
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of sterically unhindered Lewis acidic boron-doped π-conjugated polymers
Naoki
Takahashi
a and
Yuta
Nishina
 *b
*b
aGraduate School of Environmental, Life, Natural Science and Technology, Okayama University, 3-1-1 Tsushimanaka, Kita-ku, Okayama, 700-8530, Japan
bResearch Institute for Interdisciplinary Science, Okayama University 3-1-1, Tsushimanaka, Kita-ku, Okayama, 700-8530, Japan. E-mail: nisina-y@cc.okayama-u.ac.jp
First published on 17th November 2025
Abstract
We report the synthesis of sterically unhindered boron-doped π-conjugated polymers via polymerization of organo-dilithium reagents with boron trichloride. The resulting polymer exhibits Lewis acidity and catalyzes the transesterification of methyl benzoate. This performance is attributed to the electron-accepting ability, and thermally labile Lewis acid–base interactions, facilitating catalytic turnover.
Lewis acidic boron-doped π-conjugated polymers have attracted considerable attention due to their potential in a wide range of applications, including optoelectronics, gas storage, sensors, and catalysts.1–7 Owing to their high molecular weight and extended conjugation, these polymers are typically associated with advantageous properties such as improved charge transport, efficient light absorption, molecular adsorption, good thermal and chemical stability, enhanced processability, and ease of handling. Furthermore, their macromolecular nature enables the formation of robust solid-state materials, making them suitable for heterogeneous applications such as catalysis and sensing. A boron atom surrounded by aryl groups is electron-deficient due to the vacant p orbital. This characteristic endows boron-containing compounds with unique properties, particularly as a Lewis acid. Previously reported boron-doped π-conjugated polymer structures have often been composed of bulky substituents, such as mesityl or 2,4,6-tri(isopropyl)phenyl groups, and these polymers were primarily utilized for the detection of small anions, such as fluoride ions (Scheme 1a).2 Incorporating boron atoms with less bulky substituents is a promising strategy to enhance the Lewis acidity of π-conjugated polymers and expand their potential applications in catalysis and molecular recognition. For instance, triphenylborane (BPh3) exhibits significantly stronger Lewis acidity than trimesitylborane (BMes3), highlighting the effect of steric hindrance on Lewis acid strength.8 Recently, Xue et al. reported boron-containing polythiophenes bearing less hindered substituents, which showed high responsiveness to triethylamine and pyridine through Lewis acid–base interactions (Scheme 1b).5 The polymers were synthesized via transmetallation reactions between organotin monomers and boron-containing electrophiles, which are used for the construction of triarylborane-containing polymers. Although organotin reagents are commonly used for constructing triarylborane structures, their limited reactivity often requires harsh reaction conditions.9 This restricts the choice of monomers to electron-rich species. Moreover, it has been reported that Sn–C bond is further deactivated upon incorporation of an electron-deficient borane moiety,10 which would hinder the formation of the desired polymer structures.
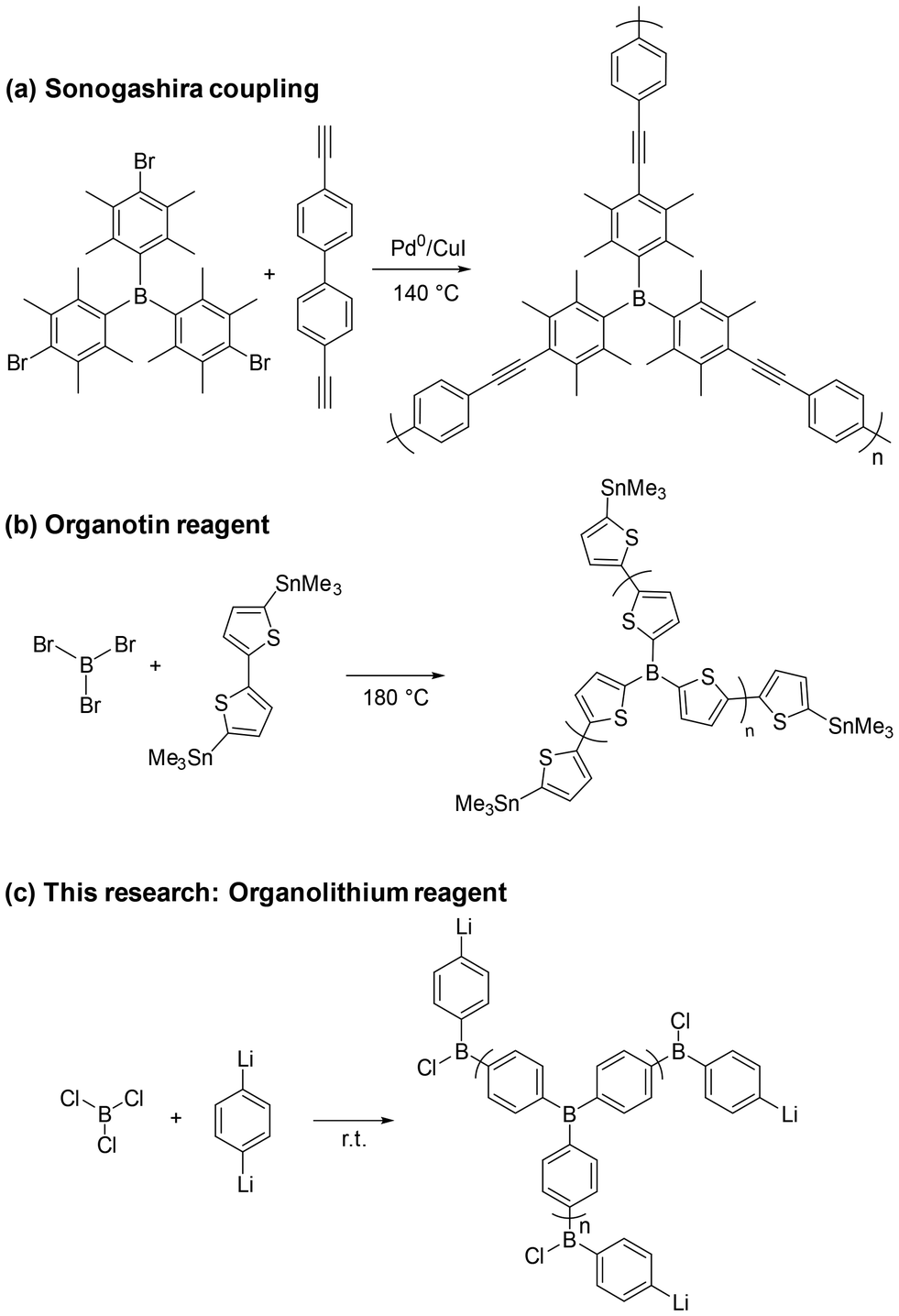 | ||
| Scheme 1 Synthesis methods of the boron-doped polymers. | ||
In this study, we report on the synthesis of Lewis acidic boron-doped π-conjugated polymers without bulky substituents such as mesityl groups. To explore suitable conditions for polymer formation, both organolithium and Grignard reagents were employed as nucleophilic coupling partners with boron trichloride. These reagents differ in their reactivity and compatibility with electron-deficient boron centers, and their comparison was expected to provide insight into the optimal synthetic route. To probe the Lewis acidity of the resulting polymer, pyridine was employed as a Lewis base, and its coordination was confirmed spectroscopically; upon heating, the coordinated pyridine was released, indicating reversible Lewis acid–base interactions. Polymerization was anticipated to influence the molecular architecture and Lewis acid–base interactions, potentially resulting in catalytic properties distinct from those of the small-molecule analogues. Arylboranes are well known to function as Lewis acid catalysts;3,4,11–14 therefore, we selected the transesterification of methyl benzoate as a model reaction to evaluate the catalytic performance of our polymer.
Several organodimetallic reagents were reacted with boron trichloride to obtain the corresponding polymers (Scheme 1c). These polymers were assumed to be unstable; therefore, their isolation, characterization, and potential applications were performed after treatment with pyridine. Initially, polymerization was attempted using 1,4-benzenediiodomagnesium and boron trichloride (Table 1, entry 1). However, no polymeric product was obtained after work-up. This might be attributed to the resulting polymer becoming electron-deficient during the reaction, rendering the intermediate insufficiently active for further propagation. For comparison, no polymer was obtained using trimethoxyborane as a boron source, possibly due to the lack of electrophilicity of the boron center. Similarly, polymerization does not proceed when dimethoxymesitylborane is reacted with a Grignard reagent derived from 1,4-dibromobenzene.15 To address this issue, we employed 1,4-dilithiobenzene, which is generally more reactive than the corresponding Grignard reagent. The reaction of this lithium reagent with boron trichloride successfully afforded an insoluble polymer (Table 1, entry 2). To investigate the polymer structure, we conducted 11B NMR analysis. The spectra indicated the presence of triarylborane units coordinated with pyridine, along with signals corresponding to B–O bonds and tetraarylborate moieties (Fig. 1a). Pyridine–borane adducts such as pyridine·BPh3 are known to be air-stable and resistant to oxidation and hydrolysis, avoiding conversion to phenylboronic acid. Based on this analogy, we infer that the B–O bonds observed are likely formed by replacement of residual B–Cl groups during the quenching process. To study the effect of stoichiometry on polymer formation, reactions were conducted using either one or two equivalents of the lithium reagent. The use of two equivalents resulted in only a small amount of polymer (Table 1, entry 3). The product exhibited poor solubility in organic solvents, and its 11B NMR spectrum did not provide meaningful information. Likewise, one equivalent of the lithium reagent also yielded a small amount of polymer (Table 1, entry 4). However, in this case, 11B NMR analysis revealed the presence of both triarylborane·pyridine complexes and B–O bonds, even though one equivalent is insufficient for stoichiometric formation of triarylboranes. These findings suggest that when 1.5 equivalents of the lithium reagent are used, substitution of B–Cl by B–Ar may proceed beyond the triarylborane stage, leading to overreaction and formation of tetraarylborates. Once tetraarylborates are formed, the remaining reagent becomes insufficient for further triarylborane formation, leaving unreacted B–Cl groups, which are subsequently converted into B–O bonds during the pyridine quenching step.
 | ||
| Fig. 1 (a) 11B NMR spectrum of the polymer synthesized using 1.5 equiv. of dilithiobenzene, measured in THF-d8, (b) IR spectra of (i) the polymer·pyridine complex and (ii) BPh3·pyridine complex, and (c) TG-MS spectrum of the polymer·pyridine complex. | ||
|
|
|||
|---|---|---|---|
| Entry | Organometal |
A![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :B molar ratio :B molar ratio |
Product (mg) |
| a Reaction conditions: A: 1 M boron trichloride solution in heptane (1.0 mL) and B: organometal reagent solution in diethyl ether were reacted under an argon atmosphere, and the mixture was stirred at room temperature for 24 hours. Then, diethyl ether (10 mL) and pyridine (18.2 mmol) were added to quench the reaction. | |||
| 1 | IMgC6H4MgI | 1:1.5 |
0 |
| 2 | LiC6H4Li | 1:1.5 |
80 |
| 3 | LiC6H4Li | 1:2.0 |
30 |
| 4 | LiC6H4Li | 1:1.0 |
30 |

The polymer synthesized using 1.5 equivalents of the lithium reagent and boron trichloride exhibited the highest degree of polymerization. Accordingly, we conducted further analyses of this polymer. First, Fourier-transform infrared (FT-IR) spectroscopy analysis was performed. The IR spectrum of the polymer displayed peaks at 1454 cm−1 and 1618 cm−1, which are also observed in the spectrum of the BPh3·pyridine complex (Fig. 1b). These peaks have been reported to correspond to pyridine coordinated to Lewis acid sites, with their positions known to shift depending on the strength of Lewis acidity.16,17 Therefore, the observed peak positions suggest that the polymer possesses moderate Lewis acidity, comparable to that of BPh3. Next, we investigated the thermal stability of the polymer. Thermogravimetry-mass spectrometry (TG-MS) was employed to directly monitor the release of pyridine associated with weight loss during heating.18,19 The TG curve indicated a weight loss of 26% up to 275 °C under a helium atmosphere (Fig. 1c). At 195 °C, the mass spectrum revealed a signal at m/z = 79, corresponding to pyridine. Given that the boiling point of free pyridine is approximately 115 °C, the observed release at higher temperatures implies strong coordination between pyridine and the Lewis acid sites in the polymer. A similar phenomenon has been reported for boron-containing polythiophenes, which exhibit weight loss associated with the release of pyridine at around 150 °C.5
Next, we evaluated its catalytic performance as a metal-free Lewis acid catalyst. The polymer was applied to the transesterification of methyl benzoate.20 To facilitate the removal of pyridine and activate the Lewis acid sites, the reaction was conducted at 180 °C. Under these conditions, the polymer catalyzed the formation of octyl benzoate with a yield of 83% using 15 wt% of the catalyst in 1 hour (Table 2, entry 1). This result suggests that pyridine was successfully removed from the polymer, thereby exposing the Lewis acid sites responsible for catalysis. For comparison, BPh3 was tested as a model of a small-molecule Lewis acid catalyst. However, the reaction proceeded with poor efficiency (Table 2, entry 2); the polymer yielded a product 6.4 times greater than that obtained with BPh3. We also tested the BPh3·pyridine complex as a model Lewis acid–base adduct. However, the reaction yield remained very low even after prolonged reaction time (Table 2, entry 3). This low reactivity can be attributed to the strong complexation between pyridine and BPh3 within the molecule, making activation of the Lewis acid site difficult under the given conditions. TG-MS analysis of the BPh3·pyridine complex revealed that pyridine (m/z = 79) was not released until approximately 200 °C, and the subsequent complete weight loss was ascribed to the vaporization or decomposition of the complex (Fig. S1).
|
|
||
|---|---|---|
| Entry | Catalyst | Yieldb (%) |
| a Reaction conditions: methyl benzoate (0.5 mmol); 1-octanol (2.5 mmol); catalysts (15 wt% of methyl benzoate). b Determined by GC using dodecane as an internal standard. c The reaction in a sealed tube. d The yield was up to 21% for 12 hours. | ||
| 1 | Polymer | 83 |
| 2 | BPh3 | 13 |
| 3 | BPh3·pyridine | 0 |
| 4c | Polymer | 3 |
| 5d | Pyridine | 0 |
| 6 | BMes3 | 60 |

To further investigate the importance of pyridine removal, the polymer was used as a catalyst in a sealed tube. Under the condition, the reaction did not proceed (Table 2, entry 4), supporting the conclusion that thermal activation and the release of pyridine from the reaction system are essential for catalytic activity. Additionally, pyridine alone was tested as a Lewis base catalyst. While a small amount of product was obtained after 12 hours, the catalytic activity was negligible (Table 2, entry 5).
Finally, trimesitylborane (BMes3) was examined as a model of a sterically hindered boron compound. Although BMes3 catalyzed the reaction with a yield of 60% (Table 2, entry 6), its efficiency was lower than that of the polymer. This is likely due to the bulky mesityl groups, which may limit the accessibility of the boron center and hinder its catalytic performance.
To elucidate the results, the reaction mixtures after catalysis using BPh3, BMes3, and the polymer were analyzed by 11B NMR in THF (Fig. S9–11). In the case of BPh3, the formation of B–O species was observed, indicating decomposition of BPh3 during the reaction. In contrast, BMes3 maintained its structure after the reaction. These results suggest that the lower yield observed with BPh3 is primarily due to its decomposition. For the polymer, B–O formation was also suggested, similar to BPh3; however, due to the potentially higher catalytic activity of the polymer, the overall yield was higher than that with BMes3.
To investigate the effect of boron atoms at the para position of the benzene ring, we calculated the Global Electrophilicity Index (GEI), a widely used quantitative and base-independent measure of Lewis acidity.21 BPh3 (Fig. 2a) and 1,4-phenylenebis(diphenylborane) (Fig. 2b) were used for the GEI calculations. The resulting GEI values for BPh3 and 1,4-phenylenebis(diphenylborane) were 2.022 eV and 2.353 eV, respectively. These results indicate that the introduction of boron at the para position enhances the molecule's electron-accepting ability.
 | ||
| Fig. 2 GEI calculations for (a) BPh3 and (b) the polymer structure with the model units highlighted. | ||
In summary, we have synthesized a sterically unhindered, boron-doped π-conjugated polymer via the direct reaction of organodilithium reagents with boron trichloride. Unlike conventional boron-containing polymers, the boron centers in our system are incorporated into the polymer backbone without bulky substituents, allowing greater accessibility of the Lewis acid sites. This structural feature enables the polymer to function as an efficient metal-free Lewis acid catalyst, as demonstrated in the transesterification of methyl benzoate. Compared to small-molecule Lewis acids such as BPh3 and BMes3, the polymer exhibited superior catalytic activity. This performance is attributed to the electron-accepting ability, and thermally labile Lewis acid–base interactions, facilitating catalytic turnover. These findings present a molecular design strategy for developing thermally stable and tunable boron-doped π-conjugated polymers as a new class of metal-free Lewis acid catalysts.
Author contributions
N. T. performed experiments and drafted the manuscript. Y. N. conceived and supervised the project and secured research funding. Both authors discussed the results and contributed to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Data availability
All data associated with this study are available in the article and supplementary information (SI). Supplementary information: experimental details, 1H NMR, 11B NMR, 13C NMR and TG-MS. See DOI: https://doi.org/10.1039/d5py00783f.Acknowledgements
We are grateful to Mr Kentaro Okura (Research Institute for Interdisciplinary Science, Okayama University, Okayama, Japan) for his help with XPS and solid-state NMR measurements. This research was supported by JST CREST (grant number JPMJCR24S6).References
- S. Kawai, S. Saito, S. Osumi, S. Yamaguchi, A. S. Foster, P. Spijker and E. Meyer, Nat. Commun., 2015, 6, 8098 CrossRef CAS PubMed.
- V. M. Suresh, A. Bandyopadhyay, S. Roy, S. K. Pati and T. K. Maji, Chem. – Eur. J., 2015, 21, 10799–10804 CrossRef CAS PubMed.
- F. Vidal, J. McQuade, R. Lalancette and F. Jäkle, J. Am. Chem. Soc., 2020, 142, 14427–14431 CrossRef CAS PubMed.
- Q. Liu, L. Yang, C. Yao, J. Geng, Y. Wu and X. Hu, Org. Lett., 2021, 23, 3685–3690 CrossRef CAS PubMed.
- C. Xue, M. Peng, Z. Zhang, X. Han, Q. Wang, C. Li, H. Liu, T. Li, N. Yu and Y. Ren, Macromolecules, 2022, 55, 3850–3859 CrossRef CAS.
- F. Zhao, G. Liao, M. Liu, T. Wang, Y. Zhao, J. Xu and X. Yin, Angew. Chem., Int. Ed., 2024, 63, e202317294 CrossRef CAS PubMed.
- J. Teotonico, D. Mantione, K. K. Hollister, H. Sardon, N. Ballard, F. Ruipérez, R. J. Gilliard and F. J. Vidal, Polym. Sci., 2025, 1–8 Search PubMed.
- R. J. Mayer, N. Hampel and A. R. Ofial, Chem. – Eur. J., 2021, 27, 4070–4080 Search PubMed.
- S. M. Berger, M. Ferger and T. B. Marder, Chem. – Eur. J., 2021, 27, 7043–7058 CrossRef CAS PubMed.
- N. Baser-Kirazli, R. A. Lalancette and F. Jäkle, Angew. Chem., Int. Ed., 2020, 59, 8689–8697 CrossRef CAS PubMed.
- D. Mukherjee, S. Shirase, K. Mashima and J. Okuda, Angew. Chem., Int. Ed., 2016, 55, 13326–13329 CrossRef CAS PubMed.
- H. Fang and M. Oestreich, Angew. Chem., Int. Ed., 2020, 59, 11394–11398 CrossRef CAS PubMed.
- J. L. Carden, A. Dasgupta and R. L. Melen, Chem. Soc. Rev., 2020, 49, 1706–1725 Search PubMed.
- M. Lamač, B. Urbán, M. Horáček, D. Bůžek, L. Leonová, A. Stýskalík, A. Vykydalová, K. Škoch, M. Kloda, A. Mahun, L. Kobera, K. Lang, M. G. S. Londesborough and J. Demel, ACS Catal., 2023, 13, 14614–14626 CrossRef PubMed.
- N. Matsumi, K. Naka and Y. Chujo, J. Am. Chem. Soc., 1998, 120, 10776–10777 Search PubMed.
- M. E. Z. Velthoen, S. Nab and B. M. Weckhuysen, Phys. Chem. Chem. Phys., 2018, 20, 21647–21659 Search PubMed.
- G. S. Szymanski, Y. Suzuki, T. Ohba, B. Sulikowski, K. Góra-Marek, K. A. Tarach, S. Koter, P. Kowalczyk, A. Ilnicka, M. ZiÈ©ba, L. Echegoyen, A. P. Terzyk and M. E. Plonska-Brzezinska, ACS Appl. Mater. Interfaces, 2021, 13, 51628–51642 CrossRef CAS PubMed.
- C. V. Hidalgo, H. Itoh, T. Hattori, M. Niwa and Y. Murakami, J. Catal., 1984, 85, 362–369 Search PubMed.
- H. G. Karge and V. Dondur, J. Phys. Chem., 1990, 94, 765–772 Search PubMed.
- Z. Zhang, P. Meng, H. Luo, Z. Pei and X. Liu, Catalysts, 2024, 14, 731–755 Search PubMed.
- A. R. Jupp, T. C. Johnstone and D. W. Stephan, Dalton Trans., 2018, 47, 7029–7035 Search PubMed.
| This journal is © The Royal Society of Chemistry 2025 |