
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence“Trading” a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h2_char_e001.gif) C moiety for four C–O bonds and a peroxide: combining ozone and O-nucleophiles for peroxidative ring expansion of cycloalkenes into medium-sized 1,2-dioxacycloalkanes
C moiety for four C–O bonds and a peroxide: combining ozone and O-nucleophiles for peroxidative ring expansion of cycloalkenes into medium-sized 1,2-dioxacycloalkanes
Roman A. Budekhin a,
Dmitri I. Fomenkova,
Alexander O. Ustyuzhanina,
Darya Yu. Sliguzovaa,
Ksenia V. Skokovaa,
Vera A. Vil'a,
Igor V. Alabugin*b and
Alexander O. Terent'ev*a
a,
Dmitri I. Fomenkova,
Alexander O. Ustyuzhanina,
Darya Yu. Sliguzovaa,
Ksenia V. Skokovaa,
Vera A. Vil'a,
Igor V. Alabugin*b and
Alexander O. Terent'ev*a
aN.D. Zelinsky Institute of Organic Chemistry, Russian Academy of Sciences, 47 Leninsky Prosp., Moscow, 119991, Russian Federation. E-mail: terentev@ioc.ac.ru
bDepartment of Chemistry and Biochemistry, Florida State University, Tallahassee, Fl 32306, USA. E-mail: alabugin@chem.fsu.edu
First published on 10th October 2025
Abstract
Despite the long and rich history of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond ozonolysis, ozonolysis of cyclic alkenes has generally been limited to the formation of ring-opening products and fragmentary evidence of unstable ozonide formation. We present a non-orthodox cycloalkene ring expansion reaction involving ozone and O-nucleophiles, which results in the formation of medium-sized peroxycycles. The mechanism is governed by a delicate balance: the cyclic structure of the substrate dictates conformational restrictions in the generated carbonyl oxide intermediate that kinetically retard the rapid [3 + 2] cycloaddition between the carbonyl and carbonyl oxide groups, enabling competitive trapping of the carbonyl oxide intermediate by an external O-nucleophile. Some of the synthesized peroxides exhibit ring-chain tautomerism, existing in either a hydroperoxide or a cyclic peroxy hemiacetal form. The use of hydroperoxides as O-nucleophiles leads to compounds containing both cyclic and exocyclic peroxide fragments. Additionally, these unique bisperoxides demonstrate intriguing reactivity, undergoing a highly selective rearrangement under basic conditions, which affects only one of the peroxide fragments. The developed peroxidative ring expansion of cycloalkenes via ozonolysis broadens the synthetic potential of classical ozonolysis and provides new pathways for constructing structurally diverse peroxides and polyfunctionalized products of their transformation.
C double bond ozonolysis, ozonolysis of cyclic alkenes has generally been limited to the formation of ring-opening products and fragmentary evidence of unstable ozonide formation. We present a non-orthodox cycloalkene ring expansion reaction involving ozone and O-nucleophiles, which results in the formation of medium-sized peroxycycles. The mechanism is governed by a delicate balance: the cyclic structure of the substrate dictates conformational restrictions in the generated carbonyl oxide intermediate that kinetically retard the rapid [3 + 2] cycloaddition between the carbonyl and carbonyl oxide groups, enabling competitive trapping of the carbonyl oxide intermediate by an external O-nucleophile. Some of the synthesized peroxides exhibit ring-chain tautomerism, existing in either a hydroperoxide or a cyclic peroxy hemiacetal form. The use of hydroperoxides as O-nucleophiles leads to compounds containing both cyclic and exocyclic peroxide fragments. Additionally, these unique bisperoxides demonstrate intriguing reactivity, undergoing a highly selective rearrangement under basic conditions, which affects only one of the peroxide fragments. The developed peroxidative ring expansion of cycloalkenes via ozonolysis broadens the synthetic potential of classical ozonolysis and provides new pathways for constructing structurally diverse peroxides and polyfunctionalized products of their transformation.
Introduction
Cyclic organic peroxides have added a new dimension to medicinal chemistry serving as a valuable source of novel therapeutic agents. A landmark example is the antimalarial compound artemisinin, whose discovery led to the Nobel Prize in 2015,1 along with its semi-synthetic analogues (Scheme 1).2–5 Today, cyclic peroxides are widely recognized for their pharmaceutical and agrochemical potential, exhibiting a broad spectrum of biological activities including antiparasitic,6 growth-regulating,7 anticancer,8 and fungicidal.9,10 The synthesis of various peroxide-containing molecular architectures remains an unresolved challenge, primarily due to the intrinsic lability of the O–O bond. This sensitivity necessitates the development of innovative synthetic strategies. A direct and versatile approach for constructing cyclic peroxides from readily available molecules such as cycloalkenes, alcohols, hydroperoxides, carboxylic acids, and ozone would be highly desirable. Such an approach would not only streamline access to structurally complex peroxides but also expand their applications across pharmaceutical, agrochemical, and materials sciences.11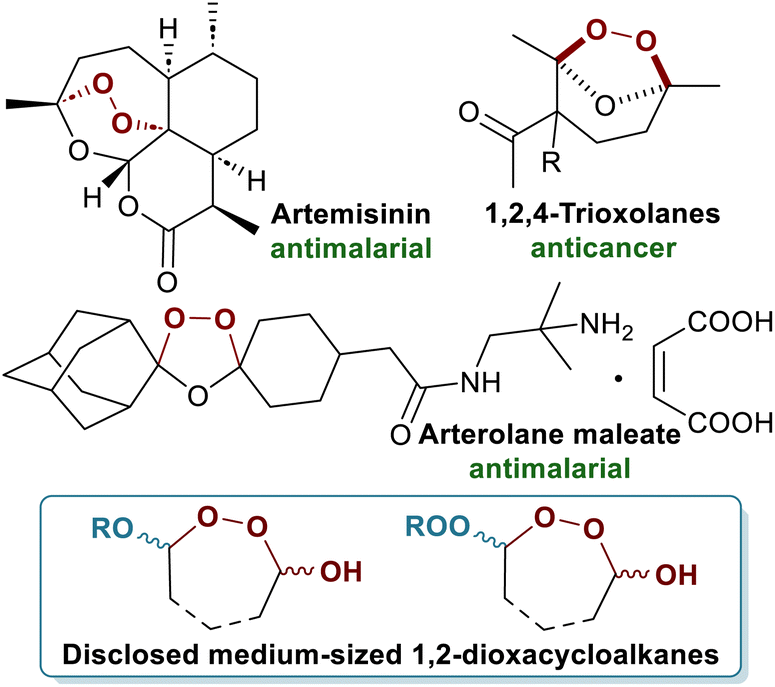 | ||
| Scheme 1 Pharmaceutically relevant cyclic peroxides and the disclosed medium-sized 1,2-dioxacycloalkanes. | ||
Ozonolysis of alkenes is a well-established method of synthesizing ozonides (1,2,4-trioxolanes), and the broad range of carbonyl derivatives including carbonyls,12,13 alcohols,14,15 acetals,16 and carboxylic acids17 (Scheme 2a).18 Recently, Kwon and co-workers developed strategies for combining alkene ozonolysis with alkoxyl radical-driven transformations of ozonides, offering access to a variety of high-value products.19–23 Ozonolysis of cyclic alkenes has generally been limited to the formation of ring-opening products, such as dicarbonyls and dicarboxylic acids, with only fragmentary evidence for the transient formation of unstable ozonides (Scheme 2b).24–26 In this work, we report a peroxidative ring expansion of cycloalkenes achieved through the combined action of ozone and O-nucleophiles, enabling the incorporation of up to four or five oxygen atoms in a one-pot transformation (Scheme 2c).
 | ||
| Scheme 2 Alkene ozonolysis. | ||
The mechanism of organic peroxide synthesis via alkene ozonolysis involves the generation and subsequent transformation of the highly reactive carbonyl O-oxide, also known as the Criegee intermediate (CI). This key species can undergo nucleophilic capture or cycloaddition reactions to yield a diverse array of peroxide-containing products.27 The Criegee intermediate is known to react with various nucleophiles, including water,28 alcohols,27,29,30 amines and ammonia,31 hydrogen peroxide32 or hydroperoxides,33–35 and carboxylic acids,36 leading to hydroperoxides with different substituents. The CI can also participate in cycloaddition processes to form cyclic peroxides, for example classic ozonides and their N-analogues (Scheme 2a).37–39 A special case involves dimerization of carbonyl O-oxides.40,41
The reactivity of carbonyl oxides, which are formed in ozonolysis of cycloalkenes, is much less predictable because these intermediates retain an additional carbonyl group in the same molecule (Scheme 2b). On one hand, one could expect that this feature will predispose the intermediate to undergo an intramolecular [3 + 2] cycloaddition, leading to the formation of a secondary cycloalkene ozonide. However, in practice, such ozonides are typically observed only as minor by-products likely due to their twisted and strained transition state (Scheme 2b). The majority of the substrate is often diverted into the formation of complex mixtures of oligomeric peroxides, making selective synthesis difficult.42
To date, selective formation of peroxides via cycloalkene ozonolysis has been reported only in a limited number of studies.43,44 Notably, McCullough and colleagues42,45,46 demonstrated the formation of well-defined peroxides, including indene ozonides, isochromanes, alkoxyhydroperoxides, and, in rare cases, dioxepanes by performing indene ozonolysis in the presence of alcohols. However, attempts to extend this methodology to substituted cyclopentenes resulted in complex mixtures of peroxidic species, with little selectivity.47
In this study, we report the development of a selective three-component reaction for the construction of structurally diverse cyclic peroxides via the ozonolysis of cycloalkenes in the presence of O-nucleophiles (Scheme 2c). A key mechanistic insight is that the intramolecular addition of the carbonyl group to the carbonyl oxide, typically a rapid and favoured pathway, is significantly slowed by conformational constraints imposed by the cyclic substrate. This kinetic suppression allows the normally fast [3 + 2] cycloaddition to be interrupted, enabling interception of the carbonyl oxide intermediate by an external O-nucleophile.
This strategy provides controlled access to peroxide frameworks featuring both endocyclic and exocyclic O–O linkages, greatly expanding the synthetic repertoire of structurally diverse cyclic peroxides that are otherwise challenging to obtain through conventional methods.
Results and discussion
Caution: although we have encountered no difficulties in working with the peroxides described below, the proper precautions, such as the use of shields, fume hoods, and the avoidance of transition metal salts, heating and shaking, should be taken whenever possible.Our initial experiments demonstrated that ozonolysis of cyclopentene 1a predominantly leads to the formation of oligomeric peroxides, accompanied by only a minor amount of secondary ozonide 1ab (Scheme 3a). The addition of tert-butylhydroperoxide (TBHP) led to the formation of a cyclic peroxide 2a with two different peroxide fragments—one endocyclic and one exocyclic—in 40% yield (Scheme 3b).
 | ||
| Scheme 3 Initial experiments on cyclopentene ozonolysis. | ||
Control experiments confirmed that 2a is not formed via transformation of the secondary ozonide 1ab or from any other peroxide species generated during the ozonolysis of cyclopentene alone (Scheme 3c), supporting a mechanistic pathway in which TBHP directly traps the carbonyl oxide intermediate.
The reaction conditions for the synthesis of cyclic peroxide 2a were optimized (Table 1). Increasing the amount of TBHP up to 5 equivalents relative to 1a led to a substantial improvement in yield, reaching 77% (entries 1–3). Further addition of TBHP had only a marginal effect (78% yield, entry 4). Substituting 70% aqueous TBHP with anhydrous TBHP (see the SI) further enhanced the yield to 88% (entry 5), likely due to the suppression of competing carbonyl oxide trapping by water.
|
|||||
|---|---|---|---|---|---|
| No. | O3, eq. min−1 | TBHP, eq. | T, °C | Solvent (mL) | Yield of 2a, % |
| a General conditions: a stream of O3 with O2 (100 mg O3 per L of gas mixture) was passed through a solution of cyclopentene 1a (1.0 mmol, 68.1 mg) and TBHP (A: 70% aq. B: anhydrous; 1.0–10.0 eq., 1.0–10.0 mmol) cooled to −70 °C in the indicated solvent (6–25 mL) for 8–24 min with stirring (1.0–4.0 mmol O3 per mmol of 1a). Isolated yield.b Conv. of 1a is 85%. | |||||
| 1 | 2/8 | A, 1 | −70 | CH2Cl2 (12.5) | 40 |
| 2 | 2/8 | A, 3 | −70 | CH2Cl2 (12.5) | 67 |
| 3 | 2/8 | A, 5 | −70 | CH2Cl2 (12.5) | 77 |
| 4 | 2/8 | A, 10 | −70 | CH2Cl2 (12.5) | 78 |
| 5 | 2/8 | B, 5 | −70 | CH2Cl2 (12.5) | 88 |
| 6 | 2/8 | B, 5 | −70 | CH2Cl2 (25) | 85 |
| 7 | 2/8 | B, 5 | −70 | CH2Cl2 (6) | 86 |
| 8 | 2/8 | B, 5 | −70 | EtOAc (12.5) | 38 |
| 9 | 2/8 | B, 5 | −70 | nBuOAc (12.5) | 47 |
| 10 | 2/8 | B, 5 | −70 | PE (12.5) | 49 |
| 11 | 2/8 | B, 5 | −70 | nHexane (12.5) | 55 |
| 12 | 2/8 | B, 5 | −70 | THF (12.5) | 34 |
| 13 | 2/8 | B, 5 | −10 | CH2Cl2 (12.5) | 52 |
| 14 | 4/8 | B, 5 | −70 | CH2Cl2 (12.5) | 89 |
| 15b | 1/8 | B, 5 | −70 | CH2Cl2 (12.5) | 69 |
| 16 | 2.5/24 | B, 5 | −70 | CH2Cl2 (12.5) | 80 |

Variation in the concentration of cycloalkene 1a had a negligible impact on the yield of 2a (entries 6–7). In contrast, changing the solvent from CH2Cl2 to ethyl acetate, butyl acetate (entries 8–9), petroleum ether (entry 10), hexane (entry 11), or THF (entry 12) resulted in significantly lower yields, with reductions of 30–50%. Performing the ozonolysis at a higher temperature also led to a decrease in the yield of 2a (entry 13).
Finally, variation of the ozonolysis parameters, including ozone quantity, ozone concentration in the gas stream, flow rate, and reaction time, did not lead to further improvements in the yield of 2a (entries 14–16), indicating that the reaction is more sensitive to solvent and nucleophile identity than to ozone delivery conditions.
With the optimal reaction conditions established (Table 1, entry 5), we investigated the scope of cycloalkenes amenable to selective cyclic peroxide assembly via ozonolysis in the presence of TBHP (Scheme 4). The cycloalkenes 1a–d have been demonstrated to be effective substrates affording the corresponding cyclic peroxides 2a–2d, including tert-butylperoxy dioxepanol (2a), dioxocanol (2b), dioxecanol (2c), and dioxacyclotetradecanol (2d), in high yields ranging from 75% to 92%.
 | ||
| Scheme 4 Aliphatic cycloalkene ozonolysis in the presence of O-nucleophiles. | ||
Substitution of TBHP with cumyl hydroperoxide (CUHP) led to the formation of three-component condensation products 3a–3d in similarly high yields (79–89%). The use of methanol as the O-nucleophile also proved effective, giving rise to peroxidative ring expansion products 4a–4d in 75–89% yields.
All compounds 2–4 exhibited ring-chain tautomerism, as determined by 1H NMR spectroscopy in CDCl3 and DMSO-d6 (Scheme 4). For example, compound 3a exists predominantly in the cyclic form (96%), with only 4% linear form, and this ratio remained unchanged in DMSO-d6, benzene-d6, and CDCl3 (see the SI). Additionally, thermal stability studies on 2c demonstrated that the cyclic-to-linear ratio remained constant after heating at 60 °C for 45 minutes (see the SI), confirming the configurational stability of the tautomeric equilibrium under these conditions.
Application of our approach to the ozonolysis of norbornene 1e resulted in the successful synthesis of bicyclic peroxides 2e and 4e in good yields of 68% and 72%, respectively (Scheme 5). The methodology also proved effective with other O-nucleophiles: AcOH afforded product 5a, and H2O2 gave 6b.
 | ||
| Scheme 5 Ozonolysis of different cycloalkenes in the presence of various O-nucleophiles. | ||
Interestingly, using a gem-bishydroperoxide as the nucleophile did not yield a cyclic peroxide but instead furnished a substituted pyran 7a with three exocyclic peroxide moieties. Ozonolysis of 1-phenylcyclohexene 1f and (+)-α-pinene 1i in the presence of O-nucleophile (TBHP and MeOH, respectively) gave hydroperoxy-substituted ketones 2f and 4i, rather than cyclic products. On the other hand, ozonolysis of indene 1g with TBHP led to two distinct cyclic products: hydroperoxyisochromane 2g in 37% yield, and tert-butylperoxy dioxepinol 2g′ in 30% yield (Scheme 5). Notably, the formation of the secondary indene ozonide or other isomeric forms of dioxepinol was not observed. As anticipated, ozonolysis of indene in MeOH led to a mixture of two structural isomers of methoxydioxepinols 4g and 4g′, confirming the structures proposed for them by McCullough and colleagues.45
In the case of indole 1h, only trace amounts of peroxide 2h were detected, likely due to its instability in solution at room temperature and its decomposition during silica gel chromatography. The major product of the ozonolysis of indole 1h in the presence of hydroperoxides was identified as N-(2-formylphenyl)formamide 2h′.
Almost all synthesized peroxide compounds 2–7 exist predominantly in their cyclic form in solution, with the exception of 2f, 3c, and 3d. Compounds 3c and 3d exhibit an approximately 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of cyclic to linear isomers, while 2f exists exclusively in the linear form. In contrast, the peroxides derived from ozonolysis of indene, i.e., 2g, 2g′, 4g, and 4g′, were found to exist entirely in the cyclic form.
:1 ratio of cyclic to linear isomers, while 2f exists exclusively in the linear form. In contrast, the peroxides derived from ozonolysis of indene, i.e., 2g, 2g′, 4g, and 4g′, were found to exist entirely in the cyclic form.
To evaluate the influence of chain length and nucleophile identity on the tautomeric equilibrium of peroxides 2a–d through 4a–d and 2′a–d through 4′a–d, the energy differences between their linear and cyclic forms were estimated (Scheme 6). We found that, as the ring size increases from 7-membered (a) to 10-membered (c) structures, the linear form becomes increasingly thermodynamically favorable. This trend is consistent with a rise in ring strain, which promotes ring opening and increases the proportion of the linear tautomer. Both theoretical predictions and experimental data indicate that 10-membered rings are among the most prone to ring-opening. Interestingly, our exploratory calculations suggest that for 14-membered rings, the cyclic form should be thermodynamically more stable. However, the experimentally observed ratios of cyclic and linear forms in 10- and 14-membered systems showed only a minor difference. This is not surprising since conformational preferences of medium size cyclic systems are known to be complex and require much more advanced modelling that would take into account the ensemble of nearly isoenergetic conformations.48–50 Such analysis goes beyond the scope of the present work.
 | ||
| Scheme 6 Ratio of cyclic and linear forms of compounds 2a–d, 3a–d, and 4a–d after column chromatography (by 1H NMR, A) and ΔG of the cycle opening (by DFT, B). | ||
In order to ascertain the influence of the acidity of the medium on the ratio of isomers, a series of experiments was conducted (Scheme 7). Initially, a substoichiometric quantity of p-TsOH (0.25 mol of p-TsOH per mol of 2c) was added to the 2c peroxide solution in DMSO-d6. Over the first ten minutes, this resulted in a notable increase in the amount of the linear form to 55%. Furthermore, it has been demonstrated that peroxide 4a treated with a small excess of BF3·Et2O (1.50 mol BF3·Et2O per mol of 4a) in CH3OH forms a mixture of 1,1,5,5-tetramethoxypentane 8a and 1-hydroperoxy-1,5,5-trimethoxypentane 8b in yields of 38 and 16%, respectively.
 | ||
| Scheme 7 Transformations of synthesized peroxides under acidic conditions. | ||
A selective rearrangement of peroxycycles 2 into ω-oxoperesters 9 was achieved under mild conditions (Scheme 8). The process proceeds smoothly under the treatment with CsCO3 in DMSO, resulting in the formation of ω-oxoperesters 9a–d, a rather rare class of compounds, in 81–99% yields. Despite the presence of two peroxide fragments in the molecules 2a–d, the rearrangement proceeds in a surprisingly selective manner, affording the ω-oxoperesters 9a–d as the sole products in near-quantitative yields.
 | ||
| Scheme 8 The selective rearrangement of peroxides 2 into ω-oxoperesters 9. | ||
Building on the successful conversion of our cyclic peroxides into ω-oxoperesters 9a–d (Scheme 8), the reactivity of this bifunctional moiety was investigated. The condensation reactions between ω-oxoperester 9a and pharmaceutically relevant 13,17-secoestra-1,3,5(10)-trien-17-oic acid hydrazide 11′ or N-phenylhydrazinecarboxamide 10′ (ref. 51) were successful and proceeded with the preservation of the peroxide fragment (Scheme 9). The condensation products 10a and 11a were successfully obtained in good yields (64 and 79%, respectively). Consequently, the developed method for the synthesis of cyclic peroxides, coupled with their transformations, provides a foundation for the synthesis of various linkers for the modification of biologically active compounds.
 | ||
| Scheme 9 Synthetic applications of cyclic peroxide 2a. | ||
Based on control experiments and literature data, we have proposed a pathway for the synthesis of cyclic peroxide 2a via cyclopentene 1a ozonolysis in the presence of TBHP (Scheme 10). The initial step of the ozonolysis process is the formation of molozonide A, which subsequently decomposes to form the Criegee intermediate (carbonyl oxide) B. The presence of a tether between the carbonyl O-oxide and the carbonyl fragment makes the synchronous cycloaddition less favourable, and the formation of the secondary ozonide E proceeds stepwise – via the formation of intermediates C or D. The nucleophilic trapping of the intermediate C by the TBHP allows the interruption of the secondary ozonide assembly, leading to the formation of the cyclic peroxide 2a. The direct nucleophilic trapping of Criegee intermediate B or, as is evidenced by the control experiments conducted, the exposure of the cyclic peroxide derivative 2a to acidic conditions results in the formation of the linear form of peroxide 2a.
 | ||
| Scheme 10 The proposed reaction mechanism for the ozonolysis of cyclopentene 1a in the presence of TBHP. | ||
An intriguing aspect of this transformation with cyclic substrates is that the cascade of three pericyclic steps involved in ozonolysis is interrupted during the transition from a primary to a secondary ozonide because of the structural shift from a fused to a bridged bicyclic framework. For small ring systems, the formation of the bridged intermediate introduces additional strain, rendering the formation of secondary ozonides less favorable.
This outcome offers a conceptually distinct yet complementary approach to the ozonolysis-diverting strategy we previously developed, which employed CN bonds in place of CC bonds to interrupt the cascade.27 In both cases, the sequence of three pericyclic transformations (cycloaddition/cycloreversion/cycloaddition) is halted before the final step, i.e., trapping of the carbonyl oxide by the carbonyl group. In our earlier work, this interruption was accomplished via electronic effects, whereas in the present study, the diversion is also assisted by using structural intramolecular constraints. Interestingly, the new pathway still initiates the formation of one of the bonds expected in the final cycloaddition step, effectively diverting rather than aborting the process. Unlike the previous approach, where interruption was induced by the presence of a heteroatom (nitrogen), the current study retains the classical CC bond, yet no secondary ozonide is formed. This subtle redirection underscores the potential of intramolecular design in steering the outcome of pericyclic cascades.
The products obtained in this study are difficult to access using traditional approaches to cyclic peroxide synthesis—particularly the acid-catalyzed addition of hydrogen peroxide to dicarbonyl compounds. Under acidic conditions, peroxidation of dicarbonyl substrates typically proceeds via peroxocarbenium ions rather than zwitterionic intermediates. The zwitterions exhibit reduced susceptibility to the inverse α-effect,52 altering the balance of stereoelectronic influences that govern ring formation. Equations in Scheme 11 illustrate that the inverse α-effect is fully relieved, enabling access to peroxide cycles that are otherwise challenging to form. In addition, the increased nucleophilicity of the carbonyl O-oxide enhances the propensity for intramolecular attack on the second carbonyl group, thereby favoring the formation of larger cyclic frameworks.
 | ||
| Scheme 11 Comparison of preferred cyclization routes for Criegee intermediate A and peroxycarbenium intermediate A′. | ||
Conclusions
The well-known and seemingly straightforward pericyclic cascade of alkene ozonolysis was redirected by introducing an external nucleophile in combination with cyclic constraints. In this system, the intramolecular [3 + 2] cycloaddition between the carbonyl O-oxide and the carbonyl group is hindered by ring strain. As a result, C–O bond formation proceeds through a stepwise mechanism, allowing the reactive intermediate to be intercepted by a nucleophile. This diversion effectively prevents the formation of secondary ozonides and instead promotes a highly selective three-component condensation, yielding 1,2-dioxacycloalkanes.Most of the products exhibit ring-chain tautomerism. A detailed study of these tautomerization patterns and the thermodynamic balance between the cyclic and linear forms of 1,2-dioxacycloalkanes revealed that the cyclic form is generally more stable. We also explored the reactivity of the resulting medium-sized cyclic peroxides and further analyzed their ring-chain equilibria. The use of unconventional nucleophiles, such as hydroperoxides, enabled the synthesis of compounds containing both cyclic and exocyclic peroxide units. Under basic conditions, these species undergo a highly selective rearrangement, affecting only one of the peroxide moieties and yielding ω-oxoperesters in excellent yields.
Based on literature data, computational studies, and control experiments, we proposed a reaction pathway that highlights key distinctions from traditional strategies for constructing peroxide rings. The study revealed important trends in how both the length and nature of the carbon bridge influence the rate of intramolecular addition during alkene ozonolysis. Notably, an external O-nucleophile successfully outcompetes an internal one. These findings offer valuable insights into the reactivity of oxycarbenium and peroxycarbenium intermediates, which play central roles in many common oxidation and condensation reactions.
Author contributions
Roman A. Budekhin: investigation, visualization, methodology; Dmitri I. Fomenkov: conceptualization, writing – original draft, methodology; Alexander O. Ustyuzhanin: formal analysis; Darya Yu. Sliguzova: investigation; Ksenia V. Skokova: investigation; Vera A. Vil': conceptualization, writing – review & editing, validation; Igor V. Alabugin: conceptualization, writing – original draft, writing – review & editing, supervision; Alexander O. Terent'ev: project administration, supervision, resources, funding acquisition.Conflicts of interest
There are no conflicts to declare.Data availability
CCDC 2483227 (3a) contains the supplementary crystallographic data for this paper.53The data supporting this article have been included as part of the supplementary information (SI). Supplementary information: procedures, compound characterization (2a–g, 2a′–e′, 2g′, 3a–d, 3a′–d′, 4a–e, 4a′–4e′, 4g, 4g′, 4i, 5a, 5a′, 6b, 7a, 8a,b, 9a–d, 10a, 11a), and NMR and HRMS spectra. All calculations were carried out using the Gaussian 16 A.03 AVX2, M062X and B3LYP functionals with the 6-311++G(d,p) basis set for all atoms and ORCA 6.1.0 (for conformational analysis). Solvation effects were modeled using the CPCM model, and the solvent is chloroform. Minimum structures were obtained from ground state optimizations. Calculation of vibrational frequencies was performed to prove that the optimized structure corresponds to a true minimum on the potential energy surface. Geometry search and conformational analysis were performed using Global Optimizer Algorithm (GOAT) XTB2 ALPB (chloroform). GaussView 6.0 and Chemcraft were used for visualization. See DOI: https://doi.org/10.1039/d5sc06768e.
Acknowledgements
The study was supported by the Russian Science Foundation (Grant No. 24-13-00310). This work, including crystal structure determination, was performed using the equipment in the Shared Research Center (Department of Structural Studies) of N.D. Zelinsky Institute of Organic Chemistry RAS, Moscow.Notes and references
- T. Efferth, S. Zacchino, M. I. Georgiev, L. Liu, H. Wagner and A. Panossian, Nobel Prize for artemisinin brings phytotherapy into the spotlight, Phytomedicine, 2015, 22, A1–A3 CrossRef.
- T. Efferth, Beyond malaria: the inhibition of viruses by artemisinin-type compounds, Biotechnol. Adv., 2018, 36, 1730–1737 CrossRef PubMed.
- L. Hou and H. Huang, Immune suppressive properties of artemisinin family drugs, Pharmacol. Ther., 2016, 166, 123–127 CrossRef.
- T. Efferth, M. R. Romero, D. G. Wolf, T. Stamminger, J. J. Marin and M. Marschall, The antiviral activities of artemisinin and artesunate, Clin. Infect. Dis., 2008, 47, 804–811 CrossRef.
- J. Li, T. Casteels, T. Frogne, C. Ingvorsen, C. Honore, M. Courtney, K. V. Huber, N. Schmitner, R. A. Kimmel and R. A. Romanov, Artemisinins target GABAA receptor signaling and impair α cell identity, Cell, 2017, 168, 86–100 CrossRef.
- A. Mendes, A. Armada, L. I. Cabral, P. S. Amado, L. Campino, M. L. Cristiano and S. Cortes, 1,2,4-Trioxolane and 1,2,4,5-tetraoxane endoperoxides against old-world Leishmania parasites: in vitro activity and mode of action, Pharmaceuticals, 2022, 15, 446 CrossRef PubMed.
- L. C. A. Barbosa, C. R. A. Maltha, R. C. Cusati, R. R. Teixeira, F. F. Rodrigues, A. A. Silva, M. G. B. Drew and F. M. D. Ismail, Synthesis and Biological Evaluation of New Ozonides with Improved Plant Growth Regulatory Activity, J. Agric. Food Chem., 2009, 57, 10107–10115 CrossRef.
- P. Coghi, I. A. Yaremenko, P. Prommana, J. N. Wu, R. L. Zhang, J. P. Ng, Y. Y. Belyakova, B. Y. K. Law, P. S. Radulov and C. Uthaipibull, Antimalarial and anticancer activity evaluation of bridged ozonides, aminoperoxides, and tetraoxanes, ChemMedChem, 2022, 17, e202200328 CrossRef PubMed.
- I. A. Yaremenko, P. S. Radulov, Y. Y. Belyakova, A. A. Demina, D. I. Fomenkov, D. V. Barsukov, I. R. Subbotina, F. Fleury and A. O. Terent'ev, Catalyst development for the synthesis of ozonides and tetraoxanes under heterogeneous conditions: disclosure of an unprecedented class of fungicides for agricultural application, Chem.–Eur. J., 2020, 26, 4734–4751 CrossRef PubMed.
- I. A. Yaremenko, P. S. Radulov, Y. Y. Belyakova, D. I. Fomenkov, V. A. Vil', M. A. Kuznetsova, V. N. Demidova, A. P. Glinushkin and A. O. Terent'ev, Cyclic Organic Peroxides as New Fungicides against Phytopathogenic Fungi, Agrochemicals, 2023, 2, 355–366 CrossRef.
- Y. A. Barsegyan, V. A. Vil' and A. O. Terent'ev, Macrocyclic Organic Peroxides: Constructing Medium and Large Cycles with O–O Bonds, Chemistry, 2024, 6, 1246–1270 CrossRef.
- C. Schwartz, J. Raible, K. Mott and P. H. Dussault, ‘Reductive ozonolysis’ via a new fragmentation of carbonyl oxides, Tetrahedron, 2006, 62, 10747–10752 CrossRef.
- R. Willand-Charnley, T. J. Fisher, B. M. Johnson and P. H. Dussault, Pyridine Is an Organocatalyst for the Reductive Ozonolysis of Alkenes, Org. Lett., 2012, 14, 2242–2245 CrossRef PubMed.
- C. E. Stivala and A. Zakarian, Total Synthesis of (+)-Pinnatoxin A, J. Am. Chem. Soc., 2008, 130, 3774–3776 CrossRef.
- A. P. Pulis, P. Fackler and V. K. Aggarwal, Short Stereoselective Synthesis of the Phytophthora Universal Mating Hormone α1 Using Lithiation/Borylation Reactions, Angew. Chem., Int. Ed., 2014, 53, 4382–4385 CrossRef.
- D. F. Taber and K. Nakajima, Unsymmetrical ozonolysis of a Diels-Alder adduct: practical preparation of a key intermediate for heme total synthesis, J. Org. Chem., 2001, 66, 2515–2517 CrossRef PubMed.
- B. A. Pearlman, A. G. Padilla, J. T. Hach, J. L. Havens and M. D. Pillai, A New Approach to the Furan Degradation Problem Involving Ozonolysis of the trans-Enedione and Its Use in a Cost-Effective Synthesis of Eplerenone, Org. Lett., 2006, 8, 2111–2113 CrossRef PubMed.
- P. Sathya Shanker and G. S. R. Subba Rao, Total synthesis of (±)-allo-cedrol [Khusiol], Tetrahedron Lett., 1994, 35, 5055–5058 CrossRef.
- B. W. Dehnert, J. H. Dworkin and O. Kwon, Dealkenylative Functionalizations: Conversion of Alkene C (sp3)–C (sp2) Bonds into C (sp3)–X Bonds via Redox-Based Radical Processes, Synthesis, 2024, 56, 71–86 CrossRef.
- M. Swain, G. Sadykhov, R. Wang and O. Kwon, Dealkenylative Alkenylation: Formal σ-Bond Metathesis of Olefins, Angew. Chem., Int. Ed., 2020, 59, 17565–17571 CrossRef PubMed.
- M. Swain, T. B. Bunnell, J. Kim and O. Kwon, Dealkenylative Alkynylation Using Catalytic FeII and Vitamin C, J. Am. Chem. Soc., 2022, 144, 14828–14837 CrossRef PubMed.
- Z. He, J. A. Moreno, M. Swain, J. Wu and O. Kwon, Aminodealkenylation: Ozonolysis and copper catalysis convert C(sp3)-C(sp2) bonds to C(3)-N bonds, Science, 2023, 381, 877–886 CrossRef PubMed.
- D. I. Fomenkov, R. A. Budekhin, P. S. Radulov, A. I. Fomenkov, L.-N. He, I. A. Yaremenko and A. O. Terent'ev, C=N Bond Ozonolysis and β-Scission: A Breakthrough Approach to the Synthesis of ω-Functionalized Compounds from Carbonyl Derivatives, Org. Lett., 2024, 26, 8095–8099 CrossRef.
- Y.-S. Hon and J.-L. Yan, Syntheses of bifunctional compounds from cycloalkenes via ozonide intermediates, Tetrahedron, 1997, 53, 5217–5232 CrossRef.
- S. L. Schreiber, R. E. Claus and J. Reagan, Ozonolytic cleavage of cycloalkenes to terminally differentiated products, Tetrahedron Lett., 1982, 23, 3867–3870 CrossRef.
- M. P. Yakovleva, A. A. Kravchenko, K. M. Saitov, I. S. Nazarov, N. M. Ishmuratova and G. Y. Ishmuratov, Reduction of peroxide products of ozonolysis of cyclooctene by sebacic acid dihydrazide, Russ. J. Org. Chem., 2025, 61, 192–196 Search PubMed.
- I. A. Yaremenko, D. I. Fomenkov, R. A. Budekhin, P. S. Radulov, M. G. Medvedev, N. V. Krivoshchapov, L.-N. He, I. V. Alabugin and A. O. Terent'ev, Interrupted Dance of Five Heteroatoms: Reinventing Ozonolysis to Make Geminal Alkoxyhydroperoxides from C=N Bonds, J. Org. Chem., 2024, 89, 5699–5714 CrossRef.
- C. E. Schiaffo and P. H. Dussault, Ozonolysis in solvent/water mixtures: direct conversion of alkenes to aldehydes and ketones, J. Org. Chem., 2008, 73, 4688–4690 CrossRef PubMed.
- P. H. Dussault and J. M. Raible, Ozonolysis in the presence of Lewis acids: directed addition to carbonyl oxides, Org. Lett., 2000, 2, 3377–3379 CrossRef PubMed.
- Z. Paryzek and U. Rychlewska, Ozonolysis of cholesterol and other Δ5-steroids in the presence of alcohols: a revised mechanism and hydroperoxide structure of the solvent-participated product, confirmed by X-ray analysis, J. Chem. Soc., Perkin Trans. 2, 1997, 2313–2318 RSC.
- K. McCullough, Synthesis of 1,2,4-dioxazolidine derivatives by the ozonolysis of indenes in the presence of primary amines, J. Chem. Soc., Perkin Trans. 1, 1997, 1939–1942 Search PubMed.
- J. C. Robertson and W. Verzino Jr, Diphenylmethyl bishydroperoxide. Anomalous product from ozonolysis of tetraphenylethylene, J. Org. Chem., 1970, 35, 545–547 CrossRef.
- D. I. Fomenkov, R. A. Budekhin, V. A. Vil' and A. O. Terent'ev, The Ozone and Hydroperoxide Teamwork: Synthesis of Unsymmetrical Geminal Bisperoxides from Alkenes, Org. Lett., 2023, 25, 4672–4676 CrossRef.
- H. J. Hamann, A. Wlosnewski, T. Greco and J. Liebscher, Novel Hydroperoxydioxolanes and Dioxanes by Hydroperoxide Rearrangement and Ozonolysis, Eur. J. Org Chem., 2006, 2006, 2174–2180 CrossRef.
- R. A. Budekhin, D. Y. Sliguzova, D. I. Fomenkov, S. V. Baranin and A. O. Terent'ev, Synthesis of unsymmetrical bisperoxides via semicarbazone ozonolysis in the presence of hydroperoxides, Org. Biomol. Chem., 2025, 23, 8259–8266 RSC.
- L. Rebrovic, The peroxidic species generated by ozonolysis of oleic acid or methyl oleate in a carboxylic acid medium, J. Am. Oil Chem. Soc., 1992, 69, 159–165 CrossRef.
- M. Mori, M. Nojima, S. Kusabayashi and K. J. McCullough, Synthesis of 1,2,4-dioxazolidines by the ozonolysis of vinyl ethers in the presence of imines. The first [3 + 2] cycloaddition of carbonyl oxide to the carbon–nitrogen double bond, J. Chem. Soc., Chem. Commun., 1988, 1550–1552 RSC.
- K. J. McCullough, M. Mori, T. Tabuchi, H. Yamakoshi, S. Kusabayashi and M. Nojima, [3+ 2] Cycloadditions of carbonyl oxides to imines: an alternative approach to the synthesis of 1,2,4-dioxazolidines, J. Chem. Soc., Perkin Trans. 1, 1995, 41–48 RSC.
- M. Mori, T. Sugiyama, M. Nojima, S. Kusabayashi and K. J. McCullough, Synthesis and x-ray analysis of dihydro-1,2,4,5-trioxazine. Evidence of a stepwise mechanism for the [3 + 3] cycloaddition of carbonyl oxides with nitrones, J. Org. Chem., 1992, 57, 2285–2294 CrossRef CAS.
- Y. Dong and J. L. Vennerstrom, Dispiro-1,2,4,5-tetraoxanes via ozonolysis of cycloalkanone O-methyl oximes: a comparison with the peroxidation of cycloalkanones in acetonitrile–sulfuric acid media, J. Org. Chem., 1998, 63, 8582–8585 CrossRef CAS.
- O. S. Kukovinets, T. I. Zvereva, N. N. Kabalnova, V. G. Kasradze, E. V. Salimova, L. R. Khalitova, M. I. Abdullin and L. V. Spirikhin, Ozonolysis of verbenone in aprotic solvents, Mendeleev Commun., 2009, 2, 106–107 CrossRef.
- I. E. DenBesten and T. H. Kinstle, Low-temperature ozonation of alkenes adsorbed on silica gel, J. Am. Chem. Soc., 1980, 102, 5968–5969 CrossRef CAS.
- R. H. Perry Jr, Ozonolysis of Norbornylene, J. Org. Chem., 1959, 24, 829–833 CrossRef.
- C. W. Jefford, J. Boukouvalas, D. Jaggi, S. Kohmoto and G. Bernardinelli, Intramolecular Oxygen Transfer on the Ozonolysis of a Diphenylcyclopentene Derivative, Helv. Chim. Acta, 1986, 69, 941–948 CrossRef.
- K. Teshima, S.-i. Kawamura, Y. Ushigoe, M. Nojima and K. J. McCullough, Ozonolyses of Indene and of 1-Alkyl- and 1,1-Dialkyl-Substituted Indenes in Protic Solvents. Remarkable Effects of the Substituent Steric Bulk and the Solvent Nucleophilicity on the Direction of Cleavage of the Primary Ozonides and on the Mode of Capture of the Carbonyl Oxide Intermediates by the Solvents, J. Org. Chem., 1995, 60, 4755–4763 CrossRef.
- K. J. McCullough, K. Teshima and M. Nojima, Unprecedented formation of a cyclic tetramer from the acidolysis of indene ozonide. Isolation and characterisation of a novel dodecaoxacycloicosane derivative, J. Chem. Soc., Chem. Commun., 1993, 931–933 RSC.
- W. H. Bunnelle and T. A. Isbell, The influence of remote heteroatom substituents on the stereoselectivity of cyclopentene ozonolysis, J. Org. Chem., 1992, 57, 729–740 CrossRef.
- V. V. Palyulin, T. Ala-Nissila and R. Metzler, Polymer translocation: the first two decades and the recent diversification, Soft Matter, 2014, 10, 9016–9037 RSC.
- K. I. Popov, V. V. Palyulin, M. Möller, A. R. Khokhlov and I. I. Potemkin, Surface induced self-organization of comb-like macromolecules, Beilstein J. Nanotechnol., 2011, 2, 569–584 CrossRef PubMed.
- I. I. Potemkin and V. V. Palyulin, Comblike macromolecules, Polym. Sci., Ser. A, 2009, 51, 123–149 CrossRef.
- A. I. Ilovaisky, V. M. Merkulova, E. I. Chernoburova, M. A. Shchetinina, D. I. Salnikova, A. M. Scherbakov, I. V. Zavarzin and A. O. Terent'ev, Secosteroidal hydrazides: Promising scaffolds for anti-breast cancer agents, J. Steroid Biochem. Mol. Biol., 2021, 214, 106000 CrossRef.
- E. Juaristi, G. dos Passos Gomes, A. O. Terent'ev, R. Notario and I. V. Alabugin, Stereoelectronic Interactions as a Probe for the Existence of the Intramolecular α-Effect, J. Am. Chem. Soc., 2017, 139, 10799–10813 CrossRef.
- CCDC 2483227 (3a): Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2pc036.
| This journal is © The Royal Society of Chemistry 2025 |