
P vacancy-induced electron redistribution and phase reconstruction of CoFeP for overall water splitting at industrial-level current density†
Xueling
Wei‡
,
Yang
Jiao‡
,
Xiangyu
Zou
 *,
Yuchen
Guo
,
Wenhu
Li
* and
Taotao
Ai
*
*,
Yuchen
Guo
,
Wenhu
Li
* and
Taotao
Ai
*
School of Materials Science and Engineering, Shaanxi University of Technology, Hanzhong 723000, Shaanxi, PR China. E-mail: zou800403@163.com; ccwhli@163.com; aitaotao016@163.com
First published on 19th February 2025
Abstract
Hydrogen production through water splitting using transition metal-based phosphide electrocatalysts represents a highly promising and sustainable energy conversion strategy. In this study, phosphating and vacancy engineering are achieved via a single-step Ar plasma-assisted process. CoFePv with phosphorus vacancies (Pv) as a bifunctional electrocatalyst effectively promotes both the oxygen evolution reaction (OER) and the hydrogen evolution reaction (HER), thereby significantly facilitating overall water splitting (OWS) in alkaline media. In the OER and HER processes, the driving overpotentials needed to attain a current density of 1 A cm−2 are only 382 and 367 mV, respectively. Furthermore, the CoFePv (+, −) OWS electrolyzer is capable of maintaining a current density of 2 A cm−2 at 1.98 V under simulated industrial settings (6 M KOH and 80 °C). It also demonstrates stable performance at a current density of 0.5 A cm−2 for a duration of 100 hours. In situ Raman spectroscopy observations show that Pv induce rapid catalyst phase reconstruction, thereby significantly enhancing the OER performance of CoFePv. Density functional theory (DFT) calculations demonstrate that phosphorus vacancies can modulate the electronic configuration of Co–Fe–P, facilitate electron transfer, and optimize the adsorption and desorption of reaction intermediates.
1. Introduction
The depletion of primary energy resources and the intensification of carbon emissions pose significant challenges to sustainable development.1,2 Leveraging renewable energy to its fullest extent and converting industrial waste and readily available raw materials into high-value, versatile products represent an effective strategy for addressing these challenges.3,4 Green-energy-driven electrochemical water splitting for hydrogen generation also represents an efficient and eco-friendly strategy to mitigate environmental pollution and alleviate the energy crisis. In the context of OWS, the OER occurs at the anode while the HER takes place at the cathode.5,6 Catalysts with bifunctional OER/HER capabilities can significantly simplify the preparation procedures.7–9Pt and Ru/Ir-oxides are well known as benchmark catalysts for water splitting.10–12 Nevertheless, the high cost, limited availability, and instability at high current densities restrict their industrial applications.13 In recent decades, researchers have developed a diverse array of cost-effective transition metal (TM) compounds,14 such as transition metal phosphides (TMPs),15 transition metal oxides (TMOs),16 transition metal sulfides (TMSs),17 transition metal nitrides (TMNs),18 and transition metal carbides (TMCs).19 Studies have shown that these materials exhibit substantial catalytic activity for water electrolysis.
Among these, TMPs exhibit excellent electrical conductivity, a flexible and tunable structure characterized by the stoichiometric ratio of phosphorus and metals, rapid charge transfer, and enhanced reaction kinetics.20 Some investigations indicate that negatively charged phosphorus atoms promote the dissociation of H2O molecules into *H or *OH species, thereby regulating the binding affinity of intermediates during the HER.21 During the OER process, phosphorus oxidation facilitates the transformation of active species into oxides/hydroxides, enhancing catalytic efficiency.22 This demonstrates the potential application of TMPs as bifunctional catalysts.
In recent years, scholars have employed a variety of strategies to conduct in-depth investigations into the dual-functional properties of TMPs. Heterogeneous structure engineering is one of the effective approaches for constructing efficient catalysts. Li et al. employed hydrothermal and chemical vapor deposition (CVD) phosphating methods to in situ construct a hybrid structure of CeO2/NiCoPx on Ni–Co foam, acting as a robust and efficient bifunctional catalyst in water splitting. The electrolyzer coupled with CeO2–NiCoPx/NCF only needs a low cell potential of 1.49 V to obtain a current density of 10 mA cm−2 and maintain stability for over 50 hours.23
Motivated by the concept that heteroatom doping can enhance electrical conductivity and expose more active sites, Ru-doped NiCoP was synthesized via etching and CVD phosphidation approaches. Notably, Ru–NiCoP@NF exhibits nearly 100% faradaic efficiency during overall water splitting.24
Defect-rich Ni-doped CoP3 was synthesized via plasma-assisted phosphorization, requiring overpotentials of only 107 mV and 306 mV to drive the HER and OER, respectively.25 Jiang et al. employed atmospheric pressure O2 plasma- and N2 plasma-assisted phosphating to develop p-VNiCoPy/NiFeOx with abundant phosphorus and oxygen vacancies.26 This catalyst demonstrated excellent durability and stability for both the HER and the OER for 100 hours of water electrolysis at 50 mA cm−2. These findings indicate that engineering defects and vacancies is an efficient approach for improving the bifunctional catalytic performance of phosphides.
However, although TMP catalysts exhibit remarkable catalytic activity and reliability under laboratory conditions (1 M KOH and ∼25 °C) to achieve industrial applications, they must exhibit reliable performance at high current densities (≥0.2 A cm−2) for over 100 hours under harsh conditions (20–30 wt% KOH and 60–90 °C). Ideal catalysts should possess superior robust bifunctional catalytic activity under industrial conditions.27,28
It is worth noting that under industrial conditions, solid–liquid–gas mass transfer kinetics and reaction rate kinetics are critical factors influencing the performance of OWS.29,30 In scenarios of large current density, the extensive production of bubbles on the surface of electrodes may obstruct mass transfer, thereby slowing down ion diffusion rates.31 Hence, constructing nanostructures with superhydrophilic and underwater superaerophobic properties can effectively promote mass transport during the OWS process at high current densities.32
In this work, for the development of facile and controllable synthesis methods for self-supported TMPs, electrochemical deposition and plasma-assisted phosphorization were employed to rapidly synthesize CoFeP with phosphorus vacancies (CoFePv). In situ Raman spectroscopy was employed to examine the dynamic evolution during the OER process, confirming that P vacancies accelerate phase reconstruction from CoFeP to CoFeOOH. DFT calculations disclosed that P vacancies modulate the electronic configuration of CoFeP, thereby lowering the energy barrier of the rate-determining steps (RDS) for the HER (*H2O → *OH + *H). Additionally, the superhydrophilic/superaerophobic nature of CoFePv facilitates efficient electrolyte penetration and expedites bubble detachment. The self-assembled CoFePv/NF (+, −) electrolyzer demonstrated excellent 100-hour stability at a current density of 500 mA cm−2 in 6 M KOH at 80 °C. This technique offers an efficient and convenient means to develop stable, high-performance bifunctional TMP catalysts, specifically tailored for industrial alkaline water splitting.
2. Experimental
2.1. Preparation of CoFe@NF
0.078 g of Co(NO3)2·6H2O and 0.028 g of FeSO4·7H2O were dissolved in 50 mL of deionized water and sonicated for 0.5 h. A platinum foil, a saturated calomel electrode (SCE), and a piece of nickel foam (NF) were utilized as the counter electrode (CE), reference electrode (RE), and working electrode (WE), respectively. Electrochemical deposition was performed at −1.0 V for 300 s. The deposited NF was then rinsed in alternating cycles with deionized water and ethanol and subsequently dried in an oven at 60 °C. This process led to the formation of CoFe-LDHs@NF, hereafter referred to as CoFe@NF.2.2. Preparation of CoFePv@NF and CoFeP@NF
2.0 g of NaH2PO2 and CoFe@NF were positioned upstream and downstream in a plasma emission tube, respectively. The argon plasma radio-frequency (RF) power was set to 200 W, with a processing duration of 6 minutes. Following natural cooling to ambient temperature, the prepared electrode was designated as CoFePv@NF. Additionally, a series of CoFePv@NF samples subjected to varying Ar plasma-assisted phosphating durations (3 minutes and 9 minutes) were also prepared. CoFeP@NF was synthesized under an Ar atmosphere without activating the RF power, heated at a rate of 2 °C min−1 up to 350 °C, and maintained at this temperature for 2 hours.2.3. Preparation of Pt/C@NF and RuO2@NF
For the fabrication of commercial Pt/C@NF and RuO2@NF electrodes, 40 mg of commercial Pt/C or RuO2 powder was mixed with a solution containing of 60 μL of Nafion, 540 μL of ethanol, and 1 mL of deionized water. The resultant suspension was subjected to ultrasonic treatment for 30 minutes to achieve a uniform ink consistency. Following this, the homogenized ink was uniformly dispensed onto the surface of a 1 × 1 cm2 NF and then dried at ambient temperature.The comprehensive details about reagents, substrate preparation, general material characterization, electrochemical characterization and density functional theory (DFT) calculations are presented in the ESI.†
3. Results and discussion
3.1. Characterization of catalysts
This research aimed to create a rapid and reliable approach for synthesizing phosphorus-vacancy TMPs. High-energy Ar plasma-assisted treatment was utilized to concurrently achieve phosphating and introduction of phosphorus vacancies in the precursor (Fig. 1a). Fig. 1b shows the SEM image of CoFePv. Compared to the precursor CoFe LDH nanosheets (Fig. S1†), the CoFePv nanosheets (Fig. S2†) and CoFeP (Fig. S3†) are noticeably thicker and rougher after phosphating. As shown in Fig. S2,† prolonged exposure to high-energy plasma results in the gradual degradation of the initial structure, which is detrimental to the formation of three-dimensional architectures. As illustrated in Fig. S4,† the optical images of electrode samples at each preparation stage highlight distinct color variations among samples derived from different processes. | ||
| Fig. 1 (a) The process for synthesizing CoFePv@NF. (b) SEM image, (c) TEM image, and (d) HRTEM image of CoFePv@NF. (e) Pseudocolored HRTEM image of CoFePv. (f) Line contours obtained from the region marked with a rectangle. (g) TEM image and the corresponding EDS mapping of CoFePv@NF. | ||
The TEM images of CoFePv revealed that the edges of the CoFePv nanosheets were rough (Fig. 1c). As evidenced by the high-resolution transmission electron microscopy (HRTEM) images presented in Fig. 1d, the images revealed characteristic interplanar distances of 0.19 and 0.18 nm, aligning with the (211) and (103) lattice planes of CoP, respectively. The lattice spacing of 0.20 nm aligned with the (112) plane of FeP. HRTEM analysis also confirmed the presence of phosphorus vacancies, as indicated by the bright red spots representing phosphorus atoms and the darker, smaller spots signifying phosphorus vacancies (Fig. 1e and 1f). The TEM image and the corresponding EDS mapping presented in Fig. 1h indicate that Co, Fe, P, and O elements were uniformly distributed throughout the structure. As shown in Fig. 2a, the main peaks in the XRD pattern of CoFePv correspond to CoP (PDF#29-0497) and FeP (PDF#65-4899), suggesting that phosphorus vacancies do not significantly alter the main crystalline phase.33 The mechanism of phosphorus vacancy generation during plasma-induced phosphating is illustrated in Fig. 2b. Under high-energy Ar plasma bombardment, NaH2PO2 rapidly decomposes into PH3, which subsequently reacts with the precursor to form CoFeP. Simultaneously, the Ar plasma induces the desorption of surface P atoms from CoFeP as PH3, thereby creating phosphorus vacancies in situ within the material.
 | ||
| Fig. 2 (a) XRD patterns of the electrodes. (b) Schematic of phosphorus vacancy formation. (c) ICP-OES mass ratios and (d) EPR spectra of CoFeP@NF and CoFePv@NF. (e) Co 2p, (f) Fe 2p, and (g) P 2p high-resolution XPS spectra of CoFeP@NF and CoFePv@NF. (h) Water contact angles and (i) water-under-gas bubble contact angles of CoFeP@NF and CoFePv@NF. | ||
The P content in the CoFeP@NF and CoFePv@NF electrodes was precisely determined using inductively coupled ICP-OES (Table S1†). The analysis shows that CoFeP@NF has a P mass percentage of 1.13% compared to 0.74% in CoFePv@NF (Fig. 2c). This confirms successful phosphating via both strategies, with the plasma-induced method yielding lower P content due to its shorter duration.
EPR spectroscopy in Fig. 2d shows that CoFePv exhibits a strong peak with a g value of 2.004, compared with the weak signal from CoFeP. This indicates the successful introduction of phosphorus vacancies during plasma-assisted phosphating.34
The full XPS spectra (Fig. S5†) of CoFeP and CoFePv verified the coexistence of Fe, Co, P, and O elements. Notably, CoFePv exhibited a lower phosphorus content compared to that of CoFeP. In the high-resolution XPS spectrum of Co 2p for CoFeP (Fig. 2e), peaks at 777.4 and 792.8 eV are indicative of the presence of Co–P bonds,35 while those at 785.3 and 802.6 eV are satellite (Sat.) peaks.36 The observed peaks at 779.8 and 792.8 eV are associated with Co2+, whereas those at 781.6 and 798.3 eV are assigned to Co3+.37 Notably, the Co2+ peak at 797.1 eV of CoFePv shifts 0.5 eV higher in energy compared to that of CoFeP, while the Co3+ peak at 798.9 eV exhibits a similar shift of approximately 0.6 eV.
In Fig. 2f, the Fe 2p spectrum of CoFeP shows that the peaks at 734.4 and 718.2 eV are satellite peaks. The peaks located at 712.2 and 721.8 eV are associated with Fe2+, whereas the peaks at 714.2 and 726.5 eV correspond to Fe3+.38,39 The peak located at 706.6 eV is the characteristic peak of the Fe–P bond,40 which is 0.3 eV higher than that of CoFePv. Compared with CoFeP, the binding energy of Fe3+ in CoFePv shifts to a lower value by 0.3 eV, while that of Fe2+ is shifted to a higher value by 0.2 eV. The introduction of P vacancies promotes electron transfer between Fe and Co. In Fig. 2g, the P 2p of CoFePv shows peaks at 130.5 eV (P 2p1/2), 129.6 eV (P 2p3/2), and 134.5 eV (P–O bond).41,42 Compared to CoFeP, the P–O peak in CoFePv exhibits a downshift of 0.4 eV and the P 2p3/2 peak shows a decrease of 0.3 eV. These shifts suggest that the introduction of phosphorus vacancies results in substantial electron accumulation around the phosphorus atoms within the catalyst.
Contact angle (CA) measurements were performed to evaluate the electrode's interaction with electrolytes and gases. The CoFePv@NF electrode showed rapid spreading upon contact with a water droplet, indicating its superhydrophilic nature (CA ≈ 0°) (Fig. 2h). The CA of CoFeP@NF was 44°, showing poorer hydrophilicity compared to the phosphorus-vacancy electrode. Additionally, the gas contact angle of CoFePv@NF was 141°, higher than that of CoFeP@NF (130°) (Fig. 2i), indicating superior hydrophobicity to CoFeP@NF. The superior hydrophilicity towards liquids and superaerophobicity towards gases exhibited by CoFePv@NF will enhance reactant diffusion, thereby promoting rapid chemical reaction kinetics.43
3.2. HER and OER catalytic performance
To investigate the electrocatalytic properties of different electrodes, the OER/HER performance of CoFePv@NF, CoFeP@NF, and CoFe@NF, as well as Pt/C@NF and RuO2@NF, was evaluated using 1 M KOH. As shown in Fig. 3a, the phosphorized electrodes achieve ampere-level current densities well within the applied potential range, indicating enhanced HER/OER activity. Impressively, the CoFePv@NF catalyst exhibits excellent HER/OER performance, reaching an industrial-level current density of 1.0 A cm−2 with overpotentials of only 367 mV for the HER and 382 mV for the OER. Simultaneously, the bifunctional activity of the electrodes prepared under varying durations of Ar plasma-assisted phosphating treatment was also investigated (Fig. S6†).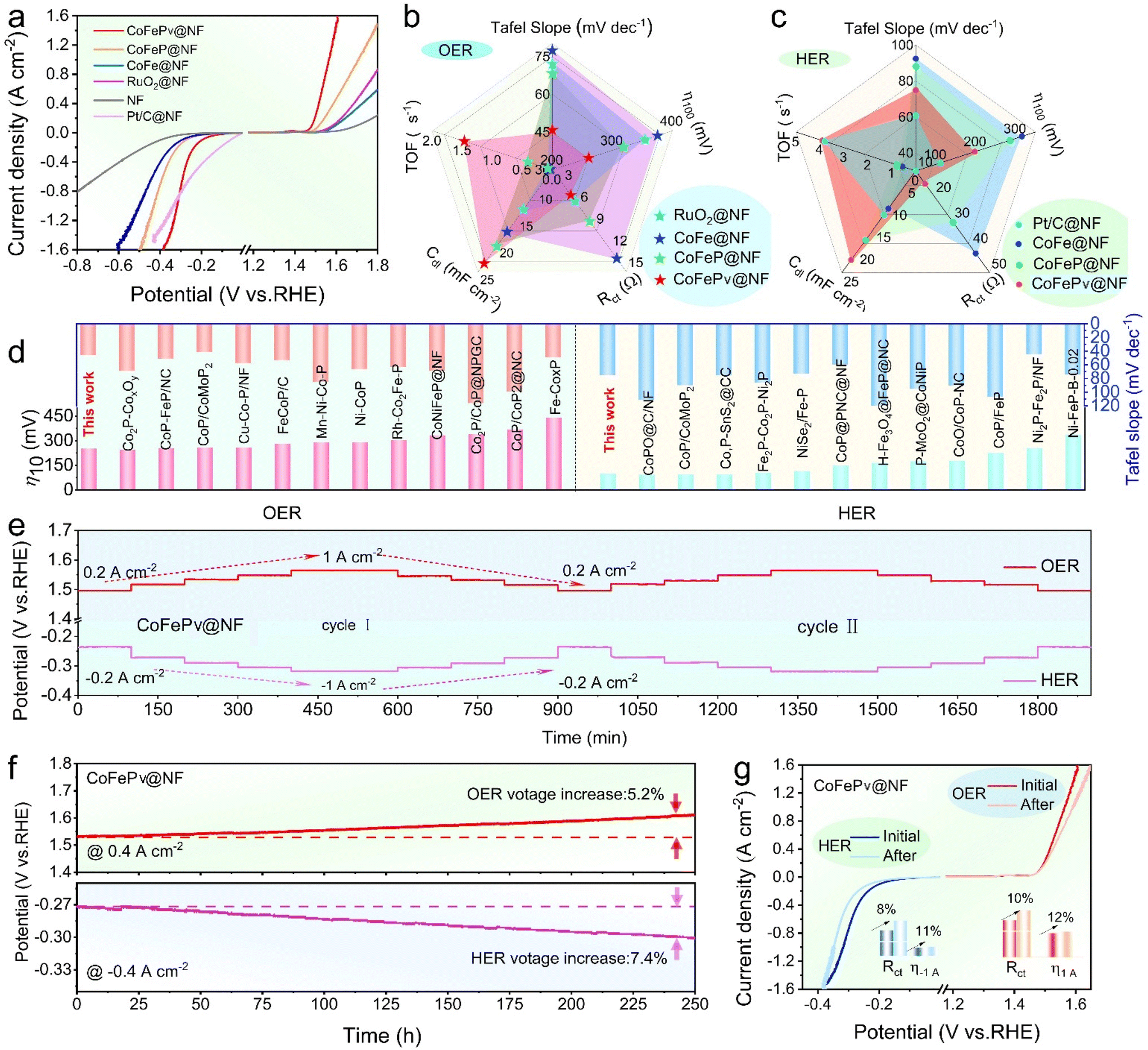 | ||
| Fig. 3 (a) LSV curves of the OER/HER for the electrodes. Comprehensive comparisons of the (b) OER and (c) HER performance of the electrodes. (d) Comparison of the overpotential and Tafel slope of CoFePv@NF with those of recently reported TMPs. (e) Multi-step current curves and (f) OER and HER duration curves of CoFePv@NF. (g) LSV curves of CoFePv@NF before and after the stability test; insets: Rct and overpotentials at 1 A cm−2. | ||
Furthermore, as shown in Fig. S7,† the catalytic kinetics of the electrodes were evaluated using Tafel slopes. During the OER process, the Tafel slope of CoFePv@NF is 45.8 mV dec−1, which is substantially lower compared to the values observed for CoFeP@NF (68.3 mV dec−1), CoFe@NF (77.4 mV dec−1), and RuO2@NF (72.0 mV dec−1). For the HER process, the Tafel slopes followed the order: Pt/C@NF (60.9 mV dec−1) < CoFePv@NF (74.9 mV dec−1) < CoFeP@NF (88.3 mV dec−1) < CoFe@NF (92.4 mV dec−1). These findings indicate that the CoFePv@NF catalyst exhibits excellent charge transfer kinetics.44
As shown in Fig. S8,† the Nyquist plots reveal that at an overpotential of 200 mV, the charge transfer resistance (Rct) for CoFePv@NF during the HER and OER is 15.1 Ω and 4.5 Ω, respectively. These values are lower than those of CoFeP@NF (30.1/8.1 Ω) and CoFe@NF (42.4/13.2 Ω). Moreover, the Rct values of CoFePv@NF are comparable to those of RuO2@NF and Pt/C@NF. This indicates that the CoFePv@NF catalyst exhibits reduced charge transfer resistance during electrochemical reactions.
The cyclic voltammetry (CV) measurements conducted in the non-faradaic region are provided in the ESI (Fig. S9 and 10†). The Cdl value of CoFePv@NF during the HER/OER is 22.0/23.5 mF cm−2, while the corresponding calculated ECSA values for the HER and OER are 55.0 cm2 and 58.8 cm2, respectively. To gain a comprehensive understanding of the inherent activity of the as-prepared electrodes, the current density (j) normalized by the ECSA and the turnover frequency (TOF) were assessed, following established methodologies in the literature.45 As illustrated in Fig. S11,† the ECSA-normalized polarization curves revealed that at an overpotential of 300 mV, CoFePv@NF reached current densities of 1.22 mA cm−2ECSA and 1.01 mA cm−2ECSA during the HER and OER, respectively. This indicates a higher intrinsic electrocatalytic activity compared to other samples.46
The TOF was calculated at an overpotential of 300 mV for the HER/OER. As shown in Fig. S12,† the TOF for CoFePv@NF in the HER process is 3.81 s−1, which is comparable to that of Pt/C@NF (4.20 s−1). For the OER, the TOF of CoFePv@NF was 2.14 s−1, approximately twice that of RuO2@NF. It is also higher than that of any other reference samples. These findings verify that CoFePv@NF possesses abundant active sites and exhibits high intrinsic catalytic activity.47
The test results were summarized to comprehensively compare the catalytic performance of CoFePv@NF, CoFeP@NF, CoFe@NF, and commercial electrodes in the OER (Fig. 3b) and HER (Fig. 3c) processes. The radar chart clearly demonstrates that the HER/OER catalytic performance of CoFePv@NF is significantly enhanced due to the synergistic effect of the components and phosphorus vacancies.
By comparing the HER/OER overpotentials at a current density of 0.2 A cm−2 (η200) and the Tafel slope with those of bifunctional phosphides, the superior alkaline water-splitting capability of CoFePv@NF is confirmed (Fig. 3d and Tables S2, S3†). Fig. 3e illustrates the multi-step current density curves of CoFePv@NF. The testing current density ranges for the HER are from −0.2 to −1.0 A cm−2, while those for the OER are from 0.2 to 1.0 A cm−2. At each current density step, the required voltage remains constant and the curves show negligible variation during the HER/OER cycling tests, indicating superior stability. Additionally, the durability of the electrode was assessed through chronopotentiometry at a fixed current density of 0.4 A cm−2. As illustrated in Fig. 3f, the final potential of CoFePv@NF exhibited only a 5.2% increase for the OER and a 7.4% increase for the HER compared to the initial values, demonstrating its stable performance over a 250-hour duration.
To further assess the stability of CoFePv@NF, its LSV curves were compared before and after the durability tests. In Fig. 3g, the LSV curves exhibit negligible changes. As evidenced by the inset in Fig. 3g, at an overpotential of 200 mV, the Rct for the HER increased by 8%, while that for the OER increased by 11%. Additionally, the overpotentials required to achieve a current density of 1 A cm−2 increased by 10% for the HER and 12% for the OER.
For unveiling the superior activity and stability, the phase composition, morphology, microstructure, and valence state of CoFePv@NF after stabilization in the HER/OER processes were comprehensively characterized. The XRD patterns of CoFePv@NF after the HER/OER still exhibit peaks for FeP (011) and (211) and CoP (011), (111), (112), and (211) planes (Fig. S13†). After the OER, new peaks appear at 20.2° and 65.4° for CoOOH (003) and (110) planes and at 14.2° for the FeOOH (020) plane.
The SEM images (Fig. S14†) indicate that CoFePv@NF retains its original nanoflake skeleton morphology after undergoing the HER/OER. The HRTEM images of CoFePv@NF show distinct lattice fringes for the (112) plane of CoP and the (212) plane of FeP (Fig. S15†). After the OER process, lattice fringes for the (101) plane of CoOOH and the (130) plane of FeOOH become evident (Fig. S16†).
The XPS spectra of CoFePv@NF after stability testing are shown in Fig. S17.† After the HER, the Co 2p, Fe 2p, and P 2p spectra exhibit a resemblance to their pre-HER states, maintaining clear Co/Fe–P peaks. However, after the OER, the Co 2p, Fe 2p, and P 2p spectra differ from those before the OER. The Co/Fe–P peaks disappear, while the intensities of the Co3+ and Fe3+ peaks increase significantly. These results suggest that CoFePv@NF undergoes surface reconstruction during the OER. Previous studies indicate that phosphides act as pre-catalysts, forming oxide layers on their surfaces in oxidative environments.25
3.3. Mechanism for the enhanced OER
To examine the interface reaction of CoFePv@NF during the OER, in situ Bode and EIS plots were utilized. As depicted in Fig. 4a–c, the in situ Bode plots illustrate the changes in the phase angle with frequency, reflecting the dynamic evolution of different catalysts under applied voltages ranging from 1.28 to 1.68 V vs. RHE. Typically, the peak phase angles at intermediate frequencies are attributed to inner-layer electron transfer, whereas those at low frequencies are indicative of surface charge conduction.48 These phenomena indicate the electrochemical oxidation reactions and the OER process occurring on the electrocatalyst. | ||
| Fig. 4 In situ phase diagrams at different applied potentials: (a) CoFePv@NF, (b) CoFeP@NF, and (c) CoFe@NF. In situ Raman spectra at different applied potentials: (d) CoFePv@NF, (e) CoFeP@NF, and (f) CoFe@NF. | ||
A smaller phase angle drop indicates faster electron transfer.49 When an electric potential was applied, CoFePv@NF showed a rapid phase angle decrease in the mid-frequency region, whereas CoFeP@NF showed a slower decline. For CoFe@NF, the phase angle decreased gradually from high to low frequencies. This suggests that the electrooxidation process of CoFePv@NF is significantly faster and more intense than that of the other catalysts.50 Once the applied voltage reached 1.42 V vs. RHE, CoFePv@NF initiated the OER, while CoFeP@NF and CoFe@NF required 1.46 and 1.50 V vs. RHE, respectively.
Nyquist plots (Fig. S19†) show that within the same potential range, CoFePv@NF has a significantly lower charge transfer resistance. This indicates that Pv enhance interfacial charge transfer and surface activation, thereby improving the OER electrocatalytic performance.
To further elucidate the role of Pv in the CoFePv@NF catalyst, in situ Raman analysis during the OER was performed (Fig. 4d and e). In the OER process, the crystal surface of Co/Fe transformed into a more defective or disordered CoFeOOH phase. For CoFePv@NF, a peak emerged between 900 and 1000 cm−1 at 1.37 V vs. RHE, suggesting the initial formation and adsorption of PO43− on the surface of the electrode in the electrolysis process.16 In contrast, for the CoFeP@NF electrode, the PO43− peak was observed at a potential of 1.42 V vs. RHE. This indicates that the presence of vacancies in CoFePv@NF promotes earlier oxidation and activation of the electrode.51
CoFePv@NF exhibits a Raman band attributed to CoOOH (450–550 cm−1)52–54 and FeOOH (600–700 cm−1),55,56 which begins to appear at 1.37 V vs. RHE and progressively intensifies with increasing voltage. In contrast, the CoFeOOH Raman bands for CoFeP@NF and CoFe@NF start to emerge at 1.42 and 1.47 V vs. RHE, respectively. This suggests that the presence of Pv significantly facilitates the local transformation of the crystal structure into active hydroxide oxides. Specifically, Pv reduce the surface electrochemical reorganization energy barrier during the OER process, thereby accelerating its oxidation and enhancing the overall water splitting catalytic performance.
3.4. Overall water splitting performance
Based on the superior bifunctional catalytic performance, an OWS electrolyzer utilizing CoFePv@NF as both electrodes (CFPE) was successfully developed. Furthermore, a commercial electrolyzer featuring RuO2@NF as an anode and Pt/C@NF as a cathode (RPE) was also constructed.The OWS performance was assessed in 1 M KOH aqueous solution at room temperature. As shown in Fig. 5a, the LSV curve of CFPE demonstrates a marked superiority compared to that of the RPE. The CFPE requires 1.90 V to achieve a current density of 0.5 A cm−2, lower than the 2.05 V needed for the RPE. Additionally, the CFPE reaches 1 A cm−2 at only 2.09 V.
 | ||
| Fig. 5 (a) LSV curves of OWS. (b) Comparative cell voltages of bifunctional catalysts for OWS at 0.5 A cm−2. (c) Photograph of oxygen collection. (d) Corresponding FE efficiency. (e) Polarization curves of CoFePv@NF (+, −) with different KOH concentrations. (f) Long-term stability of CoFePv@NF (+, −) in 6.0 M KOH at 80 °C; inset: photographs of the electrode surface during OWS. | ||
The catalytic performance of the CFPE was evaluated against recently reported bifunctional electrolyzers, as illustrated in Fig. 5b and Table S6.† The results indicate that the CFPE operates at a lower voltage in comparison with the others. The drainage method (Fig. S19†) was employed to further evaluate the Faraday efficiency (FE) of the CFPE. Fig. 5c shows detailed images documenting the time-dependent oxygen collection process. Theoretical calculations in Fig. 5d show that the average FE for OWS in the CFPE is 98.2%.
To assess the industrial application potential, the OWS performance of the CFPE was evaluated in KOH solutions ranging from 1 M to 6 M at 80 °C. As illustrated in Fig. 5e, the OWS efficiency improves with increasing KOH concentration. Notably, in 5 M KOH, with a voltage of 2.0 V, the CFPE can achieve a current density of 2.0 A cm−2, underscoring its exceptional catalytic activity and significant potential for industrial applicability.
To evaluate the durability of the CFPE under severe conditions, a chronoamperometry test was conducted at 0.5 A cm−2 in 6 M KOH at 80 °C. As illustrated in Fig. 5f, following 100 hours of testing, the cell voltage exhibited an increase of merely 9.8%. The inset image depicts the electrode surface during the test, revealing the formation and rapid overflow of bubbles. These results show that the CFPE can operate stably at high current densities under simulated industrial conditions.
3.5. DFT calculations
CoP was selected as the model and a CoFeP crystal was constructed by partially substituting Co atoms with Fe atoms. Using DFT calculations, the adsorption energy of H2O at different sites of both CoFeP and CoFePv was first investigated. As illustrated in Fig. 6a, the adsorption energies of H2O at the Fe and Co sites on the CoFePv electrode are −0.85 eV and −0.75 eV, respectively, more negative than those on CoFeP (−0.80 eV and −0.69 eV, Fig. 6b). This suggests that phosphorus vacancies augment the electrode's capacity for water adsorption, which aligns with the enhanced hydrophilicity of the CoFePv electrode observed in Fig. 2h. | ||
| Fig. 6 Adsorption energy of H2O at different sites: (a) CoFePv and (b) CoFeP. (c) Charge density difference of CoFePv (yellow and blue represent electron accumulation and electron depletion, respectively). Total projected DOSs of CoFePv (d) and CoFeP (e). (f) Free energy profiles of OER intermediates for CoFeOOH-PO43− and CoFeOOH. (g) HER pathways of CoFePv and CoFeP under alkaline conditions. | ||
To investigate the charge transfer characteristics of CoFeP with and without phosphorus vacancies, the Bader charge distributions are presented in Fig. 6c, which indicates that Pv result in a substantial redistribution of electron density. Specifically, P atoms adjacent to the vacancy display pronounced charge accumulation, whereas Fe and Co atoms exhibit charge depletion. The incorporation of Pv facilitates synergistic interactions within the crystal structure, thereby refining the electronic configuration and boosting the catalytic efficiency.
The optimization mechanism was explained using the d-band center (Ed) of the metal catalyst as an electronic structure parameter57 and the impact of P vacancies on intermediate adsorption energy was evaluated. Fig. 6d and e show the density of states (DOS) for CoFePv and CoFeP, with Ed values of −0.87 eV and −0.92 eV, respectively. According to d-band center theory,58 an Ed near the Fermi level (Ef) enhances interactions between the catalyst surface and intermediates by increasing anti-bonding orbitals. Furthermore, the spin asymmetry in the 2d orbitals of CoFePv compared to those of CoFeP enhances the adsorption capability of the active sites.
In alkaline media, the OER mechanism involves four steps with multiple intermediates (*OH, *O, *OOH, and O2).59 By DFT calculations, the Gibbs free energy change (ΔG) for each step was analyzed. Based on in situ Raman characterization results, the CoFeP(Pv) and CoFe electrodes undergo structural transformation during the OER process. Therefore, CoFeOOH–PO43− (Fig. S20†) and CoFeOOH (Fig. S21†) models were constructed to calculate the ΔG value for each intermediate on active sites. The step with the highest ΔG value is identified as the RDS. As shown in Fig. 6f, for all optimized models, the RDS corresponds to the conversion from *OOH to O2 (*OOH + OH− → O2 + H2O + e−). The ΔG value of the RDS for CoFeOOH-PO43− is 1.97 eV, which is lower than the 2.28 eV observed for CoFeOOH. This indicates that the presence of PO43− reduces the energy barrier of the RDS, thereby enhancing the catalytic activity for the OER.
For alkaline HER, CoFePv (Fig. S22†) and CoFeP (Fig. S23†) were employed as comparative models in the DFT calculations. The results depicted in Fig. 6g indicate that water adsorption is energetically favorable on both surfaces, with the dissociation of *H2O acting as the RDS. Notably, the energy barrier for the RDS on CoFePv is 0.47 eV lower than that on CoFeP (0.65 eV). This confirms that the introduction of vacancies expedites the H2O dissociation process.
The H2 adsorption energy (ΔG*H) indicates the balance between hydrogen atom adsorption and H2 desorption from active sites. A catalyst with good intrinsic activity typically has a ΔG*H value near 0 eV. Compared to CoFeP (ΔG*H = −0.31 eV), CoFePv shows closer thermodynamic neutrality (−0.10 eV), as seen in Fig. S24.† This suggests that vacancies enhance charge redistribution, improving H adsorption and accelerating HER kinetics. The introduction of vacancies not only enhances H2O adsorption and dissociation but also optimizes *H adsorption energy, thereby significantly improving the HER activity at high current densities in alkaline media.
4. Conclusion
This research presents an Ar plasma-assisted phosphating method for the synthesis of superior bifunctional CoFePv@NF catalysts, which exhibit remarkable superhydrophilicity. Under simulated industrial conditions (6 M KOH and 80 °C), the CoFePv (+, −) OWS electrolyzer can achieve a current density of 2 A cm−2 at a voltage of 1.98 V. Furthermore, it exhibits stable performance at a current density of 0.5 A cm−2 for a duration of 100 hours, highlighting its significant potential for industrial applications.The outstanding catalytic performance of the CoFePv@NF electrode can be attributed to the introduction of phosphorus vacancies. The Pv optimize the electronic structure of Co–Fe–P, thereby facilitating enhanced electron transfer and fine-tuning the adsorption/desorption of intermediates. At the same time, Pv accelerate the reconstruction of active species during the OER process, ultimately boosting intrinsic catalytic activity.
Meanwhile, the superhydrophilic/superaerophobic properties promote efficient electrolyte penetration and rapid bubble removal, accelerating reaction kinetics and significantly improving catalytic stability at high current densities for both the HER and the OER. The proposed strategy offers a simple and effective method for rapidly fabricating high-performance transition metal phosphide bifunctional electrocatalysts with significant industrial potential for hydrogen evolution.
Author contributions
Xueling Wei: data curation, formal analysis, validation, and writing – original draft. Yang Jiao: formal analysis, methodology, and validation. Xiangyu Zou: software, conceptualization, supervision, visualization, and writing – review & editing. Yuchen Guo: formal analysis and methodology. Wenhu Li: conceptualization, resources, supervision, and writing – review & editing. Taotao Ai: project administration, supervision, and writing – review & editing.Data availability
The data supporting this article have been included as part of the ESI.†Data will be made available on request.
Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This work was financially supported by the Key Research and Development Program of Shaanxi (Program No. 2023-JC-YB-404 and 2024JC-ZDXM-32). Special thanks to Youjun Huang (Xi'an Technological University) for his guidance and support on DFT.References
- P. Zhang, M. Gan, Y. Song, P. Liu, H. Liang, Y. Shen, B. Xu, T. Liao, J. Guo and Z. Sun, Bifunctional Pt dual atoms for overall water splitting, Appl. Catal., B, 2024, 355, 124214, DOI:10.1016/j.apcatb.2024.124214
.
- Y. Tian, M. Wen, A. Huang, Q. Wu, Z. Wang, Q. Zhu, T. Zhou and Y. Fu, Significantly stabilizing hydrogen evolution reaction induced by Nb-doping Pt/Co(OH)2 nanosheets, Small, 2023, 19, 2207569, DOI:10.1002/smll.202207569
- J. Xian, S. Li, H. Su, P. Liao, S. Wang, Y. Zhang, W. Yang, J. Yang, Y. Sun, Y. Jia, Q. Liu, Q. Liu and G. Li, Electrocatalytic synthesis of essential amino acids from nitric oxide using atomically dispersed Fe on N-doped carbon, Angew. Chem., Int. Ed., 2023, 62, e202304007, DOI:10.1002/anie.202304007
- P. Liao, B. Zeng, S. Li, Y. Zhang, R. Xiang, J. Kang, Q. Liu and G. Li, Cu-Bi bimetallic catalysts derived from metal-organic framework arrays on copper foam for efficient glycine electrosynthesis, Angew. Chem., 2025, 64, e202417130, DOI:10.1002/anie.202417130
- Q. Chen, X. Deng, H. He, L. Jiang, C. Liang, J. Zhang, R. Wang and J. Chen, Modulating nanostructure and electronic structure of P and W co-doped carbon encapsulated heterovalent NiCo composite for accelerating urea oxidation reaction, Chem. Eng. J., 2024, 484, 149561, DOI:10.1016/j.cej.2024.149561
- Z. Zhang, X. Li, C. Zhong, N. Zhao, Y. Deng, X. Han and W. Hu, Spontaneous synthesis of silver-nanoparticle-decorated transition-metal hydroxides for enhanced oxygen evolution reaction, Angew. Chem., Int. Ed., 2020, 59, 7245–7250, DOI:10.1002/anie.202001703
- K. Shah, R. Dai, M. Mateen, Z. Hassan, Z. Zhuang, C. Liu, M. Israr, W. C. Cheong, B. Hu and R. Tu, Cobalt single atom incorporated in ruthenium oxide sphere: a robust bifunctional electrocatalyst for HER and OER, Angew. Chem., 2022, 134, e202114951, DOI:10.1002/anie.202114951
- Y. Luo, Y. Zhang, J. Zhu, X. Tian, G. Liu, Z. Feng, L. Pan, X. Liu, N. Han and R. Tan, Material engineering strategies for efficient hydrogen evolution reaction catalysts, Small Methods, 2024, 2400158, DOI:10.1002/smtd.202400158
- R. Xu, T. Lin, S. Cao, T. Bo, Y. Liu and W. Zhou, Unveiling spin magnetic effect of TM doped SnS2 nanosheet with enhanced hydrogen evolution reaction, J. Energy Chem., 2024, 99, 636–644, DOI:10.1016/j.jechem.2024.08.007
- J. Zheng and Z. Wang, Defect engineering for surface reconstruction of metal oxide catalysts during OER, Chem Catal., 2024, 4(11), 101091, DOI:10.1016/j.checat.2024.101091
- J. Zhou, P. Li, X. Xia, Y. Zhao, Z. Hu, Y. Xie, L. Yang, Y. Liu, Y. Du and Q. Zhou, Precisely tailoring the d-band center of nickel sulfide for boosting overall water splitting, Appl. Catal., B, 2024, 359, 124461, DOI:10.1016/j.apcatb.2024.124461
- Y. Zhu, C. Han and Z. Chen, Insights into the origin of Co-based bimetallic catalysts with a para-structure exhibiting ORR and OER bifunctional activities, Inorg. Chem. Front., 2024, 11, 4590–4602, 10.1039/d4qi00793j
- P. Hu, F. F. Yang, F. Yang, F. Zhu, J. Luo, X. Chen, W. Li, J. Bian, L. Gao and K. Wang, Bimetallic organic framework derived Co-MoxN/Mo2C catalyst for HER/OER bifunctional electrocatalytic reaction, J. Colloid Interface Sci., 2025, 680, 427–436, DOI:10.1016/j.jcis.2024.11.116
- H. Zhang, A. W. Maijenburg, X. Li, S. L. Schweizer and R. B. Wehrspohn, Bifunctional heterostructured transition metal phosphides for efficient electrochemical water splitting, Adv. Funct. Mater., 2020, 30, 2003261, DOI:10.1002/adfm.202003261
- D. Xu, J. Yao, X. Ma, Y. Xiao, C. Zhang, W. Lin and H. Gao, F, N neutralizing effect induced Co-PO cleaving endows CoP nanosheets with superior HER and OER performances, J. Colloid Interface Sci., 2022, 619, 298–306, DOI:10.1016/j.jcis.2022.03.123
- X. Guo, X. He, X. Liu, S. Sun, H. Sun, K. Dong, T. Li, Y. Yao, T. Xie and D. Zheng, Arming amorphous NiMoO4 on nickel phosphide enables highly stable alkaline seawater oxidation, Small, 2024, 2400141, DOI:10.1002/smll.202400141
- P. Tan, Y. Wu, Y. Tan, Y. Xiang, L. Zhou, N. Han, Y. Jiang, S.-J. Bao and X. Zhang, In situ fast construction of Ni3S4/FeS catalysts on 3D foam structure achieving stable large-current-density water oxidation, Small, 2024, 20, 2308371, DOI:10.1002/smll.202308371
- T. Wang, P. Wang, W. Zang, X. Li, D. Chen, Z. Kou, S. Mu and J. Wang, Nanoframes of Co3O4-Mo2N heterointerfaces enable high-performance bifunctionality toward both electrocatalytic HER and OER, Adv. Funct. Mater., 2022, 32, 2107382, DOI:10.1002/adfm.202107382
- X. Ning, S. Zhang, D. Yin, P. Du and X. Lu, In operando visualization of charge transfer dynamics in transition metal compounds on water splitting photoanodes, Adv. Energy Mater., 2025, n/a, 2405137, DOI:10.1002/aenm.202405137
- Q. Zhang, X. Zeng, Z. Zhang, C. Jin, Y. Cui and Y. Gao, Electronic transfer and structural reconstruction in porous NF/FeNiP-CoP@NC heterostructure for robust overall water splitting in alkaline electrolytes, J. Colloid Interface Sci., 2024, 675, 357–368, DOI:10.1016/j.jcis.2024.07.019
- T. Zhang, T. Yang, B. Li, S. Wei and W. Gao, MOF-derived formation of ultrafine FeP nanoparticles confined by N/P Co-doped carbon as an efficient and stable electrocatalyst for hydrogen evolution reaction, Appl. Surf. Sci., 2022, 597, 153662, DOI:10.1016/j.apsusc.2022.153662
- Y. Yuan, Y. Yang, H. Xie, X. Zhong, R. Wang and Z. Xu, Trace Fe doping improved the OER and HER catalytic performance of CoP hollow nanoflower clusters, Int. J. Hydrogen Energy, 2024, 90, 1401–1410, DOI:10.1016/j.ijhydene.2024.10.122
- S. Wen, J. Huang, T. Li, W. Chen, G. Chen, Q. Zhang, X. Zhang, Q. Qian and K. Ostrikov, Multiphase nanosheet-nanowire cerium oxide and nickel-cobalt phosphide for highly-efficient electrocatalytic overall water splitting, Appl. Catal., B, 2022, 316, 121678, DOI:10.1016/j.apcatb.2022.121678
- D. Chen, R. Lu, Z. Pu, J. Zhu, H.-W. Li, F. Liu, S. Hu, X. Luo, J. Wu, Y. Zhao and S. Mu, Ru-doped 3D flower-like bimetallic phosphide with a climbing effect on overall water splitting, Appl. Catal., B, 2020, 279, 119396, DOI:10.1016/j.apcatb.2020.119396
- D. Zha, R. Wang, S. Tian, Z.-J. Jiang, Z. Xu, C. Qin, X. Tian and Z. Jiang, Defect engineering andcarbon supporting to achieve Ni-doped
CoP3 with high catalytic activities for overall water splitting, Nano-Micro Lett., 2024, 16, 250, DOI:10.1007/s40820-024-01471-9
- J. Zhang, W. Nie, R. Wang, C. Qin, Z.-J. Jiang, G.-r. Jin and Z. Jiang, Plasma technique regulates the electronic structure and dual functional catalytic performance of p-VNiCoPy/NiFeOx heterojunction catalysts for hydrogen production through overall water splitting, J. Alloys Compd., 2024, 1004, 175900, DOI:10.1016/j.jallcom.2024.175900
- H. Chen, R. Ding, B. W. Liu, F. R. Zeng and H. B. Zhao, Electrocatalytic coenhancement of bimetallic polyphthalocyanine-anchored Ru nanoclusters enabling efficient overall water splitting at ampere-level current densities, Small, 2024, 20, 2306274, DOI:10.1002/smll.202306274
- J.-T. Ren, L. Chen, H.-Y. Wang, W.-W. Tian, S.-X. Zhai, Y. Feng and Z.-Y. Yuan, Modulating interfacial charge distribution of Ni2P-NiSe2 by multiple interface engineering for accelerating water splitting with industry-level activity and stability, Appl. Catal., B, 2024, 347, 123817, DOI:10.1016/j.apcatb.2024.123817
- J. Hou, Y. Wu, B. Zhang, S. Cao, Z. Li and L. Sun, Rational design of nanoarray architectures for electrocatalytic water splitting, Adv. Funct. Mater., 2019, 29, 1808367, DOI:10.1002/adfm.201808367
- X. Shan, J. Liu, H. Mu, Y. Xiao, B. Mei, W. Liu, G. Lin, Z. Jiang, L. Wen and L. Jiang, An engineered superhydrophilic/superaerophobic electrocatalyst composed of the supported CoMoS, Angew. Chem., Int. Ed., 2020, 59, 1659–1665, DOI:10.1002/anie.201911617
- H. Sun, L. Li, M. Humayun, H. Zhang, Y. Bo, X. Ao, X. Xu, K. Chen, K. K. Ostrikov and K. Huo, Achieving highly efficient pH-universal hydrogen evolution by superhydrophilic amorphous/crystalline Rh(OH)3/NiTe coaxial nanorod array electrode, Appl. Catal., B, 2022, 305, 121088, DOI:10.1016/j.apcatb.2022.121088
- F. Diao, W. Huang, G. Ctistis, H. Wackerbarth, Y. Yang, P. Si, J. Zhang, X. Xiao and C. Engelbrekt, Bifunctional and self-supported NiFeP-layer-coated NiP rods for electrochemical water splitting in alkaline solution, ACS Appl. Mater. Interfaces, 2021, 13, 23702–23713, DOI:10.1021/acsami.1c03089
- K. Wang, X. Liu, Q. Yu, X. Wang, J. Zhu, Y. Li, J. Chi, H. Lin and L. Wang, Mn doping and P vacancy induced fast phase peconstruction of FeP for enhanced electrocatalytic oxygen evolution reaction in alkaline seawater, Small, 2024, 20, 2308613, DOI:10.1002/smll.202308613
- G. Yuan, J. Bai, L. Zhang, X. Chen and L. Ren, The effect of P vacancies on the activity of cobalt phosphide nanorods as oxygen evolution electrocatalyst in alkali, Appl. Catal., B, 2021, 284, 119693, DOI:10.1016/j.apcatb.2020.119693
- L. Guo, J. Chi, T. Cui, J. Zhu, Y. Xia, H. Guo, J. Lai and L. Wang, Phosphorus defect mediated electron redistribution to boost anion exchange membrane-based alkaline seawater electrolysis, Adv. Energy Mater., 2024, 14, 2400975, DOI:10.1002/aenm.202400975
- J.-Z. Zhang, Z. Zhang, H.-B. Zhang, Y. Mei, F. Zhang, P.-X. Hou, C. Liu, H.-M. Cheng and J.-C. Li, Prussian-blue-analogue-derived ultrathin Co2P-Fe2P nanosheets for universal-pH overall water splitting, Nano Lett., 2023, 23, 8331–8338, DOI:10.1021/acs.nanolett.3c02706
- Y. Yang, R. Zou, J. Gan, Y. Wei, Z. Chen, X. Li, S. Admassie, Y. Liu and X. Peng, Integrating electrocatalytic seawater splitting and biomass upgrading via bifunctional nickel cobalt phosphide nanorods, Green Chem., 2023, 25, 4104–4112, 10.1039/d3gc00684k
- Z. Kong, J. Chen, X. Wang, X. Long, X. She, D. Li and D. Yang, Cation vacancy driven efficient CoFe-LDH-based electrocatalysts for water splitting and Zn-air batteries, Mater. Adv., 2021, 2, 7932–7938, 10.1039/D1MA00836F
- K. Zhang, Y. Zhang, Q. Zhang, Z. Liang, L. Gu, W. Guo, B. Zhu, S. Guo and R. Zou, Metal-organic framework-derived Fe/Cu-substituted Co nanoparticles embedded in CNTs-grafted carbon polyhedron for Zn-air batteries, Carbon Energy, 2020, 2, 283–293, DOI:10.1002/cey2.35
- H. Zhang, S. Yan, W. Yi, Y. Lu, X. Ma, Y. Bin, L. Yi and X. Wang, FeP-Fe3O4 nanospheres for electrocatalytic N2 reduction to NH3 under ambient conditions, Chem. Commun., 2024, 60, 2528–2531, 10.1039/D3CC04897G
- Y. Zhao, N. Dongfang, C. A. Triana, C. Huang, R. Erni, W. Wan, J. Li, D. Stoian, L. Pan and P. Zhang, Dynamics and control of active sites in hierarchically nanostructured cobalt phosphide/chalcogenide-based electrocatalysts for water splitting, Energy Environ. Sci., 2022, 15, 727–739, 10.1039/D1EE02249K
- Z. Xiao, M. Yang, C. Liu, B. Wang, S. Zhang, J. Liu, Z. Xu, R. Gao, J.-J. Zou and A. Tang, Artificial modulated Lewis pairs for highly efficient alkaline hydrogen production, Nano Energy, 2022, 98, 107233, DOI:10.1016/j.nanoen.2022.107233
- R. Iwata, L. Zhang, K. L. Wilke, S. Gong, M. He, B. M. Gallant and E. N. Wang, Bubble growth and departure modes on wettable/non-wettable porous foams in alkaline water splitting, Joule, 2021, 5, 887–900, DOI:10.1016/j.joule.2021.02.015
- J. Wang, D. T. Tran, K. Chang, S. Prabhakaran, J. Zhao, D. H. Kim, N. H. Kim and J. H. Lee, Hierarchical Ni@CNTs-bridged MoxC/Ni2P heterostructure micro-pillars for enhanced seawater splitting and Mg/seawater battery, Nano Energy, 2023, 111, 108440, DOI:10.1016/j.nanoen.2023.108440
- P. Zhai, C. Wang, Y. Zhao, Y. Zhang, J. Gao, L. Sun and J. Hou, Regulating electronic states of nitride/hydroxide to accelerate kinetics for oxygen evolution at large current density, Nat. Commun., 2023, 14, 1873, DOI:10.1038/s41467-023-37091-x
- J. Yang, Y. Shen, Y. Sun, J. Xian, Y. Long and G. Li, Ir nanoparticles anchored on metal-organic frameworks for efficient overall water splitting under pH-universal conditions, Angew. Chem., Int. Ed., 2023, 62, e202302220, DOI:10.1002/anie.202302220
- H. Yan, Y. Xie, A. Wu, Z. Cai, L. Wang, C. Tian, X. Zhang and H. Fu, Anion-modulated HER and OER activities of 3D Ni-V-based interstitial compound heterojunctions for high-efficiency and stable overall water splitting, Adv. Mater., 2019, 31, 1901174, DOI:10.1002/adma.201901174
- X. Zheng, J. Yang, P. Li, Z. Jiang, P. Zhu, Q. Wang, J. Wu, E. Zhang, W. Sun and S. Dou, Dual-atom support boosts nickel-catalyzed urea electrooxidation, Angew. Chem., 2023, 135, e202217449, DOI:10.1002/anie.202217449
- J. He, Y. Liu, Y. Huang, H. Li, Y. Zou, C. L. Dong and S. Wang, Fe2+-induced
in situ intercalation and cation exsolution of Co80Fe20(OH)(OCH3) with rich vacancies for boosting oxygen evolution reaction, Adv. Funct. Mater., 2021, 31, 2009245, DOI:10.1002/adfm.202009245
- W. Yu, Q. Li, W. Xiao, J. Wang, B. Dong, Y. Chai, Z. Wu and L. Wang, Unique CoP Microflower decorated with phosphorous-enriched PtP2 onto nickel foam with interfacial electronic interactions to boost alkaline water-splitting, Adv. Funct. Mater., 2024, 2313935, DOI:10.1002/adfm.202313935
- J. Qi, Q. Chen, M. Chen, W. Zhang, X. Shen, J. Li, E. Shangguan and R. Cao, Promoting oxygen evolution electrocatalysis by coordination engineering in cobalt phosphate, Small, 2024, 2403310, DOI:10.1002/smll.202403310
- Y. Hu, C. Hu, A. Du, T. Xiao, L. Yu, C. Yang and W. Xie, Interfacial evolution on Co-based oxygen evolution reaction electrocatalysts probed by using in situ surface-enhanced raman spectroscopy, Anal. Chem., 2022, 95, 1703–1709, DOI:10.1021/acs.analchem.2c04931
- L. Ge, H. Yang, J. Guan, B. Ouyang, Q. Yu, H. Li and Y. Deng, Unveiling the structural self-reconstruction and identifying the reactive center of a V, Fe Co-doped cobalt precatalyst toward enhanced overall water splitting by operando raman spectroscopy, Inorg. Chem., 2023, 62, 15664–15672, DOI:10.1021/acs.inorgchem.3c02451
- W. Liu, J. Yu, T. Li, S. Li, B. Ding, X. Guo, A. Cao, Q. Sha, D. Zhou and Y. Kuang, Self-protecting CoFeAl-layered double hydroxides enable stable and efficient brine oxidation at 2 A cm−2, Nat. Commun., 2024, 15, 4712, DOI:10.1038/s41467-024-49195-z
- H. J. Liu, S. Zhang, W. Y. Yang, N. Yu, C. Y. Liu, Y. M. Chai and B. Dong, Directional reconstruction of iron oxides to active sites for superior water oxidation, Adv. Funct. Mater., 2023, 33, 2303776, DOI:10.1002/adfm.202303776
- S. Zhang, H. Jiang, L. Yan, Y. Zhao, L. Yang, Q. Fu, D. Li, J. Zhang and X. Zhao, Self-terminating surface reconstruction induced by high-index facets of delafossite for accelerating ammonia oxidation reaction involving lattice oxygen, Small, 2023, 19, 2207727, DOI:10.1002/smll.202207727
- X. Shu, D. Tan, Y. Wang, J. Ma and J. Zhang, Bimetal-bridging nitrogen coordination in carbon-based electrocatalysts for pH-universal oxygen reduction, Angew. Chem., Int. Ed., 2024, 63, e202316005, DOI:10.1002/anie.202316005
- S. Jiao, X. Fu and H. Huang, Descriptors for the evaluation of electrocatalytic reactions: d-band theory and beyond, Adv. Funct. Mater., 2022, 32, 2107651, DOI:10.1002/adfm.202107651
- S. Xu, S. Feng, Y. Yu, D. Xue, M. Liu, C. Wang, K. Zhao, B. Xu and J.-N. Zhang, Dual-site segmentally synergistic catalysis mechanism: boosting CoFeSx nanocluster for sustainable water oxidation, Nat. Commun., 2024, 15, 1720, DOI:10.1038/s41467-024-45700-6
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4qi03351e |
| ‡ These authors contributed equally to this work. |
| This journal is © the Partner Organisations 2025 |