
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Computational spectroscopy for crystalline materials: from structure to properties
Mariela M.
Nolasco
 *a,
Pedro D.
Vaz
b,
Rafael A. F.
Serrano
a,
João T.
Martins
a,
Catarina F.
Araújo
a and
Paulo
Ribeiro-Claro
a
*a,
Pedro D.
Vaz
b,
Rafael A. F.
Serrano
a,
João T.
Martins
a,
Catarina F.
Araújo
a and
Paulo
Ribeiro-Claro
a
aCICECO – Aveiro Institute of Materials, Department of Chemistry, University of Aveiro, 3810-193, Aveiro, Portugal. E-mail: mnolasco@ua.pt
bChampalimaud Foundation, Champalimaud Centre for the Unknown, 1400-038 Lisboa, Portugal
First published on 22nd May 2025
Abstract
Can computational spectroscopy predict the crystal structure of experimentally challenging systems? Once a computational model is validated, what macroscopic properties can be reliably derived from it? This review explores the potential of computational spectroscopy to address these questions by examining a few selected systems. The examples considered include inorganic cupper silicate pigment Egyptian blue, tetraalkylammonium chloride salts – key components of deep eutectic solvents – and semi-crystalline biobased furanic polyesters.
1. Introduction
As beautifully put by John Brand, “Spectra are the idiom of atoms and molecules. As we express ourselves in words and phrases, molecules announce their presence by a series of frequencies in the electromagnetic spectrum. The goal of spectroscopy is to compile the grammar by which such groups are understood.”1 Going along with this analogy, if “spectra are the idiom” then computational tools are the dictionaries that allow spectroscopists to easily interpret vibrational language.The definition, methods, and recent developments of the field known as computational spectroscopy have been thoroughly examined in an excellent primer published in Nature Reviews.2 As defined in that work, “Computational spectroscopy exploits theoretical models, provides tools and computer codes, and validates procedures for the prediction, analysis, interpretation, and understanding of spectroscopic features, properties, and phenomena.” While the primary focus is on molecular-level calculations—as suggested by the title Computational Molecular Spectroscopy—the text offers a broad and comprehensive perspective on the discipline.
In this review, we extend the scope beyond the molecular level to address materials as a whole, with a particular emphasis on crystalline solids. This shift brings forward two critical questions: first, can computational spectroscopy predict the crystal structure of experimentally challenging systems, providing insights where conventional approaches struggle? Second, once a computational model is validated, how far can we trust the macroscopic properties derived from it? These questions challenge what we can infer from simulations and demand a careful interplay between computation and experiment.
Although the term spectroscopy, in the context of computational spectroscopy, is not limited to a particular technique, vibrational spectroscopy deserves a particular focus, due to the almost direct links between the resolution of the electronic Schrödinger equation and the vibrational properties of a system. In a simple description, once the relationship between the electronic energy and the nuclear coordinates is determined, its derivatives yield the force constants, the key feature to determine the vibrational modes of the system. In fact, the comparison between calculated and experimental frequency of vibrational modes goes back to the early days of this synergic relationship between molecular orbital calculations and spectroscopic properties: the calculations assist the assignment and interpretation of vibrational spectra and vibrational spectra validate the theoretical models used.
As the reader will notice, we place great value on this methodological aspect of computational spectroscopy, particularly in the way we apply it in our work. An effective approach integrates vibrational spectroscopy and quantum chemical calculations into an iterative process, where simplified molecular models assist in vibrational assignments, which in turn guide the refinement of more realistic models. This continuous feedback loop enhances the accuracy of computational predictions, ultimately leading to better agreement with experimental data. The significance of this methodological framework will be particularly emphasized in section 4.
Once a good model is obtained, the microscopic properties can be derived from the computational results. Microscopic properties here are meant to represent the properties at the atomic or molecular level, e.g., energy of different structures, bond lengths, vibrational levels, dipole moments, and intermolecular interactions. The ultimate goal is to establish a link between microscopic and macroscopic properties, a still giant step ahead. Examples of macroscopic properties aimed at are, for instance, the heat capacity of a crystalline material, the melting point of a deep eutectic solvent, or the crystalline elastic modulus of a polymer. The following cases highlight the potentialities of computational spectroscopy in elucidating the behaviour of crystalline materials, offering new structural models and a deeper understanding of structure property relationships.
Periodic versus discrete calculations
The first component of the computational spectroscopy toolkit is the computational element. This work does not aim at a deep description of theoretical foundations and computational requirements, as these can be found in abundant literature sources,3–5 particularly in the above-mentioned work of Barone et al.2Over the years, quantum mechanical calculations have shifted from traditional ab initio methods, such as Hartree–Fock (HF), to density functional theory (DFT), which has now become the de facto standard in computational spectroscopy and materials modeling. While HF-based approaches were once the cornerstone of quantum chemistry, their limitations in accurately describing electron correlation led to their decline in favor of DFT, which offers a more balanced trade-off between accuracy and computational cost. Within the several flavours of DFT calculations, the hybrid B3LYP functional has become the most common and well-succeeded choice for discrete calculations.
Discrete DFT calculations are used to investigate the properties of molecular systems. When performed on a single “isolated” molecule (i.e., in vacuum), these calculations provide only a crude approximation of a condensed-phase system, as they neglect intermolecular interactions. To incorporate these effects, a cluster approach can be employed, where calculations consider aggregates ranging from as few as two molecules to as many as computational resources allow. The cluster-based approach remains as the best approximation for computational simulations of highly disordered or amorphous materials6 and is often quite useful as a first step in crystal structure determination, to identify the structural “motifs” required to build a model of an unknown crystal.
However, in order to model the three-dimensional ordering of crystalline materials, one needs to step beyond the cluster approximation and harness the power of periodic calculations. Periodic calculations refer to quantum mechanical simulations that account for the periodic nature of crystalline solids by applying boundary conditions that repeat the unit cell infinitely in all spatial directions. Unlike discrete DFT calculations, periodic-DFT does not have a most popular “default” functional. However, among the commonly used functionals, the Perdew–Burke–Ernzerhof (PBE) functional, based on the generalized gradient approximation (GGA), presents an interesting balance between accuracy and computational efficiency. The examples below rely on PBE, as it has been found to provide a reliable description of structural and vibrational properties in a broad range of crystalline materials. For deeper and more insightful discussions on the selection of functionals and pseudopotentials (another factor affecting the outcome of a simulation), readers are referred to other works.7–11 Since PBE is not a dispersion-corrected functional, an empirical correction scheme,12 such as the Tkatchenko–Scheffler method,13 is required to improve the description of dispersive intermolecular interactions in molecular crystals.
A key advantage of periodic-DFT calculations when used in tandem with vibrational spectroscopy is their ability to account for the entire Brillouin zone, capturing the wavevector-dependent behavior of vibrational modes. In a crystal, vibrations are not confined to a single unit cell but extend throughout the periodic framework, leading to phonon dispersion—variations in vibrational frequencies as a function of wavevector which arises from long-range interactions within the lattice. By sampling different points in the Brillouin zone, periodic DFT provides a comprehensive picture of vibrational dynamics that cannot be accessed through isolated molecular calculations. Phonon dispersion is generally less relevant for conventional infrared (IR) and Raman measurements but becomes essential in inelastic neutron scattering (INS), as discussed below.
The calculation of vibrational properties, as part of lattice dynamics, can be performed using a variety of approaches. These may vary in scope depending on the software package used and influence the ability to assess key features of the system under study, such as anharmonic effects, overtones, and combination modes.14–16 All the examples featured herein were computed using CASTEP software.14
Inelastic neutron scattering (INS)
While most vibrational spectroscopy studies rely on optical techniques—infrared, Raman, and their variations—INS presents a powerful yet underutilized alternative in computational spectroscopy.6,17–21INS, as the name suggests, takes advantage of the inelastic scattering of neutrons by atomic nuclei to generate a vibrational spectrum which provides complementary information to those of optical techniques. For instance, INS is devoid of selection rules so that all vibrational modes are INS active, and their intensity is proportional to the neutron scattering cross-section of atomic nuclei. The scattering of neutron by hydrogen is significantly stronger than by other nuclei, including deuterium, so that vibrational modes involving the movement of hydrogen nuclei dominate INS spectra, allowing the observation of vibrational motions which are difficult or impossible to detect in optical spectra (Fig. 1).
 | ||
| Fig. 1 INS (top), infrared (middle) and Raman (bottom) spectra of Egyptian blue. Reprinted with permission from ref. 22 under the terms of the CC-BY 4.0 license. | ||
Moreover, unlike photons, neutrons have mass, which makes INS inherently sensitive to phonon dispersion. In a crystalline material, vibrations are not confined to a single unit cell but propagate throughout the crystal lattice as collective excitations known as phonons. These phonons exhibit dispersion, meaning their energy depends on their wavelength and momentum. While optical techniques typically probe vibrations at or near the center of the Brillouin zone (where the phonon momentum is negligible), INS provides access to the full phonon dispersion relationship. Thus, INS is a prime tool for observing collective modes, which appear as very intense contributions below 200 cm−1. This capability is particularly valuable for understanding the dynamics of materials, as it reveals how vibrational modes evolve across the crystal and how they contribute to macroscopic properties such as thermal conductivity and phase stability.
The most compelling advantage from a computational spectroscopy perspective is that INS spectra can be reliably simulated using periodic DFT calculations. These simulations not only reproduce vibrational frequencies of the system – common to all vibrational techniques – but also provide remarkably accurate predictions of intensities, making INS a valuable tool for studying molecular vibrations in crystalline materials. In optical spectroscopy techniques (infrared and Raman), the calculated intensities are dependent on a demanding evaluation of the change of electronic properties during vibrations (dipole moment and polarizability, respectively). On the other hand, for INS spectroscopy, the intensity of a band associated with a vibrational/phonon mode, for a given atom i, is expressed by the dynamic structure factor.20,23
 | (1) |
When periodic DFT calculations are used to estimate INS spectra it is common practice not to apply scaling factors. However, depending on the quality of fit between estimated and observed spectra, each author should decide whether using scaling factors is necessary or accessory. Either way is fine, as long as the experimental details document this choice.
2. The Egyptian blue crystal
Cuprorivaite, the Egyptian blue pigment (EBlue),22 is a good first example of the simplest relation between periodic-DFT calculations and vibrational spectroscopy, with subsequent developments concerning other spectroscopic techniques and macroscopic properties. The crystal structure of cuprorivaite was already available,31 and no additional considerations are required in order to obtain the DFT optimized structure that constitutes “the model”. Geometry optimizations were performed without any constraints on the internal coordinates, including the cell parameters. The reliability of the model was confirmed through comparison with X-ray diffraction (XRD) analysis data and, within the expected adjustments and systematic errors of the PBE approach, the optimized geometry does not deviate from the experimental one. The vibrational (or phonon) modes were obtained from the optimized geometry using the standard finite displacement method.Fig. 2 compares the experimental and calculated INS spectra of EBlue, breaking down the calculated contributions from each atom type. As can be seen, the simulated spectrum is in excellent agreement with the experimental spectrum, with a nearly one-to-one match between calculated and observed bands. In addition, the analysis of atomic contributions to total INS intensities provides strong support for the assignments of EBlue's vibrational modes, allowing the elucidation of some controversial assignments in the literature.
 | ||
| Fig. 2 Comparison between the INS spectrum of Egyptian blue (top line) compared with that estimated from a periodic DFT calculation (CASTEP), averaged by the dispersion throughout the BZ (second to top line), obtained with AbINS. The individual atomic contributions to the total INS intensity are shown in the four bottom lines along with the neutron scattering cross section of each nuclei (values in barns, within brackets). Reprinted with permission from ref. 22 under the terms of the CC-BY 4.0 license. | ||
A key point in this result was the absence of a band in the calculated INS spectrum at ca. 933 cm−1. This is particularly relevant because all the remaining bands in the region – dominated by the asymmetric stretching Si–O–Si modes – are well predicted in the simulated spectrum.
The possible explanation arises from the limitations of the periodic DFT modeling: periodic calculations assume an infinite and perfect crystal, with no defects and no external boundaries. Being so, it cannot predict vibrational features arising from crystal defects or from end-groups at the surface of the crystalline particles. The origin of the experimental band at 933 cm−1 was then ascribed to the terminal Si–O units. The fact that this band is not predicted for the theoretical “infinite crystal” in periodic calculations (even when considering multi-quanta transitions, i.e., overtone and combination bands) is a pivotal argument for this assignment.
These considerations indicate that the sample under study has a polycrystalline nature, resulting in a high surface-to-bulk ratio of Si–O motifs. This is particularly significant, as both crystal defects and the reactivity of the crystal surface—primarily due to the presence of free or “dangling” Si–O units—play a crucial role in the interactions between EBlue and the matrix in a composite material, as well as in the functionalization of EBlue's surface.
The calculations also allow simulating the electronic spectrum and obtaining relevant electronic properties. The CASTEP-calculated reflectivity spectrum closely matches the experimental profile, revealing three optical transitions at 2.41, 2.98, and 3.78 eV, albeit shifted to higher energies compared to experimental values (1.56, 1.95, and 2.34 eV). Additionally, the calculations identified direct and indirect band gaps of 4.15 eV and 2.26 eV, respectively, in good agreement with experimental data (4.12 and 2.76 eV). Band structure analysis confirmed that CaCuSi4O10 is a direct band-gap semiconductor, with the valence band maximum (VBM) and conduction band minimum (CBM) located at the Γ-point for spin-up states, while for spin-down states, the indirect transition occurs between the Γ and M points. Spin-polarized DFT calculations further revealed that the spin density is primarily localized on Cu2+ ions and their neighboring oxygen atoms, with each Cu2+ ion holding a magnetic moment of 0.64 μB and a total spin magnetic moment of 4.04 μB per unit cell. The analysis also confirmed that the frontier orbitals near the Fermi level mainly originate from Cu and O contributions, with the computed band gap of 1.82 eV slightly exceeding the experimental value of 1.6 eV.
The phonon calculations in CASTEP can be used to evaluate the temperature dependence of the thermodynamic properties of EBlue, such as its enthalpy, entropy, heat capacity, and Debye temperature. Two parameters deserve particular attention, namely, heat capacity (Cp) and Debye temperature (ΘD). The Debye temperature is a parameter that is useful in the understanding of solid-state physics phenomena related to lattice vibrations and dynamics. According to the Debye theory, it corresponds to a maximum phonon frequency and also reflects the structural stability and the strength of bonds, as well as being closely linked to physical properties such as heat capacity or shear wave velocity in the crystal.
From their temperature dependence plots, the values of heat capacity and Debye temperature at 300 K can be obtained. The calculated Cp at 300 K is ca. 262 J mol−1 K−1, while the ΘD at the same temperature is ca. 911 K. To the best of our knowledge, there are no experimental or other theoretical data to directly compare these results. Nevertheless, the Cp value calculated for EBlue can be compared to the experimental value obtained for Han Blue, an isostructural pigment with barium instead of calcium. The Cp value of Han Blue at 300 K is reported to be ca. 260 J mol−1 K−1,32 and the calculated value for EBlue, ca. 262 J mol−1 K−1, is in excellent agreement with that experimental result.
3. Deep eutectic solvents and their pure components
As stated in a recent review,33 in the study of deep eutectic solvents (DES), “computational spectroscopy is often applied in a unidirectional manner, that is, either computational calculations are used to guide vibrational assignments or, less frequently, observed vibrational spectra are used to validate computational models.” However, the best results are achieved when a bidirectional strategy is followed, with information gathered from experimental data guiding the building of more accurate computational models which, when validated, serve as guides for in-depth vibrational assignments.A reliable comparison of calculated and observed frequency shifts, from pure compounds to their eutectic mixture, requires robust models of the pure crystal lattices. As aforementioned, periodic DFT calculations have great success in predicting the INS spectra of crystalline systems. This was further confirmed in a previous study which focused on the role of entropic factors in deep eutectic formation,34 where it was found that DFT calculations can very accurately predict the INS spectra of a pure crystalline system, particularly in the case of the [N1,1,1,1]Br system. Fig. 3 presents both the experimental and the periodically calculated INS spectra for this system. Notably, the calculated spectrum averaged over the Brillouin zone (Fig. 3, middle line) includes multi-quanta transitions, allowing accurate reproduction of the overall band profile whilst also providing detailed insights into spectral effects influenced by crystalline packing and multi-quanta transitions.
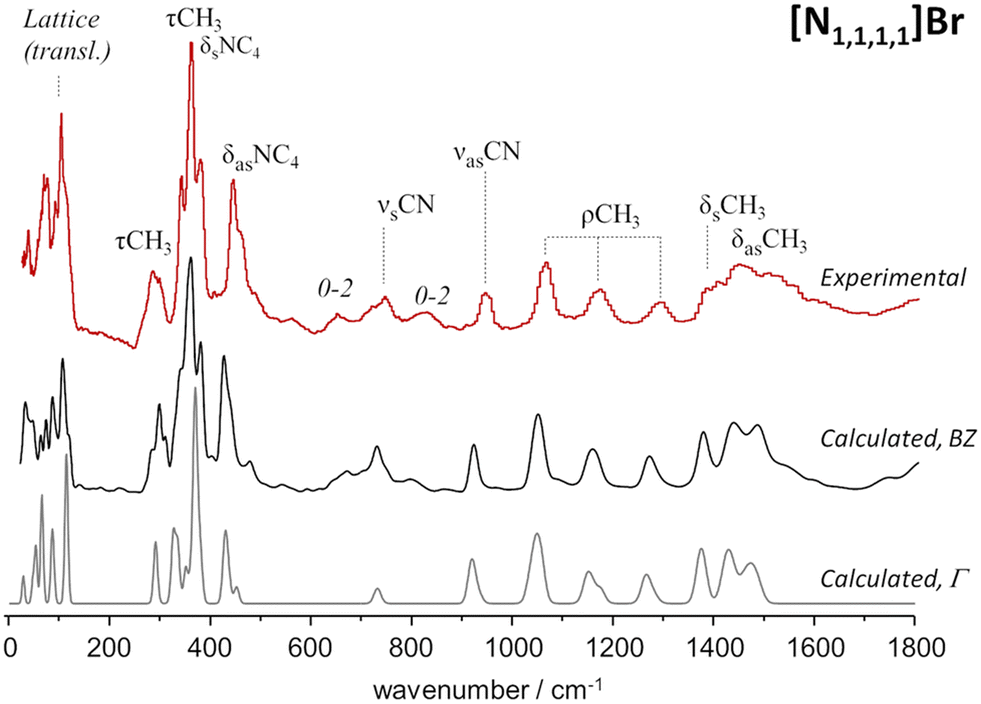 | ||
| Fig. 3 INS spectrum of tetramethylammonium bromide, [N1,1,1,1]Br (top line) and estimated INS intensities for the fundamental transitions at the Γ-point (bottom line) and including dispersion over the Brillouin zone (middle line). The assignments of some relevant vibrational modes are highlighted. Reproduced from ref. 34 with permission from the Royal Society of Chemistry. | ||
The significance of both Brillouin zone dispersion and combination modes are emphasized by the calculated spectrum for fundamental transitions at the Γ-point (Fig. 3, bottom line). This owes to the fact that dispersion influenced the overall band profile up to approximately 500 cm−1, while multi-quanta transitions became prominent in the 500–900 cm−1 range. These multi-quanta transitions were assigned as combination bands derived from CH3 torsions, which are particularly prominent in INS (due to the technique's high sensitivity to the movement of hydrogen nuclei). The intense band around 368 cm−1, a CH3 torsion band, was recognized as the source of the overtone and combination bands observed at ca. 655 cm−1 (368 + 292), 725 cm−1 (2 × 368), and 820 cm−1 (368 + 450).
The computational spectroscopy approach which combines periodic-DFT calculations with INS can also be used to select between different conformational arrangements in the cases where systems have conformational flexibility.
To illustrate this point, the periodic-DFT computed spectra for two polymorphs of [N2,2,2,2]Cl (both identified in the literature), designated HIVROT [ref] and HIVROT01 [ref] in the Cambridge Crystallographic Data Centre, are compared to the observed INS spectra of [N2,2,2,2]Cl in Fig. 4. The conformation of the ethyl groups in the cation is where the primary distinction between the two polymorphic forms resides: in the first (HIVROT) there exist two crossing C–C–N–C–C chains with trans–trans (tt) conformation, whereas in the other (HIVROT01) the conformation of the chain is trans–gauche (tg). Periodic-DFT calculations were able to differentiate between the two forms, as it is visible in Fig. 4. This is most noticeable when looking at the INS profiles in the 300–800 cm−1 region, which evidently indicates that the observed sample is in trans–trans conformation.
 | ||
| Fig. 4 INS spectrum of tetraethylammonium chloride, [N2,2,2,2]Cl (top line) and estimated INS intensities for two of its polymorphs: trans–trans (middle line) and trans–gauche (bottom line). The assignments of some relevant vibrational modes are highlighted. Reproduced from ref. 34 with permission from the Royal Society of Chemistry. | ||
Unlike the previous mentioned cases, where an experimental crystalline structure is readily available in the literature, no such structure exists for [N2,2,2,1]Cl. Therefore, in this case, it is necessary to adapt existing crystal structures through the replacement or addition/removal of certain functional groups in order to obtain the desired structure. These adaptations are necessary because a crystalline model is a pre-requisite for performing periodic-DFT calculations. In the specific case of [N2,2,2,1]Cl, two candidate crystal structures were constructed by adapting existing CIF files: tetraethylammonium chloride (CSD refcode: HIVROT), with a trans–trans conformation (tt), and triethylmethylammonium bromide (CSD refcode: AYEZOW), which features trans–gauche ethylene chains (tg).
Both structures underwent geometry optimization utilizing CASTEP, and in Fig. 5 both calculated INS spectra are displayed, as well as the experimental spectrum. The strong agreement between the observed INS spectrum and the estimated spectrum derived from the adaptation of [N2,2,2,1]Br confirms that the alkylammonium cation adopts a tg conformation in the [N2,2,2,1]Cl crystal. It was also found that the substantial differences below 600 cm−1 between the spectra estimated for the two candidate structures are caused by both distinct packing motifs and conformational variations. In particular, the gap in the 175–200 cm−1 region observed in the experimental spectrum – accurately reproduced by the tg structure but absent in the tt counterpart- was linked to different levels of crowding in the cation. More specifically, it was found that the tt structure had a greater distance between the cationic center and chloride anions, reducing steric congestion around the methyl end-groups, which in turn shifts the methyl torsional frequencies into the 175–200 cm−1 region.
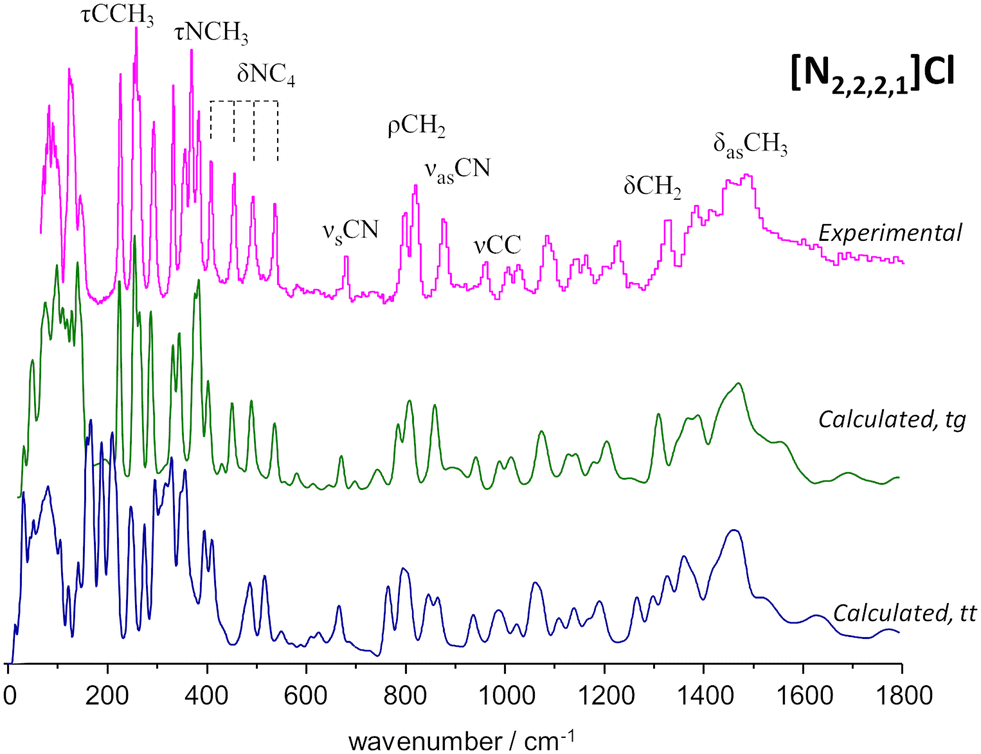 | ||
| Fig. 5 INS spectrum of triethylmethylammonium chloride, [N2,2,2,1]Cl (top line) and estimated INS intensities for two of its polymorphs: trans–gauche (middle line) and trans–trans (bottom line). The assignments of some relevant vibrational modes are highlighted. Reproduced from ref. 34 with permission from the Royal Society of Chemistry. | ||
A more recent study35 which delved into impact of cation (a) symmetry on the vibrational modes of ammonium salts and urea in eutectic mixtures – utilizing a combination of computational spectroscopy, INS and isotopic substitution – also employed a similar approach of adapting existing crystal structures due to the absence of experimental ones. Namely, the initial structures for [ChloroCh]Cl and [EthylCh]Cl were derived from the X-ray structure of [Ch]Cl named CHOCHL01 in CSD,36 where the OH group was replaced by a chlorine atom for [ChloroCh]Cl, and the H atom was substituted with a CH3 group for [EthylCh]. This modelling approach yielded a fairly reasonable match between the calculated and experimental spectrum for [ChloroCh]Cl. On the other hand, for [EthylCh]Cl two models were constructed, considering both tt and gg orientations, with the tt conformation providing a closer match to the experimental spectrum.
4. Semi-crystalline polymers
The present section will take a step further in the complexity scale to show how computational spectroscopy can be invaluable for studying polymers at the molecular level. Using furanic polyesters as case studies we will explore how to make the most of computational tools and vibrational data to determine crystal structure and explore structure–property relationships.4.1 Vibrational spectroscopy-guided crystallography
Determining the crystal structure of semi-crystalline polymers is often a challenge. Since producing large enough single crystals is often unfeasible, structural data must be retrieved from powder XRD techniques. While single-crystal XRD diagrams convey information upon atomic positions on three dimensions, in powder diffraction patterns the same information is compressed into a single dimension and a great deal of resolution is lost in the process. Besides, the co-existence of crystalline and amorphous regions often leads to powder diffraction patterns with broad intensity profiles resulting from overlapping peaks, further complicating the task of structure determination.Various strategies have been developed to address this issue,37 including the traditional structure solution approach using extracted peak intensities with direct methods or Patterson methods, as well as direct-space strategies such as Monte Carlo/simulated annealing and genetic algorithms. All of these strategies share one aspect: they look at the problem of polymer structure determination from the top-down, that is, they generate possible three-dimensional structures and then measure their ability to match the observed powder pattern.
The problem is that, due to the broad and unresolved nature of semi-crystalline powder patterns, there are often distinct crystal structure candidates, with wildly different chain conformations, that yield estimated powder patterns appearing wholly similar to the one obtained experimentally. A possible way to circumvent this hurdle is to combine powder diffraction with solid-state NMR and quantum chemistry calculations, an approach known as “NMR crystallography”.38,39 Since NMR is sensitive to relative atomic positions, trial structures may be ranked according to how well their estimated chemical shifts compare with those observed through experiment, thereby reducing ambiguity.
While, initially, NMR data was only used as a validation tool in the last step of structure determination,40,41 recently it has been proposed42 that it is much more expedient to use structural information retrieved from NMR spectra to guide the choice of trial structures, thereby reducing computational demands and increasing the chance of arriving at the correct crystal structure. Following a similar rationale, our group has combined X-ray powder diffraction, DFT calculations and vibrational spectroscopy techniques to perform de novo crystal structure determination of furanic polymers.
The proposed strategy of “vibrational spectroscopy-guided crystallography” uses computational and vibrational tools in a holistic, iterative manner where molecular models of increasing complexity are built based on the information retrieved from vibrational analysis. In this way, the problem of structure determination is viewed from the bottom-up, solving the crystal structure puzzle piece by piece, from the one-dimensional to the three-dimensional realm, as outlined in Fig. 6. Although there are examples in the literature where Raman and infrared spectroscopies aid elucidating the crystal structure of polymers, namely proteins,43–45 the strategy presented herein for “de novo” structure determination guided by vibrational spectroscopy and computational chemistry tools appears to be a novel path.
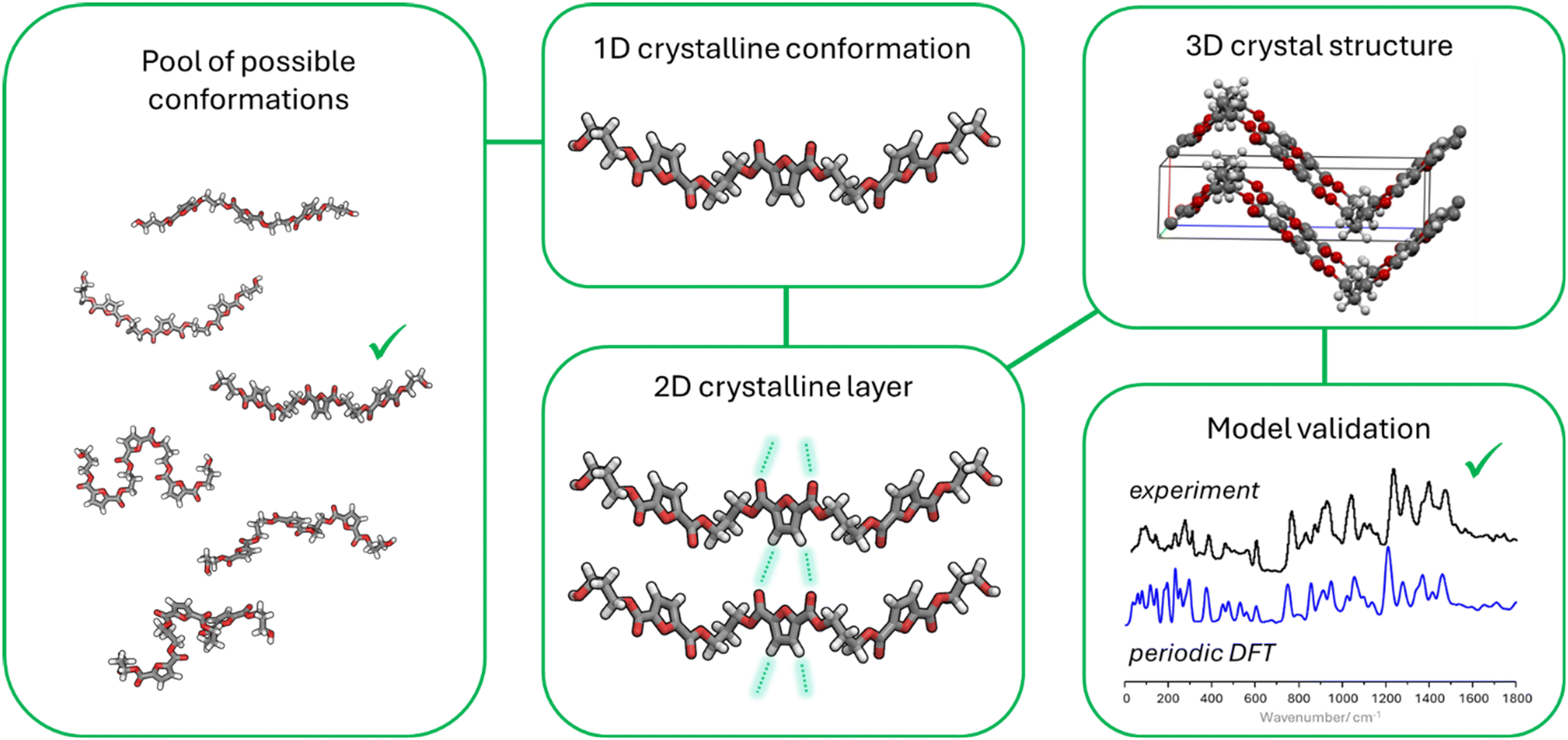 | ||
| Fig. 6 Sequence of steps for the de novo crystal structure determination of 2,5-PTF using computational spectroscopy. | ||
The first step in this strategy is to figure out which conformation is preferred in crystalline regions. To achieve this goal, several possible conformations are modelled using small oligomers and their vibrational spectra is estimated through cost-effective methods such as DFT. By comparing the spectra estimated from these simplistic models with those collected from amorphous and semi-crystalline polymer samples one may identify which conformations give rise to the appearance of novel vibrational signatures as crystallinity increases. Once the crystalline chain conformation is established larger models are built from oligomers establishing intermolecular interactions and their estimated spectra are compared with experiment to identify the two-dimensional arrangement present in the polymer crystal. At last, a three-dimensional model is built and refined by adjusting the relative positioning of stacked 2D layers and cell parameters until the estimated powder pattern and vibrational spectra resemble those observed experimentally.
To illustrate the advantage of following the vibrational spectroscopy-guided crystallography strategy over a classical structure refinement approach we will discuss the determination of the crystal structure of poly(butylene 2,5-furandicarboxylate) (2,5-PBF). There are two contrasting proposals for the crystal structure of 2,5-PBF: one, by our group,46 following the novel strategy presented herein, and another, by Zhu and colleagues,47 who used the Rietvield method to refine 2,5-PBF's structure from the powder pattern.47 The powder patterns estimated from both structures are very similar and agree with the observed pattern very well. However, the Rietvield approach leads to a structure which bears severe deficiencies. For instance, the C–C–C angles in the butanediol moiety are not realistic. Besides, the butanediol segment's conformation is gauche–gauche–gauche, in disagreement with INS and infrared evidence that favours the gauche–trans–gauche arrangement. The vibrational spectroscopy-guided strategy, in turn, identifies the correct crystalline conformer and arrives at a crystal structure that is similar to the conformation found for the crystalline poly(butylene therephtalate) (PBT) alpha polymorph.46
By following this strategy one excludes unsuitable 1D chain conformations and 2D arrangements using relatively inexpensive discrete calculations and ensure that only plausible structures reach the last stage, where computationally demanding periodic calculations are employed. Although one could test a large pool of candidate structures with alternative chain conformations using periodic DFT calculations, the reality is that due to limited computational resources the number of candidate models that undergo periodic optimization must be reduced. Hence, it is crucial to take full advantage of the information conveyed by vibrational techniques to pre-select the models that make the most sense thereby avoiding the possibility of running several expensive calculations and ending up missing the correct structure. To illustrate this pitfall we refer to the determination of the crystal structure of poly(trimethylene 2,5-furandicarboxylate) (2,5-PTF), for which two distinct models can be found in the literature, one by our own group48 and the other by Toledano et al.49 Our group followed an iterative approach to systematically eliminate conformational candidates for 2,5-PTF's crystal structure, as outlined above. By comparing the vibrational spectra of amorphous and semi-crystalline 2,5-PTF samples and identifying conformationally-sensitive vibrational modes we found that 2,5-PTF chains crystallize in the syn–syn–trans–gauche–gauche–trans conformation. Further vibrational evidence revealed a dense C–H⋯O bond network linking adjacent 2,5-PTF chains. A trial crystal structure was built based on these constraints and optimized using an expensive CASTEP calculation. Following a different route, Toledano and co-workers built five candidate crystalline models inspired on the conformations and crystal habits of related polyesters with known crystal structures.49 These trial structures were optimized by periodic-DFT calculations and ranked by lattice energy, from most to least stable. The structure with the lowest lattice energy was subsequently fine tuned to increase agreement between estimated and observed powder diffraction patterns. In contrast, our team followed the vibrational spectroscopy-guided crystallography strategy, building the model crystal structure from the 1D to the 3D and cross-validating each structural element against experimental evidence, so that expensive periodic-DFT calculations were only used in the last stage after unfeasible 1D and 2D arrangements had been ruled out. When both crystal structures are optimized using CASTEP our model appears to be more stable, by circa 20 kJ mol−1, and yields a better representation of the INS spectrum observed for a semi-crystalline 2,5-PTF sample.48 While optimizing trial 3D models inspired on related polymers is a sound and smart strategy it led Toledano et al. to miss the correct crystal structure, since none of the structural candidates featured the correct 1D chain conformation. By narrowing down candidates from the 1D to the 3D by using discrete DFT calculations in conjunction with vibrational analysis our strategy achieves better results at a lower computational cost.
The examples of vibrational spectroscopy-guided de novo crystal structure determination of polymers are still scarce, but successful. We believe that this strategy has great growth potential as either a complement or as a cheaper alternative to NMR crystallography.
4.2 Bridging amorphous and crystalline domains
Besides determining crystalline structure, computational spectroscopy is a powerful tool to study amorphous regions and to better grasp the energetic transitions required for crystallization to occur. The initial oligomer models referred in sub-section 4.1 serve not only to identify the conformational arrangement adopted in the crystal but also to assess which motifs best represent the amorphous melt. Although, in amorphous regions, polymer chain segments are disordered, adopting all possible conformations, some structural motifs prevail. For instance, in the amorphous regions of poly(ethylene 2,5-furandicarboxylate) (2,5-PEF), vibrational evidence indicates that the majority of ethylene glycol (EG) segments adopt the gauche conformation while 2,5-furandicarboxylic (FDCA) moieties tend to prefer the anti–anti and anti–syn arrangements leading to coiled polymer chains.50 Discrete DFT calculations of 2,5-PEF oligomers show that these conformational arrangements are the most stable in the absence of strong intermolecular interactions. In contrast, in crystalline regions, 2,5-PEF chains adopt an extended arrangement, with syn–syn FDCA and trans EG units. This conformation is energetically strained and a significant amount of energy is required to allow the transition from the coiled helix arrangement prevalent in the amorphous melt to the extended zig-zag chains found in the crystal. A rough estimate of the energy gap, calculated using short 2,5-PEF oligomers representing the amorphous and crystalline conformations, points to a 28 kJ mol−1 energetic penalty associated with the amorphous to crystalline transition. When a similar procedure is applied to poly(ethylene terephthalate (PET) oligomers, the resulting energy gap is merely 10 kJ mol−1, almost three times lower than for 2,5-PEF. A schematic diagram contrasting the amorphous to crystalline conformational transition for 2,5-PEF and PET is shown in Fig. 7. Although these estimates are quite crude they enable us to conclude that the amorphous to crystalline chain rearrangement is easier for PET than for 2,5-PEF, a factor which contributes to the fact that PET has a lower crystallization temperature than 2,5-PEF and requires lower stretch ratios for strain-induced crystallization. | ||
| Fig. 7 Schematic diagram representing the conformations adopted by 2,5-PEF and PET in the crystalline regions (top) and those predominant in amorphous regions (bottom). The energy required for the crystallization was estimated by calculating the difference in electronic energy between the oligomers representing amorphous and crystalline regions as described in ref. 50. | ||
The high energy penalty associated with the amorphous to crystalline conformational rearrangement is a shared characteristic of 2,5-PEF50 and its homologues with longer alkyl segments, 2,5-PTF48 and 2,5-PBF.46 For this polyester series – and, possibly, for other 2,5-FDCA based polymers – conformers bearing syn–syn FDCA units are consistently less stable than their counterparts with anti–anti or anti–syn FDCA moieties. Accordingly, syn–syn FDCA segments are the smaller population in the amorphous regions. However, in the crystal, the strained syn–syn arrangement is preferred since it allows the establishment of a vast array of intermolecular contacts among adjacent FDCA units. The formation of novel C–H⋯O bonds upon crystallization, readily observed using vibrational techniques, compensates for the energetic penalty associated with the anti–anti/anti–syn to syn–syn conformational transition. In fact, according to discrete DFT calculations, each C–H⋯O contacts contributes with circa 8 kJ mol−1 to the stabilization energy, and each FDCA unit forms 2 C–H⋯O bonds with their immediate neighbor. Thus, C–H⋯O contacts are now recognized as conformational drives in the crystallization of 2,5-FDCA based polyesters. The importance of C–H⋯O contacts in furanic polyesters is well illustrated by recent studies where they are exploited to monitor crystal formation and improve crystallization rates while a growing body of evidence suggests they play a determinant role in gas barrier properties.
Periodic DFT calculations constitute a valuable and versatile tool in the search for structure–property relationships. As mentioned in sub-section 4.1, periodic DFT calculations play a key role in validating candidate crystal structures by direct comparison among estimated and observed vibrational spectra. They also offer a comprehensive guide to vibrational assignments, allowing the visualization of the molecular movements associated with each vibrational mode. Ultimately, periodic DFT calculations may be used to estimate macroscopic properties such as the ultimate modulus of polymer crystals,51,52 electronic53 and dielectric54 properties and the thermodynamics of polymorphic transitions.55 Small variations in chemical structure may dramatically alter the energetic barriers associated with key vibrational modes which, in turn, are reflected in divergent macroscopic properties. For instance, the superior gas barrier properties of 2,5-PEF, relative to PET, have been partially attributed to differences in the ring flipping movement. While, for PET, the phenyl ring acts as a free rotor, in 2,5-PEF the corresponding movement is strained and requires the cooperative motion of the adjacent carbonyl and ethylene units. As a result, the energetic barrier to rotation is high and the ring flipping movement is hindered. Indeed, periodic DFT calculations50 of the crystal structures of PET and 2,5-PEF show that the ring flipping motion is estimated to appear at 121 cm−1 for PET, a relatively unmixed vibrational mode which essentially involves movement of the benzenic ring with subtle concomitant displacements of the adjacent COO moieties. In contrast, for 2,5-PEF, there are several mixed vibrational modes involving the torsion of the furanic ring around the Cring–C bond and the simultaneous torsion of the O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–O or C–O–CH2 segments. For 2,5-PEF these mixed ring flipping modes are scattered within the 130–150 cm−1 range. Therefore, CASTEP calculations confirm the early observations, by Burgess and colleagues,56 on the higher energy barrier to ring flipping in 2,5-PEF relative to PET as well as its mixed, cooperative nature.
C–O or C–O–CH2 segments. For 2,5-PEF these mixed ring flipping modes are scattered within the 130–150 cm−1 range. Therefore, CASTEP calculations confirm the early observations, by Burgess and colleagues,56 on the higher energy barrier to ring flipping in 2,5-PEF relative to PET as well as its mixed, cooperative nature.
5. Conclusion
In this work it becomes clear that unraveling the structure of novel, complex materials is not a straightforward task. Even less so is finding the link between their structure and observed function(s). Towards this goal, computational spectroscopy is therefore an undeniable ally which partners the potentialities of vibrational spectroscopy techniques – IR, Raman or INS – with computational tools. By deeply probing into the key phenomena occurring at the molecular level and assessing what happens at the supramolecular level this strategy provides the missing link between structure and properties, bridging the microscopic and macroscopic realms.For those materials whose structure is known, computational spectroscopy provides a direct guide to vibrational assignments and a convenient route to estimate electronic, optical and thermodynamic properties, as exemplified in section 2 for the Egyptian blue crystal.
When the structure of crystalline material is unknown, but there are known structures for very similar compounds, one can use the latter to build tentative structures and choose the most representative by comparing their estimated vibrational spectra with those obtained through experiment. Such was the strategy described in section 3, where the conformation of asymmetric tetraalkylammonium salts is determined combining INS and periodic DFT calculations.
Finally, for more complex problems, where there are many candidates for the crystalline structure, computational spectroscopy offers a viable route for de novo structure determination. The “vibrational spectroscopy-guided crystallography” framework is illustrated in section 4.1, describing its use in the crystal structure determination of furanic polyesters. Following an iterative process the crystal structure puzzle is solved from the bottom-up by first identifying the 1D chain conformation present in crystalline domains, probing its supramolecular 2D arrangement and, finally, building and fine-tuning the 3D periodic unit. By excluding unsuitable molecular candidates at an early stage (at the 1D and 2D levels) by assessing their ability to reproduce experimental data one ensures that only a small subset of options reaches the last step, where expensive periodic DFT calculations are used. In this way, computational demands are reduced, and the process is expedited. As outlined in section 4.2., the computational spectroscopy toolkit is also useful for exploring structure–property relationships in polymers, such as the energetic basis behind different crystallization temperatures, the drivers of conformational preference or the chain dynamics that contribute to barrier properties.
The faster development of computational power with more capable hardware at cheaper prices and the advent of artificial intelligence will contribute to solving complicated problems so far unreachable.
Computational spectroscopy can then set grounds by opening new perspectives in the characterization of materials and their properties, while in the mid-term future it may even evolve to develop, propose and validate new materials yet to be discovered. A good example of this was the Chemistry Nobel Prize from 2024, awarded to an AI model able to solve and predict proteins' complex structures.
It is our sincere wish that this Highlight review will inspire readers to take full advantage of the power of computational spectroscopy in their research endeavors.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was developed within the scope of the project CICECO-Aveiro Institute of Materials, UIDB/50011/2020 (DOI https://doi.org/10.54499/UIDB/50011/2020), UIDP/50011/2020 (DOI https://doi.org/10.54499/UIDP/50011/2020) & LA/P/0006/2020 (DOI https://doi.org/10.54499/LA/P/0006/2020), financed by national funds through the FCT/MCTES (PIDDAC). FCT is also acknowledged for the PhD grant to CFA (SFRH/BD/129040/2017).References
- J. C. D. Brand, in Lines of Light, ed. J. C. D. Brand, Routledge, 1st edn, 1995, pp. 1–19 Search PubMed.
- V. Barone, S. Alessandrini, M. Biczysko, J. R. Cheeseman, D. C. Clary, A. B. McCoy, R. J. DiRisio, F. Neese, M. Melosso and C. Puzzarini, Computational molecular spectroscopy, Nat. Rev. Methods Primers, 2021, 1, 38 CrossRef CAS.
- K. Lejaeghere, G. Bihlmayer, T. Bjoerkman, P. Blaha, S. Bluegel, V. Blum, D. Caliste, I. E. Castelli, S. J. Clark, A. Dal Corso, S. de Gironcoli, T. Deutsch, J. K. Dewhurst, I. Di Marco, C. Draxl, M. Dulak, O. Eriksson, J. A. Flores-Livas, K. F. Garrity, L. Genovese, P. Giannozzi, M. Giantomassi, S. Goedecker, X. Gonze, O. Granaes, E. K. U. Gross, A. Gulans, F. Gygi, D. R. Hamann, P. J. Hasnip, N. A. W. Holzwarth, D. Iusan, D. B. Jochym, F. Jollet, D. Jones, G. Kresse, K. Koepernik, E. Kuecuekbenli, Y. O. Kvashnin, I. L. M. Locht, S. Lubeck, M. Marsman, N. Marzari, U. Nitzsche, L. Nordstrom, T. Ozaki, L. Paulatto, C. J. Pickard, W. Poelmans, M. I. J. Probert, K. Refson, M. Richter, G.-M. Rignanese, S. Saha, M. Scheffler, M. Schlipf, K. Schwarz, S. Sharma, F. Tavazza, P. Thunstroem, A. Tkatchenko, M. Torrent, D. Vanderbilt, M. J. van Setten, V. Van Speybroeck, J. M. Wills, J. R. Yates, G.-X. Zhang and S. Cottenier, Reproducibility in density functional theory calculations of solids, Science, 2016, 351(6280), aad3000 CrossRef PubMed.
- N. Mardirossian and M. Head-Gordon, Thirty years of density functional theory in computational chemistry: an overview and extensive assessment of 200 density functionals, Mol. Phys., 2017, 115, 2315–2372 CrossRef CAS.
- S. McArdle, S. Endo, A. Aspuru-Guzik, S. C. Benjamin and X. Yuan, Quantum computational chemistry, Rev. Mod. Phys., 2020, 92, 015003 Search PubMed.
- H. Cavaye, Neutron Spectroscopy: An Under-Utilised Tool for Organic Electronics Research?, Angew. Chem., Int. Ed., 2019, 58, 9338–9346 Search PubMed.
- N. Mardirossian and M. Head-Gordon, Thirty years of density functional theory in computational chemistry: an overview and extensive assessment of 200 density functionals, Mol. Phys., 2017, 115, 2315–2372 CrossRef CAS.
- E. Brémond, I. Ciofini, J. C. Sancho-García and C. Adamo, Nonempirical Double-Hybrid Functionals: An Effective Tool for Chemists, Acc. Chem. Res., 2016, 49, 1503–1513 Search PubMed.
- V. Milman, K. Refson, S. J. Clark, C. J. Pickard, J. R. Yates, S.-P. Gao, P. J. Hasnip, M. I. J. Probert, A. Perlov and M. D. Segall, Electron and vibrational spectroscopies using DFT, plane waves and pseudopotentials: CASTEP implementation, J. Mol. Struct.: THEOCHEM, 2010, 954, 22–35 CrossRef CAS.
- J. Sun, R. C. Remsing, Y. Zhang, Z. Sun, A. Ruzsinszky, H. Peng, Z. Yang, A. Paul, U. Waghmare, X. Wu, M. L. Klein and J. P. Perdew, Accurate first-principles structures and energies of diversely bonded systems from an efficient density functional, Nat. Chem., 2016, 8, 831–836 CrossRef CAS PubMed.
- M. L. Cohen, in Encyclopedia of Condensed Matter Physics, ed. F. Bassani, G. L. Liedl and P. Wyder, Elsevier, Oxford, 2005, pp. 85–93 Search PubMed.
- J. Klimeš and A. Michaelides, Perspective: Advances and challenges in treating van der Waals dispersion forces in density functional theory, J. Chem. Phys., 2012, 137, 120901 Search PubMed.
- A. Tkatchenko, R. A. DiStasio, R. Car and M. Scheffler, Accurate and Efficient Method for Many-Body van der Waals Interactions, Phys. Rev. Lett., 2012, 108, 236402 CrossRef PubMed.
- S. J. Clark, M. D. Segall, C. J. Pickard, P. J. Hasnip, M. J. Probert, K. Refson and M. C. Payne, First principles methods using CASTEP, Z. Kristallogr., 2005, 220, 567–570 CAS.
- A. Erba, J. K. Desmarais, S. Casassa, B. Civalleri, L. Donà, I. J. Bush, B. Searle, L. Maschio, L. Edith-Daga, A. Cossard, C. Ribaldone, E. Ascrizzi, N. L. Marana, J.-P. Flament and B. Kirtman, CRYSTAL23: A Program for Computational Solid State Physics and Chemistry, J. Chem. Theory Comput., 2023, 19, 6891–6932 Search PubMed.
- M. L. Cohen, in Encyclopedia of Condensed Matter Physics, ed. F. Bassani, G. L. Liedl and P. Wyder, Elsevier, Oxford, 2005, pp. 85–93 Search PubMed.
- B. S. Hudson, Vibrational spectroscopy using inelastic neutron scattering: Overview and outlook, Vib. Spectrosc., 2006, 42, 25–32 Search PubMed.
- B. S. Hudson, in Frontiers of Molecular Spectroscopy, ed. J. Laane, Elsevier, Amsterdam, 2009, pp. 597–622 Search PubMed.
- S. F. Parker, A. J. Ramirez-Cuesta and L. Daemen, Vibrational spectroscopy with neutrons: Recent developments, Spectrochim. Acta, Part A, 2018, 190, 518–523 CrossRef CAS PubMed.
- S. F. Parker, D. Lennon and P. W. Albers, Vibrational Spectroscopy with Neutrons: A Review of New Directions, Appl. Spectrosc., 2011, 65, 1325–1341 CrossRef CAS.
- J. Armstrong, A. J. O'Malley, M. R. Ryder and K. T. Butler, Understanding dynamic properties of materials using neutron spectroscopy and atomistic simulation, J. Phys. Commun., 2020, 4, 072001 CrossRef CAS.
- M. M. Coimbra, I. Martins, S. M. Bruno, P. D. Vaz, P. J. A. Ribeiro-Claro, S. Rudić and M. M. Nolasco, Shedding Light on Cuprorivaite, the Egyptian Blue Pigment: Joining Neutrons and Photons for a Computational Spectroscopy Study, Cryst. Growth Des., 2023, 23, 4961–4969 Search PubMed.
- P. C. H. Mitchell, S. F. Parker, A. J. Ramirez-Cuesta and J. Tomkinson, Vibrational Spectroscopy with Neutrons: With Applications in Chemistry, Biology, Materials Science and Catalysis, World Scientific Publishing Co, Singapore, 2005 Search PubMed.
- M. M. Nolasco, C. F. Araujo, P. D. Vaz, A. M. Amado and P. Ribeiro-Claro, Vibrational dynamics of crystalline 4-phenylbenzaldehyde from INS spectra and periodic DFT calculations, Molecules, 2020, 25, 1374 CrossRef CAS PubMed.
- P. J. A. Ribeiro-Claro, P. D. Vaz, M. M. Nolasco and A. M. Amado, Understanding the vibrational spectra of crystalline isoniazid: Raman, IR and INS spectroscopy and solid-state DFT study, Spectrochim. Acta, Part A, 2018, 204, 452–459 CrossRef CAS PubMed.
- M. M. Nolasco, M. M. Coimbra, S. F. Parker, P. D. Vaz and P. J. A. Ribeiro-Claro, Structural Dynamics of Chloromethanes through Computational Spectroscopy: Combining INS and DFT, Molecules, 2022, 27(21), 7661 CrossRef CAS PubMed.
- L. Zhong and S. F. Parker, Structure and vibrational spectroscopy of methanesulfonic acid, R. Soc. Open Sci., 2018, 5, 181363 Search PubMed.
- K. Drużbicki, E. Mikuli, N. Pałka, S. Zalewski and M. D. Ossowska-Chruściel, Polymorphism of Resorcinol Explored by Complementary Vibrational Spectroscopy (FT-RS, THz-TDS, INS) and First-Principles Solid-State Computations (Plane-Wave DFT), J. Phys. Chem. B, 2015, 119, 1681–1695 CrossRef PubMed.
- A. Pawlukojć and Ł. Hetmańczyk, INS, DFT and temperature dependent IR studies on dynamical properties of acetylcholine chloride, Vib. Spectrosc., 2016, 82, 37–43 CrossRef.
- S. F. Parker and I. R. Butler, Synthesis, Computational Studies, Inelastic Neutron Scattering, Infrared and Raman Spectroscopy of Ruthenocene, Eur. J. Inorg. Chem., 2019, 1142–1146 CrossRef CAS.
- B. C. Chakoumakos, J. A. Fernandez-Baca and L. A. Boatner, Refinement of the Structures of the Layer Silicates MCuSi4O10 (M = Ca, Sr, Ba) by Rietveld Analysis of Neutron Powder Diffraction Data, J. Solid State Chem., 1993, 103, 105–113 CrossRef CAS.
- S. H. Masunaga, A. Rebello, A. T. Schye, N. Prasai, J. J. Neumeier and J. L. Cohn, Heat Capacity, Thermal Expansion and Heat Transport in the Han Blue (BaCuSi4O10): Observation of Structural Phase Transitions, J. Phys. Chem. Solids, 2015, 85, 69–74 CrossRef CAS.
- C. F. Araújo, D. O. Abranches, J. A. P. Coutinho, P. D. Vaz, P. Ribeiro-Claro and M. M. Nolasco, Good vibrations: understanding deep eutectic solvents through the lens of vibrational spectroscopy, Appl. Spectrosc. Rev., 2024, 60(2), 137–192 CrossRef.
- C. F. Araújo, P. Ribeiro-Claro, P. D. Vaz, S. Rudic, R. A. F. Serrano, L. P. Silva, J. A. P. Coutinho and M. M. Nolasco, Exploring asymmetry induced entropy in tetraalkylammonium-urea DES systems: what can be learned from inelastic neutron scattering?, Phys. Chem. Chem. Phys., 2024, 26, 5969–5977 RSC.
- R. A. F. Serrano, C. F. Araújo, P. Ribeiro-Claro, P. D. Vaz, S. Rudić, J. A. P. Coutinho and M. M. Nolasco, Decoding disorder: unravelling entropy effects in deep eutectic systems with neutron spectroscopy, Phys. Chem. Chem. Phys., 2025 Search PubMed , Advance Article.
- J. Hjortås and H. Sørum, A re-investigation of the crystal structure of choline chloride, Acta Crystallogr., Sect. B, 1971, 27, 1320–1323 CrossRef.
- K. D. M. Harris and E. Y. Cheung, How to determine structures when single crystals cannot be grown: opportunities for structure determination of molecular materials using powder diffraction data, Chem. Soc. Rev., 2004, 33, 526–538 RSC.
- R. K. Harris, NMR crystallography: the use of chemical shifts, Solid State Sci., 2004, 6, 1025–1037 CrossRef CAS.
- L. Emsley, Spiers Memorial Lecture: NMR crystallography, Faraday Discuss., 2025, 255, 9–45 RSC.
- M. Baias, J.-N. Dumez, P. H. Svensson, S. Schantz, G. M. Day and L. Emsley, De Novo Determination of the Crystal Structure of a Large Drug Molecule by Crystal Structure Prediction-Based Powder NMR Crystallography, J. Am. Chem. Soc., 2013, 135, 17501–17507 CrossRef CAS PubMed.
- T. Pawlak, M. Jaworska and M. J. Potrzebowski, NMR crystallography of α-poly(l-lactide), Phys. Chem. Chem. Phys., 2013, 15, 3137–3145 RSC.
- A. Hofstetter, M. Balodis, F. M. Paruzzo, C. M. Widdifield, G. Stevanato, A. C. Pinon, P. J. Bygrave, G. M. Day and L. Emsley, Rapid Structure Determination of Molecular Solids Using Chemical Shifts Directed by Unambiguous Prior Constraints, J. Am. Chem. Soc., 2019, 141, 16624–16634 Search PubMed.
- P. R. Carey, Raman Crystallography and other Biochemical Applications of Raman Microscopy, Annu. Rev. Phys. Chem., 2006, 57, 527–554 CrossRef CAS PubMed.
- J. T. Sage, Y. Zhang, J. McGeehan, R. B. G. Ravelli, M. Weik and J. J. van Thor, Infrared protein crystallography, Biochim. Biophys. Acta, Proteins Proteomics, 2011, 1814, 760–777 CrossRef CAS PubMed.
- J. T. Sage, Infrared Crystallography: Structural Refinement through Spectroscopy, Appl. Spectrosc., 1997, 51, 568–573 Search PubMed.
- M. M. Nolasco, L. C. Rodrigues, C. F. Araujo, M. M. Coimbra, P. Ribeiro-Claro, P. D. Vaz, S. Rudic, A. J. D. Silvestre, C. Bouyahya, M. Majdoub and A. F. Sousa, From PEF to PBF: What difference does the longer alkyl chain make a computational spectroscopy study of poly(butylene 2,5-furandicarboxylate), Front. Chem., 2022, 10, 1056286 Search PubMed.
- J. Zhu, J. Cai, W. Xie, P.-H. Chen, M. Gazzano, M. Scandola and R. A. Gross, Poly(butylene 2,5-furan dicarboxylate), a Biobased Alternative to PBT: Synthesis, Physical Properties, and Crystal Structure, Macromolecules, 2013, 46, 796–804 CrossRef CAS.
- C. F. Araújo, S. V. Pandeirada, I. M. Oliveira, G. B. Rosa, B. Agostinho, A. J. D. Silvestre, A. F. Sousa, S. Rudić, P. D. Vaz, M. M. Nolasco and P. Ribeiro-Claro, Crystal structure of poly(trimethylene 2,5-furandicarboxylate) redux – a new model supported by computational spectroscopy, Polym. Chem., 2024, 15, 4349–4363 RSC.
- O. Toledano, O. Gálvez, M. Sanz, C. Garcia Arcos, E. Rebollar, A. Nogales, M. C. García-Gutiérrez, G. Santoro, I. Irska, S. Paszkiewicz, A. Szymczyk and T. A. Ezquerra, Study of the Crystal Structure and Hydrogen Bonding during Cold Crystallization of Poly(trimethylene 2,5-furandicarboxylate), Macromolecules, 2024, 57, 2218–2229 Search PubMed.
- C. F. Araujo, M. M. Nolasco, P. J. A. Ribeiro-Claro, S. Rudić, A. J. D. Silvestre, P. D. Vaz and A. F. Sousa, Inside PEF: Chain Conformation and Dynamics in Crystalline and Amorphous Domains, Macromolecules, 2018, 51(9), 3515–3526 CrossRef CAS.
- K. Tashiro, in Structural Science of Crystalline Polymers: Microscopically-Viewed Structure-Property Relationship, ed. K. Tashiro, Springer Nature Singapore, Singapore, 2024, pp. 1–273 Search PubMed.
- T. Kurita, Y. Fukuda, M. Takahashi and Y. Sasanuma, Crystalline Moduli of Polymers, Evaluated from Density Functional Theory Calculations under Periodic Boundary Conditions, ACS Omega, 2018, 3, 4824–4835 CrossRef CAS PubMed.
- A. Alesadi, F. Fatima, W. Xia and D. Kilin, First-Principles Study on the Electronic Properties of PDPP-Based Conjugated Polymer via Density Functional Theory, J. Phys. Chem. B, 2021, 125, 8953–8964 CrossRef CAS PubMed.
- T. Lin, X.-Y. Liu and C. He, Calculation of Infrared/Raman Spectra and Dielectric Properties of Various Crystalline Poly(lactic acid)s by Density Functional Perturbation Theory (DFPT) Method, J. Phys. Chem. B, 2012, 116, 1524–1535 CrossRef CAS PubMed.
- Y. Fukuda and Y. Sasanuma, Thermal and Mechanical Properties of Poly(methylene oxide) Polymorphs Unraveled by Periodic Density Functional Theory, Macromolecules, 2018, 51, 8672–8680 Search PubMed.
- S. K. Burgess, J. E. Leisen, B. E. Kraftschik, C. R. Mubarak, R. M. Kriegel and W. J. Koros, Chain Mobility, Thermal, and Mechanical Properties of Poly(ethylene furanoate) Compared to Poly(ethylene terephthalate), Macromolecules, 2014, 47, 1383–1391 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2025 |