
Ultra-thin patchy polymer-coated graphene oxide as a novel anticancer drug carrier†
Vien T.
Huynh
 ab,
Duc
Nguyen
ab,
Liwen
Zhu
ab,
Nguyen T. H.
Pham
ab,
Pramith
Priyananda
a and
Brian S.
Hawkett
*ab
ab,
Duc
Nguyen
ab,
Liwen
Zhu
ab,
Nguyen T. H.
Pham
ab,
Pramith
Priyananda
a and
Brian S.
Hawkett
*ab
aKey Centre for Polymers and Colloids, School of Chemistry F11, The University of Sydney, NSW 2006, Australia. E-mail: brian.hawkett@sydney.edu.au
bUniversity of Sydney Nano Institute, The University of Sydney, NSW 2006, Australia
First published on 22nd July 2020
Abstract
An ultra-thin, graphene oxide (GO) based, anticancer drug carrier was developed using Reversible Addition Fragmentation chain Transfer (RAFT) mediated emulsion polymerisation. Short chain macro-RAFT copolymer, BuPATTC-(BA4-stat-AA9-stat-StS5), was used to disperse the GO in water. Subsequent free radical emulsion polymerisation produced an aqueous suspension of partially polymer-coated GO (PPC-GO). The polymer coating was unevenly distributed, forming a patchy and extremely rough surface on the GO substrate. The use of macro-RAFT copolymers greatly improved the overall colloidal stability of the GO. Furthermore, particle morphologies were found to be controllable by adjusting the amount of monomer starve fed into the polymerisation. PPC-GO was demonstrated to be an effective carrier for the anti-cancer drug doxorubicin (Dox), storing equivalent to its own original weight in Dox. A releasing mechanism using L-ascorbic acid (L-AA) as an agent was reported. Dox release can be triggered by chemical reduction using L-AA, which weakens the hydrogen bonds between Dox and GO. After release of the Dox, the remaining PPC- reduced GO (PPC-rGO) was found to be stable in aqueous suspension.
Introduction
Delivery systems for anticancer agents have been widely explored in the past few decades as a means of increasing drug efficacy and reducing systemic toxicity.1 Nanoparticles show great promise in the space, with recent findings suggesting that the size and shape of the nanoparticle have a significant effect on cellular uptake.2–5 High aspect ratio nanoparticles such as rods and tubes commonly show a higher cellular uptake efficiency than spheres.2 As such, an ultra-thin material with a very high aspect ratio would be highly desirable.Graphene, a single layer of sp2-hybridized carbon atoms arranged in a honeycomb crystal lattice, has gained enormous attention world-wide since its discovery by Novoselov et al. in 2004, with potential applications in diverse fields of nanoscience and nanotechnology.6 As an oxidised derivative of graphene, and also two-dimensional carbon allotrope with a large number of oxygen-containing groups such as hydroxyl, carbonyl, carboxyl and epoxide on its surface, graphene oxide (GO) has been widely used in wet chemistry due to its ultra-high surface area and amphipathy.7,8 The aqueous dispersibility and modifiable functional moieties impart GO as a potential candidate for drug storage and delivery systems.9–12 GO can facilitate the physical adsorption and chemical interactions of both hydrophilic and hydrophobic drugs.9,10
The biocompatibility and toxicity of GO has, however, been a controversial topic. While some studies have revealed low cell toxicity, the results in literature to date are inconsistent.13–15 Several factors could affect the toxicity of GO. One is the direct interaction between the GO sheet and cell membrane causing physical damage to the cell membrane.16 Another factor is its oxidation level where a lower oxidation degree of GO leads to faster immune cell infiltration, uptake, and clearance following both subcutaneous and peritoneal implantation.17 Dispersibility of GO also has a significant influence on toxicity. It has frequently been suggested that surface modification or coating approaches could enhance the biocompatibility of the carbon-based material.18 PEGylation of GO via covalent bonding to the functional groups is one of the most common strategies used to reduce non-specific binding to biological membranes and improve biocompatibility.9,10,19 Some studies also used functional polymers to not only reduce the toxicity of the nanomaterial but also introduce stimuli-responsive features for triggering drug release.20,21 Based on reported works, it seems that the toxicity of GO could be overcome by suitable functionalisation.
Over a decade ago, a RAFT-based and surfactant-free emulsion polymerisation approach was first employed by the Hawkett group for polymer encapsulation of inorganic pigment particles in water.22 This technique takes advantage of our early works in the field of emulsion polymerisation under RAFT control self-assembly.23,24 It was later considered as an important component of the broader polymerisation-induced self-assembly (PISA).25,26 This particle encapsulation process used living amphiphilic random macro-RAFT copolymers of acrylic acid (AA) and n-butyl acrylate (BA), adsorbed on zirconia and alumina-coated titanium dioxide (TR92) pigment, to facilitate the emulsion polymerisation to occur at the particle surface to form the polymer coating shell under starve-feeding of hydrophobic monomers. This method was found to be highly efficient, with almost all of the polymer growth within the encapsulating polymer shells. This is because chain extensions of the macro-RAFT stabilisers were found to be limited to pigment/water interface. On the other hand, the random nature of the copolymer prevented micelle formations and subsequent secondary particle nucleation occurring. Ali et al. endorsed the versatility of the particle coating technique by extending it to the encapsulation of gibbsite platelets using a similar macro-RAFT copolymer.27 Our approach was later extensively adopted by many research groups to coat a variety of materials with different sizes, shapes, and surface chemistries including carbon nanotubes,28 montmorillonite clay,29,30 lead sulfide,31 silica,32 cerium oxide,33,34 quantum dots,35 layered double hydroxides,36 and iron oxide nanoparticles.37,38
In a previous work, we demonstrated the ability to use this method to evenly coat GO sheets with uniform polymer shells.39 The stabilising charge on the surface of the GO was first altered from negative to positive by adsorption of poly(allylamine hydrochloride) (PAH). An amphiphilic macro-RAFT copolymer was then adsorbed onto the surface as a stabiliser. This was followed by RAFT controlled emulsion polymerisation leading to chain extension of the adsorbed living copolymer on the surface of the GO, generating a polymer encapsulating shell. Building on this previous study, the present work investigates the dispersion of the GO in the macro-RAFT copolymer in the absence of a switching charge polymer. The macro-RAFT copolymer, BuPATTC-(BA4-stat-AA9-stat-StS5), was directly employed to disperse the GO in an aqueous dispersion, resulting in uneven adsorption on the GO surface. RAFT-mediated emulsion polymerisation was carried out under starve-feeding MMA/BA (10/1, w/w) to form a patchy polymer coating on the GO surface. The resulting coated GO has not only a polymer coating to improve the colloidal stability but also available bare surface to adsorb Dox. This study highlighted (1) the adsorption isotherm of macro-RAFT copolymer on the GO surface in the presence and absence of an intermediate polymer, PAH, (2) the influence of macro-RAFT adsorption on polymer coating, (3) the effect of monomer feed on the polymer coating morphology, (4) the colloidal stability of the PPC-GO posterior to reduction, and finally (5) the adsorption and desorption of Dox on the synthesised PPC-GO. The overall scheme of the current study on the partial polymer coating of the GO and its Dox adsorption is depicted in Fig. 1.
 | ||
| Fig. 1 Scheme of PPC-GO, Dox loading and release. | ||
Experimental section
Materials
Acrylic acid (AA, 99%, Sigma-Aldrich) was purified by distillation under reduced pressure. 2,2′-Azobis-isobutyronitrile (AIBN, 98%, Fluka) was recrystallised from methanol (99.8%, anhydrous, Sigma-Aldrich). Graphene oxide (GO, 99%, ACS materials), n-butyl acrylate (BA, 99%, stabilised, Sigma-Aldrich), methyl methacrylate (MMA, 99%, stabilised, Sigma-Aldrich), 4-styrenesulfonic acid sodium salt hydrate (StS, Sigma-Aldrich), 1,3,5-trioxane (>99%, Sigma-Aldrich), 1,4-dioxane (>99%, Sigma-Aldrich), polyallylamine hydrochloride (PAH, 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1, Sigma-Aldrich), ammonium hydroxide (NH4OH, 99%, Sigma-Aldrich), L-ascorbic acid (L-AA, 99%, Sigma-Aldrich) and 4,4′-azobis(4-cyanovaleric acid) (ACVA, Wako) were used as received. Deionised (DI) water (ELIX 10, Reverse Osmosis, Milli-pore) was used for all experiments. 2-[(Butylsulfanyl)carbonothioyl] sulfanyl propanoic acid (BuPATTC) was synthesised as previously described23 and pure BuPATTC RAFT agent was confirmed by proton nuclear magnetic resonance (1H-NMR) characterisation as shown in Fig. S1.†
000 g mol−1, Sigma-Aldrich), ammonium hydroxide (NH4OH, 99%, Sigma-Aldrich), L-ascorbic acid (L-AA, 99%, Sigma-Aldrich) and 4,4′-azobis(4-cyanovaleric acid) (ACVA, Wako) were used as received. Deionised (DI) water (ELIX 10, Reverse Osmosis, Milli-pore) was used for all experiments. 2-[(Butylsulfanyl)carbonothioyl] sulfanyl propanoic acid (BuPATTC) was synthesised as previously described23 and pure BuPATTC RAFT agent was confirmed by proton nuclear magnetic resonance (1H-NMR) characterisation as shown in Fig. S1.†
Synthesis of short chain poly[(4-styrenesulfonic acid)-stat-(acrylic acid)-stat-(n-butyl acrylate)] macro-RAFT copolymers using BuPATTC
The amphiphilic macro-RAFT copolymers were prepared as reported in our previous work.40 BuPATTC (1.2 g, 5.0 mmol), ACVA (0.074 g, 0.3 mmol), AA (3.64 g, 50.5 mmol), StS (5.19 g, 25.2 mmol), BA (2.64 g, 20.6 mmol) in a mixture of 1,4-dioxane (10.00 g), and DI water (10.69 g) was prepared in a 50 mL round-bottomed flask. This was stirred magnetically and purged with nitrogen for 10 min. The flask was then heated at 70 °C for 7 h under constant stirring. The final copolymer solution contained 34.9% solids. The copolymers were characterised by 1H NMR (300 MHz, DMSO-d6).40 Based on 1H NMR, Mn was calculated to be 2337 (g mol−1) (Fig. S2†). 1H-NMR (300 MHz, DMSO-d6, 25 °C) δ (ppm) = 12.8–11.8 (10H + 1H, carboxylic of AA and BuPATTC end, respectively), 7.9–7.3 (2 × 5H, phenyl of StS monomer), 7.3–6.4 (2 × 5H, phenyl of StS monomer), 4.3–3.8 (2 × 4H, CH2 of BA), 3.8–2.9 (1 × 5H, CH of main chain from StS monomer unit), 2.6–2.0 (2 × 4H, CH2 of BA), 2.0–1.1 (CH and CH2 of polymer backbone), 1.1–0.6 (3 × 4H, CH3 of BA unit and 1H, BuPATT end). The macro-RAFT is, therefore, estimated to be BuPATTC-(BA4-stat-AA9-stat-StS5).Adsorption of macro-RAFT copolymer on GO
A stock solution of macro-RAFT copolymer was prepared by slowly adding ammonium hydroxide to a suspension of BuPATTC-(BA4-stat-AA9-stat-StS5) in water until full dissolution and a pH value of 8.5 was achieved. The molar extinction coefficient at 309 nm of the macro-RAFT copolymer, which is a maximum absorbance wavelength of trithiocarbonate, was then determined by UV-Visible spectroscopy from a series of standard curve dilutions prepared from the stock solution. BuPATTC-(BA4-stat-AA9-stat-StS5) adsorption onto colloidal GO was carried out using a batch method at room temperature. A series of seven macro-RAFT copolymer solutions with concentrations ranging from 0 to 0.105 mmol L−1 were prepared by appropriate dilutions of the stock solution. The solutions were vigorously stirred, and an aliquot of the GO stock dispersion was added dropwise to each prepared macro-RAFT solution. Stirring was continued for 8 h followed by 2 min of sonication at 30% power, and then centrifugation at 30000 rpm for 60 min. The supernatants were recovered and the concentration of the free macro-RAFT copolymer in supernatants were analysed by measuring absorbance at 309 nm after appropriate dilutions. The amount of BuPATTC-(BA4-stat-AA9-stat-StS5) adsorbed on the GO surface was calculated by subtracting the free macro-RAFT copolymer in the supernatant from the total amount of the macro-RAFT copolymer used in each sample. As a comparison, a series of GO/PAH dispersions were prepared and the macro-RAFT copolymer adsorptions were similarly carried out. The adsorption isotherms were obtained by plotting the adsorbed amount versus the total macro-RAFT copolymer.
Dispersion of GO in water using amphiphilic macro-RAFT copolymers as stabilisers
The GO dispersion in water was prepared using the following procedure. In a 100 mL beaker, GO (0.010 g) was first mixed with water (30 g) using a magnetic stirrer. While under stirring, the dispersion pH was adjusted to 8.5 by 2.8% (w/w) NH4OH to produce a black dispersion. This was further dispersed using a Vibra-Cell Ultrasonic Processor (Sonics and Materials) standard probe at amplitude of 30% for 10 min. A BuPATTC-(BA4-stat-AA9-stat-StS5) stabiliser solution was prepared by mixing the macro-RAFT solution (0.10 g, 0.010 mmol) with 20.5 g DI water. This was followed by pH adjustment to 8.5 using NH4OH (2.8%, w/w). To this macro-RAFT solution, the previously prepared GO dispersion was slowly added under constant stirring. It was then followed by ultrasonication for 10 min (at the amplitude of 30%) to ensure thorough dispersion of GO.Synthesis of the PPC-GO using amphiphilic macro-RAFT copolymers as stabilisers
After sonication, the dispersion was transferred to a 100 mL round-bottomed flask (RBF) containing ACVA (0.028 g, 0.10 mmol), which was subsequently sealed and purged with nitrogen for 10 min. The whole flask was then immersed in an oil bath with a temperature setting of 70 °C and was magnetically stirred. A deoxygenated 10:1 (weight ratio) mixture of MMA and BA (1.0 mL) was injected into the flask, while in the 70 °C oil bath, at a rate of 1.0 mL h−1. After monomer addition, the heating was continued for another hour to produce a black stable latex containing 2.7% solids. The final latex was characterised by transmission electron microscopy (TEM JEOL 1400). For better observation of the coated graphene oxide, free polymer particles that contained no graphene oxide were removed from the samples by centrifugation prior to TEM sample preparation. Zeta potential measurements were performed on a Zetasizer (nano series, Malvern Instruments). Polymer content in the shells of the coated graphene oxide was analysed by thermogravimetric analysis (TGA) (TA Instruments Discovery TGA) ramping from ambient temperature to 600 °C at 20 °C min−1 under nitrogen gas.
Synthesis of poly(MMA-co-BA) particle latex using amphiphilic macro-RAFT copolymers as stabilisers for GO dispersion
The macro-RAFT copolymer of BuPATTC-(BA4-stat-AA9-stat-StS5) (0.10 g, 0.010 mmol) was dissolved in water (70 g) followed by pH adjustment to 8.5 using NH4OH (2.8%, w/w) to yield a clear yellow solution. The solution was transferred to a 100 mL RBF containing ACVA (0.031 g, 0.11 mmol), which was subsequently sealed and purged with nitrogen for 10 min. The whole flask was then immersed in an oil bath with a temperature setting of 70 °C and was magnetically stirred. A deoxygenated 10:1 (w/w) mixture of MMA and BA (1 mL) was injected into the flask, while in the 70 °C oil bath, at a rate of 1 mL h−1. After monomer addition, the heating was continued for another hour to produce a yellowish stable latex. By TEM and DLS, it was found to contain spherical polymer particles with an average size of 65 nm.
Polymer latex was employed to disperse GO to compare preformed particle surface adsorption and previously described partial polymer coating. In a typical experiment, the latex (2.0 g, 1.6% in water) was mixed with GO dispersion (2.0 mL, 0.015% in water) at pH 8.5 producing a black dispersion. The dispersion was left stirring for 8 h to facilitate particle adsorption onto the GO surface. The particle/GO interaction was then characterised by TEM. For this purpose, un-adsorbed polymer particle latex in the supernatant was removed by centrifugation at 14500 rpm for 30 min. The black deposit was washed, then redispersed in water prior to TEM preparation.
Reduction of GO or PPC-GO using L-ascorbic acid (L-AA)
The GO reduction method reported by Zhang et al.41 was adopted to reduce our PPC-GO to PPC-rGO. Briefly, the reduction of GO was conducted in water at room temperature (∼25 °C). L-AA (2.0 mg) was added to 2.0 ml of each GO solution to make up 1.0 mg mL−1 under consistent magnetic stirring. The reduction progress was monitored by UV-Vis spectroscopy over time. TGA was carried out on GO and rGO to confirm formation of the reduced products. In order to attain PPC-rGO, the PPC-GO was first purified from all homopolymer latex by centrifugation at 14500 rpm for 10 min. After supernatant removal, the black deposit was washed then redispersed in water. The process was repeated three times. Final redispersion in water produced a dark brown sample which contained the PPC-GO. To this dispersion, L-AA solution (6.0 mM) was added and the whole sample was left stirring for 24 h at room temperature. Once complete, the excess L-AA was removed by centrifugation and the black deposit was redispersed in water to yield the PPC-rGO.
Dox loading onto PPC-GO
100 μL Dox 1.0 mM in phosphate buffer solution (PBS) was added to 3.0 mL of PPC-GO dispersion (0.24 mg mL−1) at pH 7.0. The sample was placed in a foil-wrapped container and mixed over 48 h at room temperature using a shaker. After mixing, unadsorbed Dox was removed from the sample by centrifugation at 14500 rpm for 10 min and washed three times with water to ensure removal of the unadsorbed Dox. Finally, Dox-adsorbed PPC-GO (Dox-PPC-GO) was redispersed in water to yield a stable pinkish black dispersion. The sample was stored in a foil-wrapped container at 5 °C for further testing. During centrifugation, the concentration of unbound Dox in the supernatant was quantified by fluorescence or UV-Vis spectroscopy to calculate the Dox loading.
Dox release from Dox-PPC-GO
The Dox-PPC-GO was incubated at room temperature for 24 h in different L-AA concentrations varying from 0 to 11 mM. The samples were then centrifuged at 14500 rpm for 10 min and supernatants were collected for UV-Vis spectroscopy to quantify the amount of Dox released from PPC-rGO.
Results and discussion
Macro-RAFT copolymer adsorption onto GO
In previous work, the polymer chain extension of adsorbed macro-RAFT copolymers was found to govern the final surface morphology of the coated GO. Therefore, understanding the adsorption capacity of macro-RAFT copolymer on the GO is of great importance. As described in the Experimental section, the macro-RAFT used in this work was a short chain statistical copolymer with an estimated molecular structure of BuPATTC-(BA4-stat-AA9-stat-StS5). For the adsorption study, the same amount of GO was dispersed in different polymer solutions at various concentrations. Fig. 2 shows the copolymer adsorption isotherm on bare GO. For comparison purposes, adsorption on the PAH-modified GO (PAH@GO) was also displayed to highlight the importance of surface charge on polymer/GO interactions.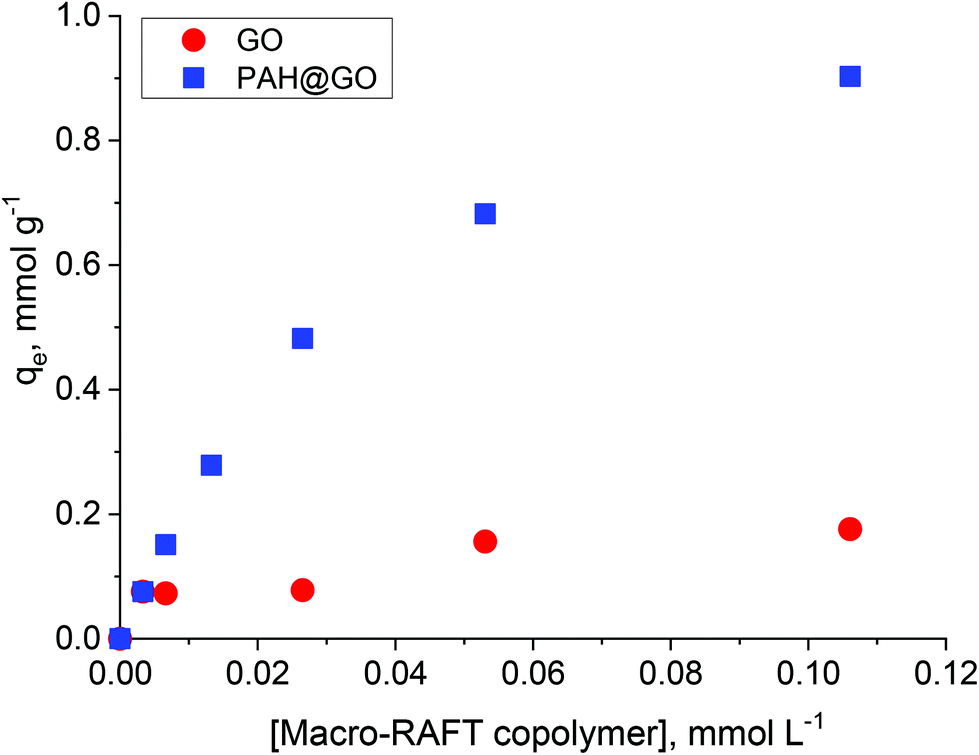 | ||
| Fig. 2 Adsorption isotherms of macro-RAFT copolymer onto GO and PAH@GO. | ||
As shown, the amount of macro-RAFT copolymer adsorbed onto PAH@GO increased from 0 to 0.95 mmol g−1 when increasing the concentration of macro-RAFT copolymer in the suspension until a plateau was reached. The macro-RAFT copolymer, in fact, progressively saturated the PAH@GO surface as the macro-RAFT agent concentration was increased. In contrast, the amount of BuPATTC-(BA4-stat-AA9-stat-StS5) adsorbed onto the bare GO was reduced by a factor of six. The result shows that up to 0.18 mmol of the macro-RAFT copolymer adsorbed on 1 gram of the bare GO, although both the macro-RAFT and the GO are negatively charged under the basic pH conditions used to disperse the GO in macro-RAFT solution. This fortuitous adsorption is thought to be the result of non-electrostatic (van der Waals and hydrophobic) interactions between hydrophobic moieties of the copolymer (BA units) and the hydrophobic region of GO surface, which has been previously reported in literature. Tu et al. observed that hydrophobic interactions between lipids and hydrophobic patches on GO causes lipid adsorption on to GO sheets through nano-scale dewetting.42 This nano-scale dewetting of the GO surface is made possible by the presence of a significant amount of unoxidised graphene-like regions.42,43 Sumaryada et al. also reported the strong glucose oxidase-graphene oxide affinity due to hydrophobic interactions.44 Dispersing the GO in an aqueous solution further promoted the macro-RAFT copolymer adsorption because water-mediated interactions drive the polymer to adsorb strongly at a hydrophobic interface.45 However, distribution of the adsorbed macro-RAFT on the GO surface is not expected to be even. This is because of the random presence of negative charges on the substrate, which prevents the polymer/surface interactions via same charge repulsion. As a result, macro-RAFT copolymer coverage was only patchy (as depicted in Fig. 1) even at high concentrations. In the case of PAH@GO, both the positive charge and hydrophobic domains facilitated strong and even adsorption of the copolymer on the surface.
PPC-GO synthesis
The PPC-GO was synthesised by starve-fed monomer conditions with the RAFT mediated emulsion polymerisation. In this process, polymer growth on the GO was only limited to chain extension of adsorbed macro-RAFT. Since the amphiphilic copolymers also acted as stabilisers, their negatively-charged carboxylate and sulfonate functional groups were always at the polymer/water interface to maintain colloidal stability as the polymerisation progressed. TEM micrographs in Fig. 3A showed that the spherical domains were randomly distributed on all GO sheets. By atomic force microscopy (AFM) (Fig. 3B and D), polymer domains were mostly spherical caps in shapes with diameters ranging from 20–80 nm and heights from 10–25 nm. The height profile in Fig. 3D showed polymer cap positions could be as far apart as several hundred nanometres with bare GO surface between them. However, some regions contained a higher density of such domains, which merged together due to the relatively low Tg of the synthesised polymers to produce very rough morphologies on top of the underlying GO substrate. This contrasted with the smooth surface of polymer-coated PAH@GO presented in Fig. 3B. Distinct morphologies between the two cases reconfirmed uneven macro-RAFT adsorption onto GO in the absence of PAH. This was instrumental to the successful formation of PPC-GO due to selective polymer growth. Analysis of the area fraction on the AFM micro-structure using ImageJ software showed around 43% polymer coverage compared to the total area of GO (Fig. S6†).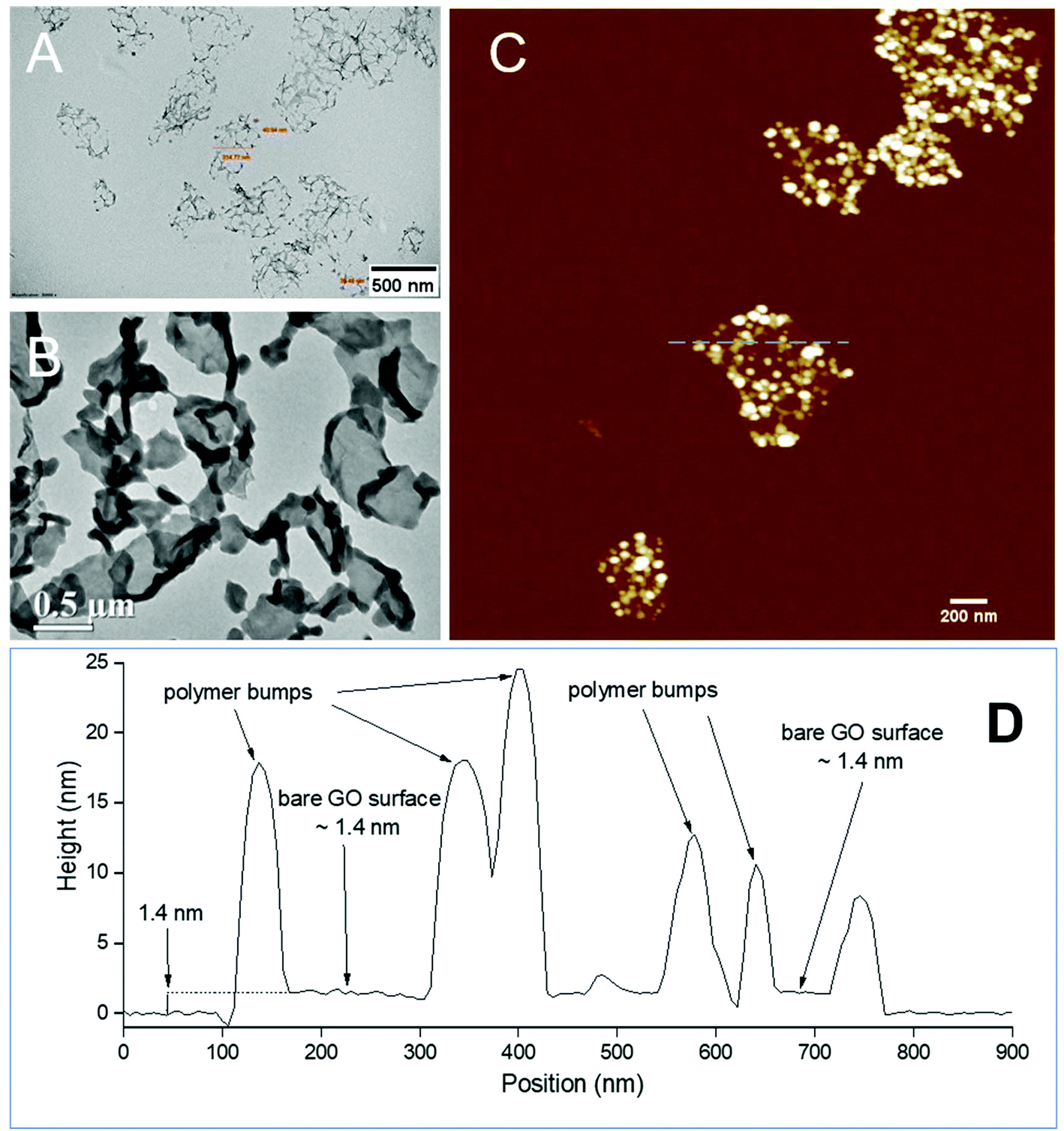 | ||
| Fig. 3 (A) and (C) TEM and AFM micrographs of the PPC-GO coated by MMA/BA (10/1, w/w) with a mass ratio of monomer/GO = 35 (w/w), (D) cross section profile of the PPC-GO marked by a blue dash line in (C), (B) TEM image of even polymer coated GO. | ||
In contrast to previous studies focusing on polymer nanoparticles armoured with graphene oxide sheets46,47 or clay nanosheets,36 the currently investigated PPC-GO revealed a totally different scale. While the armoured polymer particles were large and could accommodate several sheets on one particle, many polymer bumps and nodes, several tens of nanometres in size, in this work were located on both sides of a single GO sheet, which produced a unique nanomaterial structure. It should be noted that other studies have also carried out the functionalisation of GO with synthesised polymers by taking advantage of available functional groups or inducing π–π interactions with the graphene lattice.48,49 These studies did not, however, investigate possible changes in the modified GO surface morphologies.
During the emulsion polymerisation, the formation of homopolymer particles from labile macro-RAFT copolymer was also observed. To confirm that PPC-GO was not formed due to the adsorption of free polymer particles, a polymer latex was synthesised under the same conditions in the absence of GO. This latex was then mixed with GO to verify any changes in surface morphology. By TEM (Fig. 4B) and DLS, the latex was found to contain spherical particles with an average particle size of 64 nm. Particles were negatively charged, with the zeta potential found to be −64.5 mV at pH 6.8 (Fig. S5†). The negative charge was attributed to the carboxylate and sulfonate groups on the macro-RAFT and the ACVA initiator. It was also observed that the polymer particles had an overall negative charge at all investigated pH values (Fig. S5†), possibly due to the high ratio of sulfonate groups present on their surface. In this process, the final particles were colloidally stable without the need of any additional surfactants. Once synthesised, the particles were mixed with the GO at pH 8.5. The mixture was subjected to ultrasonication under the same conditions as in the case of GO dispersion in the macro-RAFT solution. Extensive centrifugations and washings were subsequently carried out to ensure removal of the majority of unadsorbed particles. As shown by TEM in Fig. 4C, there was little interaction between the polymer particles and the GO. This was because both materials were negatively charged and repelled each other (Fig. S3 and S5†). Most particles settled on the GO edges, with almost none on the sheet surface. There was apparent presence of free latex, probably due to entrapments between the GO sheets during centrifugation. However, clear differences between Fig. 3 and 4 confirmed that the patchy morphology observed for PPC-GO was due to in situ polymer growth on the graphene surface and not due to particle adsorption.
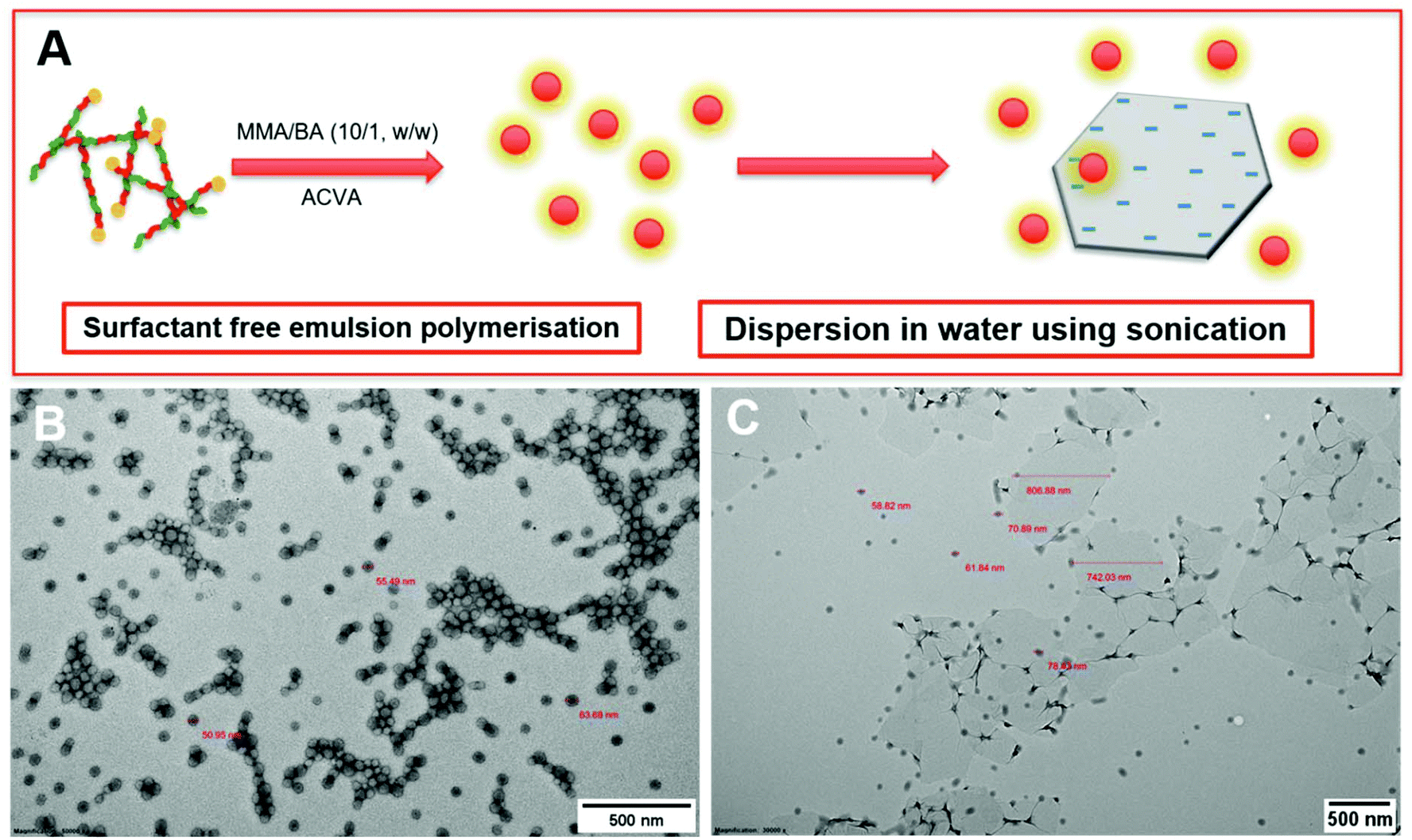 | ||
| Fig. 4 (A) Scheme of polymer latex particle preparation by ab initio emulsion polymerisation via RAFT-controlled self-assembly and starve-fed monomer, (B) TEM micrograph of the resulting polymer particles from the experiment above, and (C) TEM micrograph of the synthesised particles dispersed with GO at pH 8.5 using sonication. | ||
Morphological control of PP-GO by monomer feeding
Continuous addition in this work offered excellent flexibility over the amount and composition of the monomers used in the encapsulation. This allowed possible fine-tuning of PPC-GO morphology to maximise the drug adsorption capacity while still maintaining overall colloidal stability. In order to study the influence of monomer feed on the surface morphology, a series of RAFT-mediated emulsion polymerisations were carried out with various amounts of monomer while GO concentration and other conditions were maintained the same. As shown by the TEM micrographs in Fig. 5 and AFM micrographs in Fig. 6, there was a consistent increase in the size and density of polymer domains present on the PPC-GO with an increase of monomer feed. The surface started very flat and featureless with the original GO (Fig. 5A and 6A) and after macro-RAFT adsorption (Fig. 5B and 6B). Polymer domains in the form of spherical caps were observable at a mass ratio of monomer/GO of 10 (up to 25 nm height (H); 30 nm in radius (R)) (Fig. 5C and 6C), becoming apparent at the ratio of 35 (35 nm H; 36 nm R) (Fig. 5D and 6D). With the addition of more monomer (mass ratios of monomer/GO from 70 to 140), such features not only grew in size (70 nm H; 50 nm R) but also formed clusters occupying most of the surface (Fig. 5E, F and 6E, F). It is important to note that the polymer domains are firmly attached to the underlying GO template despite monomer presence and size growth during encapsulation. Since the aim of this work was to take advantage of the GO surface for drug storage and delivery, the condition in Fig. 5D and 6D with the mass ratio of monomer/GO of 35 was chosen for subsequent reduction with L-AA and Dox loading.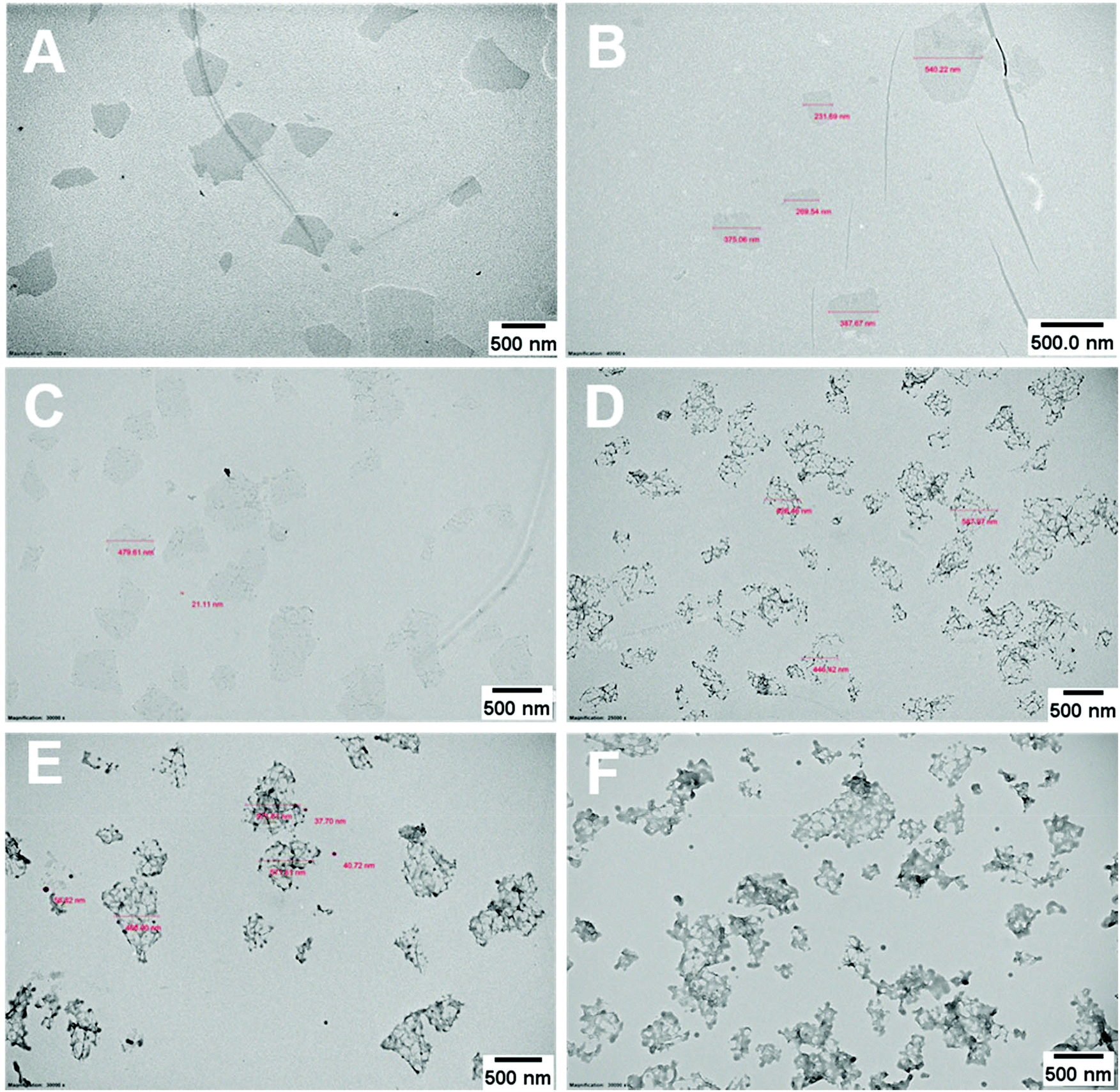 | ||
| Fig. 5 TEM micrographs (A) bare GO dispersed in DI water at pH 9 with the assistance of sonication, (B) GO dispersed in macro-RAFT copolymer, BuPATTC-(BA4-stat-AA9-stat-StS5) at pH 8.5, partial poly(MMA-co-BA) coated GO using RAFT-mediated emulsion polymerisation under starve-feeding of MMA/BA (10/1, w/w) with mass ratio of monomer/GO = (C) 10, (D) 35, (E) 75, and (F) 140. | ||
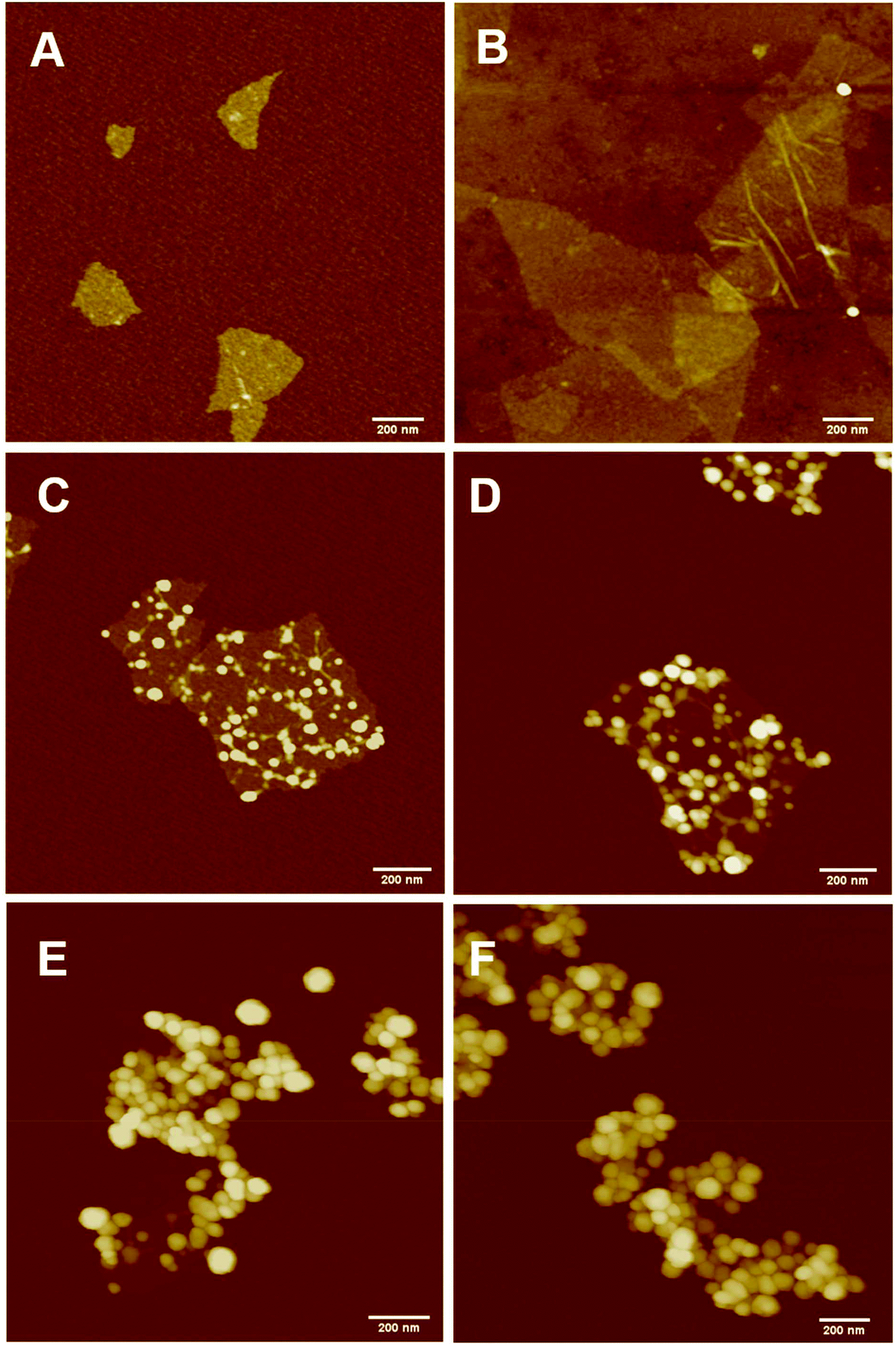 | ||
| Fig. 6 AFM micrographs (A) bare GO dispersed in DI water at pH 9 with sonication assistance, (B) GO dispersed in macro-RAFT copolymer, BuPATTC-(BA4-stat-AA9-stat-StS5) at pH 8.5, partial poly(MMA-co-BA) coated GO using RAFT-mediated emulsion polymerisation under starve-feeding of MMA/BA (10/1, w/w) with mass ratio of monomer/GO = (C) 10, (D) 35, (E) 75, and (F) 140. | ||
Reduction of PPC-GO with L-ascorbic acid (L-AA)
Reduction of bare GO with L-AA was first tested prior to application on the PPC-GO. L-AA was chosen as the reducing agent for this work due to its biocompatibility. It is a common vitamin, which is non-toxic and used as an antioxidant in supplements due to its mild reductive ability. L-AA has also been used as a primary reductant in the laboratory.41,50 In Fig. S7,† the TGA study clearly shows the thermal instability of the GO prior to reduction. It displayed significant weight loss in the 160 to 250 °C range, possibly due to its oxygen-containing groups. On the other hand, the material was observed to be very thermally stable up to 550 °C after the reduction reaction with L-AA. This thermal stability suggested a graphene-like chemical structure of the rGO as previously reported in literature.51,52 As shown in Fig. 7F, rGO by itself was not dispersible in water after reactions with L-AA. This was because the reduction process removed most of hydrophilic stabilising groups. Reduced surface charge also facilitated interactions between more hydrophobic rGO sheets, resulting in inseparable black aggregates, as shown in Fig. 7F. | ||
| Fig. 7 TEM micrographs of the PPC-GO before (A) and after (B) reducing with L-AA; AFM micrographs of the PPC-GO before (C) and after (D) reducing with L-AA; (E) GO was dispersed in DI water at pH 8; (F) GO reduced by L-AA in DI water; (G) the PPC-GO dispersed in DI water at pH 8; (H) the PPC-rGO in DI water at pH 8. | ||
Once validated, the reduction by L-AA was similarly applied to PPC-GO to produce PPC-rGO. As shown by the TEM and AFM micrographs in Fig. 7, PPC-rGO maintained similar surface morphologies with unchanged polymer domains compared to those of PPC-GO (Fig. 7A–D), while still retaining its colloidal stability (Fig. 7H). The PPC-rGO dispersion became dark, which might indicate a successful reducing reaction. However, the colloidal stability of PPC-rGO in water (compared to rGO) clearly showed the benefit of having attached stabilising groups. This feature is particularly important because aggregation limits the ability of a drug carrier to travel to tumour sites, greatly reducing its efficiency and biological clearance.
Adsorption and desorption of Dox on the PPC-GO
The PPC-GO was synthesised for a balance between colloidal stability and available bare GO surface where the adsorption of Dox can be expedited. The coated GO used in this exercise had around 43% polymer coverage, calculated from the AFM micrograph and contained about 73.5% polymer based on TGA (Fig. 8). To further probe Dox adsorption on the PPC-GO, a series of adsorption experiments were conducted varying only the Dox concentration. The removal of free polymer particles from the final PPC-GO latex was carefully carried out to ensure that there was no impact on Dox adsorption. This was done by washing three times with DI water using centrifugation at 14500 rpm for 10 min. Various concentrations of Dox, from 0.20 to 1.2 mM, were then added to 800 μL of the washed PPC-GO (114 μg based on pure GO). Note that the pH had a significant influence on Dox loading because it directly affected the surface charge of GO and the degree of ionisation of Dox.53 All the investigated adsorption reactions were conducted at pH 7.0, which was optimised for the highest Dox loading. The Dox adsorption reaction was incubated at room temperature with consistent shaking for 24 h to allow the adsorption to reach equilibrium prior to removal of excess Dox. The adsorbed Dox on the PPC-GO was determined by measuring the residual Dox in the supernatant using UV-Vis.
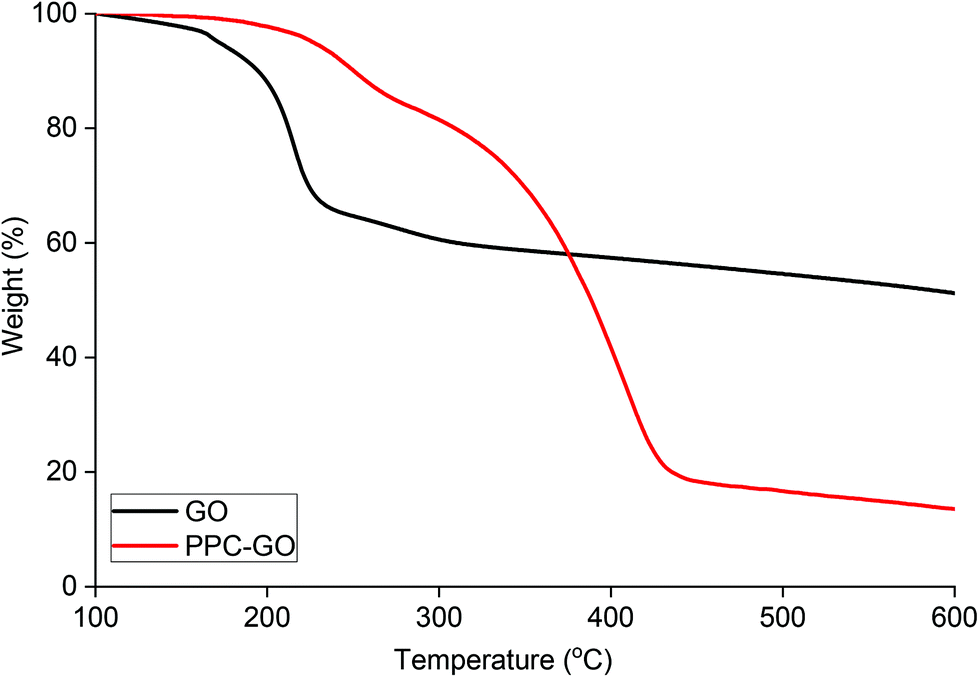 | ||
| Fig. 8 TGA of bare GO and PPC-GO with a mass ratio of MMA/BA (10/1, w/w)/GO = 35. | ||
As can be seen in Fig. 9, a substantial amount of Dox was able to physically adsorb on the available surface of the PPC-GO. As a qualitative comparison, Fig. 9A clearly shows that most of the Dox adsorbed onto the material to leave almost clear supernatants, even when 150% (weight percentage of Dox compared to GO mass) was employed. Indeed, the adsorbed Dox quantitatively increased with increasing Dox concentration (Fig. 9B). High Dox loading efficiency was observed and reached a maximum at above 90%, with a GO:Dox (w/w) of as much as 1:1, whilst the loading efficiency was reduced to around 70% with a GO:Dox (w/w) of 1:1.5 (Fig. 9B). As stated in the introduction, GO is a two-dimensional thin nanosheet with a large surface area that can facilitate drug adsorption on its surfaces and edges and thus it was not surprising to obtain very high Dox loading.54 Dox adsorption on the GO surface is generally promoted by non-covalent interactions, including hydrogen bonding, electrostatic interactions, and π–π stacking.54,55 The available uncoated surface of the PPC-GO was highly hydrophilic with a large number of oxygen functional groups, which inhibited π–π stacking but facilitated hydrogen bonding and electrostatic interactions between Dox and the investigated material. The hydrogen bonding and electrostatic interactions should, therefore, be dominant in the investigated system. Interestingly, the stability of PPC-GO was well retained after Dox adsorption as seen in Fig. S8.†
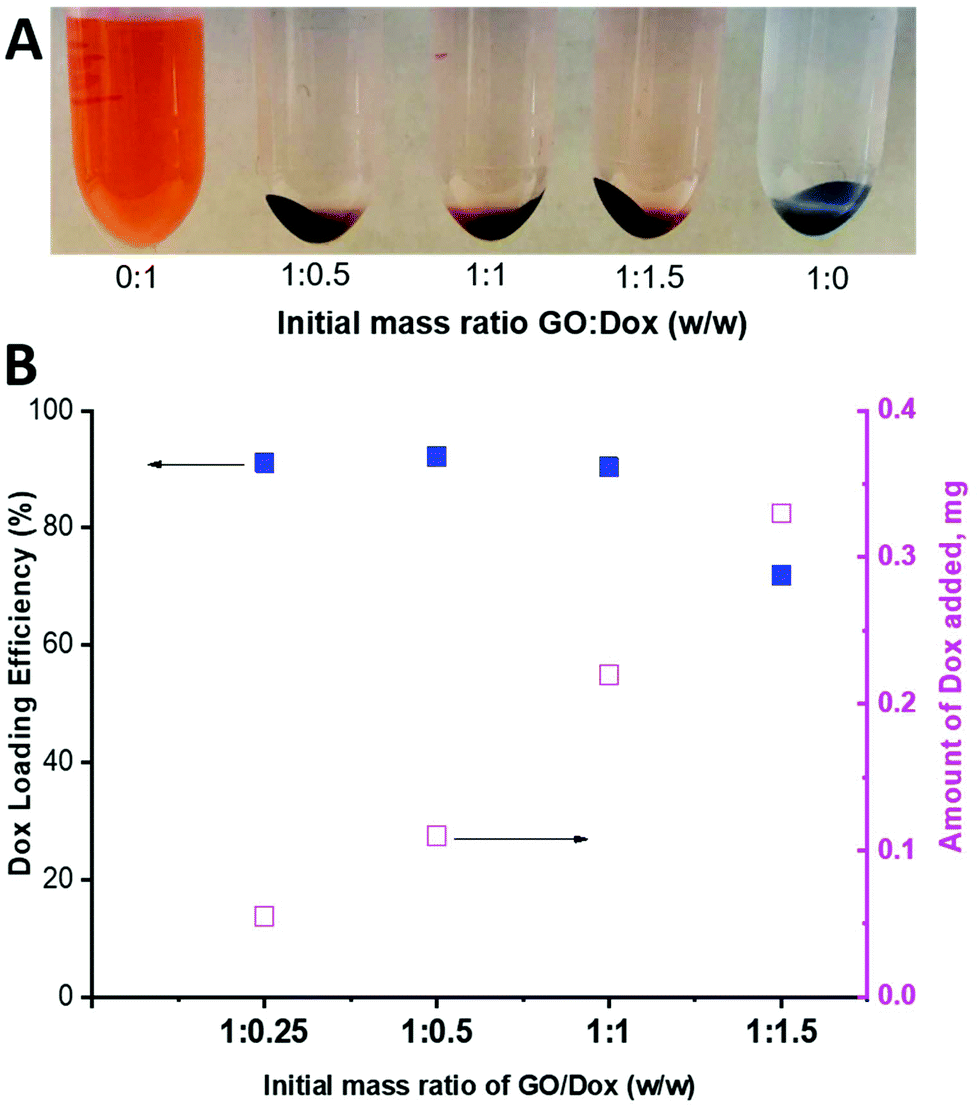 | ||
| Fig. 9 (A) Photo of Dox-loaded PPC-GO at different GO/Dox ratios and (B) Dox loading efficiency and the amount of Dox adsorbed on the PPC-GO surface. | ||
This also means that chemical reduction of the PPC-GO with Dox adsorbed, can trigger drug release because the removal of the functional groups via reduction weakens the hydrogen bonding between Dox and PPC-GO. The release of the adsorbed Dox on the PPC-GO was significantly influenced by the concentration of L-AA. The Dox-PPC-GO was incubated at room temperature for 24 h in different L-AA concentrations varying from 0 to 11 mM. Fig. 10 shows that around 30% of Dox was released from Dox-PPC-GO in 0.7 mM L-AA, almost 6 times as much as that in DI water, while there was not significant difference in Dox releasing at higher L-AA concentrations. Triggering Dox release by L-AA is a feasible approach as such a reducing agent is existed in intra- and extra-cellular media56,57 or could be leveraged via dietary intake. A release via reduction mechanism could be more selective for tumour environments, which often have highly reducing microenvironments.58 It is worth noting that the low Dox release from the material could be due to the re-adsorption of Dox on the PPC-rGO caused by L-AA.55
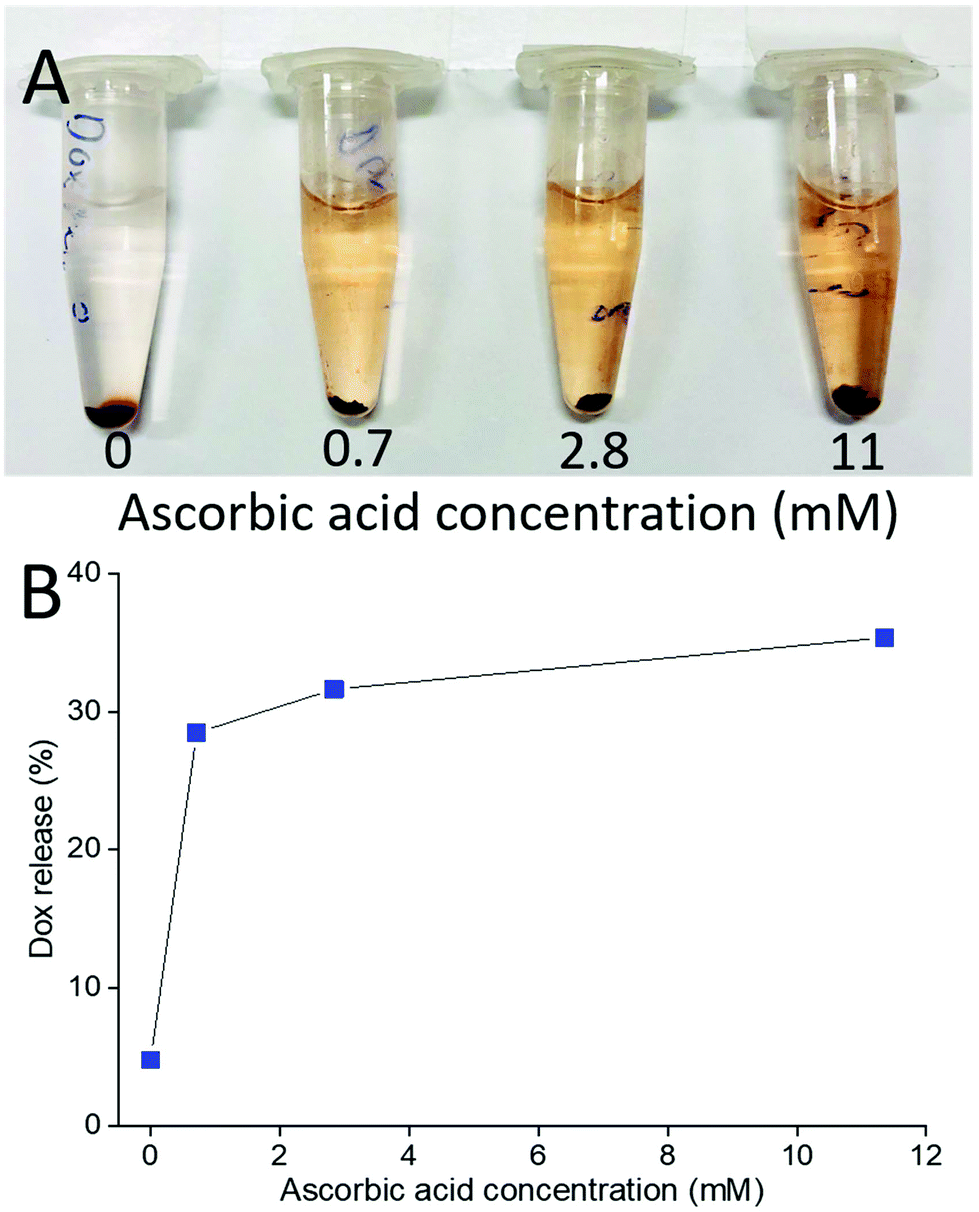 | ||
| Fig. 10 (A) Photo of Dox release at different L-AA concentrations and (B) Dox release versusL-AA concentration. | ||
Designing a drug nanocarrier system with high drug loading capacity and colloidal stability under physiological conditions is crucial in clinical application. Unstable materials could cause aggregation which hinders their biological clearance from body and causes toxicity. Up to date, the issue with stability of Dox-loaded GO systems have been modestly overcome, especially after Dox is released. Liu et al. stated that chemical reduction to increase Dox release can result in aggregation of GO due to van der Waals forces and restacking between nanosheets.55 In our work, the Dox-PPC-GO possessed an excellent colloidal stability which could make it a potential drug carrier. In addition to the very high Dox loading and the nano-sized material, the patchy polymer coating on the GO surface facilitated the colloidal stability of Dox-PPC-GO and, more importantly, the stability remained after chemical reduction using L-AA (releasing the adsorbed Dox), as discussed in the earlier section (Fig. 7).
Conclusions
The PPC-GO was successfully obtained by employing a starved-feed emulsion process using RAFT polymerisation. Unlike most comparable material encapsulations reported in the literature, the PPC-GO had not only the polymer bumps and nodes to stabilise it in the aqueous phase, but also the available bare GO surface for Dox adsorption. The ratio of the polymer-coated and uncoated areas on the GO nanosheet could be easily tuned by varying the amount of monomer fed in order to maximise Dox loading while keeping the material colloidal stability. The size of the polymer bumps significantly increased from 10 to 67 nm with an increase of the MMA/BA (10/1, w/w) feeding. A high Dox loading on the PPC-GO (carrying a 1:1 ratio of Dox by weight) was observed to yield a colloidally stable system. Chemical reduction of the Dox-loaded PPC-GO using L-AA led to Dox release, and the reduced PPC-GO having a similar microstructure to the PPC-GO and being stable in an aqueous dispersion. Taking into account all of these features, the PPC-GO is a promising candidate for drug delivery which may offer several advantages over previously reported carriers. Biological studies to further ascertain its suitability as a carrier will be carried out as part of a separate publication.
Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
VTH and BSH thank Syngenta Crop Protection for financial support. The authors also thank the Australian Research Council Linkage scheme (LP170100090). A/Prof Ronald Clarke is acknowledged for the fluorescence spectrometry and Prof Chiara Neto was thanked for AFM access.Notes and references
- C. Li, J. Wang, Y. Wang, H. Gao, G. Wei, Y. Huang, H. Yu, Y. Gan, Y. Wang, L. Mei, H. Chen, H. Hu, Z. Zhang and Y. Jin, Acta Pharm. Sin. B, 2019, 9, 1145–1162 CrossRef PubMed.
- W. Wang, K. Gaus, R. D. Tilley and J. J. Gooding, Mater. Horiz., 2019, 6, 1538–1547 RSC.
- V. T. Cong, K. Gaus, R. D. Tilley and J. J. Gooding, Expert Opin. Drug Delivery, 2018, 15, 881–892 CrossRef CAS PubMed.
- N. P. Truong, M. R. Whittaker, C. W. Mak and T. P. Davis, Expert Opin. Drug Delivery, 2015, 12, 129–142 CrossRef CAS PubMed.
- C. Kinnear, T. L. Moore, L. Rodriguez-Lorenzo, B. Rothen-Rutishauser and A. Petri-Fink, Chem. Rev., 2017, 117, 11476–11521 CrossRef CAS PubMed.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666–669 CrossRef CAS PubMed.
- M. Mathesh, J. Liu, N. D. Nam, S. K. H. Lam, R. Zheng, C. J. Barrow and W. Yang, J. Mater. Chem. C, 2013, 1, 3084–3090 RSC.
- V. C. Sanchez, A. Jachak, R. H. Hurt and A. B. Kane, Chem. Res. Toxicol., 2012, 25, 15–34 Search PubMed.
- Z. Liu, J. T. Robinson, X. Sun and H. Dai, J. Am. Chem. Soc., 2008, 130, 10876–10877 CrossRef CAS PubMed.
- X. Pei, Z. Zhu, Z. Gan, J. Chen, X. Zhang, X. Cheng, Q. Wan and J. Wang, Sci. Rep., 2020, 10, 2717 CrossRef CAS PubMed.
- J.-H. Jiang, J. Pi, H. Jin and J.-Y. Cai, Artif. Cells, Nanomed., Biotechnol., 2018, 46, S297–S307 CrossRef CAS PubMed.
- X. Yang, Y. Wang, X. Huang, Y. Ma, Y. Huang, R. Yang, H. Duan and Y. Chen, J. Mater. Chem., 2011, 21, 3448–3454 RSC.
- Y. Chang, S.-T. Yang, J.-H. Liu, E. Dong, Y. Wang, A. Cao, Y. Liu and H. Wang, Toxicol. Lett., 2011, 200, 201–210 CrossRef CAS PubMed.
- S.-R. Ryoo, Y.-K. Kim, M.-H. Kim and D.-H. Min, ACS Nano, 2010, 4, 6587–6598 CrossRef CAS PubMed.
- A. Bianco, Angew. Chem., Int. Ed., 2013, 52, 4986–4997 CrossRef CAS PubMed.
- W. Hu, C. Peng, M. Lv, X. Li, Y. Zhang, N. Chen, C. Fan and Q. Huang, ACS Nano, 2011, 5, 3693–3700 CrossRef CAS PubMed.
- S. A. Sydlik, S. Jhunjhunwala, M. J. Webber, D. G. Anderson and R. Langer, ACS Nano, 2015, 9, 3866–3874 CrossRef CAS PubMed.
- Z. Liu, S. Tabakman, K. Welsher and H. Dai, Nano Res., 2009, 2, 85–120 CrossRef CAS PubMed.
- X. Sun, Z. Liu, K. Welsher, J. T. Robinson, A. Goodwin, S. Zaric and H. Dai, Nano Res., 2008, 1, 203–212 CrossRef CAS PubMed.
- M. Pooresmaeil and H. Namazi, Colloids Surf., B, 2018, 172, 17–25 CrossRef CAS PubMed.
- C.-H. Zhu, Y. Lu, J. Peng, J.-F. Chen and S.-H. Yu, Adv. Funct. Mater., 2012, 22, 4017–4022 CrossRef CAS.
- D. Nguyen, H. S. Zondanos, J. M. Farrugia, A. K. Serelis, C. H. Such and B. S. Hawkett, Langmuir, 2008, 24, 2140–2150 CrossRef CAS PubMed.
- C. J. Ferguson, R. J. Hughes, D. Nguyen, B. T. T. Pham, R. G. Gilbert, A. K. Serelis, C. H. Such and B. S. Hawkett, Macromolecules, 2005, 38, 2191–2204 CrossRef CAS.
- C. J. Ferguson, R. J. Hughes, B. T. T. Pham, B. S. Hawkett, R. G. Gilbert, A. K. Serelis and C. H. Such, Macromolecules, 2002, 35, 9243–9245 CrossRef CAS.
- N. J. Warren and S. P. Armes, J. Am. Chem. Soc., 2014, 136, 10174–10185 CrossRef CAS PubMed.
- H. Tanaka, K. Yamauchi, H. Hasegawa, N. Miyamoto, S. Koizumi and T. Hashimoto, Phys. B, 2006, 385–386, 742–744 CrossRef CAS.
- S. I. Ali, J. P. A. Heuts, B. S. Hawkett and A. M. van Herk, Langmuir, 2009, 25, 10523–10533 CrossRef CAS PubMed.
- D. Nguyen, C. H. Such and B. S. Hawkett, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 250–257 CrossRef CAS.
- M. A. Mballa Mballa, S. I. Ali, J. P. A. Heuts and A. M. van Herk, Polym. Int., 2012, 61, 861–865 CrossRef CAS.
- M. M. Mballa, J. A. Heuts and A. Herk, Colloid Polym. Sci., 2013, 291, 501–513 CrossRef CAS.
- P. Das and J. Claverie, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 2802–2808 CrossRef CAS.
- E. Bourgeat-Lami, A. J. P. G. Franca, T. C. Chaparro, R. D. Silva, P. Y. Dugas, G. M. Alves and A. M. Santos, Macromolecules, 2016, 49, 4431–4440 CrossRef CAS.
- J. Garnier, J. Warnant, P. Lacroix-Desmazes, P.-E. Dufils, J. Vinas, Y. Vanderveken and A. M. van Herk, Macromol. Rapid Commun., 2012, 33, 1388–1392 CrossRef CAS PubMed.
- N. Zgheib, J. L. Putaux, A. Thill, E. Bourgeat-Lami, F. D'Agosto and M. Lansalot, Polym. Chem., 2013, 4, 607–614 RSC.
- P. Das, W. Zhong and J. P. Claverie, Colloid Polym. Sci., 2011, 289, 1519 CrossRef CAS.
- S. Pearson, M. Pavlovic, T. Augé, V. Torregrossa, I. Szilagyi, F. D'Agosto, M. Lansalot, E. Bourgeat-Lami and V. Prévot, Macromolecules, 2018, 51, 3953–3966 CrossRef CAS.
- D. Nguyen, B. T. T. Pham, V. Huynh, B. J. Kim, N. T. H. Pham, S. A. Bickley, S. K. Jones, A. Serelis, T. Davey, C. Such and B. S. Hawkett, Polymer, 2016, 106, 238–248 CrossRef CAS.
- T. R. Guimarães, M. Lansalot and E. Bourgeat-Lami, J. Mater. Chem. B, 2020, 8, 4917–4929 RSC.
- V. T. Huynh, D. Nguyen, C. H. Such and B. S. Hawkett, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 1413–1421 CrossRef CAS.
- D. Nguyen, C. Such and B. Hawkett, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 346–352 CrossRef CAS.
- J. Zhang, H. Yang, G. Shen, P. Cheng, J. Zhang and S. Guo, Chem. Commun., 2010, 46, 1112–1114 RSC.
- Y. Tu, M. Lv, P. Xiu, T. Huynh, M. Zhang, M. Castelli, Z. Liu, Q. Huang, C. Fan, H. Fang and R. Zhou, Nat. Nanotechnol., 2013, 8, 594–601 CrossRef CAS.
- A. Ganguly, S. Sharma, P. Papakonstantinou and J. Hamilton, J. Phys. Chem. C, 2011, 115, 17009–17019 CrossRef CAS.
- T. Sumaryada, M. Sandy Gunawan, S. Perdana, S. Arjo and A. Maddu, Biosensors, 2019, 9, 18 CrossRef CAS PubMed.
- S. N. Jamadagni, R. Godawat and S. Garde, Langmuir, 2009, 25, 13092–13099 CrossRef CAS PubMed.
- Y. Fadil, F. Jasinski, T. Wing Guok, S. C. Thickett, H. Minami and P. B. Zetterlund, Polym. Chem., 2018, 9, 3368–3378 RSC.
- S. H. Che Man, S. C. Thickett, M. R. Whittaker and P. B. Zetterlund, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 47–58 CrossRef CAS.
- J. Liu, W. Yang, L. Tao, D. Li, C. Boyer and T. P. Davis, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 425–433 CrossRef CAS.
- B. Wang, X. Su, J. Liang, L. Yang, Q. Hu, X. Shan, J. Wan and Z. Hu, Mater. Sci. Eng., C, 2018, 90, 514–522 CrossRef CAS.
- K. K. H. De Silva, H.-H. Huang and M. Yoshimura, Appl. Surf. Sci., 2018, 447, 338–346 CrossRef CAS.
- S. Jin, Q. Gao, X. Zeng, R. Zhang, K. Liu, X. Shao and M. Jin, Diamond Relat. Mater., 2015, 58, 54–61 CrossRef CAS.
- P. Cui, J. Lee, E. Hwang and H. Lee, Chem. Commun., 2011, 47, 12370–12372 RSC.
- S. L. Wu, X. D. Zhao, Y. H. Li, Q. J. Du, J. K. Sun, Y. H. Wang, X. Wang, Y. Z. Xia, Z. H. Wang and L. H. Xia, Materials, 2013, 6, 2026–2042 CrossRef CAS PubMed.
- M. Mahdavi, F. Rahmani and S. Nouranian, J. Mater. Chem. B, 2016, 4, 7441–7451 RSC.
- Z. Liu, J. Liu, T. Wang, Q. Li, P. S. Francis, C. J. Barrow, W. Duan and W. Yang, J. Mater. Chem. B, 2018, 6, 1251–1259 RSC.
- R. M. Evans, L. Currie and A. Campbell, Br. J. Nutr., 1982, 47, 473–482 CrossRef CAS PubMed.
- J. M. May and Z.-C. Qu, Mol. Cell. Biochem., 2009, 325, 79–88 CrossRef CAS PubMed.
- P. Kuppusamy, H. Li, G. Ilangovan, A. J. Cardounel, J. L. Zweier, K. Yamada, M. C. Krishna and J. B. Mitchell, Cancer Res., 2002, 62, 307–312 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0py00769b |
| This journal is © The Royal Society of Chemistry 2021 |