
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOvercoming antibiotic resistance in Pseudomonas aeruginosa biofilms using glycopeptide dendrimers†
Gaëlle
Michaud
a,
Ricardo
Visini
a,
Myriam
Bergmann
a,
Gianluca
Salerno
b,
Rosa
Bosco
b,
Emilie
Gillon
c,
Barbara
Richichi
b,
Cristina
Nativi
b,
Anne
Imberty
c,
Achim
Stocker
a,
Tamis
Darbre
*a and
Jean-Louis
Reymond
*a
aDepartment of Chemistry and Biochemistry, University of Berne, Freiestrasse 3, 3012 Berne, Switzerland. E-mail: jean-louis.reymond@dcb.unibe.ch
bDipartimento di Chimica, Polo Scientifico e Tecnologico, Universita' degli Studi di Firenze, Via della Lastruccia 3, 13, I-50019 Sesto Fiorentino, Firenze, Italy
cCentre de Recherches sur les Macromolécules Végétales, UPR5301, CNRS and Université Grenoble Alpes, 601 rue de la Chimie, F38041 Grenoble, France
First published on 25th November 2015
Abstract
Antibiotic resistance in the opportunistic pathogen Pseudomonas aeruginosa is partly caused by biofilms forming a physical barrier to antibiotic penetration. Here we focused on modifying tetravalent glycopeptide dendrimer ligands of P. aeruginosa lectins LecB or LecA to increase their biofilm inhibition activity. First heteroglycoclusters were investigated displaying one pair each of LecB specific fucosyl groups and LecA specific galactosyl groups and binding simultaneously to both lectins, one of which gave the first fully resolved crystal structure of a peptide dendrimer as LecB complex providing a structural model for dendrimer–lectin interactions (PDB 5D2A). Biofilm inhibition was increased by introducing additional cationic residues in these dendrimers but resulted in bactericidal effects similar to those of non-glycosylated polycationic antimicrobial peptide dendrimers. In a second approach dendrimers displaying four copies of the natural LecB ligand Lewisa were prepared leading to slightly stronger LecB binding and biofilm inhibition. Finally synergistic application of a LecB specific non-bactericidal antibiofilm dendrimer with the antibiotic tobramycin at sub-inhibitory concentrations of both compounds allowed effective biofilm inhibition and dispersal.
Introduction
The appearance of widespread antibiotic resistance in pathogenic bacteria is a global public health threat. One approach to overcome antibiotic resistance focuses on preventing the formation of biofilms, by which bacteria escape antibiotics by forming a physical barrier to their penetration. Biofilm inhibitors should not be toxic by themselves to avoid the possibility of an evolutionary pressure towards resistance.1 One potential application of this approach concerns Pseudomonas aeruginosa, a gram negative bacterium causing lethal airway infections in immunocompromised and cystic fibrosis patients by forming antibiotic resistant biofilms.2 It has been shown that two P. aeruginosa lectins, namely LecA binding specifically to galactosides, and LecB binding specifically to fucosides, are implicated in biofilm formation by this bacterium,3–7 as evidenced by the impaired biofilm formation in deletion mutants8,9 and the successful treatment of P. aeruginosa infections with lectin-binding saccharide solutions.10–12Recently we reported glycopeptide dendrimers targeting these lectins and capable of both biofilm inhibition and dispersal of already established biofilms. The first dendrimer FD2, a tetravalent fucosylated dendrimer with sequence (fucose-α-CH2CO-Lys-Pro-Leu)4(Lys-Phe-Lys-Ile)2Lys-His-Ile-NH2 (Lys = branching lysine residue), was initially identified in a glycopeptide dendrimer combinatorial library13,14 by screening for binding to a fucose specific plant lectin,15–18 and showed tight binding to LecB.19–21 Two additional dendrimers were subsequently developed by appending galactosyl groups to the peptide backbone of FD2, yielding tight binding LecA ligands GalAG2 and GalBG2 (Scheme 1).22 These glycopeptide dendrimers belong to the very few LecA or LecB ligands thus far reported to display P. aeruginosa biofilm inhibition properties,23 among a large number of related multivalent lectin ligands.24–30 Structure–activity relationship studies with FD2 and GalAG2 showed that multivalency and the nature of the amino acid sequence were critical for biological activity, however in both cases no significant activity increase was achieved beyond the level of a minimal biofilm inhibitory concentration (MBIC) value of 20 μM, calling for alternative approaches for activity improvement.20,21,31–34
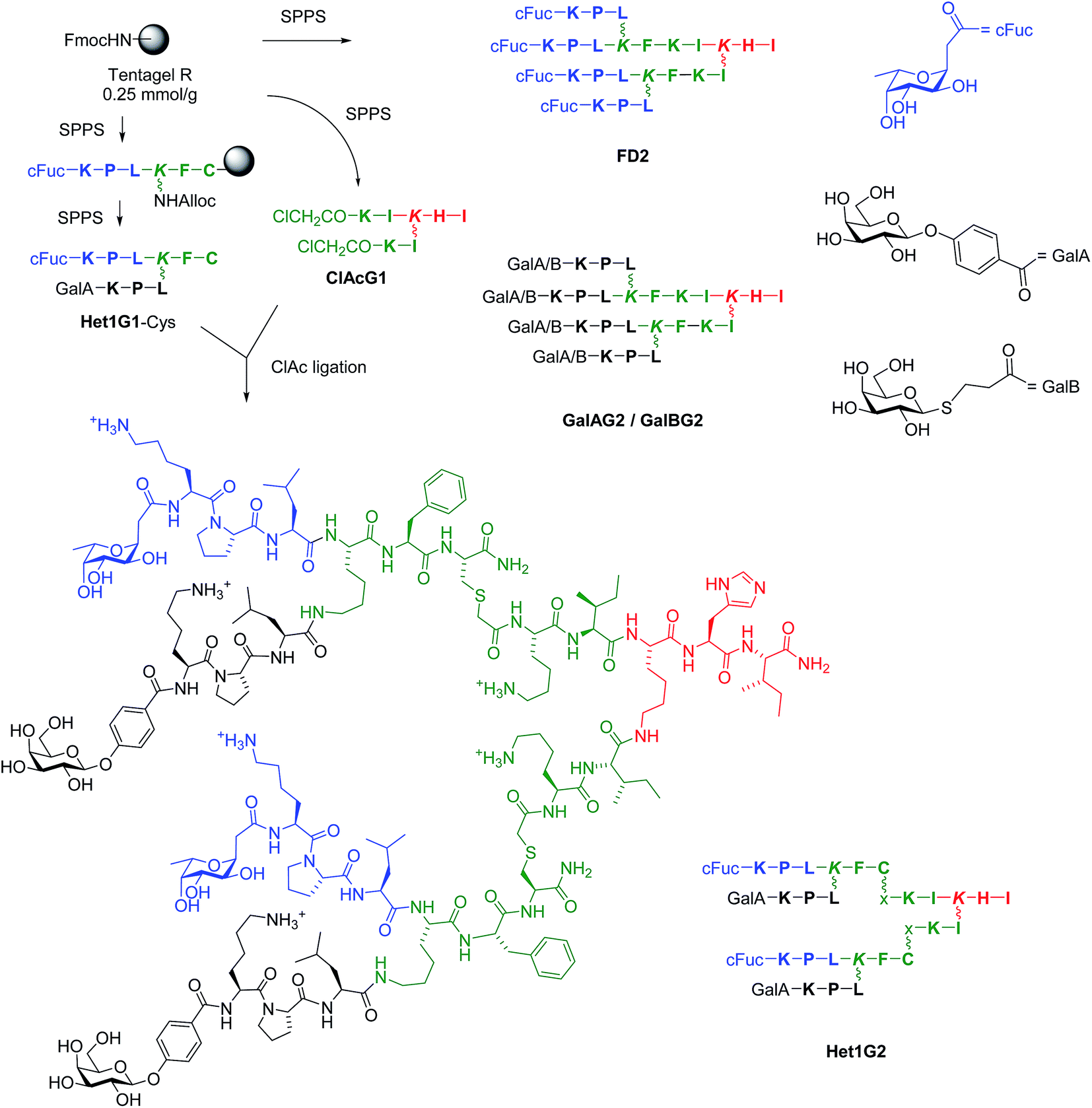 | ||
Scheme 1 Structure and synthesis of glycopeptide dendrimers. Conditions: SPPS: (a) Fmoc deprotection with piperidine/NMP 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 (v/v), 20 min; alloc deprotection: Pd(PPh3)4, PhSiH3, CH2Cl2; amino acid coupling (3 eq. Fmoc-aa-OH, 3 eq. PyBOP, 5 eq. DIEA in NMP), 2–4 hours; carbohydrate coupling: 4 eq. carbohydrate building block, 3 eq. HATU, 5 eq. DIPEA in NMP, overnight; (b) deacetylation: MeOH/25% NH3/H2O (8:1:1, v/v/v); (c) cleavage: TFA/TIS/H2O (95:2.5:2.5, v/v/v) or TFA/TIS/H2O/1,2-ethandithiol (92.5/2.5/2.5/2.5) for Cys containing peptide; (d) RP-HPLC purification, ClAc ligation: ClAcG1 (1 eq.), Het1G1-Cys (3 eq.), KI (20 eq.) DIPEA (55 eq.) in NMP/H2O (1:1, v/v), RT, overnight under Argon atmosphere. One letter codes for L-amino acids, x = –CH2–CO–, wobbly bonds mark the side-chain lysine connectivity. The color codes are used to differentiate G0 (red), G1 (green) and G2 (black/blue) branches. :4 (v/v), 20 min; alloc deprotection: Pd(PPh3)4, PhSiH3, CH2Cl2; amino acid coupling (3 eq. Fmoc-aa-OH, 3 eq. PyBOP, 5 eq. DIEA in NMP), 2–4 hours; carbohydrate coupling: 4 eq. carbohydrate building block, 3 eq. HATU, 5 eq. DIPEA in NMP, overnight; (b) deacetylation: MeOH/25% NH3/H2O (8:1:1, v/v/v); (c) cleavage: TFA/TIS/H2O (95:2.5:2.5, v/v/v) or TFA/TIS/H2O/1,2-ethandithiol (92.5/2.5/2.5/2.5) for Cys containing peptide; (d) RP-HPLC purification, ClAc ligation: ClAcG1 (1 eq.), Het1G1-Cys (3 eq.), KI (20 eq.) DIPEA (55 eq.) in NMP/H2O (1:1, v/v), RT, overnight under Argon atmosphere. One letter codes for L-amino acids, x = –CH2–CO–, wobbly bonds mark the side-chain lysine connectivity. The color codes are used to differentiate G0 (red), G1 (green) and G2 (black/blue) branches. | ||
Herein we report the extension of our search towards more potent biofilm inhibitors through further synthetic variations and activity combination approaches. The multivalent chloroacetyl cysteine thioether (ClAc) ligation35 was used as an efficient method to access heteroglycoclusters such as Het1G2 targeting both LecA and LecB (Scheme 1). One of the heteroglycoclusters provided the first fully resolved crystal structure of a peptide dendrimer as a LecB complex and a structural model for dendrimer–LecB interactions. Heteroglycoclusters incorporating cationic residues showed enhanced biofilm inhibition, however the activity reflected a bactericidal effect similar to that of membrane disrupting polycationic dendrimers.36 In a second approach LecB targeting glycopeptide dendrimers were investigated displaying analogs of the Lewisa antigen, which is the probable natural LecB ligand,37 resulting in stronger LecB binding but only a small increase in biofilm inhibition. Finally synergistic application of the LecB specific non-bactericidal antibiofilm dendrimer FD2 with the antibiotic tobramycin at sub-inhibitory concentrations of both compounds allowed effective biofilm inhibition and dispersal.
Results and discussion
Synthesis and lectin binding of heteroglycoclusters targeting LecA and LecB
Initial biofilm inhibition experiments using the LecB specific dendrimer FD2 and the LecA specific dendrimer GalAG2 simultaneously showed simple additive effects between both dendrimers (see ESI Fig. S109†). Heteroglycocluster analogs of these dendrimers displaying fucosyl and galactosyl groups were prepared to test if a compound targeting both lectins might show enhanced activity compared to the co-application. Due to difficulties in obtaining 8-fold glycosylated G3 peptide dendrimers we focused on the preparation of 4-fold glycosylated G2 peptide dendrimers carrying one pair each of fucosyl and galactosyl groups, which should enable binding to LecB respectively LecA by means of a chelate binding mode on two adjacent carbohydrate binding sites of the same lectin tetramer.33,38The synthesis of G2 glycoclusters was carried out by ClAc ligation between the bis-chloroacetylated G1 dendrimer ClAcG1 and bis-glycosylated G1 peptide dendrimers with a cysteine residue at their core (Scheme 1).35 This convergent synthesis was more economical than the previously used direct SPPS of G2 glycopeptide dendrimers, for which the four-fold glycoside coupling consumed large amounts of carbohydrate building block.34 The method was used to prepare analogs of FD2, GalAG2 and GalBG2, namely FD2x, GalAG2x and GalBG2x, by ligation of the G1 intermediates FD1-Cys, GalAG1-Cys and GalBG1-Cys to the bis-chloroacetylated G1 dendrimer ClAcG1. Heteroglycoclusters were prepared by the same ligation approach using bis-glycosylated G1 dendrimers bearing a different carbohydrate on each branch, which were obtained using an alloc protected lysine branching residue allowing sequential growth of each G1 branch. All combinations of α-L-fucosylacetic acid (cFuc) with 4-(β-D-galactosyloxy)-benzoic acid (GalA) or 3-(β-D-thiogalactosyl)-propionic acid (GalB) on the main chain tripeptide versus side chain tripeptide were realized, yielding intermediates Het1G1-Cys to Het4G1-Cys and G2 heteroglycoclusters Het1G2–Het4G2. The same synthesis was carried out using the tripeptide Lys-Lys-Leu instead of Lys-Pro-Leu in the outer branch of the peptide dendrimer to obtain more polycationic glycoclusters combining lectin binding with a membrane directed antimicrobial effect, yielding G1 intermediates Het5G1–Het8G1 and the G2 ligation products Het5G2–Het8G2 (Table 1).
| Compound | Sequencea | MS calc./obs.b | Yieldc (mg/%) |
|---|---|---|---|
| a One letter code for amino acids. Branching Lys in italics. GalB is carboxpropyl β-thiogalactoside. GalA is 4-carboxyphenyl β-galactoside. cFuc is α-c-fucoside. x = –CH2CO–. The peptide C-terminus is carboxamide –CONH2. See structural formula of Het1G2 in Scheme 1 for correspondence between line notation and structure. b MS calc. for [M + H]+/MS obs. determined by ESI+. c Yields are reported for the RP-HPLC purified product after SPPS or chloroacetyl cysteine (ClAc) ligation. d From ref. 15. e From ref. 22. | |||
| FD0 | cFuc-KPL | 544.34/544.33 | 19.5/29d |
| GalAG0 | GalA-KPL | 638.33/638.2 | 20.9/55e |
| GalBG0 | GalB-KPL | 606.31/606.2 | 11/30e |
| FD1 | (cFuc-KPL)2KFKI | 1586.97/1586.98 | 123.3/63d |
| GalAG1 | (GalA-KPL)2KFKI | 1774.98/1774.8 | 45.7/43e |
| GalBG1 | (GalB-KPL)2KFKI | 1710.93/1710.8 | 39.5/39e |
| FD1-Cys | (cFuc-KPL)2KFC | 1448.81/1447.80 | 33.5/18 |
| GalAG1-Cys | (GalA-KPL)2KFC | 1636.82/1637.82 | 79/39 |
| GalBG1-Cys | (GalB-KPL)2KFC | 1572.77/1573.78 | 29.5/15 |
| Het1G1-Cys | cFuc-KPLK(GalA-KPL)FC | 1542.82/1543.81 | 63/33 |
| Het2G1-Cys | GalA-KPLK(cFuc-KPL)FC | 1542.82/1543.81 | 50/26 |
| Het3G1-Cys | cFuc-KPLK(GalB-KPL)FC | 1510.79/1510.79 | 52/28 |
| Het4G1-Cys | GalB-KPLK(cFuc-KPL)FC | 1510.79/1510.78 | 50/26 |
| Het5G1-Cys | cFuc-KKLK(GalA-KKL)FC | 1604.90/1604.90 | 30/15 |
| Het6G1-Cys | GalA-KKLK(cFuc-KKL)FC | 1604.90/1604.90 | 32.5/16 |
| Het7G1-Cys | cFuc-KKLK(GalB-KKL)FC | 1572.88/1572.88 | 66/34 |
| Het8G1-Cys | GalB-KKLK(cFuc-KKL)FC | 1572.88/1572.87 | 52/26 |
| ClAcG1 | (ClAc-KI)2KHI | 1030.58/1029.57 | 89/70 |
| FD2 | (cFuc-KPL)4(KFKI)2KHI | 3534.16/3534.16 | 31/8d |
| GalAG2 | (GalA-KPL)4(KFKI)2KHI | 3909.17/3911 | 18.2/7e |
| GalBG2 | (GalB-KPL)4(KFKI)2KHI | 3783.07/3783 | 22.1/10e |
| FD2x | (cFuc-KPL)4(KFCxKI)2KHI | 3854.22/3854.23 | 7.9/70 |
| GalAG2x | (GalA-KPL)4(KFCxKI)2KHI | 4230.24/4231.24 | 2.9/24 |
| GalBG2x | (GalB-KPL)4(KFCxKI)2KHI | 4102.15/4103.15 | 3.8/32 |
| Het1G2 | (cFuc-KPLK(GalA-KPL)FCxKI)2KHI | 4042.23/4043.23 | 10.3/88 |
| Het2G2 | (GalA-KPLK(cFuc-KPL)FCxKI)2KHI | 4042.23/4042.23 | 9.3/79 |
| Het3G2 | (cFuc-KPLK(GalB-KPL)FCxKI)2KHI | 3978.19/3979.18 | 3.6/31 |
| Het4G2 | (GalB-KPLK(cFuc-KPL)FCxKI)2KHI | 3978.19/3978.19 | 7.3/63 |
| Het5G2 | (cFuc-KKLK(GalA-KKL)FCxKI)2KHI | 4166.40/4166.40 | 6.6/54 |
| Het6G2 | (GalA-KKLK(cFuc-KKL)FCxKI)2KHI | 4166.40/4166.40 | 7.1/59 |
| Het7G2 | (cFuc-KKLK(GalB-KKL)FCxKI)2KHI | 4102.36/4102.36 | 11.1/69 |
| Het8G2 | (GalB-KKLK(cFuc-KKL)FCxKI)2KHI | 4102.36/4102.36 | 10.3/57 |
The binding affinities of the different compounds to P. aeruginosa lectins LecB and LecA was investigated by isothermal titration calorimetry (ITC). Binding to the fucose specific lectin LecB ranged between KD = 430 nM for the reference carbohydrate methyl-α-L-fucoside39 and KD = 66 nM for the tetravalent dendrimer FD2 (Table 2). At the level of the monovalent ligands, the c-fucosylated tripeptide FD0 was a poorer ligand than methyl-α-L-fucoside, while the monovalent Het1G1-Cys and Het2G1-Cys bound even less tightly and showed a too high n value, which might indicate aggregation via disulfide bond formation and/or intermolecular β-sheets as observed in the crystal structure (see below), possibly hindering lectin access for part of the glycosyl groups. Binding was not influenced by multivalency, as shown by the relative potency of binding per carbohydrate (r.p./nFuc < 2 in all cases), or by additional positive charges in the peptide sequences (Het7G2,Het8G2). Nevertheless the tetravalent fucosides FD2 and FD2x were the tightest LecB ligands in the series.
| Ligand | n Fuc | n | ΔH [kcal mol−1] | −TΔS [kcal mol−1] | ΔG [kcal mol−1] | K D [nM] | r.p./nFucc |
|---|---|---|---|---|---|---|---|
| a Thermodynamic parameters and dissociation constants KD are reported as an average of two independent runs from ITC in 20 mM Tris, 100 mM NaCl, 100 μM CaCl2, pH = 7.5. Isothermal Titration Calorimetry (ITC) data are for LecB binding. Titration concentrations for Ligand/LecB are indicated in brackets: Me-α-fucose (0.44 mM/0.0558 mM), FD0 (0.5 mM/0.044 mM), Het1G1-Cys (0.0457 mM/0.5 mM), Het2G1-Cys (0.0497 mM/0.95 mM), FD1 (0.25 mM/0.0496 mM), FD1-Cys (0.0497 mM/0.5 mM), Het1G2 (0.42 mM/0.042 mM), Het2G2 (0.42 mM/0.042 mM), Het3G2 (0.15 mM/0.042 mM), Het4G2 (0.24 mM/0.042 mM), Het7G2 (0.24 mM/0.042 mM), Het8G2 (0.24 mM/0.042 mM), FD2 (0.1 mM/0.028 mM), FD2x (0.052 mM/0.5 mM). b n defines the stoichiometry of the binding. c Relative potency per fucoside r.p./nFuc = KD(FD0)/KD(ligand)/n, nFuc = number of copies of fucosides in the glycopeptide dendrimers. See ESI Fig. S107 for ITC plots. | |||||||
| FucOCH3 | 1 | 0.77 ± 0.03 | −9.87 ± 0.24 | 1.17 ± 0.24 | −8.70 ± 0.02 | 430 ± 10 | 1.8 |
| FD0 | 1 | 0.80 ± 0.01 | −9.93 ± 0.33 | 0.99 ± 0.05 | −8.34 ± 0.02 | 770 ± 20 | 1 |
| Het1G1-Cys | 1 | 1.64 ± 0.00 | −4.82 ± 0.01 | −2.79 ± 0.03 | −7.61 ± 0.024 | 2630 ± 110 | 0.30 |
| Het2G1-Cys | 1 | 2.14 ± 0.03 | −3.53 ± 0.024 | −4.11 ± 0.02 | −7.63 ± 0.09 | 2570 ± 400 | 0.30 |
| FD1 | 2 | 0.28 ± 0.01 | −23.87 ± 1.21 | 14.43 ± 1.10 | −9.43 ± 0.10 | 123 ± 2 | 3.1 |
| FD1-Cys | 2 | 0.65 ± 0.01 | −10.71 ± 0.01 | 1.81 ± 0.02 | −8.89 ± 0.05 | 300 ± 25 | 1.3 |
| Het1G2 | 2 | 0.40 ± 0.05 | −15.9 ± 0.05 | 6.89 ± 0.30 | −8.94 ± 0.01 | 276 ± 1.5 | 1.4 |
| Het2G2 | 2 | 0.66 ± 0.05 | −15.2 ± 0.02 | 6.28 ± 0.03 | −8.92 ± 0.01 | 292 ± 57 | 1.3 |
| Het3G2 | 2 | 0.31 ± 0.01 | −25.15 ± 2.75 | 15.91 ± 2.82 | −9.23 ± 0.09 | 179 ± 27 | 2.1 |
| Het4G2 | 2 | 0.21 ± 0.01 | −35.25 ± 0.02 | 25.79 ± 0.15 | −9.43 ± 0.03 | 121 ± 5 | 3.2 |
| Het7G2 | 2 | 0.30 ± 0.02 | −25.1 ± 0.02 | 16.14 ± 0.24 | −8.96 ± 0.02 | 327 ± 6 | 1.2 |
| Het8G2 | 2 | 0.28 ± 0.02 | −24.8 ± 0.06 | 15.61 ± 0.07 | −9.17 ± 0.02 | 193 ± 4 | 2.0 |
| FD2 | 4 | 0.16 ± 0.01 | −40.9 ± 1.25 | 30.83 ± 1.7 | −9.80 ± 0.09 | 66 ± 1 | 2.9 |
| FD2x | 4 | 0.59 ± 0.02 | −9.72 ± 0.9 | 0.99 ± 0.9 | −9.62 ± 0.02 | 88 ± 2 | 2.2 |
The range of binding affinities was much broader in the case of the galactose specific lectin LecA ranging from KD = 37 μM for the monovalent galactopeptide GalBG0 down to KD = 2.5 nM for the tetravalent galactoside GalAG2 (Table 3). Binding was first influenced by the nature of the glycosidic group. All compounds carrying an aromatic aglycone (the reference nitrophenyl-β-galactoside NPG and all GalA containing products) showed a 10–20 fold stronger binding than their GalB analogs containing an aliphatic chain as aglycone, reflecting the previously documented favourable CH–π interaction between the aromatic group and the ε-CH of residue His50 on the lectin.32 As for LecB, the monovalent Het1G1-Cys and Het2G1-Cys bound poorly and gave high n values which might be caused by disulfide bond formation and/or aggregation. Furthermore LecA behaved as a typical lectin by showing enhanced binding to multivalent ligands.40 For the aromatic galactosides, the relative potency of binding per galactoside reached r.p./nGal = 20 in divalent galactosides GalAG1, Het1G2 and Het2G2, and r.p./nGal = 300 in tetravalent galactoside GalAG2 showing the strongest binding. However GalAG2x only reached r.p./nGal = 32, showing that the ClAc ligated peptide dendrimer backbone was less optimal for LecA binding compared to the original G2 peptide dendrimer. In the weaker binding aliphatic thiogalactosides of the GalB series divalent dendrimers had comparable relative potencies in the range 18 < r.p./nGal < 75, including the more cationic heteroglycoclusters Het7G2 and Het8G2. The tetravalent dendrimer GalBG2 reached a relative potency of r.p./nGal = 230 corresponding to KD = 40 nM of comparable value to aromatic galactosides.
| Ligand | n Gal | n | ΔH [kcal mol−1] | −TΔS [kcal mol−1] | ΔG [kcal mol−1] | K D [nM] | r.p./nGald |
|---|---|---|---|---|---|---|---|
| a Thermodynamic parameters and dissociation constants KD are reported as an average of two independent runs from ITC in 20 mM Tris, 100 mM NaCl, 100 μM CaCl2, pH = 7.5. Isothermal Titration Calorimetry (ITC) data are for LecA binding. Titration concentrations for Ligand/LecA are indicated in brackets: NPG (3.0 mM/0.3 mM), GalAG0 (0.5 mM/0.0516 mM), Het1G1-Cys (1.2 mM/0.051 mM), Het2G1-Cys (0.57 mM/0.05 mM), GalAG1 (0.25 mM/0.0486 mM), GalAG1-Cys (0.255 mM/0.05 mM), Het1G2 (0.25 mM/0.05 mM), Het2G2 (0.25 mM/0.05 mM), GalAG2 (0.03 mM/0.018 mM), GalAG2x (0.0909 mM/0.0166 mM), GalBG0 (1.0 mM/0.091 mM), GalBG1 (0.25 mM/0.049 mM), GalBG1-Cys (0.4 mM/0.0526 mM), Het3G2 (0.5 mM/0.1 mM), Het4G2 (0.1 mM/0.05 mM), Het7G2 (0.175 mM/0.05 mM), Het8G2 (0.175 mM/0.05 mM), GalBG2 (0.03 mM/0.08 mM). b n defined the stoichiometry of the binding. c NPG is 4-nitrophenyl-β-D-galactopyranoside (reference used in the ITC measurements with LecA), (3 mM/0.3 mM, Ligand/LecA). d Relative potency per galactoside. r.p./nGal = KD(GalAG0 or GalBG0)/KD(ligand)/nGal. nGal = number of copies of galactosides in the glycopeptide dendrimer. See ESI Fig. S108 for ITC plots. | |||||||
| NPG | 1 | 0.91 ± 0.03 | −10.6 ± 0.45 | 4.0 | 6.5 | 16000 ± 500 |
0.2 |
| GalAG0 | 1 | 0.65 ± 0.02 | −17.8 ± 0.3 | 10.2 | −7.5 | 3000 ± 50 | 1 |
| Het1G1-Cys | 1 | 1.17 ± 0.04 | −7.83 ± 0.26 | 0.85 | −7.0 | 7800 ± 1200 | 0.4 |
| Het2G1-Cys | 1 | 1.37 ± 0.09 | −7.36 ± 0.093 | 0.35 | −7.0 | 7340 ± 300 | 0.4 |
| GalAG1 | 2 | 0.30 ± 0.003 | −29 ± 0.5 | 19.3 | −9.7 | 83 ± 12 | 18 |
| GalAG1-Cys | 2 | 0.66 ± 0.06 | −14.2 ± 0.05 | 4.9 | −9.3 | 164 ± 0.2 | 9 |
| Het1G2 | 2 | 0.44 ± 0.01 | −23.0 ± 0.14 | 13.3 | −9.7 | 75 ± 17 | 20 |
| Het2G2 | 2 | 0.36 ± 0.03 | −25.0 ± 0.15 | 15.2 | −9.7 | 75 ± 18 | 20 |
| GalAG2 | 4 | 0.14 ± 0.01 | −68.6 ± 1.10 | 57 | −11.7 | 2.5 ± 0.1 | 300 |
| GalAG2x | 4 | 0.21 ± 0.02 | −32.4 ± 0.47 | 22 | −10.4 | 23 ± 0.3 | 32 |
| GalBG0 | 1 | 0.71 ± 0.01 | −14 ± 0.04 | 7.8 | −6.0 | 37000 ± 800 |
1 |
| GalBG1 | 2 | 0.37 ± 0.02 | −20 ± 0.1 | 12 | −8.2 | 1060 ± 160 | 18 |
| GalBG1-Cys | 2 | 0.53 ± 0.06 | −13.9 ± 0.82 | 5.4 | −8.5 | 600 ± 1 | 31 |
| Het3G2 | 2 | 0.25 ± 0.02 | −30.3 ± 0.40 | 21 | −9.0 | 255 ± 14 | 73 |
| Het4G2 | 2 | 0.15 ± 0.00 | −42.0 ± 0.21 | 36 | −9.4 | 120 ± 2 | 153 |
| Het7G2 | 2 | 0.25 ± 0.02 | −28.4 ± 0.69 | 20 | −8.8 | 340 ± 70 | 55 |
| Het8G2 | 2 | 0.25 ± 0.00 | −25.5 ± 0.25 | 16.4 | −9.0 | 250 ± 16 | 75 |
| GalBG2 | 4 | 0.18 ± 0.02 | −43 ± 1 | 33 | −10.1 | 40 ± 1 | 230 |
Overall the ITC data showed that G2 heteroglycoclusters achieved a relatively potent binding of both LecB and LecA at levels comparable to those of G1 glycopeptide dendrimers carrying only one type of carbohydrate. Binding affinities remained 2–10 fold weaker than that of the purely fucose or purely galactose containing dendrimers FD2, FD2x, GalAG2, GalAG2x and GalBG2, highlighting that the higher binding affinities of these G2 dendrimers required the tetravalent display of four carbohydrates of the same type.
Crystal structure of peptide dendrimer (Het1G1-Cys)2 as LecB complex
A crystallography screen was undertaken to gain a structural insight into the glycopeptide dendrimer lectin interaction. G2 heteroglycoclusters Het1G2 and Het2G2 and the parent G1 dendrimers Het1G1-Cys and Het2G1-Cys were tested as complexes with either LecB or LecA by direct co-crystallization or by soaking preformed lectin crystals with ligand. A well diffracting crystal was obtained for the Het2G1-Cys·LecB complex, which provided a dataset at 2.13 Å resolution. In this structure each of the four carbohydrate binding sites on each LecB tetramer is occupied by a fucose ligand. Two binding sites are occupied by fucosyl groups attached to a fully visible Het2G1-Cys ligand, whereby this ligand is oxidatively dimerized to a disulfide bridged (Het2G1-Cys)2 peptide dendrimer (Fig. 1A, grey stick model and electron density). The other two LecB binding sites are occupied by fucosyl groups where only the terminal fucosylated tripeptide dendrimer arm is visible (Fig. 1A, cyan stick model). This tripeptide adopts a stretched conformation very similar to that of the fully resolved ligand, and distinct from the more folded conformation previously observed in a LecB complex with the fucosylated tripeptide FD0 (Fig. 1A: red stick model),19 consistent with attachment to the dendrimer. Het2G1-Cys is possibly disordered or not oxidatively dimerized and flexible and therefore not visible at this crystal position. All fucosyl groups adopt the typical position seen in other fucose·LecB complexes, with binding interactions between the C2(OH), C3(OH) and C4(OH) group of fucose and the pair of Ca2+ ions and Asp99 on LecB. A tight (2.3 Å) hydrogen bond between the backbone amide of Asn70 and the phenylalanyl–cysteine amide bond in the fully resolved ligand represents the only visible additional protein dendrimer contact. | ||
| Fig. 1 Crystal structure of dendrimer·LecB complex (PDB 5D2A, see also ESI† for crystal data table). A. Overlay view of a single LecB binding site bound with (i) grey model and electron density: fully resolved (Het2G1-Cys)2 dendrimer; (ii) cyan model: terminal tripeptide at the partially resolved crystal position; (iii) red model: tripeptide FD0, from ref. 19 in each case the peptide backbone is fully visible, but several side chains are disordered and not visible, which is typical for peptides and loop residues in proteins B. Detail of the β-sheet interaction connecting two different (Het2G1-Cys)2 dimers. Dashed lines indicate H-bonded atoms that are 2.7 Å apart. C–F. Side and top view of the supramolecular macrocycle [(Het2G1-Cys)2]3 binding to six LecB tetramers. Each of the three dendrimers and the two LecB tetramers to which it is bound have the same color: orange, red, or grey. | ||
The electron density of (Het2G1-Cys)2 observed at the fully resolved crystal position is remarkable since it features the first example of a complete crystal structure of a G2 peptide dendrimer. A closer analysis shows that this dendrimer bridges binding sites on two different LecB tetramers. Furthermore, each (Het2G1-Cys)2 interacts with two further symmetrically positioned dendrimers via antiparallel β-sheets involving the C-terminal tetrapeptides Leu-Lys-Phe-Cys (Fig. 1B). The arrangement is repeated around the C3 symmetry axis such that three dendrimers form a C3-symmetrical [(Het2G1-Cys)2]3 supramolecular macrocycle with three fucosyl groups pointing above the ring and three fucosyl groups below the ring and connecting six different LecB tetramers (Fig. 1C–F). The six galactosyl groups are located within the supramolecular macrocycle and are somewhat hidden from solution. Although this supramolecular macrocycle is probably specific of the crystal lattice, one can imagine that similar intermolecular β-sheet interactions might occur in solution and cause sterical hindrance around single carbohydrate groups, which could explain the unusually high stoichiometry of binding observed in ITC for some of the dendrimers. The absence of chelate binding mode with any of the LecB tetramers in the present structure might be a general feature of dendrimer LecB interactions, as suggested by the absence of significant multivalency effect with all glycopeptide dendrimers including the tightest binder FD2.
Biofilm inhibition by glycodendrimers and antimicrobial compounds
Biofilm inhibition activities were investigated next. In view of a possible direct antimicrobial effect of Het5G2–Het8G2 incorporating additional cationic residues in their sequences, control non-glycosylated dendrimers were prepared to test the effect of the peptide backbone alone. These were the N-terminally acetylated peptide dendrimer AcG2 and its ClAc ligation analog AcG2x, as well as the more cationic sequence AcG2xK and peptide dendrimer NG2 with four free N-termini, each carrying four additional positive charges (Table 4). Two recently reported polycationic antimicrobial peptide dendrimers with strong activity against P. aeruginosa presumably by a membrane disrupting effect, G3KL and G3LK,36 were also included in the study, as well as the reference antibiotics polymyxin B and tobramycin (Table 5).| Compound | Sequence | MS calc./obs.b | Yieldc (mg/%) |
|---|---|---|---|
| a One letter code for L-amino acids. Lys = branching lysine residue, x = –S–CH2–CO–. Ac = acetyl. C-termini are carboxamides CONH2. b MS calc. for [M + H]+/MS obs. determined by ESI+. c Yields are reported for the RP-HPLC purified products. d From ref. 36. | |||
| AcG2 | (Ac-KPL)4(KFKI)2KHI | 2949.92/2949.92 | 26/7 |
| AcG1-Cys | (Ac-KPL)2KFC | 1156.69/1156.69 | 38/19 |
| AcG1K-Cys | (Ac-KKL)2KFC | 1217.78/1218.78 | 76/50 |
| AcG2x | (Ac-KPL)4(KFCxKI)2KHI | 3269.99/3270.99 | 4.8/50 |
| AcG2xK | (Ac-KKL)4(KFCxKI)2KHI | 3392.43/3393.18 | 5.9/60 |
| NG2 | (KPL)4(KFKI)2KHI | 2781.88/2781.88 | 8/44 |
| G3KL | (KL)(KKL)4(KKL)2KKL | 4534.19/4535.10 | 52.2/8 |
| G3LK | (LK)(KLK)4(KLK)2KLK | 4534.19/4534.84 | 7.8/1 |
| Compound | MBICa[μM] | MBCb[μM] | Dispersalc | MICd |
|---|---|---|---|---|
| a MBIC is minimal biofilm inhibitory concentration. b MBC = minimal bactericidal concentration under biofilm forming conditions, determined by CFU counts in the biofilm supernatant and growth test in full medium. c Effect of the compounds at 50 μM or concentration stated on dispersal of already established biofilms. d MIC = minimal inhibitory concentration in a standard 2-fold dilution assay for antibacterial activity in full LB medium. n.i. = no inhibition in a growth assay using 50 μM compound, nd = not determined. e From ref. 19. f From ref. 36. g From ref. 41. See also ESI Fig. S110–S114 (biofilm inhibition), S116 (biofilm dispersal), S117–119 (MIC data), Table S1 (cell viability data) and Table S2 (control growth curves). | ||||
| FD2 | 20 | >30 | 100% dispersal | n.i.e |
| GalAG2 | 20 | >30 | 50% dispersal | n.i. |
| GalBG2 | 20 | nd | 60% dispersal | nd |
| FD2x | 45 | >45 | ∼35% dispersal | nd |
| GalAG2x | 30 | 30 | 100% dispersal | nd |
| GalBG2x | 30 | >45 | Increase | nd |
| Het1G2 | 30 | >45 | ∼30% dispersal | nd |
| Het2G2 | 30 | >45 | ∼35% dispersal | nd |
| Het3G2 | >45 | >45 | ∼85% dispersal | nd |
| Het4G2 | >45 | >45 | No dispersal | nd |
| (Het2G1-Cys)2 | >45 | nd | No dispersal | nd |
| Het5G2 | 20 | 20 | 100% dispersal | >64 μM |
| Het6G2 | 20 | 20 | 100% dispersal | >64 μM |
| Het7G2 | 13 | 13 | 100% dispersal | >64 μM |
| Het8G2 | 20 | 20 | 100% dispersal | >64 μM |
| AcG2 | 30 | 30 | 100% dispersal | n.i. |
| AcG2x | 30 | 30 | Increase | nd |
| AcG2xK | 13 | 13 | 100% dispersal | >64 μM |
| NG2 | 2.5 | 2.5 | 100% dispersal @ 22 μM | >64 μM |
| G3KL | 4 | 6 | 95% dispersal @ 10 μM | 0.44 μM |
| G3LK | 6 | 9 | 95% dispersal @ 10 μM | 2 μM |
| Polymyxin B | 6 | 6 | ∼70% dispersal @ 10 μM | 2 μg mL−1f |
| Tobramycin | 0.5 | 0.5 | 100% dispersal @ 4 μM | 1 μg mL−1g |
Biofilm inhibition was measured by determining the amount of biofilm formed after 24 h of growth with or without compound at different concentrations. The biofilm was quantified using the WST-8 assay indicating the amount of viable cells as previously described.31 To distinguish between biofilm inhibition and a direct bactericidal effect, the number of viable cells in the supernatant was also determined by CFU (colony forming unit) counts and by a growth test in an inoculated full medium (OD600 measurement after 7–8 h growth). In a third assay, the ability of the compounds to disperse already established biofilms was tested by preincubating the bacteria under biofilm forming conditions for 24 h, followed by addition of the test compound at a fixed concentration, incubation for an additional 24 h, and quantification of the remaining biofilm as above. Finally, biofilm inhibitory compounds were also tested for their direct bactericidal activity under full medium either by determination of the minimal inhibitory concentration (MIC) by standard serial dilution series, or with a measurement of their effect on bacterial growth in full medium.
All dendrimers except the GalB containing heteroglycoclusters Het3G2 and Het4G2 showed substantial biofilm inhibition activity at a level comparable to the reference biofilm inhibitor FD2 (MBIC = 20 μM). Most of these dendrimers were also able to disperse already established biofilms, the most potent compound still being the parent fucosylated dendrimer FD2. Except for GalAG2x all dendrimers derived from the parent peptide sequence of FD2 were not bactericidal at their MBIC concentration, showing that their biofilm inhibition activity was not caused by a simple bactericidal effect. The related disulfide-bridged heterodimer (Het2G1-Cys)2 described above as a LecB complex was not active as a biofilm inhibitor.
Polycationic heteroglycoclusters Het5G2–Het8G2 also showed good activities, however these compounds were toxic at their biofilm inhibitory concentration as evidenced by a comparable minimal bactericidal concentration (MBC) value determined by CFU counts of the supernatant and growth test. The control, non-glycosylated dendrimers AcG2, AcG2x and their analogs AcG2xK and NG2 with an additional four positive charges were also bactericidal at their biofilm inhibition concentration. A similar bactericidal antibiofilm effect was detected with the polycationic antimicrobial peptide dendrimers G3KL and G3LK as well as with antibiotics polymyxin B and tobramycin. All of these bactericidal compounds were furthermore able to disperse already established biofilms, although the effect was not as complete as observed with glycodendrimer FD2, and at least 10-fold higher concentrations were necessary compared to their MIC values determined in full medium, where bacteria grow in suspension (planktonic state). The biofilm inhibition study highlighted the remarkable non-toxic biofilm inhibition and dispersal effect of glycopeptide dendrimers such as FD2 in contrast to the direct bactericidal effect of polycationic dendrimers and antibiotics.
Multivalent Lewisa dendrimers
The above study showed that tetrafucosylated dendrimer FD2 remained the most potent non-toxic biofilm inhibitor among the different glycopeptide dendrimers tested. A second variation study was therefore undertaken focusing on variation of the fucosyl group of this dendrimer. Analogs of FD2 were prepared by the ClAc ligation approach using carbohydrate building blocks appended with a nucleophilic hexylthiol tether. The ligation was performed onto a tetrachloroacetylated dendrimer ClAcD2 derived from FD2 in which the phenylalanine residue in the G1 branch had been removed to compensate for the introduction of the alkylthiol tether in terms of hydrophobicity and overall arm length of the dendrimers. This approach did not require excess of non-peptidic reagents and therefore allowed to consider more complex carbohydrates. We focused on preparing a tetravalent peptide dendrimer version of the Lewisa trisaccharide, which is the tightest monovalent binder known for LecB and its probable natural ligand.37To obtain the desired Lea trisaccharide, L-fucosyl donor 1 and the selectively protected disaccharide 2 with a single free hydroxyl group at position C(4) of its glucosamine component were synthesized by published procedures,42,43 and coupled by glycosylation to give a protected Lewisa trisaccharide. Removal of benzyl ethers and azide reduction by catalytic hydrogenation, acetylation, desilylation of the anomeric hydroxyl group, formation of the trichloroacetimidate and Schmidt glycosydation of 6-hydroxyhexyl-thioacetate then gave the protected thiol tethered Lewisa trisaccharide 3. Finally, deacetylation and ligation with dendrimer ClAcD2 gave the desired G2 dendrimer LeaC6G2. The tethered protected β-fucoside 5 was similarly obtained from intermediate 4.43 Deacetylation and ligation with ClAcD2 as above gave G2 dendrimer FucC6G2 (Scheme 2, Table 6).
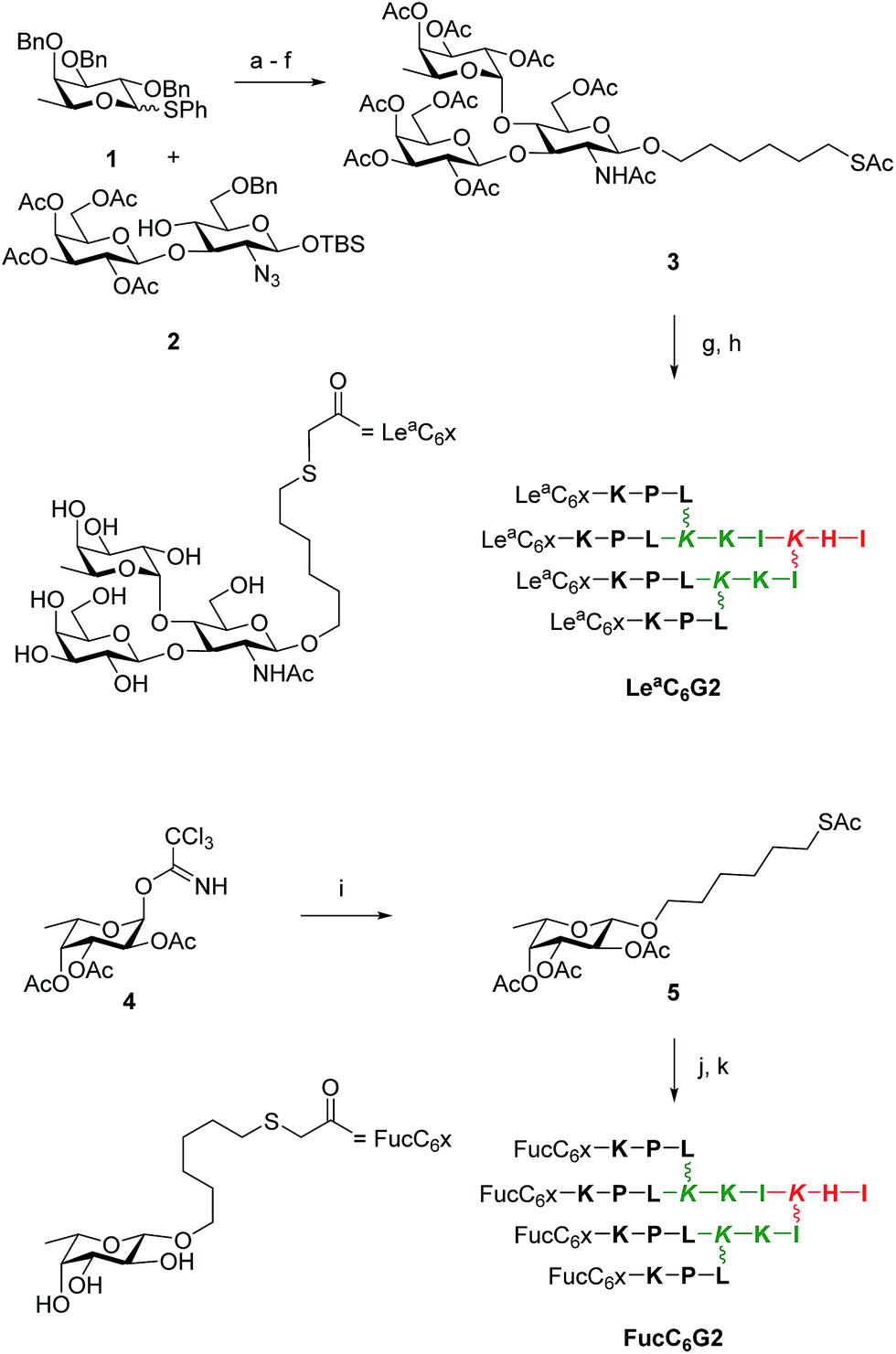 | ||
| Scheme 2 Synthesis of LeaC6G2 and FucC6G2. Conditions: (a) N-Iodosuccinimide, AgOTf, CH2Cl2, 0 °C, 30 min (quant.); (b) H2, Pd(OH)2, THF, AcOH, 17 h, 25 °C; (c) pyridine/Ac2O, cat. DMAP/NMM, CH2Cl2, 25 °C, 4 h (55% over 2 steps); (d) TBAF, AcOH, 25 °C, 17 h (60%); (e) Cl3CCN, DBU, CH2Cl2, 0 °C, 40 min (80%); (f) AcS(CH2)6OH, TMSOTf, CH2Cl2, 0 °C, 20 min (64%); (g) K2CO3, MeOH, 25 °C, 1 h (75%); (h) ClAcD2, KI, DIPEA, H2O/CH3CN, 25 °C, 17 h, then prep. HPLC (63%); (i) step f (84%); (j) step g (75%); (k) step h (31%). See ESI† for synthesis details. | ||
| Compound | Sequence | MS calc./obs.a | Yield (mg/%) |
|---|---|---|---|
| a One letter code for amino acids. Branching Lys in italics. The peptide C-terminus is carboxamide CONH2. aMS calc. for [M + H]+/MS obs. determined by ESI+. Yield is reported for the RP-HPLC purified product after bSPPS or cClAc ligation. | |||
| ClAcD2 | (ClCH2CO–KPL)4(KKI)2KHI | 2791.64/2791.64 | 43/17b |
| FucC6G2 | (Fuc–O–(CH2)6–S–CH2CO–KPL)4(KKI)2KHI | 3768.27/3768.27 | 2.5/31c |
| LeaC6G2 | (Lea–O–(CH2)6–S–CH2CO–KPL)4(KKI)2KHI | 5228.80/5228.80 | 8/63c |
The tetravalent dendrimers LeaC6G2 and FucC6G2 showed the expected binding affinities to LecB as measured by ITC, with simply additive but no multivalency effect on binding compared to their parent monovalent ligand. Nevertheless the slightly tighter LecB binding of Lewisa compared to simple fucosyl groups and a slightly more than additive effect on LecB binding in the dendrimer resulted in KD = 39 nM for LeaC6G2, such that this first multivalent version of the Lewisa antigen represents one of the tightest LecB ligands reported thus far (Table 7).
| Ligand | n Fuc | n | ΔH [kcal mol−1] | −TΔS [kcal mol−1] | ΔG [kcal mol−1] | K D[nM] | r.p./nsugar |
|---|---|---|---|---|---|---|---|
| a Thermodynamic parameters and dissociation constants KD are reported as an average of two independent runs from ITC in 20 mM Tris, 100 mM NaCl, 100 μM CaCl2, pH = 7.5. Isothermal Titration Calorimetry (ITC) data are for LecB binding. Titration concentrations for Ligand/LecB are indicated in brackets: Lea (0.324 mM/0.0327), FucC6G2 (0.3 mM/0.08 mM), LeaC6G2 (0.07 mM/0.042 mM). b n defined the stoichiometry of the binding. c Relative potency per fucoside. r.p./nFuc = KD(FucOCH3)/KD(ligand)/nFuc. See ESI Fig. S107 for ITC plots. | |||||||
| FucOCH3 | 1 | 0.77 ± 0.03 | −9.87 ± 0.24 | 1.17 ± 0.24 | −8.70 ± 0.02 | 430 ± 10 | 1 |
| Lea | 1 | 1.08 ± 0.01 | −8.35 ± 0.02 | −0.74 ± 0.002 | −9.10 ± 0.02 | 213 ± 2.0 | 2 |
| FucC6G2 | 4 | 0.22 ± 0.01 | −14.7 ± 0.38 | 5.20 ± 0.28 | −9.44 ± 0.09 | 121 ± 20 | 0.9c |
| LeaC6G2 | 4 | 0.12 ± 0.01 | −58.5 ± 1.17 | 48.5 ± 1.65 | −10.1 ± 0.06 | 39 ± 0.8 | 2.8c |
Both compounds showed biofilm inhibition and dispersal activities in the same range as the parent FD2, with a slightly stronger biofilm inhibition in the case of FucC6G2 with MBIC = 9 μM. In both cases the compounds were not bactericidal, confirming the absence of toxicity for this type of glycosylated peptide dendrimer biofilm inhibitors. Considering its relatively straightforward and efficient synthesis, FucC6G2 stood out as an improved version of biofilm inhibitor FD2 (Table 8).
Synergistic effects between glycopeptide dendrimer biofilm inhibitors and tobramycin
The MBIC value of 9 μM reached with FucC6G2, although the best value among non-toxic glycopeptide dendrimers, still represented an activity of only 34 μg mL−1 due to the high molecular weight of the compound (MW = 3.7 kDa), implying that further improvement were necessary to reach a practically useful compound. To search for further activity improvements, possible synergistic effects between our glycopeptide dendrimers and antimicrobial compounds were investigated. Synergistic effects between different antibiotics are well known, and combination therapies with multiple antibiotics are commonly used in the clinic.44–47| Ligand | MBICa [μM] | MBCb [μM] | Dispersal | MICc |
|---|---|---|---|---|
| a MBIC is minimal biofilm inhibitory concentration. b MBC = minimal bactericidal concentration under biofilm forming conditions, determined by CFU counts in the biofilm supernatant. c MIC = minimal inhibitory concentration in a standard 2-fold dilution assay for antibacterial activity in full LB medium. n.i. = no inhibition in a growth assay using 50 μM compound, nd = not determined. d From ref. 19. See also ESI Fig. S115/S116 (biofilm data), S120 (MIC data), and Table S1 (cell viability data). | ||||
| FD2 | 20 | >30 | 100% dispersal at 50 μM | n.i.d |
| FucC6G2 | 9 | >20 | 100% dispersal at 30 μM | >64 μM |
| LeaxG2 | 30 | nd | ∼88% dispersal at 50 μM | nd |
Here we focused on synergistic effect with tobramycin, a classical aminoglycoside antibiotic targeting the ribosome, against which P. aeruginosa readily acquires resistance. Indeed synergistic effects have been previously reported between tobramycin and iron chelators, which act as P. aeruginosa biofilm inhibitors in airway epithelial cystic fibrosis cells.48,49 A survey with various glycopeptide dendrimers showed that co-application of FD2 and tobramycin effected a synergistic biofilm inhibition effect, with both compounds acting at sub-inhibitory concentration (Fig. 2A), allowing to use only 5 μM of FD2, which is 4-fold lower than its MBIC value, and 0.1 μM tobramycin, which is 5-fold lower than its MBIC value, thus achieving a remarkable synergistic effect (Table 9). A similar co-application of subinhibitory concentrations of tobramycin with FucC6G2 also acted as toxic biofilm inhibitors, however without decreasing the MBIC of FucC6G2 itself (Fig. 2E). In terms of biofilm dispersal application of subinhibitory concentrations of tobramycin also lowered the dispersal concentration of FD2 by 4-fold (Fig. 2B) but not that of FucC6G2 (Fig. 2F). The effects can be interpreted in terms of triggering a non-toxic dispersal of the biofilm by the glycopeptide dendrimers, allowing facilitated entry of the toxic tobramycin into the cells.
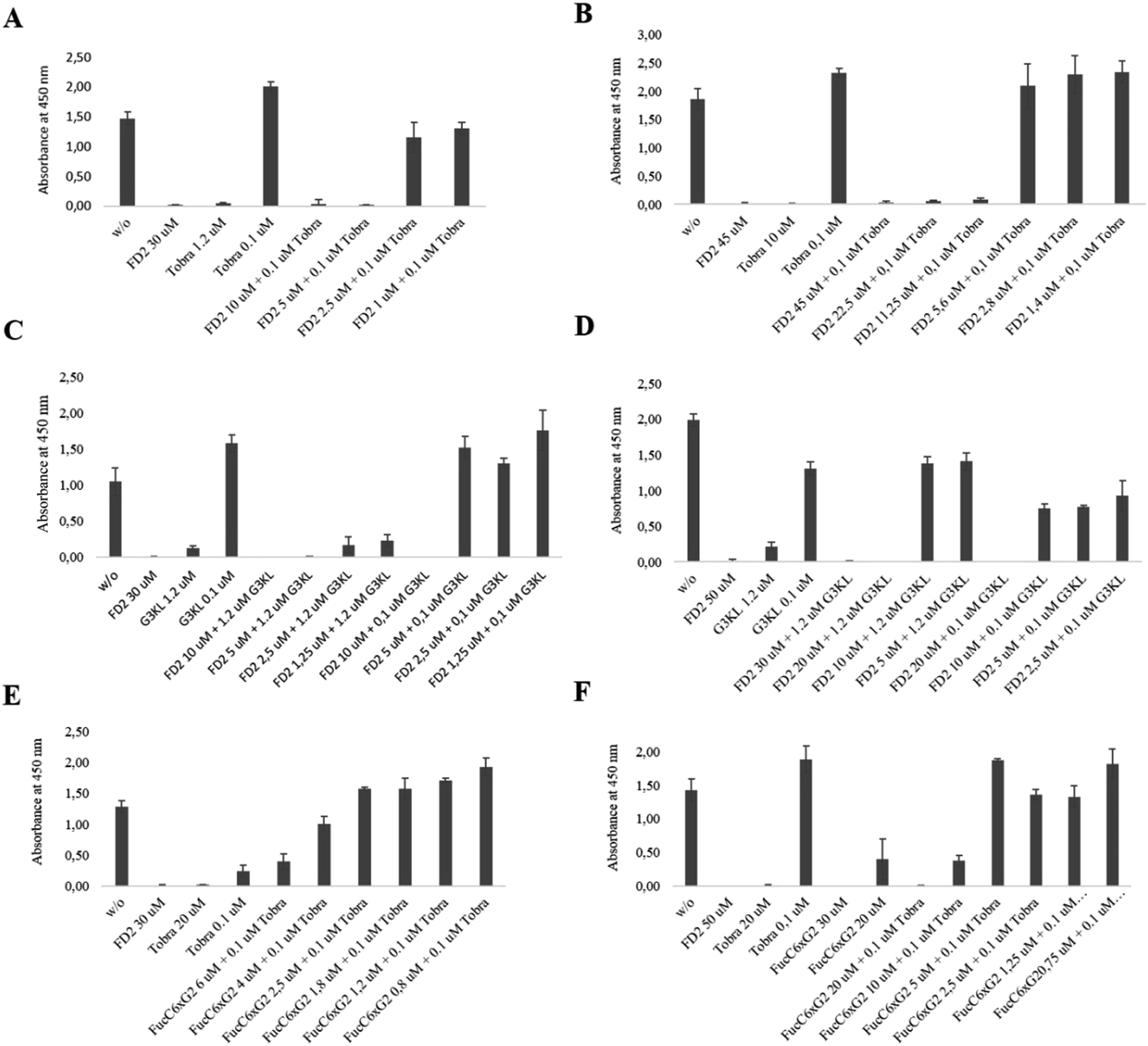 | ||
| Fig. 2 Inhibition and dispersal of Pseudomonas aeruginosa strain PA01 biofilms by synergy. (A) Biofilm inhibition with FD2/Tobramycin. (B) Biofilm dispersal with FD2/Tobramycin. (C) Biofilm inhibition with FD2/G3KL. (D) Biofilm dispersal with FD2/G3KL. (E) Biofilm inhibition with FucC6G2/Tobramycin. (F) Biofilm dispersal with FucC6G2/Tobramycin. For inhibition, biofilms were grown on microtiter plates for 24 hours at 37 °C in the presence of the indicated compounds, and viable biofilms were stained with WST-8/PE. For dispersal, biofilms were first grown on microtiter plates for 24 hours at 37 °C in the absence of any compounds, planktonic bacteria were removed, and the biofilms were incubated with compounds for another 24 hours. Viable biofilms were stained with WST-8/PES. See also Table 9. | ||
A similar synergistic effect was obtained by combining FD2 with the antimicrobial peptide dendrimer G3KL lowering of the MBIC value of each compound by 4 to 5-fold. Most strikingly, even a 0.1 μM application of G3KL induced cytotoxic biofilm inhibition at only 10 μM FD2 (Fig. 2C), and dispersal at only 20 μM (Fig. 2D). As for tobramycin, the biofilm disrupting effect of FD2 probably facilitates access of G3KL to the bacteria. Overall these experiments suggest that the non-toxic biofilm inhibitor FD2 might be useful to increase the sensitivity of P. aeruginosa to standard antibiotics.
Conclusion
In summary an extensive search for improved analogs of our previously reported glycopeptide dendrimer inhibitors of P. aeruginosa biofilms FD2 and GalAG2 was carried out exploiting the multivalent chloroacetyl cysteine thioether (ClAc) ligation. G2 heteroglycoclusters displaying a pair of fucosyl groups targeting the fucose specific P. aeruginosa lectin LecB and a pair of galactosyl groups targeting the galactose specific lectin LecA achieved binding to both lectins at the level of G1 peptide dendrimers. One of the heteroglycoclusters provided the first fully resolved crystal structure of a peptide dendrimer as a LecB complex, revealing a supramolecular arrangement of three dendrimers connected via β-sheets and cross-linking six different LecB tetramers without chelate binding, consistent with the absence of multivalency effects by multivalent ligands of this lectin. Hetereoglycoclusters Het1G2–Het4G2 inhibited P. aeruginosa biofilms at levels comparable to the parent glycodendrimers FD2 and GalAG2. Analogs Het5G2–Het8G2 with additional positive charges showed stronger biofilm inhibition and dispersal, however the gain in activity reflected a direct bactericidal effect of the dendrimers comparable to other polycationic dendrimers such as the antimicrobial peptide dendrimer G3KL and the control, non-glycosylated dendrimer backbone NG2. In a second approach G2 glycopeptide dendrimers displaying four copies of the Lewisa antigen, which is the natural LecB ligand, yielded slightly stronger LecB ligands and biofilm inhibitors. Finally a search for synergistic effects between non-toxic glycopeptide dendrimer biofilm inhibitors and classical antibiotics revealed that biofilm inhibition and dispersal could be achieved by co-application of dendrimer FD2 and the antibiotic tobramycin at sub-inhibitory concentrations of both compounds. A similar synergistic effect was observed by combining FD2 with the antimicrobial peptide dendrimer G3KL, allowing to inhibit biofilm with both compound significantly below their MBIC value. The synergistic use of non-toxic glycopeptide dendrimer biofilm inhibitors to restore sensitivity to antibiotics might offer new opportunities to address antibiotic resistance in P. aeruginosa.Materials and methods
Synthesis
Amino acids were used as the following derivatives: Fmoc-His(Trt)-OH, Fmoc-Leu-OH, Fmoc-Phe-OH, Fmoc-Ile-OH, Fmoc-Pro-OH, Fmoc-Lys(Boc)-OH, Fmoc-Lys(Fmoc)-OH, Fmoc-Cys(Trt)-OH, Fmoc-Ser(tBu)-OH. Chemicals were used as supplied and solvents were of analytical grade. Analytical RP-UHPLC was performed in Dionex ULTIMATE 3000 RS chromatography system (ULTIMATE 3000 RS photo diode array detector) using a Dionex Acclaim® RSLC 120 C18, 3.0 × 50 mm, particle size 2.2 μm, 120 Å pore size, flow rate 1.2 mL min−1 column. Eluent A contained water with 0.1% TFA; eluent D contained acetonitrile and water (90:10) with 0.1% TFA. Compounds were detected by UV absorption at 214 nm. Preparative RP-HPLC was performed with a Waters PrepLC4000 chromatography system using a Dr Maish GmbH Reprospher Column (C18-DE, 5 μm, 100 × 30 mm, pore size 100 Å, flow rate of 40 mL min−1); compounds were detected by UV absorption at 214 nm using a Waters 486 Tunable Absorbance detector or with Waters automatic PrepLC with the four following modules: Waters2489 UV/Vis detector, Waters2545 pump, Waters Fraction Collector III and Waters 2707 Autosampler. For the automatic system, data recording and processing was performed with Waters ChromScope version 1.40 from Waters Corporation. All RP-HPLC were using HPLC-grade acetonitrile and Milli-Q deionized water. The elution solutions were: (A) MilliQ deionized water containing 0.1% TFA; (B) MilliQ deinoized water/acetonitrile (50:50, v/v); (D) MilliQ deionized water/acetonitrile (10:90, v/v) containing 0.1% TFA. MS spectra were provided by the Service of Mass Spectrometry of the Department of Chemistry and Biochemistry, University of Bern. See ESI† for images of analytical HPLC and MS spectra of all compounds.
Solid-phase peptide synthesis
TentaGel S RAM resin (500 mg, loading: 0.22–0.26 mmol g−1) was loaded in a 10 mL polypropylene syringe fitted with a polypropylene frit, a Teflon stopcock, and a stopper. The resin was swollen in DCM (6 mL, 20 min). After removal of the DCM, the Fmoc-protecting group of the resin was removed using a solution of 20% piperidine in NMP. Stirring of the reaction mixture was performed by attaching the closed syringes to a rotating axis. The completion of the reaction and the removal of the Fmoc protecting group were checked using the TNBS test (1 drop of 1% TNBS in DMF and 1 drop 10% DIPEA in DMF). Orange or red beads indicated successful deprotection, colorless beads indicated a complete coupling reaction. In case of proline, the chloranil-test (1 drop of 2% chloranil in DMF and 1 drop 2% acetaldehyde in DMF) for secondary amines was used. In this case, violet or dark green beads indicated successful deprotection as colorless beads indicated complete coupling reaction. Removal of the Fmoc protecting group was performed by using a solution of 20% piperidine in NMP (6 mL, 20 min). After filtration, the resin was washed with NMP (2 × 6 mL), MeOH (2 × 6 mL) and DCM (2 × 6 mL).Coupling of the Fmoc-protected amino acids was performed using Fmoc-protected amino acids (3 eq.), PyBOP (3 eq.) and DIPEA (6 eq.) in NMP (6 mL). The reaction times for linear peptides were 1 hour, for G1 dendrimers 1.5 hours and for G2 dendrimers 2 hours. The resin was then washed with NMP (2 × 6 mL), MeOH (2 × 6 mL) and DCM (2 × 6 mL). Capping of free amine groups was performed after each amino acid coupling using a solution of acetic anhydride:dichloromethane (1:1, v/v).
For allyl/alloc deprotection, the polypropylene syringe was equipped with a septum and dried under high vacuum for one hour. Then it was swollen in dry DCM for 10 min under argon. After removal of the solvent, Pd(PPh3)4 (0.15 eq.) and PhSiH3 (15 eq.) were diluted in 4 mL of dry DCM and added to the resin under argon. The reaction was stirred under argon bubbling for 20 min. The reagents were then removed by filtration and the resin washed with dry DCM. The alloc deprotection procedure was repeated for another 20 min. Finally, the resin was washed with sodium diethylcarbamate solution (0.02 M in DMF) and then with DCM (8 mL, 2 × 10 min).
N-Chloroacetylation of the resin was performed with a solution of chloracetic acid anhydride (10 eq. per free N-terminus) in NMP (5 mL) for 2 × 10 min. The resin was then washed with NMP (2 × 6 mL), MeOH (2 × 6 mL) and DCM (2 × 6 mL).
Carbohydrate coupling was performed by using the protected peracetylated sugar derivative (4 eq. per N-terminus for GalA and GalB, 5 eq. per N-terminus for c-fucoside), HATU (3 eq. per arm) and DIPEA (5 eq. per arm) in NMP (6 mL). The resin was stirred overnight. The resin was washed with NMP (2 × 6 mL), MeOH (2 × 6 mL) and DCM (2 × 6 mL). The deprotection of the carbohydrates was performed by basic methanolysis with a solution of MeOH/25% NH3/H2O (8:1:1, v/v/v). The resin was washed with NMP (2 × 6 mL), MeOH (2 × 6 mL) and DCM (2 × 6 mL) and dried.
Acetylation of the N-termini was performed by using acetic anhydride:dichloromethane (1:1, v/v) for 20 min. The resin was washed with NMP (2 × 6 mL), MeOH (2 × 6 mL) and DCM (2 × 6 mL).
TFA cleavage was performed by adding a solution of TFA/TIS/H2O (95:2.5:2.5, v/v/v) to the resin for 2.5–3 hours. Peptide dendrimers containing Cys residues were cleaved with TFA/TIS/H2O/1,2-ethanedithiol (92.5:2.5:2.5:2.5, v/v/v/v). The peptide dendrimers were then precipitated with ice-cold TBME and removed from TBME by centrifugation. The crude product was dried at high-vacuum to remove residual TFA and TBME.
For purification, the crude was dissolved in MilliQ deionized water containing 0.1% TFA or in a mixture of MilliQ deionized water and acetonitrile containing 0.1% TFA. The purification was performed by HPLC. The obtained products were analysed by analytical RP-UHPLC, ESI-MS and amino acid analysis.
Chloroacetyl cysteine (ClAc) ligation
A solution of core dendrimer (sequence containing chloroacetyl endgroups, around 3 mg, 1 eq.) and KI (20 eq.) in MeCN/H2O (1:1, v/v) (300 μL) was prepared in a 2 mL glass vial. The mixture was flushed with Ar during 10 min. In another 2 mL glass vial arm dendrimer (Cys containing sequence, 1.5 eq. per chloroacetyl endgroup in core sequence) was weighed and flushed with Ar during 10 min. The core dendrimer/KI solution was transferred to the glass vial containing the arm dendrimer via a gas-tight syringe. DIPEA (55 eq.) was added and the solution stirred overnight at room temperature under argon atmosphere. The reaction was followed by analytical RP-UHPLC. After 16 to 23 hours, the reaction was quenched by the addition of 3.5 mL of eluent A (MilliQ deionized water containing 0.1% TFA). After filtration, the solution was directly purified by preparative RP-HPLC. x denotes the –CH2–CO– bridge between the Cys side chain of the arm and the N-termini of the dendritic cores. Yields are given for the chloroacetyl cysteine (ClAc) ligation reaction.
Chloroacetyl cysteine (ClAc) ligation of FucC6 and Lea
A solution of core dendrimer (sequence containing chloroacetyl endgroups), around 3 mg (1 eq.) and KI (20 eq.) in 130 μL of H2O/MeCN (1:1, v/v) was prepared in a 5 mL pointed round-bottom flask. The solution was degassed under high vacuum for 10 min and pressurized under Ar. In a second 5 mL pointed round-bottom flask, peracetylated 6-mercaptohexyl-α-fucoside or β-mercaptohexyl-Lewis A (10 eq.) and K2CO3 (0.3 eq.) were loaded, degassed under high vacuum for 10 min, pressurized under Ar, and dissolved in 200 μL of dry MeOH. The reaction mixture was stirred for 1 hour at RT under an argon atmosphere, which effected complete deacetylation. The previously prepared solution of the core dendrimer and DIPEA (55 eq.) was then added to the reaction mixture which was stirred for 16 to 23 hours at RT under an argon atmosphere. The reaction was followed by RP-UHPLC. After completion the reaction was quenched by adding 10 mL of eluent A (MilliQ deionized water containing 0.1% TFA) and directly purified by RP-HPLC. Yields are given for the chloroacetyl cysteine (ClAc) ligation reaction.
Homodimerisation of Het2G1-Cys
G1 dendrimer Het2G1-Cys (4.5 mg, 1.0 eq.) was dissolved in 200 μL of degassed mQ-deionized water and flushed for 5 min with argon. Then, Aldrithiol (2,2-dithiodipyridine) (45.5 mM in MeOH, 0.45 eq.) was added. The thiol activation was followed by analytical RP-HPLC. When the reaction was completed, MeOH was evaporated and excess Aldrithiol was removed by extraction with DCM (4 times, 300 μL). The water phase was transferred to a new 5 mL pointed round-bottomed flask and flushed with Ar for 5 min. The pH was then adjusted to 8–9 with (NH4)HCO3 buffer solution (400 mM). A second portion of the monomeric dendrimer Het2G1-Cys (4.5 mg, 1.0 eq.) was added to the reaction mixture. The reaction mixture was stirred for 18 hours under argon at room temperature and followed by analytical RP-HPLC. After completion of the reaction, the mixture was acidified with 3 mL of water containing 0.1% of TFA. The homodimer (Het2G1-Cys)2 was purified by preparative RP-HPLC.Isothermal titration calorimetry (ITC)
Recombinant PA-IIL was purified from Escherichia coli BL21(DE3) containing the plasmid pET25pa2l as described previously.50 Lyophilised LecB was dissolved in buffer (20 mM Tris, 100 mM NaCl, 100 μM CaCl2, pH = 7.5). Protein concentration was checked and adjusted by measurement of absorbance at 280 nm using 1 Abs = 0.576 mg mL−1 (MW = 11863 g mol−1) using a NanoDrop instrument. Ligands were dissolved using the same buffer. ITC were performed with iTC200 calorimeter (MicroCal Inc.). Titration was performed on 0.028–0.50 mM LecB in the 200 μL sample cell using 2 μL injections of ligand every 180 s at 25 °C. The data were fitted with MicroCal Origin 8 software, according to standard procedures using a single-site model. Change in free energy ΔG was calculated from the equation: ΔG = ΔH − TΔS where T is the absolute temperature, ΔH and ΔS are the change in enthalpy and entropy, respectively. Two independent titrations were performed for each ligand tested. Effective concentration has been determined using α-Me-fucose, a high affinity ligand that gives precise determination of the stoichiometry in active sites. The effective concentration that has been determined for this batch of LecB was of 70% (of mass concentration). The concentrations of LecB were then corrected based on this value.
Recombinant PA-IL was purified from Escherichia coli BL21(DE3) containing the plasmid pET25paIL as described previously.51 Lyophilised LecA was dissolved in buffer (Tris 20 mM, NaCl 100 mM, CaCl2 100 μM, pH = 7.5). Protein concentration was checked and adjusted by measurement of absorbance at 280 nm using 1 Abs = 2.116 mg mL−1 (MW = 12893 g mol−1) using a NanoDrop instrument. Ligands were dissolved using the same buffer. ITC were performed with iTC200 calorimeter (MicroCal Inc.). Titration was performed on 0.0166–0.526 mM LecA in the 200 μL sample cell using 2 μL injections of 200 μM ligand every 120 s at 25 °C. The data were fitted with MicroCal Origin 8 software, according to standard procedures using a single-site model. Change in free energy ΔG was calculated from the equation: ΔG = ΔH − TΔS where T is the absolute temperature, ΔH and ΔS are the change in enthalpy and entropy, respectively. Two independent titrations were performed for each ligand tested.
Biofilm inhibition and dispersal
A modified version of the method described by Diggle et al.9 was employed.31 96-well sterile, U-bottomed polystyrene microtiter plates (TPP Switzerland) were prepared by adding 200 μL of sterile deionized water to the peripheral wells to decrease evaporation from test wells. Aliquots of 180 μL of culture medium (0.25% (w/v) nutrient broth no. 2, Oxoid) containing desired concentration of the test compound were added to the internal wells. Compound containing solutions were sterile filtered (pore size 0.22 μm) prior to addition to the wells. Inoculum of Pseudomonas aeruginosa strain PAO1 was prepared from 5 mL overnight culture grown in LB broth at 37 °C and 180 rpm shaking. Aliquots of 20 μL of overnight cultures, pre-washed in 0.25% (w/v) nutrient broth and normalised to an OD600 of 1, were inoculated into the test wells. Plates were incubated in a humid environment for 24–25 hours at 37 °C under static conditions. Wells were washed twice with 200 μL sterile deionized water before staining with 200 μL 0.25% (w/v) nutrient broth containing 0.5 mM WST-8 and 20 μM phenazine ethosulfate for 2.5–3 hours at 37 °C under static conditions. Afterwards, the well supernatants were transferred to a polystyrene flat bottomed 96-well plate (TPP Switzerland) and the absorbance was measured at 450 nm with a plate reader (SpectraMax250 from Molecular Devices).For biofilm dispersal, biofilm was being formed as described above but in the absence of compound for 24 hours. Wells were washed twice with 200 μL sterile deionized water before adding 200 μL 0.25% (w/v) nutrient broth containing the desired concentration of compound. Compound containing solutions were sterile filtered (pore size 0.22 μm) prior to addition to the wells. After another 24 hours of incubation at 37 °C under static conditions, the well supernatants were removed and the wells were washed twice with 200 μL sterile deionized water. The biofilm was stained with 200 μL of 0.25% (w/v) nutrient broth containing 0.5 mM WST-8 and 20 μM phenazine ethosulfate for 2.5–3 hours at 37 °C under static conditions. The resulting absorbance was measured as in the biofilm inhibition experiment.
Broth microdilution method
Glycopeptide dendrimers cytotoxicity was assayed against Pseudomonas aeruginosa PAO1. To determine the Minimal Inhibitory Concentration (MIC), broth microdilution method was used. A colony of bacteria from glycerol stock was grown in LB medium overnight at 37 °C and 180 rpm shaking. The compounds were prepared as stock solutions of 64 μM in LB medium, added to the first well of 96-well sterile, F-bottomed polystyrene microtiter plates (TPP, untreated) and diluted serially by 1/2. Compound containing solutions were sterile filtered (pore size 0.22 μm) prior to addition to the wells. The concentration of the bacteria was quantified by measuring absorbance at 600 nm and diluted to an OD600 of 0.022 in LB medium. The sample solutions (150 μL) were mixed with 4 μL diluted bacterial suspension with a final inoculation of about 5 × 105 CFU. For each test, two columns of the plate were kept for sterility control (LB medium only), growth control (LB medium with bacterial inoculum, no compound). The positive control, polymyxin B (starting with a concentration of 64 μg mL−1) in LB medium with bacterial inoculums, was introduced in the two first lines of the plate. The plates were incubated at 37 °C for ∼18 hours under static conditions. 15 μL of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) (1 mg mL−1 in sterilized milliQ deionized water) were added to each well and the plates were incubated for 10 minutes at room temperature. The minimal inhibitory concentration (MIC) was defined as the lowest concentration of the glycopeptide dendrimer that inhibits the visible growth of the tested bacteria (yellow) with the unaided eye. For broth microdilution assay, polymyxin B was used as references.17Quantification of bacterial cell viability in biofilm inhibition and dispersal assays
96-well sterile, U-bottomed polystyrene microtiter plates (TPP Switzerland) were prepared by adding 200 μL of sterile deionized water to the peripheral wells to decrease evaporation from test wells. Aliquots of 180 μL of culture medium (0.25% (w/v) nutrient broth no. 2, Oxoid) containing desired concentration of the test compound were added to the internal wells. Compound containing solutions were sterile filtered (pore size 0.22 μm) prior to addition to the wells. Inoculum of Pseudomonas aeruginosa strain PAO1 was prepared from 5 mL overnight culture grown in LB broth at 37 °C and 180 rpm shaking. Aliquots of 20 μL of overnight cultures, pre-washed in 0.25% (w/v) nutrient broth and normalised to an OD600 of 1, were inoculated into the test wells. Plates were incubated in a humid environment for 24–25 hours at 37 °C under static conditions. The supernatants were used to investigate the bacterial cell viability by two different methods: CFU plating and OD600 measurement.For OD600 measurement, the 200 μL of the supernatant were added to 5 mL of LB medium and shaken at 37 °C for 7–8 hours. The OD600 was measured with an Ultrospec 10 cell density meter from GE Health Care Life Sciences and compared to the w/o supernatant. For CFU plating, the supernatant was added to the first well of a 96-well sterile, F-bottomed microtiter plate (TPP, untreated) and diluted serially by 1/2 (raw 1 to 6) in 180 μL NaCl 0.9%. 4 μL of raw 6 were plated on LB agar and incubated at 37 °C for 24 hours under static conditions. Wells were washed twice with 200 μL sterile deionized water before staining with 200 μL 0.25% (w/v) nutrient broth containing 0.5 mM WST-8 and 20 μM phenazine ethosulfate for 2.5–3 hours at 37 °C under static conditions. Afterwards, the well supernatants were transferred to a polystyrene flat bottomed 96-well plate (TPP Switzerland) and the absorbance was measured at 450 nm with a plate reader (SpectraMax250 from Molecular Devices).
For biofilm dispersal, biofilm was being formed as described above but in the absence of compound for 24 hours. Wells were washed twice with 200 μL sterile deionized water before adding 200 μL 0.25% (w/v) nutrient broth containing the desired concentration of compound. After another 24 hours of incubation at 37 °C under static conditions, the well supernatants were used to check the bacterial cell viability by CFU plating and OD600 measurement. For OD600 measurement, the 200 μL of the supernatant were added to 5 mL of LB medium and shaken at 37 °C for 7–8 hours. The OD600 was measured and compared to the w/o supernatant. For CFU plating, the supernatant was added to the first well of a 96-well sterile, F-bottomed microtiter plate (TPP, untreated) and diluted serially by 1/2 (raw 1 to 6) in 180 μL NaCl 0.9%. 4 μL of the raw 6 were plated on LB agar and incubated at 37 °C for 24 hours under static conditions. The wells were washed twice with 200 μL sterile deionized water. The biofilm was stained with 200 μL of 0.25% (w/v) nutrient broth containing 0.5 mM WST-8 and 20 μM phenazine ethosulfate for 2.5–3 hours at 37 °C under static conditions. The resulting absorbance was measured as in the biofilm inhibition experiment.
Growth curve control studies
180 μL of LB medium were added to wells of a 96-well sterile, F-bottomed polystyrene microtiter plate. Compound containing solutions were sterile filtered (pore size 0.22 μm) prior to addition to the wells. An overnight culture of Pseudomonas aeruginosa strain PA01 grown in LB was standardised to an OD600 of 0.5 and 20 μL aliquots were inoculated into test wells. The microtiter plate was shaken at 37 °C and 180 rpm. Growth was followed at several time points over a period of 24 hours with a plate reader at 600 nm (SpectraMax250 from Molecular Devices).Crystallization of Het1G1·LecB
LecB lectin was expressed, cleaned up and dialyzed as previously described.52 Co-crystallization of Het1G1 with LecB lectin was carried out by the sitting drop method. In brief, lyophilized protein was dissolved in water (5 mg mL−1) in the presence of salts (1 mM CaCl2 and MgCl2). The compound Het1G1 was added to the LecB homo-tetramer at a 16:1 molar excess. Crystals were obtained within five days after mixing 1.5 μL of LecB ligand-complex with 1.5 μL of reservoir solution at 18 °C. Primary crystallization condition was found in Crystal Screens I/II (Hampton Research, Laguna Niguel, CA, USA). Crystals of highest diffraction were obtained from condition 0.1 M sodium acetate trihydrate, 2 M sodium chloride pH 4.6, Crystal Screen II-9. The structures were solved using the XDS,53 CCP4,54 the phenix55 program suite and the coot56 graphical program. Pictures were done with the help of pymol.
Acknowledgements
This work was supported financially by the University of Bern, the Swiss National Science Foundation, and the COST Actions D34 and CM1102 MultiGlycoNano. We thank the staff at the Swiss Light Source, Beamline X06DA (PXIII), Villigen, Switzerland, for support during data collection.References
- M. N. Hurley, M. Camara and A. R. Smyth, Eur. Respir. J., 2012, 40, 1014–1023 CrossRef CAS PubMed.
- V. E. Wagner and B. H. Iglewski, Clin. Rev. Allergy Immunol., 2008, 35, 124–134 CrossRef CAS PubMed.
- N. Garber, U. Guempel, A. Belz, N. Gilboa-Garber and R. J. Doyle, Biochim. Biophys. Acta, 1992, 1116, 331–333 CrossRef CAS.
- G. Cioci, E. P. Mitchell, C. Gautier, M. Wimmerova, D. Sudakevitz, S. Perez, N. Gilboa-Garber and A. Imberty, FEBS Lett., 2003, 555, 297–301 CrossRef CAS PubMed.
- A. Imberty, M. Wimmerova, E. P. Mitchell and N. Gilboa-Garber, Microbes Infect., 2004, 6, 221–228 CrossRef CAS PubMed.
- E. Mitchell, C. Houles, D. Sudakevitz, M. Wimmerova, C. Gautier, S. Perez, A. M. Wu, N. Gilboa-Garber and A. Imberty, Nat. Struct. Biol., 2002, 9, 918–921 CrossRef CAS PubMed.
- R. Loris, D. Tielker, K. E. Jaeger and L. Wyns, J. Mol. Biol., 2003, 331, 861–870 CrossRef CAS PubMed.
- M. Mewe, D. Tielker, R. Schonberg, M. Schachner, K. E. Jaeger and U. Schumacher, J. Laryngol. Otol., 2005, 119, 595–599 Search PubMed.
- S. P. Diggle, R. E. Stacey, C. Dodd, M. Camara, P. Williams and K. Winzer, Environ. Microbiol., 2006, 8, 1095–1104 CrossRef CAS PubMed.
- H. P. Hauber, M. Schulz, A. Pforte, D. Mack, P. Zabel and U. Schumacher, Int. J. Med. Sci., 2008, 5, 371–376 CrossRef CAS PubMed.
- C. Chemani, A. Imberty, S. de Bentzmann, M. Pierre, M. Wimmerova, B. P. Guery and K. Faure, Infect. Immun., 2009, 77, 2065–2075 CrossRef CAS PubMed.
- A. Bernardi, J. Jimenez-Barbero, A. Casnati, C. de Castro, T. Darbre, F. Fieschi, J. Finne, H. Funken, K. E. Jaeger, M. Lahmann, T. K. Lindhorst, M. Marradi, P. Messner, A. Molinaro, P. V. Murphy, C. Nativi, S. Oscarson, S. Penades, F. Peri, R. J. Pieters, O. Renaudet, J. L. Reymond, B. Richichi, J. Rojo, F. Sansone, C. Schaffer, W. B. Turnbull, T. Velasco-Torrijos, S. Vidal, S. Vincent, T. Wennekes, H. Zuilhof and A. Imberty, Chem. Soc. Rev., 2013, 42, 4709–4727 RSC.
- A. Clouet, T. Darbre and J. L. Reymond, Angew. Chem., Int. Ed., 2004, 43, 4612–4615 CrossRef CAS PubMed.
- E. M. V. Johansson, E. Kolomiets, F. Rosenau, K.-E. Jaeger, T. Darbre and J.-L. Reymond, New J. Chem., 2007, 31, 1291–1299 RSC.
- E. Kolomiets, E. M. Johansson, O. Renaudet, T. Darbre and J. L. Reymond, Org. Lett., 2007, 9, 1465–1468 CrossRef CAS PubMed.
- T. Darbre and J. L. Reymond, Curr. Top. Med. Chem., 2008, 8, 1286–1293 CrossRef CAS PubMed.
- J.-L. Reymond and T. Darbre, Org. Biomol. Chem., 2012, 10, 1483–1492 CAS.
- J. L. Reymond, M. Bergmann and T. Darbre, Chem. Soc. Rev., 2013, 42, 4814–4822 RSC.
- E. M. Johansson, S. A. Crusz, E. Kolomiets, L. Buts, R. U. Kadam, M. Cacciarini, K. M. Bartels, S. P. Diggle, M. Camara, P. Williams, R. Loris, C. Nativi, F. Rosenau, K. E. Jaeger, T. Darbre and J. L. Reymond, Chem. Biol., 2008, 15, 1249–1257 CrossRef CAS PubMed.
- E. Kolomiets, M. A. Swiderska, R. U. Kadam, E. M. Johansson, K. E. Jaeger, T. Darbre and J. L. Reymond, ChemMedChem, 2009, 4, 562–569 CrossRef CAS PubMed.
- E. M. V. Johansson, R. U. Kadam, G. Rispoli, S. A. Crusz, K.-M. Bartels, S. P. Diggle, M. Camara, P. Williams, K.-E. Jaeger, T. Darbre and J.-L. Reymond, MedChemComm, 2011, 2, 418–420 RSC.
- R. U. Kadam, M. Bergmann, M. Hurley, D. Garg, M. Cacciarini, M. A. Swiderska, C. Nativi, M. Sattler, A. R. Smyth, P. Williams, M. Camara, A. Stocker, T. Darbre and J. L. Reymond, Angew. Chem., Int. Ed., 2011, 50, 10631–10635 CrossRef CAS PubMed.
- G. M. L. Consoli, G. Granata, V. Cafiso, S. Stefani and C. Geraci, Tetrahedron Lett., 2011, 52, 5831–5834 CrossRef CAS.
- J. J. Lundquist and E. J. Toone, Chem. Rev., 2002, 102, 555–578 CrossRef CAS PubMed.
- C. C. Lee, J. A. MacKay, J. M. J. Frechet and F. C. Szoka, Nat. Biotechnol., 2005, 23, 1517–1526 CrossRef CAS PubMed.
- K. Marotte, C. Preville, C. Sabin, M. Moume-Pymbock, A. Imberty and R. Roy, Org. Biomol. Chem., 2007, 5, 2953–2961 CAS.
- B. Gerland, A. Goudot, G. Pourceau, A. Meyer, S. Vidal, E. Souteyrand, J. J. Vasseur, Y. Chevolot and F. Morvan, J. Org. Chem., 2012, 77, 7620–7626 CrossRef CAS PubMed.
- E. L. Dane, A. E. Ballok, G. A. O'Toole and M. W. Grinstaff, Chem. Sci., 2014, 5, 551–557 RSC.
- S. Zhang, R. O. Moussodia, C. Murzeau, H. J. Sun, M. L. Klein, S. Vertesy, S. Andre, R. Roy, H. J. Gabius and V. Percec, Angew. Chem., Int. Ed., 2015, 54, 4036–4040 CrossRef CAS PubMed.
- S. Cecioni, A. Imberty and S. Vidal, Chem. Rev., 2015, 115, 525–561 CrossRef CAS PubMed.
- R. U. Kadam, M. Bergmann, D. Garg, G. Gabrieli, A. Stocker, T. Darbre and J.-L. Reymond, Chem.–Eur. J., 2013, 18, 17054–17063 CrossRef PubMed.
- R. U. Kadam, D. Garg, J. Schwartz, R. Visini, M. Sattler, A. Stocker, T. Darbre and J. L. Reymond, ACS Chem. Biol., 2013, 8, 1925–1930 CrossRef CAS PubMed.
- R. Visini, X. Jin, M. Bergmann, G. Michaud, F. Pertici, O. Fu, A. Pukin, T. R. Branson, D. M. Thies-Weesie, J. Kemmink, E. Gillon, A. Imberty, A. Stocker, T. Darbre, R. J. Pieters and J. L. Reymond, ACS Chem. Biol., 2015, 10, 2455–2462 CrossRef CAS PubMed.
- M. Bergmann, G. Michaud, R. Visini, X. Jin, E. Gillon, A. Stocker, A. Imberty, T. Darbre and J. L. Reymond, Org. Biomol. Chem., 2015 10.1039/c1035ob01682g.
- N. A. Uhlich, T. Darbre and J. L. Reymond, Org. Biomol. Chem., 2011, 9, 7071–7084 CAS.
- M. Stach, T. N. Siriwardena, T. Kohler, C. van Delden, T. Darbre and J. L. Reymond, Angew. Chem., Int. Ed., 2014, 53, 12827–12831 CrossRef CAS PubMed.
- S. Perret, C. Sabin, C. Dumon, M. Pokorna, C. Gautier, O. Galanina, S. Ilia, N. Bovin, M. Nicaise, M. Desmadril, N. Gilboa-Garber, M. Wimmerova, E. P. Mitchell and A. Imberty, Biochem. J., 2005, 389, 325–332 CrossRef CAS PubMed.
- F. Pertici, N. J. de Mol, J. Kemmink and R. J. Pieters, Chem.–Eur. J., 2013, 19, 16923–16927 CrossRef CAS PubMed.
- C. Sabin, E. P. Mitchell, M. Pokorna, C. Gautier, J. P. Utille, M. Wimmerova and A. Imberty, FEBS Lett., 2006, 580, 982–987 CrossRef CAS PubMed.
- Y. C. Lee and R. T. Lee, Acc. Chem. Res., 1995, 28, 321–327 CrossRef CAS.
- L. R. Hoffman, D. A. D'Argenio, M. J. MacCoss, Z. Zhang, R. A. Jones and S. I. Miller, Nature, 2005, 436, 1171–1175 CrossRef CAS PubMed.
- S. Komba, H. Ishida, M. Kiso and A. Hasegawa, Bioorg. Med. Chem., 1996, 4, 1833–1847 CrossRef CAS PubMed.
- A. Toepfer and R. R. Schmidt, J. Carbohydr. Chem., 1993, 12, 809–822 CrossRef CAS.
- H. Anwar and J. W. Costerton, Antimicrob. Agents Chemother., 1990, 34, 1666–1671 CrossRef CAS PubMed.
- S. D. Aaron, W. Ferris, K. Ramotar, K. Vandemheen, F. Chan and R. Saginur, J. Clin. Microbiol., 2002, 40, 4172–4179 CrossRef CAS PubMed.
- R. Eckert, K. M. Brady, E. P. Greenberg, F. Qi, D. K. Yarbrough, J. He, I. McHardy, M. H. Anderson and W. Shi, Antimicrob. Agents Chemother., 2006, 50, 3833–3838 CrossRef CAS PubMed.
- M. Tre-Hardy, F. Vanderbist, H. Traore and M. J. Devleeschouwer, Int. J. Antimicrob. Agents, 2008, 31, 329–336 CrossRef CAS PubMed.
- S. Moreau-Marquis, G. A. O'Toole and B. A. Stanton, Am. J. Respir. Cell Mol. Biol., 2009, 41, 305–313 CrossRef CAS PubMed.
- S. Moreau-Marquis, B. Coutermarsh and B. A. Stanton, J. Antimicrob. Chemother., 2015, 70, 160–166 CrossRef CAS PubMed.
- E. P. Mitchell, C. Sabin, L. Snajdrova, M. Pokorna, S. Perret, C. Gautier, C. Hofr, N. Gilboa-Garber, J. Koca, M. Wimmerova and A. Imberty, Proteins, 2005, 58, 735–746 CrossRef CAS PubMed.
- B. Blanchard, A. Nurisso, E. Hollville, C. Tetaud, J. Wiels, M. Pokorna, M. Wimmerova, A. Varrot and A. Imberty, J. Mol. Biol., 2008, 383, 837–853 CrossRef CAS PubMed.
- H. Funken, K. M. Bartels, S. Wilhelm, M. Brocker, M. Bott, M. Bains, R. E. Hancock, F. Rosenau and K. E. Jaeger, PLoS One, 2012, 7, e46857 CAS.
- W. Kabsch, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 125–132 CrossRef CAS PubMed.
- M. D. Winn, C. C. Ballard, K. D. Cowtan, E. J. Dodson, P. Emsley, P. R. Evans, R. M. Keegan, E. B. Krissinel, A. G. W. Leslie, A. McCoy, S. J. McNicholas, G. N. Murshudov, N. S. Pannu, E. A. Potterton, H. R. Powell, R. J. Read, A. Vagin and K. S. Wilson, Acta Crystallogr., 2011, 67, 235–242 CrossRef CAS PubMed.
- P. D. Adams, P. V. Afonine, G. Bunkoczi, V. B. Chen, I. W. Davis, N. Echols, J. J. Headd, L.-W. Hung, G. J. Kapral, R. W. Grosse-Kunstleve, A. J. McCoy, N. W. Moriarty, R. Oeffner, R. J. Read, D. C. Richardson, J. S. Richardson, T. C. Terwilliger and P. H. Zwart, Acta Crystallogr., 2010, 66, 213–221 CrossRef CAS PubMed.
- P. Emsley, B. Lohkamp, W. G. Scott and K. Cowtan, Acta Crystallogr., 2010, 66, 486–501 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Details of carbohydrate synthesis building blocks and peptide dendrimer synthesis, ITC plots, biofilm inhibition and dispersal assay, MIC determination, cell viability assays and control growth curves, and crystallographic data table for 5D2A. See DOI: 10.1039/c5sc03635f |
| This journal is © The Royal Society of Chemistry 2016 |