Bioelectrochemical systems (BES) for sustainable energy production and product recovery from organic wastes and industrial wastewaters
Deepak
Pant
*a,
Anoop
Singh
b,
Gilbert
Van Bogaert
a,
Stig
Irving Olsen
b,
Poonam
Singh Nigam
c,
Ludo
Diels
a and
Karolien
Vanbroekhoven
a
aSeparation & Conversion Technologies, VITO-Flemish Institute for Technological Research, Boeretang 200, 2400 Mol, Belgium. E-mail: pantonline@gmail.com; deepak.pant@vito.be; Tel: +32 1433 6969; Fax: +32 1432 6586
bQuantitative Sustainability Assessment, Department of Management Engineering, Technical University of Denmark, Lyngby, Denmark
cFaculty of Life and Health Sciences, University of Ulster, Coleraine, Northern Ireland, United Kingdom BT52 1SA
First published on 23rd December 2011
Abstract
Bioelectrochemical systems (BESs) are unique systems capable of converting the chemical energy of organic waste including low-strength wastewaters and lignocellulosic biomass into electricity or hydrogen/chemical products in microbial fuel cells (MFCs) or microbial electrolysis cells (MECs) respectively, or other products formed at the cathode by an electrochemical reduction process. As compared to conventional fuel cells, BESs operate under relatively mild conditions, use a wide variety of organic substrates and mostly do not use expensive precious metals as catalysts. The recently discovered use of BES for product synthesis via microbial electrosynthesis have greatly expanded the horizon for these systems. Newer concepts in application as well as development of alternative materials for electrodes, separators, and catalysts, along with innovative designs have made BESs very promising technologies. This article discusses the recent developments that have been made in BESs so far, with an emphasis on their various applications beyond electricity generation, resulting performances and current limitations.
 Deepak Pant | Dr Deepak Pant is a research scientist at the Flemish Institute for Technological Research (VITO) currently working on bioenergy, specifically, the design and optimization of bioelectrochemical cells for energy recovery from wastewaters and microbial electrosynthesis. He has a PhD degree in environmental biotechnology from TERI University, New Delhi (India) and has 17 peer-reviewed publications (h-Index 9) and 8 book chapters to his credit. His research experience lies in industrial wastewater treatment, wasteland reclamation and restoration, biofertilizers, sustainable agriculture, biofuels and bioenergy, life cycle analysis (LCA). |
 Anoop Singh | Dr Anoop Singh, completed his Doctoral Degree in Botany in 2004. Before joining Technical University of Denmark, he worked at the University College Cork, Ireland, The Energy and Resources Institute (TERI), New Delhi, India, Indian Agricultural Research Institute (IARI), New Delhi, India, Banaras Hindu University, Varanasi, India and VBS Purvanchal University, Jaunpur, India. He has published more than forty research articles in scientific journals and is a member of several scientific communities. His research interests are focused on sustainable agriculture, the utilization of industrial, agricultural and household waste for eco-friendly energy production, renewable energy and their life cycle assessment. |
 Gilbert Van Bogaert | Ir. Gilbert Van Bogaert is a well-known fuel cell developer with more than 25 years experience in PEM and alkaline fuel cells and provides guidance in design and operation of bioelectrochemical systems. He was the Belgian delegate in the executive committee (EXCO) of the IEA Advanced Fuel Cells implementing agreement, and was the Belgian representative in IEA Task 40 on Advanced Fuel Cells. |
 Stig Irving Olsen | Dr Stig Irving Olsen is associate professor in sustainable production at the section for Quantitative Sustainability Assessment, Department of Management Engineering at the Technical University of Denmark. He obtained his Ph.D. in LCA from Technical University of Denmark in 1997 and a Master of Science in Biology from University of Copenhagen in 1988. Since his PhD his main research area has been in methodology development in LCA, particularly in the life cycle impact assessment of human health impact. During the last years his research has focused more on application of LCA in several technology areas, including renewable energy and nanotechnology. |
 Ludo Diels | Prof. Dr Ludo Diels is Scientific Manager for Sustainable Chemistry. He has a background in microbiology and biotechnology and is professor at the University of Antwerp, Belgium (environmental stress, environmental engineering, separation technology, Green chemistry and sustainable development). He has more than 30 years experience in environmental biotechnology, biotechnology, engineering and chemistry. He has large experience in environmental technology and has more than 150 publications and 7 patents. He was for 15 years, the manager of the Environmental Technology Department at VITO. |
 Karolien Vanbroekhoven | Dr ir. Karolien Vanbroekhoven, Programme manager at VITO, is responsible for the team of about 30 people involved in the programme of industrial biotechnology and renewable chemicals in the unit of SCT. She has been involved as partner/coordinator in several EU projects, national research projects and feasibility tests. She is an expert in bioconversion (like dark fermentation processes) and bioelectrochemical systems. She is responsible for the scientific programme in the team as well as people and financial management. |
1. Preface
The year 2010–2011 marks the 100th anniversary of the discovery of the fact that certain bacteria can transfer their electron extracellularly while degrading organic waste. It was in year 1911 when M. C. Potter published his seminal work titled ‘Electrical effects accompanying the decomposition of organic compounds’.1 This was a follow up of the paper in 1910 where he mentioned that the disintegration of organic compounds by microorganisms is accompanied by the liberation of electrical energy.2 Using Saccharomyces cerevisiae as the test organism, platinum as electrode and glucose as substrate, a maximum voltage of 0.3 to 0.5 voltage was recorded.1 He later followed it up with some other research papers dealing with ionisation of the gases produced during fermentation3 and electrical effects accompanying the decomposition of organic compounds considered in relation to photosynthesis and plant nutrition.4 An important feature which marked all his research was the emphasis on electrical effects accompanying fermentation or putrefaction under the influence of microorganisms such as yeast or bacteria. Later, in 1931, Cohen studied the potential differences arising between various cultures and sterile media; he also built a bacterial battery which produced a small current for a short period of time.5 He observed that the potential of a vigorously growing bacterial culture amounted to 0.5–1 V over the control medium. After these initial efforts, interest in biofuel cells was renewed in early 1960s with the onset of manned space travel due to the potential of these cells to convert biowaste to energy in spacecraft.6 The first patent to describe microbial fuel cell (MFC) technology was issued to John Davis from Mobil Corporation in 1967 which described an externally mediated MFC using Nocardia salmonicolor isolated from sludge oxidizing hydrocarbons to alcohols, aldehydes and carboxylic acids.7 However, it was only in late 1990's and the decade of 2000 that research in this domain began in right earnest and has led to remarkable improvements and several potential applications. The power density of MFCs have increased from 0.001 to 0.01 milliwatts per square meter (mW m−2) of projected surface area of the anode in 1999 to 787 mW m−2 in 2003 and finally to levels of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 770 mW m−2 in 2008.8–10 The power demand for electrical devices have decreased significantly with recent advances in microelectronics, as a result of which the interest in microbe-catalyzed small fuel cells have emerged again as an alternative to fuel cells employing inorganic catalysts.11
770 mW m−2 in 2008.8–10 The power demand for electrical devices have decreased significantly with recent advances in microelectronics, as a result of which the interest in microbe-catalyzed small fuel cells have emerged again as an alternative to fuel cells employing inorganic catalysts.11
This article is dedicated to 100 years of research on bioelectrochemical systems. The research in this area was carried out intermittently over the years and some of these efforts have been documented recently in detail.12
2. Energy from wastewater—Introduction to BES
It has been universally accepted that energy is the currency that will drive the global economy of the future. According to Lewis,13 taking the number of joules of energy consumed by humans in a typical year and dividing that by the number of seconds in a year yields an average burn rate of about 13 trillion watts, or 13 TW. This is the amount of power consumed worldwide to run our planet. It was recently reported that energy use limits economic activity directly.14 This study concluded that an enormous increase in energy supply will be required to meet the demands of projected world population growth and lift the developing world out of poverty without jeopardizing standards of living in most developed countries. It was further added that the possibilities for substantially increasing energy supplies are highly uncertain. Electrochemical energy production is under serious consideration as an alternative energy/power source, as long as this energy consumption is designated to be more sustainable and more environment friendly.15 Energy generation from “negative-value” waste streams can simultaneously help meet the world's energy needs, reduce pollution, and reduce costs associated with water and wastewater treatment. For over a century, anaerobic digestion has been used for methane recovery from solid and liquid waste streams. Methane fermentation has several intrinsic advantages over aerobic treatment processes including renewable energy (methane) generation, reduced energy costs through elimination of aeration, and reduced sludge treatment and disposal expenses.16 Anaerobic technology has been successfully commercialized for the treatment of waste, and several full-scale anaerobic treatment plants are in operation worldwide.17 In recent years, biohydrogen production from waste and wastewater through dark fermentation has also drawn considerable attention due to interest in clean energy production using hydrogen fuel cells. Despite a stoichiometric potential of 12 mol H2/mol glucose, current fermentation techniques can unfortunately produce a maximum of only 2–3 mol H2/mol glucose, because most organic matter remains mired as volatile fatty acids and alcohols. The process is thus limited to feedstocks with suitable fermentation substrates, that is, those rich in carbohydrates, such as glucose.18,19Several metal reducing bacteria such as Geobacter sulfurreducens and Shewanella oneidensis catalyze the transfer of electrons from reduced electron donors to a solid electrode material, called an anode (mostly graphite), that serve as electron acceptor.20 When combined with a cathode through an external circuit to provide a path for the electron flow, bacterial respiration can be utilized to generate power in a fuel cell.21 This capability of certain bacteria to use insoluble electrode surfaces as a terminal electron acceptor creates an opportunity to induce biofilm growth, and thus electricity, from bacteria using controlled potential or electrical voltage. Biofilms of such electroactive bacteria (EAB) can facilitate proficient organic carbon removal from wastewater while producing biological renewable energy in the form of electricity in a particular type of BES, the MFC.
3. Types of BES
Depending on the bioctalayst, BESs can be classified as MFCs and enzymatic fuel cells (EFCs).22 Based on their mode of application, BESs can be further sub-divided into MFCs, microbial electrolysis cell (MEC), microbial desalination cells (MDCs) and microbial solar cells (MSC). The concept of MSC have been described in detail.23,24 The idea of using BESs as a mode of simultaneous desalination as well as energy/hydrogen recovery in the form of MDC was introduced recently25 and further explained by other researchers later.26,27 Stacked MDCs were described recently in which desalination chambers and concentrated chambers were spaced by compartmental anion exchange membranes (AEMs) and cation exchange membrane (CEMs).28 The maximum total desalination rate (TDR) of 0.0252 g h−1 was obtained using a two desalination-chambered SMDC with an external resistance of 10 Ω, which was 1.4 times that of single-desalination-chambered MDC. In fact, the term MXC was recently coined for these systems, the X standing for the different types and applications.29,30 Very recently, the concept of the microbial electrochemical snorkel (MES), which is a simplified design of a “shortcircuited” MFC was introduced for the treatment of urban wastewaters.31 Unlike MFCs, an MES does not divert energy to produce electricity but it ensures maximum efficiency for the oxidation of organic matter. Thus, a MES does not provide current but enhances the treatment efficiency. Several operational differences among these BES types can be identified and are discussed in detail further. The book “Bioelectrochemical Systems” published last year covers the fundamentals, microbiology, electrochemistry, technology, materials development and application aspect of these systems.323.1 Microbial fuel cell
In MFCs, bacteria convert chemical energy to electrical energy via the catalytic breakdown of organic substrates.33 The oxidation of organics by certain bacteria takes place in anode compartment as a result of which electrons and protons are generated. The electrons are then transferred through an external electric circuit to a terminal electron acceptor (TEA) which is reduced by the electrons. At the same time protons generated at the anode are transferred to the cathode through a membrane separating the anode form the cathode or through the electrolyte. TEA's such as oxygen, nitrate, and sulfate can diffuse into the cell and accept electrons to form new products that can then leave the cell. However, certain exoelectrogenic bacteria can transfer their electrons outside the cell (exogenously) to the awaiting TEA. These are the bacteria that produce power within an MFC system.34 As an emerging technology, MFCs are receiving increasing scientific,35 and more recently commercial,36 attention as their potential for alternative energy production, wastewater treatment and bioremediation of contaminated environments is steadily realized. A number of reviews have shown the versatility of MFCs to utilize a wide variety of substrate materials.35,37 Further, the power outputs of MFCs have improved rapidly over the last decade by altering their designs, optimizing configurations, operating conditions and choice of biocatalyst.38The fundamental aspects, working principle, terminology and measurements associated with MFCs have already been described in detail.16,39,40 A book published on the subject of MFCs gives a detailed description on the exoelectrogens, voltage and power generation, materials and architecture, and application aspect of these systems.41 A big advantage of MFCs is that these systems can operate at low loading rates.42 Other bioprocesses are seldom operated at very low COD concentrations. Anaerobic digestion would expect to receive influent organic concentrations of the order of 20000 mg COD/L or higher before delivering net energy, while aerobic processes are typically used below this for municipal or industrial waste streams with concentrations.43 However, aerobic processes require forced aeration which consumes considerable energy (∼0.5 kWh m-3), and typically volatilizes part of the COD to atmosphere.44 The use of BES will allow biological reduction of low COD concentrations ∼20 mg COD/L,42 which acts as a effluent polishing process, extracts the chemical energy, and converts residuals to electricity (MFCs), hydrogen (MEC) or other reduced products such as hydrogen peroxide,45 caustic.46 Given the current state-of-the-art, in near-term though, MFCs that produce enough electricity from organic wastes are unlikely to act as a perpetual source of electric power. However, they may prove practical sooner for some relatively high-energy liquid wastes, such as those from food processing and milk, where electricity generation could help to convert treatment costs.47
3.2 Microbial electrolysis cell (MEC)
MECs are a relatively new method for generating hydrogen from acetate and other fermentation end products by electrohydrogenesis. In MEC, which is a modified MFC, bacteria referred to as exoelectrogens48 oxidize a substrate and release electrons to the anode. Normally, in a MFC, in presence of oxygen at cathode, current is produced by oxygen reduction but in MEC, cathode is anaerobic and thus in absence of oxygen, no spontaneous current generation is possible. Thus, a small voltage is applied externally to the circuit, allowing hydrogen production at the cathode through the reduction of protons.49 When acetate is used as a substrate, a voltage of >0.2 V in practice is required for hydrogen evolution,50 which is substantially less than the 1.8–2.0 V used in practice for hydrogen production via water electrolysis in low temperature electrolysis.51 The anodic reaction, therefore, is the same as in the microbial generation of electricity in MFC, while the cathodic reaction proceeds in absence of oxygen. These systems were also referred to as bio-electrochemically assisted microbial reactors (BEAMR).52 The concept, operating principles and state of the art for this technology has been described earlier.49,53Compared with the fermentative reactor producing hydrogen from wastes, the MEC has a higher hydrogen recovery and a wider substrate diversity.54 However, when compared to MFCs, where a number of substrates have been evaluated,35,37 most MEC studies so far have relied on the use of pure chemical compounds (primarily acetate) as the substrate. When other substrates such as domestic or animal wastewaters were used,55,56 the hydrogen yields were low or there was substantial methane production. Table 1 presents a comprehensive list of substrates that have been used in MEC studies. Hydrogen production from cellulose was demonstrated in a two-chamber MEC at hydrogen yields (63%) similar to that obtained with glucose (64%) but less than that with acetic acid (82%), indicating that hydrogen recovery was not achieved for the fermentation step in the process.50 Recently, Lu et al.58 reported on the use of effluent after buffering from a ethanol-type dark-fermentation reactor producing hydrogen in a MEC for further hydrogen production. This two stage process resulted in an electrical energy demand of only 1.12 kWh/m3 H2, which was much lower than that needed for water electrolysis (5.6 kWh/m3 H2).
| Type of substrate | Concentration | Source inoculum | Type of MEC (with applied voltage, Eap) | Maximum H2 recovered (m³ H2/m³/d) | Reference |
|---|---|---|---|---|---|
| Cellulose | 1 g L−1 | Bacteria derived from a soil used to start up the cell in MFC mode | Two-chamber MEC with graphite granules as anode (1320 m²/m³) and a platinized carbon cloth as cathode (1 cm²); Eap = 0.6 V | 0.11 | 50 |
| Cellobiose dark-fermentation effluent | 4 g L−1 COD | Domestic wastewater used to start up the cell in MFC mode | Single-chamber air cathode MEC with graphite fiber brush as anode and carbon cloth air cathode (7 cm²); Eap = 0.5 V | 1.0 | 57 |
| Corn-stover dark-fermentation effluent | 5 g L−1 COD | Domestic wastewater used to start up the cell in MFC mode | Single-chamber air cathode MEC with graphite fiber brush as anode and carbon cloth air cathode (7 cm²); Eap = 0.5 V | 0.96 | 57 |
| Effluent from an ethanol-type fermentation CSTR used for hydrogen production | 6500 mg L−1 COD | Domestic wastewater used to start up the cell in MFC mode | Single-chamber membraneless MEC with carbon fiber brush anode and carbon cloth air cathode (7 cm²); Eap = 0.6 V | 1.41 | 58 |
| Glucose | 1 g L−1 | Domestic wastewater used to start up the cell in MFC mode | Single-chamber membraneless MEC with carbon brush anode and carbon cloth with Pt catalyst as cathode; Eap = 0.9 V | 1.87 | 59 |
| P-Glycerol | 1160 mg L−1 COD | Domestic wastewater used to start up the cell in MFC mode | Single-chamber membraneless MEC with carbon brush anode and carbon cloth with Pt catalyst as cathode; Eap = 0.9 V | 2 | 59 |
| Lactate | 1 g L−1 | Pure culture of Shewanella oneidensis MR-1 | Single-chamber membraneless MEC with carbon cloth anode (12.25 cm²) and platinized carbon cloth as cathode (20 cm²); Eap = 0.6 V | 0.025 | 60 |
| Sodium acetate | 10 mM | Effluent from an active bio-electrochemical cell | Two-chambered MEC operated in single chamber configuration with only anode (3.3 L) filled with medium and cathode for gas collection; graphite felt as anode (400 cm²) | 0.3 | 61 |
| Sodium acetate | 20 mM | Domestic wastewater used to start up the cell in MFC mode | Single-chamber membraneless MEC with carbon cloth anode (12.25 cm²) and platinized carbon cloth as cathode (20 cm²); Eap = 0.6 V | 0.69 | 60 |
| Sodium acetate | 1 g L−1 | Effluent from an active MFC | Single-chamber MEC with graphite fiber brush anode and stainless steel brush cathode; Eap = 0.5 V | 1.7 | 62 |
| Sodium acetate and yeast extract | 960 mg L−1 COD | Anaerobic sludge | Membraneless MEC with carbon felt anode (50 cm²); Eap = 1.0 V | 6.3 | 63 |
| Swine wastewater | 12000–17000 mg L−1 COD |
Diluted swine wastewater (2000 mg L−1 COD) used to start up the cell in MFC mode | Single-chamber MEC with graphite fiber brush anode; Eap = 0.5 V | 0.9–1.0 | 56 |
MECs are also an effective method for hydrogen recovery from swine wastewater treatment, although the process needs to be further evaluated for reducing methane production, increasing the efficiency of converting the organic matter into current, and increasing recovery of hydrogen gas produced at the cathode.56 These authors reported treatment efficiencies in MEC tests with swine wastewater ranging from 19 to 72% based on COD reduction. Further, the coulombic efficiency (CE) was also low indicating that a large percentage of electrons were not successfully transferred into current. Glycerol, which is now being produced in abundance as a byproduct of biodiesel production, has also been tried as a substrate in MECs64 though a higher applied voltage (0.9 V) than that typically used for acetate (0.5 V) was needed for consistent electrolysis operation and methane reduction.59 Very recently, the performance of a pilot-scale (1000 L) continuous flow MEC fed with winery wastewater was reported.65 Peak reactor performance was 7.4 A m−3, or 0.41 A m−2 based on the cathode surface area (18.1 m²/m³) which was 44% less than that estimated from the laboratory set up.
3.3 Enzymatic biofuel cell (EFC)
EFCs employ enzymes as catalysts for anodic and/or cathodic processes, and use biofuels that are already available in nature like sugars and alcohols.66 In comparison to MFCs, EFCs typically possess orders of magnitude higher power densities (although still lower than conventional fuel cells), but can only partially oxidize the fuel and have limited lifetimes (typically 7–10 days) owing to the fragile nature of the enzyme.67 Though in recent years, this life time has been extended by use of novel polymers to immobilize and stabilize enzymes, significantly extend enzyme operating lifetimes.68 A system employing surface display technique whereby microorganisms at the anode surface display redox enzymes that are used as catalysts for the oxidation of glucose was also demonstrated for increasing the operational time of EFC.69 Further, enzymes are much more specific thus eliminating the need for a membrane separator.70 The use of single enzyme (or enzyme cascades) allows to have defined reaction pathways on the electrode surface as well as to overcome the limited output performance of microbial biofuel cells, which is considered to be due to mass transfer resistances across the cell membranes.71Redox enzymes (also known as oxidoreductases) are extensively used to construct amperometric enzyme electrodes. They usually lack direct electron transfer communication between their active redox centres and electrode support.72 For biological cathodes, the main enzymes employed are the multi-copper oxidases, which are capable of a four-electron reduction of oxygen to water and have a high specificity for this reaction.73 Current enzymatic biofuel cells have low efficiency, as only a single type of enzyme is employed and can only partially oxidize the fuel. This is in direct contrast to living cells that can completely oxidize biofuels (e.g. ethanol, lactate and glucose) to carbon dioxide and water.70 In recent years, immobilization of enzymes on electrode surfaces have led to improvement in the performance of these systems by way of increased selectivity, improved mass transfer and long-term stability. The various immobilization strategies such as physical adsorption, entrapment in conducting polymers and nanostructured electrodes have been described previously.22,66
4. Electron transfer mechanisms in BES
The electron transfer mechanisms so far observed in BESs resemble closely to the mechanisms investigated for dissimilatory metal reducing microorganisms. So far, 3 main possible strategies have been identified for facilitating electron transfer. These are 1) direct electron transfer (DET) involving proteins located on cell surfaces, 2) mediated electron transfer (MET) through use of small, redox reactive molecules that ‘shuttle’ electrons from bacteria to the electrode surfaces by a diffusion-limited process and 3) electrically conductive appendages known as microbial or bacterial nanowires.74 Yet, the mechanism of electron transfer between microbes and electrodes, which could ultimately limit power extraction remain controversial.75 The several proposed electron transfer mechanisms in BESs have been illustrated in Fig. 1. The established models include indirect electron transfer by externally added mediators or self-produced mediators and direct electron transfer by a single outer membrane cytochrome and/or by ‘nanowires. The other proposed models include indirect electron transfer of non-electroactive species achieved by using mediators that are produced by electroactive species and direct electron transfer by a layer of assembled outer membrane cytochromes.76 The most used microorganisms in the MFCs belong to Geobacter, Shewanella, Proteobactor and Pseudomonas families. All these biocatalysts used in both anode and cathode of a BES have been discussed recently.78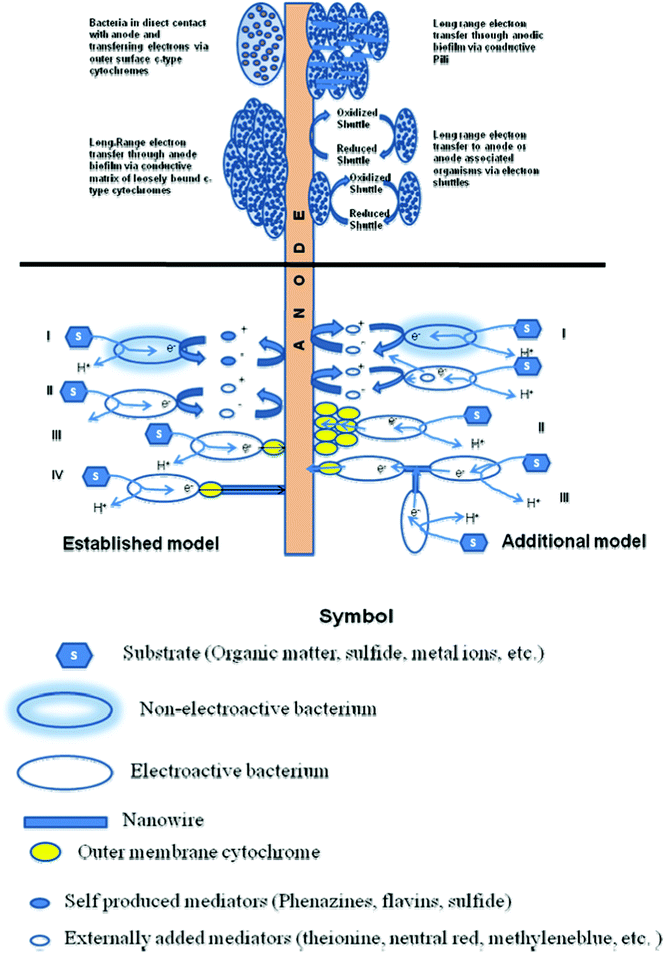 | ||
| Fig. 1 Various proposed electron transfer mechanisms in bioelectrochemical systems and interactions between bacteria and solid electrode. The established model shows indirect electron transfer by (i) external mediators, (ii) self-produced mediators; direct electron transfer by (iii) single outer membrane cytochrome; and (iv) ‘nanowires’. The other proposed models show (i) indirect electron transfer of non-electroactive species achieved by using mediators produced by electroactive species; (ii) direct electron transfer by a layer of assembled outer membrane cytochromes; and (iii) electron transfer from cell to cell through ‘nanowires’. (Adapted from ref. 34, 76 and 77). | ||
Most research concerning the composition, conductivity and roles of bacterial nanowires have focused on those produced by the metal reducing bacteria Geobacter and Shewanella. Nanowires produced by G. sulfurreducens are reported to contain no conventional electron transport proteins, such as cytochromes, and are presumed to be conductive as a result of amino acid sequence and tertiary structure of the type IV pilin protein, PilA.79 Nanowires from S. oneidensis MR-1 are complex assemblages of proteins believed to contain both structural (pilin) and electron transport (multiheme cytochrome) proteins.80 The mechanism of electron transfer in G. sulfurreducens and S. oneidensis have been discussed in detail.77,81,82 In this regard two recent different experiments on S. oneidensis with contrasting findings must be discussed here. In one experiment, researchers measured for the first time electron transport along the wires in S. oneidensis at micrometre distances with electron transport rates up to 109 electrons/s at 100 mV of applied voltage.83S. oneidensis was grown under conditions that promote the production of lots of nanowires, namely by limiting the number of available electron acceptors. Platinum rods were then rested at each end of a nanowire and an external voltage applied leading to a measurable electrical current response. After the nanowire was cut, there was no measurable current response to applied voltage, confirming that the observed conduction path was indeed through the nanowire. Another set of researchers investigated Shewanella's electron transfer with a miniature fuel-cell experiment. An array of gold–titanium composite nanoelectrodes on a glass chip was fashioned, to which a microbial culture was exposed. The access of microbes to the nanoelectrodes was carefully controlled by covering the nanoelectrode array with a 400-nm-thick layer of insulating silicon nitride. They then etched through the insulating layer to expose alternating electrodes with either a grid of holes, each just a few hundred nanometres across, or a single window of 6 × 10 μm. The total exposed area was the same for both types of electrodes, but whereas the windowed electrodes would allow free access to several microbes at a time, the nanoholes would preclude any direct contact between the electrode and the cell membrane. Following addition of Shewanella cells, short-circuit current measurements showed similar amplitude and temporal response for both electrode configurations, while in situ optical imaging demonstrates that the measured currents were uncorrelated with the cell number on the electrodes. Both types of electrodes yielded similar currents at longer times in dense cell layers and exhibited a rapid drop in current upon removal of diffusible mediators thus showing that electron transfer occurs predominantly by mediated mechanism.75 With these developments, it is expected that in the future a better understanding of how microbes transfer electrons could help researchers identify ways to extract stronger currents from them.
In the case of EFCs, the two main electron transfer mechanisms are: (a) direct electron transfer (tunnelling mechanism) from electrode surface to the active site of an enzyme, and (b) electron transfer via redox mediator.66
5. Bioenergy production potential from the global organic waste and wastewater resource
The production of renewable biomass often involves generation of co-products, by-products or wastes. Lignocellulosic biomass is available in massive quantities and provides enormous potential for bioethanol production.84 Together these could potentially constitute a rich source of substrate to be used in BESs. Biomass is the fourth largest energy source after coal, oil and natural gas, and is found common at global scale. It is the most important renewable energy option presently that can be transformed in to different forms of energy. Therefore, it is capable of providing all the energy services required in a modern society.85 The annual global primary production of biomass is equivalent to the 4500 EJ of solar energy captured each year that is equivalent to 10 times of world's present total primary energy demand. The global biomass energy potentials were estimated recently between 200–500 EJ/a for 2050.86
Extra cellulose fuel is always available in the form of crop residue left behind after harvest, and manure is plentiful. Rumen contents, which generally are discarded, are available each time ruminants (sheep, goats, llamas, camels and cattle) are slaughtered. Organic wastes that can be utilized for energy production are mentioned in Fig. 2. While discussing the feedstocks for BES conversions, Hawkes et al.37 mentioned cellulosic feedstocks and chitin as possible candidates as BES substrate. Previously, it has been reported that electricity generation from cellulose is possible in an MFC using a defined coculture of the cellulolytic fermenter Clostridium cellulolyticum and the electrochemically active G. sulfurreducens.87 In fed-batch tests using two-chamber MFCs with ferricyanide as the catholyte, the coculture achieved maximum power densities of 143 mW m−1² (anode area) and 59.2 mW m−1² from 1 g L−1 carboxymethyl cellulose (CMC) and MN301 cellulose, respectively. Neither pure culture alone produced electricity from these substrates. Another approach for utilizing lignocellulosics in BESs is to first convert them to volatile fatty acids (VFAs) such as acetic, formic, succinic, and lactic acids, followed by using these VFAs as substrate in MFCs or electrohydrogenesis to convert into hydrogen gas.57,88
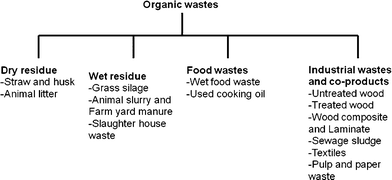 | ||
| Fig. 2 Potential organic wastes suitable for energy production in BES. | ||
In a study, the wastes generated by the Russian agro-industrial complex were estimated and it was reported that Russia generated about 773 million ton waste annually that can be converted to 62.5 billion m3 of biogas, equivalent to 31 billion L of gasoline/diesel, or 106 GWh of electricity and 1 billion GJ of heat.89 This energy is sufficient to become energetically autonomous through a rational utilization of its wastes. Moreover, the electroenergy generated will also be sufficient for supplying electricity to the entire rural population (39 million inhabitants) in the country and also create autonomy for fertilizers. In another study, it was estimated that total, technical and economic potential of bioenergy is 467, 129 and 69 tons coal equivalent/annum, respectively in Russia. The evaluated economic potential of bioenergy only is equivalent to 561 TWh.90
The embedded energy in food wastes in US was estimated on the bases of 2007 data and it was concluded that food wasted in the U.S. represents approximately 2030 trillion BTU of embedded energy, i.e. equivalent to 2142 PJ energy.91 The wasted energy calculated in the study is a conservative estimate both because the food waste data are incomplete and outdated and the energy consumption data for food service and sales are incomplete. The recoverable bioenergy potential in Turkey is estimated to be 17.2 Mtoe based on the recoverable energy potential from the main agricultural residues, livestock farming wastes, forestry, wood processing residues and municipal wastes.92 Switzerland has a sustainable potential of 82 PJ bioenergy production annually from organic residues.93 The energy potential of EU-27 from organic residues are presented in Table 2.
| Feed stock | Energy potential (ktoe/a) | |
|---|---|---|
| 2000 | 2020 | |
| Agricultural Biomass (Solid agricultural residues, wet and dry manure) | 49100 |
59912 |
| Forest biomass (Forest by-products and refined wood fuels) | 42086 |
51352 |
| Industrial biomass (Solid industrial residues, black liquor, sewage sludges) | 25650 |
31302 |
| Waste Biomass (Biodegradable municipal waste, demolition wood) | 18029 |
43324 |
| Total |
134![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) 865 865
|
185890
|
Increasing scarcity of freshwater resources and growing environmental awareness give rise to the use of reclaimed wastewater as an additional source of water supply.95 BESs are one of the treatment options for such wastewater that also provide some electricity in addition to pollutant removal. The consumption of fresh water by domestic usage takes up to 70–80% of the total volume of wastewater globally.96,97 The global wastewater production is increasing due to increase in population, industrialization and urbanization. The wastewater can be used for energy production using anaerobic digestion, algal biomass cultivation, BES, biohydrogen production, etc.35,98,99,100 Many species of microalgae are able to effectively grow in wastewater conditions through their ability to utilise abundant organic carbon and inorganic N and P in the wastewater.101 The algal biomass production using wastewater provides dual benefits at one hand it remove pollutants from wastewater and at another hand provide biomass for energy production or as food.
The Lawrence Berkeley National Laboratory estimated that the energy potential in municipal wastewater, in the United States, was equivalent to generating 7.2 billion kilowatt hours of electricity, annually in 2005.102 In a study Meggers and Leibundgut103 concluded that there is great potential in higher temperature extraction from wastewater (especially industrial which have high temperature) when the recovery is combined with a low exergy system that incorporates a high performance, low temperature-lift heat pump.
6. Recalcitrant pollutant degradation in BES
Apart from their role in electric power generation in MFCs and hydrogen production in MECs, BESs have also been used in various forms for the treatment of certain recalcitrant pollutants. These include industrial wastewaters (such as from breweries, paper, municipal, food, and animal wastewaters).35,104 Some of the representative examples of such treatment process are discussed below.6.1 Dye decolorization and removal
Dyes are widely used in different industries, especially in the textile manufacturing. The treatment of effluents containing dyes is indispensable due to their toxicity, carcinogenic impact and pollution effect on environment.105 In recent years, several studies have focused on the treatment of dye containing wastewaters in the BESs. Simultaneous azo dye degradation and bioelectricity generation utilizing a single chamber microbial fuel cell have been investigated.106 Effective decolorization of a metal azo dye at the cathode of a BES was achieved while oxidizing acetate in the anode.107 Yet another study concentrated on reduction of azo dyes in cathode of the MFC harnessing electrons produced from metabolic oxidation of Klebsiella pneumoniae strain L17 in the anode.108 Similarly, MFC systems have been used for the decolorization of Congo red by providing various co-substrates such as acetate.109 MFCs operated at higher power densities could simultaneously increase COD removal efficiency as well as the rate of dye decolorization, even though bioelectricity generation seemed to be competitive to dye decolorization. Glucose, acetate sodium and ethanol have also been used as energy substrates for simultaneous decolorization and bioelectricity generation.110 It was reported that >98% of Congo red could be decolorized in 36 h using a proton exchange membrane (PEM) air-cathode single-chamber MFC.Table 3 shows the various dyes and other colorants that have been treated in BESs for decolorization of the wastewater.
| Type of dye/colorant | Type of BES used | Decolorization achieved | Remark | Reference |
|---|---|---|---|---|
| Active brilliant red X-3B (Azo dye) | Air-cathode single-chamber MFC with glucose as co-substrate | 90% (initial dye concentration of 100 mg L−1) 77% (initial dye concentration of 1500 mg L−1) | Electricity generation in the MFCs was affected by dye reduction due to the competition between the anode and the azo dye for electrons from carbon sources | 106 |
| Acid orange 7 (Azo dye) | Two-chamber BES with acetate oxidation at anode and dye decolorization at cathode | 78.7% (0.19 mM original dye concentration) 35% (0.7 mM original dye concentration) | Cathodic decolorization increased on controlling the cathodic potential in the range of −350 to −550 mV vs. standard hydrogen electrode (SHE) | 107 |
| Methyl orange (Azo dye) | Two-chamber MFC with glucose oxidation at anode by Klebsiella pneumoniae and dye decolorization at cathode | 100% (0.05 mM of original MO solution) in 3 H | Controlling the redox potentials of pollutant-containing catholyte is important for the control of power output as well as the degradation rate | 108 |
| Congo Red (Azo dye) | Two-chamber MFC with a loop to convey the effluent of anode to the cathode. Cell operated initially with glucose (1 g L−1) and later added with artificial wastewater containing 100 mg L−1 of dye | 69.3% (with original glucose concentration of 100 mg L−1) 92.7% (with original glucose concentration of 4000 mg L−1) | Recovering electricity during a sequential aerobic–anaerobic azo dye treatment process enhanced chemical oxygen demand removal and did not decrease azo dye removal | 109 |
| C.I. reactive blue 160 (Azo dye) | Single chamber air-cathode mediator-less, membrane-less MFCs (ML-MFCs) with activated carbon cloth as anode and hydrophobic carbon cloth as cathode | 97.2% (specific decolorization rate of 14.62 which was significantly higher than other azo-free, mono-azo and triazo dyes tested) | Chemical structures of azo dyes significantly affected the performance of dye decolorization | 111 |
6.2 Organochlorine removal
Chlorinated solvents or chlorinated aliphatic hydrocarbons (CAH) such as trichloroethylene (TCE) and 1,2-dichloroethane (1,2-DCA) are used as industrial solvents and degreasing agents that enter and contaminate the soil and groundwater through leakage from storage tanks and poor storage and disposal practices.112 Recently, BESs, in which solid-state (e.g., graphite based) electrodes are employed as direct electron donors (in place of organic electron donors) in the reductive dechlorination of chlorinated solvents, have been proposed.113 Development of bioelectrochemical remediation technologies for TCE, have revealed that certain dechlorinating bacteria are capable of “picking” electrons from the surface of potentiostatically controlled electrodes and using them to metabolically reduce CAHs.114,115 The degradation of 1,2-DCA by anodophilic bacteria enriched in MFCs at the rate of up to 102 mg per litre reactor volume per day has been demonstrated.116 Further, energy released from this degradation could be partially recovered (up to 43%) as electricity. The reduction current resulting from the microbial reductive dechlorination process could be continuously measured with a potentiostat. Recently, it was reported that the use of redox mediators such as the humic acid analogue anthraquinone-2,6-disulfonate (AQDS) was a key to achieving fast and highly selective electron transfer between the dechlorinating bacteria and the electrode surface.1176.3 Leachate treatment
Leachates are discharges from landfills with high concentration of complex organic matters [e.g., chemical oxygen demand (COD) of 5000–20000 mg L−1] and ammonium-nitrogen (e.g., 3000–5000 mg L−1), distinguishing it from municipal wastewater, which may cause some serious problems such as contaminations to ambient ground water, eutrophication of water bodies or odor release.118 The main problem with leachate treatment is that the ammonium concentrations at these levels persist for many years after a landfill has closed and levels of other pollutants, such as COD and biological oxygen demand (BOD), have long since dropped. Leachate is considered a well-matched substrate for use in a MFC because of its relatively high amount of organics, conductivity, and buffering capacity, yet minimal solids.119 An attempt to treat landfill leachate in bio fuel cell for COD removal was made as early as 1991.120 The results obtained in different studies on leachate treatment in BES are shown in Table 4. The greater power density obtained in one of the studies118 can be attributed to the smaller scale MFC and the amended leachate substrate.
| Type of leachate | Type of BES used | Treatment efficiency | Remark | Reference |
|---|---|---|---|---|
| Landfill leachate (9810 mg L−1) | Both dual chamber and single chamber MFC | 69.54%–98% COD removal depending on initial COD | Maximum power density of 2060 mW m−3 for dual-chamber MFC and 6817 mW m−3 for single chamber MFC were obtained | 118 |
| Landfill leachate (908–3200 mg L−1 COD) | Three designs, a square (995 mL), circle (934 mL) and a large scale MFC (18.3 L) using graphite as anode) and carbon cloth with Pt catalyst as cathode | BOD, TOC, and Ammonia were removed at 50–72%, 17–53%, and 7–69%, respectively | Leachate was used as both the substrate and inoculum and no additional anaerobic bacteria or nutrient were added. Maximum power density of 669–844 mW m−3 for circular MFC was obtained. | 119 |
| Landfill leachate (6000 mg L−1) | Two-chambered tubular MFC with carbon veil electrode (360 cm²) | MFC columns showed low BOD5 removal (< 34%BOD5) as compared to biological aerated filter (66%). | A maximum current density of 0.0004 mA cm−1² was obtained and power density increased linearly with the leachate strength | 121 |
| Landfill leachate (7050 mg L−1) | Two-chambered tubular MFC with carbon veil electrode (1080 cm²) | 79.4% of the COD and 81.6% of the BOD5 removed after 4 days of continuous recirculation of effluent | Increasing the electrode surface area led to increase in power output | 122 |
| Landfill leachate (3480 mg L−1 COD) | An air-cathode MFC with graphite granules as anode and carbon cloth with Pt catalyst as cathode with net anodic volume of 167 mL | Up to 8.5 kgCODm−3 d−1 of biodegradable organic matter was removed at the same time as electricity (344 mWm−3) was produced. | High free ammonia concentrations inhibited the activity of nitrifier microorganisms | 123 |
6.4 Sulfide removal
Apart from organics, wastewaters often contain inorganic matters, such as sulfide. Sulfide is a hazardous substance that needs to be removed from wastewater before discharge into the environment. Sulfides can function as a mobile carrier of electrons from bacteria to electron acceptors such as Fe(III) (hydr)oxides.124 The multivalence states of sulfur, coupled with their facile interconversion and the multiplicity of sulfur compounds, have made sulfide oxidation in MFCs very complex and diverse.125The MFC system has been found to be effective for simultaneous sulfide removal and electricity generation.126 The sulfide oxidation in the anodic compartment resulted in electricity generation with power outputs up to 47 W m−3 total anode compartment. Also by controlling the anode potential, the corresponding efflux of sulfide was decreased. Later it was shown that the microbe-assisted sulfide oxidation generated a higher persistent current density than the sulfide oxidation via single electrochemical reactions only.125 SO42−, S2O32−, polythionates, S0, Sx2−, and sulfide (H2S/HS−/S2−) were the potential sulfur compounds present in the anode and microbe-assisted production of S2O32− and SO42− resulted in a persistent current (115 mA m−2) from the MFC. Further elucidation of the microbial diversity in a sulfide-fed MFC anode showed the presence of exoelectrogenic bacteria in both on the anode and in the solution. The sulfur-oxidizing bacteria were present in greater abundance on the anode (dominant genera Pseudomonas and Acinetobacter) than in the solution, while the sulfate-reducing bacteria preferably lived in the solution (dominant genera Comamonas and Acinetobacter).127 Synergistic association between the anode-attached and planktonic bacteria was proposed to play an important role in the electricity generation from the sulfide oxidation process in the MFC. In another study, 91% and 86% sulfite and thiosulfate removal conversions respectively, were reported using a pure culture of Desulfovibrio desulfuricans.128 At an anode open circuit potential of −0.24 V vs. Ag/AgCl reference, the sulfide was rapidly oxidized at the anode, causing a sharp decrease in its concentration, allowing sulfite and thiosulfate to be continuously biologically reduced and to be finally removed from the wastewater.
7. Product formation and recovery in BES (Microbial electrosynthesis)
It has been known for quite some time that BESs can have applications other than wastewater treatment and electric power generation.76,129 Recently, using life cycle assessment it was shown that a MEC provides significant environmental benefits over MFCs through the displacement of chemical production by conventional means.130 The term ‘microbial electrosynthesis’ was coined in 2010 for the reduction of carbon dioxide to multicarbon compounds with electrons donated from an electrode as the electron donor.131 It was shown that showed that biofilms of Sporomusa ovata growing on graphite cathode surfaces consumed electrons with the reduction of carbon dioxide to acetate and small amounts of 2-oxobutyrate. This field addresses the use of microorganisms as catalysts on cathodes (i.e. biocathodes) to achieve electricity-driven synthesis of chemicals and fuels.132 Some of the products that have been explored in BES are described below.7.1 Methane
Initially, the production of methane in the cathode of a MEC was considered as a nuisance and it is only recently that electrochemical production of methane at cathode of a MEC is being considered as an attractive option.129 The conversion of acetate to hydrogen is thermodynamically unfavorable under standard conditions (+13.8 kJ mol−1 electrons or 0.48 kWh kg−1 COD) and therefore an additional voltage of at least 0.14 V needs to be applied to the microbial electrolysis cell. In practice, at least 0.20 V needs to be applied to start the current production.49 Provided that the electron donor is ‘free’ (in form of organic compounds in wastewater) and the applied electrical energy is lower than the specific energy content of the produced product, a positive energy balance can theoretically be obtained in a MEC. The specific energy content of hydrogen and methane, based on the change in Gibbs free energy, is 4.12 kWh kg−1 COD equivalents (or −119 kJ mol−1 electrons) and 3.52 kWh kg−1 COD equivalents (or −101 kJ mol−1 electrons) respectively, while the electrical energy demand is 3.35 kWh kg−1 COD per Volt applied implying that hydrogen and methane production becomes energetically unfavorable at applied voltages higher than 1.23 and 1.05 V respectively.133As far as to the specific energy content is concerned, hydrogen production is preferred over methane production, because methanogenic conversion of hydrogen to methane results in a specific thermodynamic energy loss of approximately 15%. However, due to unavoidable methane production, at present hydrogen cannot be produced as high grade pure hydrogen in MECs, which makes it not applicable as a chemical for some purposes. Hydrogen purification might be energy intensive, thus increasing its energy and production costs. Since high membrane costs, high ohmic cell resistances and unsustainable pH operation can easily be avoided by removing the ion selective membrane in MECs, several researchers have focused on the operation of membraneless MECs.56,60,63 A dual-chamber MEC using a membrane to separate the anode from the cathode can present a unique concentration loss due to [H+] or [OH−] accumulation in a chamber, since they are net produced at half reactions on the electrodes.134 The high concentrations of other ions in the liquid supplied to an MEC (e.g., Na+), compared to [H+] or [OH−], means that charge neutrality can be achieved with little transport of H+ or OH− ions through membrane, and a strong pH gradient can develop across the membrane, causing a substantial concentration energy loss.135 In all these cases, the presence of methane could not be avoided. Methanogens in cathodic biofilms might be protected from oxygen, high proton concentrations and wash-out due to short hydraulic retention times.
The advantage of methane is that it can easily be stored or transported. Compression, transport in pipes and storage involves mature technologies and could rapidly be integrated into an existing infrastructure.136 Methane producing MECs have been suggested as an energy friendly effluent polishing step for digester effluents, most likely entailing low sludge production rates and no aeration costs.137 Production of methane by reduction of carbon dioxide at the biocathode of a MEC with a pure culture of Methanobacterium palustre through electromethanogenesis have already been shown.136 Though it was suggested that there is a possibility of direct electron transfer to methanogens, it needs to be conclusively proven.138 Previously, methane production in MEC have been reported from acetate via acetoclastic methanogenesis and hydrogenotrophic methanogensis using hydrogen gas produced in the process.63,139 The disadvantage of methane production at moderate temperatures is the higher methane solubility (approximately 25–50% higher versus mesophilic conditions, depending on the salinity). The discharge of methane from the effluent into the environment needs to be avoided. Also for methane production at higher temperatures, there is a need for methane removal from digesters effluents. Table 5 shows the methane production in BESs as reported in literature.
| Type of BES used | Methane recovered | Remark | Reference |
|---|---|---|---|
| Single- and two-chamber MEC containing a single graphite fiber brush anode and several carbon cloth cathodes | Methane was produced at an overall energy efficiency of 80% at a set voltage of–1 V, | Carbon dioxide was reduced to methane electromethanogenesis even with an abiotic anode with cathode covered by an archaea, Methanobacterium palustre | 136 |
| Membraneless MECs with high specific surface area granular graphite electrodes | 0.33 ± 0.07 L CH4 L−1 MEC day−1 (65% of the acetate removed) | Maximum current density of 223 A m−3 MEC were obtained at an applied cell voltage of -0.8 V; methane production was possible even in acidified, carbonate-limited continuous systems | 137 |
| Single chamber MEC with graphite fiber brush anode and carbon cloth with Pt catalyst as cathode | 28% of the gas at an applied voltage of 0.2 V and a longer retention time | Maximum current density of 292 A m−3 MEC for 28 mL MECs and hydrogen was the main product (87%) | 140 |
| Single chamber MECs graphite fiber brush anode and carbon cloth with Pt catalyst as cathode | 3.1% at an applied voltage of 0.7 V with Methane production being higher than H2 gas production at an applied voltage of 0.4 V | Methane production primarily occurred in the latter part of reaction cycle when H2 gas concentrations were high, and not in the beginning when acetate concentrations were highest | 141 |
7.2 Ethanol
Biological acetate reduction with hydrogen is a potential method to convert wet biomass waste into ethanol. Recently, the reduction of acetate to ethanol with methyl viologen (MV) as mediator in the cathode compartment of a BES was demonstrated.142 Ethanol production had a CE of 49% and apart from ethanol, hydrogen, n-butyrate, and the non-reversible reduced MV2+ were produced in the cathode. MV inhibited side reactions such as methanogenesis and enhanced ethanol production, however, it was depleted rapidly owing to irreversible reduction at the cathode, and in its absence high yields of butyrate (an undesired end product) were found and methanogenesis started. Previously, these authors demonstrated the reduction of butyrate to butanol using H2 at low overall alcohol yields.143 It has been suggested that if this could be achieved effectively in the earlier mentioned set-up converting acetate to ethanol, then the butyrate formation could lead to butanol as a more attractive end product.12 Also, in order to improve the ethanol production process in a BES, further research should focus on non-mediated reduction of acetate at the cathode itself by growing microorganisms at the electrode or on immobilization of methyl viologen on the electrode.114,1427.3 Hydrogen peroxide
The production of hydrogen peroxide (H2O2), an important industrial chemical in BES was reported based on the bioelectrochemical oxidation of wastewater organics at an anode coupled to the cathodic reduction of oxygen to H2O2.45 At an applied voltage of 0.5 V, the system was capable of producing approximately 1.9 kg H2O2/m3 day−1 from acetate at an overall efficiency of 83.1%. As most of the required energy was derived from the acetate, the system had a low energy requirement of around 0.93 kWh/kg H2O2. However, H2O2 was produced in very low concentrations (0.13%) making any useful recovery very difficult.Besides the aforementioned applications of BESs as a tool for bioremediation and product synthesis, they can also be used as biosensors. In fact in last decade, several researchers have reported the development of BES based biosensors.144,145
An overall microbial biorefinery concept based on BESs with different potential reactions occurring at the anode and cathode are shown in Fig. 3. Though most of the processes depicted in this scheme are proven at lab-scale, a detailed economic and cost benefit assessment is yet to be done.
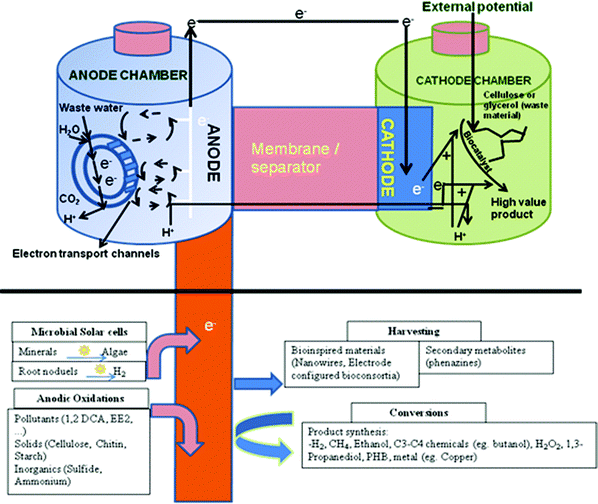 | ||
| Fig. 3 A microbial biorefinery concept involving a bioelectrochemical system based on different possible reactions at the anode and cathode for energy production, bioremediation and/or high-value product synthesis (Adapted from ref. 12 and 76). | ||
8. Current bottlenecks and challenges for BES
The progress in the field of BESs in terms material and engineering science and microbial and biotechnological perspective have gone hand in hand. While the advances in design engineering have yielded higher power output and increased efficiency, there is increased understanding of the components and mechanisms involved in electron transfer from bacteria to the electrode surfaces. Though a consensus is yet to emerge on the final electron transfer mechanism, researchers are convinced of a breakthrough soon.Right from the beginning, the main limitation in bio fuel cells is the low power densities, the power generated per unit electrode surface area, due to several major limitations such as slow transport across cellular membranes.146 Other major losses associated with these systems are ohmic voltage losses (caused due to resistance to charge transport and including both ionic and electronic resistances), activation overpotentials (caused due to energy barriers to charge transfer from bacteria to electrode), concentration overpotentials (caused by resistance to mass transport) and finally the coulombic losses. Coulombic losses are defined as the ratio of coulombs transferred from the substrate to the anode to the maximum coulombs produced theoretically from the complete oxidation of substrate (×100) and is caused due to biomass build up, occurrence of side-reactions not contributing to current production and crossover of substrate from cathode to anode and vice versa. All of them have been described in detail earlier.22,147 pH issues, a high ohmic cell resistance and high overpotentials are the factors that prevent the industrial implementation of BESs. Environmental factors like oxidant and proton flux towards the biocathodes are of major importance in the development of well performing biocathodes.
Another important challenge pertaining to these systems is related to up-scaling. There is a general recognition that the issue of scale-up is an important and difficult barrier,148 and at present few plausible options for efficient and economic increase in scale exist. The large scale reactors need to achieve at least a similar performance as bench-scale reactors nowadays, while the production costs need to be economically and environmentally feasible. It has been suggested that for energizing real world applications, a plurality of MFC units must be employed as a stack. However, operating biocatalyzed reactions in a stacked configuration is extremely vulnerable to cell reversal. Moreover, when both the anode and the cathode would be biologically catalyzed, a stacked operation will be challenging. Unfortunately all these strategies are believed to further increase the reactor costs and up-scaling BESs might be a long term quest. Previously, it has been reported that the maximum power density generated by an MFC is not directly proportional to the surface area of the anode, but is instead proportional to the logarithm of the surface area of the anode.149 In other words, in MFCs power density decreases with increasing surface area of the current-limiting electrode and that when scaling up these systems, it cannot be assumed that power density will remain constant with the increased electrode surface area. However, it was later reported that enlarging surface area of electrode increases the total reaction rate, hence increases the amount of collected current.150 Later, it was demonstrated that in a single chamber MFC, with anode made of a packed bed of irregular graphite granules, the current output was found to increase with increase in thickness of the anode bed and with the approximate anode area. However, scaling up from a flat sheet to a higher surface area packed bed did not produce a corresponding increase in current due to issues of current distribution and also mass transport limitations.151 Fornero et al. recently discussed some of the challenges associated with the reactor scale up for MFCs. They suggested three main challenges while scaling up the MFC reactors. These include maintaining low internal resistance while increasing the levels of electrochemically-active biomass, optimization of reactor design and developing newer ways of separating anode from cathode.104
Besides the above mentioned limitations, a common and most frequently mentioned challenge with BESs is the comparison of results reported, as sometimes key experimental parameters are not provided or critical comparative measurements of electrical output are not reported.11 Besides this, a wide variety of designs ranging from two-chambered to single chamber, mediator or without mediator, membrane or membrane-less makes a comparison difficult. Apart from the design itself, a range of materials used such as electrodes ranging from graphite foil, rods, granules, and fibre brush, carbon paper, cloth, felt, and foam, activated carbon cloth, reticulated vitreous carbon, electrodes modified with conductive polymers, and metals such as aluminum, nickel or stainless steel makes it practically difficult to compare the performances of the set ups used by researchers across the globe.152 Several approaches have been described through which these drawbacks can be overcome. Some of these include background experiments to identify and clarify the electrochemical reaction mechanisms, the effects of the electrode materials, biofilm, substrate and metabolites, experiments to measure reproducibility and repeatability, inclusion of a reference electrode and evaluation of the surface chemistry of the electrode material from different suppliers.152
In recent years, it has been proposed that the growth in power densities in terms of biocatalyst has hit a plateau and the next big growth will come from improved materials used in these systems.12 This includes improved electrodes for anode and cathodes,153–155 separators156,157 and newer designs of the cells.158 The role of new materials in developing next generation of bioelectrochemical systems was recently discussed by Logan.148 The recent progress of anode/cathode materials and filling materials as three-dimensional electrodes for MFCs was also reviewed recently.159 It demonstrated that different electrodes exhibited different behaviors and electrode modification proved to be a good alternative for enhancing the performance of MFCs.
The use of electrodes with precious metal catalyst and a membrane as separator have been identified as the most expensive components of a BES.160 It is known that Pt is the most commonly used catalyst on the cathode, but its high cost prohibits its use for commercial MFC applications.161 With improvements in designs and development of novel and cheaper materials, the costs associated with these systems is also expected to go down. Already some figures have been mentioned36,160 for MFCs for electric power production from wastewaters which are expected to be even better for MECs if hydrogen production is taken into account.130 Based on the LCA study, Foley et al. suggested that for MFCs to be commercially viable and environmentally competitive with existing anaerobic treatment technology, their performance definitely needs to exceed 500 W m−3. Though it has been suggested that present bioelectrochemical reactors are cost intensive due to the need for electrode materials, current collectors, membranes, etc., the advantage of microbial electrosynthesis lies in the on-site use of electricity for bioproduction and its independence from arable land availability.162
9. Future outlook
Electricity recovery from wastewater remains an attractive option because it provides the possibility of decreasing overall treatment costs while reducing the production of biomass. From the perspective of electric current and power production, the exploration of novel materials and cell components is becoming more important as attractive price and superior performance will greatly expand the applicability of MFCs. In addition to the benefit of providing sustainable and logistically easily accessible fuels with high energy density, BESs can be built for portable applications. While the initial research focus has been the development of MFCs with bioanodes, the research field of BESs is rapidly expanding due to the interesting developments in the fields of biocathodes and MXCs as well. The critical factors for bringing BESs to a commercial level are the pH issues, the high ohmic resistance and the high overpotentials. For wastewater treatment, the integration of MFCs with the present treatment technologies seems to be more realistic, cost-efficient and feasible. The recent emergence of microbial electrosynthesis provides an alternative option for sustainable production via bioelectrochemical route by either extracting from or supplying electric current to microorganisms in order to stimulate chemical production.Acknowledgements
The research on bioelectrochemical systems at VITO is supported by a grant from VITO's strategic research fund.References
- M. C. Potter, Proc. R. Soc. London, Ser. B, 1911, 84, 260–276 CrossRef.
- M. C. Potter, Proc. Univ. Durham Phil. Soc., 1910, 3, 245–249 Search PubMed.
- M. C. Potter, Proc. R. Soc. London, Ser. A, 1915, 91, 465–480 CrossRef CAS.
- M. C. Potter, Zentralbl. F. Bakt., Paras u. Infekt. (II), 1929, 78, 56–65 Search PubMed.
- B. Cohen, J. Bacteriol., 1931, 21, 18–19 CAS.
- D. Sell, Biotechnology, vol. 10 Special Processes, 2001, 5–10, Wiley VCH Search PubMed.
- J. C. Biffinger and B. R. Ringeisen, Recent Pat. Biotechnol., 2008, 2, 150–155 CrossRef CAS.
- B. H. Kim, H. J. Kim, M. S. Hyun and D. H. Park, Shewanella putrefaciens, J. Microbiol.Biotechnol., 1999, 9, 127–131 CAS.
- D. H. Park and J. G. Zeikus, Biotechnol. Bioeng., 2003, 81, 348–355 CrossRef CAS.
- D. Xing, Y. Zuo, S. Cheng, J. M. Regan and B. E. Logan, Environ. Sci. Technol., 2008, 42, 4146–4151 CrossRef CAS.
- K. Noll, In: Fuel cell technology: Reaching towards commercialization, Ed. N. Sammes, 2006, 277–296 Search PubMed.
- K. Rabaey and R. A. Rozendal, Nat. Rev. Microbiol., 2010, 8, 706–716 CrossRef CAS.
- N. S. Lewis, MRS Bull., 2007, 32, 808–820 CrossRef.
- J. H. Brown, W. R. Burnside, A. D. Davidson, J. P. DeLong, W. C. Dunn, M. J. Hamilton, N. Mercado-Silva, J. C. Nekola, J. G. Okie, W. H. Woodruff and W. Zuo, BioScience, 2011, 61, 19–26 CrossRef.
- M. Winter and R. J. Brood, Chem. Rev., 2004, 104, 4245–4269 CrossRef CAS.
- H. Liu, In: Anaerobic Biotechnology for Bioenergy Production: Principles and Applications (ed. S. K. Khanal), Wiley-Blackwell, Oxford, UK., 2008, 221–246 Search PubMed.
- C. Gallert, A. Henning and J. Winter, Water Res., 2003, 37, 1433–1441 CrossRef CAS.
- C. Li and H. H. P. Fang, Environ. Sci. Technol., 2007, 37, 1–39 CrossRef CAS.
- B. E. Logan, Environ. Sci. Technol., 2004, 38, 160A–167A CrossRef CAS.
- D. P. Lies, M. E. Hernandez, A. Kappler, R. E. Mielke, J. A. Gralnick and D. K. Newman, Appl. Environ. Microbiol., 2005, 71, 4414–4426 CrossRef CAS.
- J. C. Biffinger, J. Pietron, R. Ray, B. Little and B. R. Ringeisen, Biosens. Bioelectron., 2007, 22, 1672–1679 CrossRef CAS.
- M. H. Osman, A. A. Shah and F. C. Walsh, Biosens. Bioelectron., 2011, 26, 3087–3102 CrossRef CAS.
- M. Rosenbaum, Z. He and L.T. Angenent, Curr. Opin. Biotechnol., 2010, 21, 259–264 CrossRef CAS.
- D. P. B. T. B. Strik, R. A. Timmers, M. Helder, K. J. J. Steinbusch, H. V. M. Hamelers and C. J. N. Buisman, Trends Biotechnol., 2011, 29, 41–49 CrossRef CAS.
- X. Cao, X. Huang, P. Liang, K. Xiao, Y. Zhou, X. Zhang and B. E. Logan, Environ. Sci. Technol., 2009, 43, 7148–7152 CrossRef CAS.
- M. Mehanna, P. D. Kiely, D. F. Call and B. E. Logan, Environ. Sci. Technol., 2010, 44, 9578–9583 CrossRef CAS.
- K. S. Jacobson, D. M. Drew and Z. He, Bioresour. Technol., 2011, 102, 376–380 CrossRef CAS.
- X. Chen, X. Xia, P. Liang, X. Cao, H. Sun and X. Huang, Environ. Sci. Technol., 2011, 45, 2465–2470 CrossRef CAS.
- F. Harnisch and U. Schröder, Chem. Soc. Rev., 2010, 39, 4433–4448 RSC.
- A.K. Marcus, C.I. Torres and B.E. Rittmann, Bioresour. Technol., 2011, 102, 253–262 CrossRef CAS.
- B. Erable, L. Etcheverry and A. Bergel, Biofouling, 2011, 27, 319–326 CrossRef CAS.
- K. Rabaey, L. T. Angenent, U. Schröder, J. Keller, London: IWA Publishing, 2009, pp. 524.
- D. R. Lovley, Nat. Rev. Microbiol., 2006, 4, 497–508 CrossRef CAS.
- D. R. Lovley, Curr. Opin. Biotechnol., 2008, 19, 564–571 CrossRef CAS.
- D. Pant, G. Van Bogaert, L. Diels and K. Vanbroekhoven, Bioresour. Technol., 2010, 101, 1533–1543 CrossRef CAS.
- D. Pant, A. Singh, G. Van Bogaert, Y. Alvarez Gallego, L. Diels and K. Vanbroekhoven, Renewable Sustainable Energy Rev., 2011, 15, 1305–1313 CrossRef CAS.
- F. R. Hawkes, J. R. Kim and G. Premier, in: Bioelectrochemical systems: From extracellular electron transfer to biotechnological application, K. Rabaey, L. T. Angenent, U. Schroder and J. Keller (editors), Integrated Environmental Technology Series, 2010, London, New York: IWA Publishing Search PubMed.
- I. S. Kim, K. J. Chae, M. J. Choi and W. Verstraete, Environ. Eng. Res., 2008, 13, 51–65 CrossRef.
- B. E. Logan, B. Hamelers, R. Rozendal, U. Schrorder, J. Keller, S. Freguia, P. Aelterman, W. Verstraete and K. Rabaey, Environ. Sci. Technol., 2006, 40, 5181–5192 CrossRef CAS.
- M. H. Osman, A. A. Shah and F. C. Walsh, Biosens. Bioelectron., 2010, 26, 953–963 CrossRef CAS.
- B. E. Logan, Microbial Fuel Cells. John Wiley and Sons, Inc., Hoboken, New Jersey. 2008, pp. 200 Search PubMed.
- J. R. Kim, G. C. Premier, F. R. Hawkes, J. Rodriguez, R. M. Dinsdale and A. J. Guwy, Bioresour. Technol., 2010, 101, 1190–1198 CrossRef CAS.
- S. Ke, Z. Shi and H. H. P. Fang, Int. J. Environ. Pollut., 2005, 23, 65–80 CrossRef CAS.
- Y. Kadra and G. Sibioni, 17th World energy Congress held in Houston, Texas, USA, September 1998. http://87.224.35.44/tech_papers/17th_congress/2_3_09.asp (Last accessed on 19 April, 2010) Search PubMed.
- R. A. Rozendal, E. Leone, J. Keller and K. Rabaey, Electrochem. Commun., 2009, 11, 1752–1755 CrossRef CAS.
- K. Rabaey, S. Bützer, S. Brown, J. Keller and R. A. Rozendal, Environ. Sci. Technol., 2010, 44, 4315–4321 CrossRef CAS.
- D. R. Lovley, Microbe, 2006, 1, 323–329 Search PubMed.
- B. E. Logan and J. M. Regan, Trends Biotechnol., 2006, 14, 512–518 CAS.
- B. E. Logan, D. Call, S. Cheng, H. V. M. Hamelers, T. H. J. A. Sleutels, A. W. Jeremiasse and R.A. Rozendal, Environ. Sci. Technol., 2008, 42, 8630–8640 CrossRef CAS.
- S. Cheng and B. E. Logan, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 18871–18873 CrossRef CAS.
- H. J. Kim, H. S. Park, M. S. Hyun, I. S. Chang, M. Kim and B. H. Kim, Enzyme Microb. Technol., 2002, 30, 145–152 CrossRef CAS.
- B. E. Logan and S. Grot, US patent application. 2006, 2006.588.022 Search PubMed.
- R. A. Rozendal, H. V. M. Hamelers, G. J. W. Euverink, S. J. Metz and C. J. N. Buisman, Int. J. Hydrogen Energy, 2006, 31, 1632–1640 CrossRef CAS.
- M. Sun, G. -P. Sheng, Z. -X. Mu, X. -W. Liu, Y. -Z. Chen, H. -L. Wang and H. -Q. Yu, J. Power Sources, 2009, 191, 338–343 CrossRef CAS.
- J. Ditzig, H. Liu and B. E. Logan, Int. J. Hydrogen Energy, 2007, 32, 2296–2304 CrossRef CAS.
- R. C. Wagner, J. M. Regan, S. -E. Oh, Y. Zuo and B. E. Logan, Water Res., 2009, 43, 1480–1488 CrossRef CAS.
- E. Lalaurette, S. Thammannagowda, A. Mohagheghi, P. -C. Maness and B. E. Logan, Int. J. Hydrogen Energy, 2009, 34, 6201–6210 CrossRef CAS.
- L. Lu, N. Ren, D. Xing and B. E. Logan, Biosens. Bioelectron., 2009, 24, 3055–3060 CrossRef CAS.
- P. A. Selembo, J. M. Perez, W. A. Lloyd and B. E. Logan, Int. J. Hydrogen Energy, 2009, 34, 5373–5381 CrossRef CAS.
- H. Hu, Y. Fan and H. Liu, Water Res., 2008, 42, 4172–4178 CrossRef CAS.
- R. A. Rozendal, H. V. M. Hamelers, R. J. Molenkamp and C. J. N. Buisman, Water Res., 2007, 41, 1984–1994 CrossRef CAS.
- D. F. Call, M. D. Merrill and B. E. Logan, Environ. Sci. Technol., 2009, 43, 2179–2183 CrossRef CAS.
- B. Tartakovsky, M. -F. Manuel, H. Wang and S. R. Guiot, Int. J. Hydrogen Energy, 2009, 34, 672–677 CrossRef CAS.
- S. Sakai and T. Yagishita, Biotechnol. Bioeng., 2007, 98, 340–348 CrossRef CAS.
- R. D. Cusick, B. Bryan, D. S. Parker, M. D. Merrill, M. Mehanna, P. D. Kiely, G. Liu and B. E. Logan, Appl. Microbiol. Biotechnol., 2011, 89, 2053–2063 CrossRef CAS.
- S. C. Barton, J. Gallaway and P. Atanassov, Chem. Rev., 2004, 104, 4867–4886 CrossRef CAS.
- J. Kim, H. Jia and P. Wang, Biotechnol. Adv., 2006, 24, 296–308 CrossRef CAS.
- W. Gellett, M. Kesmez, J. Schumacher, N. Akers and S. D. Minteer, Electroanalysis, 2010, 22, 727–731 CAS.
- S. Fishilevich, L. Amir, Y. Fridman, A. Aharoni and L. Alfonta, J. Am. Chem. Soc., 2009, 131, 12052–12053 CrossRef CAS.
- S. D. Minteer, B. Y. Liaw and M. J. Cooney, Curr. Opin. Biotechnol., 2007, 18, 228–234 CrossRef CAS.
- I. Ivanov, T. Vidaković-Koch and K. Sundmacher, Energies, 2010, 3, 803–846 CrossRef CAS.
- I. Willner, Y. -M. Yan, B. Willner and R. Tel-Vered, Fuel Cells, 2009, 9, 7–24 CrossRef CAS.
- E. I. Solomon, U. M. Sundaram and T. E. Machonkin, Chem. Rev., 2006, 96, 2563–2606 CrossRef.
- P. Aelterman, K. Rabaey, L. De Schamphelaire, P. Clauwaert, N. Boon and W. Verstraete, In: Bioenergy (ed. J.D. Wall, C.S. Harwood, A.L. Demain). Washington, D.C.ASM Press, 2008, 307–322.
- X. Jiang J. Hu, L. A. Fittzgerald, J. C. Biffinger, P. Xie, B. R. Ringeisen and C. M. Lieber, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 16806–16810 CrossRef.
- T. H. Pham, P. Aelterman and W. Verstraete, Trends Biotechnol., 2009, 27, 168–178 CrossRef CAS.
- B. E. Logan, Nat. Rev. Microbiol., 2009, 7, 375–381 CrossRef CAS.
- V. Sharma and P. P. Kundu, Enzyme Microb. Technol., 2010, 47, 179–188 CrossRef CAS.
- G. Reguera, K. D. McCarthy, T. Mehta, J. S. Nicoll, M. T. Tuominen and D. R. Lovley, Nature, 2005, 435, 1098–1101 CrossRef CAS.
- Y. A. Gorby, S. Yanina, J. S. McLean, K. M. Rosso, D. Moyles, A. Dohnalkova, T. J. Beveridge, I. S. Chang, B. H. Kim, K. S. Kim, D. E. Culley, S. B. Reed, M. F. Romine, D. A. Saffarini, E. A. Hill, L. Shi, D. A. Elias, D. W. Kennedy, G. Pinchuk, K. Watanabe, S. Ishii, B. Logan, K. H. Nealson and J. K. Fredrickson, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 11358–11363 CrossRef CAS.
- V. G. Debabov, Mikrobiologiya, 2008, 77, 149–157 CAS.
- A. E. Franks, N. Malvankar and K. P. Nevin, Biofuels, 2010, 1, 589–604 CrossRef CAS.
- M. Y. El-Naggar, G. Wanger, K. M. Leung, T. D. Yuzvinsky, G. Southam, J. Yang, W. M. Lau, K. H. Nealson and Y. A. Gorby, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 18127–18131 CrossRef CAS.
- A. Singh, D. Pant, N. E. Korres, A. S. Nizami, S. Prasad and J. D. Murphy, Bioresour. Technol., 2010, 101, 5003–5012 CrossRef CAS.
- S. Ladanai and J. Vinterbäck, Report 013, SLU, Institutionen för energi och teknik, Swedish University of Agricultural Sciences, Department of Energy and Technology, 2009, ISSN 1654–9406, Uppsala Search PubMed.
- V. Dornburg, D. van Vuuren, G. van de Ven, H. Langeveld, M. Meeusen, M. Banse, M. van Oorschot, J. Ros, G. J. van den Born, H. Aiking, M. Londo, H. Mozaffarian, P. Verweij, E. Lysen and A. Faaij, Energy Environ. Sci., 2010, 3, 258–267 Search PubMed.
- Z. Ren, T. E. Ward and J. M. Regan, Environ. Sci. Technol., 2007, 41, 4781–4786 CrossRef CAS.
- G. Mohanakrishna, S. Venkata Mohan and P.N. Sarma, Int. J. Hydrogen Energy, 2010, 35, 3440–3449 CrossRef CAS.
- S. Kalyuzhnyi, Pure Appl. Chem., 2008, 80, 2115–2124 CrossRef CAS.
- E. Tveritinova, M.Sc. Thesis. University of Jyväskylä/School of Business and Economics, 2008 Search PubMed.
- A. D. Cuéllar and M. E. Webber, Environ. Sci. Technol., 2010, 44, 6464–6469 CrossRef.
- M. Balat, Biomass Bioenergy, 2005, 29, 32–41 CrossRef.
- B. Steubing, R. Zah, P. Waeger and C. Ludwig, Renewable Sustainable Energy Rev., 2010, 14, 2256–2265 CrossRef.
- C. Panoutsou, J. Eleftheriadis and A. Nikolaou, Energy Policy, 2009, 37, 5675–5686 CrossRef.
- H. Yang and K. C. Abbaspour, Desalination, 2007, 212, 238–250 CrossRef CAS.
- R. M. Harrison, The Royal Society of Chemistry, Redwood Books Ltd., UK, 1999.
- T. Asano, Water Sci. Technol., 2002, 43, 24–33 Search PubMed.
- A. Singh, B. M. Smyth and J. D. Murphy, Renewable Sustainable Energy Rev., 2010, 14, 277–288 CrossRef CAS.
- P. S. Nigam and A. Singh, Prog. Energy Combust. Sci., 2011, 37, 52–68 CrossRef CAS.
- A. Singh, P. S. Nigam and J. D. Murphy, Bioresour. Technol., 2011, 102, 10–16 CrossRef CAS.
- J. K. Pittman, A. P. Dean and O. Osundeko, Bioresour. Technol., 2011, 102, 17–25 CrossRef CAS.
- O. Bailey and E. Worrell, Lawrence Berkeley National Laboratory Study, 2005, 43 Search PubMed , http://www.filtrationdynamics.com/7.2_Billion_kWh.pdf..
- F. Meggers and H. Leibundgut, Energy Build., 2011, 43, 879–886 CrossRef.
- J. J. Fornero, M. Rosenbaum and L. T. Angenent, Electroanalysis, 2010, 22, 832–843 CAS.
- D. Pant, A. Singh, Y. Satyawali and R. K. Gupta, J. Environ. Biol., 2008, 29, 79–84 CAS.
- J. Sun, Y. -Y. Hu, Z. Bi and Y. -Q. Cao, Bioresour. Technol., 2009, 100, 3185–3192 CrossRef CAS.
- Y. Mu, K. Rabaey, R. A. Rozendal, Z. Yuan and J. Keller, Environ. Sci. Technol., 2009, 43, 5137–5143 CrossRef CAS.
- L. Liu, F. -B. Li, C. -H. Feng and L. Xiang-zhong, Appl. Microbiol. Biotechnol., 2009, 85, 175–183 CrossRef CAS.
- Z. Li, X. Zhang, J. Lin, S. Han and L. Lei, Bioresour. Technol., 2010, 101, 4440–4445 CrossRef CAS.
- Y. Cao, Y. Hu, J. Sun and B. Hou, Bioelectrochemistry, 2010, 79, 71–76 CrossRef CAS.
- B. Y. Chen, M. M. Zhang, C. T. Chang, Y. ding, K. L. Lin, C. S. chiou, C. C. Hsueh and H. Xu, Bioresour. Technol., 2010, 101, 4737–4741 CrossRef CAS.
- P. Bhatt, M. S. Kumar, S. Mudliar and T. Chakrabarti, Crit. Rev. Environ. Sci. Technol., 2007, 37, 165–198 CrossRef CAS.
- S. M. Strycharz, S. M. Gannon, A. R. Boles, A. E. Franks, K. P. Nevin and D. R. Lovley, Environ. Microbiol. Rep., 2010, 2, 289–294 CrossRef CAS.
- F. Aulenta, A. Catervi, M. Majone, S. Panero, P. Reale and S. Rossetti, Environ. Sci. Technol., 2007, 41, 2554–2559 CrossRef CAS.
- F. Aulenta, A. Canosa, P. Reale, S. Rossetti, S. Panero and M. Majone, Biotechnol. Bioeng., 2009, 103, 85–91 CrossRef CAS.
- H. Pham, N. Boon, M. Marzorati and W. Verstraete, Water Res., 2009, 43, 2936–2946 CrossRef CAS.
- F. Aulenta, V. D. Maio, T. Ferri and M. Majone, Bioresour. Technol., 2010, 101, 9728–9733 CrossRef CAS.
- S. J. You, Q. L. Zhao, J. Q. Jiang, J. N. Zhang and S. Q. Zhao, J. Environ. Sci. Health. Part A, 2006, 41, 2721–2734 CAS.
- J. R. Jambeck and L. Damiano, Environmental Research and Education Foundation (EREF) project report, 2010, 112 Search PubMed http://erefdn.org/publications/uploads/EREF+MFC+Report+2-25-10_FINAL.pdf (Accessed on 11 April 2011).
- W. Habermann and E. H. Pommer, Appl. Microbiol. Biotechnol., 1991, 35, 128–133 CrossRef CAS.
- J. Greenman, A. Gálvez, L. Giusti and I. Ieropoulos, Enzyme Microb. Technol., 2009, 44, 112–119 CrossRef CAS.
- A. Gálvez, J. Greenman and I. Ieropoulos, Bioresour. Technol., 2009, 100, 5085–5091 CrossRef.
- S. Puig, M. Serra, M. coma, M. Cabre, M. D. Balaguer and J. Colprim, J. Hazard. Mater., 2011, 185, 763–767 CrossRef CAS.
- K. L. Straub and B. Schink, Appl. Environ. Microbiol., 2004, 70, 5744–5749 CrossRef CAS.
- M. Sun, Z. X. Mu, Y. P. Chen, G. P. Sheng, X. W. Liu, Y. Z. Chen, Y. Zhao, H. L. Wang, H. Q. Yu, L. Wei and F. Ma, Environ. Sci. Technol., 2009, 43, 3372–3377 CrossRef CAS.
- K. Rabaey, K. van de Sompel, L. Maignien, N. Boon, P. Aelterman, P. Clauwaert, L. de Schamphelaire, H.T. Pham, J. Vermeulen, M. Verhaege, P. Lens and W. Verstraete, Environ. Sci. Technol., 2006, 40, 5218–5224 CrossRef CAS.
- M. Sun, Z. H. Tong, G. P. Sheng, Y. Z. Chen, F. Zhang, Z. X. Mu, H. L. Wang, R. J. Zeng, H. Q. Yu, L. Wei and F. Ma, Biosens. Bioelectron., 2010, 26, 470–476 CrossRef CAS.
- F. Zhao, N. Rahunen, J. R. Varcoe, A. J. Roberts, C. Avignone-Rossa, A. E. Thumser and R. C. T. Slade, Biosens. Bioelectron., 2009, 24, 1931–1936 CrossRef CAS.
- H. V. M. Hamelers, A. Ter Heijne, T. H. J. A. Sleutels, A. W. Jeremiasse, D. P. B. T. B. Strik and C. J. N. Buisman, Appl. Microbiol. Biotechnol., 2010, 85, 1673–1685 CrossRef CAS.
- J. M. Foley, R. A. Rozendal, C. K. Hertle, P. A. Lant and K. Rabaey, Environ. Sci. Technol., 2010, 44, 3629–3637 CrossRef CAS.
- K. P. Nevin, T. L. Woodard, A. E. Franks, Z. M. Summers and D. R. Lovley, mBio, 2010, 1(2) DOI:10.1128/mBio.00103-10 e00103-10.
- K. Rabaey, A. Johnstone, A. Wise, S. Read and R. A. Rozendal, Biotech. International, 2010, 7, 6–8 Search PubMed.
- P. Clauwaert, J. Desloover, C. Thoeye, G. De Gueldre and W. Verstraete, 2010, IWA, World Water Conference, Montreal, http://iwawaterwiki.org/xwiki/bin/view/EventsExtra/FullTitle (Accessed on 15 March 2011).
- R. A. Rozendal, H. V. M. Hamelers, G. J. W. Euverink, S. J. Metz and C. J. N. Buisman, Environ. Sci. Technol., 2006, 40, 5206–5211 CrossRef CAS.
- H. S. Lee and B. E. Rittman, Int. J. Hydrogen Energy, 2010, 35, 920–927 CrossRef CAS.
- S. Cheng, D. Xing, D. F. Call and B. E. Logan, Environ. Sci. Technol., 2009, 43, 3953–3958 CrossRef CAS.
- P. Clauwaert and W. Verstraete, Appl. Microbiol. Biotechnol., 2009, 82, 829–836 CrossRef CAS.
- D. Lovley, Environ. Microbiol. Rep., 2010, 3, 27–35 CrossRef.
- P. Clauwaert, R. Toledo, D. v. d. Ha, R. Crab, W. Verstraete, H. Hu, K. M. Udert and K. Rabaey, Water Sci. Technol., 2008, 57, 575–579 CrossRef CAS.
- D. Call and B. E. Logan, Environ. Sci. Technol., 2008, 42, 3401–3406 CrossRef CAS.
- A. Wang, W. Liu, S. Cheng, D. Xing, J. Zhou and B. E. Logan, Int. J. Hydrogen Energy, 2009, 34, 3653–3658 CrossRef CAS.
- K. J. J. Steinbusch, H. V. M. Hamelers, J. D. Schaap, C. Kampman and C. J. N. Buisman, Environ. Sci. Technol., 2010, 44, 513–517 CrossRef CAS.
- K. J. J.Steinbusch, H. V. M. Hamelers and C. J. N. Buisman, Water Res., 2008, 42, 4059–4066 CrossRef.
- I. S. Kim, J. K. Jang, G. C. Gil, M. Kim, H. J. Kim, B. W. Cho and B. H. Kim, Biosens. Bioelectron., 2004, 19, 607–613 CrossRef.
- J. M. Tront, J. D. Fortner, M. Plötze, J. B. Hughes and A. M. Puzrin, Biosens. Bioelectron., 2008, 24, 586–590 CrossRef CAS.
- G. T. R. Palmore and G. M. Whitesides, In: Enzymatic conversion of biomass for fuels production. (ed. E. Himmel, J. O. Baker and R. P. Overend), American Chemical Society, 1994, 271–290.
- P. Clauwaert, P. Aelterman, T. H. Pham, L. De Schamphelaire, M. Carballa, K. Rabaey and W. Verstraete, Appl. Microbiol. Biotechnol., 2008, 79, 901–913 CrossRef CAS.
- B. E. Logan, Appl. Microbiol. Biotechnol., 2010, 85, 16665–21671 CrossRef.
- A. Dewan, H. Beyenal and Z. Lewandowski, Environ. Sci. Technol., 2008, 42, 7643–7648 CrossRef CAS.
- M. Di Lorenzo, T. P. Curtis, I. M. Head and K. Scott, Water Res., 2009, 43, 3145–3154 CrossRef CAS.
- M. Di Lorenzo, K. Scott, T. P. Curtis and I. M. Head, Chem. Eng. J., 2010, 156, 40–48 CrossRef CAS.
- F. Zhao, R. C. T. Slade and J. R. Varcoe, Chem. Soc. Rev., 2009, 38, 1926–1939 RSC.
- F. Zhang, S. A. Cheng, D. Pant, G. Van Bogaert and B. E. Logan, Electrochem. Commun., 2009, 11, 2177–2179 CrossRef CAS.
- D. Pant, G. Van Bogaert, M. De Smet, L. Diels and K. Vanbroekhoven, Electrochim. Acta, 2010, 55, 7709–7715 CrossRef.
- F. Zhang, D. Pant and B. E. Logan, Biosens. Bioelectron., 2011, 30, 49–55 CAS.
- F. Harnisch and U. Schröder, ChemSusChem, 2009, 2, 921–926 CrossRef CAS.
- W. -W. Li, G. -P. Guo-Ping Sheng, X. -W. Liu and H. Q. Yu, Bioresour. Technol., 2011, 102, 244–252 CrossRef CAS.
- H. -Y. Wang, A. Bernarda, C.-Y. Huang, D.-J. Lee and J.-S. Chang, Bioresour. Technol., 2011, 102, 235–243 CrossRef CAS.
- M. Zhou, M. Chi, J. Luo, H. He and T. Jin, J. Power Sources, 2011, 196, 427–4435 Search PubMed.
- R. A. Rozendal, H. V. M. Hamelers, K. Rabaey, J. Keller and C. J. N. Buisman, Trends Biotechnol., 2008, 26, 450–459 CrossRef CAS.
- E. H. Yu, S. Cheng, K. Scott and B. Logan, J. Power Sources, 2007, 171, 275–281 CrossRef.
- K. Rabaey, P. Girguis and L. K. Nielson, Curr. Opin. Biotechnol., 2011, 22, 371–377 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |