
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Protein crystallisation with gas microbubbles as soft template in a microfluidic device
Wenqing
Tian
,
Oladayo
Ogunyinka
,
Charlie
Oretti
,
H. C. Hemaka
Bandulasena
 ,
Chris
Rielly
and
Huaiyu
Yang
*
,
Chris
Rielly
and
Huaiyu
Yang
*
Department of Chemical Engineering, Loughborough University, Loughborough, UK. E-mail: h.yang3@lboro.ac.uk
First published on 18th August 2023
Abstract
Lysozyme crystallisation was first-time performed in a microfluidic device in the presence of different gases: helium, nitrogen, oxygen, and carbon dioxide microbubbles. It was found that protein adsorbed on the gas–liquid interface stabilised the gas bubbles in the aqueous solution, and bubble stability increased with the protein concentration in the solution. The heterogeneous nucleation of protein on the gas–liquid interface was preferred than on the capillary glass wall, limiting the fouling inside the capillary. The crystals formed with curved surfaces, and the crystals floated in the solution with gas bubbles. The population density of lysozyme crystals increased with an increase in the solubility of four types of gases. Three stages of the protein crystallisation on the gas–liquid, gas–solid and liquid–solid interfaces were discussed.
Design, System, ApplicationThe work has investigated the molecules interactions, between gases (four types of gases), solvent (water and salt) and protein (lysozyme) molecules on the gas–liquid, gas–solid and liquid–solid interfaces. The microfluidic system has been designed to generate the gas bubbles and to observe the crystallisation process with gas bubbles, which can be used in screening and optimisation of the crystallisation conditions for biomaterials. There are advantages to use microfluidic for optimisation the crystallisation process with limited expensive biomaterials, and it is also able to observe the crystal growth process under optical microscope, the restriction of the microfluidic is the scalability, as the conditions explored in the microfluidic system need to be transferred to larger-scale crystallization processes. This study has the potential to be applied in both chemical and biopharmaceutical manufacturing. By using gas templates during protein crystallization, this system offers several advantages, including shortened nucleation time, controlled crystal size, and optimized crystal quality, all without introducing other solid templates that may cause impurities. These advantages are particularly important in the manufacturing of biopharmaceuticals, where increasing efficiency and minimizing impurities are critical factors. |
Introduction
In recent decades, protein-based biological products played an increasingly significant role in treating many illnesses like diabetes, cancer and SARS-CoV-2,1–3 due to the advancements in pharmacodynamics and pharmacokinetics. The biopharmaceuticals have become one of the fastest growing therapeutics and is predicted to reach a global market value of $337 billion by 2025, with an annual growth rate of 7.4%.4 With the rapid growth in demand, protein purification could become vital. However, manufacturing the biopharmaceuticals still presents significant challenges, due to the complexity of the macro molecules and cost of purification.5 The macro molecules have inherent complex structure and purifying them while maintaining the bioactivity requires long and complex process.6,7 The current protein purification usually employed chromatography, which requires large amount of resins, making the process costly.3,8,9 Crystallisation has been an important unit operation in downstream processing in pharmaceutical industry with advantages of stability, storage, and delivery. Recent research on protein crystallisation has thus focused from crystallography to developing an efficient technology to obtain purified protein crystals.10–13Microfluidic systems can be designed by introduction of different components and different phases into crystallisation solution at microscale. Microfluidics is an ideal platform to study crystallisation due to its unique advantages such as minimum usage of reagents14–16 and ability to achieve precise control15,17 of pharmaceuticals like paracetamol16,18 and alanine.19,20 The microfluidic devices often require only a minimum amount of reagents, a clear advantage for protein crystallisation as most of the protein reactants are quite expensive and only available in low quantity. Nano-litre crystallisation droplets mixed with a carrier fluid in a microfluidic system was reported for thaumatin protein crystallisation.21 The system only required ∼1 μL protein solution that was separated into hundreds of nanolitre droplets, which were examined by analysing the nucleation rate. Another advantage of using microfluidic devices is its capability of mixing multiphase flow and miniaturisation of crystallisation reactors.20 A microfluidic device enabled injecting microbubbles into a flowing solution for studying heterogeneous nucleation of paracetamol.16 A two-phase flow regime was generated by the microfluidic device with different gas and liquid flowrates to identify the optimal conditions for microbubble generation. The nucleation rate was found to increase in the presence of microbubbles and the manipulation of flowrates allowed control the crystallisation process of paracetamol. Two immiscible liquid phases could be introduced in a microfluidic device for protein crystallisation to improve mixing and reduce the axial dispersion.22 A microfluidic device can enhance heat and mass transfer rates due to high surface/volume ratio for protein nucleation and crystal growth.23 Microfluidic systems thus provides a unique opportunity and insight to study protein crystallisation at μL or mL scale,21 and enables the introduction of multiphase mixing.
Gassing (i.e., introduction of gas bubbles) in crystallisation could facilitate the crystallisation process such as in an airlift crystalliser.24 Protein interactions at the gas–liquid interfaces are complicated and the process often includes adsorption, rearrangement, relaxation and unfolding.25–28 The proteins like bovine serum albumin, β-lactoglobulin, and lysozyme adsorbed at the gas–liquid interface at the bulk concentration as low as 0.001 mg mL−1.28 The transient surface pressure, an indicator for the level of protein adsorption, can change within 1 h of adsorption time. The globular protein like lysozyme was found to hold strong conformational stability and resisted the unfolding at the gas–liquid interface,29 which can crystallise on the gas–liquid interface.30 However, the mechanism of crystallisation on the interface between gas–liquid–solid are still not fully understood, the influences of the surface properties of the gas bubbles on protein molecules or the protein crystals require further investigations.
In this work, the gas bubbles containing helium, nitrogen, oxygen, and carbon dioxide were injected into the crystallisation solution by a microfluidic device. The size of the gas bubbles was controlled to be consistent. The initial lysozyme concentration in the crystallisation solution was in the range of 25–60 mg mL−1. The stability of gas bubbles in protein solutions with time and the protein crystallisation process on the different gas-bubble surfaces were observed.
Experimental section
Materials
Hen egg white lysozyme (∼70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 units per mg protein), sodium acetate (purity >99%), sodium chloride (purity >99.5%), and glacial acetic acid (purity >99.5%) were purchased from Sigma-Aldrich and used without further purification. Buffer solution was prepared as 0.1 M sodium acetate in deionized water at pH 4.2 by addition of acetic acid. Lysozyme solutions with concentrations of 50, 70, 100 and 120 mg mL−1 were prepared by dissolving lysozyme in sodium acetate buffer solution, and NaCl solutions were prepared in the same method to reach a concentration of 1.1 M. All solutions were filtered through a 0.2 μm cellulose acetate filter and stored at 20 °C in an incubator (VWR INCU-Line 150R) for experiments. All experiments were carried out at room temperature (20 °C) to avoid temperature changes in preparing the samples and crystallisation experiments.
000 units per mg protein), sodium acetate (purity >99%), sodium chloride (purity >99.5%), and glacial acetic acid (purity >99.5%) were purchased from Sigma-Aldrich and used without further purification. Buffer solution was prepared as 0.1 M sodium acetate in deionized water at pH 4.2 by addition of acetic acid. Lysozyme solutions with concentrations of 50, 70, 100 and 120 mg mL−1 were prepared by dissolving lysozyme in sodium acetate buffer solution, and NaCl solutions were prepared in the same method to reach a concentration of 1.1 M. All solutions were filtered through a 0.2 μm cellulose acetate filter and stored at 20 °C in an incubator (VWR INCU-Line 150R) for experiments. All experiments were carried out at room temperature (20 °C) to avoid temperature changes in preparing the samples and crystallisation experiments.
Experimental
The microfluidic device mainly consisted of two parts: the microfluidic chip for microbubble production and the capillary tube for crystallisation. As shown in Fig. 1(a), the gas was supplied to the microchip by a compressed gas cylinder and controlled by a mass flow controller (Alicat MC 100 sccm); the aqueous solution was injected using a syringe pump (KD scientific 100 legacy). The microfluidic chip was made using standard photolithography and polydimethylsiloxane (PDMS) replica moulding techniques and details of manufacturing this microfluidic channel were provided in a previous study.31 A 150 μm glass cover slide was bonded to a cured 500 μm thick PDMS layer with 150 μm deep channels moulded by photolithography. The gas and liquid inlet were 200 μm wide, where the microbubbles were produced in a flow-focusing junction and moved into a 1.6 mm wide bubbly flow channel shown in Fig. 1(b). The mechanism of microbubble formation was reported by Garstecki et al.32 The second part of the device was a borosilicate glass capillary with an internal diameter of 0.58 mm and OD 1 mm (World Precision Instruments), which were cleaned by an ultrasonic cleaner, and thoroughly rinsed with filtered DI water using a syringe pump, ensuring clean environment and maintained flow stability. The liquid inflow containing crystallisation solution was introduced into the microfluidic chip by a syringe pump, whereas the gas flow was supplied by a gas cylinder and controlled by a mass flow controller as shown in Fig. 1(a). | ||
| Fig. 1 Schematic set-up for protein crystallisation (a) and microscopic image of bubble formation inside the microfluidic chip (b). | ||
A white light LED source (Kern Dual Fiber Unit LED), and a high-speed camera (Photon Fastcam, M2.1) with a microscope lens (INFINITY KC IF-3) were focused on the microfluidic channel to capture the bubble formation in the channel. The bubbles generated within the chip had a narrow size distribution as the bubbling process was tightly controlled. The outlet of the microfluidic chip was connected to the capillary by a 5 cm long polytetrafluoroethylene tube (Cole Palmer) with an inner diameter of 1.5 mm.
The protein solution, with concentration of 50 mg mL−1 to 120 mg mL−1, and the precipitant solution of 1.1 M NaCl with pH 4.2 sodium acetate buffer were mixed at 1:1 ratio in a clean beaker, and the premixed solution was filled into a syringe pump of 5 mL. After mixing, the crystallisation solution had 0.55 M NaCl and 25–60 mg mL−1 lysozyme. The period between mixing and injecting into the microfluidic device was under 2 min, which was very short compared to the nucleation time at the equal crystallisation condition,30 to ensure there was no nucleation before the mixed solution was pumped into the capillary. The liquid flowrate to the microfluidic chip was set at 60 ml h−1. The variation of protein concentrations was tested with the four types of gas (helium, nitrogen, oxygen or carbon dioxide), supplied by a compressed gas cylinder and controlled by the mass flow meter at a constant flowrate of 0.4 sccm. Since the volumetric flowrate for gas and liquid are constant, the ratio between gas and liquid phase is constantly 3:7 across all the experimental conditions. The initial bubble diameter (400 ± 10 μm) and the number of bubbles per unit volume were constant for all types of gasses. After filling the solutions containing stable bubbles in the capillaries, the capillaries were disconnected from the microfluidic device and were immediately sealed at both ends with paraffin oil to avoid evaporation of the solution. The capillaries were stored at 20 °C in an incubator (VMR, INCU-Line 150R) and observed periodically under an optical microscope (GT Vision, GTC-20) with an observation period of 56 h.30 All the experiments were repeated at least 5 times.
ImageJ software (Wayne Rasband, National Institute of Health, USA) was used to examine the microscope images of the crystals inside the capillary. With the same magnification (×40), the section of capillary captured in the microscope was consistent to be 1.5 μL. For each condition, microscope images at least 5 fixed locations distributed along the whole capillary were taken. The number of crystals and number of bubbles were counted individually in the ImageJ software and crystal sizes and bubble sizes per section were measured and recorded. For each condition, all the recorded sizes were used to find the number average and calculate the standard deviation of the size distribution according to the descriptive statistical model. The population density (P), defined as the number of crystals per unit volume, and calculated as the average value of the crystal numbers divided by 1.5 μL solution in each microscope image for each condition. P was used to compare the influence on crystallisation progress of different conditions. The minimum diameter circle was drawn in ImageJ to enclose the crystals edges in the images for estimation of the crystal facets, which was then converted into crystal size. The crystal size and bubble size were estimated by using the circular equivalent diameter:
 | (1) |
 | (2) |
| m = V × ρ × P | (3) |
Result and discussion
Gas–solid interface during crystallisation
Fig. 2(a) shows that many crystals formed on the bubble surfaces, and Fig. 2(b) shows that many crystals formed on the wall surfaces and some crystal formed in the solution and sank on the bottom of the capillary due to gravity. Fig. 2(a) shows that the crystals formed on the interface of the gas bubbles, which were much larger than the crystals formed on the wall surfaces in the solution without gas bubbles, shown as Fig. 2(b). It was consistent with report that the presence of gas bubbles could improve the protein crystal growth.34 The first observation of crystals in the solution without gas bubbles were always on the inside capillary wall, while the first observation of crystals in the solution with gas bubbles were always on the gas bubbles. In the experiments without gas bubbles, the formation of tiny crystals on the capillary wall, as fouling, due to heterogeneous nucleation on the capillary wall.11 In the experiments with gas bubbles, crystals preferred to nucleate on the gas bubble surface. Fouling could lead to troubles in heat transferring and process control,35 and, especially for continuous crystallization of proteins,36 with much longer operation period to reduce the risk of blockage and maintain the mixing efficiency. As shown in this work, gas bubbles as template could be a possible technology to limit the fouling. | ||
| Fig. 2 Microscope images and schematic images of crystal formation in the solution: (a) 60 mg mL−1 lysozyme, 0.55 M NaCl with (a) and without (b) helium gas bubbles. (c) Schematic diagram for floating crystals grown on the bubbles. (d) and (e) two examples of the curved crystals initially grew on the bubble template obtained in this solution with helium gas bubbles. | ||
The SEM images of the lysozyme crystals show a curved surface of each crystal in Fig. 2(d) and (e) instead of normal tetragonal shape without the gas bubble. The curved surface were observed in nearly all the crystal nucleated on the gas bubbles, but the curved surface areas were different. Crystals with different part-spherical curved surfaces resulted by the nucleation on the gas surface and crystal growth blocked by the air bubble template, which were dependant on the crystal growth direction. Some of the curved surfaces were porous surface and some were smooth surface, but no correlations were found with concentrations of lysozyme and the salts. It is worth to mention that due to the strong attachment between the curved crystal with gas bubble, at early stage of crystal growth, there was limited chance the gaps appeared between the curved crystals with gas bubble surfaces. As shown in our previous work,30 the gas bubble surface attached on the crystals maintained the shape while the gas bubble surface without attaching on the crystals shrank much faster. It is very interesting to observe that crystals formed on the bubble surface were able to float, as shown in Fig. 2(a) and (c), proving the strong attachment of the crystals on the gas bubbles. The crystals that floated with bubbles or those sank to the bottom can be easily distinguished by rotating the capillary. When the buoyant force overcame the gravity of the crystals (the volume of crystals was estimated to be smaller than 1.2 times of the bubble), the bubbles floated with crystal. Oversized crystals crystallised on the air bubble surface dragged the bubble together to sink to the bottom, which was observed in a few cases. The curved crystal surface and the floating crystals in this work, as well as the rotation of the crystals in previous work,30 all revealed the strong attachment of crystal on the gas bubble surface and the interactions between the protein molecules with the gas molecules on the solid-air interface.
The yield increased with increase in the lysozyme concentrations as expected. The solubility of lysozyme was 7.9 mg mL−1 at the experimental temperature of 20 °C with 1.1 M NaCl precipitation solution (0.55 M in the mixed solution).12 The lysozyme concentration raised from 25 mg mL−1 to 60 mg m−1 and supersaturation increased from 3.2 to 7.6, leading to the higher yield at the end of experiment. Larger crystals were obtained with high concentration of the lysozyme at the end, due to longer growth period,49 but much higher of the lysozyme concentration may decrease the average crystal size due to the dominating effect of the crystal nucleation over the crystal growth as reported in literatures.30 The mean crystal size at 55 h increased from 149.6 μm in 25 mg mL−1 lysozyme solution to 268.3 μm in 50 mg mL−1 lysozyme solution. In the 60 mg mL−1 lysozyme experiment, the mean crystal size reduced to 203.4 μm due to formation of much more crystals.
Four different gases, He, N2, O2 and CO2, were tested in the microfluidic device with solution of the same volume at 50 mg mL−1 lysozyme and 0.55 M NaCl. The same size of the gas bubbles was injected shown in Fig. 1, and different size of the gas bubbles remained at final stage of the experiment, shown in Fig. 3(a–d). All the bubbles of different gases shrank (decrease in average size), and the shrinking rates were different. Fig. 3(d) shows CO2 bubbles shrank most than the bubbles of the other gases in the solution, due to its highest solubility in the solution. Despite the different degrees of shrinkage for all the gas bubbles, the crystals formed on the bubble surfaces were still strongly attached to the gas bubble surface. Some crystals on the bottom of the capillary were observed in the solutions with He, N2, and O2 bubbles, but there were all above 80% of crystals formed on bubble surfaces of all types, and almost 100% of the crystals formed on the CO2 bubble surface.
 | ||
| Fig. 3 Crystallisation at 56 h in helium (a), nitrogen (b), oxygen (c) carbon dioxide (d) with scale bar of 100 μm. (e) Population densities of lysozyme crystals obtained with 50 mg mL−1 lysozyme and 0.55 M NaCl vs. gas solubilities37 for different gases. (f) Percentage of crystals formed on the gas bubble surface vs. the gas polarizability.38 Error bars for the variations in repeated experiments and dash lines for a guiding. | ||
Fig. 3(e) shows a higher solubility of the gas linked to formation of more crystals. There was a highest possibility of the crystal forming on the CO2 bubble surfaces, crystals on CO2 bubbles were most, and the lowest possibility and lest crystals were on He bubbles. It is noted that CO2 is an acidic gas, but it was found that pH in the solution did not change within the range of CO2 addition in this work (tested in experiments), due to the acetate buffer solution used in protein crystallisation. Based on the experimental condition that 0.04 mol of carbon dioxide in 1 L solution,38 and the maximum carbon dioxide concentration that could be dissolved in the solution was about 0.01 M estimated with its solubility of 0.878 mL mL−1 in water. The maximum amount of dissolved CO2 was much smaller than the acetate buffer concentration. In addition, the pKα of carbonic acid was about 6.4,39 which was within the buffer range of acetate buffer. Despite no change of pH in bulk solution, but local pH changes near the CO2 gas–liquid interfaces may influence the nucleation on the air bubble surfaces.40,41
The mean lysozyme crystal sizes at 56 h were 268.3 μm, 207.5 μm, 349.6 μm and 325.2 μm in the solution with bubbles of He, N2, O2 and CO2, respectively. The mean size of crystals obtained in the solution with He and N2 bubbles was smaller than the crystals obtained in the solution with bubbles of oxygen and carbon dioxide. Solubilities of He, N2, O2 and CO2 in 1 ml fresh water at 20 °C and under atmospheric pressure are 0.0138, 0.0164, 0.031, and 0.878 ml, respectively.37 The trend in mean crystal size was similar to the trend of gas solubility. The yield estimated by eqn (3) in the solution with CO2 bubbles was the highest, 48.1 mg mL−1, more than four times than that in the solution with the N2 bubbles.
Fig. 3(f) shows with increase of the gas polarizability, the percentage of crystals formed on the bubble surfaces tended to increase. The covariance between these two variables were found to be 0.05, and the square of the correlation coefficient (R2) was calculated as 0.80, which indicate a moderately positive linear correlation between the polarisability of the gas and the percentage of crystals formed on the bubble surfaces. The gas polarizability could impact the interactions with protein molecules in the solution. The isoelectric point of lysozyme was pH 11 and the lysozyme was partially charged under the acidic buffer.42 There were possible two kinds of interactions43 and attractive forces. A stronger interaction was electrostatic force due to partially charged molecules and van der Waals force was the weaker interaction between lysozyme molecules and gas due to their induced dipoles. These interactions could induce adsorption of more protein molecules on the bubble surface, contributing to nucleation process. It is noted that the percentages of crystals forming on the bubble surface in the solution with different gases followed the same trend of zeta potentials (in the pH neutral environment) of these gases, oxygen < nitrogen < carbon dioxide.44,45 The links between crystal products and gas properties could provide guidance for selectively protein crystallization, and it is worth in further investigations on other gas types, which could contribute to better understanding of the complex system and progress involving gas–solid–liquid interfaces. With these new phenomena and detailed analysation, we want to explore a new and complex and multidisciplinary (surface, crystallisation, colloid, gas) system and inspire more further research to have a fully understanding the underpinning mechanism.
Gas–liquid interface before crystallisation
After the crystallisation solution with equal size bubble injected into capillary, shown in Fig. 1(b), before nucleation the bubbles average size of the gas bubbles in capillaries gradually reduced, shown in Fig. 4(a). Fig. 4(c) shows that the bubbles of the gas with high solubilities had a smaller average bubble size at the final stage. The bubbles of all kinds of gases had an obvious reduction of average bubble size due to dissolution, and the percentage reduction of the average gas bubble diameters was in the order of He < N2 < O2 < CO2, which was consistent with the order of their solubility in the solution. The average bubble sizes of helium, nitrogen, oxygen and carbon dioxide were 372.3 ± 29.8 μm, 328.8 ± 5.0 μm, 317.6 ± 15.7 μm, and 153.8 ± 9.8 μm at the end of experiments, respectively. | ||
| Fig. 4 The change of the nitrogen bubble size (a) in the solution of 35 mg mL−1 lysozyme and 0.55 M NaCl, schematic diffusion process (b) during bubble size splitting, average bubble diameters (c) at 1 h and 56 h for four types of gases in the solution, and change in bubble size against gas solubilities (d) with standard deviations and guiding line. | ||
Despite the decrease in average size of the air bubbles (overall gas bubble shrinkage), many bubbles became smaller with time but many bubbles became larger than initial stage, shown in Fig. 4(a). Therefore, the variations of the bubble sizes highly increased, and the gas bubbles can be easily sorted into groups of either large or small diameter. The phenomenon was due to the Ostwald ripening as a result of Laplace pressures difference among bubbles,46 shown in Fig. 4(b). Laplace pressure, ΔP, measures the pressure difference between inside and outside of a curved gas surface in a liquid, and for a spherical bubble it is47
 | (4) |
 | (5) |
Fig. 4(d) shows that for He, N2, and O2 gas bubbles, the average size of large-bubble group was higher than the initial size of the bubbles. The size splitting of gas bubbles between the small-bubble and large-bubble groups of N2 and O2 were about 350–410 μm, and the size splitting for He and CO2 were much less, below 200 μm. The He bubbles were relatively stable with less diffusion, but, for CO2 bubbles, the large size bubbles tended to dissolve due to high solubility, resulting in less variations of the large-bubble and small-bubble group bubbles.
The shrinkage was dependent on the gas solubility and was also influenced by the concentration of the protein in the solution. With same gas, the change of the average bubble size decreased in higher protein concentration solution, shown in Fig. 5(a) as an example of solutions with different concentration of protein and all with same helium gas. The surfactant can lower the surface tension of the bubbles, increase the elasticity of the bubble surface, promoting the gas bubble stability in the solution.49,50 The gas bubble could adsorb lysozyme molecules, covering the gas–liquid interface, due to specific geometric distribution of the hydrophilic and hydrophobic function groups in the protein. This created an accumulation of proteins onto the bubble surface and acted as a surfactant46 to reduce the interfacial tension between the gas and the solution, to reduce the gas dissolution rate in the solution,51 and, therefore, to stabilise the gas bubbles in the solution. With higher protein concentration in the solution, more lysozyme molecules can be adsorbed on the bubble surface, enhancing the stability of the gas bubbles, shown in Fig. 5(b).
 | ||
| Fig. 5 Change in average bubble size between 5 h and 56 h in the lysozyme solution of concentrations 25–60 mg mL−1 with helium gas microbubbles (a) and corresponding schematic diagrams of lysozyme adsorption on the bubble surface during the bubble shrinkage (b). | ||
It is proposed that before nucleation, the protein attached onto the bubble surface, as reported in literatures.52,53 The adsorption was likely to induce a local supersaturation around the gas bubble and the higher level of supersaturation at the gas–liquid interface could promote the protein nucleation. The phenomena were consistent with the previous work and the literature that indomethacin accumulated near the minute gas–liquid interface leading to a local supersaturation increase during antisolvent crystallisation.54,55 The shrinkage of gas bubbles could drag protein molecules attached on the gas bubbles surface toward closer, shown in Fig. 5(b), leading to a higher local concentration of protein molecules and higher local supersaturation near the interface to accelerate the nucleation. The promotion of the nucleation was consistent with the previous work that the nucleation preferred to occur on the gas bubble with a larger shrinkage.30 The nucleation time were mainly in the range of 12–24 h, ensure the nucleation occurred after injection of the crystallization solution into the capillary. However, in this work, the nucleation times were not systematically compared, because it was difficult to determine the accurate nucleation times. In the hanging drop experiments, one microscope image was able to observe the whole droplet, but in this work for the long capillary, more than five locates of each capillary were used to observe the crystallization process, which still not covered the whole capillary. It was not possible to predict where the nucleation occurred firstly, and it would take long time to check the whole capillary, inducing variations of the temperature inside the capillary and the nucleation process.
Three stages in crystallisation with gas bubbles
The protein crystallisation is proposed with three stages: adsorption of protein on gas–liquid interface, nucleation on gas–liquid–solid interface, and crystal growth on gas–solid interface. Fig. 6(a) shows a schematic diagram for free energy change during the crystallisation process with and without gas bubble templates. Without gas bubble templates, the nucleation occurred after across the free energy barrier ΔGhomonuc for homogeneous nucleation, shown as dashed curve in Fig. 6(a).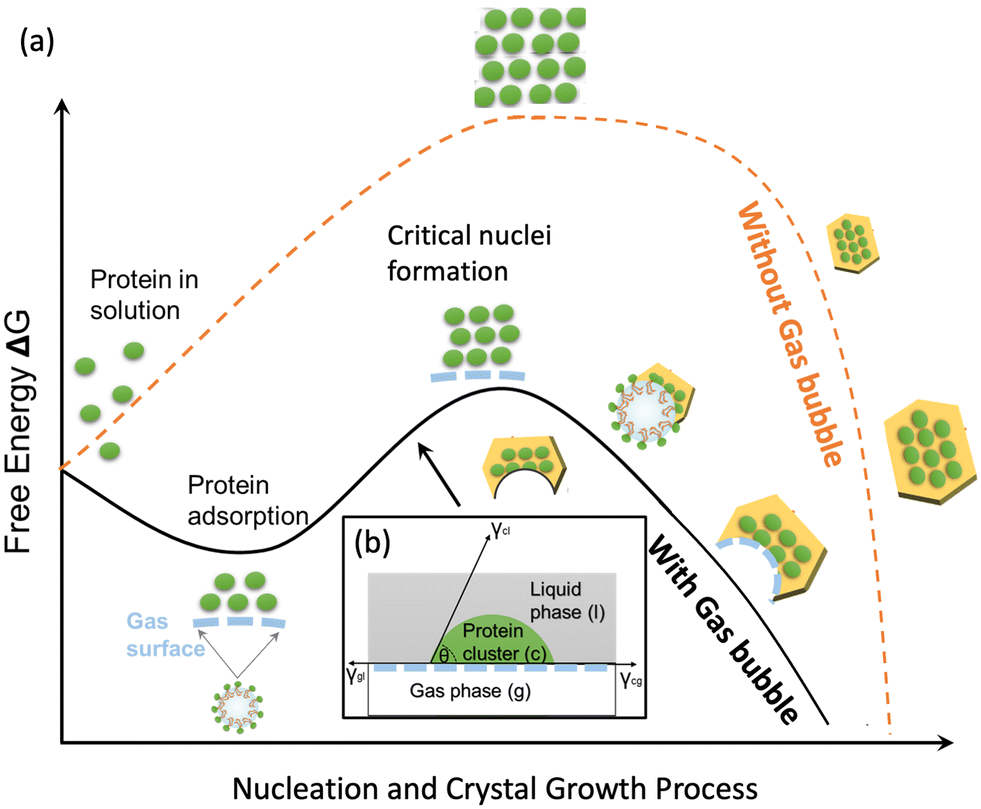 | ||
| Fig. 6 (a) Schematic diagram of the protein nucleation and crystal growth processes with and without gas bubbles and (b) contact of protein molecules with gas molecules at the gas–liquid interface. | ||
With the gas bubble templates, in the first stage, adsorption stage, the protein molecules in the solution were adsorbed rapidly at the gas–liquid interface, and the protein molecules were assumed to form multiple layer structures outside the gas bubbles during the adsorption process, forming a network of protein molecules via hydrogen bonds and van de Waals forces.28 The Gibbs free energy change (ΔG) for the nucleation of a protein nucleus at the gas–liquid interface after the adsorption process is:
| ΔG = ΔGads + ΔGnuc | (6) |
| ΔGads = −ΓAΔμads | (7) |
In the second stage, nucleation stage, heterogeneous nucleation started on the gas–liquid interface and completed with forming new gas–solid interface and gas–liquid–solid interface. The protein molecules adsorbed on the gas surface or the protein molecules very closed to the gas bubbles started to form cluster (based on the classical nucleation theory) or form concentrated liquid phase (based on the secondary nucleation theory). With a foreign gas surface in the solution, the heterogeneous nucleation free energy is lower than the homogeneous nucleation free energy due to the factor φ, which is always below 1. The φ can be estimated from contact angle, which is dependent on the interactions of gas–liquid phase, gas–solid phase and liquid–solid phases, shown in Fig. 6(b).
| ΔGnuc = ΔGheteronuc = φΔGhomonuc | (8) |
In the third stages, crystal growth stage, the crystals grew larger on the gas–solid interface with the decreasing free energy. The crystals nucleated on the air bubble surface continued to grow larger, but the growth direction towards the gas bubbles was blocked, forming curved face on one direction and normal crystal face on the opposite direction. The shape and size of the part-spherical curved face were dependent on the nucleation location on the gas bubble, size of the bubble, and the crystal growth directions. The crystals nucleated on the gas–liquid interface strongly attached on the gas bubbles, and many of the crystals could float with gas bubbles in the solution, but some very large crystals sank together with gas bubbles, dependent on the buoyant force of the gas bubbles and the gravity force of the crystals.
Conclusions
Protein crystallisation was performed in a microfluidic device with the helium, oxygen, nitrogen, and carbon dioxide gas bubbles as soft template. The crystallisation process was proposed with three stages: adsorption of protein on gas–liquid interface, nucleation on gas–liquid–solid interface, and crystal growth on gas–solid interface. The protein molecules adsorbed on the gas bubble surface and stabilised the gas bubbles. The heterogenous nucleation occurred on the gas bubble surface, which highly prevented the crystallisation fouling inside the capillary wall. The gas bubble shrinkage pulled the molecules closer, would contribute to the nucleation preference. With equal concentrations, there were most proportion of crystals on CO2 bubbles and least on He bubbles, and the total number of crystals followed the same trend as the solubility and polarizability of the gases. The crystals grown on the gas bubbles were strongly attached and able to float in the solution. Part of the crystal growth was blocked by the spherical gas bubble templates, and, therefore, the part-spherical curved crystal surfaces were formed with other crystal surfaces remained flat. This work demonstrated the influences of the interactions among the gas, solid and liquid phases on the protein crystallisation, which would lead to potential applications of new systems and platforms to design and control the protein crystal products and the crystallisation processes.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
We thank Loughborough Materials Characterisation Centre for the support in SEM analysis. WT would like to acknowledge school of AACME, Loughborough University, and HY is grateful to the UK EPSRC (Engineering and Physical Sciences Research Council) for support (EP/T005378/1).References
- G. Walsh, Nat. Biotechnol., 2018, 36, 1136–1145 CrossRef CAS PubMed.
- D. E. Gordon, G. M. Jang, M. Bouhaddou, J. Xu, K. Obernier, K. M. White, M. J. O'Meara, V. V. Rezelj, J. Z. Guo, D. L. Swaney, T. A. Tummino, R. Hüttenhain, R. M. Kaake, A. L. Richards, B. Tutuncuoglu, H. Foussard, J. Batra, K. Haas, M. Modak, M. Kim, P. Haas, B. J. Polacco, H. Braberg, J. M. Fabius, M. Eckhardt, M. Soucheray, M. J. Bennett, M. Cakir, M. J. McGregor, Q. Li, B. Meyer, F. Roesch, T. Vallet, A. Mac Kain, L. Miorin, E. Moreno, Z. Z. C. Naing, Y. Zhou, S. Peng, Y. Shi, Z. Zhang, W. Shen, I. T. Kirby, J. E. Melnyk, J. S. Chorba, K. Lou, S. A. Dai, I. Barrio-Hernandez, D. Memon, C. Hernandez-Armenta, J. Lyu, C. J. P. Mathy, T. Perica, K. B. Pilla, S. J. Ganesan, D. J. Saltzberg, R. Rakesh, X. Liu, S. B. Rosenthal, L. Calviello, S. Venkataramanan, J. Liboy-Lugo, Y. Lin, X. P. Huang, Y. F. Liu, S. A. Wankowicz, M. Bohn, M. Safari, F. S. Ugur, C. Koh, N. S. Savar, Q. D. Tran, D. Shengjuler, S. J. Fletcher, M. C. O'Neal, Y. Cai, J. C. J. Chang, D. J. Broadhurst, S. Klippsten, P. P. Sharp, N. A. Wenzell, D. Kuzuoglu-Ozturk, H. Y. Wang, R. Trenker, J. M. Young, D. A. Cavero, J. Hiatt, T. L. Roth, U. Rathore, A. Subramanian, J. Noack, M. Hubert, R. M. Stroud, A. D. Frankel, O. S. Rosenberg, K. A. Verba, D. A. Agard, M. Ott, M. Emerman, N. Jura, M. von Zastrow, E. Verdin, A. Ashworth, O. Schwartz, C. d'Enfert, S. Mukherjee, M. Jacobson, H. S. Malik, D. G. Fujimori, T. Ideker, C. S. Craik, S. N. Floor, J. S. Fraser, J. D. Gross, A. Sali, B. L. Roth, D. Ruggero, J. Taunton, T. Kortemme, P. Beltrao, M. Vignuzzi, A. García-Sastre, K. M. Shokat, B. K. Shoichet and N. J. Krogan, Nature, 2020, 583, 459–468 CrossRef CAS PubMed.
- D. S. Dimitrov, in Methods in Molecular Biology, ed. J. M. Walker, Springer, 2012, vol. 899, pp. 1–26 Search PubMed.
- Transparency Market Research, Bioengineered Protein Drugs Market, 2017.
- A. McPherson and J. A. Gavira, Acta Crystallogr., Sect. F: Struct. Biol. Commun., 2014, 70, 2–20 CrossRef CAS PubMed.
- R. G. Harrison, P. Todd, S. R. Rudge and D. P. Petrides, Bioseparations Science and Engineering, 2nd edn, 2015 Search PubMed.
- S. K. Basu, C. P. Govardhan, C. W. Jung and A. L. Margolin, Expert Opin. Biol. Ther., 2005, 4, 301–317 CrossRef.
- C. Morrison, Nat. Biotechnol., 2017, 35, 108–112 CrossRef CAS PubMed.
- D. Hekmat, Bioprocess Biosyst. Eng., 2015, 38, 1209–1231 CrossRef CAS PubMed.
- H. Yang, W. Chen, P. Peczulis and J. Y. Y. Heng, Cryst. Growth Des., 2019, 19, 983–991 CrossRef CAS.
- M. M. Roberts, J. Y. Y. Heng and D. R. Williams, Cryst. Growth Des., 2010, 10, 1074–1083 CrossRef CAS.
- W. Tian, W. Li and H. Yang, Cryst. Growth Des., 2023, 23, 5181–5193 CrossRef CAS.
- W. Chen, T. N. H. Cheng, L. F. Khaw, X. Li, H. Yang, J. Ouyang and J. Y. Y. Heng, Sep. Purif. Technol., 2021, 255, 117384 CrossRef CAS.
- L. Li and R. F. Ismagilov, Annu. Rev. Biophys., 2010, 39, 139–158 CrossRef CAS PubMed.
- L. Li, D. Mustafi, Q. Fu, V. Tereshko, D. L. Chen, J. D. Tice and R. F. Ismagilov, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 19243–19248 CrossRef CAS PubMed.
- N. Fatemi, Z. Dong, T. van Gerven and S. Kuhn, Langmuir, 2019, 35, 60–69 CrossRef CAS PubMed.
- J. Ferreira, F. Castro, F. Rocha and S. Kuhn, Chem. Eng. Sci., 2018, 191, 232–244 CrossRef CAS.
- N. Fatemi, C. Devos, T. van Gerven and S. Kuhn, Chem. Eng. Sci., 2022, 248, 117095 CrossRef CAS.
- X. Zhang, Z. Wei, H. Choi, H. Hao and H. Yang, Adv. Mater. Interfaces, 2021, 8, 2001200 CrossRef CAS.
- B. Zheng and R. F. Ismagilov, Angew. Chem., Int. Ed., 2005, 44, 2520–2523 CrossRef CAS PubMed.
- D. L. Chen, C. J. Gerdts and R. F. Ismagilov, J. Am. Chem. Soc., 2005, 127(27), 9672–9673 CrossRef CAS PubMed.
- B. Zheng, A. L. S. Roach and R. F. Ismagilov, J. Am. Chem. Soc., 2003, 125(37), 11170–11171 CrossRef CAS PubMed.
- A. Günther and K. F. Jensen, Lab Chip, 2006, 6, 1487–1503 RSC.
- K. Mathew Thomas and R. Lakerveld, Ind. Eng. Chem. Res., 2019, 58, 20381–20391 CrossRef CAS.
- B. Noskov and A. Mikhailovskaya, Soft Matter, 2013, 9, 9392–9402 RSC.
- M. M. Ouberai, K. Xu and M. E. Welland, Biomaterials, 2014, 35, 6157–6163 CrossRef CAS PubMed.
- R. Z. Guzman, R. G. Carbonell and P. K. Kilpatrick, J. Colloid Interface Sci., 1986, 114, 536–547 CrossRef CAS.
- V. Mitropoulos, A. Mütze and P. Fischer, Adv. Colloid Interface Sci., 2014, 206, 195–206 CrossRef CAS PubMed.
- Y. Desfougères, A. Saint-Jalmes, A. Salonen, V. Vié, S. Beaufils, S. Pezennec, B. Desbat, V. Lechevalier and F. Nau, Langmuir, 2011, 27, 14947–14957 CrossRef PubMed.
- W. Tian, C. Rielly and H. Yang, CrystEngComm, 2021, 23, 8159–8168 RSC.
- O. Ogunyinka, A. Wright, G. Bolognesi, F. Iza and H. C. H. Bandulasena, Microfluid. Nanofluid., 2020, 24(2), 1–16 CrossRef.
- P. Garstecki, H. A. Stone and G. M. Whitesides, Phys. Rev. Lett., 2005, 94, 164501 CrossRef.
- A. K. W. Leung, M. M. V. Park and D. W. Borhani, J. Appl. Crystallogr., 1999, 32, 1006–1009 CrossRef CAS.
- Y. Yu, X. Wang, D. Oberthür, A. Meyer, M. Perbandt, L. Duan and Q. Kang, J. Appl. Crystallogr., 2012, 45, 53–60 CrossRef CAS.
- J. W. Mullin, Crystallization, Butterworth-Heinemann, 2001 Search PubMed.
- W. Chen, H. Yang and J. Y. Y. Heng, in The Handbook of Continuous Crystallization, 2020, pp. 372–392 Search PubMed.
- J. Colt, Computation of Dissolved Gas Concentration in Water as Functions of Temperature, Salinity and Pressure, Elsevier, 2012 Search PubMed.
- R. H. Perry and G. D. W. Green, Perry's Chemical Engineers' Handbook, McGraw-Hill, New York, 2007 Search PubMed.
- J. H. Hildebrand and R. E. Powel, Principles of Chemistry, Macmillan, NY, 1964 Search PubMed.
- R. A. Judge, R. S. Jacobs, T. Frazier, E. H. Snell and M. L. Pusey, Biophys. J., 1999, 77, 1585–1593 CrossRef CAS PubMed.
- J. Huang, Q. Yin and J. Ulrich, Chem. Eng. Technol., 2016, 39, 1213–1218 CrossRef CAS.
- A. J. Sopmmopoulos, C. K. Rhodes, D. X. Holcomb and K. E. V. Holde, J. Biol. Chem., 1962, 237, 1107–1112 CrossRef.
- M. Tagliabue, D. Farrusseng, S. Valencia, S. Aguado, U. Ravon, C. Rizzo, A. Corma and C. Mirodatos, Chem. Eng. J., 2009, 155, 553–566 CrossRef CAS.
- F. Y. Ushikubo, M. Enari, T. Furukawa, R. Nakagawa, Y. Makino, Y. Kawagoe and S. Oshita, IFAC Proc. Vol., 2010, 43, 283–288 CrossRef.
- M. Takahashi, J. Phys. Chem. B, 2005, 109, 21858–21864 CrossRef CAS.
- M. Lee, E. Y. Lee, D. Lee and B. J. Park, Soft Matter, 2015, 11, 2067–2079 RSC.
- H.-J. Butt, K. Graf and M. Kappl, Physics and Chemistry of Interfaces, 2003 Search PubMed.
- X. Zeng, X. Lan, H. Zhu, H. Liu, H. A. Umar, Y. Xie, G. Long and C. Ma, Materials, 2020, 13, 1820 CrossRef CAS.
- J. Hanwright, J. Zhou, G. M. Evans and K. P. Galvin, Langmuir, 2005, 21, 4912–4920 CrossRef CAS.
- P. S. Epstein and M. S. Plesset, J. Chem. Phys., 1950, 18, 1505–1509 CrossRef CAS.
- L. Yu, L. Zhang and Y. Sun, J. Chromatogr. A, 2015, 1382, 118–134 CrossRef CAS PubMed.
- Q. Wang, C. Xue, H. Zhao, Y. Qin, X. Zhang and Y. Li, Colloids Surf., A, 2020, 589, 124471 CrossRef CAS.
- L. S. Roach, H. Song and R. F. Ismagilov, Anal. Chem., 2005, 77, 785–796 CrossRef CAS PubMed.
- M. Matsumoto, M. Ohno, Y. Wada, T. Sato, M. Okada and T. Hiaki, J. Cryst. Growth, 2017, 469, 91–96 CrossRef CAS.
- T. Tadros, Encyclopedia of Colloid and Interface Science, 2013, pp. 626–626 Search PubMed.
- P. G. Vekilov, Cryst. Growth Des., 2010, 10, 5007–5019 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2023 |