözinger, F. Schüth and J. Weitkamp, 2008, vol. 13, pp. 2920–2949 Search PubMed
- M. Jitaru, D. A. Lowy, M. Toma, B. C. Toma and L. Oniciu, J. Appl. Electrochem., 1997, 27, 875–889 CrossRef CAS
- L. H. Ou, F. Yang, Y. W. Liu and S. L. Chen, J. Phys. Chem. C, 2009, 113, 20657–20665 CAS
- J. Onsgaard, L. Bech, C. Svensgaard, P. J. Godowski and S. V. Hoffmann, Prog. Surf. Sci., 2001, 67, 205–216 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra09294a |
| This journal is © The Royal Society of Chemistry 2015 |