
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Comprehensive elucidation of the effect of residual lignin on the physical, barrier, mechanical and surface properties of nanocellulose films†
Ester
Rojo
*a,
Maria Soledad
Peresin
b,
William W.
Sampson
c,
Ingrid C.
Hoeger
d,
Jari
Vartiainen
b,
Janne
Laine
a and
Orlando J.
Rojas
*ad
aBiobased Colloids and Materials group (BiCMat), Department of Forest Products Technology, Aalto University, School of Chemical Technology, P.O. Box 16300, 00076 Aalto, Espoo, Finland. E-mail: erojorecio@gmail.com; orlando.rojas@aalto.fi; Tel: +358 505124225
bVTT Technical Research Centre of Finland, P.O. Box 1000, 02044 Espoo, Finland
cSchool of Materials, University of Manchester, Manchester, M13 9PL, UK
dDepartments of Forest Biomaterials and Chemical and Biomolecular Engineering, North Carolina State University, Campus Box 8005, Raleigh, NC 27695-8005, USA
First published on 7th January 2015
Abstract
We elucidate the effect of residual lignin on the interfacial, physical and mechanical properties of lignocellulose nanofibrils (LCNF) and respective nanopapers. Fibers containing ∼0, 2, 4, and 14 wt% residual lignin were microfluidized into LCNF aqueous suspensions and were processed into dry films (nanopapers). A systematic decrease in fibril diameter with increasing residual lignin was observed upon fibrillation, consistent with the radical scavenging ability of the lignin that results in better cell wall deconstruction. The stiff nature of the lignin-containing fibrils made them less able to conform during filtration and improved extensively dewatering, owing to a more open structure. However, the softening of the lignin during hot-pressing of the nanopapers and its amorphous nature enabled a binding effect, filling the voids between the nanofibers (thus reducing the number of micropores) and making the surface of the nanopapers smoother. The interfacial free energy of interaction changed drastically with the increased lignin content: the corresponding water contact angles were 35° and 78° for the lignin-free and for the (14%) lignin-containing nanopaper, respectively, revealing the increase in hydrophobicity. Together with the significantly less porous structure of LCNF nanopapers, lower water absorbency was observed with increased lignin content. Lignin in the nanopapers reduced the oxygen permeability by up to 200-fold. Water vapor permeability, in turn, did not correlate linearly with lignin content but depended most significantly on material density. The tensile strength, modulus, and strain for the LCNF nanopapers were found to be in the range 116–164 MPa, 10.5–14.3 GPa, and 1.7–3.5%, respectively. To a good degree of approximation, these mechanical properties were rather insensitive to lignin content and comparable to those of nanopapers derived from fully bleached CNF. Whilst it might be expected that lignin interferes in hydrogen bonding between fibrils, this was apparently counteracted by the uniform distribution of lignin seemingly aiding stress-transfer between fibrils and thus preserving mechanical properties. Overall, LCNF is demonstrated to be a suitable precursor of nanopaper, especially when reduced polarity and low hydrophilicity are desirable in related bio-products.
Introduction
Residual biomass can be valorized in the form of engineered materials (paper, composites, etc.) and for the production of cellulose nanofibrils (CNF). CNF are obtained after disintegrating cellulosic fibers into their sub-structural fibrils by mechanical treatment or by its combination with chemical1,2 or enzymatic processes.3 Refining and high-pressure homogenization are generally used in the production of cellulose nanofibrils.4 The small width and large aspect ratio (length to width ratio) provide CNF with a very large specific surface area, which is beneficial for the development of high strength in CNF webs as well as other properties. Because of its abundance, biodegradability, renewability and unique physical and structural characteristics, CNF has been gradually gaining attention as a green alternative in composites, coatings, films, membranes and packaging materials. In addition, CNF is an excellent stabilizer or thickener of cosmetic, food, paint, and pharmaceutical emulsions and suspensions and can offer promising opportunities based on its rheological properties.Most studies on cellulose nanofibrils use lignin-free (bleached) fibers for CNF production. Thus, different delignification methods have been tested as possible initial steps for the preparation of nanocellulose. However, unbleached fibers containing residual lignin, hemicelluloses and extractives represent an alternative raw material to produce lignocellulose nanofibrils (LCNF) with the benefits of high yield, low production costs and low environmental impact. Moreover, lignin can be used to adjust the polarity and hydrophilicity of cellulose, which for some applications are challenges that need to be addressed. Hydroxyl groups in CNF facilitate dispersion of the fibrils in dilute water suspensions but promote aggregation in non-polar solvents; therefore, the CNF dispersion ability in non-polar media is dramatically compromised. To avoid aggregation, CNF are usually stored as water suspensions at low solid contents (typically below 2 wt%), which limits large-scale utilization.5
Depending on the process used in fiber production, LCNF can be less polar and more hydrophobic than CNF, allowing better dispersion in non-polar media, for example in composites using hydrophobic matrices or in aqueous dispersions at solid contents higher than 2 wt%. Although some reports discuss LCNF production and utilization, the effect of lignin on the properties of the nanofibrils and their films (or “nanopapers”) remains unclear. Spence et al.6–9 studied the energy and chemical requirements in the production of lignin-containing nanopapers, which showed promising properties for packaging applications. The precise effect of lignin on the performance of LCNF was not clear, since different wood sources, pulping and bleaching methods were used in the respective investigations, making systematic analysis difficult. Ferrer et al.10,11 addressed this issue by selecting fibers from a single wood source containing various lignin amounts to explain their impact on LCNF and nanopaper properties. However, these studies were limited since very small lignin concentrations were used. Today, kraft pulp dominates the paper and board product market, as well as the production of CNF. In the latter case, however, SO2–ethanol–water (SEW) fibers may be an attractive raw material for LCNF production. This stems from the fact that SEW pulping is a promising fractionation process for lignocellulose within the biorefinery concept. The SEW pulping chemistry is employed by American Process Inc. in a biorefinery process termed AVAP® (American Value Added Pulping), which consists of the treatment of lignocellulosic materials for producing biofuels and other bioproducts.12 Various patent applications for the SEW pulping chemistry disclose variations of this process.13–16 Compared to kraft pulps, SEW fibers have lower fiber strength and a higher fiber swelling capacity, which is associated with internal fibrillation. Moreover, the SEW process enables better fractionation of wood components than conventional processes and allows a better control of the composition of the resulting fibers. More important to the present investigation, considering LCNF production, SEW fibers may require low energy for deconstruction because of the easier breaking of loosened internal fiber wall structures.17 Sulfonation of lignin occurs during the SEW process. The main reactions of lignin follow the monomolecular nucleophilic substitution mechanisms summarized in Fig. 1.18
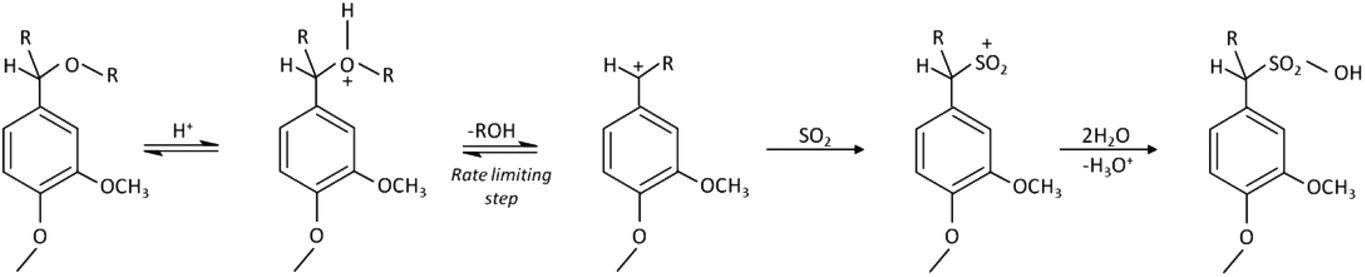 | ||
| Fig. 1 Mechanism for the sulphonation of lignin that occurs during the SEW process. R: alkyl groups. | ||
In this study, SEW fibers were obtained with full control of their composition in terms of residual lignin and heteropolysaccharide contents; these fibers were used to produce LCNF and nanopapers. Norway spruce wood chips were subjected to SEW processing to yield fibers with varying degree of delignification, covering a wide range of residual lignin values (1.7, 3.7, and 13.5 wt% lignin). Fully bleached Norway spruce kraft pulp was used as the reference system. Dewatering during nanopaper production was compared for the different SEW LCNF. Morphological, tensile, and barrier properties of the respective nanopapers were evaluated. In addition, the porous structure, surface free energy as well as properties related to interactions of the nanopapers with water were elucidated. Thus, this investigation reports on a systematic evaluation to elucidate the effect of lignin on the properties of nanopaper.
Experimental
Lignocellulosic fibers, LCNF and nanopapers
SEW fibers were produced from Norway spruce using 12% (w/w) SO2 at temperatures ranging from 135 to 155 °C and fractionation times ranging from 25 to 160 min. The temperature and fractionation time were varied in order to obtain fibers with different degrees of delignification. Thus, variations related to fiber source and morphology, digestion process, and related aspects were minimized. The SEW fibers containing 1.7, 3.7, and 13.5 wt% residual lignin were used in the production of LCNF and nanopapers. Samples were named according to their approximate residual lignin content as 2L (1.7 wt%), 4L (3.7 wt%), and 14L (13.5 wt%). Table S1 of the ESI† includes the conditions used in SEW reactions and the detailed compositions of the obtained SEW fibers. Fully bleached Norway spruce kraft pulp (lignin content <0.5%) was used as the reference.Preparation of LCNF consisted of two steps: mechanical pre-treatment and high shear disintegration via microfluidization. A refining treatment before microfluidization was used in order to improve fiber accessibility and fibrillation efficiency. Shortly, mechanical pretreatment of the fiber suspensions consisted of an initial grinding step in a small Wiley mill (30 mesh screen) and refining with a Polytron PT 10/35 homogenizer. The obtained aqueous fiber suspensions (solids contents <2 wt%) were processed in a high-pressure microfluidizer (Microfluidizer M-110 P, Microfluidics Corp., 2010) using six passes. The resulting LCNF suspensions were collected and stored at 4 °C until use.
To manufacture nanopapers, the respective LCNF suspension was diluted to 0.8 wt% solids content in deionized water and stirred overnight at room temperature. Each suspension was poured slowly into an over-pressurized filtration device and drainage was carried out at 2.5 bar for 15 minutes. The device was equipped with a qualitative filter paper with particle retention of 12–15 μm to remove water and retain the fibrils. In addition to the paper, an open mesh fabric screen (Sefar Nitex 03-10/2, a mesh opening of 10 μm with an open area of 2%) was used in order to improve the retention of fibrils and enhance the surface finish. The volume of filtrate from the diluted LCNF suspensions was measured at different times for a total of 15 min and the dewatering time was determined for the different samples. This dewatering behavior is considered to be a useful indicator of the potential for ‘easy’ production of nanopapers. The webs obtained after filtering were first cold-pressed between two blotting papers (4 bar, 4 min) and then hot-pressed (100 °C, 220 bar, 2 h). This last step, besides drying the films, also promoted the formation of inter-fiber bonds because of the simultaneous applications of heat and pressure. Nanopapers with a target dry basis weight of 80 g m−2 were obtained and stored in a conditioned room at 23 °C and 50% relative humidity prior to testing.
LCNF morphology
Atomic force microscopy (AFM) (Nanoscope IIIa multimode scanning probe instrument from Digital Instruments Inc., Santa Barbara, USA) was used to assess the morphology of the dispersed LCNF and the roughness of the respective nanopapers. The gel-like suspensions obtained after the microfluidization process were extensively diluted to obtain images of individual fibrils. Scanning was performed in tapping mode in air using silicon cantilevers (NSC 15/AIBS, MicroMasch, Tallinn, Estonia). The nominal resonance frequency of the cantilevers was around 300 kHz. The scanned areas (2 × 2 μm2) were imaged in at least three different locations of the respective samples. Images were taken in height, amplitude error, and phase modes. No image processing except flattening was carried out.Scanning electron microscopy (SEM, JEOL JSM-6400F) was carried out to obtain cross section images and to study the microstructure of the films. The cross-section samples were prepared by fracturing the nanopapers in liquid nitrogen to avoid structural deformations of the materials. Samples were sputtered with graphite and gold to enhance the conductivity. The operation voltage was 5 kV and the working distance was set to 20 mm.
Density and mechanical properties of LCNF nanopapers
The apparent density of the nanopapers was calculated by dividing sample basis weight by the apparent thickness. The basis weight was determined according to TAPPI standard T410. Film thickness was obtained using TAPPI Method T411 by means of a Lorentzen & Wettre Micrometer 51 instrument. Each sample was measured 10 times.Tensile tests were carried out at 23 °C and 50% relative humidity, in accordance with ISO 527-2, using an MTS 400/M Vertical Tensile Tester equipped with a 200 N load cell. Specimens of 5 mm wide and 75 mm length were cut out from the nanopapers using a dog bone shape cutting die. The tests were performed with a cross-head speed of 5 mm min−1 and the clamp span was set to 50 mm. The tensile strength, elongation, elastic modulus and tensile energy absorption were obtained from the stress–strain curves. The specific tensile strength, or ‘tensile index’, was calculated as the failure load divided by the basis weight, allowing comparison between tensile strengths of samples with small differences in basis weight.19 The specific elastic modulus, i.e. weight-corrected Young's modulus, was obtained as the slope of the initial linear region of the stress–strain curve. The specific Tensile Energy Absorption (TEA) was calculated as the area under the load-elongation curve divided by the basis weight and measures the work done to break the sample per unit sample weight (J g−1). The average and standard deviation of five measurements were reported.
Porous structure of LCNF nanopapers
The porous structure of the nanopapers was evaluated by thermoporosimetry20 using differential scanning calorimetry (DSC). There are various methods to determine the pore size distribution, but DSC allows the use of non-dried samples and prevents the “hornification” phenomenon, i.e., physical and chemical changes occurring in the cellulose fibers during drying and wetting. Hornification might significantly alter the pores in the cellulose fibers and may possibly lead to inaccurate determination of the pore size distribution in cellulose nanofibers and related nanopapers. The DSC method can be used to measure the pore size distribution of samples at different moisture ratios and has been successfully applied to cellulose fibers.20–22 Moreover, DSC has been found to be in good agreement with other methods commonly applied in cellulosic materials,23e.g., NMR cryoporosimetry23,24 or inverse size-exclusion chromatography.25Nanopaper samples of 3 × 3 mm were placed in 40 μl pre-weighed aluminum pans and 2 mg of distilled water per mg of solid was added using a microsyringe. The pore size distribution obtained with thermoporosimetry is nearly independent of moisture content above 1 g g−1.21 After sealing, hermetic pans were kept at room temperature overnight in order to reach equilibrium moisture content. Measurements were carried out on a Mettler DSC 30 equipped with a cooling apparatus under a nitrogen gas flow rate of 100 mL min−1. Two different DSC programs were applied for evaluating the pore size distribution and the bound water content of the nanopapers.
For the pore size distribution the isothermal method was used. The basic principle of the method is the fact that water contained within pores is at an elevated pressure compared to free water and therefore has a depressed melting temperature. Measurements of the energy absorbed when water melts in the frozen nanopaper at given temperatures approaching 0 °C give information about the number of pores of respective size within the nanopaper. The samples were frozen to −35 °C at 1 °C min−1 and maintained at this temperature for 4 min. Then the temperature was raised to −33 °C and the sensible heat of the wet nanopapers was determined by assuming the absence of melting. The cycle was repeated with the isothermal melting point at −20, −17, −14, −11, −9, −7, −5, −3.5, −2.5, −1.6, −0.8, −0.4 and −0.2 °C with enough time in each step to complete the melting transition. For every melting temperature (Tm), a representative pore diameter (D) was obtained using the Gibbs–Thomson equation (eqn (1)) and the calculated values are shown in Table S2 of the ESI.†
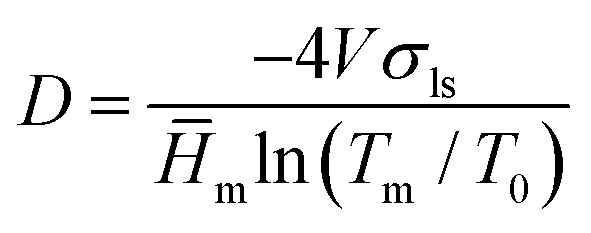 | (1) |
![[H with combining macron]](https://www.rsc.org/images/entities/i_char_0048_0304.gif) m is the specific melting enthalpy of water (334 J g−1), and T0 is the melting point of water at 1 atmosphere pressure (273.15 K). Although we expect inter-fiber voids to be irregular, eqn (1) was derived for cylindrical pores; accordingly, the method provides a measure of an effective pore diameter.
m is the specific melting enthalpy of water (334 J g−1), and T0 is the melting point of water at 1 atmosphere pressure (273.15 K). Although we expect inter-fiber voids to be irregular, eqn (1) was derived for cylindrical pores; accordingly, the method provides a measure of an effective pore diameter.
The heat absorbed during melting in each isothermal period, Ht, was obtained by integrating the endotherm peak. Subsequently, the melting enthalpy, Hm, was calculated by subtracting a sensible heat (CpΔT) according to eqn (2). The melting enthalpy was then used to determine the amount of water melted within the temperature step by means of the specific melting enthalpy of water (334 J g−1). After the experiments, pan lids were pierced and the samples were dried overnight at 105 °C to subsequently obtain total water content in the sample as well as the mass of dry solid. The melting water fraction at different isothermal temperatures was expressed as a mass fraction and was used with eqn (1) to provide an effective pore diameter distribution.
| Hm = Ht − CpΔT | (2) |
The bound water content of the nanopapers was measured independently to avoid hysteresis phenomena related to consecutive heating and cooling cycles. To this end, the sample pan was frozen to −30 °C and ramped at 1 °C min−1 to 25 °C. Two overlapping peaks were obtained (related to freezing bound and free water) and were separated by splitting the integrated areas of heat flow at the inflection temperature between the peaks. The integration of the complete overlapping peaks provided the total amount of freezing water. Finally, non-freezing bound water was obtained by subtracting the total freezing water from the total amount of water in the sample. All of the calculated water fractions were expressed in mass of water per mass of dried solid.
The porosity, ε, of the nanopapers was calculated from their densities (eqn (3)) by assuming the density of cellulose and lignin to be 1.5 and 1.3 g cm−3, respectively;26 applying a simple rule of mixtures yields a very narrow range of densities for solid lignocellulose in the ratios investigated, i.e. between 1.47 and 1.5 g cm−3; accordingly, we may be confident that differences in density arise almost entirely from differences in porosity.
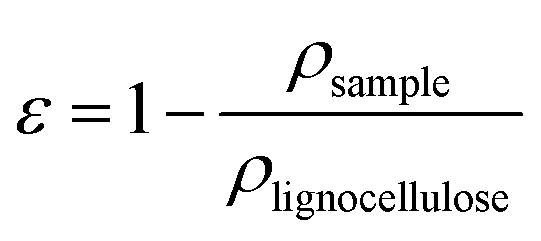 | (3) |
Surface and interfacial free energies
Nanopaper surface free energy and surface energy components were determined from contact angle (CA) measurements with four pure liquids of different polarities, namely water, formamide, diiodomethane, and ethylene glycol. The CA of sessile drops was determined using a KSV CAM200 optical contact angle goniometer (KSV Instruments). Surface free energies of the nanopapers and its components were obtained with the CAM2008 software. A more detailed explanation of the surface energy calculation is provided in the ESI.† Briefly, the total surface energy of the solid (γS) was calculated from the disperse (γLWS) and polar (acid–base) (γABS) components. The polar component was further divided into γ+S, the electron-acceptor or Lewis acid parameter, and γ−S, the electron-donor or Lewis base parameter. The solid/liquid interfacial energy (γSL) was also calculated and the surface free energy components of the solid were obtained by combining Young's equation (CA data) and the solid/liquid interfacial energy. The calculation used the known surface tension components of the probing liquid (γLWL, γ+L, γ−L). In addition, the interfacial free energy of the nanopapers (ΔGIFsws) was obtained in order to define the hydrophilic or hydrophobic nature of the nanopapers.Interactions with water and barrier properties
The advancing and receding water CAs were directly measured on sessile drops with an optical contact angle meter (KSV Instruments Ltd, CAM 200). Water drops with a volume of around 4 μl were placed on the nanopaper surface from a micro-syringe. Then water was continuously added to the droplet from the syringe up to 6.7 μl to advance the contact line, and advancing contact angles (θa) were determined. For receding contact angles (θr) the liquid was withdrawn with the syringe needle until the contact line retracted. Measurements were made on three or more drops and averaged. CA hysteresis was obtained as the difference between advancing and receding angles.Film water absorption was determined by immersing 5 × 20 mm strips in deionized water for 2 h and recording the sample weight after 1.5, 5, 10, 30, 60, and 120 min using a top pan balance. Care was taken before weighing the samples to remove the excess of water by means of a standard roller using blotting paper on both sides of the nanopaper samples. Relative water absorption (RWA) was determined using eqn (4):
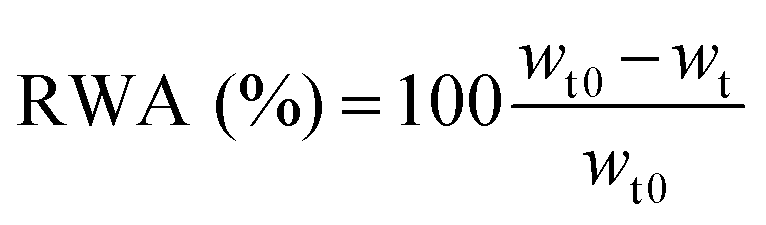 | (4) |
The water vapor and oxygen transmission rates, WVTR and OTR, were measured. WVTR of the nanopapers was determined gravimetrically using a “wet cup” ASTM E96A procedure. Nanopapers were cut into circles of 25 cm2 and mounted on aluminum dishes containing 30 mL of water. Dishes were stored at 23 °C and 50% relative humidity and weighed periodically until a constant rate of weight reduction was attained. The relative humidity gradient across the samples was 100–50%. OTR measurements were performed with Oxygen Permeation Analyzer Models 8001 and 8011 (Systech Instruments Ltd, UK) according to ASTM D3985. Tests were carried out at 23 °C and varying relative humidity (RH: ≤50% and 80%) using 100% oxygen as the test gas. Aluminum foil masks, with an inner diameter area of 5 cm2, were used to mount nanopaper samples in the test cells. Transmission rates (WVTR and OTR) were normalized to sample thicknesses and expressed as permeabilities (WVP and OP). The average and the standard deviation of three measurements were reported.
Results and discussion
Effect of lignin content on filtration time of LCNF suspensions during nanopaper manufacture
Energy consumption during the production of cellulose nanofibrils and nanopapers is important in assessing the environmental impact of CNF and their applications. Energy reductions have been reported before for the production of CNF containing lignin (LCNF), in comparison to fully bleached nanofibers.7,8 In preparing films, an important aspect of energy consumption is the dewatering resistance of the fibril network evolving from the suspension. Here, we recorded the cumulative filtrate volume during dewatering of LCNF suspensions; Fig. 2 shows the cumulative filtrate volume vs. filtration time of the diluted suspensions for LCNF of different lignin contents. Due to small differences in suspension concentration, the cumulative volume is normalized to 100% of that at 15 minutes; this time corresponds to that for complete filtration of the reference sample. We note that filtration of LCNF suspensions was complete in less than 6 minutes in all cases; further the filtration rate increased with increasing lignin content, though the difference between samples 4L and 14L was less than that between other samples. Now, to a reasonable approximation, and guided by classical models such as, e.g., Kozeny–Carman,27,28 we can expect the flow rate during filtration to be indicative of the porosity of the evolving filter cake. So, here we observe a more open structure formed from fibrils with increasing lignin content; this is consistent with the expectation that these fibrils are stiffer and thus less able to conform to each other when packed under gravity filtration. We return to this observation in our discussion of mechanical properties.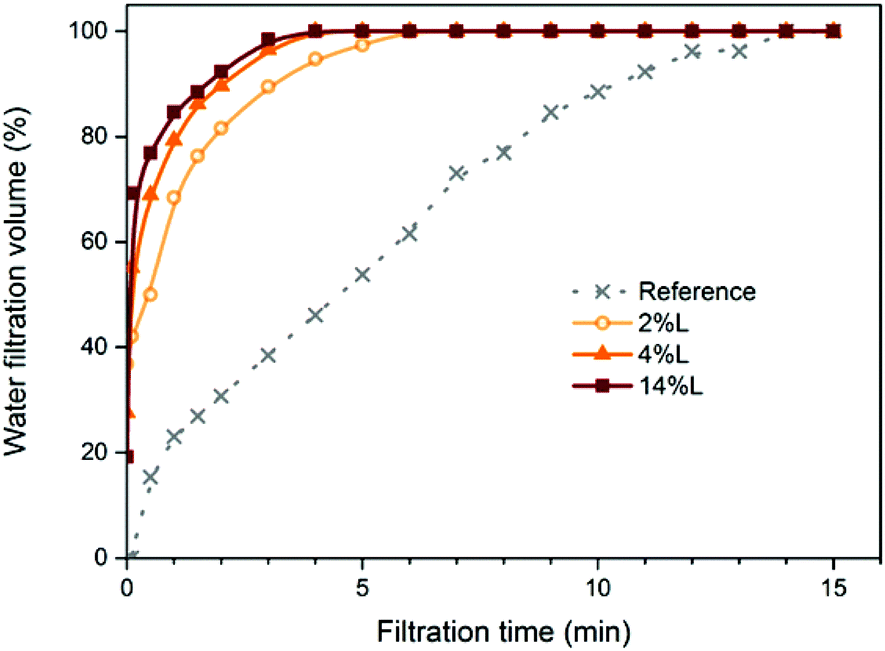 | ||
| Fig. 2 Water filtration volume vs. filtration time of the CNF (obtained from fully bleached fibers) and LCNF suspensions with different lignin contents, as indicated. | ||
Morphology
The morphology of the dispersed nanofibrils was evaluated by AFM using height and phase imaging (Fig. 3). Some fibrils with widths of several nanometers were observed in the images of the reference sample (R), which is typical for fully bleached CNF. AFM images of the 2L fibrils appeared to be quite similar to those from the reference sample. However, some small, globular-shaped particles could be identified in addition to the fibrils in the 2L sample, especially in the phase image. Similar particles were more clearly distinguished in sample 4L. It is likely that these globular features correspond to lignin nanoparticles. In the case of the sample with the highest lignin concentration (14L), these particles are predominantly located between the cellulosic nanofibers, forming complex composite structures with the fibrils. These observations are consistent with the role of lignin in the native wood cell wall, where it exists as a stiff phase between cellulosic fibers.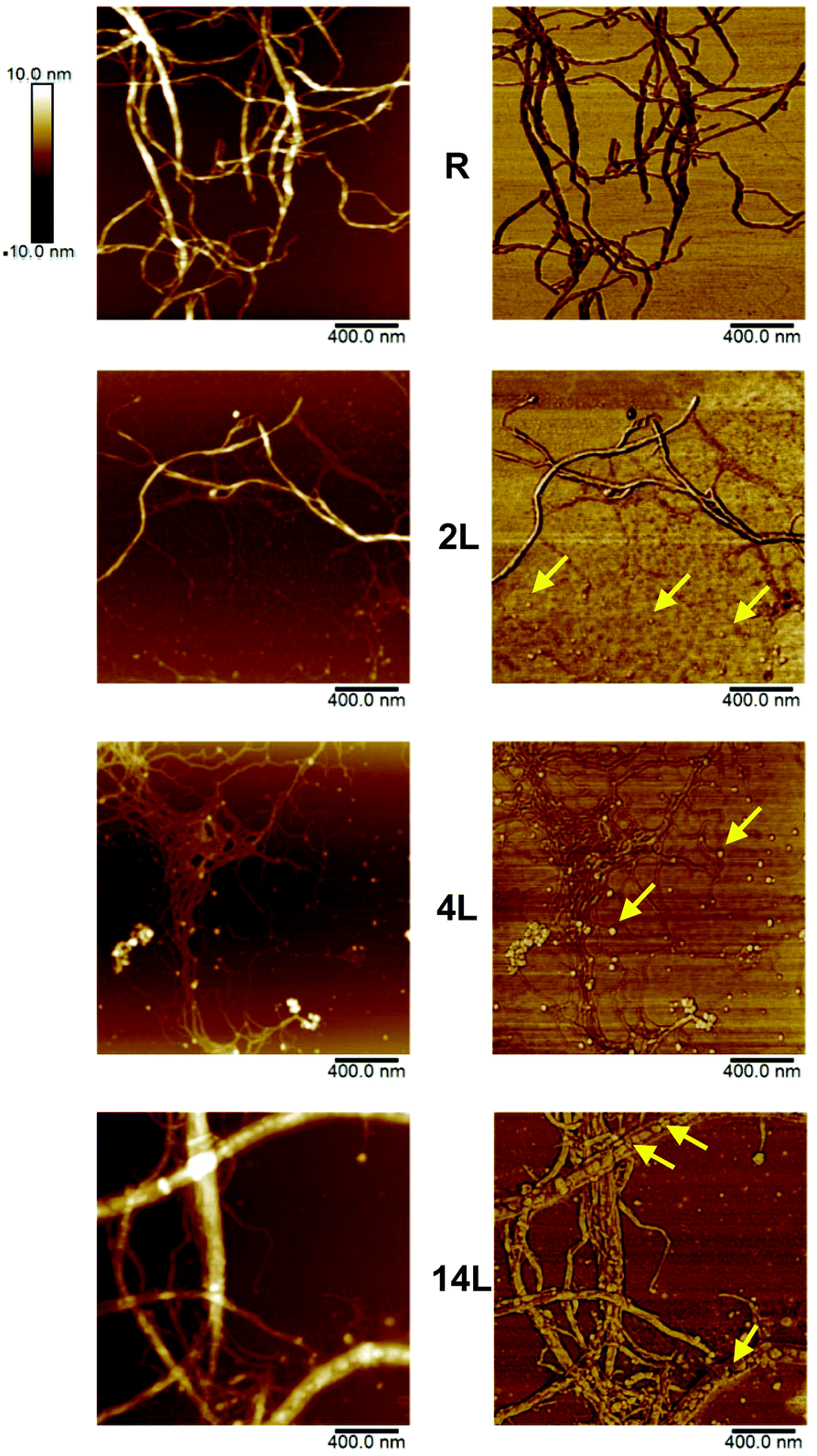 | ||
| Fig. 3 AFM images of 4 μm2 areas of LCNF (samples 2L, 4L and 14L) as well as the reference CNF (R), deposited on mica after air-drying from diluted aqueous suspensions. The height and phase images are presented in the left and right columns, respectively. The arrows point to some of the lignin nanoparticles, which act as a cementing material between the cellulose nanofibrils. | ||
From the micrographs in Fig. 3, it is apparent that the fibril diameter reduces with increased lignin content; the mean fibril diameters, as obtained from AFM measurements, were 44 ± 3, 25 ± 1, 20 ± 2, and 16 ± 2 nm for the reference, 2L, 4L, and 14L samples, respectively. Thus, we observe a systematic decrease in fibril diameter with increasing lignin content. This reduction in fibril diameter is related to lignin's ability to promote fibrillation. Lignin is a known antioxidant and thus it stabilizes cellulosic mechano-radicals formed during the microfluidization.29 Cellulosic radicals are extremely reactive and can participate in recombination reactions (crosslinking) that counteract the fibrillation at reduced lignin content. At higher lignin content, the radical scavenging ability of the lignin results in less pronounced crosslinking of cellulose, which allows better deconstruction of the fibrils. In sum, compared with lignin-depleted fibers, lignin-containing fibers are fibrillated more extensively.
The plane view and the cross section of LCNF nanopapers were accessed by AFM and SEM, respectively. Representative images are provided in Fig. 4, where AFM amplitude error images of nanopaper surfaces and SEM images of the cross-sections of the reference and the 14L samples are compared. The AFM roughness (Rq) values of 17.1 ± 1.5, 10.9 ± 1, 10 ± 0.8, and 8.6 ± 1.6 nm were measured for the reference CNF and 2L, 4L and 14L LCNF nanopapers from the AFM height profiles (not shown). The roughness is related to the diameter of the fibrils and the degree of consolidation on the surface upon drying. As can be seen in the AFM images in Fig. 4, fibrils with higher residual lignin content presented smaller widths and formed smoother surfaces, compared to the reference nanopaper; indeed, regression of the roughness data against the measured fibril diameter yields a straight line with the coefficient of determination, r2 = 0.995 (eqn (5)).
| Rq = 0.3df + 3.7 | (5) |
 | ||
| Fig. 4 AFM amplitude error images of 4 μm2 areas of nanopaper surfaces (at the top) and SEM images of the cross-sections (at the bottom) of the reference and 14L samples. | ||
Lignin acts as cementing material between the fibrils, making the surface of the nanopapers smoother. This binder effect of lignin is possibly owing to its softening during the hot-pressing (100 °C) of the nanopapers, as evidenced in the AFM image of the 14L nanopaper (Fig. 4). In theory, the softening temperature for softwood lignin is about 135 °C but when water is present in the medium it has a plasticizing effect and can reduce the softening temperature down to 80–90 °C.18 The observation of a reduced nanopaper roughness with increased lignin content was previously reported by Ferrer et al.,11 although in their study no physical evidence for the location of the lignin in the suspensions and nanopapers was possible due to the limited range of lignin concentrations tested (0.5–2.7%) in contrast to the present, wider range of lignin concentrations (0–14%).
Since the nanopapers were prepared by over-pressurized filtration of the LCNF suspensions and over a timescale of several minutes, we anticipate that nanofibrils were primarily organized with their principal axis in the plane of the film yielding layered structures. Further, we expect this in-plane structural characteristic to persist after the nanofibrils were dried and pressed into a stiff and strong film. The anticipated layered structures formed by the nanofibrils are observed in the SEM cross-section images of the nanopapers (Fig. 4). The laminar structures were more apparent in the nanopapers with high lignin content. Ferrer et al.11 reported that the layer spacing was less in nanopapers containing more residual lignin.
Density and tensile properties
The density and tensile properties of the nanopapers with different lignin content are summarized in Table 1. The densities of the LCNF nanopapers were in the range between 1.1 and 1.2 g cm−3; with the exception of sample 2L, the densities were not significantly different at the 95% level, so we do not observe the systematic dependencies of lignin content reported previously.6,11 Recall from our discussion of Fig. 2 that the filtration behaviors of the suspensions indicated that the structures of networks containing lignin were more open than those of the reference material and showed a dependence on lignin content. The densities of the dry nanopapers reveal no such dependence, suggesting that the processes of hot-pressing and drying densify the structures through mechanical compression and perhaps by allowing lignin to fill the voids between cellulosic nanofibrils. Analyses of AFM and SEM images of the dispersed nanofibrils and nanopapers support this hypothesis and allow us to propose a model system describing the location and form of the lignin within the samples. We elucidate this by reference to Fig. 5: lignin nanoparticles are detached from the cellulosic nanofibrils during the deconstruction in the microfluidizer and, due to their sticky nature, they adhere to the cellulosic nanofibrils in the diluted LCNF suspensions (Fig. 5a). During filtering, a more concentrated cake of cellulose nanofibrils and lignin nanoparticles is formed, and the amorphous lignin is able to begin filling some voids between the fibers (Fig. 5b). Subsequently, during pressing at 100 °C and 220 bar, softening of the lignin occurs and lignin completely fills the voids between the cellulosic nanofibrils acting as a binder, effectively recreating the fiber–lignin composite of wood with fibres deconstructed into their constituent fibrils (Fig. 5c).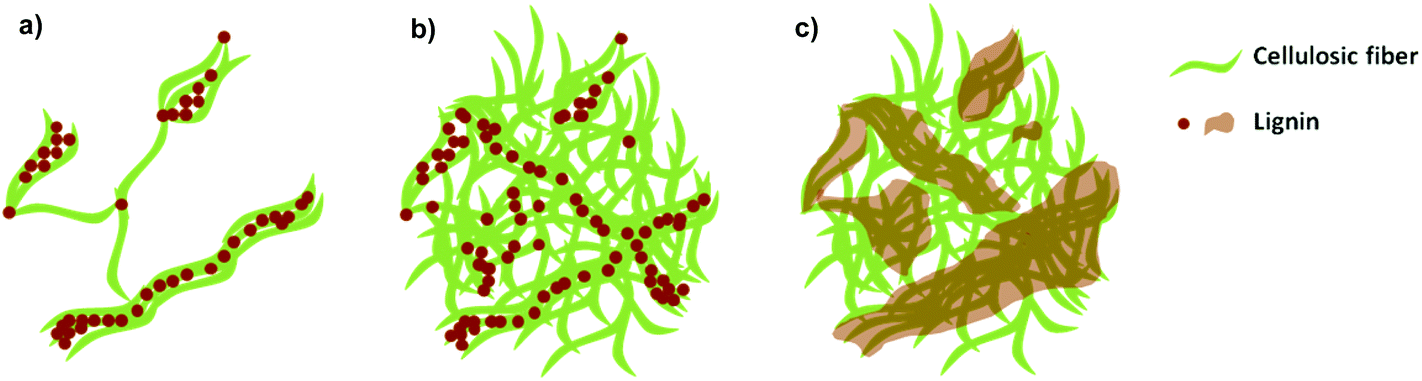 | ||
| Fig. 5 Proposed model to describe the location of the lignin within nanofibril suspensions and nanopapers. (a) LCNF suspension, (b) LCNF suspension after filtering, (c) LCNF nanopaper after pressing. | ||
| Reference | 2L | 4L | 14L | |
|---|---|---|---|---|
| Density (g cm−3) | 1.24 ± 0.03 | 1.10 ± 0.03 | 1.18 ± 0.05 | 1.20 ± 0.02 |
| Tensile strength (MPa) | 164 ± 17 | 123 ± 8 | 156 ± 17 | 116 ± 7 |
| Tensile index (kN m kg−1) | 132 ± 14 | 112 ± 7 | 132 ± 15 | 97 ± 6 |
| Breaking strain (%) | 2.9 ± 0.1 | 3.5 ± 0.5 | 2.8 ± 0.4 | 1.7 ± 0.3 |
| Elastic modulus (GPa) | 14.3 ± 0.5 | 10.5 ± 0.1 | 13.4 ± 0.9 | 12.2 ± 0.2 |
| Specific elastic modulus (MN m kg−1) | 11.5 ± 0.4 | 9.5 ± 0.1 | 11.4 ± 0.8 | 10.1 ± 0.2 |
| Specific TEA (kJ g−1) | 1.9± 0.3 | 2.0 ± 0.4 | 1.6 ± 0.3 | 0.7 ± 0.2 |
The tensile properties of the nanopapers are given in Table 1. Here we discuss the data in terms of the specific strength (or the tensile index) and the specific elastic modulus as obtained by dividing the tensile strength and Young's modulus by density, respectively. In general, nanopapers 4L, containing approximately 4% lignin, exhibited mechanical properties close to those of the reference sample. For the lignin-containing nanopapers, the tensile index exhibited a maximum for sample 4L; although differences were significant at the 95% level, we note that the coefficient of variation of the tensile index across all samples is only 14%. Although the maximum is observed also in the data for the specific elastic modulus, the coefficient of variation of the specific elastic modulus across all samples is only 9%. Accordingly, we may state that to a good degree of approximation, the tensile strength and the elastic modulus of our nanopapers containing up to 14% lignin are rather insensitive to lignin content. This fact is also illustrated in Fig. S1 of the ESI.† We have already noted that there is limited effect of lignin on the density of our nanopapers and, although we might expect lignin to interfere in hydrogen bonding between fibrils, this is apparently counteracted by the uniform distribution of lignin within the hot-pressed nanopapers seemingly aiding stress-transfer between fibrils and thus preserving mechanical properties.
It should be noted however that the breaking strain, and hence TEA, decreased with increasing lignin content. The values measured for these properties were plotted against lignin content in the nanopapers (Fig. S1, ESI†) and linear (inverse) relationships were found. Thus, it may be speculated that the retention of strength at increased lignin content in the nanopapers is associated with a loss of ductility, which could limit applications where such a property is required. The reduction in breaking strain with the lignin content is probably related to the faster filtration during the manufacture of the nanopapers, which limits component dispersion. Thus, while a reduced filtration time is beneficial (reduced energy consumption), the elongation properties of the resulting nanopapers can be, to some degree, compromised.
The tensile strength, modulus, and strain of the nanopapers studied in the present investigation were compared with others reported in the literature for softwood, hardwood, and non-wood resources (see Fig. 6). Most of the values reported correspond to nanopapers from fully bleached CNF. The mechanical property data set determined in this work is displayed in Fig. 6 (group “A”) and includes the range of values determined for LCNF with different lignin contents. The literature data include individual property values as well as ranges of tensile properties (for example, when reported by authors who tested different sources of cellulose, pectin content, solvent used). The data, mostly for nanopapers from fully bleached fibers, include tensile strength, modulus, and strain within the ranges 75–250 MPa, 6–18 GPa, and 1–10%, respectively. The values for the same properties in the present investigation are 116–164 MPa for tensile strength, 10.5–14.3 GPa for tensile modulus, and 1.7–3.5% for tensile strain, all of which are within the ranges reported for fully bleached CNF. Therefore, it is possible to conclude, as a first approximation, that the presence of lignin in the nanopapers resulted in mechanical properties comparable to those of nanopapers derived from fully bleached CNF.
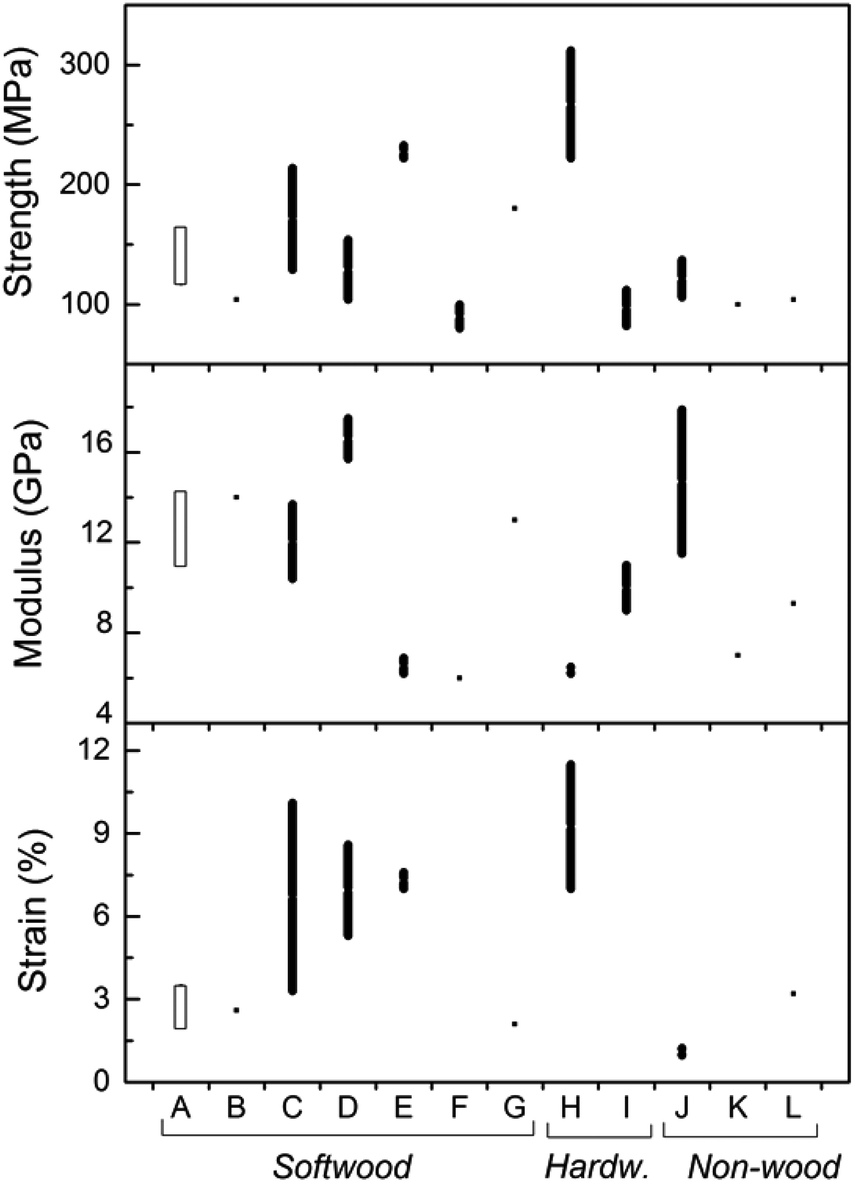 | ||
| Fig. 6 Range of values for tensile properties of nanopapers investigated (A) compared with those reported in the literature from different sources of fibers (B–L). Softwoods: nanopapers from SEW fibers from Norway spruce of varying lignin content (present work) correspond to group A; B30,31 (sulfite); C32 (dissolving grade fibers); D33 (bleached sulfite); E34 (dissolving grade and bleached sulfite fibers); F35 (bleached kraft); G36 (bleached spruce sulfite). Hardwoods: H37 (bleached kraft pulp); I11 (birch kraft pulp). Non-wood: J38 (swede root pulp); K39 (sugar beet pulp chips); L10 (palm fruit bunch fibers). | ||
Porous structure of LCNF nanopapers
The effect of lignin content on the pore size distribution of LCNF nanopapers was investigated by DSC. While the effect of hemicelluloses has been reported in the literature and was proved to increase the amount of micropore water in pulps,20 the extent to which lignin contributes to the porous structure remains unclear, in both, papers from fibers or nanopapers from (L)CNF. The water absorbed in the nanopapers includes non-freezing bound water, freezing bound water, and free water. Non-freezing bound water (NFBW) corresponds to the first 1–3 layers of water adjacent to the surface of the fiber wall and corresponds to water with limited motion. Freezing bound water (FBW) corresponds to water within pores that has a depressed melting temperature (below 0 °C). Free or freezing non-bound water (FNBW) freezes at the same temperature as pure bulk water. NFBW and FBW are accommodated in the micropores within the nanopapers, while FNBW is held in the macropores between the nanofibrils. Total bound water (TBW) represents the total amount of micropores that can be detected by thermoporosimetry, which are voids within the cell wall where the water is subject to thermodynamic effects as explained before, including both NFBW and FBW.The influence of lignin content on the amount of micropores (total bound water) is shown in Fig. 7. TBW decreased from 1.05 to 0.81 g g−1 with increasing LCNF lignin content, mainly because of the reduction in FBW (from 0.61 to 0.36 g g−1). As can be observed in Fig. 4 and 5, residual lignin is located between the cellulose nanofibrils in the nanopapers, acting as an inter-fibril binder and reducing the amount of micropores in the nanopaper (reduction in TBW). Linear correlations between TBW and FBW vs. lignin content in the nanopapers were observed (Fig. S2 of the ESI†). It is also interesting to note that the total amount of bound water exceeded the dry mass of nanocellulose (TBW > 1) for the samples with no lignin (reference) or with low lignin content (2L). Common TBW values for cellulose fibers are around 0.63 g g−1.22 The large amount of pore water measured in this work may be associated either with a particular pore structure in the nanopapers or with the high water holding capacity of CNF, associated with the large fibril surface area. NFBW was independent of the lignin content of the samples, with a value close to 45% based on dry solid mass. Since NFBW is closely related to the number and type of accessible hydration sites,40 it can be stated that lignin did not affect the number of hydration sites in the nanopapers (Fig. S2 of the ESI†). Some studies indicate that NFBW content of dried wood specimens are within 20 and 40%.20–22,41,42 The higher value of NFBW obtained in this work is related to the large surface area of the cellulose nanofibrils, which enables a larger number of hydroxyl groups on the fiber surface to come into contact with individual water molecules. A similar trend was found by Hatakeyama,43 who also reported NFBW values above 40% in the case of CNF.
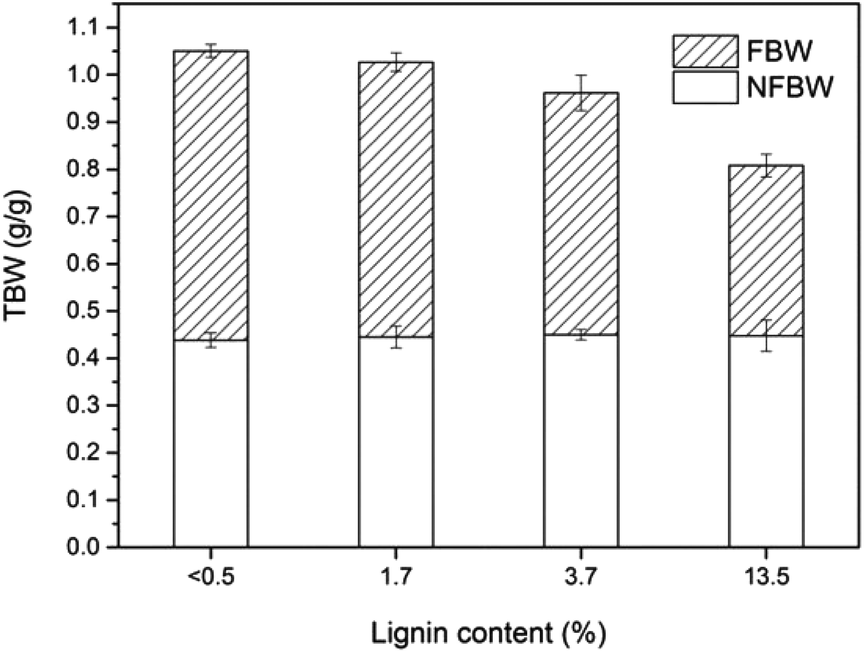 | ||
| Fig. 7 Total bound water (TBW) determined as the sum of freezing bound water (FBW) and non-freezing bound water (NFBW) of nanopapers with different lignin content. | ||
The voids in layered fibrous materials are anisotropic with a mean in-plane dimension about double that perpendicular to the plane.44 We expect the mean pore size determined by DSC to be dominated by the larger in-plane voids. The theory for random fiber networks45 states that this mean pore dimension, ![[d with combining macron]](https://www.rsc.org/images/entities/i_char_0064_0304.gif) , depends only on the porosity of the network, ε, and the diameter of fibers, df (eqn (6)).
, depends only on the porosity of the network, ε, and the diameter of fibers, df (eqn (6)).
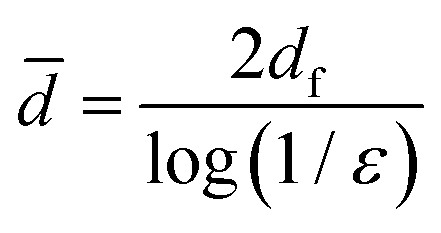 | (6) |
So, for our networks of approximately constant porosity formed from fibrils of smaller diameter with increasing lignin content, eqn (6) yields the expectation that the mean pore size will decrease with increasing lignin content.
Cumulative TBW values are plotted as a function of pore size in Fig. 8a. The largest pore diameter that was considered in this work was ∼215 nm and was calculated from the depressed melting temperature of −0.2 °C (Table S2†). All the samples considered appeared to have a similar pore size distribution up to 8.5 nm pore size. However, compared with the samples with small amounts or no lignin present, nanopapers with larger lignin content presented a smaller fraction of the large pores. This information supports earlier findings about lignin acting as a cementing material between the fibrils, thereby reducing the porosity of the nanopapers, especially in the region 17 to 54 nm. No plateau in the cumulative TBW profile versus pore size was observed in the region 100–215 nm, indicating that the pores did not collapse upon drying. Additionally, this implies that a significant fraction of pores larger than 215 nm were still present in the nanopapers, which probably originated from the consolidation of the fiber network and represent inter-fiber pores. Then, as represented by cumulative TBW in Fig. 8a, with the increased lignin content, micropores in the nanopapers were progressively reduced in number. Meanwhile, the porosity of the samples, which includes both micropores (TBW) and macropores (not measured), were 27, 21 and 20%, for 2L, 4L and 14L nanopapers, respectively, as calculated by eqn (3). The reduction in porosity with the increment of lignin content is related to the effect of lignin as adhesive between the fibrils.
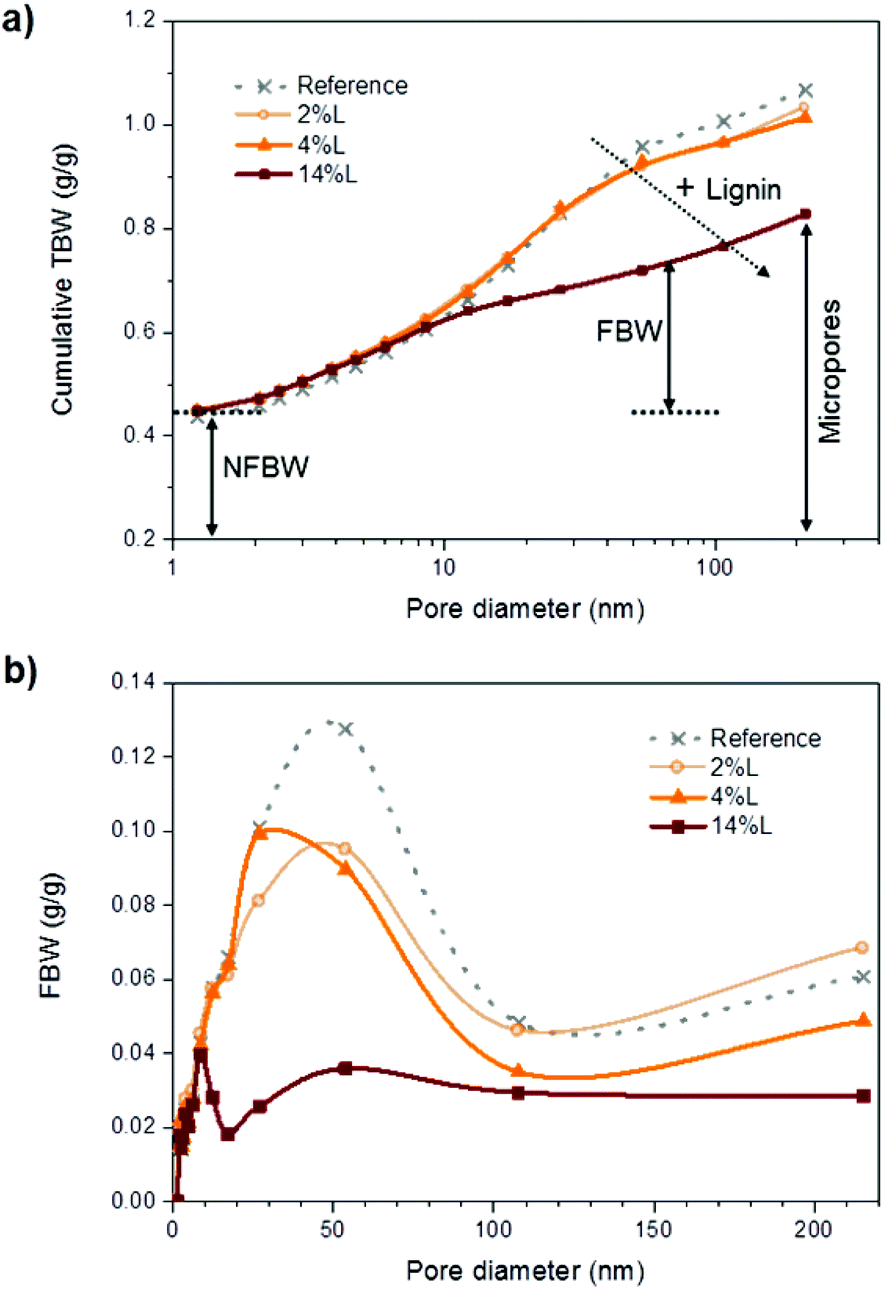 | ||
| Fig. 8 (a) Cumulative total bound water and (b) freezing bound water vs. pore diameter for the nanopapers with different lignin content. | ||
Pore size distribution was further evaluated in terms of FBW vs. pore diameter in Fig. 8b. Given that pores greater than 214 nm were excluded from the analysis, we have not calculated a mean pore size, but consider instead the modal value of pore size in the nanopapers. In agreement with the prediction of eqn (6), this reduced with lignin content, from 54 nm in the nanopapers without and with low lignin content (reference and 2L) to 27 and 9 nm in the nanopapers with medium and high lignin contents (4L and 14L, respectively). Indeed, these values are close to the mean pore sizes predicted using eqn (6): 50 and 58 nm for the reference and sample 2L, respectively, and 26 and 20 nm for samples 4L and 14L, respectively. Accordingly, we may state that the pore size in the nanopapers depends strongly on the diameters of the nanofibrils, which decrease with increasing lignin content. The relationship between pores size and lignin content in the nanopapers can be evaluated in more detail in Fig. S2 of the ESI.† The entire range of dominant pore sizes measured in this study (9–54 nm range) was lower than average pore size values reported in the literature for wood fibers, typically ranging from 80 to 100 nm.22 To our knowledge, no previous reports on the pore structure in nanopapers from CNF exist. Overall, it can be concluded that micropores were reduced in size and number with the presence of lignin, due to the reduction of the diameter of the fibrils and the cementing effect of lignin, respectively.
Surface free energy and hydrophilicity of LCNF nanopapers
The average contact angles (CA) for four probing liquids were measured on nanopapers with different lignin contents and were used in the surface energy evaluation, as summarized in Table 2. The highest CA were measured with water, whereas ethylene glycol yielded, in most of the cases, the lowest CA. For a given liquid, CA increased with nanopaper lignin content. The surface energy components were calculated from CA values according to the acid–base theory (see ESI†). The acid–base framework to describe surface energy has been found to be most suitable to explain the properties of wood surfaces, and gives detailed information about their surface chemistry.46 In addition to the disperse component, this theory allows calculation of the acid and base components of the surface free energy.| Liquid | Contact angle (°) of different nanopapers | |||
|---|---|---|---|---|
| Reference | 2L | 4L | 14L | |
| a Standard deviation of the contact angle values was less than 2°. | ||||
| Water | 35 | 49 | 61 | 78 |
| Formamide | 15 | 16 | 21 | 36 |
| Diiodomethane | 20 | 29 | 31 | 34 |
| Ethylene glycol | 18 | 13 | 20 | 34 |
Surface free energy of a solid is a characteristic parameter that has a large effect on many interfacial processes such as absorption, wetting and adhesion. Literature surface free energy parameters for the test liquids, as well as surface energy components calculated from the acid–base approach for the nanopapers in this work, are included in Table 3. The total surface free energy of the nanopapers (γS) ranged from 46.7 to 53.9 mJ m−2, which are values consistent with the range 43.1–53.7 mJ m−2 previously reported by Peng et al.48 for cellulose nanofibrils. Moreover, values of the total surface energy and their components found in this work for the Norway spruce nanopapers were in agreement with those recorded by other authors for spruce wood.46 The total surface energy was reduced with the nanopaper lignin content, from 53.9 mJ m−2 in the 2L sample to 46.7 mJ m−2 in the 14L nanopaper. Thus, lignin decreases the surface energy of the fibrils, as can be expected from the larger percentage of C–C and C–H bonds and the lower O/C ratio compared to cellulose.49–51
| γ TOT | γ LW | γ AB | γ − | γ + | γ −/γ+ | ΔGIFSWS | |
|---|---|---|---|---|---|---|---|
| a γ TOT = γL for the test liquids and γTOT = γS for the nanopapers. | |||||||
| Test liquids | |||||||
| Water | 72.8 | 21.8 | 51 | 25.5 | 25.5 | — | — |
| Formamide | 58 | 39 | 19 | 39.6 | 2.28 | — | — |
| Diiodomethane | 50.8 | 50.8 | 0 | 0 | 0 | — | — |
| Ethylene glycol | 48 | 29 | 19 | 47 | 1.9 | — | — |
| Nanopaper | |||||||
| Reference | 52.6 | 48.6 | 4 | 40.2 | 0.1 | 400 | 13.8 |
| 2L | 53.9 | 45.5 | 8.5 | 24 | 0.7 | 32 | −11.0 |
| 4L | 52.3 | 44.5 | 7.8 | 11.8 | 1.3 | 9 | −33.3 |
| 14L | 46.7 | 43.2 | 3.5 | 2.3 | 1.4 | 2 | −62.2 |
The dispersive component (γLW) accounts for the capacity of the surface to take part in London or dispersive interactions, the so-called nonspecific component of the surface energy. The contribution of the disperse component to the total surface free energy of the nanopapers was larger than the contribution of the acid–base component (γAB). For example, in the 2L nanopaper, γLW was 45.5 mJ m−2, which represents 84% of the total surface free energy, while γAB was 8.5 mJ m−2, which represents the remaining 16% of the total energy. The disperse component showed a similar trend to that of the total surface energy; it decreased with the lignin content of the samples from 45.5 mJ m−2 in 2L to 43.2 mJ m−2 in 14L nanopaper. The disperse component in cellulosic materials was observed to depend mostly on the presence and concentration of free hydroxyl groups on the surface.52 By increasing the lignin content in the nanopapers, lower values of the disperse component were achieved due to the reduction in the relative amount of cellulose and thus in the hydroxyl groups exposed on the surface. The trend correlates with the lower dispersive component of lignin with respect to cellulose.49 It may be predicted that the wettability between the lignin-containing nanopapers and polar liquids such as water would be weaker than that in the case of the reference CNF due to the lower dispersive component of the surface energy.
The acid–base component (γAB), also called the specific component of the surface energy, refers to all the other possibilities of interactions (induction, dipole, and hydrogen bond). The electron-donor (Lewis-base) component of the polar surface energy dominated over the acid one (γ−/γ+ > 1), leading to a strong electron donating capacity of the nanopapers and the ability to participate in polar interactions with acid species. Lignin content in the nanopapers significantly reduced γ− from 24 to 2.3 mJ m−2 in the nanopapers and therefore also reduced the γAB, accounting for 59% from 8.5 mJ m−2 in the 2L to 3.5 mJ m−2 in the 14L nanopapers. On the other hand, the acid component was increased with the lignin content and the relationship between the base and acid components (γ−/γ+) was strongly reduced. Thus, while the reference nanopaper exhibited predominantly monopolar electron-donicity (γ− ≫ γ+ ≈ 0), 2L, 4L and 14L showed a certain amphoteric behavior (acid and base) that was more pronounced with the increase in lignin content. In the reference sample the large number of electron donating sites neutralized the few accepting sites through hydrogen bonding (Lewis neutralization).53 The presence of fewer γ− sites in the nanopapers with high lignin content could cause that more γ+ sites remained without neutralization and this would justify the certain acid character of these nanopapers (amphoteric character). The existence of a non-zero γ+ parameter in the nanopapers with higher lignin contents would allow interactions with other bi-polar polymers.
The affinity to water of the nanopapers was assessed from the interfacial free energy of interaction, ΔGIFsws (Table 3). The terms “hydrophobic” and “hydrophilic” are used loosely and, as such, they have a limited utility. Surface thermodynamic theory, however, allows a natural and potentially powerful definition of these terms. The boundary between hydrophobicity and hydrophilicity occurs when the difference between the apolar attraction and the polar repulsion between molecules or particles immersed in water is equal to the cohesive polar attraction between the water molecules. Under these conditions, the interfacial free energy of interaction between materials immersed in water, ΔGIFsws, is exactly zero. Then, the thermodynamic convention is followed. When ΔGIFsws is positive, the interaction of the material with water dominates and the surface of the material is hydrophilic. When ΔGIFsws is negative, the surfaces of the material immersed in water prefer to be in contact with each other (cohesive attraction) rather than forming an interface with water; hence, this material is hydrophobic.54 Thus, the sign of ΔGIFsws defines the nature of the surface and the magnitude of ΔGIFsws may be used as a quantitative measure of the surface hydrophilicity or hydrophobicity. ΔGIFsws had a positive value for the reference nanopaper, the one without lignin, indicating a dominant hydrophilic character. However, ΔGIFsws was negative for all the nanopapers from SEW fibers containing different lignin contents (2, 4, and 14L). Therefore, these nanopapers could be defined as hydrophobic with the magnitude of ΔGIFsws being a quantitative measure of the surface hydrophobicity of these materials. The hydrophobic character of the nanopapers was found to increase with the presence of lignin, according to a six-fold increase in ΔGIFsws from 2L to the 14L nanopaper. Hydrophobicity may be a desirable attribute for enhancing compatibility with hydrophobic polymer materials.
Interactions with water and barrier properties
Advancing and receding water CA and hysteresis values of the nanopapers with different lignin contents are summarized in Table 4. Since the lignin is a highly complex, condensed macromolecule rich in aromatic groups, less hydrophilic than cellulose, it was expected that the advancing water contact angle increased with the lignin content. In fact, θa increased from 35° in the reference nanopaper to 78° in the 14L sample. Meanwhile, the receding contact angle was not significantly affected by lignin. Thus, the hysteresis increased with lignin content as a result of the advancing contact angle increment. The significance of hysteresis arises from roughness and/or chemical heterogeneity of the surface.55–57 In either water-wettable or water-repellent surfaces the effect of a roughened surface is to magnify the wetting properties of the solid.58 Thus, a water-wettable solid will further enhance the wetting with increased roughness, and for a water-repelling one, roughness will increase water repellency. All the nanopapers in this work displayed certain wettability (θa < 90°), in such cases an increase in roughness will increase wettability. AFM roughness of the nanopapers varied from 17.1 nm in the reference to 8.6 nm in the 14L sample. The roughest surface (reference) showed stronger wettability (lower advancing contact angle) and hence lower hysteresis. Thus, the increment in hysteresis with increasing lignin content does not derive from the roughness of the nanopapers, but it arises from the chemical heterogeneity introduced by lignin.| Reference | 2L | 4L | 14L | |
|---|---|---|---|---|
| θ a | 35.4 ± 0.5 | 48.6 ± 1.1 | 60.9 ± 4.1 | 77.7 ± 3 |
| θ r | 25.8 ± 1 | 25.7 ± 0.7 | 25.9 ± 1.7 | 25.8 ± 1.8 |
| (θa − θr) | 9.6 | 22.9 | 35.1 | 51.9 |
Relative water absorption (RWA) of the nanopapers derived from CNF and LCNF measured after given water immersion times is shown in Fig. 9. Water absorption increased with time especially during the first 10 min. For example, RWA of the 4L nanopaper increased from 16 to 24% during the first 10 min. After this time, RWA was increased moderately or remained constant, depending on the sample. RWA decreased with the lignin content in the nanopapers for any given immersion time. For a better understanding of the relationship between this property and lignin content, RWA (%) and water absorption (g m−2) of the nanopapers after 2 h of immersion in water are plotted vs. lignin content in the nanopaper (Fig. 10), which yielded a linear relationship with negative slope. Similar behavior was observed previously in the literature.7,10,11 Water absorption and water contact angle are known to be dependent on the pore structure near the surface as well as on the fiber surface energy. Thus, lower RWA values obtained for the nanopapers with higher lignin can be explained by the significantly less porous nanopaper, which prevented water penetration. In addition, the lower surface energy and specifically the lower electron donor parameter of the nanopapers with higher lignin content also contributed to the decrease in RWA. Reduction on Lewis-base component of the surface energy of the nanopapers with higher lignin content was also responsible for the higher water CA and related lower hydrophilicity.
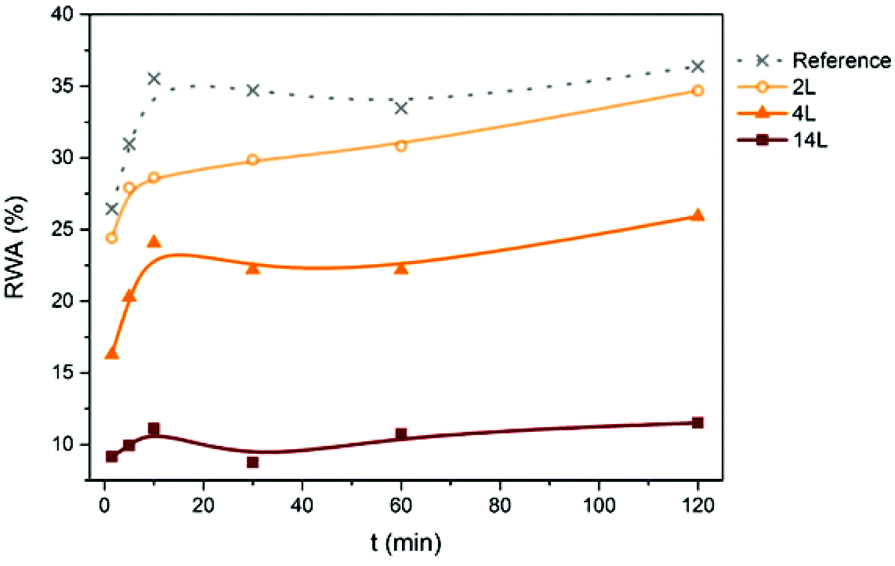 | ||
| Fig. 9 Relative water absorption (RWA) of the nanopapers with different lignin content measured after different immersion times in water. | ||
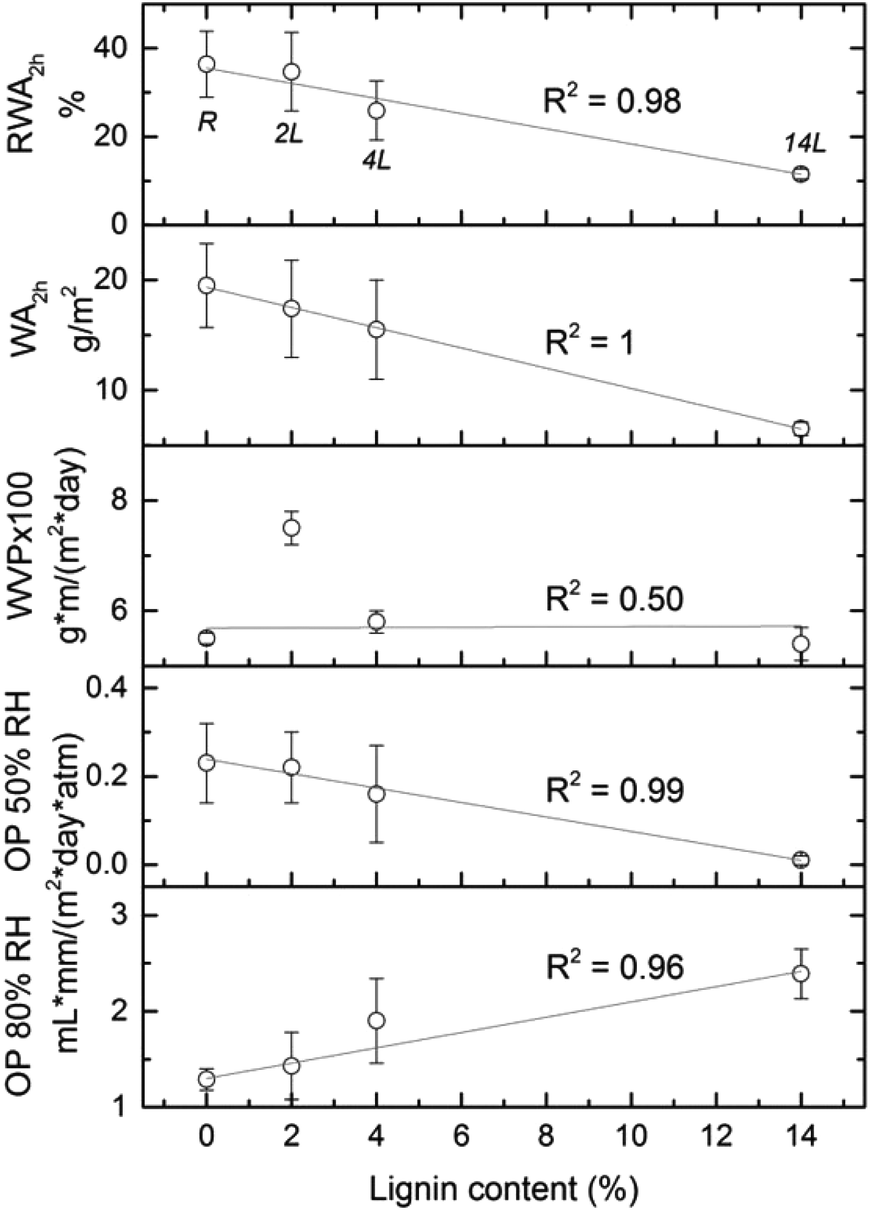 | ||
| Fig. 10 Relative water absorption of the nanopapers after 2 h of immersion in water (RWA2h), water absorption (WA2h) and barrier properties (WVP and OP) of the nanopapers evaluated as a function of the lignin content. | ||
Barrier properties of the nanopapers were evaluated in terms of water vapor and oxygen permeabilities (WVP and OP, respectively). Good barrier properties of the crystalline components of cellulose fibrils in combination with the ability of the nanofibers to form a dense network by strong inter-fibrillar bonds make the nanopapers a suitable barrier material,36 especially against oxygen transmission. The relationship between the barrier properties of the nanopapers and the lignin content can be evaluated in Fig. 10. Oxygen permeabilities at two relative humidities showed a strong linear relationship with the nanopaper's lignin content, which was not the case for WVP. At lower relative humidity (50%), lignin in the nanopapers reduced OP from 0.23 in the reference nanopaper, to 0.01 mL mm/ (m2 day atm) in the 14L sample. At relative humidity of 80%, lignin slightly increased OP, but this increment was not significant compared to the more than 15-fold decrease observed at relative humidity of 50%. Improved oxygen barrier properties of the nanopapers with higher lignin content are a consequence of the smaller pore structure of these samples. Water vapor permeability, in turn, did not correlate linearly with lignin content; in fact WVP depended most significantly on material density.7 Samples with higher density (reference CNF and 14L LCNF nanopapers) have a more compact structure and are less water vapor permeable, regardless of the lignin content. 14L nanopaper provided the best water vapor and oxygen barrier properties.
Lignin in its native state is more hydrophobic than cellulose. However, the residual lignin in fibers after pulping can be hydrophilic, depending on the digestion process used. It should be noted that SEW fractionation produces hydrophilic, low-sulfonated “organosolv-like” lignin.59 Moreover, all the SEW pulps were obtained using 12% SO2 and therefore the lignin in all the samples is expected to have similar levels of hydrophilicity. In the present work, water CA increased with the content of SEW residual lignin in the nanopapers, reaching a value of 78° in the 14L sample. Meanwhile, water absorption was gradually decreased at increasing the lignin content. These results greatly differ from those reported by Ferrer et al.,11 who found higher water absorptions for nanopapers with higher residual kraft lignin contents and did not find important differences in water CA between nanopapers with different lignin contents. In order to have a first approximation of the effect of the nature of the residual lignin on the properties of the nanopapers, water CA has been measured on nanopapers obtained from kraft pulps, chemithermo-mechanical pulp (CTMP) and thermo-mechanical pulp (TMP), containing 10, 21, and 26 wt% of residual lignin, respectively. Water CA were 53° for the kraft-pulp derived nanopaper, 54° for CTMP and 57° for the TMP, all of them much lower than the water CA measured for the 14L SEW derived nanopaper (78°). It can be concluded that SEW residual lignin are less hydrophilic than kraft, CTMP and TMP residual lignins and for this reason lower water absorptions were obtained in this work for SEW nanopapers in comparison with kraft-derived nanopapers reported in the literature.11
Conclusions
This work dealt with an exhaustive analysis of the variables affecting LCNF properties and a detailed characterization of the products to elucidate the effect of the lignin on the properties of cellulose nanofibrils and nanopapers. The presence of lignin improved extensively dewatering during the filtration process of the nanopaper manufacture. Lignin was found to act as a cementing material between the cellulose nanofibrils in LCNF nanopapers, similarly to lignin in native wood. Thus, fibrils with higher residual lignin content formed nanopapers with smoother surfaces and reduced micropores in number and size. Mechanical properties of the lignin-containing nanopapers were comparable to those derived from fully bleached materials. The lower surface energy and specifically the lower electron donating capacity of the nanopapers with higher lignin content were responsible for the lower water wettability measured for these samples (higher water contact angle and lower water absorption capability). The less porous and more compact structure of the lignin-containing nanopapers also contributed to improve oxygen barrier properties of these materials and to prevent water penetration. In summary, the use of lignin-containing cellulose nanofibrils not only leads to lower production cost by reducing bleaching chemicals but also reduces the polarity and hydrophilicity of the nanocellulose, which are nowadays an important challenge limiting the large-scale utilization of such valuable product.Acknowledgements
We are grateful for funding support by the Academy of Finland through its Centers of Excellence Program (2014–2019), under project “Molecular Engineering of Biosynthetic Hybrid Materials Research” (HYBER), the project “Nanocellulose Assemblies for Microsensing and Fluidics” and “Design Driven Value Chains in the World of Cellulose” (DWoC), a strategic opening by Finland's Tekes. Dr Mikhail Iakovlev kindly supplied the SEW pulps. Anu Anttila, Ritva Kivelä, and Leena Nolvi are thanked for their laboratory assistance and Dr Juan José Valle for helping with AFM imaging.Notes and references
- T. Saito, S. Kimura, Y. Nishiyama and A. Isogai, Biomacromolecules, 2007, 8, 2485 CrossRef CAS PubMed.
- L. Wågberg, G. Decher, M. Norgren, T. Lindström, M. Ankerfors and K. Axnäs, Langmuir, 2008, 24, 784 CrossRef PubMed.
- M. Pääkkö, M. Ankerfors, H. Kosonen, A. Nykänen, S. Ahola, M. Österberg, J. Ruokolanen, J. Laine, P. T. Larsson, O. Ikkala and T. Lindström, Biomacromolecules, 2007, 8, 1934 CrossRef PubMed.
- I. Siró and D. Plackett, Cellulose, 2010, 17, 459 CrossRef.
- L. Berglund, Natural fibers, biopolymers, and biocomposites, CRC Press, Boca Raton, USA, 2005, pp. 807–832 Search PubMed.
- K. L. Spence, R. A. Venditti, Y. Habibi, O. J. Rojas and J. J. Pawlak, Bioresour. Technol., 2010, 101, 5961 CrossRef CAS PubMed.
- K. L. Spence, R. A. Venditti, O. J. Rojas, Y. Habibi and J. J. Pawlak, Cellulose, 2010, 17, 835 CrossRef CAS.
- K. L. Spence, R. A. Venditti, O. J. Rojas, Y. Habibi and J. J. Pawlak, Cellulose, 2011, 18, 1097 CrossRef CAS.
- K. L. Spence, R. A. Venditti, O. J. Rojas, J. J. Pawlak and M. A. Hubbe, BioResources, 2011, 6, 4370 CAS.
- A. Ferrer, I. Filpponen, A. Rodríguez, J. Laine and O. J. Rojas, Bioresour. Technol., 2012, 125, 249 CrossRef CAS PubMed.
- A. Ferrer, E. Quintana, I. Filpponen, I. Solala, T. Vidal, A. Rodríguez, J. Laine and O. J. Rojas, Cellulose, 2012, 19, 2179 CrossRef CAS.
- T. Retsina and V. Pylkkänen, Paper 360°, 2007, February issue, 18.
- V. Pylkkänen, T. Retsina and R. Ryham, Patent Application, WO 2007146245 A2, 2007 Search PubMed.
- T. Retsina and V. Pylkkänen, Patent Application, WO 2010151536 A1, 2010 Search PubMed.
- T. Retsina and V. Pylkkänen, Patent Application, WO 2011044378 A1, 2011 Search PubMed.
- T. Retsina and V. Pylkkänen, US Patent, 8030039 B1, 2011 Search PubMed.
- M. Iakovlev, E. Hiltunen and A. van Heiningen, Nord. Pulp Pap. Res. J., 2010, 25, 428 CAS.
- H. Sixta, Handbook of pulp, Wiley-VCH, Weinheim, Germany, 2006 Search PubMed.
- S. J. I'Anson and W. W. Sampson, Compos. Sci. Technol., 2007, 67, 1650 CrossRef PubMed.
- T. C. Maloney, P. Stenius and H. Paulapuro, Nord. Pulp Pap. Res. J., 1998, 13, 31 CAS.
- T. C. Maloney and H. Paulapuro, J. Pulp Pap. Sci., 1999, 25, 430 CAS.
- S. Park, R. A. Venditti, H. Jameel and J. J. Pawlak, Carbohydr. Polym., 2006, 66, 97 CrossRef CAS PubMed.
- P. A. C. Gane, C. J. Ridgway, E. Lehtinen, R. Valiullin, I. Furó, J. Schoelkopf, H. Paulapuro and J. Daicic, Ind. Eng. Chem. Res., 2004, 43, 7920 CrossRef CAS.
- Å. Östlund, T. Köhnke, L. Nordstierna and M. Nydén, Cellulose, 2010, 17, 321 CrossRef PubMed.
- J. Berthold and L. Salmén, Holzforschung, 1997, 51, 361 CrossRef CAS.
- T. Q. Hu, Chemical modification, properties, and usage of lignin, Springer Science & Business Media, New York, USA, 2002 Search PubMed.
- J. Kozeny, Sitzungsber Akad. Wiss. Wien, 1927, 136, 271 Search PubMed.
- P. Carman, Trans. Inst. Chem. Eng., 1937, 15, 150 CAS.
- I. Solala, A. Volperts, A. Andersone, T. Dizhbite, N. Mironova-Ulmane, A. Vehniainen, J. Pere and T. Vuorinen, Holzforschung, 2012, 66, 477 CrossRef CAS.
- T. Zimmermann, E. Pohler and T. Geiger, Adv. Eng. Mater., 2004, 4, 754 CrossRef.
- T. Zimmermann, E. Pohler and P. Schwaller, Adv. Eng. Mater., 2005, 7, 1156 CrossRef CAS.
- M. Henrikson and L. A. Berglund, J. Appl. Polym. Sci., 2007, 106, 2817 CrossRef.
- A. J. Svagan, M. A. S. A. Samir and L. A. Berglund, Biomacromolecules, 2007, 8, 2556 CrossRef CAS PubMed.
- M. Henrikson, L. A. Berglund, P. Isaksson, T. Lindström and T. Nishino, Biomacromolecules, 2008, 9, 1579 CrossRef PubMed.
- H. Fukuzumi, T. Saito, T. Wata, Y. Kumamoto and A. Isogai, Biomacromolecules, 2009, 10, 162 CrossRef CAS PubMed.
- K. Syverud and P. Stenius, Cellulose, 2009, 16, 75 CrossRef CAS.
- T. Saito, M. Hirota, N. Tamura, S. Kimura, H. Fukuzumi, L. Heux and A. Isogai, Biomacromolecules, 2009, 10, 1992 CrossRef CAS PubMed.
- D. M. Bruce, R. N. Hobson, J. W. Farrent and D. G. Hepworth, Composites, Part A, 2005, 36, 1486 CrossRef PubMed.
- J. Leitner, B. Hinterstoisser, M. Wastyn, J. Keckes and W. Gindl, Cellulose, 2007, 14, 419 CrossRef CAS PubMed.
- J. Berthold, Water adsorption and uptake in the fiber cell wall as affected by polar groups and structure, Doctoral Thesis, Royal Inst. Technology, Stockholm, Sweden, 1996 Search PubMed.
- P. P. Kärenlampi, P. Tynjälä and P. Ström, J. Wood Sci., 2005, 51, 118 CrossRef.
- M. Borrega and P. P. Kärenlampi, Wood Fiber Sci., 2011, 43, 206 CAS.
- T. Hatakeyama, Y. Inui, M. Iijima and H. Hatakeyama, J. Therm. Anal. Calorim., 2013, 113, 1019 CrossRef CAS PubMed.
- W. W. Sampson and S. J. Urquhart, J. Porous Mater., 2008, 15, 411 CrossRef.
- S. J. Eichhorn and W. W. Sampson, J. R. Soc., Interface, 2010, 7, 641 CrossRef CAS PubMed.
- M. Gindl, G. Sinn, W. Gindl, A. Reiterer and S. Tschegg, Colloids Surf., A, 2001, 181, 279 CrossRef CAS.
- C. J. van Oss, R. J. Good and M. K. Chaundhury, Langmuir, 1988, 4, 884 CrossRef CAS.
- Y. Peng, D. J. Gardner, Y. Han, Z. Cai and M. A. Tshabalala, J. Colloid Interface Sci., 2013, 405, 85 CrossRef CAS PubMed.
- W. Shen, I. H. Parker and Y. J. Sheng, J. Adhes. Sci. Technol., 1998, 12, 161 CrossRef CAS PubMed.
- J. Laine, P. Stenius, G. Carlsson and G. Ström, Cellulose, 1994, 1, 145 CrossRef CAS.
- P. Stenius and J. Laine, Appl. Surf. Sci., 1994, 75, 213 CrossRef CAS.
- G. Garnier and W. G. Glasser, Polym. Eng. Sci., 2006, 36, 885 Search PubMed.
- C. J. van Oss, R. F. Giese and W. Wu, J. Adhes., 1997, 63, 71 CrossRef CAS.
- C. J. van Oss and R. F. Giese, Clays Clay Miner., 1995, 43, 474 CAS.
- E. Chibowski and F. González-Caballero, J. Adhes. Sci. Technol., 1993, 7, 1195 CrossRef CAS PubMed.
- D. Li, Colloids Surf., A, 1996, 116, 1 CrossRef CAS.
- D. Y. Kwok, Contact angles and surface energetics, Doctoral Thesis, University of Toronto, Toronto, Canada, 1998 Search PubMed.
- R. N. Wenzel, Ind. Eng. Chem., 1936, 28, 988 CrossRef CAS.
- M. Iakovlev and A. van Heiningen, RSC Adv., 2012, 2, 3057 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4gc02398f |
| This journal is © The Royal Society of Chemistry 2015 |