
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Cooperative Lewis acid–onium salt catalysis as tool for the desymmetrization of meso-epoxides†
Florian
Broghammer
,
Daniel
Brodbeck
,
Thorsten
Junge
and
René
Peters
 *
*
Universität Stuttgart, Institut für Organische Chemie, Pfaffenwaldring 55, 70569 Stuttgart, Germany. E-mail: rene.peters@oc.uni-stuttgart.de
First published on 23rd December 2016
Abstract
Epoxide desymmetrizations by bromide are very rare despite the large synthetic potential of chiral bromohydrins. Herein we present a new concept for epoxide desymmetrizations in which a bifunctional Lewis acid/ammonium salt catalyst allows for efficient enantioselective epoxide ring openings by Br−. With acetylbromide as a Br− source bromohydrin esters are formed.
Desymmetrizations of meso-epoxides via nucleophilic ring opening reactions provide an attractive synthetic tool towards enantiopure β-functionalized alcohols possessing two adjacent stereocenters.1 Different methods for O,2 N,3 and Cl4 centred nucleophiles are established. In comparison, the use of bromide and iodide has been described in a single study so far. Nugent (DuPont) reported in 1998 that a chiral zirconium complex is capable of catalyzing the asymmetric ring opening of primarily cycloolefin derived epoxides using a combination of TMSN3 and an excess allyl iodide or bromide.5‡ A common feature of this and many other epoxide desymmetrizations1 is the simultaneous silyl protective group installation on the generated alcohol functions. The direct installation of alternative protecting group types is much more unusual.6
In this communication we report the development of an enantioselective method to form O-acetyl protected bromohydrins starting from achiral epoxides and acetylbromide. The use of an ester protecting group is synthetically appealing, because it might be later readily removed by mild enzymatic or chemical protocols.6,7 Bromohydrin esters are interesting building blocks which allow for a range of chemical manipulations. In this context the ester group does not only serve as protective group, but has also been further functionalized as a part of the targeted skeleton.8
Ether cleavages9 including those of epoxides by acylhalides10 are assumed to usually proceed via O-acylation of the ethers followed by a nucleophilic halide attack. For the present study we strived for a different scenario: a chiral Lewis acid should trigger an enantioselective nucleophilic attack at the epoxide by Br− which is directed by an onium moiety connected to the ligand (Fig. 1, right). The generated metal alcoholate might then be acylated by acetylbromide to release the product and regenerate the catalytically active Lewis acid and Br−.
 | ||
| Fig. 1 Desymmetrization of meso-epoxides by cooperative catalysis. | ||
For epoxide desymmetrizations cooperative bimetallic catalysis has been successfully employed as dual activation strategy1,11 (Fig. 1, left).3–5,12 The here reported approach is conceptually different as it capitalizes on the cooperation of an aprotic onium salt ion pair with a Lewis acid. Delivering an anionic nucleophile via an aprotic ion pair was expected to offer a reactivity advantage, because metal coordination of a nucleophile is likely to considerably decrease its electron density.13 Our research group has previously introduced the concept of cooperative intramolecular Lewis acid/aprotic onium salt catalysis to asymmetric synthesis.14,15 In these earlier studies a halide counterion was an active part of the catalyst. In the present case it is also a part of the final product and thus needs to be regenerated to enable catalytic turnover.
At the outset of the present investigation the reaction of stilbene oxide 1a and AcBr 2 was examined (Table 1). The Al-salen/bispyridinium complex 3a was initially chosen, because it provided the highest stereoselectivities for the asymmetric synthesis of trans-configured β-lactones.14c Gratifyingly, the envisioned reaction proceeded in quantitative yield at −20 °C, albeit with low enantioselectivity (entry 1).
|
|
|||||||
|---|---|---|---|---|---|---|---|
| No. | 3 (X) | Y | Additive | T [°C] | Yielda [%] | drb | erc [%] |
| a Yield determined by 1H-NMR of the crude product using an internal standard. b Diastereomeric ratio determined by 1H-NMR of the crude product. c Enantiomeric ratio of determined by HPLC. d t = 48 h. | |||||||
| 1 | 3a (10) | Me | — | −20 | >99 | 37![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 :1 |
64.5:35.5 |
| 2 | 3b (10) | Me | — | −20 | >99 | 29:1 |
80.5:19.5 |
| 3 | 3c (10) | Me | — | −20 | >99 | 10:1 |
85:15 |
| 4 | 3c (10) | Me | HOAc | −20 | 99 | 14:1 |
89:11 |
| 5 | 3c (5) | Cl | HOAc | −20 | 99 | 14:1 |
89.5:10.5 |
| 6d | 3c (5) | Cl | HOAc | −40 → RT | >99 | 33:1 |
91.5:8.5 |
| 7d | 3d (5) | Cl | HOAc | −40 → RT | 99 | 31:1 |
92:8 |
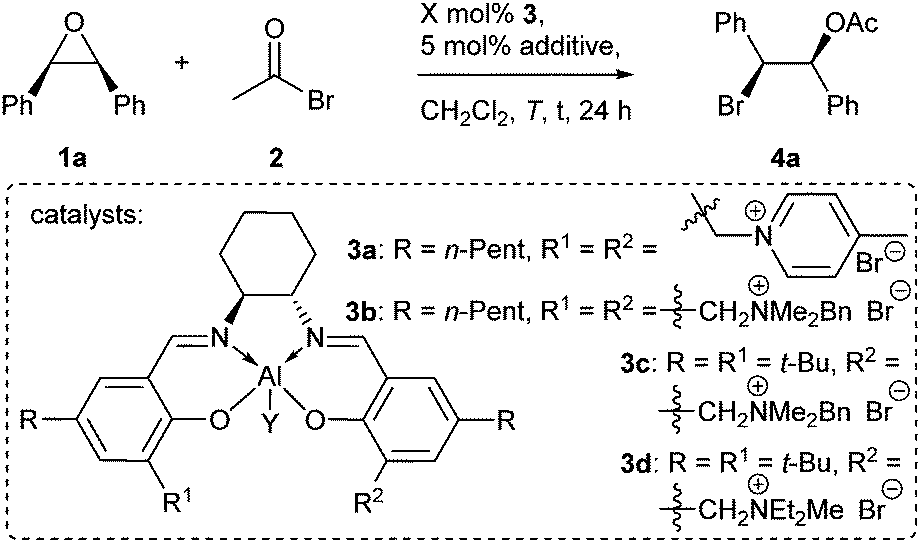
During the subsequent optimization it was found that quaternary ammonium residues like in 3b allowed for a better enantiotopos differentiation than pyridinium moieties (entry 2). Another improvement was made by the use of catalysts equipped with only one ammonium residue like in 3c (entry 3) and further by using acetic acid as a catalytic additive (entry 4). With Me and Cl substituents Y bound to the Al center nearly identical results were obtained (entries 4 and 5). Since the Al–Cl containing catalysts are more robust, they were preferentially used in this study. A decrease in the reaction temperature to −40 °C resulted in further improvements in stereoselectivity (entry 6). The best enantioselectivity was obtained with catalyst 3d possessing a diethylmethylammonium bromide moiety (entry 7).
The optimized reaction conditions were applied to aromatic meso-epoxides with different substitution patterns (Table 2). Although some product was always lost upon chromatographic purification as a result of the high reactivity of the benzylic bromides 4 (compare Table 1/entry 7 to Table 2/entry 1), the yields of isolated products were high for all examples (82–92%). Regarding the diastereomeric ratios there is a tendency. Whereas electron donors like methyl groups caused somewhat lower diastereomeric ratios (entries 2 and 3), electron withdrawing substituents led to diastereomerically pure products. This might be due to a lower tendency for a competing SN1-pathway in the latter case. The highest enantioselectivities were noticed for a CF3 and fluoro substituted substrates (entries 4–6), although in these cases the reactions were found to be a little slower. Good results were also attained with chloro (entry 8) and methoxy (entry 9) substituents on the aromatic residues R.
|
|
||||||
|---|---|---|---|---|---|---|
| No. | 4 | R | Conversiona [%] | Yieldb [%] | drc | erd [%] |
| a Conversion of 1 determined by 1H-NMR of the crude product. b Yield of isolated 4 after column chromatography. c Diastereomeric ratio determined by 1H-NMR of the crude product. d Enantiomeric ratio determined by HPLC. | ||||||
| 1 | 4a | Ph | >99 | 88 | 31:1 |
92:8 |
| 2 | 4b | 4-Me-C6H4 | >99 | 88 | 22:1 |
91:9 |
| 3 | 4c | 3-Me-C6H4 | >99 | 85 | 25:1 |
91:9 |
| 4 | 4d | 4-F-C6H4 | >99 | 91 | 50:1 |
95:5 |
| 5 | 4e | 3-F-C6H4 | >99 | 92 | >50:1 |
94:6 |
| 6 | 4f | 4-F3C-C6H4 | >99 | 89 | >50:1 |
95.5:4.5 |
| 7 | 4g | 3-F3C-C6H4 | >99 | 82 | >50:1 |
88:12 |
| 8 | 4h | 3-Cl-C6H4 | >99 | 89 | >50:1 |
90.5:9.5 |
| 9 | 4i | 3-MeO-C6H4 | >99 | 85 | >50:1 |
91:9 |

The scope is complementary to Nugent's Zr-catalyzed reaction which was not described for substrates with aromatic residues.5 In contrast, our method provided lower enantioselectivity for aliphatic substrates, while reactivity was comparable (e.g., with cyclohexenoxide 99% yield under standard conditions, dr > 50:1, nearly racemic product; for R = CH2OBn: 80% yield after 72 h, dr > 50:1, ee = 40%).
A reaction monitoring was conducted by 1H-NMR (Fig. 2) which shows that in the preferred pathway the acylated product 4a is not directly formed from the expected alkoxide intermediate II generated in the ring opening step (Scheme 1). Instead alcohol 5 is first released. Fig. 2 shows that the reaction reaches 92% conversion after 24 h. Additional reaction time is mainly needed for acylation of the alcohol intermediate. Conversion of 1a is practically complete after 48 h (99%). To form the ester product in a quantitative yield it is then necessary to warm the reaction mixture to room temperature to ensure that unreacted OH groups (8% after 48 h) are acylated.
 | ||
| Fig. 2 1H-NMR monitoring of the reaction described in Table 1/entry 7. | ||
 | ||
| Scheme 1 Mechanistic proposal. | ||
We propose a major mechanistic pathway, in which the epoxide is activated by coordination to the catalyst (I, Scheme 1). Br− is delivered as nucleophile by the internal ammonium moiety in I providing Al alkoxide II. For protonation of II HOAc may act as a proton source, but also HBr, which is present in acetylbromide as an impurity and difficult to remove. HBr is also released during the acylation step and could serve to regenerate HOAc plus a bromide anion for the next turnover.
A number of experiments were also performed with Jacobsen's Al salen catalyst 3e16 as control system (Scheme 2).§ By optimizing the reaction conditions, an er of 84:16 could be achieved with this catalyst. Comparison of 3e with the bifunctional catalyst 3d by kinetic monitoring (see ESI†) showed that the latter is considerably more active. Moreover, addition of nBu4NBr had a beneficial effect on both reactivity and enantioselectivity using 3e (Scheme 2) thus supporting a cooperative mechanism. Dual activation by Lewis acid catalysts like 3e and onium salts might therefore also allow for useful catalyst systems in the future. Nevertheless, yield and enantioselectivity were significantly lower than with bifunctional 3d. Since 3d is readily prepared in high yields over 3 steps starting from commercial 1,2-diaminocyclohexane (see ESI†), its use is also operationally similarly appealing.
 | ||
| Scheme 2 Control experiments. | ||
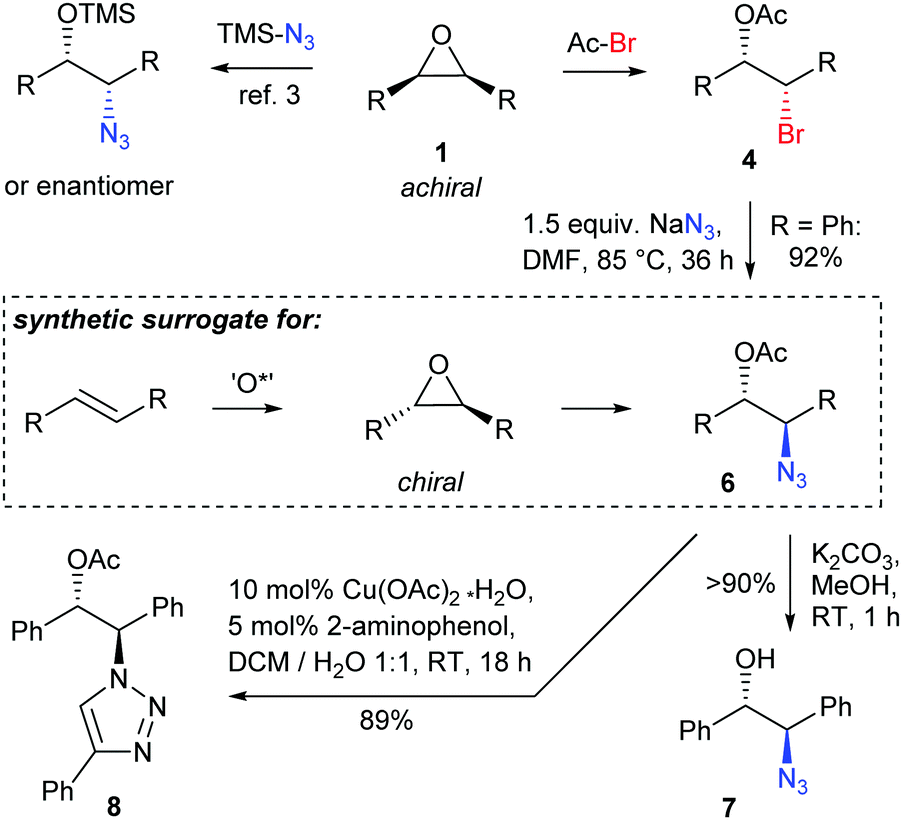
Like the meso-epoxides the bromohydrin esters 4 serve as alkylating agents, yet provide access to the complementary set of product diastereomers compared to the direct use of meso-epoxides. The reported products thus represent synthetic equivalents for enantioenriched trans-epoxides (Scheme 3) for which only few protocols have been developed.5,17
 | ||
| Scheme 3 Examples for derivatizations. | ||
To showcase the value of the acetyl protected bromohydrins, 4a was treated with NaN3 under SN2 conditions providing azide 6 by smooth inversion of the configuration at the reacting C-atom. As all known desymmetrizations using TMSN3 have a limitation for alkyl substituted epoxides,3 the here reported method is also complementary in that aspect to the existing methods. As expected, deprotection of the acetyl group was readily achieved under mild conditions. The acetyl group should also be interesting for mild enzymatic deprotections allowing for the formation of almost enantiopure products often being required for pharmaceutical applications. The absolute configuration of 4a could be assigned by the optical rotation of literature known derivative 7.18 Azide 6 was also applied to a click reaction to form a (1R,2S)-configured 2-(triazol-1-yl)ethyl acetate 8, a motif which might, e.g., be of potential pharmacological interest.
A catalyst concept is described for the desymmetrization of epoxides which exploits the cooperation of a Lewis acid and an aprotic onium salt moiety within a bifunctional chiral catalyst framework. This catalyst type allowed for the development of a reaction in which achiral epoxides react with AcBr to deliver enantioenriched acetyl protected bromohydrins. The title reaction was shown to proceed via the intermediate formation of the bromohydrin which is then acylated. Preliminary studies show that the combination of a Lewis acid catalyst plus a cooperating external ammonium salt cocatalyst is also an attractive option for future developments.
This work was financially supported by the Deutsche Forschungsgemeinschaft (DFG, PE 818/3-1) and the Carl-Zeiss-Stiftung (PhD fellowship to DB).
Notes and references
- (a) D. M. Hodgson, A. R. Gibbs and G. P. Lee, Tetrahedron, 1996, 52, 14361 CrossRef CAS; (b) P. A. Wang, Beilstein J. Org. Chem., 2013, 9, 1677 CrossRef PubMed; (c) S. Meninno and A. Lattanzi, Chem. – Eur. J., 2016, 22, 3632 CrossRef CAS PubMed; (d) L. P. C. Nielsen and E. N. Jacobsen, in Aziridines and Epoxides in Organic Synthesis, ed. A. K. Yudin, Wiley-VCH, Weinheim, 2006, pp. 229–269 Search PubMed; (e) E. N. Jacobsen, Acc. Chem. Res., 2000, 33, 421 CrossRef CAS PubMed; (f) A. Borissov, T. Q. Davies, S. R. Ellis, T. A. Fleming, M. S. W. Richardson and D. J. Dixon, Chem. Soc. Rev., 2016, 45, 5474 RSC.
- (a) E. N. Jacobsen, F. Kakiuchi, R. G. Konsler, J. F. Larrow and M. Tokunaga, Tetrahedron Lett., 1997, 38, 773 CrossRef CAS; (b) T. Iida, N. Yamamoto, S. Matsunaga, H.-G. Woo and M. Shibasaki, Angew. Chem., Int. Ed., 1998, 37, 2223 CrossRef CAS; (c) S. Matsunaga, J. Das, J. Roels, E. M. Vogl, N. Yamamoto, T. Iida, K. Yamaguchi and M. Shibasaki, J. Am. Chem. Soc., 2000, 122, 2252 CrossRef CAS.
- (a) W. A. Nugent, J. Am. Chem. Soc., 1992, 114, 2768 CrossRef CAS; (b) L. E. Martínez, J. L. Leighton, D. H. Carsten and E. N. Jacobsen, J. Am. Chem. Soc., 1995, 117, 5897 CrossRef; (c) K. B. Hansen, J. L. Leighton and E. N. Jacobsen, J. Am. Chem. Soc., 1996, 118, 10924 CrossRef CAS; (d) B. W. McCleland, W. A. Nugent and M. G. Finn, J. Org. Chem., 1998, 63, 6656 CrossRef CAS; (e) A. Sekine, T. Ohshima and M. Shibasaki, Tetrahedron, 2002, 58, 75 CrossRef CAS; (f) J. A. Birrell and E. N. Jacobsen, Org. Lett., 2013, 15, 2895 CrossRef CAS PubMed.
- (a) E. Denmark, P. A. Barsanti, K.-T. Wong and R. A. Stavenger, J. Org. Chem., 1998, 63, 2428 CrossRef PubMed; (b) B. Tao, M. M.-C. Lo and G. C. Fu, J. Am. Chem. Soc., 2001, 123, 353 CrossRef CAS PubMed; (c) S. E. Denmark, P. A. Barsanti and G. L. Beutner, Adv. Synth. Catal., 2007, 349, 567 CrossRef CAS; (d) E. Gnanamani, N. Someshwar, J. Sanjeevi and C. R. Ramanathan, Adv. Synth. Catal., 2014, 356, 2219 CrossRef CAS and cited references; with fluoride: ; (e) J. A. Kalow and A. G. Doyle, J. Am. Chem. Soc., 2011, 133, 16001 CrossRef CAS PubMed.
- W. A. Nugent, J. Am. Chem. Soc., 1998, 120, 7139 CrossRef CAS.
- M. R. Monaco, S. Prévost and B. List, Angew. Chem., Int. Ed., 2014, 53, 8142 CrossRef CAS PubMed.
- P. G. M. Wuts and T. W. Greene, Greene's Protective Groups in Organic Synthesis, Wiley-Interscience, 4th edn, 2006 Search PubMed.
- (a) K. G. Watson, Y. M. Fung, M. Gredley, G. J. Bird, W. R. Jackson, H. Gountzos and B. R. Matthews, J. Chem. Soc., Chem. Commun., 1990, 1018 RSC; (b) H. C. Kolb, Y. L. Bennani and K. B. Sharpless, Tetrahedron: Asymmetry, 1993, 4, 133 CrossRef CAS; (c) Y. Iwasaki and T. Yamane, J. Mol. Catal. B: Enzym., 2000, 10, 129 CrossRef CAS; (d) J. Prades, S. S. Funari, P. V. Escriba and F. Barcelo, J. Lipid Res., 2003, 44, 1720 CrossRef CAS PubMed; (e) K. Parang, L. I. Wiebe and E. E. Knaus, Curr. Med. Chem., 2000, 7, 995 CrossRef CAS PubMed.
- R. L. Burwell, Jr., Chem. Rev., 1954, 54, 615 CrossRef.
- Examples: (a) H. Kotsuki, Y. Ichikawa and H. Nishizawa, Chem. Lett., 1988, 673 CrossRef CAS; (b) T. Oriyama, A. Ishiwata, Y. Hori, T. Yatabe, N. Hasumi and G. Koga, Synlett, 1995, 1004 CrossRef CAS; (c) D. J. Goldsmith, E. Kennedy and R. G. Campbell, J. Org. Chem., 1975, 40, 3571 CrossRef CAS; (d) K. Nakano, S. Kodama, Y. Permana and K. Nozaki, Chem. Commun., 2009, 6970 RSC; (e) G. Aghapour and R. Hatefipour, Synth. Commun., 2013, 43, 1030 CrossRef CAS.
- Cooperative Catalysis – Designing Efficient Catalysts for Synthesis, ed. R. Peters, Wiley-VCH, Weinheim, 2015 Search PubMed.
- (a) T. Iida, N. Yamamoto, H. Sasai and M. Shibasaki, J. Am. Chem. Soc., 1997, 119, 4783 CrossRef CAS; (b) S. Matsunaga, J. Das, J. Roels, E. M. Vogl, N. Yamamoto, T. Iida, K. Yamaguchi and M. Shibasaki, J. Am. Chem. Soc., 2000, 122, 2252 CrossRef CAS.
- Selected reviews on asymmetric onium salt catalysis: (a) S. Shirakawa and K. Maruoka, Angew. Chem., Int. Ed., 2013, 52, 4312 CrossRef CAS PubMed; (b) K. Maruoka, Org. Process Res. Dev., 2008, 12, 679 CrossRef CAS.
- (a) T. Kull and R. Peters, Angew. Chem., Int. Ed., 2008, 47, 5461 CrossRef CAS PubMed; (b) T. Kull, J. Cabrera and R. Peters, Chem. – Eur. J., 2010, 16, 9132 CrossRef CAS PubMed; (c) P. Meier, F. Broghammer, K. Latendorf, G. Rauhut and R. Peters, Molecules, 2012, 17, 7121 CrossRef CAS PubMed; (d) M. Mechler and R. Peters, Angew. Chem., Int. Ed., 2015, 54, 10303 CrossRef CAS PubMed.
- The concept is also useful in macromolecular chemistry. See, e.g.: (a) M. Winnacker, S. Vagin and B. Rieger, Cooperative Catalysis in Polymerization Reactions, in Cooperative Catalysis – Designing Efficient Catalysts for Synthesis, ed. R. Peters, Wiley-VCH, Weinheim, 2015 Search PubMed; (b) T. Ema, Y. Miyazaki, J. Shimonishi, C. Maeda and J.-y. Hasegawa, J. Am. Chem. Soc., 2014, 136, 15270 CrossRef CAS PubMed; (c) W.-M. Ren, Y. Liu and X.-B. Lu, J. Org. Chem., 2014, 79, 9771 CrossRef CAS PubMed; (d) D. J. Darensbourg, W.-C. Chung and S. J. Wilson, ACS Catal., 2013, 3, 3050 CrossRef CAS ; it has also been recently used by other groups for small molecules: ; (e) D. Tian, B. Liu, Q. Gan, H. Li and D. J. Darensbourg, ACS Catal., 2012, 2, 2029 CrossRef CAS; (f) K. Ohmatsu, S. Kawai, N. Imagawa and T. Ooi, ACS Catal., 2014, 4, 4304 CrossRef CAS.
- M. S. Sigman and E. N. Jacobsen, J. Am. Chem. Soc., 1998, 120, 5315 CrossRef CAS.
- (a) S. Chang, J. M. Galvin and E. N. Jacobsen, J. Am. Chem. Soc., 1996, 116, 6937 CrossRef; (b) V. K. Aggarwal, E. Alonso, G. Hynd, K. M. Lydon, M. J. Palmer, M. Porcelloni and J. R. Studley, Angew. Chem., Int. Ed., 2001, 40, 1430 CrossRef CAS.
- (a) P. Lupattelli, C. Bonini, L. Caruso and A. Gambacorta, J. Org. Chem., 2003, 68, 3360 CrossRef CAS PubMed; (b) B. B. Lohray and J. R. Ahuja, J. Chem. Soc., Chem. Commun., 1991, 95 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterization data of new compounds. See DOI: 10.1039/c6cc09774j |
| ‡ In contrast, the direct use of TMSBr results in a very rapid epoxide ring opening even in the absence of a catalyst thus hampering useful enantioselectivity as our own studies revealed. |
| § In no case using bifunctional catalysts 3a–d or Jacobsen's catalyst 3e a pinacol type rearrangement product was observed as side product. |
| This journal is © The Royal Society of Chemistry 2017 |