
Confined PEO crystallisation in immiscible PEO/PLLA blends†
Thiago do Carmo Rufino and
Maria Isabel Felisberti*
Institute of Chemistry, University of Campinas, PO Box 6154, Campinas, São Paulo 13083-970, Brazil. E-mail: misabel@iqm.unicamp.br
First published on 18th March 2016
Abstract
The thermal and morphological properties of PEO/PLLA blends have been widely studied, and the kinetics of PLLA crystallisation have been very well described. However, their miscibility is still a subject of controversy and PEO crystallisation behaviour in these blends is not completely understood. In this paper, the miscibility of PEO/PLLA blends was studied over a wide molar range (5 to 163 kDa and 11 to 199 kDa, for PEO and PLLA, respectively). DSC, DMA, and POM results indicate that PEO/PLLA blends are immiscible over the entire composition range of the molar masses studied. Moreover, the PLLA content of the blends crystallises under cooling at a constant rate from melting to form a continuous crystalline phase, such as a crystal network, even for blends with low PLLA content. Therefore, the PEO phase is confined in the interlamellae and interspherulite regions and a confined and fractional type of crystallisation occurs as the density of the PLLA crystalline phase increases (with increasing PLLA content in the blends). XRD and SAXS data showed that both PLLA and PEO influence each other's crystallisation. While the presence of PEO induces PLLA crystallisation in the α-form, PLLA acts as a template for PEO crystallisation.
1. Introduction
Blends of poly(ethylene oxide) and poly(L-lactide) (PEO/PLLA blends) have been extensively studied for decades.1–18 PLLA is a semi-crystalline hydrophobic polymer that combines properties such as biodegradability and biocompatibility that render it highly useful for a wide range of applications.2,3,19–21 On the other hand, PEO is a semi-crystalline, water-soluble polymer that also presents good biocompatibility and holds much potential for applications in several health areas.8–10 The combination of PLLA and PEO in blends allows for the control of the hydrophobic/hydrophilic balance, which is a very important property to be considered in the design of biomaterials.Fig. 1 shows a map of the various molar masses of PEO and PLLA explored in many studies, usually concerning the miscibility, the kinetics of crystallisation, and the morphology of the PLLA crystalline phase. Despite these studies, the miscibility of PEO/PLLA miscibility remains controversial. The partial-miscibility of these blends is commonly reported, and this conclusion is usually based on the melting point depression observed in the compounds4,5,7,10 and on morphological changes of the PLLA spherulites.1,22 However, recent studies indicate that PEO and PLLA are immiscible.16–18
 | ||
| Fig. 1 Graphical representation of studies carried out on PEO/PLLA blends. | ||
For many applications, a concentration of up to 20 wt% of PEO is added to PLLA in order to adjust the properties of the PLLA for specific purposes such as modifying PLLA mechanical strength, hydrophilicity, degradation rate, and crystallisation kinetics. In the most cases, low molar mass PEO is blended with PLLA, while the molar mass of PLLA varies from 2 × 103 g mol−1 to 800 × 103 g mol−1 (Fig. 1).
The influence of PEO on the morphology of the PLLA crystalline phase was studied by Woo et al.23,24 and Sun et al.17 Woo et al.23,24 reported that the morphology of the PLLA crystalline phase changes from ringless spherulites for PLLA alone to ring-banded spherulites for blends containing more than 50 wt% of PEO. While the characteristics of PLLA in PLLA/PEO blends are well established, little is known about PEO crystallisation.
The fractional crystallisation of PEO has been reported for blends with poly(butylene succinate), PBS,25–27 poly(3-hydroxybutyrate), PHB,28 and poly(ethylene succinate), PES,29 containing up to 30 wt% of PEO. Fractional crystallisation occurs when a polymer crystallises by multiple stages under distinct undercooling. Theses stages can be observed by differential scanning calorimetry (DSC) in the form of several exothermic peaks. This phenomenon has been observed for immiscible blends30,31 and might also occur for miscible ones.25–29 In all cases, one of the compounds should be dispersed into the lamellae, fibrils or spherulites of the other one, and the heterogeneity of the bulk should be low, thereby the second compound would crystallise by a homogeneous nucleation mechanism.25,30
The purpose of this work was to investigate the phase behaviour of PEO/PLLA blends in a wide range of molar masses for both polymers, covering a huge-unexplored region, as denoted by the dashed rectangle in Fig. 1. Furthermore, the non-isothermic crystallisation behaviour of PLLA and PEO as a function of the blends composition was investigated.
Commercial PEO and commercial and synthesised PLLA were used to prepare PEO/PLLA blends by freeze-drying from benzene solutions. The blends were characterised by DSC, dynamic-mechanical analysis (DMA), polarized optical microscopy (POM), X-ray diffraction (XRD) and small angle X-ray scattering (SAXS).
2. Experimental
PLLA (Mw = 199 kg mol−1, ĐM = 2.3) and PEO (Mw = 5 kg mol−1, ĐM = 1.1; 51 kg mol−1 ĐM = 1.1; and 163 kg mol−1, ĐM = 4.0), were purchased from Sigma-Aldrich. PLLA (Mw = 11 kg mol−1, ĐM = 1.3; 46 kg mol−1, ĐM = 1.4; and 87 kg mol−1, ĐM = 1.9) were synthesised via ring-opening polymerization (ROP).32 A typical FTIR spectrum of PLLA is shown in the ESI – SI 01.† The molar masses were determined by gel permeation chromatography (GPC) using a Viscotek chromatograph, model GPC max, with a refractive index detector, model VE3580, tetrahydrofuran as the eluent heated to 40 °C, a flow rate of 1 mL min−1, a solution concentration of 6 mg mL−1, and polystyrene standards. The GPC chromatograms are shown in the ESI – SI 02.† The homopolymers are denoted as PEO-X and PLLA-Y, where X and Y are the molar masses of these polymers expressed in kg mol−1. They were blended in solution in benzene, 5 wt% of polymer, at 60 °C at various PEO/PLLA mass ratios, Table 1. After 24 h of stirring, the benzene was removed by freeze-drying from the ternary homogeneous solution to form sponge-like blends. To avoid physical aging, the blends were then stored at −80 °C. Thermal characterisation of the blends was performed through DSC analysis, using MDSC 2910-TA Instruments equipment. Films of the blends were compression moulded under 0.7 GPa pressure in a hydraulic Marconi press, model MA 098, at approximately 25 °C and cut up to the sample holder dimensions of the DSC, the sample mass range was between 5 to 10 mg. The analyses were carried out under argon at a flow rate of 50 mL min−1, in a temperature range of −100 °C to 200 °C and at a heating rate of 10 °C min−1, followed by cooling and a second heating cycle conducted at the same rate. The DMA was performed using a DMTA V apparatus, Rheometric Scientific Ltd., at a frequency of 1 Hz, an amplitude of 0.05% and a heating rate of 2 °C min−1 from −80 °C to 200 °C. Rectangular shaped specimens, 5.0 × 15.0 × 0.33 mm3 in dimension, were pre-compressed and moulded under 0.7 GPa pressure at room temperature, as described above.| Polymers | PEO-5a | PEO-51a | PEO-163a |
|---|---|---|---|
| a Purchased from Sigma-Aldrich.b Synthesised via ring-opening polymerization. | |||
| PLLA-11b | 20%; 40% | 20%; 40% | 20%; 40% |
| 60%; 80% | 60%; 80% | 60%; 80% | |
| PLLA-46b | 20%; 40% | 20%; 40% | 20%; 40% |
| 60%; 80% | 60%; 80% | 60%; 80% | |
| PLLA-87b | 20%; 40% | 20%; 40% | 20%; 40% |
| 60%; 80% | 60%; 80% | 60%; 80% | |
| PLLA-199a | 20%; 40% | 20%; 40% | 20%; 40% |
| 60%; 80% | 60%; 80% | 60%; 80% | |
A Nikon optical microscope, model Eclipse 80i, was used for POM analysis. Thin films of the blends prepared by compression moulding at room temperature were placed between glass dishes and then onto a Linkam hot-stage, model CSS-450. The samples were subjected to heating and cooling in the temperature range from 30 °C to 200 °C at a rate of 10 °C min−1.
XRD was performed at room temperature on thin films, which were previously subjected to thermal treatment in the DSC oven by the same heating/cooling program used for thermal characterisation of the polymers and their blends. XRD patterns were recorded on a diffractometer (Shimadzu, XRD-7000) in the range of 2θ = 5–35°, operating in reflection mode at 40 kV and 30 mA with a CuKα radiation beam (λ = 0.154 nm).
SAXS experiments were performed on the D01ASAXS2 beamline of the Brazilian Synchrotron Light Laboratory (LNLS) using thin films which were previously treated in the DSC oven using the same program described previously (heating and cooling at a rate of 10 °C min−1), to guarantee total correlation between the SAXS, XRD and DSC results. The scattered beam (λ = 0.1488 nm) was detected on a Pilatus 300k area detector placed 1300 mm from the scattering vector range (q = (4π/λ)sin(θ)), from 0.07 to 3.00 nm−1. DRX and SAXS analyses were performed at 20 °C.
3. Results
PEO and PLLA are soluble in many solvents such as chloroform, tetrahydrofuran and benzene. PEO/PLLA films, prepared by solvent casting from these solvent solutions, present macroscopic phase segregation, especially in blends richer in PLLA. The influence of the solvent on the phase behaviour of blends was reported 40 years ago by Bank and co-authors for polystyrene (PS), and poly(vinyl-methyl ether) (PVME) blends.33 Dragging the solvent out of the ternary solution produces homogeneous and heterogeneous PS/PVME blends, depending on the solvent nature. This phenomenon occurs due to the preferential solvatation of one polymer over the other, leading to non-equilibrium in the mixture. In order to avoid any such solvent influence, we adopted the freeze-drying technique as the PEO/PLLA blend preparation method. Films prepared by compression moulding of the freeze-drying blends are macroscopically homogeneous.3.1. Phase behaviour of PEO/PLLA blends
The glass transition temperatures (Tg) of PEO and PLLA are around −49 °C and 55 °C, respectively. This practically does not vary for the molar mass ranges studied. On the other hand, the melting points (Tm) of PEO are 63 °C, 67 °C and 67 °C for molar masses of 5 kg mol−1, 51 kg mol−1 and 163 kg mol−1, respectively. For PLLA, the Tm are 150 °C, 172 °C, 174 °C, and 176 °C for molar masses of 11 kg mol−1, 46 kg mol−1, 87 kg mol−1 and 199 kg mol−1, respectively.DSC curves corresponding to the 1st heating, cooling, and 2nd heating scan for PEO-51/PLLA-46 blends are shown in Fig. 2. All DSC curves were normalized in regards to the mass of the sample. DSC curves for the other PEO/PLLA pairs (see ESI – SI 03†) are similar to those shown in Fig. 2. Cooling curves show peaks of PLLA and PEO crystallisation while the 1st and the 2nd heating scan curves show the melting peaks of both polymers. The PLLA glass transition is overlapped by the melting of PEO while the glass transition of PEO is hard to observe in Fig. 2a and c. However, Fig. 2d shows the expanded DSC curves in the PEO glass transition range, in which the transition can be observed. The Tg of PEO is not affected by the presence of PLLA in any amount, indicating immiscibility. Besides the Tg criterion for miscibility, another criterion for miscibility is the melting point depression. For this purpose we used the data from the 2nd heating scan (Fig. 2c), since the crystalline phases were formed during the controlled cooling rate (10 °C min−1).
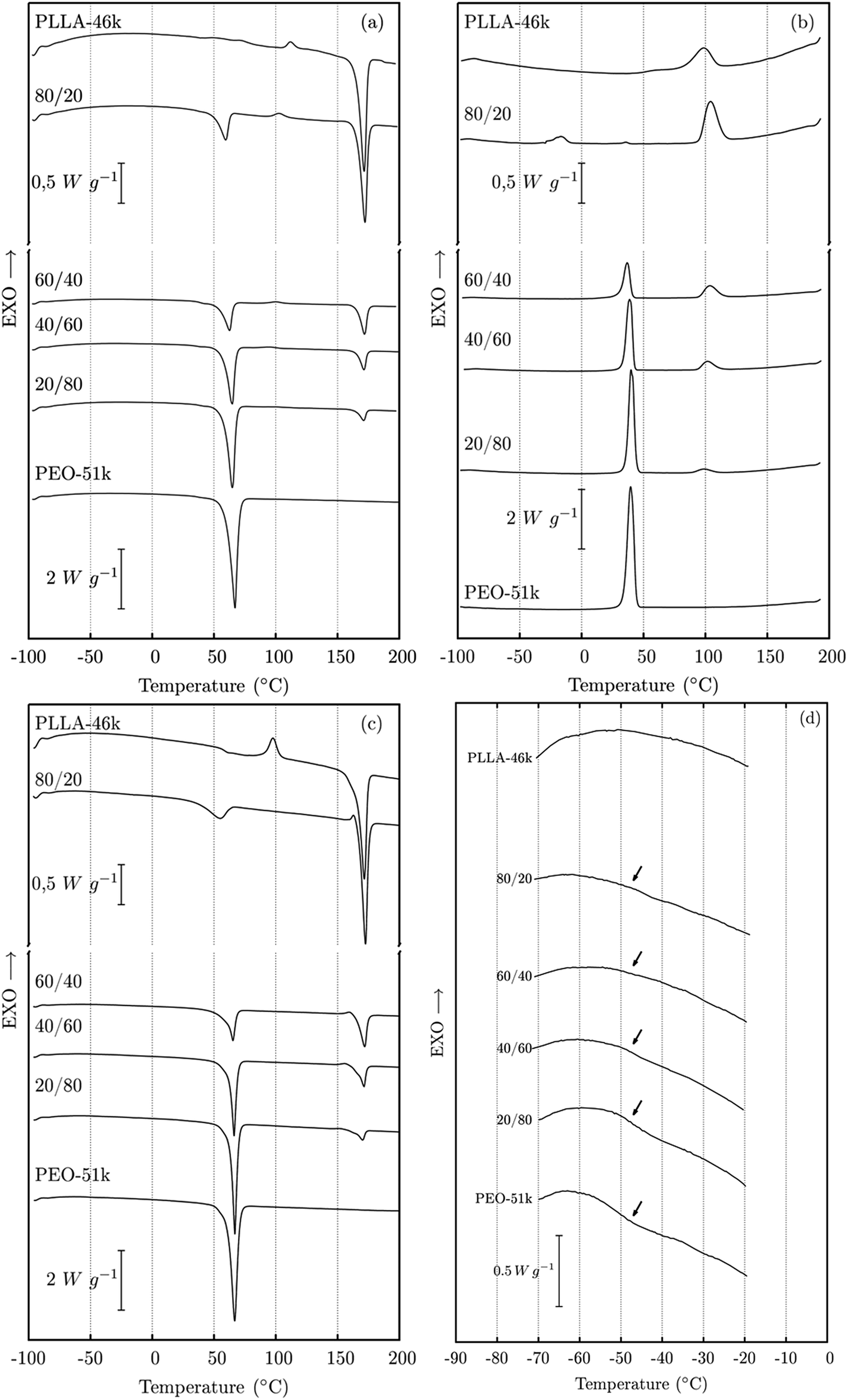 | ||
| Fig. 2 DSC curves taken at a rate of 10 °C min−1 for PEO-51/PLLA-46 blends: (a) 1st heating, (b) cooling, (c) 2nd heating, and (d) zoomed-in view of the PEO glass transition range. The curves were normalized with respect to the sample mass. | ||
Fig. 3a shows the PLLA melting point, TmPLLA (taken as the minimum of the endothermic peak), as a function of the weight average molar mass, MwPLLA, and the blend composition in terms of the mass fraction of PLLA, XPLLA. On analysis of Fig. 3a it can be seen that if one value is picked for MwPLLA, the TmPLLA does not vary along with XPLLA. This indicates that TmPLLA does not depend on the amount of PEO in the blend.
 | ||
| Fig. 3 (a) 3D-plot of PLLA melting temperature as a function of its molar mass and mass fraction in PEO-51/PLLA blends. (b) 3D-plot of PLLA melting temperature in PEO/PLLA 20/80 blends as a function of both homopolymers' molar masses. (c) PEO-51 melting temperature (TmPEO) in PEO/PLLA blends as a function of blend composition, (XPLLA), and PLLA molar mass (MwPLLA). Data was taken from the 2nd DSC heating curves obtained at a heating rate of 10 °C min−1. | ||
On the other hand, when one value of XPLLA is picked, TmPLLA varies with MwPLLA in an asymptotic way, a behaviour typical of homopolymers.34 Fig. 3b shows that TmPLLA is not affected by the molar mass of PEO. These results are indicative of PEO–PLLA immiscibility. This hypothesis is reinforced by analysing the melting point of PEO in the blends. Fig. 3c shows that the melting temperatures for PEO-51, TmPEO (taken as the minimum of the endothermic peak) does not vary in the composition range of 0.2 ≤ XPLLA ≤ 0.6. This is also true for the other blends.
For all of the PEO/PLLA blends studied, only where XPLLA was 0.8 a decrease of in TmPEO was observed. The decrease in TmPEO could be attributed to the miscibility of PEO with PLLA. However, this hypothesis is not corroborated by the thermal properties of PLLA in these blends, since TmPLLA does not change in comparison with neat PLLA, and there is no evidence of the presence of a glass transition in the DSC curves that could be assigned to an amorphous and miscible PEO/PLLA phase. DMA data reinforce the hypothesis of immiscibility. Fig. 4a and b show the storage (E′) and loss (E′′) modulus curves for PEO/PLLA blends containing 20 wt% of PEO. The E′ × T curves (Fig. 4a) show two main events: the melting of the PEO and PLLA phases starting around 30 °C and 175 °C, respectively. Further, cold crystallisation of the PLLA phase is observed at approximately 115 °C for PEO-51/PLLA-46. These events are observed as peaks in the E′′ × T curves (Fig. 4b). Furthermore, a peak of lower intensity is observed in the E′′ × T curves at −50 °C which can be assigned to the glass transition of PEO. A shoulder around 75 °C (indicated by an arrow in Fig. 4b) can be assigned to the glass transition of PLLA. Cold crystallisation is also observed as a peak in the E′′ × T curves above 100 °C.
 | ||
| Fig. 4 (a) Storage and (b) loss modulus curves for: (●) PEO-163/PLLA-199 20/80 and (△) PEO-51/PLLA-46 20/80 blends. | ||
Another hypothesis to explain the depression of the TmPEO in blends containing 20 wt% of PEO could be a phenomenon known as fractional crystallisation.
Based on DSC and rheological data, Xu et al.16 concluded that PEO/PLLA blends present an upper critical solution temperature (UCST) behaviour. Xue et al.35 reported a microphase separation in symmetric PEO-b-PLLA block copolymer. According to the authors, both blocks influence each other's crystallization and the crystallization can induce phase separation. For example, the isothermal crystallization of PLLA block from a miscible mixture in the melting state results in a heterogeneous system composed by a crystalline and an amorphous phase of PLLA and a PEO phase located between the PLLA crystalline lamellae. The entrapped PEO phase crystallizes under confined conditions. Based on these researches and on our results, we believe that PEO crystallization under confined condition also occurs in PEO/PLLA blends, as will be discussed.
3.2. Fractional crystallisation
The DSC cooling curves for PEO/PLLA 20/80 blends prepared using polymers with different molar masses, are shown in Fig. 5. All DSC curves were normalized to the mass of the sample and are shown in the same scale. The exceptions are the DSC curves for pure PEO, which were additionally multiplied by 0.2, to allow for better visual comparison with the results for blends containing only 20 wt% PEO. PLLA crystallises from 125 °C to 80 °C under cooling at 10 °C min−1, depending on the molar mass (see Fig. 2 and ESI – SI.03†). Therefore, the exothermic events shown in Fig. 5, between −50 °C and 50 °C, correspond only to PEO crystallisation. In general, all DSC curves for PEO/PLLA 20/80 blends (Fig. 5a–c) present at least two exothermic peaks over the temperature range: (1) similar to pure PEO and (2) shifted about 50 °C lower. The multiple exothermic peaks indicate the fractional crystallisation of PEO in the PEO/PLLA 20/80 blends. | ||
| Fig. 5 DSC cooling curves taken at a rate of 10 °C min−1 for all PEO/PLLA 20/80 blends containing (a) PEO-5, (b) PEO-51, and (c) PEO-163 blends. DSC curves for pure PEO (+) were multiplied by 0.2 in order to show adequate peak intensity allowing for a better visual comparison with the blend peaks containing 20 wt% in PEO. (d) DSC cooling curves for PEO-51/PLLA 80/20 blends. | ||
Dalnoki-Veress and co-workers36–38 performed very elegant experiments on the nucleation within discrete PEO drops of different diameters. They verified that homogeneous crystallisation occurs in drops with the smallest diameters, due to the absence of impurities. In contrast, the probability of finding impurities in PEO drops increases with increasing diameter and in-turn the probability of heterogeneous crystallisation. A similar phenomenon has also been described for PEO confined in porous structures.39–43 Our results suggest that PEO should crystallise in different environments in the PEO/PLLA blends. Thus, the peak observed at higher temperatures can be attributed to crystallisation from heterogeneous nucleation, while the peaks shifted to lower temperatures can be assigned to crystallisation from homogeneous nucleation, which occurs in confined conditions, as will be discussed.
The profile of the DSC curves for PEO/PLLA 20/80 blends varies with the molar mass of both polymers, as can be seen in Fig. 5. However, some features are common to all blends. For example, for blends containing PEO-5 (Fig. 5a), a PEO crystallisation peak at lower temperatures (around −20 °C) is observed for all of the DSC curves. This is also true for blends with PEO-51 (Fig. 5b) and PEO-163 (Fig. 5c). On the other hand, the events at higher temperatures vary in quantity, temperature range and intensity with the molar masses of both polymers in a complex way. This means that the spatial distribution of PEO in different environments is determined by the molar mass and molar mass distribution of the polymers. This is not a surprise given that the diffusion coefficient of PLLA chains toward the front of crystal growth and the diffusion coefficient of PEO chains in the opposite direction, depend on the molar mass and molar mass distribution of the polymers.
Although the location of PEO in the blends seems to be an important and determining factor in the crystallisation profile of blends containing 20 wt% in PEO, this is not true for PEO/PLLA 80/20 blends. For these blends (Fig. 5d) and for other compositions (ESI – SI 03†) only one exothermic peak is observed for PEO crystallisation. Moreover, this peak appears in the same temperature range as for pure PEO.
Fig. 6 shows POM micrographs taken during the dynamic crystallisation of PEO in the PEO-51/PLLA-46 80/20 blend, which was subjected to cooling at a rate of 10 °C min−1 from 200 °C. The complete video of this experiment can be found in ESI – SI 04†. The micrographs were taken just before (39 °C), at the beginning (38 °C) and at the end (30 °C), of the PEO crystallisation process. Although PLLA is the minor component in this blend, its crystallisation takes place all over the sample (Fig. 6a). This behaviour is entirely different when compared with the isothermal crystallisation of PLLA in PEO/PLLA 80/20 blends previously reported in the literature, for which isolated and dispersed spherulites were observed.44 Moreover, the PLLA spherulites in the PEO/PLLA blends were ringless, which differs completely from the results reported by Woo and co-workers. These authors found that spherulites of PLLA in PEO/PLLA blends with similar composition were ring banded, independent of whether the film surface was free or covered with a glass slide. The direction of the PEO crystallisation front can clearly be observed in the micrograph taken at 38 °C Fig. 6b, where the crystallisation can be seen beginning in the top, left corner from where it propagates throughout the polymer film (the white arrow in Fig. 6b shows the crystallisation direction). PEO crystallisation enhances the optical birefringence, making the PLLA spherulites in the micrograph more defined and the interface between them conveniently sharp. As can be seen in Fig. 6c, at 30 °C the process was complete and the spherulites observed are ringless with some Maltese cross extinction evident. The emergence of a dark region between the PLLA spherulites and the increase in the contrast of the pre-existent PLLA spherulites, suggest PEO crystallisation in different environments: interspherulites and interlamellae, respectively. Scanning electron microscopy (SEM) analyses were performed for PEO-51/PLLA-46 blends containing 40 and 60 wt% of PEO before and after the extraction of the PEO. The images revealed empty spaces inside and around PLLA spherulites, as can be seen in the Fig. SI 05 in the ESI.† This reinforces our conclusion about the PEO location in the blends. The dark interspherulite regions disappeared completely when the samples were further heated at temperatures above PEO melting (see video shown in ESI SI 06†). Therefore, in contrast to the work of Woo et al., the crystallisation of PEO does not result in spherulite cracks.24
 | ||
| Fig. 6 POM micrographs taken during the dynamic crystallisation of PEO in a PEO-51/PLLA-46 80/20 blend at a rate of 10 °C min−1 from 200 °C to: (a) 39 °C, (b) 38 °C and (c) 30 °C. The scale bar size is 200 μm. The video of the experiment is available in the ESI – SI 04.† | ||
POM micrographs of PEO-51/PLLA-46 20/80 blends were also taken at different temperatures, when the samples were subjected to cooling at a rate of −10 °C min−1 from 200 °C to 0 °C. At 50 °C, only the PLLA spherulites are present and the PEO phase remains in its liquid phase. The PLLA spherulites are ring banded and the ridges and the valleys of the bands present opposite birefringence (blue-yellow colours), as also observed for blends of similar compositions, crystallised under isothermic conditions.24 With further cooling to 30 °C, the PEO crystallisation temperature was reached and the formation of a dark inter-spherulitic region can be observed (Fig. 7b). It is important to note that the emergence of the dark inter-spherulitic region occurs in the same temperature range of the pure PEO crystallisation and this process is reversible and there is no evidence of sample cracks. For this blend, fractional crystallisation was not observed.
 | ||
| Fig. 7 POM micrographs for PEO-5/PLLA-11 20/80 subjected to cooling at a rate of 10 °C min−1 from 200 °C to: (a) 50 °C and (b) 30 °C. The scale bar size is 200 μm. | ||
The 2nd DSC heating curves for PEO-51/PLLA 20/80 blends, showing the melting of the PEO phase, are presented in Fig. 8. Although multiple crystallisation peaks are observed for PEO in the cooling step (Fig. 5), only one melting peak is observed in the 2nd heating scan curves for PEO/PLLA blends containing 20 wt% in PEO. Moreover, PEO-51 blended with PLLA-87 crystallises predominantly below −10 °C (Fig. 5b). Nevertheless, it melts in the same temperature range as PEO-51 blended with PLLA-199, for which PEO-51 crystallises between 0 °C and 40 °C. TmPEO, taken as the temperature corresponding to the minimum of the melting peak, varies with the PLLA molar mass in these blends in the following order: PLLA-46/PEO-51 < PLLA-87/PEO-51 < PLLA-11/PEO-51 < PLLA-199/PEO-51. The decrease in miscibility in the polymer pairing with increasing molar mass of the polymers is well established in the literature.45 Therefore, these results reinforce the hypothesis of immiscibility also for PEO/PLLA blends containing 20 wt% of PEO, because TmPEO should be lower for a miscible blend with lower molar mass PLLA (PEO-11/PLLA-51). On the other hand, the same results reinforce the hypothesis of confined crystallisation, which affects the morphology of the PEO crystalline phase and consequently its melting. Besides, the width at half height of the melting peak, expressed as ΔTh/2, and the asymmetry of the melting peak of PEO-51 in PLLA blends with different molar mass, also changes.
 | ||
| Fig. 8 2nd DSC heating curves captured at a heating rate of 10 °C min−1 for PEO-51/PLLA 20/80 blends and pure PEO-51 multiplied by 0.2. | ||
The 3D-plot shown in Fig. 9 illustrates how ΔTh/2PEO varies with the PLLA mass fraction XPLLA and PLLA molar mass MwPLLA. Choosing one composition, for example XPLLA = 0.4, in Fig. 9, the ΔTh/2PEO does not vary with PLLA molar mass, and this is valid for all of the compositions studied. This means that ΔTh/2PEO does not depend on the PLLA molar mass. Choosing one PLLA molar mass, for example MWPLLA = 46 kg mol−1 in Fig. 9, the ΔTh/2PEO does not vary with PLLA mass ratio in the interval of 0.2 ≤ XPLLA ≤ 0.6, and this is valid for all of the PLLA molar masses studied. Actually, ΔTh/2PEO values for blends containing up to 60 wt% of PLLA are similar to pure PEO, which complies with the hypothesis of an immiscible blend, as the pure component property is maintained in the blend.
 | ||
| Fig. 9 Width at half height of the melting peak of PEO-51, ΔTh/2PEO, in PEO-51/PLLA blends. Data extracted from the 2nd DSC heating curves captured at a heating rate of 10 °C min−1. | ||
At XPLLA = 0.8 (20 wt% of PEO), ΔTh/2PEO reaches the highest values over the whole range of PLLA molar masses studied, Fig. 9, which means that the melting occurs over a broader temperature range. The broader the peak is, the more crystallite families of different lamellar thickness are present in the PEO crystalline phase.34 In other words, ΔTh/2 reflects a morphological issue. Similar results were reported by Duran and co-workers46 for the homogeneous nucleation of isotactic polypropylene (iPP) confined in nanoporous alumina. These authors suggested that the enlargement and asymmetry of the melting peaks of iPP crystallised in confined conditions reflects the melting of crystals resulting from homogeneous and heterogeneous nucleation. Moreover, the characteristic peaks may also appear as a result of reorganisation and recrystallisation processes that could occur during the heating of the DSC experiments. It is noteworthy that PEO-5 and PEO-163 blends followed the same trend as PEO-51 blends (ESI – SI 03†).
The diffractograms for PEO-51, PLLA-46 and their blends, with the same thermal history as the samples analysed by DMA and DSC, are shown in Fig. 10. PLLA crystallises into different crystal structures which depend on the crystallisation conditions. However, the α-form is the most stable and common form and presents as an orthorhombic unit cell. The α-form is observed for PLLA crystallised from melting and from solution, the α′-form is a “disordered-crystal” form, which is also reported for PLLA crystallised below 120 °C, and presents the same chain conformation as the α-form, in a pseudo-hexagonal unit cell however.47,48 The diffractogram for PLLA-46 presents characteristic peaks of the α′-form: the main peaks appear at 2θ = 16.4°, 18.7° and 24.3° and they are related to (200/110), to 203 and to 116 reflections, respectively.47,48 For neat PEO-51, the more intense peaks are located at 2θ = 19.3° and 2θ = 23.5° due to (120) and (112/004) reflections, respectively.49 The (120) plane is perpendicular to the extended chain direction in the crystal. Moreover, the (120) plane follows the direction of the fastest crystal growth.40
 | ||
| Fig. 10 Diffractograms for PEO-51/PLLA-46 blends. All samples were thermally treated using the same heating–cooling program as DSC analysis. 0% diffractogram stands for PLLA-46. | ||
In general, the diffractograms of all of the blends combine the diffraction patterns of both homopolymers. However, some peculiarities should be discussed. The peak related to the (200/110) reflection of the PLLA crystals is shifted from 2θ = 16.4° to 2θ = 16.7° as the PEO content in the blend increases (see ESI – SI 07†). This means that the PLLA chain packing becomes denser. Zhang et al.47 reported a similar shift of the (200/110) reflection when PLLA was isothermally crystallised at temperatures above 120 °C due to the formation of α-form crystals. Moreover, they concluded that a mixture of α and α′ crystals were obtained when the crystallisation was conducted between 120 °C and 100 °C and at temperatures below 100 °C the crystals presented in the α′-form. Since neat PLLA crystallises under cooling at a constant rate of 10 °C min−1 from 110 °C to 75 °C, and in the blends between 120 °C and 100 °C (Fig. 2 and ESI – SI 03†), we believe that the increase in PEO content in the blend subjected to PLLA dynamic crystallization and the increase of the temperature in which neat PLLA is isothermally crystallized have an similar effect on PLLA crystallisation: both favour the formation of α-form crystals. If the blends are miscible in the melting, as proposed by Xue et al.35 for symmetric PEO-b-PLLA block copolymer, then PEO could reduce the viscosity favouring the PLLA crystallization, while inducing α-form crystal. The (203) reflection of the neat PLLA is also shifted to larger angles, from 2θ = 18.7° to 2θ = 19.1° with increasing PEO content, which could reflect the gradual substitution of the α′ form with α-form crystals, since α-form is characterised by a (203) reflection at 2θ = 19.1°. However, PEO presents a reflection peak at 19.3° that could overlap the (203) reflection of PLLA, making the analysis complex. The peak at 22.1°, attributed to the reflection of (210) of the α-form, is quite weak for PLLA. However, its intensity increases with increasing PEO content in the blend (ESI – SI 07†). The appearance of this reflection was also reported by Zhang and co-workers47 for PLLA crystallised at temperatures above 120 °C and was attributed to the presence of α-form crystals. The relative intensity of the peaks related to the (200/110) and (210) reflections tend to even out as the PEO content in the blend increases, which reinforces the hypothesis of a progressive replacement of the α′-form by α-form crystals with increasing PEO. These results show that PEO affects the crystallisation of PLLA under non-isothermic conditions. Recently, Zhou and co-workers50 found that PLLA blocks crystallise in an α-form crystal, independent of the PEO/PLLA copolymers' architecture and molar mass. Yang and co-workers51 studied the confined crystallisation of a poly(L-lactide)-b-poly(ethylene glycol) copolymer series. Time-resolved wide angle X-ray scattering (WAXS) profiles of the copolymer during isothermal crystallisation at 110 °C and 30 °C, revealed reflection peaks characteristics of α-form crystals and the appearance of reflections of PEO crystals, respectively. Moreover, they observed a shift of PLLA reflections to larger angles, concluding that a transition of the PLLA α-form to α′-form occurs during PEO crystallisation at 30 °C. The authors proposed that the PLLA crystals may be stretched or sheared by the PEO blocks during crystallisation.
As described above, PLLA crystallises throughout the sample independent of the blend composition, forming a network in which PEO crystallises. Consequently, a strong influence of the PLLA template on the PEO crystallisation is observed. For instance, the peak at 2θ = 23.5° (120) for neat PEO is shifted to 23.1° in the blends, probably due to the increasing imperfections in the crystals. Our DSC and POM results indicate that PEO crystallises in inter-lamella and inter-spherulite locations and that confinement should occur in the inter-lamella ones.
SAXS profiles of PEO-51/PLLA-46 and PEO-5/PLLA-11 blends with different compositions are shown in Fig. 11. The samples analysed by XRD and SAXS were thermally treated (see Experimental section) in order to reproduce the same thermal history. Both analyses were conducted at approximately 20 °C. The DSC curves do not present any evidence of melting in the temperature range from −40 °C to 20 °C, Fig. 8.
 | ||
| Fig. 11 SAXS profiles of (a) PEO-51/PLLA-46 and (b) PEO-5/PLLA-11 blends. Average long period, L, was calculated by L = 2π/qmax. | ||
The curves I(q2) vs. q for the semicrystalline PLLA present no peaks in the q range analysed. This is probably due to the occurrence of the interference SAXS peak at a scattering vector too low to be detected within the SAXS experimental conditions adopted in this work. This hypothesis is reinforced by the work of Mano and co-workers,52 who reported a scattering peak at approximately q = 0.033 nm−1 for PLLA with Mw of 151![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1 and 30000 g mol−1.
000 g mol−1 and 30000 g mol−1.
On the other hand, the I(q2) vs. q curves for PEO-51 and PEO-5 present peaks centred at qmax = 0.2938 nm−1 and 0.5632 nm−1, respectively, corresponding to the average long period (L = 2π/qmax) of 21.4 nm and 11.2 nm. The L value for PEO-5 is close to the value reported by Yang et al.51 for the PEO/PLLA copolymer, in which the PEO block molar mass was 5000 g mol−1. The increase of the long period with increasing PEO molar mass is in agreement with the literature34 and our DSC data: Tm is higher for PEO-51.
The scattering peak maximum of PEO-51 shifts to higher q values and the long period L decreases from 21.4 nm to 15.2 nm for PEO, PEO-51/PLLA-46 40/60 and 20/80 blends (Fig. 11a). POM experiments show that PLLA crystallises from the blends under cooling at a constant rate forming a crystalline network. The density of this network, defined as the number of lamellae per volume,53 increases as the PLLA contents in the blends increases. Thus, the interlamellar space, in which PEO is entrapped, decreases as the blends become richer in PLLA. Consequently, the PEO long period decreases, as observed. On the other hand, the long period for PEO-5 in PEO-5/PLLA-11 does not depend on the blend composition, possibly because the maximum long period that PEO-5 could reach is still smaller than the PLLA interlamellar space.
The long period L is the sum of the crystalline lamellar thickness (lc) and the amorphous thickness (la). The crystalline period, lc, and amorphous period, la, were calculated as reported elsewhere54 using the following equations:
| L = lc + la |

The following data were used in the equations above: density of 1.123 g cm−3 and 1.235 g cm−3 for amorphous (ρa) and crystalline (ρc) phases of PEO, respectively.55 The crystalline degree wc was calculated from DSC data (melting enthalpy) using a melting enthalpy value for 100% crystalline PEO of 203 J g−1.56
A decrease of the L and lc value is observed for blends containing up to 40 wt% of PEO-51. A decrease of lc was also observed for PEO-5, however the values of L do not vary significantly between the blends, Fig. 12b. These results support the hypothesis that PEO phase is entrapped between PLLA lamellae and due to this confinement, the conformational changes of PEO chains for developing its crystalline phase are hindered.
 | ||
| Fig. 12 Crystalline, lc, and amorphous, la, periods for PEO in (a) PEO-51/PLLA-46 and (b) PEO-5/PLLA-11 blends. Average long period, L, is defined as L = lc + la. | ||
The PEO lamella thickness decreases as the PLLA amount in the blends increases Fig. 12, this effect is more pronounced for blends containing 20 wt% of PEO. Therefore, the decrease of Tm for the PEO crystalline phase in the immiscible blend is due to a morphology issue, which is imposed by the confinement of the PEO phase in the PLLA crystalline network.
The inclusion of PEO between lamellae of a second polymer, and the consequence of this on the PEO fractional crystallisation in miscible blends with PBS,25 PHB,28 and PES,29 has been reported. For PEO/PBS blends containing 20 wt% of PEO, Inoue et al.25 demonstrated the complete incorporation of PEO in the interlamellar region of PBS using SAXS experiments. Pan et al.26 concluded that the crystallisation temperature of PBS influences the crystallisation of PEO in PEO/PBS blends when they are cooling from the melting point, because PBS crystallises first and PEO crystallises into PBS interlamellar regions. Wang et al.29 showed that the history of the crystallisation of PES strongly influences the PEO crystallisation in the PEO/PES blends. DSC curves for blends subject to PES isothermal crystallisation at 65 °C followed by cooling to −50 °C at a rate of 10 °C min−1, showed two crystallisations peaks for PEO due to the fractional crystallisation of PEO. Moreover, the authors concluded that fractional crystallisation arises from PEO located in different environments: the interlamellar region and outside this region (interspherulitic). Further, Inoue et al.28 showed that the PEO crystallisation behaviour in blends with PHB changes with aging. The authors concluded that under cooling, PEO crystallises mainly via a homogeneous nucleation mechanism for blends in which PHB was previously isothermally crystallised at 90 °C. After storage at 25 °C for 6 months, a fraction of PEO is expelled from the interlamellar region and its crystallisation occurs by active heterogeneous nucleation, in the bulk.28
Our DSC, DMA and POM results confirmed the immiscibility of PEO/PLLA over a wide molar mass and over a wide range of relative polymer compositions. Hence, based on the literature and on the results presented herein, the presence of PEO between PLLA spherulites and PLLA lamellae can be explained based on the following model:
1. PLLA crystallises between 120 °C to 75 °C when the blends are cooled from the melting state at a constant rate. PLLA crystals spread throughout the sample forming a continuous phase like a network, in which the PEO phase is entrapped;
2. An increase in PEO content in the blend results in a decrease of the PLLA crystal density. In this case, PEO crystallisation in the blends is close to neat PEO;
3. In contrast, the PLLA crystalline network density increases as the PEO content decreases, and PEO crystallises in restricted spaces (interlamellar spaces), confined crystallisation.
4. Conclusion
The immiscibility of PEO/PLLA blends have been proven over a wide range of molar mass and blend compositions using DSC, DMA and POM analyses. All of the blends that were crystallised at a constant cooling rate from melting, presented PLLA crystalline phase morphology throughout, like a crystalline network. The PLLA crystal form is influenced by the presence of PEO, changing from α′-form to α-form as the amount of PEO is increased. Furthermore, the PEO phase is entrapped in the PLLA crystals, which leads to the confined fractional crystallisation of PEO in blends containing 20 wt% of this polymer. This fractional crystallisation occurs due the distribution of the PEO phase in different micro- and nano-environments: the interspherulites and interlamellae of PLLA, respectively. The distribution of PEO in these environments and the blend properties can be tailored by controlling the molar mass and molar mass distribution of the polymers. The main factor in determining the PEO confinement seems to be the density of the PLLA crystalline network, not the molar mass of the polymers. The PLLA interlamellae effect on the PEO crystallisation vanishes as the interlamellae distance increases, due to PLLA dilution in the blends richer in PEO.Acknowledgements
The authors acknowledge CAPES for the scholarship, FAPESP (Proc. No. 2010/17804-7) and CNPq for the financial support, and LNLS for the beanline time (Proc. No. 17044).References
- D. Shin, K. Shin, K. A. Aamer, G. N. Tew, T. P. Russell, J. H. Lee and J. Y. Jho, A morphological study of a semicrystalline poly(L-lactic acid-b-ethylene oxide-b-L-lactic acid) triblock copolymer, Macromolecules, 2005, 38, 104–109, DOI:10.1021/ma0481712
.
- Y. Agari, K. Sakai, Y. Kano and R. Nomura, Preparation and properties of the biodegradable graded blend of poly(L-lactic acid) and poly(ethylene oxide), J. Polym. Sci., Part B: Polym. Phys., 2007, 45(21), 2972–2981, DOI:10.1002/polb.21294
- M. Sheth, R. A. A. Kumar, V. Davé, R. A. Gross and S. P. McCarthy, Biodegradable polymer blends of poly(lactic acid) and poly(ethylene glycol), J. Appl. Polym. Sci., 1997, 66(8), 1495–1505, DOI:10.1002/(SICI)1097-4628(19971121)66:8<1495:AID-APP10>3.0.CO;2–3
- W.-C. C. Lai, W.-B. B. Liau and T.-T. T. Lin, The effect of end groups of PEG on the crystallization behaviors of binary crystalline polymer blends PEG/PLLA, Polymer, 2004, 45(9), 3073–3080, DOI:10.1016/j.polymer.2004.03.003
- A. J. Nijenhuis, E. Colstee, D. W. Grijpma and A. J. Pennings, High molecular weight poly(L-lactide) and poly(ethylene oxide) blends: thermal characterization and physical properties, Polymer, 1996, 37(26), 5849–5857, DOI:10.1016/s0032-3861(96)00455-7
- C. Nakafuku, Effects of molecular weight on the melting and crystallization of poly(L-lactic acid) in a mixture with poly(ethylene oxide), Polym. J., 1996, 28(7), 568–575, DOI:10.1295/polymj.28.568
- C. Nakafuku and M. Sakoda, Melting and Crystallization of Poly(L-lactic acid) and Poly(ethylene oxide) Binary Mixture, Polym. J., 1993, 25(9), 909–917, DOI:10.1295/polymj.25.909
- H. Younes and D. Cohn, Phase separation in poly(ethylene glycol)/poly(lactic acid) blends, Eur. Polym. J., 1988, 24(8), 765–773, DOI:10.1016/0014-3057(88)90013-4
- F. von Burkersroda, R. Gref and A. Göpferich, Erosion of biodegradable block copolymers made of poly(d,l-lactic acid) and poly(ethylene glycol), Biomaterials, 1997, 18(24), 1599–1607, DOI:10.1016/S0142-9612(97)00098-7
- W. Lai, W. Liau and L. Yang, The effect of ionic interaction on the miscibility and crystallization behaviors of poly(ethylene glycol)/poly(L-lactic acid) blends, J. Appl. Polym. Sci., 2008, 110(6), 3616–3623, DOI:10.1002/app.28878
- E. Piorkowska, Z. Kulinski, A. Galeski and R. Masirek, Plasticization of semicrystalline poly(L-lactide) with poly(propylene glycol), Polymer, 2006, 47(20), 7178–7188, DOI:10.1016/j.polymer.2006.03.115
- C. Nakafuku, High Pressure Crystallization of Poly(L-lactic acid) in a Binary Mixture with Poly(ethylene oxide), Polym. J., 1994, 26(6), 680–687, DOI:10.1295/polymj.26.680
- K. Nakane, Y. Hata, K. Morita, T. Ogihara and N. Ogata, Porous poly(L-lactic acid)/poly(ethylene glycol) blend films, J. Appl. Polym. Sci., 2004, 94(3), 965–970, DOI:10.1002/app.20959
- Y. S. Hu, M. Rogunova, V. Topolkaraev, A. Hiltner and E. Baer, Aging of poly(lactide)/poly(ethylene glycol) blends. Part 1. Poly(lactide) with low stereoregularity, Polymer, 2003, 44(19), 5701–5710, DOI:10.1016/S0032-3861(03)00614-1
- J.-M. Yang, H.-L. Chen, J.-W. You and J. C. Hwang, Miscibility and Crystallization of Poly(L-lactide)/Poly(ethylene glycol) and Poly(L-lactide)/Poly(ε-caprolactone) Blends, Polym. J., 1997, 29(8), 657–662, DOI:10.1295/polymj.29.657
- Y. Xu, W. Yu and C. Zhou, Liquid–liquid phase separation and its effect on the crystallization in polylactic acid/poly(ethylene glycol) blends, RSC Adv., 2014, 4(98), 55435–55444, 10.1039/c4ra08985e
- G. Sun, L.-T. Weng, J. M. Schultz and C.-M. Chan, Formation of banded and non-banded poly(l-lactic acid) spherulites during crystallization of films of poly(l-lactic acid)/poly(ethylene
oxide) blends, Polymer, 2014, 55(7), 1829–1836, DOI:10.1016/j.polymer.2014.02.036
- R. Bao, W. Yang, X. Wei, B. Xie and M. Yang, Enhanced Formation of Stereocomplex Crystallites of High Molecular Weight Poly(l-lactide)/Poly(d-lactide) Blends from Melt by Using Poly(ethylene glycol), ACS Sustainable Chem. Eng., 2014, 2(10), 2301–2309, DOI:10.1021/sc500464c
- M. P. Shaver and D. J. a. Cameron, Tacticity control in the synthesis of poly(lactic acid) polymer stars with dipentaerythritol cores, Biomacromolecules, 2010, 11(12), 3673–3679, DOI:10.1021/bm101140d
- Y.-L. Peng, Y. Huang, H.-J. Chuang, C.-Y. Y. Kuo and C.-C. C. Lin, Synthesis and characterization of biodegradable polylactides and polylactide-block-poly(Z-lysine) copolymers, Polymer, 2010, 51(19), 4329–4335, DOI:10.1016/j.polymer.2010.07.016
- P. Pan, Z. Liang, B. Zhu, T. Dong and Y. Inoue, Blending Effects on Polymorphic Crystallization of Poly(L-lactide), Macromolecules, 2009, 42, 3374–3380, DOI:10.1021/ma8024943
- F.-C. Chiu, C.-Y. Kan and J.-C. Yang, The effects of melt annealing and counterpart's molecular weight on the thermal properties and phase morphology of poly(L-lactide)-based blends, J. Polym. Sci., Part B: Polym. Phys., 2009, 47(15), 1497–1510, DOI:10.1002/polb.21751
- E. M. Woo, G. Lugito, Y.-T. Hsieh and S. Nurkhamidah, Unusual large-pitch banding in poly(L-lactic acid): effects of composition and geometry confinement, AIP Conf. Proc., 2014, 7, 7–13, DOI:10.1063/1.4866721
- E. M. Woo, G. Lugito and J.-H. Tsai, Effects of top confinement and diluents on morphology in crystallization of poly(L-lactic acid) interacting with poly(ethylene oxide), J. Polym. Sci., Part B: Polym. Phys., 2015, 53(16), 1160–1170, DOI:10.1002/polb.23756
- Y. He, B. Zhu, W. Kai and Y. Inoue, Nanoscale-Confined and Fractional Crystallization of Poly(ethylene oxide) in the Interlamellar Region of Poly(butylene succinate), Macromolecules, 2004, 37(9), 3337–3345, DOI:10.1021/ma035886g
- P. Pan, L. Zhao, J. Yang and Y. Inoue, Fractional Crystallization and Phase Segregation in Binary Miscible Poly(butylene succinate)/Poly(ethylene oxide) Crystalline Blends: Effect of Crystallization Temperature, Macromol. Mater. Eng., 2013, 298(2), 201–209, DOI:10.1002/mame.201200006
- Y. He, B. Zhu, W. Kai and Y. Inoue, Effects of crystallization condition of poly(butylene succinate) component on the crystallization of poly(ethylene oxide) component in their miscible blends, Macromolecules, 2004, 37, 8050–8056, DOI:10.1021/ma049482f
- L. Zhao, W. Kai, Y. He, B. Zhu and Y. Inoue, Effect of aging on fractional crystallization of poly(ethylene oxide) component in poly(ethylene oxide)/poly(3-hydroxybutyrate) blends, J. Polym. Sci., Part B: Polym. Phys., 2005, 43(19), 2665–2676, DOI:10.1002/polb.20552
- H. Wang, T. Zhao, X. Wang, P. Guo, L. Ren, T. Qiang, X. Luo and X. Qiang, Effects of crystallization condition of poly(ethylene succinate) on the crystallization of poly(ethylene oxide) in their blends, Polym. Bull., 2012, 69(8), 955–965, DOI:10.1007/s00289-012-0806-y
- H. Frensch and B.-J. Jungnickel, Some novel crystallization kinetic peculiarities in finely dispersing polymer blends, Colloid Polym. Sci., 1989, 267(1), 16–27, DOI:10.1007/BF01410144
- S. Bose, A. R. Bhattacharyya, P. V. Kodgire and A. Misra, Fractionated crystallization in PA6/ABS blends: influence of a reactive compatibilizer and multiwall carbon nanotubes, Polymer, 2007, 48(1), 356–362, DOI:10.1016/j.polymer.2006.11.019
- M. Jalabert, C. Fraschini and R. E. Prud’homme, Synthesis and characterization of poly(L-lactide)s and poly(D-lactide)s of controlled molecular weight, J. Polym. Sci., Part A: Polym. Chem., 2007, 45(10), 1944–1955, DOI:10.1002/pola.21960
- M. Bank, J. Leffingwell and C. Thies, The Influence of Solvent upon the Compatibility of Polystyrene and Poly(vinyl methyl ether), Macromolecules, 1971, 4(1), 43–46, DOI:10.1021/ma60019a010
- L. Mandelkern, Crystallization of Polymers, Cambridge University Press, Nova York, 2nd edn, 2002 Search PubMed
- F. Xue, X. Chen, L. An, S. S. Funari and S. Jiang, Crystallization induced layer-to-layer transitions in symmetric PEO-b-PLLA block copolymer with synchrotron simultaneous SAXS/WAXS investigations, RSC Adv., 2014, 4(99), 56346–56354, 10.1039/C4RA09284H
- M. V. Massa, J. L. Carvalho and K. Dalnoki-Veress, Direct visualisation of homogeneous and heterogeneous crystallisation in an ensemble of confined domains of poly(ethylene oxide), Eur. Phys. J. E: Soft Matter Biol. Phys., 2003, 12(1), 111–117, DOI:10.1140/epje/i2003-10045-3
- M. V. Massa and K. Dalnoki-Veress, Homogeneous Crystallization of Poly(ethylene oxide) Confined to Droplets: The Dependence of the Crystal Nucleation Rate on Length Scale and Temperature, Phys. Rev. Lett., 2004, 92(25), 255509, DOI:10.1103/PhysRevLett.92.255509
- M. V. Massa, J. L. Carvalho and K. Dalnoki-Veress, Confinement effects in polymer crystal nucleation from the bulk to few-chain systems, Phys. Rev. Lett., 2006, 97(24), 1–4, DOI:10.1103/PhysRevLett.97.247802
- S. Jiang, D. Yu, X. Ji, L. An and B. Jiang, Confined crystallization behavior of PEO in silica networks, Polymer, 2000, 41(6), 2041–2046, DOI:10.1016/S0032-3861(99)00342-0
- J. Maiz, J. Martin and C. Mijangos, Confinement effects on the crystallization of poly(ethylene oxide) Nanotubes, Langmuir, 2012, 28(33), 12296–12303, DOI:10.1021/la302675k
- Y. Guan, G. Liu, P. Gao, L. Li, G. Ding and D. Wang, Manipulating crystal orientation of poly(ethylene oxide) by nanopores, ACS Macro Lett., 2013, 2, 181–184, DOI:10.1021/mz300592v
- Y. Suzuki, H. Duran, M. Steinhart, H.-J. Butt and G. Floudas, Homogeneous crystallization and local dynamics of poly(ethylene oxide) (PEO) confined to nanoporous alumina, Soft Matter, 2013, 9(207890), 2621–2628, 10.1039/c2sm27618f
- R. M. Michell, I. Blaszczyk-Lezak, C. Mijangos and A. J. Müller, Confined crystallization of polymers within anodic aluminum oxide templates, J. Polym. Sci., Part B: Polym. Phys., 2014, 52(18), 1179–1194, DOI:10.1002/polb.23553
- Y.-T. Hsieh, S. Nurkhamidah and E. M. Woo, Lamellar orientation and interlamellar cracks in co-crystallized poly(ethylene oxide)/poly(L-lactic acid) blend, Polym. J., 2011, 43, 762–769, DOI:10.1038/pj.2011.63
- O. Olabisi, L. M. Robeson and M. T. Shaw, Polymer–Polymer Miscibility, Academic Press, New York, 1979 Search PubMed
- H. Duran, M. Steinhart, H.-J. Butt and G. Floudas, From Heterogeneous to Homogeneous Nucleation of Isotactic Poly(propylene) Confined to Nanoporous Alumina, Nano Lett., 2011, 11(4), 1671–1675, DOI:10.1021/nl200153c
- J. Zhang, K. Tashiro, H. Tsuji and A. J. Domb, Disorder-to-Order Phase Transition and Multiple Melting Behavior of Poly(L-lactide) Investigated by Simultaneous Measurements of WAXD and DSC, Macromolecules, 2008, 41(4), 1352–1357, DOI:10.1021/ma0706071
- S. Saeidlou, M. A. Huneault, H. Li and C. B. Park, Poly(lactic acid) crystallization, Prog. Polym. Sci., 2012, 37(12), 1657–1677, DOI:10.1016/j.progpolymsci.2012.07.005
- S. Radhakrishnan and P. D. Venkatachalapathy, Effect of casting solvent on the crystallization in PEO/PMMA blends, Polymer, 1996, 37(16), 3749–3752, DOI:10.1016/0032-3861(96)00192-9
- D. Zhou, J. Shao, G. Li, J. Sun, X. Bian and X. Chen, Crystallization behavior of PEG/PLLA block copolymers: effect of the different architectures and molecular weights, Polymer, 2015, 62, 70–76, DOI:10.1016/j.polymer.2015.02.019
- J. Yang, Y. Liang, J. Luo, C. Zhao and C. C. Han, Multilength Scale Studies of the Confined Crystallization in Poly(L-lactide)-Block-Poly(ethylene glycol) Copolymer, Macromolecules, 2012, 45(10), 4254–4261, DOI:10.1021/ma202505f
- J. F. Mano, Y. Wang, J. C. Viana, Z. Denchev and M. J. Oliveira, Cold Crystallization of PLLA Studied by Simultaneous SAXS and WAXS, Macromol. Mater. Eng., 2004, 289(10), 910–915, DOI:10.1002/mame.200400097
- Z. Qiu, T. Ikehara and T. Nishi, Unique morphology of poly(ethylene succinate)/poly(ethylene oxide) blends, Macromolecules, 2002, 35(22), 8251–8254, DOI:10.1021/ma025599x
- R. S. Kurusu, N. R. Demarquette, C. Gauthier and J.-M. Chenal, Effect of ageing and annealing on the mechanical behaviour and biodegradability of a poly(3-hydroxybutyrate) and poly(ethylene-co-methyl-acrylate-co-glycidyl-methacrylate) blend, Polym. Int., 2014, 63(6), 1085–1093, DOI:10.1002/pi.4616
- S. A. Liberman, A. D. S. Gomes and E. M. Macchi, Compatibility in poly(ethylene oxide)–poly(methyl methacrylate) blends, J. Polym. Sci., Polym. Chem. Ed., 1984, 22, 2809–2815, DOI:10.1002/pol.1984.170221107
- Z. Qiu, T. Ikehara and T. Nishi, Miscibility and crystallization of poly(ethylene oxide) and poly(ε-caprolactone) blends, Polymer, 2003, 44(10), 3101–3106, DOI:10.1016/S0032-3861(03)00167-8
Footnote |
| † Electronic supplementary information (ESI) available: (SI 01) FTIR spectra, (SI 02) GPC chromatograms, (SI 03) DSC curves for all studied PEO/PLLA blends, (SI 04) POM cooling video, (SI 05) SEM images, (SI 06) POM 2nd heating video, and (SI 07) XRD zoom, is supplied. See DOI: 10.1039/c6ra02406h |
| This journal is © The Royal Society of Chemistry 2016 |