
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The reluctant polymorph: investigation into the effect of self-association on the solvent mediated phase transformation and nucleation of theophylline†
Raitis
Bobrovs
*ab,
Linda
Seton
*a and
Nicola
Dempster
a
aDrug Delivery and Materials Science Research Group, School of Pharmacy and Biomolecular Sciences, Liverpool John Moores University, Liverpool, L3 3AF, UK. E-mail: raitis.bobrovs@gmail.com; L.Seton@ljmu.ac.uk
bFaculty of Chemistry, University of Latvia, Kr. Valdemāra iela 48, Rīga, LV-1013, Latvia
First published on 17th February 2015
Abstract
Little is known concerning the pathway of the crystallization of the thermodynamically stable polymorph of theophylline, form IV. Here we study the reasons why the thermodynamically stable theophylline form IV can be obtained only by slow, solvent mediated phase transformation (SMPT) in specific solvents, and whether the presence of prenucleation aggregates affect the polymorphic outcome. Solution concentration, polymorphic composition and morphology were monitored over time during the transformation from form II to form IV in several solvents. NMR and FTIR spectroscopy were used to detect prenucleation molecular aggregates present in the solutions. It was determined that theophylline self-associates in solvents which are good H-bond donors and the presence of these aggregates hinder the nucleation and phase transformation. SMPT from form II to form IV is a nucleation-growth controlled polymorphic transformation, nucleation is most likely homogenous, and form IV crystals grow along the (001) plane, forming plate-like crystals.
1. Introduction
Polymorphism is a well-known phenomenon whereby a chemical compound may exist in more than one crystalline form, and each of these forms displays different physical characteristics such as density, melting point and solubility. Polymorphism is of great importance in the pharmaceutical industry, because these differences in physical properties among the polymorphs can sometimes lead to apparent differences in drug processing, formulation, and bioavailability. Therefore, it is a common requirement in Pharmacopeia for active pharmaceutical ingredients to be produced in one fixed crystalline form, and for this reason the thermodynamically stable form is generally preferred for pharmaceutical production. However, a metastable polymorph may be preferred if the solubility and/or bioavailability of the thermodynamically stable polymorph does not meet required criteria.1–3 In such cases knowledge of the possible phase transformation in a given polymorphic system is essential.Theophylline – a methyl xanthine derivative (3,7 dihydro-1,3-dimethyl-1H-purine-2,6-dione) is an example of a pharmaceutically active compound that has been manufactured and used in a metastable crystalline phase (clearly, because of the lack of comprehensive knowledge of the polymorphic landscape of this compound) (Scheme 1). Theophylline is known to exist in four polymorphic forms (I, II, III and IV), as a monohydrate and a dimethyl sulfoxide solvate.4 Theophylline form II is a metastable polymorph crystallized from most non-aqueous solution at room temperature. It is kinetically stable and was historically considered as the thermodynamically stable polymorph, until form IV was presented5 and proved to be more stable. The fact that the thermodynamically stable theophylline polymorph was discovered only recently, is because it does not crystallize directly from solution, and is obtained only by slow, solvent mediated transformation (SMPT) from form II in contact with solvent e.g. methanol, 2-propanol or chloroform.5 Theophylline form I has been reported as the stable polymorph at higher temperatures, whereas form III is highly metastable and has been obtained only during the dehydration of monohydrate.6,7 Theophylline monohydrate (referred to as form M) is a monoclinic channel type hydrate which has been shown to lose water, either in low humidity or at temperatures above 353 K, to produce form II.8,9 Theophylline has been screened for cocrystallization and shown to produce co-crystals with a range of coformers; many complexes formed between theophylline and acids (e.g. oxalic acid, malonic acid, salicylic acid, sulfathiazole, acetaminophen, etc.) as well as bases (e.g. urea, benzylamine, phenobarbital, etc.) have been summarized by Childs et al.10
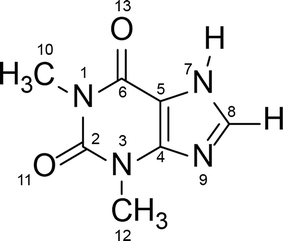 | ||
| Scheme 1 Molecular structure of theophylline (3,7 dihydro-1,3-dimethyl-1H-purine-2,6-dione). | ||
The aim of this work is to understand why the thermodynamically stable theophylline form IV can be obtained only by solvent mediated transformation in specific solvents, and to investigate the presence of prenucleation aggregates which direct the polymorphic outcome of crystallization.
Hunter et al.11–13 and Davey et al.14,15 have shown that there is a correlation between supramolecular aggregates in solution and the solid state structure which subsequently crystallizes. Early experiments16–19 on theophylline self-association provide evidence that theophylline does self-associate in aqueous solution and the proposed aggregate is the theophylline dimer. The theophylline dimer discussed in these studies is present in the thermodynamically stable form IV, and theophylline monohydrate, form M, which crystallizes from aqueous solutions. Theophylline form II, the polymorph commonly crystallized from most non-aqueous solutions, does not contain this dimer motif (crystal structures of theophylline polymorphs are discussed in detail in section 2.1). Such behaviour raises two questions: why does metastable form II crystallize from non-aqueous solvents; what prenucleation aggregates are present in non-aqueous solvents? In order to answer these questions, we need to determine (a) does theophylline self-associate in other solvents besides water, and, if it does, what is the nature of the association, and (b) since the nucleation of form IV is kinetically slow, does solution aggregation change over time?
2. Background
2.1. Polymorphism of theophylline
Crystal structures of theophylline crystalline forms relevant for this research are shown in Fig. 1. In the form II structure the best hydrogen bond donor (N7–H) bonds to the best hydrogen bond acceptor (N9),20,21 consistent with Etter's rules,22 forming an R22(8) motif. The structure has two weak, bifurcated C8–H⋯O13 hydrogen bonds. Theophylline molecules are linked in chains and stacked along (010). This catemer arrangement promotes crystal growth along the molecular chains leading to an elongated crystal morphology. Form IV has two molecules in its asymmetric unit,23 forming a dimer with the R22(10) motif and connected via (N7–H⋯O13) hydrogen bonds. The dimer is discrete and only links to other dimers by weak interactions: C8–H⋯N9, C8–H⋯O11, and by π–π stacking, forming a two-dimensional network parallel to the (001). The dimer is similar to the motif observed in the monohydrate and in a number of theophylline cocrystals.10,21 The presence of this dimer motif in the thermodynamically stable form IV and theophylline monohydrate, considered to be the most stable structure in an aqueous environment, may account for the thermodynamic stability of this structural motif compared to the chain motif of form II. In form M, two centrosymmetrically related theophylline molecules form a dimer through two hydrogen bonds (N7–H⋯O13).24 Theophylline dimers are connected by water molecules through hydrogen bonds, forming parallel, crosslinked chains, leading to two-dimensional hydrogen bonded layers, parallel to (10−1) plane. Water molecules are situated in channels along the a axis, where they form hydrogen bonds to the theophylline N9 atom. In theophylline dimethyl sulfoxide solvate theophylline is hydrogen bonded to the dimethyl sulfoxide molecule through an N7–H⋯O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S hydrogen bond.4 The packing consists of molecular chains lying parallel to the (010), stacked by π–π interaction between pyrimidine and imidazole rings, and weak hydrogen bonds between dimethyl sulfoxide methyl groups and theophylline carbonyl group (CDMSO–H⋯O13).
S hydrogen bond.4 The packing consists of molecular chains lying parallel to the (010), stacked by π–π interaction between pyrimidine and imidazole rings, and weak hydrogen bonds between dimethyl sulfoxide methyl groups and theophylline carbonyl group (CDMSO–H⋯O13).
 | ||
| Fig. 1 Crystal packing motifs in theophylline form II, form IV, theophylline monohydrate and DMSO solvate. | ||
2.2. Solvent mediated phase transformations
Solvent mediated phase transformations (SMPTs) are common in polymorphic materials which often follow Ostwald's rule of stages.25 The transformation proceeds by three stages: firstly, the metastable polymorph in contact with solution dissolves into the bulk. Secondly, the stable form nucleates, and thirdly undergoes crystal growth. When complete, only the stable form remains in solution. The thermodynamic driving force is the Gibbs free energy difference between the two solid structures, whereas the kinetic driving force is the difference in the solubilities of the two polymorphs and thus the level of supersaturation with respect to the stable form.26–30SMPTs are characterized by the phase transformation time, induction time, and active phase transformation time. The term “phase transformation” refers to the whole process from the beginning of the experiment until the phase transformation is complete; the initial period during which no phase transformation product is observed is called “induction”; and the time from the point when thermodynamically stable phase appears until the sample has been fully converted is called the “active transformation phase”.31 All of these parameters can be influenced by choice of solvent and solution composition.
2.3. Spectroscopic self-association studies
NMR spectroscopy is mainly used as a method for organic compound identification and characterization; however NMR methods have been demonstrated to be useful for the study of molecular aggregation and self-association in solution. It is possible to use solution NMR measurements to study molecular association because 1H chemical shifts are sensitive to changes in the local environment. Hydrogen bonding between solute molecules and associate formation affects the 1H local environment and a result of this interaction is a chemical shift displacement in the NMR spectrum.32–34It is generally recognized that dimer and other associate formation and their concentration are affected by solution concentration – the more concentrated the solution, the more dimers and other associates are present, therefore dependence of 1H chemical shifts on concentration is analysed. This approach has been used in numerous studies.35–44 Most of the previous studies show a decrease in the 1H chemical shifts with concentration increase, suggesting that the analysed compounds are involved in self-association processes. Upon self-association, 1H chemical shifts are displaced to lower field due to proton deshielding – the bond of the proton involved in self-association weakens, bond length increases, stretching frequency decreases.45–49 The limitations for 1H NMR self-association studies are that 1H NMR shifts for nonexchangeable hydrogens only can be measured, and the NMR method has a limited sensitivity at the low concentrations often necessary for initial self-association studies.
In recent years, 1H NMR chemical shift displacement measurements have been used to provide information on the structure of prenucleation aggregates in the solution.50 Hunter et al.13,51 showed that the predictions from concentration-dependent changes in 1H NMR chemical shifts agreed with the structures of dimers found in the corresponding X-ray crystal structures.
Other magnetically active nuclei (13C, 15N and others) involved in hydrogen bonding or aromatic stacking also show displacement in chemical shifts upon concentration changes.35,52–55
Theophylline self-association in aqueous solution has been previously studied using NMR spectroscopy,17,18 partitioning between water and chloroform–isooctane mixture,19 analytical ultracentrifuge;16 and in chloroform-D solution using IR spectroscopy.56 These studies suggest that theophylline does self-associate in aqueous solutions and chloroform-D solution; however there is no consensus between these studies on the degree of self-association and nature of aggregates present in the solution.
3. Results and discussion
3.1. Characterisation of solvent mediated phase transformation
The SMPT of unprocessed commercial anhydrous theophylline form II in methanol was investigated. Three parallel experiments were performed, but for the sake of clarity only one case is taken as an example of the whole set of experiments to describe and discuss in detail the results obtained.During the solvent mediated phase transformation qualitative and quantitative analysis of the solid phase were performed using powder X-ray diffractometry (PXRD), Fourier transform infrared spectroscopy (FTIR) and scanning electron microscopy (SEM) methods; while the solution concentration throughout the transformation was monitored using ultraviolet-visible (UV/Vis) spectroscopy. The composition of solid phase determined by PXRD method and solution concentration throughout SMPT is presented in Fig. 2. Results of all three parallel experiments showed that the phase transformation from theophylline form II to form IV took ~6 to 8 days. Phase transformation had a considerable induction time with a relatively high dispersion (4 to 6 days), while the active phase transition (actual form IV increase in the sample) consistently took ~2 days. Such behaviour, that induction time for parallel experiments had a great variation, while the active phase transition time in all cases was approximately the same, suggests that the limiting step for this SMPT is the nucleation of form IV. The main factors that might affect the nucleation and hence the induction time are discussed further.
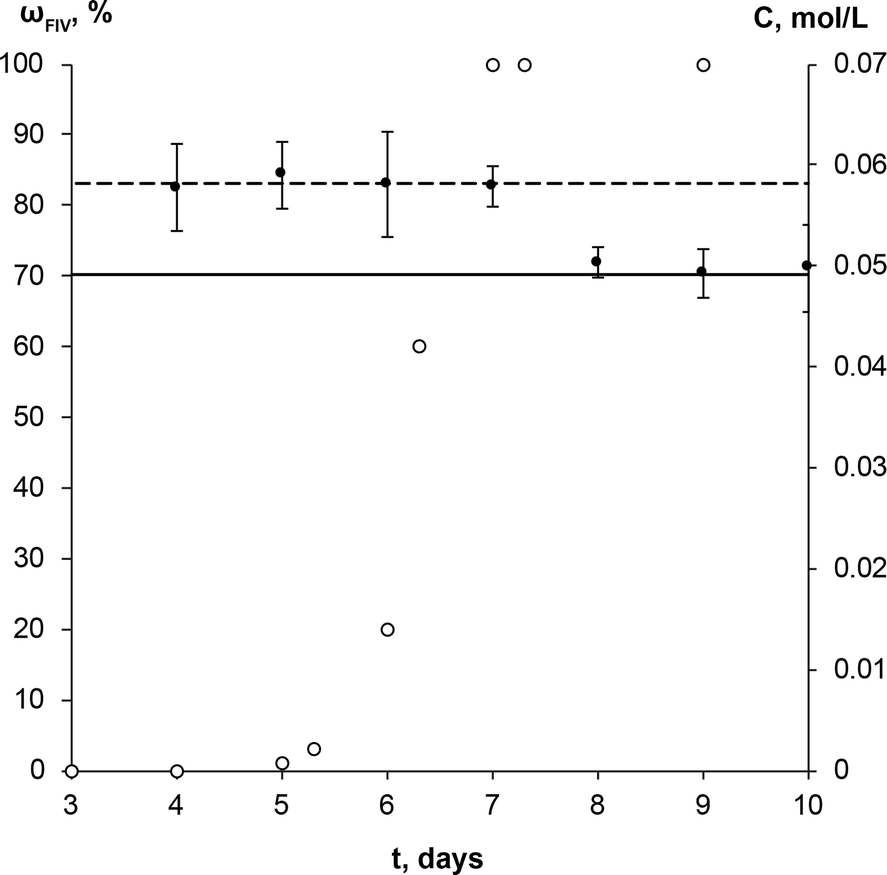 | ||
| Fig. 2 Solution concentration (●), and weight fraction of the theophylline form IV in the solid phase (○) during the solution mediated transformation from theophylline form II to form IV in methanol at 23 °C. The dashed line (---) represents the solubility of theophylline form II, continuous line (—) – solubility of theophylline form IV. | ||
The equilibrium saturation of the metastable form II was reached ~3 h after the theophylline addition to methanol. The concentration of the solution remained the same until phase transformation was completed. Solution concentration dropped to the equilibrium saturation of the thermodynamically stable form IV within a few hours of the phase transformation being completed, indicating that, once all of the form II in the sample had dissolved, form IV continued to grow until the solution reached the form IV equilibrium concentration. This shows that the dissolution rate of theophylline form II was faster than the growth of form IV and dissolution was not the limiting step in the phase transformation. Such a solution concentration profile and pronounced induction period indicates that the SMPT was a ‘nucleation-growth controlled polymorphic transformation’.57 The fact that phase transformation is also growth limited was confirmed by the time scale of the studied SMPT – most of the SMPTs described in the literature take place within a few hours,29,57–65 but here phase transformation took ~2 days from the moment when form IV nucleated. This might be due to low supersaturation, which provided the necessary driving force to overcome the energy barrier and promoted form IV crystallization.3 In this case, supersaturation is the difference between the solubilities of form II and form IV.
The increase of the form IV content in the sample exhibited an exponential nature – the amount of form IV increased slowly in the initial stage and then accelerated. Such behaviour suggests that the growth of form IV might be limited by the surface area of form IV in the sample; hence, the rate of SMPT increased as the crystal size (surface area) in the sample increased. This assumption was consistent with SEM imaging data, shown in Fig. 3.
 | ||
| Fig. 3 SEM images throughout SMPT from theophylline form II to form IV in methanol at 23 °C. Inset: time of sample gathering (days); 40 μm scale bar, crystals of theophylline form IV indicated with blue colour. | ||
It is known5 that theophylline form II exhibits a needle-like morphology, while form IV crystallizes in hexagonal plate-like crystals. These distinct differences in the crystal shape allowed phase transition monitoring using microscopy methods. SEM imaging of the solid phase throughout SMPT (Fig. 3) revealed that during the first days, while no phase transformation was observed, the only apparent change in the solid phase was the agitation and Ostwald's ripening of the theophylline form II crystals – small theophylline form II crystals dissolved as growth occurred on the larger form II crystal side planes (see Fig. 3, day 1 and day 3). Thus, larger theophylline form II crystals grew at the expense of the small particles. The reason for this process was the difference in dissolution rate between small and large particles.66 Theophylline form IV crystals were first observed after 5 days, and all observed hexagonal form IV crystals were approximately the same size: ~40 μm in diameter and 1–2 μm thick. A similar size for all observed form IV crystals suggests that they nucleated simultaneously or in a very short time interval. Recent studies60,65,67,68 show that surface nucleation dominates in most of SMPTs, however in our case there was no clear evidence of such behaviour. We believe that theophylline form IV nucleated by homogeneous primary nucleation from saturated solution because none of the observed form IV crystals were clearly merged together with metastable form II. Moreover, taking in to account that theophylline form IV nucleated only after several days, homogeneous nucleation is more likely as heterogeneous nucleation usually takes place in shorter time scale due to lower energy barrier. Over time form IV crystals grew significantly in 2 dimensions, forming large, plate-like crystals. This growth pattern indicates that the growth of the form IV was governed by the surface of the crystal edges. Eventually, breakage of form IV crystals was observed (day 6), which would lead to increased surface area of the edge faces, where crystal growth was fastest, thereby accelerating the crystallization rate of form IV.
Preferred form IV crystal growth directions were determined by the PXRD method. It is observed that the PXRD pattern of form IV crystals after the SMPT have two very intensive peaks at 12 and 23° 2θ (Fig. 4), suggesting that the sample exhibits preferred orientation. Comparison of the pattern with that simulated from the crystal structure revealed that these intensive diffraction peaks arise from crystal planes (002) and (004) – the multiple planes of the {001} face family. Since plate-like crystals tend to lay down with the dominant faces parallel to the PXRD sample holder, the most intense diffraction should occur from this plane. It can therefore be concluded that the dominant face in the form IV crystals obtained during SMPT is (001) and crystal growth occurred almost exclusively along this plane. The reason for such crystal growth behaviour is that there are no significant intermolecular interactions in the c-direction of theophylline form IV, whereas crystal growth along (001) plane is favoured by hydrogen bonding and π–π stacking (Fig. 5).
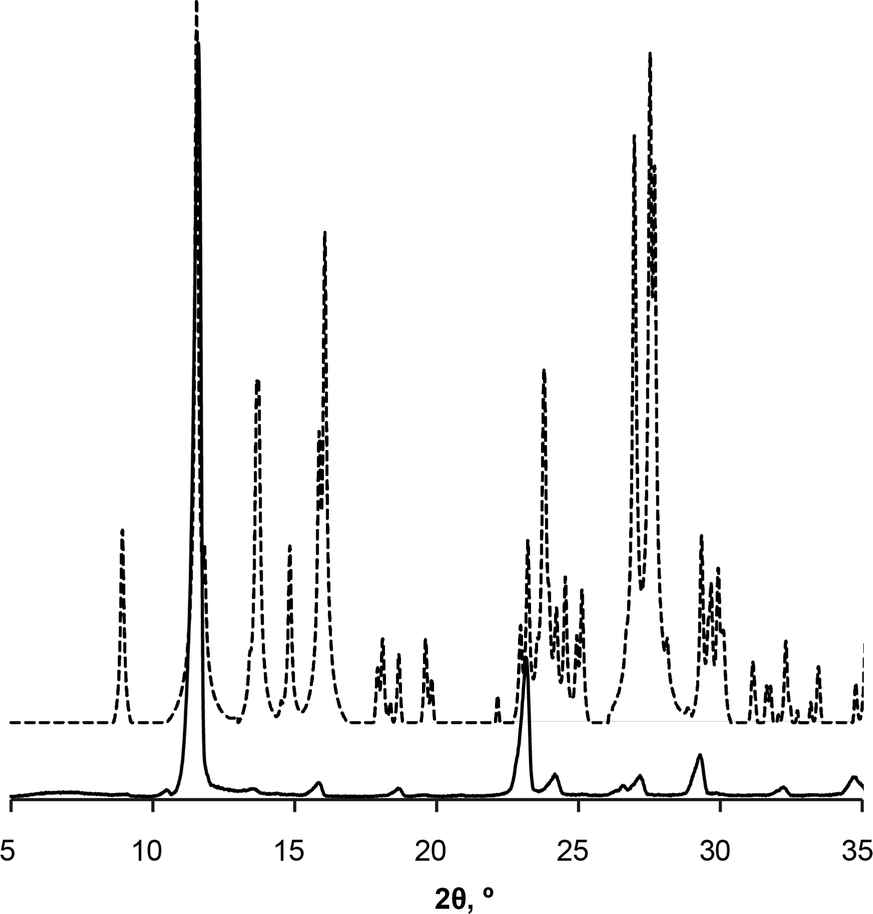 | ||
| Fig. 4 PXRD patterns of form IV obtained by SMPT in methanol (solid line); and simulated from crystal structure (dashed line). | ||
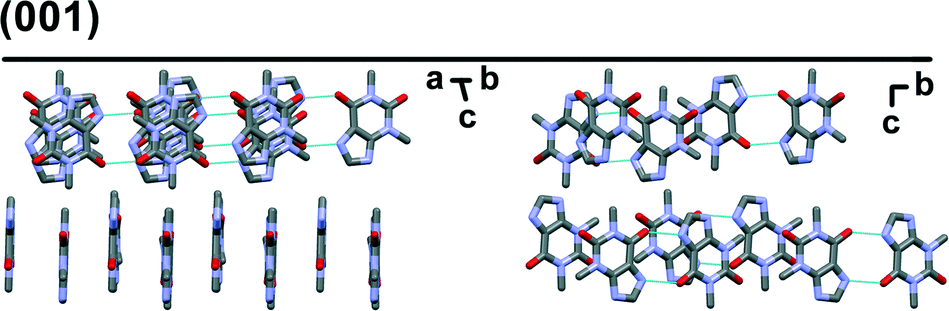 | ||
| Fig. 5 Crystal packing in theophylline form IV along (001) plane. Hydrogens are omitted for clarity. | ||
Based on morphological observation, form IV crystal growth perpendicular to (001) was observed only when phase transformation was finished – during the SMPT the surfaces of form IV crystal plane (001) were flat (Fig. 3, day 8.3), whereas 2 days after the SMPT elevations on the surface of the form IV were observed because of crystal agitation (Fig. 3, day 10). The crystal growth rate along (001) was considerably higher than growth rate perpendicular to (001) during phase transformation, likely to be because energy released on solution molecule attachment to growing crystal edge surface was higher.69 When phase transformation was complete, Ostwald's ripening66 took place and crystals tended to achieve minimum total surface energy by reducing the crystal surface area. Here, this means that crystals grew perpendicular to the large (001) plane at the expense of the crystal edges, which dissolved more easily.66
Surface nucleation dominates in the majority of SMPTs, therefore experiments with ground and lyophilized theophylline as a starting material were performed to evaluate the effect of the form II surface on the nucleation and crystallization of form IV. There were no significant differences in the phase transformation behaviour when ground and unground theophylline was used. PXRD and SEM data showed that when the ground form II was agitated, Ostwald's ripening took place, and crystallization of form IV followed only after 6 days. If theophylline form IV nucleated on the surface of the form II, the increase of the form II surface area in the ground sample should reduce the induction time. However, this was not observed, suggesting that form IV did not nucleate on the surface of form II. When lyophilized theophylline, confirmed as a mixture of amorphous theophylline and microcrystalline form II, was used, no phase transition to form IV was observed within the studied time (90 days). The crystallinity of lyophilized material increased due to transformation of amorphous material to form II (Fig. 6), but no phase transformation to form IV was detected. It is not clear why crystallinity of the form II starting material should influence the nucleation of form IV since this is not a surface nucleation process. Later experiments on water content (see below) indicate that the ability of amorphous material to absorb water might be more important than crystallinity.
 | ||
| Fig. 6 SEM images throughout lyophilized theophylline slurrying in methanol at 23 °C. Inset: time of sample gathering (days) and 20 μm scale bar. | ||
In order to ascertain whether theophylline aggregates present in the methanol solution affect the phase transformation, an experiment using methanol solution saturated with respect to form IV as a solution medium was performed. The SMPT held approximately the same induction and active phase transformation time (5 and 2 days, respectively) as experiments in methanol.
Similar experiments with theophylline form II and form IV mixture (wII/wIV; 90/10) as a starting material were performed to exclude induction time and observe only phase transformation. No induction times were observed in either case, and phase transformation rates were the same regardless of solution composition (Fig. 7). This means the molecular aggregates, if there were any at all, in both saturated solutions were the same and/or they did not play a significant role in the phase transformation. If we compare theophylline form IV weight fraction change over the time in these experiments and those performed with commercial form II as a starting material, we see that active phase transformation with theophylline form II and IV mixture as a starting material occurs three times faster. It is possible that this phase transformation rate mismatch is due to differences in the form IV crystal active edge surface areas for the samples. SEM imaging confirmed that form IV edge surface area in prepared polymorphic mixtures were larger, than in the sample where the same amount of form IV was generated by SMPT. Since this is the region in which growth of form IV dominates, larger surface area leads to faster rate of growth, therefore a faster transformation. It is also possible that in the studied SMPT the initial nucleation is homogeneous, but, once form IV is present, then secondary nucleation may subsequently occur, leading to increasing transformation rate as observed. By seeding with form IV in this experiment, the initial phase was skipped and form IV nucleated by secondary nucleation. Secondary nucleation promoted faster phase transformation by detachment of weak outgrowths of form IV seed crystals or fragmentation of a weak polycrystalline form IV mass.66
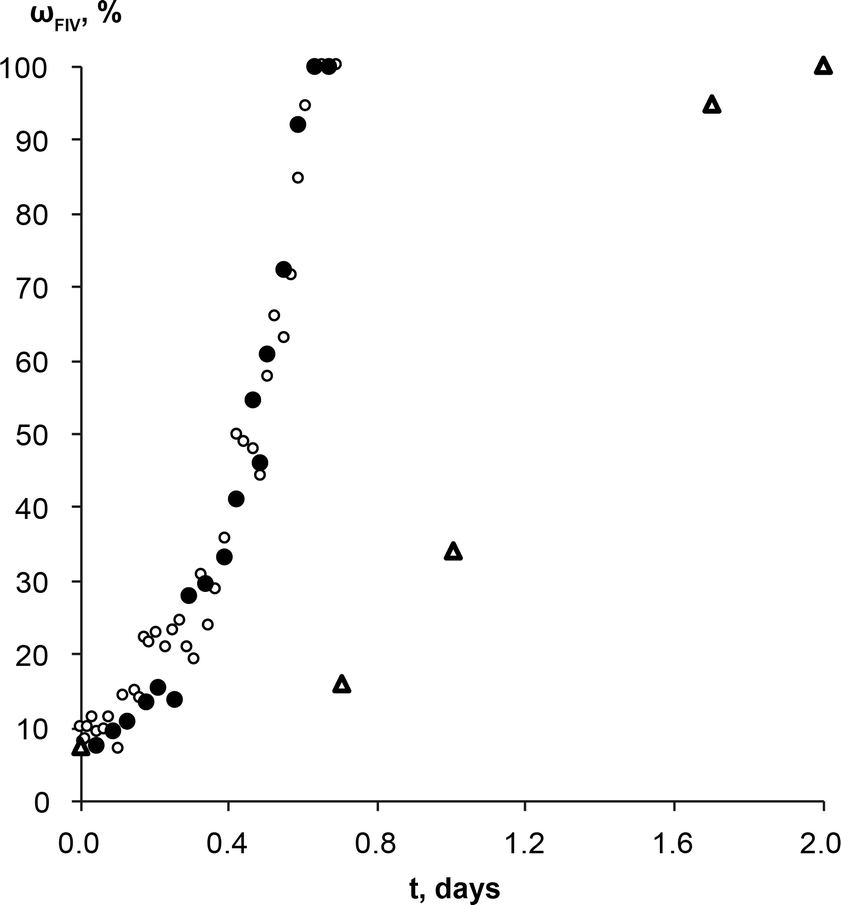 | ||
| Fig. 7 Weight fraction of the theophylline form IV in the SMPT from theophylline form II/form IV mixture (wII/wIV; 90/10) to form IV, in saturated (○) form IV and (●) form II methanol solutions at 23 °C. Δ represents the weight fraction of the theophylline form IV in the solid phase during the SMPT from theophylline form II in methanol at 23 °C. All kinetic curves are aligned to the point when solid phase of respective SMPT contained 10% of form IV. | ||
Seton et al.5 reported that theophylline equilibrium solid phase depends on the water activity in the solution, and that form IV is preferred when water activity in the solution is below 0.69 (VMeOH/VH2O, 55/45). To investigate the possibility that water activity also influences the phase transformation rate to form IV, SMPT was observed in methanol samples with different water contents. SMPT from theophylline form II to form IV, performed in dried methanol and methanol/water mixtures with volume ratios (VMeOH/VH2O) 99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 95:5 and 80:20, clearly showed that increased water in the solution increased the induction time (Fig. 8A). Given the stochastic nature of nucleation, it can be difficult to draw conclusions from a few experiments, however, the three parallel SMPT showed relatively good reproducibility and a clear trend. Active phase transformation time also increased but with a lesser effect – from 1.5 days in dried methanol to 5 days in methanol/water mixture (VMeOH/VH2O; 95/5).
:1, 95:5 and 80:20, clearly showed that increased water in the solution increased the induction time (Fig. 8A). Given the stochastic nature of nucleation, it can be difficult to draw conclusions from a few experiments, however, the three parallel SMPT showed relatively good reproducibility and a clear trend. Active phase transformation time also increased but with a lesser effect – from 1.5 days in dried methanol to 5 days in methanol/water mixture (VMeOH/VH2O; 95/5).
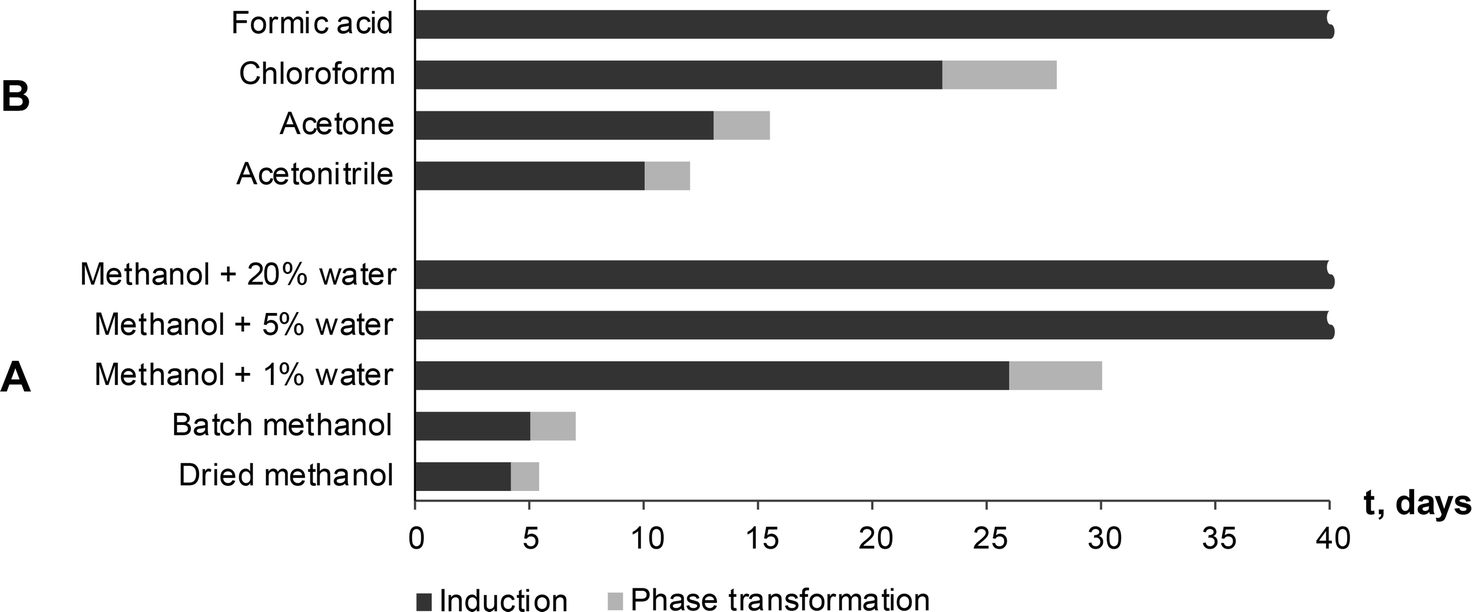 | ||
| Fig. 8 Induction and phase transformation times of SMPT from theophylline form II to form IV at 23 °C depending on used solvent environment. No phase transformation to form IV was observed in formic acid and methanol/water mixtures with 5% and 20% of water within the studied period (40 days). | ||
These results might explain why fluctuation of induction time were observed in the earlier SMPTs. Methanol used in initial experiments was used as received and was taken from different batches, therefore the water content (adsorbed from the air) was not the same in all samples. Karl Fisher titration showed that batch methanol contained ~0.2% of water, whereas methanol dried with anhydrous NaSO4 contained less than 0.1% water. These minor water impurities affected the induction time of the SMPT. This might be the reason why phase transformation with lyophilized theophylline as starting material was hindered. Theophylline lyophilisation resulted in partially amorphous theophylline, which tends to absorb more water from the air than crystalline phases,70 and it is possible that the phase transition in this case was slowed down by adsorbed water.
The induction times and transformation times in a range of solvents were measured and compared (Fig. 8B). We see that phase transition times correlated to induction times – the longer the induction time, the slower the phase transformation. This might indicate that nucleation and form IV growth were limited by the same factors. The solvents were used as received and were from newly opened bottles. Water doping experiments were not performed in the other solvents, so further investigation would show whether induction and transformation times were similarly affected in all solvents.
It has been shown previously30 that in the nucleation-growth controlled SMPT between α and β tegafur, the phase transformation rate and induction time linearly depends on supersaturation level, i.e. difference between solubilities of the polymorphs. The difference in solubility of the two forms in methanol is low, so supersaturation with respect to form IV is always low. In formic acid, the solubility of form II is high, (30 times that in methanol, see ESI†) which might favour the metastable form according to Ostwald's rule.
Crystallization always gave form II, including crystallization from solutions with form IV equilibrium concentration. Even seeding with form IV, gave form II and form IV mixture.
Induction and phase transition times were longer in solvents (solvent mixtures) which had good proton donor groups. Fastest phase transformation and shortest induction time were observed in dried methanol, followed by stock methanol, acetonitrile and acetone. Longest induction times were observed in proton donor solvents – chloroform and formic acid, and previously discussed methanol/water mixtures. In fact, no form IV has been detected in formic acid and methanol/water mixtures 95/5 and 80/20 within the studied period of 90 days. An influencing factor might be behaviour of solvated/aggregated theophylline molecules in solution. Theophylline molecules are solvated in the solution, but in order to nucleate form IV and continue its growth, theophylline molecules have to be desolvated, and solvent molecules on the nuclei or crystal surface be replaced by incoming theophylline molecules. Taking in to account that induction time was several days and phase transformation was very slow, we can assume that solute–solvent interaction in studied SMPT was strong compared to solute–solute interaction and thus the desolvation process inhibited form IV nucleation and growth.
In order to further understand how the solvent environment affects SMPT rate and induction time, NMR spectroscopy was used to investigate for prenucleation aggregates in the solution.
3.2. Theophylline self-association studies
To understand solution chemistry and ascertain possible aggregation of theophylline molecules in solution, the influence of theophylline solution concentration on 1H NMR chemical shift displacement was analysed. 1H NMR experiments were carried out in seven solvents (methanol-D4, chloroform-D, acetone-D6, dimethyl sulfoxide-D6, deuterium oxide, acetonitrile-D3 and formic acid-D2). These solvents were chosen because: (a) it is known that in dimethyl sulfoxide and water theophylline crystallizes as solvates and the crystal structures of these solvates are not similar; (b) they have different H-bond donor/acceptor properties: acetone, acetonitrile and dimethyl sulfoxide are H-bond acceptor; chloroform is H-bond donor; water, methanol and formic acid are both – H-bond donor and acceptor; and (c) from formic acid theophylline crystallizes into form II with no subsequent transformation observed.The concentration range covered in this experiment was the same for all solvents – from saturated to 1 μM theophylline form II solution. The number of scans for NMR spectra acquisition was adjusted depending on solution concentration. Theophylline solutions with lower concentrations were not studied because the NMR spectra acquisition time, necessary to obtain spectra with acceptable signal/noise ratio, would be unreasonably long.
The theophylline used in the experiment had natural 1H/2H abundances and therefore the most acidic imidazolium group proton (N7–H) took part in proton exchange with the deuterated solvent deuterium atoms. As a result, the imidazolium group proton was visible in the NMR spectra in solvents where only partial proton exchange took place – chloroform-D, dimethyl sulfoxide-D6 and acetonitrile-D3. Methyl group protons and alkene group proton (C8–H) did not showed any effect on proton exchange.
If there are multiple structures that have different thermodynamic stabilities and perturb the 1H NMR chemical shifts in different ways, the shapes of the dilution curves differ for different signals. While this does not rule out the possibility of multiple structures, the data can be treated as a simple two state equilibrium, and the complexation-induced changes in chemical shift are assumed to relate to a single specific aggregate structure.51 Since the solubility of the theophylline in all selected solvents except formic acid is low (>0.06 M) multiple aggregate structures are not expected.
No chemical shift displacement upon concentration changes were observed in methanol-D4, acetone-D6, dimethyl sulfoxide-D6 and acetonitrile-D3. This means that no self-association takes place in these solvents, or theophylline undergoes dimerization or other self-association at concentrations lower than those covered in the experiment, i.e. below 1 μM, and other agglomerates do not form in the concentration region studied. It is unlikely that self-association occurs at such low concentrations, and it is probable that solvated theophylline monomers were the main species in the solution.
1H NMR dilution studies in chloroform-D, deuterium oxide and formic acid-D2 showed large concentration-dependent changes in chemical shifts. In the chloroform-D all chemical shifts were displaced to lower fields as concentration increased (Fig. 9A). In deuterium oxide methyl groups 1H chemical shifts were displaced to higher field while the alkene group proton (C8–H) chemical shift was displaced to the lower field (Fig. 9B). Imidazolium group proton (N7–H) chemical shift was not observed in the deuterium oxide due to proton exchange. In formic acid-D2 alkene group proton (C8–H) chemical shift was displaced to higher field, whereas imidazolium group proton (N7–H) chemical shift was displaced to lower field (Fig. 9C). Minor methyl groups 1H chemical shift displacement to higher and field were observed as concentration increased. The pattern and magnitude of chemical shift changes are completely different in all solvents, indicating that aggregates present in these solutions are different. It is likely the associate existing in the deuterium oxide is the theophylline dimer which is also present in the crystal structure of theophylline monohydrate (Fig. 10A). The associates in chloroform-D solution might be the asymmetric dimer corresponding to Etter’s rule (Fig. 10B), π–π stacked dimer or some associate involving solvent molecules. It is also possible, that chemical shift displacement is reflecting the average structure of multiple aggregates. Since theophylline solubility in formic acid is noticeably higher than in other solvents (more than 30 times), it is possible that associates present in formic acid-D2 were oligomers and not dimers. Data shows that theophylline concentration at which associates were formed were different in each solvent. In deuterium oxide theophylline formed self-associates at 10−4 M solution, in chloroform at 10−3 M solution, and in formic acid-D2 at 10−2 M solution.
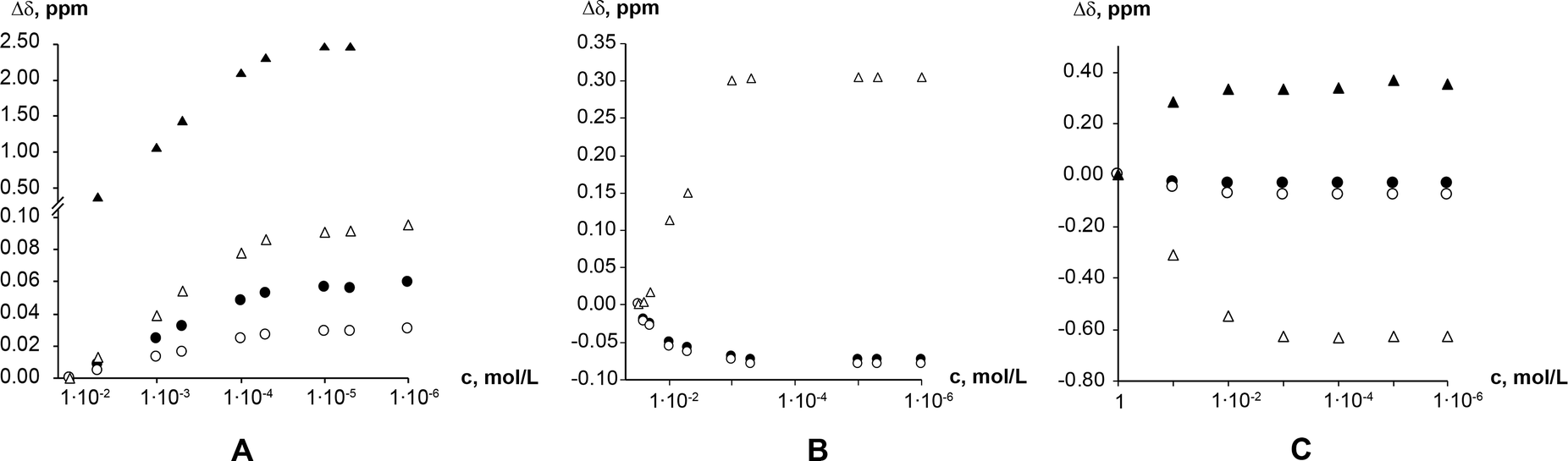 | ||
| Fig. 9 Theophylline chemical shift displacement depending on chloroform-D (A); deuterium oxide (B); and formic acid-D2 (C) concentration. Chemical shifts are indicated as: methyl group C10 protons (●); methyl group C12 protons (○); alkene group proton (C8–H) (Δ); and imidazolium group proton (N7–H) (▲). | ||
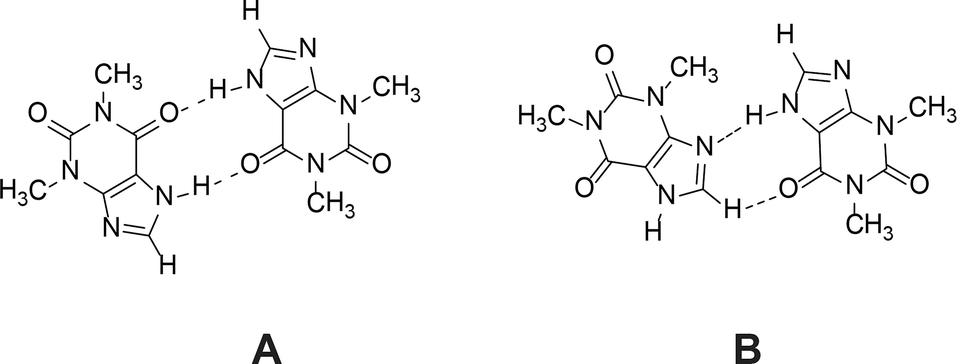 | ||
| Fig. 10 Theophylline dimer present in crystal structures of monohydrate, form IV and most cocrystals (A); and the preferred theophylline dimer according to Etter’s rules (B). | ||
No change in chemical shift displacement was observed with time (>20 days) in any studied solvents. Small scale SMPT performed in methanol-D4 showed the same – solution composition did not change over time; hence, no aggregates are formed or disarranged during slurrying indicating that this is not the reason for long induction times.
Theophylline aggregates formed in solvents which are good proton donors.71 These were the solvents where SMPT exhibited longest induction times and phase transition rates (we should mention that SMPT in water does not occur, because theophylline monohydrate is the most stable crystalline form in aqueous environments). Such correlation implies that the presence of theophylline dimers or aggregates in the solution hinders the nucleation and growth of form IV. It is likely that the reason why good proton donors inhibit phase transformation and extend induction time is the formation of solvent molecule stabilized theophylline aggregates. Trask et al.21 have suggested that the theophylline dimer motif is favoured by the presence of a competing strong hydrogen bond donor in the system (in this case formic acid, chloroform or water in methanol/water mixture). The strongest hydrogen bond donor in theophylline solution forms a hydrogen bond with the system's most basic group – theophylline imidazole group nitrogen (N9). This bond fulfils the ‘best-donor–best-acceptor’ rule and consequently permits theophylline dimer formation by secondary hydrogen bonding (N7–H⋯O13). Such solvated theophylline dimer should be the most stable aggregate in the solution, since it uses the system's best donors and acceptors. This explains why increasing water in the methanol sample increased induction time and extended phase transformation time. The increase of water in methanol/water mixture increased the level of hydrated aggregates in the solution, and the more solution theophylline molecules were bound in these dimers, the more formation of form IV was hindered.
Solvated theophylline aggregates inhibit nucleation and growth of form IV, either because they are not the correct structure to nucleate form IV or because these associates need to be desolvated or disarranged in order to crystallize. If the solute solvent bonds are stronger than the weak inter dimer interactions in the solid structure, then the desolvation process is unfavourable and therefore phase transition is slow. Recent work by Sullivan et al.72 shows that desolvation is a significant factor in the nucleation process and can dominate nucleation kinetics.
3.3. FTIR studies of theophylline crystallization from saturated solutions
It is known2,3,73–75 that FTIR spectra of polymorphs and their solutions are different. Parveen et al.76 have shown that FTIR spectroscopy can be used to show a direct relationship between molecular self-associates in solution and motifs in the subsequently crystallized solid phases. Here we use FTIR spectroscopy to monitor theophylline crystallization from acetone, acetonitrile, chloroform, methanol and water. FTIR spectra of saturated theophylline solution were continuously recorded during solvent evaporation and subsequent theophylline crystallization. FTIR spectra of both theophylline polymorphs studied in this work are clearly different and therefore suitable for such an experiment (Fig. 11). However, due to low theophylline solubility in the selected solvents, only the strongest carbonyl group stretching bands were visible in the initial spectra.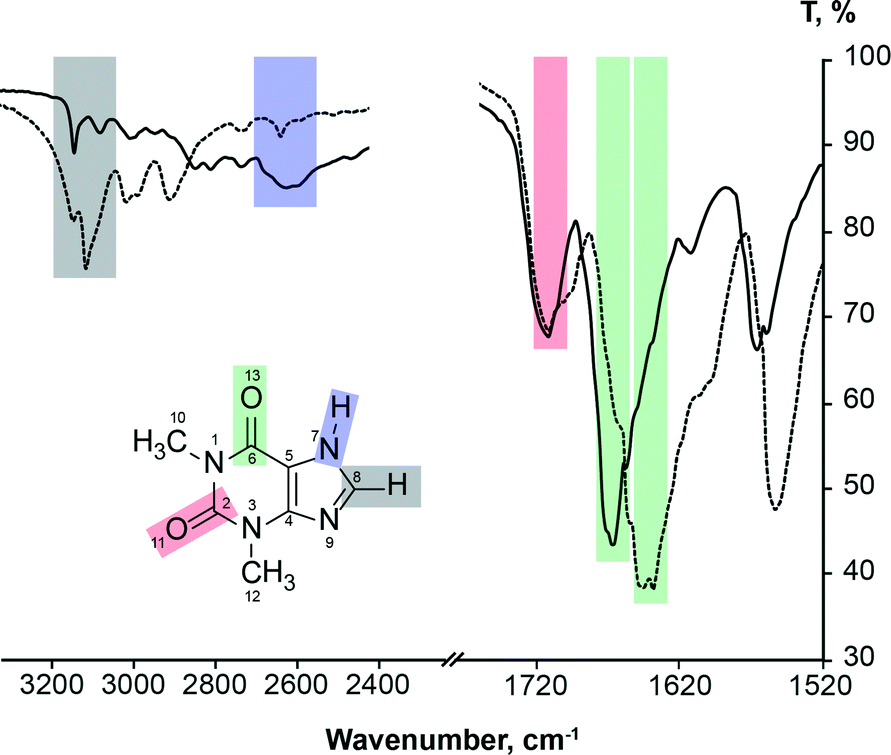 | ||
| Fig. 11 FTIR spectra of theophylline form II (solid line) and form IV (dashed line), showing carbonyl group, alkene group hydrogen bond and imidazolium group hydrogen bond stretching band assignment. | ||
It is known77 that hydrogen bonding lengthens and weakens the CO bond, therefore the carbonyl group stretching band is observed at a lower frequency; thus the stronger the hydrogen bonding, the lower the stretching frequency. FTIR can be used to show hydrogen bonding of the carbonyl group.78 This rule can be clearly seen in the case of theophylline. Carbonyl group C2O11, which is not involved in the hydrogen bonding in either polymorph (there are only weak interactions with theophylline methyl group hydrogens), has an identical band position in the FTIR spectra (1706 cm−1) and the stretching frequency is higher than that of C6O13 carbonyl group. In form IV, the carbonyl group C6O13 is involved in strong hydrogen bonding with the best hydrogen bond donor (N7–H) and this results in a stretching frequency of 1640 cm−1; while in form II, the C6O13 group is involved in two weak bifurcated C8–H⋯O13 hydrogen bonds, stretching band is observed at 1664 cm−1.
Fig. 12 illustrates the carbonyl group stretching band position and intensity changes during theophylline crystallization from saturated form II solutions during solvent evaporation.
 | ||
| Fig. 12 FTIR spectra of carbonyl group stretching band region during theophylline crystallization from saturated solutions upon solvent evaporation at room temperature. Colour gradient shows FTIR spectra changes throughout crystallization (red – saturated solution, blue – crystallization product). FTIR spectra of solid theophylline form II (dashed line) and IV (continuous line) are added for comparison purpose. Acetone carbonyl group stretching band is marked with ▲. | ||
The stretching bands of other groups/bonds expected to take part in self-association – alkene group hydrogen bond and imidazolium group hydrogen bond – were not visible in solution FTIR spectra due to low theophylline solubility in the chosen solvents, and they were observed only when all the solvent had evaporated. During the solvent evaporation the intensities of all theophylline bands increased because of increasing theophylline concentration in the solution. An exception to this was the final part of crystallization from saturated methanol solution, where theophylline carbonyl group intensities decreased. For clarity these spectra are shifted by y axis. Crystallization from acetone, acetonitrile, chloroform, methanol and formic acid gave theophylline form II, and water produced theophylline monohydrate. Intensive water OH bending band was overlapping with both theophylline carbonyl group stretching bands, therefore the crystallization experiment from saturated water solution was not suitable for studies.
The strongest carbonyl group stretching bands in theophylline solution spectra were observed in concentrated solution, whereas weaker spectral bands appeared only when theophylline crystals emerged. In all solvents the most distinct changes in theophylline band positions and intensities were observed at the moment when solid theophylline emerged. This was due to fact that, before the crystallization FTIR spectra represented mostly solvated theophylline and theophylline associates, whereas during the crystallization, spectra were reflecting the average spectra of solvated theophylline, multiple theophylline associates and crystalline theophylline. The ratio of these species changed during the evaporation/crystallization and therefore, the band position of functional groups involved in these processes also changed. Finally, when all solvent have evaporated, FTIR spectra of crystalline theophylline was observed.
The position of C2O11 carbonyl group stretching band in methanol, acetonitrile and chloroform solutions were the same (1706 cm−1) and it coincided with the position of this group in both polymorphs, therefore we can conclude that this group was not involved in strong hydrogen bonding in the solution (similar to crystalline theophylline) and the nearby environment of this group was similar to that in both polymorphs. The position of this stretching band in acetone solution cannot be determined, as acetone carbonyl group stretching bands overlaps with this carbonyl group stretching band. The position of C2O11 carbonyl group stretching band does not change during the crystallization from methanol solution, whereas during the crystallization from acetonitrile and chloroform solutions the band shifted to 1720 cm−1 indicating a change in bonding. The final position of this group stretching band does not match that in the solid form II. FTIR spectra did not change within 5 h. Such band shift to higher frequency indicates that hydrogen bonding was weaker in the crystallized material than in solution. Crystallizations from both solvents were repeated in larger scale. PXRD showed that crystallization products were form II, and FTIR spectra of obtained material coincided with form II spectra. The reason why C2O11 carbonyl group stretching band position in FTIR in situ experiment does not match the position of this band in form II spectra might be related to theophylline crystallization manner from acetonitrile and chloroform. No distinct theophylline crystals were observed on the FTIR spectrometer liquids plate after solvent evaporation, meaning that theophylline crystallized as a thin film. Such crystallization behaviour might promote monolayer product along (100) plane, where this carbonyl group is on the surface and it is not involved in any hydrogen bonding. Consequently, C2O11 carbonyl group stretching band would be shifted to higher frequency for such crystallization product.
In saturated formic acid solution position of C2O11 carbonyl group stretching band was 1696 cm−1 and during the crystallization it gradually shifted to the position of this carbonyl group in crystalline theophylline. This happened because in the solution this carbonyl group was involved in hydrogen bonding with formic acid proton, whereas during the crystallization this hydrogen bond was disarranged.
Unlike C2O11 carbonyl group, the position of the C6O13 carbonyl group stretching bands were not the same in the methanol solution and crystallized material (Fig. 12A). In the methanol solution the carbonyl group C6O13 stretching band was observed at 1656 cm−1, i.e. in between the stretching bands of this carbonyl group in solid form II and form IV. This suggest that the associates (dimers, aggregates, solvated entities) in the solution were not the same as in the solid phases and that the hydrogen bonding in the solution was stronger than that in the form II and weaker than that in form IV. From 1H NMR experiment we know that there was no theophylline self-association in methanol, therefore we can conclude that the association causing band shift must be hydrogen bonding between the carbonyl group and methanol –OH group proton. The changes of the C6O13 stretching band position were observed at the final part of the crystallization and the band position shifted from 1656 to 1664 cm−1, which is the position of this band in theophylline form II.
In chloroform, analogous C6O13 carbonyl group stretching band shift were observed (Fig. 12D), with the only difference that initially this band had two maxima – at 1656 cm−1 and 1648 cm−1. The band at 1656 cm−1 corresponds to solvated theophylline species, analogous to those in methanol, whereas the band at 1648 cm−1 most likely corresponds to the dimer detected by NMR measurements. A similar situation was observed when theophylline crystallized from formic acid; with the difference that in saturated formic acid solution a wide band in this region was observed. The wider carbonyl group stretching band must be a result of multiple associates. The concentration of saturated formic acid solution was ~100 times higher than the concentration of saturated chloroform solution, therefore it was expected that associates formed were more complex than dimers and with some diversity. Upon crystallization these agglomerates were disarranged and desolvated, and the final position of C6O13 carbonyl group stretching band matched the position of form II.
Minor changes in the position of the C6O13 carbonyl group stretching band were observed when theophylline crystallized from acetone solution. This suggests that theophylline hydrogen bonding did not change significantly upon crystallization from acetone, at least as regards to this carbonyl group. The position of C6O13 carbonyl group stretching band remained the same during the crystallization from acetonitrile. Such behaviour points out, that this theophylline carbonyl group does not form hydrogen bonds in acetone and acetonitrile solutions, neither to solvent molecules, nor to other theophylline molecules. It is understandable – acetone and acetonitrile are not proton donor solvents. However it was expected that these solvents would compete with the basic theophylline imidazole group nitrogen (N9) to form a hydrogen bond with the most acidic theophylline proton (N7–H). NMR experiments showed that theophylline molecules in the acetone and acetonitrile solution are monomeric and self-association does not occur in these solvents, meaning that species with solvated (N7–H) group dominated in acetone and acetonitrile.
The same FTIR in situ crystallization experiment was repeated with saturated form IV solution in methanol and identical results were obtained, suggesting that there are the same associates in the solution regardless of to which polymorph the solution is saturated.
4. Conclusions
The SMPT from theophylline form II to form IV is a very slow nucleation-growth controlled polymorphic transformation. Form IV nucleation is most likely homogenous and is slow. Nucleation of and therefore induction of the phase transformation is hindered by solution aggregates. Form IV crystals grow along the (001) direction, forming plate-like crystals. SMPT induction times correlate to phase transition times in studied solvents, indicating that nucleation and form IV growth is limited by the same factors. Theophylline forms associates in solvents which are good H-bond donors i.e. chloroform, water and formic acid. There are the same molecular aggregates in the solutions saturated with respect to form II and form IV. The theophylline aggregates present in solution do not change over time and therefore the long induction time of form IV is not dependent on aggregation kinetics. NMR and FTIR data suggest that the nature of solution aggregates is solvent dependent, most likely linked to hydrogen bonding character of the solvent.5. Experimental section
5.1. Materials
Anhydrous theophylline was obtained from Sigma-Aldrich and was certified >99% pure. Anhydrous theophylline was confirmed to be form II and was used as received. Methanol (HPLC grade), acetonitrile (HPLC grade), acetone (ARC grade) and chloroform (HPLC grade) were obtained from Fischer Scientific, formic acid (>98%) from Sigma-Aldrich. Solvents were used without further purification. Distilled water was used. Methanol-D4 (CD3OD, 99.8%), acetone-D6 ((CD3)2O, 99.9%), dimethyl sulfoxide-D6 ((CD3)2SO, 99.9%) and formic acid-D2 (98%; <5% D2O) were obtained from Cambridge Isotope Laboratories, deuterium oxide (D2O, 99.96%) from BDH Chemicals, chloroform-D (CCl3D, 99.8%) from Euriso-top, and acetonitrile-D3 (CD3CN, 99.8%) from Apollo Scientific.5.2. Preparation of theophylline crystalline forms
5.3. Examination of solution mediated phase transformation
An excess amount (2.5 g) of anhydrous theophylline form II (used as received) was added to 100 mL of methanol. The suspension was stirred at 600 rpm for 14 days at room temperature (23 ± 1 °C) and the following measurements were performed every 7 to 24 h.(a) Solution concentration monitoring. Theophylline concentration in the solution was measured every 1 h to 1 day. The solution (~1 mL) was filtered through a syringe filter of 0.20 μm size, and solution concentration was determined as described in section 5.7. Three parallel solution concentration determination experiments were performed.
(b) Crystallization product from solution. 2 mL of the solution was filtered through 0.20 μm syringe filter, transferred to a Petri dish and was left to evaporate at room temperature. Crystallized dry residue of three parallel experiments was combined and phase composition was examined using PXRD.
(c) FTIR spectra of the solution. ~0.3 mL of the filtered solution was gathered as described above, and the FTIR spectra of solution were recorded immediately after sample gathering.
(d) Phase composition of the solid phase. The stirring of the slurry was stopped 30 seconds prior to sample gathering, to allow the suspended solid particles to settle. Solid phase sample (10–20 mg) was collected with a metal spoon from the suspension and was quickly filtered through Buhner funnel filter under reduced pressure. The solid phase was examined using PXRD, FTIR and SEM.
SMPT was repeated in triplicate.
SMPT was additionally performed with different starting materials and solvents (given in Table 1). Single runs were performed for these experiments. The phase composition of the solid phase was monitored every 1 to 7 days, except experiments where form II and form IV mixture was used; the phase composition in these experiments was monitored every 10 min to 1 h. The solvent and theophylline ratio in all SMPT experiments were the same (2.5 g of theophylline and 100 mL of solvent), except SMPT in formic acid, where 15.0 g of theophylline were added to 25 ml of formic acid due to high theophylline solubility in formic acid.
| Starting material | Solvent |
|---|---|
| a Prepared as described in section 5.2. b Small scale experiment, Vtotal = 4.0 Ml. | |
| Commercial form II | Methanol |
| Form II, ground in mortar with pestle for 3 min | Methanol |
| Lyophilized form IIa | Methanol |
| Commercial form II | Methanol saturated to theophylline form IV |
| Commercial form II/form IVamixture (wII/wIV; 90/10) | Methanol saturated to theophylline form II |
| Commercial form II/form IVamixture (wII/wIV; 90/10) | Methanol saturated to theophylline form IV |
| Commercial form II | Methanol/water mixture (VMeOH/VH2O; 99/1) |
| Commercial form II | Methanol/water mixture (VMeOH/VH2O; 95/5) |
| Commercial form II | Methanol/water mixture (VMeOH/VH2O; 80/20) |
| Commercial form II | Acetone |
| Commercial form II | Acetonitrile |
| Commercial form II | Chloroform |
| Commercial form II | Formic acid |
| Commercial form II | Methanol-D4b |
SMPT in methanol-D4 was performed for H1 NMR studies. An excess amount (0.5 g) of commercial theophylline form II was added to 4.0 mL of methanol-D. The suspension was stirred at 600 rpm for 10 days at room temperature (23 ± 1 °C). PXRD patterns of solid phase and solution H1 NMR spectra (see section 5.9) were recorded each day. Stirring was stopped 1 min prior to sample collection, and: (a) clear solution (~1 mL) was transferred to NMR tube; (b) solid phase (10–20 mg) was collected with a metal spoon and quickly filtered through a Buchner funnel filter under reduced pressure. PXRD and H1 NMR measurements were performed immediately after sample collection. Solution from NMR experiment was returned back to the reaction vial immediately after recording the NMR spectrum.
5.4. Determination of theophylline solubility
Theophylline form II solubility in acetone, acetonitrile, methanol, chloroform and formic acid and form IV solubility in methanol were determined as follows. An excess amount of theophylline was added to 15 mL of solvent and was left to stir overnight at 23 ± 1 °C. The saturated solution was filtered through a 0.20 μm syringe filter and then 10.0 mL of clear solution was transferred to a preweighed vial. Solution was left to evaporate at room temperature, weighed and theophylline solubility was calculated. The PXRD pattern of the filtrate was recorded to ensure that the solubility of the desired polymorph was determined. Two parallel experiments were performed.5.5. Powder X-ray diffraction (PXRD)
Samples were analysed with a Rigaku Miniflex powder X-ray diffractometer. Diffraction patterns within the 2θ range of 5° to 40° were recorded at room temperature using Cu Kα radiation at 1.54180 Å, with the following measurement conditions: tube voltage 30 kV, tube current 15 mA, step-scan mode with the step size 2θ = 0.02°, and the counting time 2 s per step. Diffractometer slits were set as follows: divergence slit – variable; scattering slit – 4.2°, receiving slit – 0.3 mm. Powder samples were packed into aluminium sample holders and pressed by a glass slide to ensure co-planarity of the powder surface with the surface of the holder.5.6. FTIR spectroscopy
FTIR spectra were recorded using a Perkin-Elmer Spectrum BX spectrometer fitted with a PIKE Technologies MIRacle sampling accessory. MIRacle liquids plate was used when spectra of solutions were recorded. The samples were scanned at a resolution of 4 cm−1 between 4000 cm−1 and 600 cm−1. Each spectrum consisted of 16 co-added scans if not otherwise stated.5.7. UV/Vis spectroscopic solution concentration monitoring
Solution concentration throughout SPMTs in methanol was monitored by measurements of the UV/Vis absorption at 272 nm using a Perkin-Elmer Lambda 25 UV/Vis spectrophotometer. Calibration was performed in the concentration region between 0.2 mM and 0.01 mM (R2 = 0.99990). For solution concentration determination 20 μL aliquot of filtered reaction medium was diluted with 40 mL of methanol.5.8. Scanning electron microscopy (SEM)
SEM imaging was performed using Inspect S SEM (FEI, Holland) system. Samples were initially gold coated using a K550X sputter coater (EMITECH, UK) and subsequently scanned using an acceleration voltage of 5.0 kV at a working distance of approximately 10 mm.5.9. NMR spectroscopic self-association studies
1H NMR spectra were recorded as a function of theophylline concentration in the solution. Experiments were performed in methanol-D4, chloroform-D, acetone-D6, dimethyl sulfoxide-D6, deuterium oxide, acetonitrile-D3 and formic acid-D2 concentration region from nearly saturated solutions (1.0 M in formic acid-D2, 0.050 M in methanol-D4, 0.015 M in acetone-D6, 0.10 M in dimethyl sulfoxide-D6, deuterium oxide, chloroform-D and acetonitrile-D3) to 1 μM solutions was covered. Ground anhydrous theophylline form II was used to prepare the most concentrated solution in each solvent and the rest of the solutions were prepared by subsequent dilution. Additional samples, where an excess amount of ground theophylline was added to deuterated solvents, were prepared to simulate suspensions similar to that examined in SMPT. The mass of theophylline added was 120% of the mass necessary to prepare saturated solution in the respective solvent. An analytical balance (±0.1 mg) and micropipettes (±1 μL) were used for solution preparation. NMR spectra of prepared solutions were recorded right after solution preparation, and repeated after 1 and 2 weeks. NMR tubes of prepared solutions were closed with lids and sealed with parafilm. Samples were stored at 20.0 ± 0.5 °C between measurements.1H NMR spectra were recorded on a Bruker Avance DRX 300 MHz spectrometer using residual solvent as an internal standard. NMR spectra were recorded at 26.8 ± 0.5 °C (300.0 ± 0.5 K).
Theophylline 1H chemical shifts were allocated by 1H-13C HSQC according to literature assignments.811H-13C HSQC experiments were carried out using the standard Bruker program hsqcetgpsi2.82,83
5.10. In situ monitoring of crystallization process
Two drops (~0.07 mL) of saturated theophylline methanol, acetone, acetonitrile, chloroform, water and formic acid solutions from solubility determination experiments (section 5.4) were placed on a FTIR spectrometer liquids plate and spectra of the solution were continuously recorded during solvent evaporation/theophylline crystallization. Each FTIR spectra showed the average of 16 co-added scans, recorded in 75 s. For acetone solution 4 co-added scans (recorded in 17 s) were averaged. Spectra were recorded until three continuous spectra were identical and no peaks of solvents were visible. The experiment with each solvent was repeated in triplicate.Acknowledgements
This work has been supported by the European Social Fund within the project “Support for Doctoral Studies at University of Latvia”. The authors gratefully acknowledge the support of Erasmus Student Mobility programme. We thank Dace Rasiņa and Alessandro Pozzoli for fruitful discussions.Notes and references
- S. R. Vippagunta, H. G. Brittain and D. J. W. Grant, Adv. Drug Delivery Rev., 2001, 48, 3–26 CrossRef CAS.
- H. G. Brittain, Polymorphism in Pharmaceutical Solids, Informa Healthcare USA, Inc., New York, NY, 2nd edn, 2009, vol. 192 Search PubMed.
- R. Hilfiker, Polymorphism: in the Pharmaceutical Industry, Wiley-VCH, Weinheim, 2006 Search PubMed.
- C. Cardin, Y. Gan and T. Lewis, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2007, 63, o3175 CAS.
- L. Seton, D. Khamar, I. J. Bradshaw and G. A. Hutcheon, Cryst. Growth Des., 2010, 10, 3879–3886 CAS.
- K. Matsuo and M. Matsuoka, Cryst. Growth Des., 2007, 7, 411–415 CAS.
- C. Nunes, A. Mahendrasingam and R. Suryanarayanan, Pharm. Res., 2006, 23, 2393–2404 CrossRef CAS PubMed.
- H. Zhu, Int. J. Pharm., 1996, 135, 151–160 CrossRef CAS.
- M. D. Ticehurst, R. A. Storey and C. Watt, Int. J. Pharm., 2002, 247, 1–10 CrossRef CAS.
- S. L. Childs, G. P. Stahly and A. Park, Mol. Pharmaceutics, 2007, 4, 323–338 CrossRef CAS PubMed.
- M. Cioffi, C. A. Hunter, M. J. Packer, M. J. Pandya and M. P. Williamson, J. Biomol. NMR, 2009, 43, 11–19 CrossRef CAS PubMed.
- D. Musumeci, C. A. Hunter and J. F. McCabe, Cryst. Growth Des., 2010, 10, 1661–1664 CAS.
- C. A. Hunter, J. F. McCabe and A. Spitaleri, CrystEngComm, 2012, 14, 7115–7117 RSC.
- K. Chadwick, R. J. Davey, G. Dent, R. G. Pritchard, C. A. Hunter and D. Musumeci, Cryst. Growth Des., 2009, 9, 1990–1999 CAS.
- R. J. Davey, G. Dent, R. K. Mughal and S. Parveen, Cryst. Growth Des., 2006, 6, 1788–1796 CAS.
- J. Kirschbaum, J. Pharm. Sci., 1973, 62, 168–169 CrossRef CAS PubMed.
- D. Guttman and T. Higuchi, J. Pharm. Sci., 1971, 60, 1269–1270 CrossRef CAS PubMed.
- A. L. Thakkar, L. G. Tensmeyer and W. L. Wilham, J. Pharm. Sci., 1971, 60, 1267–1269 CrossRef CAS PubMed.
- D. Guttman and T. Higuchi, J. Am. Pharm. Assoc., 1957, 46, 4–10 CrossRef CAS PubMed.
- Y. Ebisuzaki, P. D. Boyle and J. A. Smith, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1997, 53, 777–779 Search PubMed.
- A. V. Trask, W. D. S. Motherwell and W. Jones, Int. J. Pharm., 2006, 320, 114–123 CrossRef CAS PubMed.
- M. C. Etter, Acc. Chem. Res., 1990, 23, 120–126 CrossRef CAS.
- D. Khamar, R. G. Pritchard, I. J. Bradshaw, G. A. Hutcheon and L. Seton, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2011, 67, o496–o499 CAS.
- C. Sun, D. Zhou, D. J. W. Grant and V. G. Young, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2002, 58, o368–o370 CAS.
- W. Ostwald, Z. Phys. Chem., 1897, 22, 289 CAS.
- P. T. Cardew and R. J. Davey, Proc. R. Soc. London, Ser. A, 1985, 398, 415–428 CrossRef CAS.
- R. J. Davey, P. T. Cardew, D. McEwan and D. E. Sadler, J. Cryst. Growth, 1986, 79, 648–653 CrossRef CAS.
- R. J. Davey, N. Blagden, S. Righini, H. Alison and E. S. Ferrari, J. Phys. Chem. B, 2002, 106, 1954–1959 CrossRef CAS.
- R. J. Davey, N. Blagden, S. Righini, H. Alison and E. S. Ferrari, J. Phys. Chem. B, 2002, 106, 1954–1959 CrossRef CAS.
- R. Bobrovs, L. Seton and A. Actiņš, CrystEngComm, 2014, 16, 10581–10591 RSC.
- J. E. House, Principles of chemical kinetics, Academic Press, 2007 Search PubMed.
- E. D. Becker, J. Chem. Phys., 1959, 31, 269 CrossRef CAS PubMed.
- C. M. Huggins, G. C. Pimentel and J. N. Shoolery, J. Phys. Chem., 1956, 60, 1311–1315 CrossRef CAS.
- E. D. Becker, U. Liddel and J. N. Shoolery, J. Mol. Spectrosc., 1958, 2, 1–8 CrossRef CAS.
- Y. Lam and G. Kotowycz, Can. J. Chem., 1977, 55, 3620–3630 CrossRef CAS.
- I. Horman and B. Dreux, Helv. Chim. Acta, 1984, 67, 754–764 CrossRef CAS PubMed.
- I. J. McLennan, R. E. Lenkinski and Y. Yanuka, Can. J. Chem., 1985, 63, 1233–1238 CrossRef CAS PubMed.
- N. Marchettini, G. Valensin and E. Gaggelli, Biophys. Chem., 1990, 36, 65–70 CrossRef CAS.
- A. N. Veselkov, L. N. Dymant, S. F. Baranovskii, P. A. Bolotin, O. S. Zav'yalova, D. A. Veselkov, H. Parkes and D. Davies, J. Struct. Chem., 1995, 36, 69–75 CrossRef.
- D. B. Davies, L. N. Djimant and A. N. Veselkov, J. Chem. Soc., Faraday Trans., 1996, 92, 383 RSC.
- M. Bastian and H. Sigel, Biophys. Chem., 1997, 67, 27–34 CrossRef CAS.
- M. P. Evstigneev, V. P. Evstigneev and D. B. Davies, J. Mol. Struct., 2006, 784, 162–168 CrossRef CAS PubMed.
- M. Bogdan, C. G. Floare and A. Pîrnău, J. Phys.: Conf. Ser., 2009, 182, 012002 CrossRef.
- J. S. Chen and R. B. Shirts, J. Phys. Chem., 1985, 89, 1643–1646 CrossRef CAS.
- E. D. Becker, Spectrochim. Acta, 1961, 17, 436–447 CrossRef CAS.
- L. P. Kuhn and R. E. Bowman, Spectrochim. Acta, 1961, 17, 650–660 CrossRef CAS.
- U. Liddel and E. D. Becker, Spectrochim. Acta, 1957, 10, 70–84 CrossRef CAS.
- R. J. Abraham and M. Mobli, Magn. Reson. Chem., 2007, 45, 865–877 CrossRef CAS PubMed.
- F. Hibbert and J. Emsley, Adv. Phys. Org. Chem., 1990, 26, 255–379 CAS.
- C. A. Hunter and M. J. Packer, Chem. – Eur. J., 1999, 5, 1891–1897 CrossRef CAS.
- A. Spitaleri, C. A. Hunter, J. F. McCabe, M. J. Packer and S. L. Cockroft, CrystEngComm, 2004, 6, 489–493 RSC.
- W. H. Gmeiner and C. D. Poulter, J. Am. Chem. Soc., 1988, 110, 7640–7647 CrossRef CAS.
- D. V. Waterhous and D. D. Muccio, Magn. Reson. Chem., 1990, 28, 223–226 CrossRef CAS PubMed.
- S. L. Cockroft, J. Perkins, C. Zonta, H. Adams, S. E. Spey, C. M. R. Low, J. G. Vinter, K. R. Lawson, C. J. Urch and C. A. Hunter, Org. Biomol. Chem., 2007, 5, 1062–1080 CAS.
- C. E. Hughes, S. Hamad, K. D. M. Harris, C. R. A. Catlow and P. C. Griffiths, Faraday Discuss., 2007, 136, 71 RSC.
- S. Ng, Mol. Pharmacol., 1971, 7, 177–182 CAS.
- A. Maher, D. M. Croker, Å. C. Rasmuson and B. K. Hodnett, Cryst. Growth Des., 2012, 12, 6151–6157 CAS.
- P. Lehto, J. Aaltonen, M. Tenho, J. Rantanen, J. Hirvonen, V. P. Tanninen and L. Peltonen, J. Pharm. Sci., 2009, 98, 985–996 CrossRef CAS PubMed.
- N. Rodríguez-Hornedo and D. Lechuga-Ballesteros, Int. J. Pharm., 1992, 85, 149–162 CrossRef.
- J. Schöll, D. Bonalumi, L. Vicum, M. Mazzotti and M. Müller, Cryst. Growth Des., 2006, 6, 881–891 Search PubMed.
- S. Dharmayat, J. Calderon De Anda, R. B. Hammond, X. Lai, K. J. Roberts and X. Z. Wang, J. Cryst. Growth, 2006, 294, 35–40 CrossRef CAS PubMed.
- F. Tian, T. Rades and N. Sandler, AAPS PharmSciTech, 2008, 9, 390–394 CrossRef CAS PubMed.
- S. Dharmayat, R. B. Hammond, X. Lai, C. Ma, E. Purba, K. J. Roberts, Z. Chen, E. Martin, J. Morris and R. Bytheway, Cryst. Growth Des., 2008, 8, 2205–2216 CAS.
- A. Maher, D. M. Croker, Å. C. Rasmuson, B. K. Hodnett and M. A. O'Mahony, Cryst. Growth Des., 2012, 12, 1925–1932 Search PubMed.
- M. A. O'Mahony, C. C. Seaton, D. M. Croker, S. Veesler, Å. C. Rasmuson and B. K. Hodnett, Cryst. Growth Des., 2013, 13, 1861–1871 Search PubMed.
- J. W. Mullin, Crystallization, Butterworth-Heinemann, Oxford, 4th edn, 2001 Search PubMed.
- D. Croker and B. K. Hodnett, Cryst. Growth Des., 2010, 10, 2806–2816 CAS.
- E. S. Ferrari and R. J. Davey, Cryst. Growth Des., 2004, 4, 1061–1068 CAS.
- R. Docherty, G. Clydesdale, K. J. Roberts and P. Bennema, J. Phys. D: Appl. Phys., 1991, 24, 89–99 CrossRef CAS.
- B. C. Hancock and G. Zografi, J. Pharm. Sci., 1997, 86, 1–12 CrossRef CAS PubMed.
- L. R. Snyder, J. Chromatogr., 1974, 92, 223–230 CrossRef CAS.
- R. A. Sullivan, R. J. Davey, G. Sadiq, G. Dent, K. R. Back, J. H. Ter Horst, D. Toroz and R. B. Hammond, Cryst. Growth Des., 2014, 14, 2689–2696 CAS.
- H. G. Brittain, J. Pharm. Sci., 1997, 86, 405–412 CrossRef CAS PubMed.
- G. A. Stephenson, R. A. Forbes and S. M. Reutzel-Edens, Adv. Drug Delivery Rev., 2001, 48, 67–90 CrossRef CAS.
- D. E. Bugay, Adv. Drug Delivery Rev., 2001, 48, 43–65 CrossRef CAS.
- S. Parveen, R. J. Davey, G. Dent and R. G. Pritchard, Chem. Commun., 2005, 1531–1533 RSC.
- P. B. Coleman, Practical sampling techniques for infrared analysis, CRC Press, London, 1993 Search PubMed.
- S. M. Kashid and S. Bagchi, J. Phys. Chem. Lett., 2014, 5, 3211–3215 CrossRef CAS.
- C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, J. van de Streek and P. A. Wood, J. Appl. Crystallogr., 2008, 41, 466–470 CrossRef CAS.
- J. Bergmann, P. Friedel and R. Kleeberg, CPD Newsl., 1998, vol. 20, pp. 5–8 Search PubMed.
- E. D. L. Smith, R. B. Hammond, M. J. Jones, K. J. Roberts, J. B. O. Mitchell, S. L. Price, R. K. Harris, D. C. Apperley, J. C. Cherryman and R. Docherty, J. Phys. Chem. B, 2001, 105, 5818–5826 CrossRef CAS.
- L. Kay, P. Keifer and T. Saarinen, J. Am. Chem. Soc., 1992, 114, 10663–10665 CrossRef CAS.
- A. G. Palmer, J. Cavanagh, P. E. Wright and M. Rance, J. Magn. Reson., 1991, 93, 151–170 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ce02484b |
| This journal is © The Royal Society of Chemistry 2015 |