
Catalytic performance and deoxygenation path of methyl palmitate on Ni2P/SiO2 synthesized using the thermal decomposition of nickel hypophosphite
Qingxin Guana,
Fei Hana and
Wei Li*ab
aCollege of Chemistry, Key Laboratory of Advanced Energy Materials Chemistry (Ministry of Education), Nankai University, Tianjin, 300071, China. E-mail: weili@nankai.edu.cn; Fax: +86-22-23508662; Tel: +86-22-23508662
bCollaborative Innovation Center of Chemical Science and Engineering (Tianjin), Nankai University, Tianjin, 300071, China
First published on 22nd March 2016
Abstract
In this paper, the catalytic performance and deoxygenation path of methyl palmitate on Ni2P/SiO2 catalysts were systematically studied in a continuous flow fixed-bed reactor. A series of Ni2P/SiO2 catalysts (with different molar ratios of P/Ni and Ni2P loadings) were synthesized at 300 °C using the thermal decomposition of nickel hypophosphite. The increased molar ratio of P/Ni generates phosphate-rich nickel phosphide catalysts and increasing conversion. Interestingly, Ni2P/SiO2 showed significantly higher conversion of methyl palmitate in comparison with Ni/SiO2. Furthermore, an activation temperature higher than 500 °C would significantly reduce the catalytic activity, as a result of the sintering of Ni2P. The pressure in a range of 3.0 to 0.5 MPa almost has no effect on the deoxygenation of methyl palmitate, but significantly affects the reaction path and product distribution. Finally, a possible deoxygenation path over Ni2P/SiO2 was proposed based on a GC-MS investigation.
1. Introduction
In recent times, biomass has attracted much attention as a sustainable resource with advantages over fossil fuels such as renewability, lower expense, a neutral carbon cycle, and protection of the environment.1–5 In the 1980s, first-generation biodiesel, its main component being a fatty acid methyl ester (FAME), was reported. However, there are too many problems when used in the engine. Second-generation biodiesel, being composed of alkanes, can be obtained by hydrodeoxygenation (HDO) and isomerization of FAME.6–10 The second-generation biodiesel does not contain oxygen and sulfur, has a lower density and viscosity, with a high cetane number and lower cloud point. Hence, the second-generation biodiesel can also be termed as “renewable diesel” and resembles petrodiesel in terms of hydrocarbon ingredient, and thus has been gaining great attention in recent years. The renewable diesel can be synthesized from a number of plant oils, such as jatropha oil, palm oil, castor oil, and algae oil. These bio-oils usually contain large amounts of oxygen and an upgrading process is necessary to reduce oxygen content to improve heat value and stability. Among several strategies, catalytic HDO has been considered as a very promising route.9Similar to hydrodesulfurization and hydrodenitrogenation, HDO mainly involves the cleavage of C–O and C–C bond.10 Several catalysts have been developed for HDO.11,12 However, conventional transition metal sulfide catalysts (such as CoMo/Al2O3 and NiMo/Al2O3) does not have sufficient activity with enough stability under HDO reaction conditions. On the other hand, sulfur content will be gradually reduced during the reaction.13 Therefore, more and more studies focused on seeking for noble metal catalyst and other novel catalytic materials to promote HDO efficiently. In this context, the transition metal phosphide catalyst showed excellent activity and selectivity.14–18 Y. Yang and coworkers studied the Ni2P/SBA-15 as a HDO catalyst with enhanced selectivity for converting methyl oleate into n-octadecane. They found that the Ni/SBA-15 displays a slightly higher activity than Ni2P/SBA-15. Overall, higher pressure and lower temperature and space velocity favors the formation of C18 hydrocarbon.14 J. A. Cecilia and coworkers studied the HDO of dibenzofuran over silica-supported nickel phosphide with different initial P/Ni ratios. As a result, nickel phosphide based catalysts showed a high resistance to deactivation by coke formation.17 J. Moon and coworkers studied the active sites of Ni2P/SiO2 catalyst for HDO of guaiacol by using XAFS and DFT study. They revealed that the active site of Ni2P is composed of threefold hollow Ni and P sites which lead to adsorption of H or OH groups.18
In previous studies, different metal phosphides were synthesized using the thermal decomposition of metal hypophosphite and metal phosphate precursors.19–23 As shown in Scheme 1, Ni2P/SiO2 was synthesized for converting methyl palmitate to pentadecane and hexadecane. Compared with palmitic acid, methyl palmitate as a model compound for triglyceride feedstock having a lower melting point (30 °C), is more suitable for laboratory studies. However, all of the published papers on the HDO of methyl palmitate were used supported nickel catalysts and carried out in a batch stainless steel autoclave.24–26 Therefore, it would be interesting to study the Ni2P/SiO2 for the HDO of methyl palmitate in a continuous flow fixed-bed reactor. In our recent studies, we have carried out kinetic investigation of deoxygenation of methyl palmitate over SiO2-supported nickel phosphide catalysts.27 Herein, a series of Ni2P/SiO2 catalysts (with different molar ratio of P/Ni and Ni2P loadings) were synthesized at 300 °C using the thermal decomposition of nickel hypophosphite. Catalytic performance and deoxygenation path of methyl palmitate on Ni2P/SiO2 catalysts were systematically studied in a continuous flow fixed-bed reactor. Different with Yang and co-workers published results,14 Ni2P/SiO2 showed significantly higher conversion in comparison with Ni/SiO2. Interestingly, the activation temperatures and reaction pressures significantly affect the conversion and product distribution. Finally, a possible HDO mechanism for the present HDO over Ni2P/SiO2 was proposed based on GC-MS investigation.
 | ||
| Scheme 1 HDO reaction of methyl palmitate over Ni2P/SiO2. | ||
2. Experimental methods
SiO2 was purchased from Tianjin Chemist Scientific Ltd., China. It has a specific surface area of 206 m2 g−1, a pore volume of 0.8 cm3 g−1. Other reagents were analytical pure grade and purchased from Tianjin Guangfu Fine Chemical Research Institute, China.2.1. Synthesis of bulk and supported nickel phosphides
The preparation of bulk Ni2P is illustrated as follows. Nickel chloride (NiCl2) and sodium hypophosphite (NaH2PO2) were dissolved in deionized water under stirring with a mole ratio of P/Ni = 1.5/1. After stirring for 1 h, the solution was evaporated slowly to dehydrate, and was dried at 80 °C for 8 h to obtain the precursor. At the beginning of the thermal decomposition, the air in the reactor was removed by flowing Ar, after which the precursor was treated at 300 °C for 0.5 h in a static Ar atmosphere (heating rate: 5–20 °C min−1). The product was cooled to ambient temperature under Ar and was washed several times with deionized water to remove ion impurities, after which the wet material was dried at 120 °C for 3 h.The preparation of Ni2P/SiO2 is illustrated as follows. NiCl2 and NaH2PO2 were dissolved in deionized water under stirring with a mole ratio of P/Ni = 1.5/1. After stirring for 1 h, the solution was impregnated onto the SiO2 using incipient wetness impregnation. Then, the wet solid was evaporated slowly to dehydrate, and was dried at 80 °C for 8 h to obtain the precursor. At the beginning of the thermal decomposition, the air in the reactor was removed by flowing Ar, after which the precursor was treated at 300 °C for 0.5 h in a static Ar atmosphere (heating rate: 5–20 °C min−1). The product was cooled to ambient temperature under Ar and was washed several times with deionized water to remove ion impurities, after which the wet material was dried at 120 °C for 3 h. Nickel phosphide catalysts with different mole ratio of P/Ni and different Ni loading have similar synthesis procedure.
The Ni/SiO2 was synthesized using the thermal decomposition of nickel nitrate precursor, the precursor being prepared using incipient wetness impregnation. The wet precursors were evaporated slowly to dehydrate, and were dried at 120 °C for 5 h. Subsequently, the precursor was calcined in a muffle furnace at 350 °C for 3 h, after which the sample was reduced at 350 °C for 3 h in flowing H2 atmosphere. Finally, the product was cooled to ambient temperature under flowing H2 and was passivated for 1 h under flowing 1% O2/N2.
For comparison, the bulk Ni2P was synthesized using the temperature-programmed reduction (TPR) method. Initially, nickel nitrate and diammonium hydrogen phosphate, with a mole of P/Ni = 0.5, were dissolved in deionized water and stirred for 1 h. Then, the solution was evaporated at 120 °C for 3 h and calcined at 550 °C for 2 h to obtain the oxidic precursor. Subsequently, the precursor was loaded into the quartz tube reactor. The temperature program of the TPR method was as follows. The flow rate of H2 in the whole procedure was 200 mL min−1. A heating rate of 5 °C min−1 was used from room temperature to 400 °C, and maintained at 400 °C for 1 h. Then, a heating rate of 1–2 °C min−1 was used from 400 to 600 °C, and maintained at 600 °C for 3 h. Subsequently, the product was cooled to ambient temperature under flowing H2, and was passivated for 2 h under flowing 1% O2/N2.
2.2. Characterization
The characterization was performed for passivated sample, which was exposed to the air and without any reduction treatment. Powder X-ray diffraction (XRD) was performed on a Bruker D8 focus diffractometer, with Cu Kα radiation at 40 kV and 40 mA. The transmission electron microscope (TEM) images were acquired using a Philips Tecnai G2 F-20 field emission gun TEM. Scanning electron microscope (SEM) images were acquired using a JEOL JSM-7500F SEM equipped with an energy dispersive spectrometer (EDS). The compositions of the sample were measured by inductively coupled plasma-atomic emission spectrometry (ICP-AES). The Brunauer–Emmett–Teller (BET) specific surface areas were obtained using nitrogen adsorption/desorption measurements at 77 K with a BELSORP-Mini instrument. Samples were degassed at 300 °C for 3 h before measurement. The surface area was calculated using a multi-point BET model. X-ray photoelectron spectroscopy (XPS) was carried out using a Kratos Axis Ultra DLD spectrometer employing a monochromated Al-Kα X-ray source (hν = 1486.6 eV), hybrid (magnetic/electrostatic) optics and a multi-channel plate and delay line detector (DLD). All XPS spectra were recorded using an aperture slot of 300 × 700 microns, survey spectra were recorded with a pass energy of 80 eV, and high-resolution spectra with a pass energy of 40 eV. In order to subtract the surface charge effect, the C 1s peak was fixed at a binding energy of 284.6 eV. The CO chemisorption was performed with Micromeritics Chemisorb 2750 gas-adsorption equipment. The sample was loaded into a quartz reactor and pretreated in 10% H2/Ar at 450 °C for 3 h. After cooling in He, pulses of 10% CO/He in a He carrier (25 mL (NTP) min−1) were injected at 30 °C through a loop tube.2.3. Catalytic activity test
The HDO catalytic activities were evaluated using 50 wt% methyl palmitate in decalin. The HDO reaction was carried out in a fixed-bed microreactor. The catalyst was pelleted, crushed, and sieved with 20–40 mesh; 2.0 g of the catalyst was diluted with SiO2 to a volume of 10.0 mL in the reactor. Prior to the reaction, catalysts were pretreated in situ with flowing H2 (100 mL min−1) for 3 h. The testing conditions for the HDO reaction were 3 MPa, weight hourly space velocity (WHSV) = 3 h−1, and H2/oil = 1000. Liquid products were collected every hour after a stabilization period of 6 h. Both feed and products were analyzed with an Agilent 7890A/5975C GC-MS equipped with a flame ionization detector and an HP-5 column. The same catalyst was used to investigate the effects of different temperatures. The reaction temperature is being investigated in descending order, and each temperature is maintained 3 h. The methyl palmitate conversion and turnover frequency (TOF) were used to evaluate the catalytic activity. The TOF was calculated using eqn (1):
 | (1) |
3. Results and discussion
3.1. Synthesis of bulk and supported nickel phosphides
In our previous published paper, Ni2P was synthesized from nickel hypophosphite in the mole ratio of P/Ni range of 1.5 to 1.75.23 To further study the influence of the mole ratio of P/Ni on NixP crystal forms, a series of NixP were prepared using the thermal decomposition of a nickel hypophosphite precursor with different mole ratios of P/Ni. As shown in Fig. 1, we can clearly see that Ni2P can be synthesized in the mole ratio of P/Ni range of 1.5 to 1.75. The weak diffraction peaks of Ni5P4 were detected when the mole ratio of P/Ni is 2.0, which indicates that P is a slight excess for the synthesis of bulk Ni2P. Not surprisingly, XRD detected more diffraction peaks of Ni5P4 when the mole ratio of P/Ni is 2.5. | ||
| Fig. 1 The XRD patterns of bulk nickel phosphides prepared using the thermal decomposition of a nickel hypophosphite precursor with different mole ratios of P/Ni. | ||
To study the effect of preparation method (hypophosphite method and TPR method) on bulk Ni2P particles, SEM images and XRD patterns of the samples before and after washing were given in Fig. 2. The typical diffraction peaks of NaCl were detected in unwashed sample, and which were disappeared in washed sample. As shown in the SEM images, Ni2P nanoparticles (about 200 nm) are embedded in sodium chloride in unwashed sample (Fig. 2a). This embedded coexistence can effectively prevent sintering of particles. Particle size of Ni2P obtained using hypophosphite method is smaller than that of using TPR method (500–1000 nm) in Fig. 2d and e.
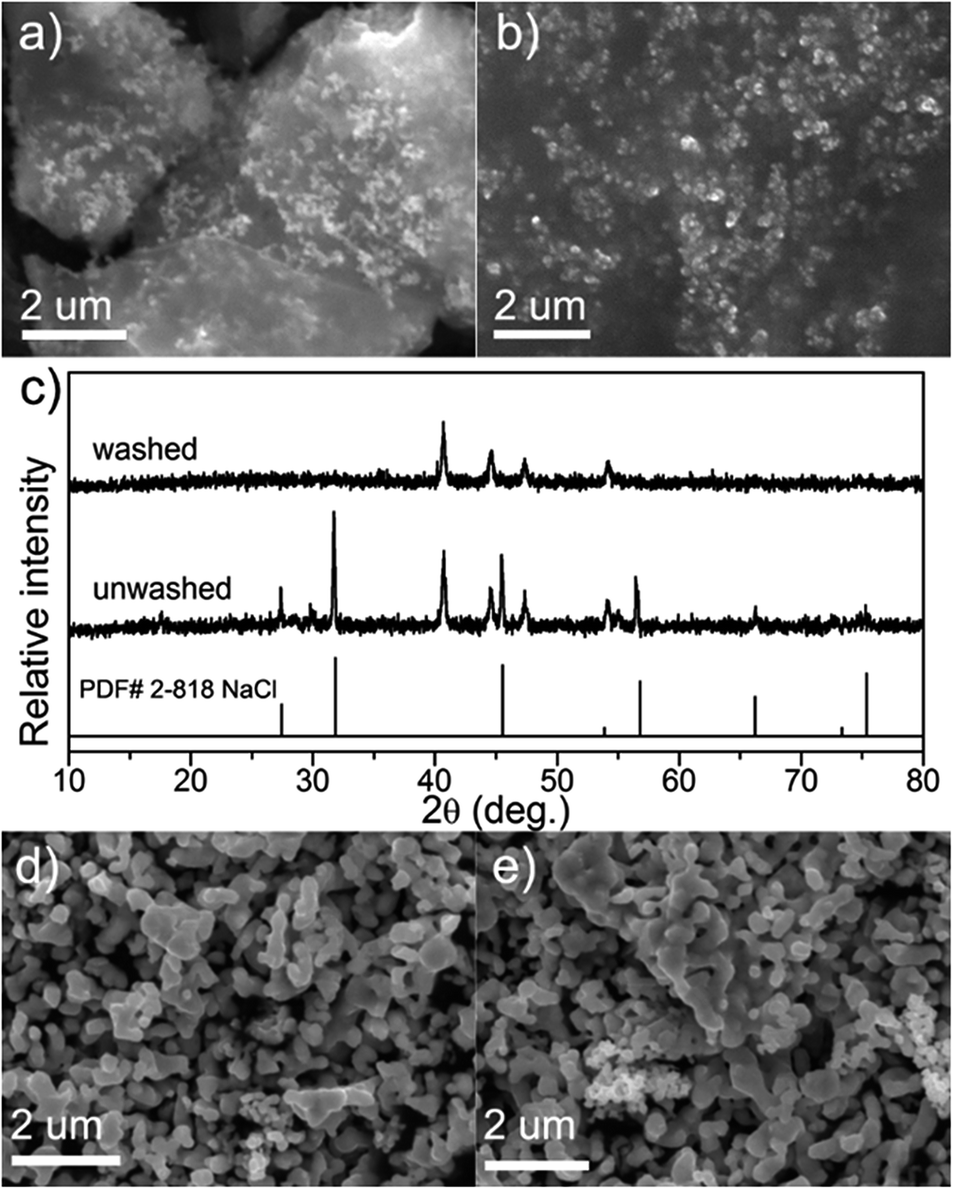 | ||
| Fig. 2 The SEM images and XRD patterns of bulk nickel phosphides prepared using the thermal decomposition of a nickel hypophosphite precursor with a mole ratio of P/Ni = 1.5; (a) unwashed sample, (b) washed sample, (c) XRD patterns of unwashed and washed samples, (d) and (e) bulk Ni2P synthesized using TPR method. | ||
Fig. 3 shows the XRD patterns of 10 wt% Ni2P/SiO2 catalysts prepared using the thermal decomposition of a nickel hypophosphite precursor with various mole ratios of P/Ni. No typical diffraction peaks of Ni2P were detected, which indicates that the crystal of nickel phosphide is smaller than the detection limit of XRD and has a good dispersion on the SiO2 support. Low calcination temperature can significantly reduce the sintering of crystal grain. Fig. 4 shows the XRD patterns of Ni2P/SiO2 catalysts prepared using the thermal decomposition of a nickel hypophosphite precursor at a mole ratio of P/Ni = 1.5 with different loadings. No typical diffraction peaks of Ni2P were detected when the loading was less than 15 wt%. The crystal size of Ni2P gradually becomes large with increasing loading. As shown in Fig. 4, weak diffraction peaks of Ni2P were detected in 15 wt% Ni2P/SiO2, and diffraction peaks of Ni2P increase as the loading increases from 15 to 25 wt%. The result shows that the increased loading could cause the sintering of nickel phosphide crystal grain. In order to observe the distribution of nickel phosphide particles, we performed TEM characterization.
 | ||
| Fig. 3 The XRD patterns of 10 wt% Ni2P/SiO2 catalysts prepared using the thermal decomposition of a nickel hypophosphite precursor with different mole ratios of P/Ni. | ||
 | ||
| Fig. 4 The XRD patterns of Ni2P/SiO2 catalysts prepared using the thermal decomposition of a nickel hypophosphite precursor at a mole ratio of P/Ni = 1.5 with different loadings. | ||
Fig. 5 shows the TEM images of Ni2P/SiO2 prepared using the thermal decomposition of a nickel hypophosphite precursor at a mole ratio of P/Ni = 1.5 with different loadings. The TEM images show fairly uniform nanoparticles with a size of 2–4 nm, indicating Ni2P has good dispersion on the SiO2 support. The weak diffraction peaks of Ni2P for 15, 20, and 25 wt% Ni2P/SiO2 catalysts may be caused by a few larger sintered Ni2P crystals. As shown in Fig. 5b, a magnified high resolution TEM image of 10 wt% Ni2P/SiO2, yields d-spacing values of 0.221 nm, for the (111) crystallographic planes of Ni2P, in good agreement with the calculated values (PDF# 65-1989). It shows the formation and good dispersion of nanocrystalline Ni2P on the SiO2 support.
 | ||
| Fig. 5 The TEM images of Ni2P/SiO2 catalysts prepared using the thermal decomposition of a nickel hypophosphite precursor at a mole ratio of P/Ni = 1.5 with different loadings, (a) 5 wt%, (b) 10 wt%, (c) 15 wt%, and (d) 20 wt%. | ||
Fig. 6 shows the SEM and EDS mapping images of different catalysts (red dot is nickel atom and blue dot is phosphorus). Fig. 6a and b show that the Ni/SiO2 has better metal dispersion than the Ni2P/SiO2 with the same Ni loading. Fig. 6c gives the mole ratio of P/Ni = 0.71 measured by EDS, which is slightly higher than the ICP-AES result, P/Ni = 0.62. Considering that EDS is a semiquantitative analysis method, and a small amount of phosphate ion is not washed away, the mole ratio of P/Ni = 0.71 is a reasonable value. Fig. 6b and d show that Ni and P dispersion increased significantly with the increase in Ni loading.
 | ||
| Fig. 6 The SEM and EDS mapping images of different catalysts (red dot refer to nickel atom, and blue dot refer to phosphorus atom), (a) 10 wt% Ni/SiO2, (b) 10 wt% Ni2P/SiO2, P/Ni = 1.5, (c) 10 wt% Ni2P/SiO2, P/Ni = 1.5, and (d) 25 wt% Ni2P/SiO2, P/Ni = 1.5. | ||
To observe of the valence of Ni and P elements on 10 wt% Ni2P/SiO2 surface, we carried out XPS analysis. Fig. 7 shows the XPS spectra of 10 wt% Ni2P/SiO2 prepared using the thermal decomposition of a nickel hypophosphite precursor with a mole ratio of P/Ni = 1.5. According to the most of the published XPS results,17,19 the Ni 2p spectrum of Ni2P mainly involves two peaks, the first one is assigned to Niδ+ of Ni2P and centered at 852.9 eV, and the second one is at 856.5 eV, corresponding to Ni2+ ions interacting possibly with phosphate ions as a consequence of a superficial passivation. For P 2p, the binding energy at 129.9 eV is attributed to Pδ− of Ni2P and 134.0 eV is attributed to surface nickel phosphate species. As a result of the XPS characterization sample is 10 wt% Ni2P/SiO2, therefore, the intensity of the peaks of Ni 2p and P 2p are relatively low.
 | ||
| Fig. 7 The XPS spectra of 10 wt% Ni2P/SiO2 prepared using the thermal decomposition of a nickel hypophosphite precursor with a mole ratio of P/Ni = 1.5. | ||
3.2. Catalytic activity
The HDO catalytic activities were evaluated using 50 wt% methyl palmitate in decalin.In order to investigate the influence of hydrogen activation, different catalyst activation temperatures were first studied. Fig. 8 shows the catalytic activities of 10 wt% Ni/SiO2 and 10 wt% Ni2P/SiO2 catalysts after different pretreatment temperatures (testing conditions: 3 MPa, 290 °C, WHSV = 3 h−1, H2/oil = 1000). The result indicates that Ni2P/SiO2 showed significantly higher activity than Ni/SiO2 after different pretreatment temperatures. In particular, the catalytic activity of Ni2P/SiO2 after being pretreated at 330 or 400 °C was more than twice that of Ni/SiO2. To find out why activation temperature affect activity significantly, we measured the CO uptakes for catalysts after different pretreatment temperatures. The CO uptakes of catalysts after pretreated at 330, 400, 500, and 600 °C is 6.1, 6.2, 3.9, 3.5 μmol g−1, respectively. These results indicate that the high pretreatment temperature might cause sintering of the Ni2P nanoparticles. Therefore, HDO activity decreased when the activation temperature was increased to 500, up to 600 °C.
 | ||
| Fig. 8 The catalytic activities of 10 wt% Ni/SiO2 and 10 wt% Ni2P/SiO2 (P/Ni = 1.5) catalysts after different pretreatment temperatures (testing conditions: 3 MPa, 290 °C, WHSV = 3 h−1, H2/oil = 1000). | ||
To study the detailed reaction pathway of methyl palmitate HDO, different WHSVs, reaction temperatures, reaction pressures, and loadings were investigated in detail. Fig. 9 shows the catalytic activities of 10 wt% Ni2P/SiO2 (P/Ni = 1.5) at different WHSVs (testing conditions: 3 MPa, 290 °C, H2/oil = 1000). Not surprisingly, HDO conversion and selectivity of pentadecane (C15) and hexadecane (C16) were gradually decreased as the WHSV increased. Meanwhile, the amount of intermediates (such as pentadecene, palmitic acid, hexadecanal, and hexadecanol) gradually increased. In order to better observe the changes of intermediates, subsequent tests were carried out at high WHSV. Fig. 10 shows the catalytic activities of 10 wt% Ni2P/SiO2 (P/Ni = 1.5) at different reaction temperatures (testing conditions: 3 MPa, WHSV = 9 h−1, H2/oil = 1000). HDO conversion and selectivity of C15 and C16 gradually increased as the reaction temperature increased. Meanwhile, the amount of intermediates (such as pentadecene, palmitic acid, hexadecanal, and hexadecanol) gradually decreased.
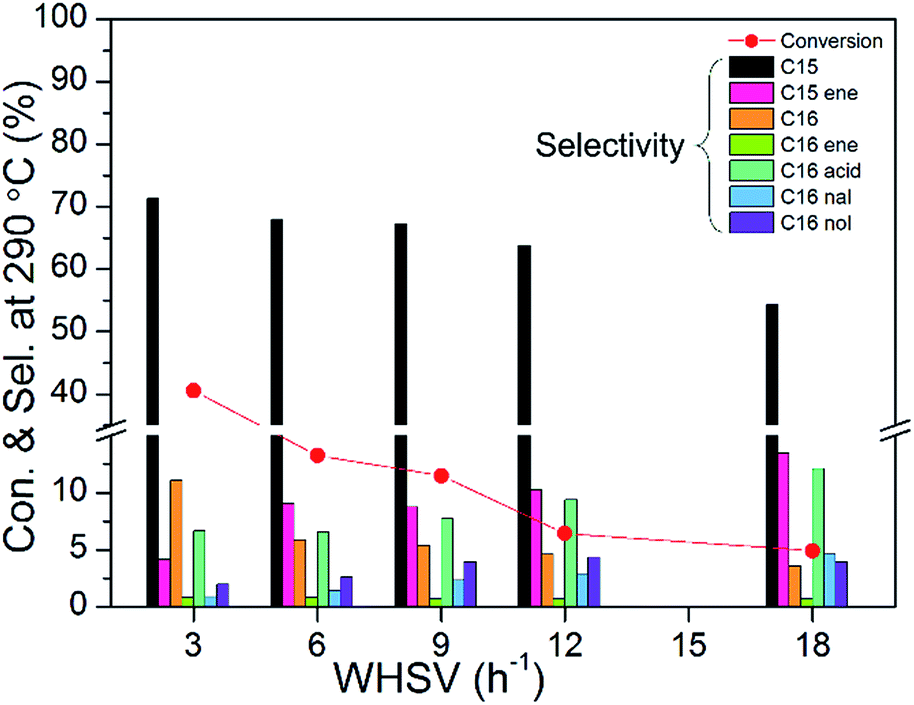 | ||
| Fig. 9 The catalytic activities of 10 wt% Ni2P/SiO2 (P/Ni = 1.5) at different WHSVs (testing conditions: 3 MPa, 290 °C, H2/oil = 1000). | ||
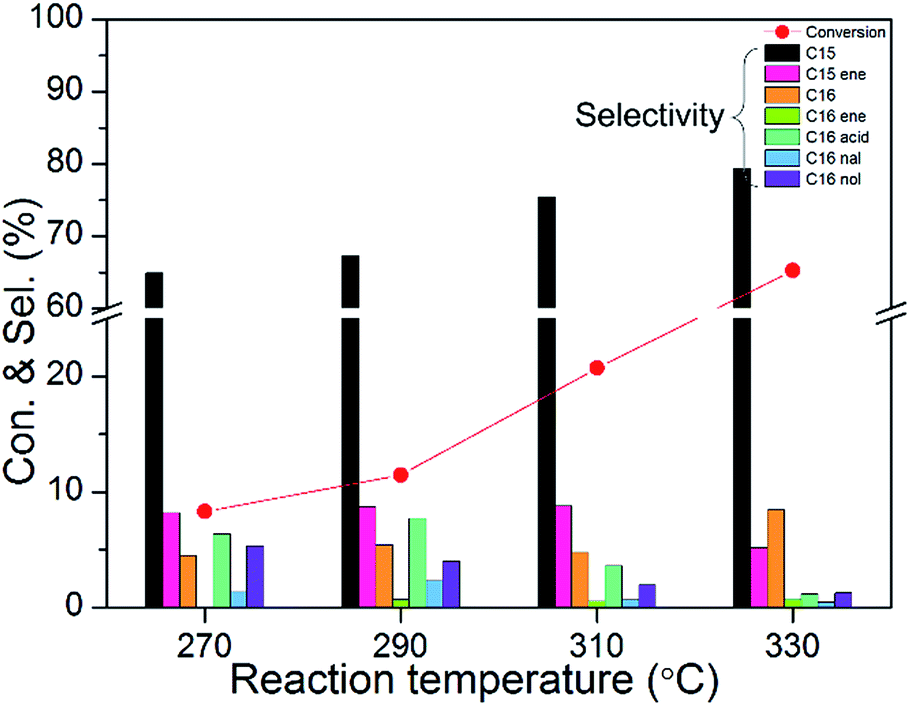 | ||
| Fig. 10 The catalytic activities of 10 wt% Ni2P/SiO2 (P/Ni = 1.5) at different reaction temperatures (testing conditions: 3 MPa, WHSV = 9 h−1, H2/oil = 1000). | ||
Fig. 11 shows the catalytic activities of 10 wt% Ni2P/SiO2 (P/Ni = 1.5) at different reaction pressures (testing conditions: 290 °C, WHSV = 9 h−1, H2/oil = 1000). We can clearly see that the pressures in a range of 3.0 to 0.5 MPa almost has no effect on the conversion of methyl palmitate. However, selectivity of C15, C16, palmitic acid, hexadecanal, and hexadecanol gradually decreased as the reaction pressure decreased, and the content of pentadecene and hexadecene gradually increased. The decreasing amount of C16 indicates that the cleavage of C–O bond is unfavorable at lower pressure. The increased contents of pentadecene and hexadecene show that the olefin hydrogenation reaction at low pressures is unfavorable.
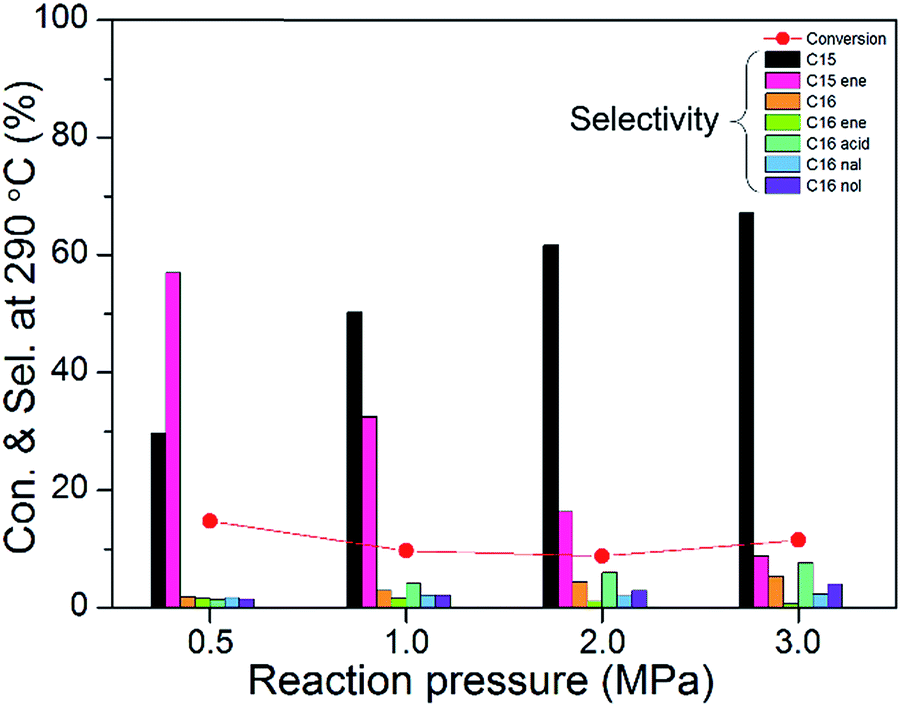 | ||
| Fig. 11 The catalytic activities of 10 wt% Ni2P/SiO2 (P/Ni = 1.5) at different reaction pressures (testing conditions: 290 °C, WHSV = 9 h−1, H2/oil = 1000). | ||
Table 1 shows the catalytic activities of Ni/SiO2 and Ni2P/SiO2 at different loadings (testing conditions: 3 MPa, 290 °C, WHSV = 3 h−1, H2/oil = 1000), which indicates that the HDO conversion gradually increased as the Ni2P loading increased. The result also indicates that Ni2P/SiO2 showed significantly higher activity than the Ni/SiO2 catalyst. The TOF was calculated using eqn (1). Irreversible CO uptake measurements were used to titrate the surface metal atoms and to provide an estimate of the active sites on the catalysts. We can see that 10 wt% Ni2P/SiO2 and 10 wt% Ni/SiO2 have CO uptakes are 6.1, and 8.0 μmol g−1, and TOF are 0.10, and 0.04 s−1, respectively. The CO uptakes and TOF values indicate that Ni2P/SiO2 having lower metal dispersion and higher catalytic activity than Ni/SiO2.
| Catalysts | S (m2 g−1) | CO uptake (μmol g−1) | Conversion (%) | TOF (s−1) | Carbon balance (%) |
|---|---|---|---|---|---|
| Ni/SiO2 10 wt% | 179 | 8.0 | 20 | 0.04 | 98 |
| Ni2P/SiO2 5 wt% | 189 | 3.8 | 28 | 0.11 | 97 |
| Ni2P/SiO2 10 wt% | 175 | 6.1 | 39 | 0.10 | 97 |
| Ni2P/SiO2 15 wt% | 156 | 7.8 | 80 | 0.15 | 97 |
| Ni2P/SiO2 20 wt% | 132 | 8.4 | 74 | 0.15 | 97 |
| Ni2P/SiO2 25 wt% | 116 | 7.5 | 74 | 0.15 | 97 |
In our previous studies, increasing mole ratios of P/Ni of the precursor increases the phosphorus content of the product (Fig. 1), which might affect the catalytic activity. Fig. 12 shows the catalytic activities of 10 wt% Ni2P/SiO2 prepared using the thermal decomposition of a metal hypophosphite precursor with different mole ratios of P/Ni (testing conditions: 3 MPa, 290 °C, WHSV = 3 h−1, H2/oil = 1000). The result indicates that conversion and selectivity of C16 were gradually increased with the increasing mole ratios of P/Ni in the precursor. We speculate that phosphorus-rich nickel phosphide could be more favorable for HDO reaction.
 | ||
| Fig. 12 The catalytic activities of 10 wt% Ni2P/SiO2 prepared using the thermal decomposition of a metal hypophosphite precursor with different mole ratios of P/Ni (testing conditions: 3 MPa, 290 °C, WHSV = 3 h−1, H2/oil = 1000). | ||
Finally, a possible HDO reaction path for HDO of methyl palmitate on Ni2P/SiO2 was proposed based on GC-MS investigation. As shown in Fig. 13, the reaction pathway of the HDO of methyl palmitate mainly includes 10 steps. Methyl palmitate could produce palmitic acid via hydrogenation with methane removal (step 1). At the same time, methyl palmitate could produce hexadecanal through methanol abstraction (step 2). A similar result has been reported by Chen et al.28 Palmitic acid could directly generate pentadecane by decarbonation (step 3),26 and also could generate hexadecanal by dehydration (step 4).29,30 Similar to Mavrikakis et al.,31 we give an adsorption scheme of palmitic acid on the Ni2P surface (red rectangle). Hexadecanal could directly generate pentadecene by take-off of the carbon monoxide (step 5),32 which can be hydrogenated to produce pentadecane (step 8). As shown in Fig. 11, pentadecene gradually increased and C15 decreased as the reaction pressure decreased. The number of molecules in step 5 is increased, and decreasing pressure more favorable promote the positive reaction. Hence, the content of pentadecene is increased as the reaction pressure decreased. Similarly, the number of molecules in step 3 is also increased, but the content of C15 is decreased, which shows that the C15 generated not only from step 3 but also from step 8. Meanwhile, hexadecanal could produce hexadecene from dehydration (step 6) or produce hexadecanol by hydrogenation (step 7), which are parallel reactions.33 The number of molecules in step 6 is not changed, so the content of hexadecene almost is not changed as the reaction pressure decreased. The number of molecules in step 7 is decreased, and decreasing pressure unfavorable promote the positive reaction. Hence, the content of hexadecanol is decreased as the reaction pressure decreased. C16 could be generated from hexadecene (step 9) and hexadecanol (step 10).34 The number of molecules is decreased in the two reaction pathway (step 6 + 9 and step 7 + 10), not surprisingly, content of C16 is also decreased as the reaction pressure decreased. The above results indicate that the pressures in a range of 3.0 to 0.5 MPa significantly affect the intermediate products attribute number of molecules in the different reaction paths are different.
 | ||
| Fig. 13 Reaction pathway of the HDO of methyl palmitate on Ni2P/SiO2 catalysts. | ||
4. Conclusions
In this paper, a series of SiO2 supported Ni2P catalysts were synthesized using the thermal decomposition of nickel hypophosphite precursors. The XRD and TEM results indicate that the crystal of nickel phosphide is smaller than the detection limit of XRD and has a good dispersion on the SiO2 support. The increased molar ratio of P/Ni (>1.75) gave phosphate-rich nickel phosphide. The Ni2P/SiO2 catalysts were characterized using XRD, TEM, SEM, EDS, ICP-AES, BET, and carbon monoxide chemisorption and catalytic performance was evaluated for HDO of methyl palmitate in a continuous flow fixed-bed reactor. Notably, Ni2P/SiO2 showed significantly higher conversion of methyl palmitate in comparison with Ni/SiO2. The selectivity of C15 is much higher than that of C16 at different reaction conditions. Furthermore, the catalytic activity of Ni2P/SiO2 after being pretreated at 330 or 400 °C was more than twice that of Ni/SiO2. Activation temperature at higher than 500 °C would significantly reduce the catalytic activity, as a result of the sintering of Ni2P. However, the pressures in a range of 3.0 to 0.5 MPa almost does not affect conversion of methyl palmitate, significantly affect the intermediate products attribute number of molecules in the different reaction paths are different. Finally, a possible HDO reaction path for HDO of methyl palmitate on Ni2P/SiO2 was proposed based on GC-MS investigation.Acknowledgements
This work was financially supported by the NSFC (21376123, U1403293), MOE (IRT-13R30 and 113016A), NSFT (15JCQNJC05500), and the Research Fund for 111 Project (B12015).References
- X. Zhang, T. Wang, L. Ma, Q. Zhang, X. Huang and Y. Yu, Appl. Energy, 2013, 112, 533–538 CrossRef CAS.
- N. Sharma, H. Ojha, A. Bharadwaj, D. P. Pathak and R. K. Sharma, RSC Adv., 2015, 5, 53381–53403 RSC.
- K. L. Luska, P. Migowski and W. Leitner, Green Chem., 2015, 17, 3195–3206 RSC.
- C. Li, X. Zhao, A. Wang, G. W. Huber and T. Zhang, Chem. Rev., 2015, 115, 11559–11624 CrossRef CAS PubMed.
- N. Yan and X. Chen, Nature, 2015, 524, 155–157 CrossRef CAS PubMed.
- S. C. De Vries, G. W. J. van de Ven and M. K. van Ittersum, Eur. J. Agron., 2014, 52, 166–179 CrossRef.
- M. Mohammadi, G. D. Najafpour, H. Younesi, P. Lahijani, M. H. Uzir and A. R. Mohamed, Renewable Sustainable Energy Rev., 2011, 15, 4255–4273 CrossRef CAS.
- S. N. Naik, V. V. Goud, P. K. Rout and A. K. Dalai, Renewable Sustainable Energy Rev., 2010, 14, 578–597 CrossRef CAS.
- Z. Zhu, H. Tan, J. Wang, S. Yu and K. Zhou, Green Chem., 2014, 16, 2636–2643 RSC.
- B. Van de Beld, E. Holle and J. Florijn, Appl. Energy, 2013, 102, 190–197 CrossRef CAS.
- P. M. Mortensen, J. D. Grunwaldt, P. A. Jensen, K. G. Knudsen and A. D. Jensen, Appl. Catal., A, 2011, 407, 1–19 CrossRef CAS.
- A. H. Zacher, M. V. Olarte, D. M. Santosa, D. C. Elliott and S. B. Jones, Green Chem., 2014, 16, 491–515 RSC.
- D. A. Ruddy, J. A. Schaidle, J. R. Ferrell, J. Wang, L. Moens and J. E. Hensley, Green Chem., 2014, 16, 454–490 RSC.
- Y. Yang, C. Ochoa-Hernández, V. A. De La Peña O'Shea, J. M. Coronado and D. P. Serrano, ACS Catal., 2012, 2, 592–598 CrossRef CAS.
- Y. X. Yang, C. Ochoa-Hernandez, P. Pizarro, V. A. D. O'Shea, J. M. Coronado and D. P. Serrano, Fuel, 2015, 144, 60–70 CrossRef CAS.
- Y. Yang, J. Chen and H. Shi, Energy Fuels, 2013, 27, 3400–3409 CrossRef CAS.
- J. A. Cecilia, A. Infantes-Molina, E. Rodríguez-Castellón, A. Jiménez-López and S. T. Oyama, Appl. Catal., B, 2013, 136–137, 140–149 CrossRef CAS.
- J. S. Moon, E. G. Kim and Y. K. Lee, J. Catal., 2014, 311, 144–152 CrossRef CAS.
- Q. Guan, X. Cheng, R. Li and W. Li, J. Catal., 2013, 299, 1–9 CrossRef CAS.
- Q. Guan and W. Li, Catal. Sci. Technol., 2012, 2, 2356–2360 CAS.
- Q. Guan, C. Sun, R. Li and W. Li, Catal. Commun., 2011, 14, 114–117 CrossRef CAS.
- Q. Guan and W. Li, J. Catal., 2010, 271, 413–415 CrossRef CAS.
- Q. Guan, W. Li, M. Zhang and K. Tao, J. Catal., 2009, 263, 1–3 CrossRef CAS.
- I. V. Deliy, E. N. Vlasova, A. L. Nuzhdin, E. Y. Gerasimov and G. A. Bukhtiyarova, RSC Adv., 2014, 4, 2242–2250 RSC.
- H. L. Zuo, Q. Y. Liu, T. J. Wang, N. Shi, J. G. Liu and L. L. Ma, J. Fuel Chem. Technol., 2012, 40, 1067–1073 CAS.
- H. Zuo, Q. Liu, T. Wang, L. Ma, Q. Zhang and Q. Zhang, Energy Fuels, 2012, 26, 3747–3755 CrossRef CAS.
- F. Han, Q. Guan and W. Li, RSC Adv., 2015, 5, 107533–107539 RSC.
- J. Chen, H. Shi, L. Li and K. Li, Appl. Catal., B, 2014, 144, 870–884 CrossRef CAS.
- B. X. Peng, X. G. Yuan, C. Zhao and J. A. Lercher, J. Am. Chem. Soc., 2012, 134, 9400–9405 CrossRef CAS PubMed.
- S. Brillouet, E. Baltag, S. Brunet and F. Richard, Appl. Catal., B, 2014, 148–149, 201–211 CrossRef CAS.
- M. Mavrikakis and M. A. Barteau, J. Mol. Catal. A: Chem., 1998, 131, 135–147 CrossRef CAS.
- O. I. Şenol, E. M. Ryymin, T. R. Viljava and A. O. I. Krause, J. Mol. Catal. A: Chem., 2007, 268, 1–8 CrossRef.
- D. Li, P. Bui, H. Y. Zhao, S. T. Oyama, T. Dou and Z. H. Shen, J. Catal., 2012, 290, 1–12 CrossRef CAS.
- R. W. Gosselink, S. A. W. Hollak, S.-W. Chang, J. Haveren, K. P. Jong, J. H. Bitter and D. S. van Es, ChemSusChem, 2013, 6, 1576–1594 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |