
The synthesis and ring-opening metathesis polymerization of glycomonomers†
Lucy G. Weaver*a,
Yogendra Singha,
Paul L. Burnb and
Joanne T. Blanchfield*a
aThe School of Chemistry & Molecular Biosciences, University of Queensland, St Lucia, QLD 4072, Australia. E-mail: lucy.weaver@uqconnect.edu.au; j.blanchfield@uq.edu.au
bCentre for Organic Photonics & Electronics, University of Queensland, St Lucia, QLD 4072, Australia
First published on 22nd March 2016
Abstract
The synthesis of a series of short poly(norbornene)s displaying pendant disaccharides is reported. para-(Propargyloxy)benzyl moieties were attached to a norbornenyl group via an ester or amide linkage, giving two different pre-monomers. A set of protected β-(1→6)-linked glucosamine-based disaccharides, structurally similar to the bacterial biofilm constituent poly-N-acetylglucosamine (PNAG), were attached to the pre-monomers via a Huisgen 1,3-dipolar cycloaddition reaction to generate a series of ‘glycomonomers’. In the presence of a Grubbs' 3rd generation catalyst, the glycomonomers displayed variable reactivities that were dependent on the type of linkage (ester or amide) between the norbornenyl and benzyl moieties. In general, the amide-linked glycomonomers polymerized at a much slower rate and had a comparatively lower degree of polymerization than the corresponding ester-linked constructs. All the materials displayed a relatively narrow molecular weight distribution (Đ = 1.2–1.5).
Introduction
The side-chain density, multiplicity of architectures and biocompatibility of polymers are key properties that have been exploited in the burgeoning glyconanotechnology field.1,2 Polymer backbones with pendant oligosaccharide chains, so-called ‘glycopolymers’, have found particular prominence in the synthesis of well-defined chemical entities that are able to mimic naturally derived polysaccharides.3–7 Given the role polysaccharides play in biological systems such as cell–cell adhesion, structure–function recognition and signal transduction, the ability to synthesize multivalent biomacromolecules that are able to augment function in place of the native polysaccharide is highly desirable.To date, synthetic glycopolymers have been investigated for ligand–receptor binding (e.g., carbohydrate lectins), and in the mimicry of glycosaminoglycans (GAG) and mucins.5,6,8–13 In addition to other ‘living’ radical polymerization techniques such as RAFT and ATRP, many of the strategies investigated for the above applications have employed ring-opening metathesis polymerization (ROMP) for the synthesis of glycopolymers,14,15 as this method usually produces well-defined products in a controlled manner and in high yields, with no post-polymerization modifications required. In addition, this method is reported to encompass a high degree of functional group tolerance during polymerization reactions, making it compatible with carbohydrate entities both in their protected and unprotected forms.6
Synthesis of glycopolymers via ROMP is typically achieved by polymerizing a carbohydrate monomer (glycomonomer) or by synthesis of a functionalized polymer backbone followed by attachment of the oligosaccharide chains.12,16 Both methodologies have distinct advantages and disadvantages. Glycopolymer synthesis from a glycomonomer usually requires multistep synthetic routes that can be lengthy and inefficient, but generally produces better-defined products overall, as the composition of each repeat unit is predetermined. On the other hand, attaching multiple carbohydrate units to a polymer backbone in one step can be achieved using selective and efficient reactions (e.g. the Huisgen 1,3-dipolar cycloaddition reaction) but this can result in a mixture of products that are only partially functionalized.17–19
Drawing upon the past successes in this field, we herein describe the application of ROMP in the development of glycopolymers aimed at emulating the polysaccharide, poly-N-acetylglucosamine (PNAG), produced by Staphylococcus aureus and Staphylococcus epidermidis.20–22 We describe the preparation of functionalised pre-monomers (1 and 2; Fig. 1) containing para-(propargyloxy)benzyl moieties connected to norbornenyl units, and show they can be attached to protected disaccharides. ROMP then results in glycopolymers depending on the structure of the monomer. This research presents the first application of ROMP in the synthesis of polymers containing β-(1→6)-linked glucosamine-based carbohydrates with a view to provide a clustered repetitious display of disaccharides to enable research exploring the immunochemistry associated with biofilm-producing bacterial specimens.23–27
 | ||
| Fig. 1 The two norbornenyl-based pre-monomer units 1 and 2. | ||
Experimental
Materials and methods
All chemicals were obtained from Sigma-Aldrich. All solvents were obtained from either Sigma-Aldrich or Merck, and were reagent grade, unless otherwise stated. Tetrahydrofuran was distilled from sodium-benzophenone ketyl prior to use. Anhydrous dichloromethane and toluene were obtained by distillation from calcium hydride. Thin layer chromatography (TLC) was performed on aluminium-backed silica plates and were visualized in either anisidine phthalate dip or under a UV lamp. Flash column chromatography was performed using silica gel 60 (0.04–0.06 mm, 230–400 mesh, Scharlau or Merck) unless otherwise stated. High-resolution mass spectrometry (HR-ESIMS) was performed in positive electrospray ionization mode. UV-Visible absorption measurements were recorded on a Perkin Elmer Lambda 35 Spectrometer. Gel Permeation Chromatography for all compounds was carried out on a Waters 2695 Separations Module, fitted with two Ultrastyragel linear columns (7.8 × 300 mm) and one Styragel linear column connected in series and at a constant temperature of 40 °C for all analyses. Tetrahydrofuran was used as the eluent, under a flow rate of 1.0 mL min−1. Calibration was carried out using narrow molecular weight polystyrene standards (PDI < 1.1) ranging between 500 and 2 million g mol−1. Sample detection was achieved using a Waters 410 refractive index detector and a Waters 996 Photodiode Array Detector, which were connected in series. NMR spectra were recorded on either a 400 MHz or 500 MHz spectrometer. Proton and carbon assignments were made using COSY, TOCSY, HSQC and HMBC experiments. Spectra of experiments run in CDCl3 or a mixture of CDCl3/CD3OD were referenced to residual CHCl3 at δ 7.26 ppm for the 1H NMR and to CDCl3 at δ 77.0 ppm for the 13C NMR.Pre-monomer synthesis
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–CH2), 3.84 (s, 3H, OCOCH3), 4.71 (d, 2H, 4JCH2,CH = 2.4 Hz, HCC–CH2), 6.96 and 7.97 (AA′BB′, 4H, Ph-H); 13C NMR (100 MHz; CDCl3): δ 51.8 (OCOCH3), 55.7 (HCC–CH2), 76.0 (HCC–CH2), 77.7 (HCC–CH2), 114.4 (Ph-CH), 123.3 (Ph-C–), 131.4 (Ph-CH), 161.0 (Ph-C–), 166.5 (C
C–CH2), 3.84 (s, 3H, OCOCH3), 4.71 (d, 2H, 4JCH2,CH = 2.4 Hz, HCC–CH2), 6.96 and 7.97 (AA′BB′, 4H, Ph-H); 13C NMR (100 MHz; CDCl3): δ 51.8 (OCOCH3), 55.7 (HCC–CH2), 76.0 (HCC–CH2), 77.7 (HCC–CH2), 114.4 (Ph-CH), 123.3 (Ph-C–), 131.4 (Ph-CH), 161.0 (Ph-C–), 166.5 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O); HRESIMS: calc. for C11H10NaO3+ [M + Na]+ m/z = 213.0522, found: 213.0529. The data is in agreement with that previously reported.28C–CH2), 4.62 (s, 2H, OH–CH2–Ph), 4.69 (d, 2H, 4JCH2,CH = 2.4 Hz, HCC–CH2), 6.97 and 7.30 (AA′BB′, 4H, Ph-H); 13C NMR (100 MHz; CDCl3): δ 55.9 (HCC–CH2), 64.9 (OH–CH2–Ph), 75.5 (HCC–CH2), 78.5 (HCC–CH2), 115.0 (Ph-CH), 128.6 (Ph-CH), 134.1 (Ph-C–), 157.1 (Ph-C–); HRESIMS: calc. for C10H10NaO2+ [M + Na]+ m/z = 185.0573, found: 185.0578. The data is in agreement with that previously reported.29
O); HRESIMS: calc. for C11H10NaO3+ [M + Na]+ m/z = 213.0522, found: 213.0529. The data is in agreement with that previously reported.28C–CH2), 4.62 (s, 2H, OH–CH2–Ph), 4.69 (d, 2H, 4JCH2,CH = 2.4 Hz, HCC–CH2), 6.97 and 7.30 (AA′BB′, 4H, Ph-H); 13C NMR (100 MHz; CDCl3): δ 55.9 (HCC–CH2), 64.9 (OH–CH2–Ph), 75.5 (HCC–CH2), 78.5 (HCC–CH2), 115.0 (Ph-CH), 128.6 (Ph-CH), 134.1 (Ph-C–), 157.1 (Ph-C–); HRESIMS: calc. for C10H10NaO2+ [M + Na]+ m/z = 185.0573, found: 185.0578. The data is in agreement with that previously reported.29![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ε/M−1 cm−1, 4.05), 273 (3.19), 279 (3.11); 1H NMR (400 MHz; CDCl3): δ 1.35–1.39 (m, 2H, CH2), 1.52–1.54 (m, 1H, CH2), 1.93 (dt, 1H, 2JCH,CH = 11.8, JCH,CH = 4.0 Hz, CH2), 2.25 (q, 1H, JCH,CH = 5.6, JCH,CH2 = 4.5 Hz, CH), 2.53 (t, 1H, 4JCH,CH2 = 2.4 Hz, HCC–CH2), 2.91 (br s, 1H, CH), 3.04–3.05 (br m, 1H, CH), 4.69 (d, 2H, 4JCH2,CH = 2.4 Hz, HCC–CH2), 5.07 (s, 2H, O–CH2–Ph), 6.09 (dd, 1H, JCH,CH = 5.6, JCH,CH = 2.9 Hz, HCCH), 6.13 (dd, 1H, JCH,CH = 5.6, JCH,CH = 2.9 Hz, HCCH), 6.97 and 7.31 (AA′BB′, 4H, Ph-H); 13C NMR (100 MHz; CDCl3): δ 30.3 (CH2), 41.6 (CH), 43.1 (CH), 46.3 (bridge CH2), 46.6 (CH), 55.7 (HCC–CH2), 65.8 (O–CH2–Ph), 75.6 (HCC–CH2), 78.4 (HCC–CH2), 114.9 (Ph-CH), 129.3 (Ph-C–), 129.8 (Ph-CH), 135.7 (HCCH), 138.0 (HCCH), 157.4 (Ph-C–), 176.0 (CO); HRESIMS: calc. for C18H18NaO3+ [M + Na]+ m/z = 305.1148, found: 305.1141; IR (cm−1): 3290, 2974, 2122, 1722, 1512, 1217, 1165, 1151, 1027, 822, 718.C–CH2), 4.28 (s, 2H, N3–CH2–Ph), 4.70 (d, 2H, 4JCH2,CH = 2.4 Hz, HCC–CH2), 6.99 and 7.26 (AA′BB′, 4H, Ph-H); 13C NMR (100 MHz; CDCl3): δ 54.2 (N3–CH2–Ph), 55.8 (HCC–CH2), 75.6 (HCC–CH2), 78.3 (HCC–CH2), 115.1 (Ph-CH), 128.4 (Ph-C–), 129.6 (Ph-CH), 157.5 (Ph-C–). The data is in agreement with that previously reported.30:1 v/v) with the exclusion of oxygen, and triphenylphosphine (1.70 g, 6.49 mmol) was added. The reaction mixture was stirred at room temperature for 24 h. The solvent was removed under reduced pressure and the residue was dried further under high vacuum. The crude product was purified using flash silica gel column chromatography, first with 90% ethyl acetate/petroleum spirit (40–60 °C), followed by ethyl acetate containing increasing quantities of methanol as the eluent. The title compound 7 (ref. 31) eluted in 10–25% methanol/ethyl acetate and was dried to give a white solid (0.39 g, 82%). Rf 0.08 (20% methanol/ethyl acetate); 1H NMR (400 MHz; CDCl3): δ 1.69 (br s, 2H, NH2), 2.51 (t, 1H, 4JCH,CH2 = 2.4 Hz, HCC–CH2), 3.81 (s, 2H, NH2–CH2–Ph), 4.67 (d, 2H, 4JCH2,CH = 2.4 Hz, HCC–CH2), 6.94 and 7.24 (AA′BB′, 4H, Ph-H); 13C NMR (100 MHz; CDCl3): δ 45.8 (NH2–CH2–Ph), 55.8 (HCC–CH2), 75.4 (HCC–CH2), 78.6 (HCC–CH2), 114.9 (Ph-CH), 128.2 (Ph-CH), 136.4 (Ph-C–), 156.4 (Ph-C–). The data is in agreement with that previously reported.31ε/M−1 cm−1, 4.00), 275 (3.18), 282 (3.12); 1H NMR (400 MHz; CDCl3): δ 1.30–1.34 (m, 1H, CH2), 1.35–1.39 (m, 1H, CH2), 1.75 (br dt, 1H, CH2), 1.92–1.97 (m, 1H, CH2), 2.00 (dq, 1H, JCH,CH = 5.4, JCH,CH2 = 4.4, JCH,CH2 = 1.3 Hz, CH), 2.51 (t, 1H, 4JCH,CH2 = 2.4 Hz, HCC–CH2), 2.91–2.96 (br m, 2H, CH), 4.40 (ABX, 2H, NH–CH2–Ph), 4.69 (d, 2H, 4JCH2,CH = 2.4 Hz, HCC–CH2), 5.70 (br s, 1H, NH), 6.08 (dd, 1H, JCH,CH = 5.6, JCH,CH = 2.9 Hz, HCCH), 6.14 (dd, 1H, JCH,CH = 5.6, JCH,CH = 2.9 Hz, HCCH), 6.95 and 7.23 (AA′BB′, 4H, Ph-H); 13C NMR (100 MHz; CDCl3): δ 30.6 (CH2), 41.6 (CH), 43.2 (NH–CH2–Ph), 44.8 (CH), 46.4 (bridge CH2), 47.2 (CH), 55.9 (HCC–CH2), 75.5 (HCC–CH2), 78.5 (HCC–CH2), 115.2 (Ph-CH), 129.1 (Ph-CH), 131.7 (Ph-C–), 136.0 (HCCH), 138.3 (HCCH), 157.0 (Ph-C–), 175.3 (CO); HRESIMS: calc. for C18H19NaNO2+ [M + Na]+ m/z = 304.1308, found: 304.1316; IR (cm−1): 3279, 3059, 2918, 2135, 1635, 1511, 1236, 1023.
ε/M−1 cm−1, 4.05), 273 (3.19), 279 (3.11); 1H NMR (400 MHz; CDCl3): δ 1.35–1.39 (m, 2H, CH2), 1.52–1.54 (m, 1H, CH2), 1.93 (dt, 1H, 2JCH,CH = 11.8, JCH,CH = 4.0 Hz, CH2), 2.25 (q, 1H, JCH,CH = 5.6, JCH,CH2 = 4.5 Hz, CH), 2.53 (t, 1H, 4JCH,CH2 = 2.4 Hz, HCC–CH2), 2.91 (br s, 1H, CH), 3.04–3.05 (br m, 1H, CH), 4.69 (d, 2H, 4JCH2,CH = 2.4 Hz, HCC–CH2), 5.07 (s, 2H, O–CH2–Ph), 6.09 (dd, 1H, JCH,CH = 5.6, JCH,CH = 2.9 Hz, HCCH), 6.13 (dd, 1H, JCH,CH = 5.6, JCH,CH = 2.9 Hz, HCCH), 6.97 and 7.31 (AA′BB′, 4H, Ph-H); 13C NMR (100 MHz; CDCl3): δ 30.3 (CH2), 41.6 (CH), 43.1 (CH), 46.3 (bridge CH2), 46.6 (CH), 55.7 (HCC–CH2), 65.8 (O–CH2–Ph), 75.6 (HCC–CH2), 78.4 (HCC–CH2), 114.9 (Ph-CH), 129.3 (Ph-C–), 129.8 (Ph-CH), 135.7 (HCCH), 138.0 (HCCH), 157.4 (Ph-C–), 176.0 (CO); HRESIMS: calc. for C18H18NaO3+ [M + Na]+ m/z = 305.1148, found: 305.1141; IR (cm−1): 3290, 2974, 2122, 1722, 1512, 1217, 1165, 1151, 1027, 822, 718.C–CH2), 4.28 (s, 2H, N3–CH2–Ph), 4.70 (d, 2H, 4JCH2,CH = 2.4 Hz, HCC–CH2), 6.99 and 7.26 (AA′BB′, 4H, Ph-H); 13C NMR (100 MHz; CDCl3): δ 54.2 (N3–CH2–Ph), 55.8 (HCC–CH2), 75.6 (HCC–CH2), 78.3 (HCC–CH2), 115.1 (Ph-CH), 128.4 (Ph-C–), 129.6 (Ph-CH), 157.5 (Ph-C–). The data is in agreement with that previously reported.30:1 v/v) with the exclusion of oxygen, and triphenylphosphine (1.70 g, 6.49 mmol) was added. The reaction mixture was stirred at room temperature for 24 h. The solvent was removed under reduced pressure and the residue was dried further under high vacuum. The crude product was purified using flash silica gel column chromatography, first with 90% ethyl acetate/petroleum spirit (40–60 °C), followed by ethyl acetate containing increasing quantities of methanol as the eluent. The title compound 7 (ref. 31) eluted in 10–25% methanol/ethyl acetate and was dried to give a white solid (0.39 g, 82%). Rf 0.08 (20% methanol/ethyl acetate); 1H NMR (400 MHz; CDCl3): δ 1.69 (br s, 2H, NH2), 2.51 (t, 1H, 4JCH,CH2 = 2.4 Hz, HCC–CH2), 3.81 (s, 2H, NH2–CH2–Ph), 4.67 (d, 2H, 4JCH2,CH = 2.4 Hz, HCC–CH2), 6.94 and 7.24 (AA′BB′, 4H, Ph-H); 13C NMR (100 MHz; CDCl3): δ 45.8 (NH2–CH2–Ph), 55.8 (HCC–CH2), 75.4 (HCC–CH2), 78.6 (HCC–CH2), 114.9 (Ph-CH), 128.2 (Ph-CH), 136.4 (Ph-C–), 156.4 (Ph-C–). The data is in agreement with that previously reported.31ε/M−1 cm−1, 4.00), 275 (3.18), 282 (3.12); 1H NMR (400 MHz; CDCl3): δ 1.30–1.34 (m, 1H, CH2), 1.35–1.39 (m, 1H, CH2), 1.75 (br dt, 1H, CH2), 1.92–1.97 (m, 1H, CH2), 2.00 (dq, 1H, JCH,CH = 5.4, JCH,CH2 = 4.4, JCH,CH2 = 1.3 Hz, CH), 2.51 (t, 1H, 4JCH,CH2 = 2.4 Hz, HCC–CH2), 2.91–2.96 (br m, 2H, CH), 4.40 (ABX, 2H, NH–CH2–Ph), 4.69 (d, 2H, 4JCH2,CH = 2.4 Hz, HCC–CH2), 5.70 (br s, 1H, NH), 6.08 (dd, 1H, JCH,CH = 5.6, JCH,CH = 2.9 Hz, HCCH), 6.14 (dd, 1H, JCH,CH = 5.6, JCH,CH = 2.9 Hz, HCCH), 6.95 and 7.23 (AA′BB′, 4H, Ph-H); 13C NMR (100 MHz; CDCl3): δ 30.6 (CH2), 41.6 (CH), 43.2 (NH–CH2–Ph), 44.8 (CH), 46.4 (bridge CH2), 47.2 (CH), 55.9 (HCC–CH2), 75.5 (HCC–CH2), 78.5 (HCC–CH2), 115.2 (Ph-CH), 129.1 (Ph-CH), 131.7 (Ph-C–), 136.0 (HCCH), 138.3 (HCCH), 157.0 (Ph-C–), 175.3 (CO); HRESIMS: calc. for C18H19NaNO2+ [M + Na]+ m/z = 304.1308, found: 304.1316; IR (cm−1): 3279, 3059, 2918, 2135, 1635, 1511, 1236, 1023.General cycloaddition procedure
Detailed experimental procedures for the synthesis of glycomonomers 15, 16, and 17 can be found in the ESI† section. All cycloaddition experiments were performed in sealed reaction vessels under inert conditions and using freshly distilled solvents. The general procedure involved dissolving a mixture of pre-monomer (1 or 2), disaccharide (8, 9 or 10) and N,N,N′,N′′,N′′-pentamethyldiethylenetriamine in dry toluene (3 mL; with the addition of 0.5 mL of N,N-dimethylformamide to solubilize the components if necessary) and the solutions were purged with argon for 20 min. Copper(I) bromide was then added and the reaction mixture was purged with argon for a further 3 min, before the reaction vessel was sealed, and the mixture stirred at room temperature for 4 d, protected from light. After this time, air was slowly bubbled through the dark green solution for 10 min. The solvent was then removed under reduced pressure. The crude glycomonomers were purified using flash silica gel column chromatography using different mixtures of petroleum spirit (40–60 °C), ethyl acetate and methanol as the eluents. The isolated fractions were dried to give the products as white solids.General polymerization procedure
Detailed experimental procedures for the synthesis of polymers 18, 19, and 20 can be found in the ESI† section. All polymerization experiments were performed in sealed reaction vessels under inert conditions and using freshly distilled solvents. The general procedure involved dissolving a solution of glycomonomer in dry solvent (1 mL) followed by the addition of Grubbs' catalyst in 0.5 mL of solvent under anhydrous conditions. The reaction vessel was sealed and the mixture was stirred either at room temperature or 60 °C for 1–2 d, protected from light. After this time, ethylvinyl ether (0.5 mL) was added to quench the reaction. The solvent was then removed under reduced pressure. GPC analysis was undertaken of the crude polymers, which were then purified using flash silica gel column chromatography (Davisil® LC150 Å 35–70 μm) with ethyl acetate containing increasing quantities of methanol as the eluent. The desired products eluted in 10% methanol/ethyl acetate and were dried to give glassy solids. The solids were redissolved in dichloromethane (1 mL) and precipitated by pouring into diethyl ether (40 mL). The precipitates were isolated by centrifugation (3000 rpm, 3 min) and the process was repeated twice more before the solids were dried under high vacuum.Results and discussion
Synthesis and characterization of the pre-monomers 1 and 2
Pre-monomers 1 and 2 were synthesized from methyl-4-hydroxybenzoate (3) in three and five steps, respectively (Scheme 1). Compound 3 was treated with propargyl bromide in the presence of a base to attach an alkyne moiety to the para-hydroxyl group, giving 4 in quantitative yield. Reduction of the methyl ester of 4 using lithium aluminum hydride generated the benzyl alcohol 5, again in quantitative yield. The primary alcohol of 5 was then esterified with (1R,2S,4R)-bicyclo[2.2.1]hept-5-ene-2-carboxylic acid (also called exo-5-norbornene carboxylic acid) to give pre-monomer 1 in 87% yield. Synthesis of pre-monomer 2 was achieved following conversion of the benzyl alcohol intermediate 5 to the corresponding azide 6 in the presence of diphenyl phosphoryl azide (DPPA) and a base (1,8-diazabicyclo-undec-7-ene; DBU). The azide of 6 was reduced to the primary amine, giving 7 in 82% yield. Reaction of 7 with exo-5-norbornene carboxylic acid produced pre-monomer 2 in 46% yield, which had an amide linkage between the norbornene and benzyl moieties, rather than an ester linkage as in 1.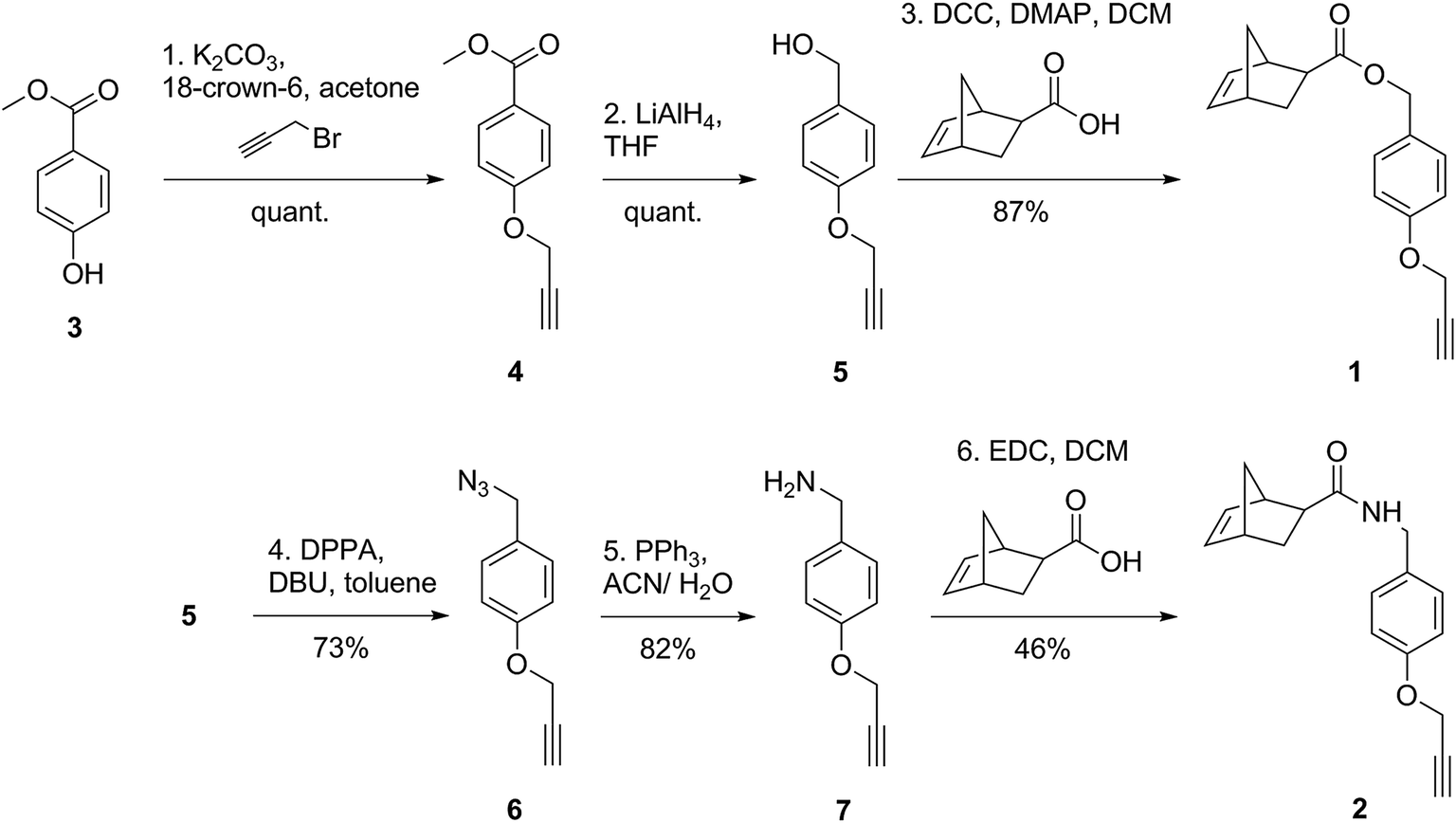 | ||
| Scheme 1 Synthesis of the ester-linked pre-monomer 1 and the amide-linked pre-monomer 2. | ||
Although the attachment of the norbornenyl moiety in compound 1 was achieved in high yield using Steglich esterification conditions (DCC/DMAP mixture),32 the optimal conditions for the synthesis of compound 2 utilized EDC instead. This stemmed from the fact that, during the purification of compound 7, it was evident that the presence of the amine group caused a significant increase in its polarity compared to the hydroxyl derivative 5. Thus, due to the expected higher polarity of the desired amide product 2 compared to ester 1, it was anticipated that the N,N′-dicyclohexylurea by-product from the DCC coupling reagent would be difficult to separate from compound 2 by chromatography. EDC on the other hand generates a water-soluble by-product that was easily removed from the crude reaction mixture by extraction with water prior to chromatography.
In addition to NMR analysis, infrared spectroscopy provided structural confirmation of the functional groups present in compounds 1 and 2. The spectra depicted characteristic bands of unsaturation at approximately 3100 cm−1 and 718 cm−1 corresponding to the norbornenyl alkene and benzyl groups, and a propargyl C–H stretch at ∼3290 cm−1. In addition, 1 had a carbonyl stretch at 1722 cm−1 corresponding to the ester, while 2 had the amide carbonyl stretch at 1635 cm−1.
Carbohydrate synthesis
Three different disaccharides, 8, 9 and 10, were used during the glycopolymer synthesis as shown in Fig. 2. The purpose of using differently protected disaccharides (8–10) was to directly assess whether the protecting group strategy had any effects on the ROMP reactions. It is important to note that the critical protecting groups with respect to the polymerization are those on the glycoside ring with the azide moiety, as these are the ones that will be closest to the reactive component of the monomer during the ROMP reaction. Therefore, in this work we compare the effects of the acetate (Ac) and benzoate (Bz) protecting groups.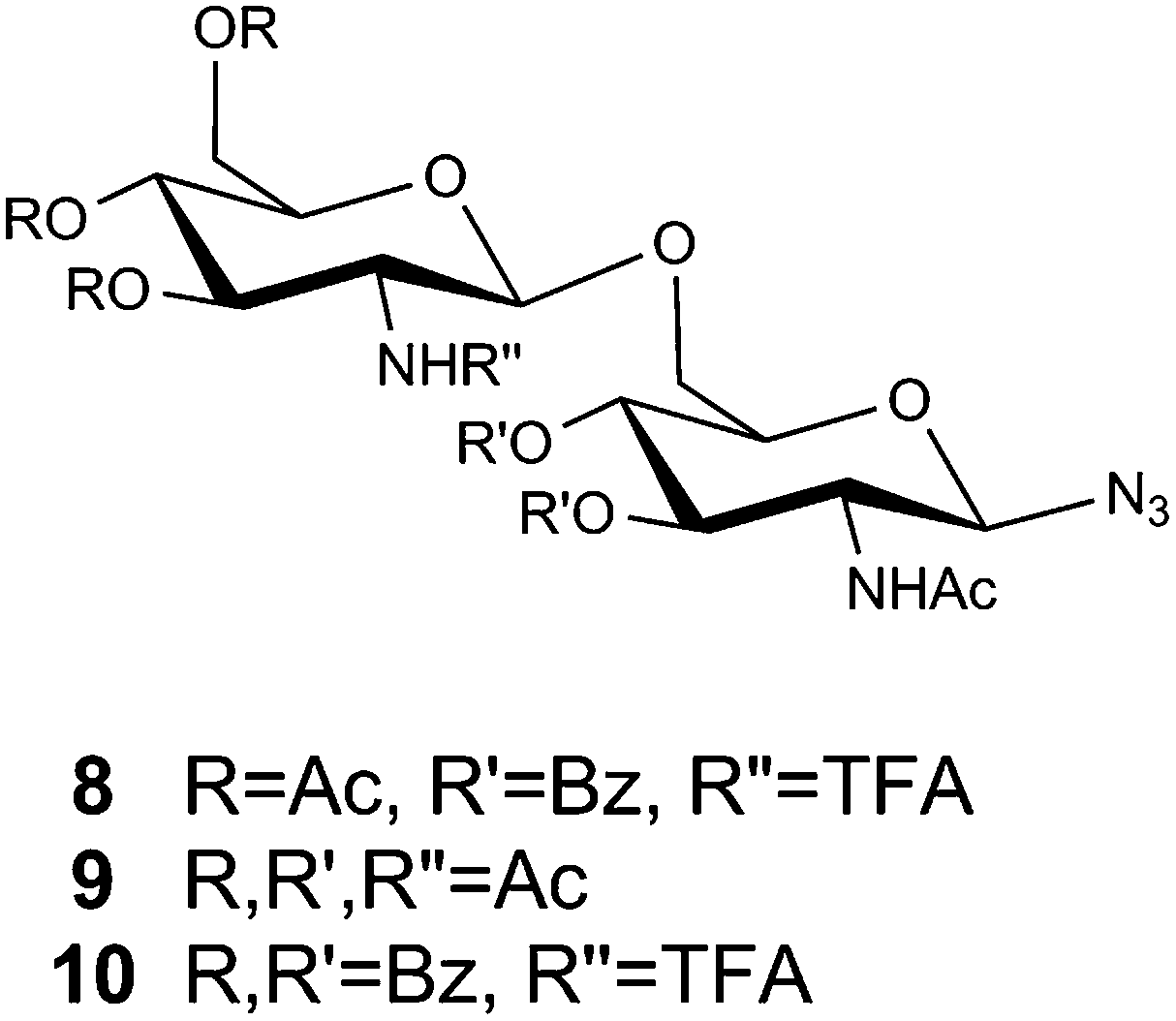 | ||
| Fig. 2 The disaccharides 8, 9 and 10 used in the synthesis of the glycopolymers. | ||
The synthesis of disaccharides 8 and 9 is shown in Scheme 2, with the synthesis of disaccharide 10 reported previously.33 Briefly, 8 was synthesized following glycosylation of acceptor 11 with donor 12 under NIS/triflate promoted conditions. Compound 9 was synthesized via condensation of 13 and 14 under benzyltriethylammonium chloride catalyzed conditions, generating the per-acetylated disaccharide in 10% isolated yield.34 The low yield from this reaction stems primarily from a competing intramolecular acyl migration of the C-4 acetyl group to the C-6 hydroxyl position, which generated a significant amount of the β-(1→4) disaccharide product that was isolated via HPLC. A secondary by-product that was also isolated was 3,4,6-tri-O-acetyl-2-acetamido-2-deoxy-α-D-glucopyranose, which was produced upon hydrolysis of the glycosyl chloride donor and any activated intermediates that were present when the reaction was worked up.
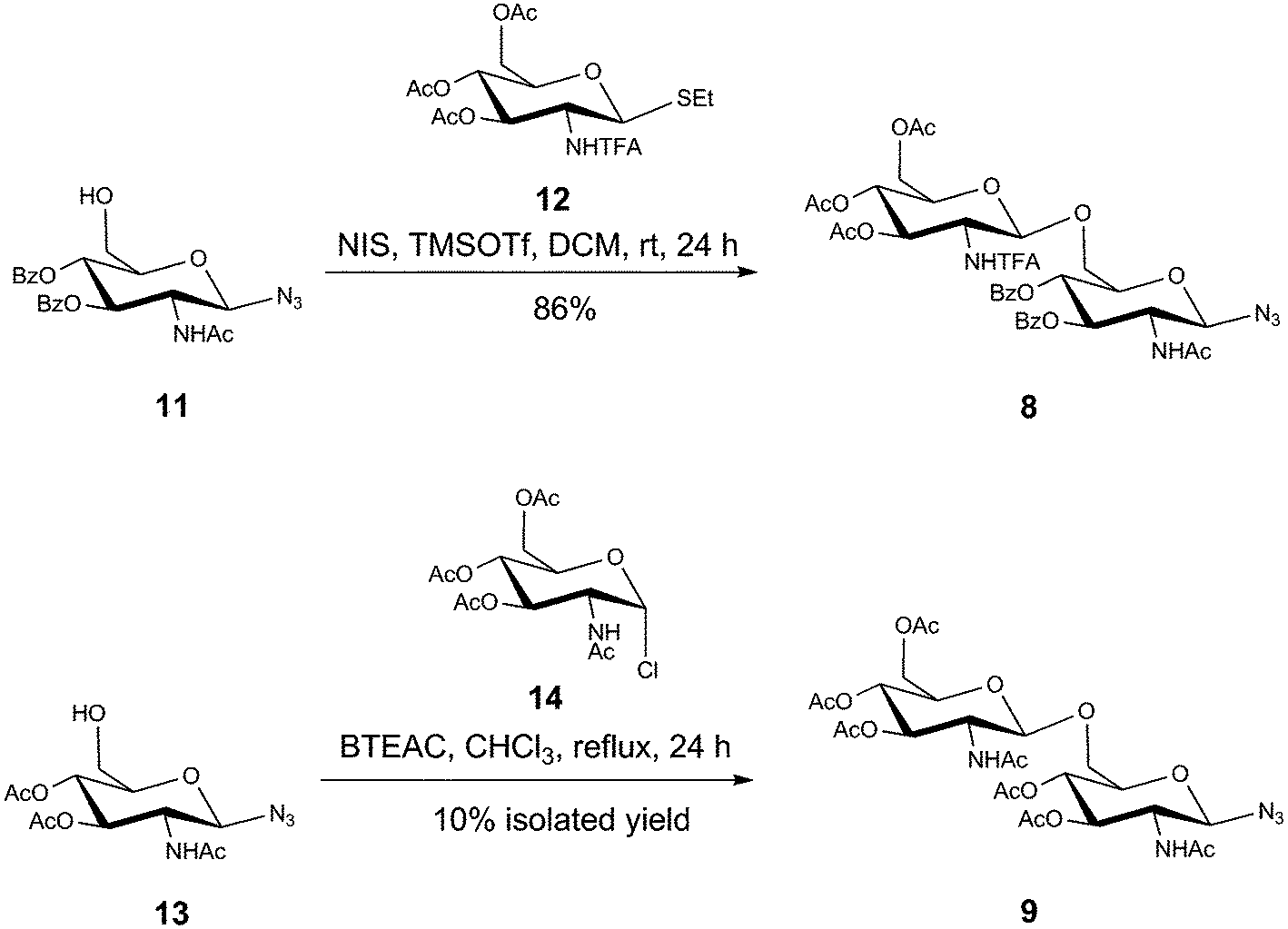 | ||
| Scheme 2 Synthesis of the disaccharides 8 and 9. | ||
Synthesis and characterization of the glycomonomers
The synthesis of glycomonomers 15, 16 and 17 was performed under Huisgen 1,3-dipolar cycloaddition reaction conditions (Scheme 3) that installed a triazole ring between the monomer (1 or 2) and the relevant disaccharide unit (8, 9 or 10). By treating a solution of the two starting materials (monomer and carbohydrate) with copper(I) bromide and N,N,N′,N′′,N′′-pentamethyldiethylenetriamine (PMDETA) in toluene (plus N,N-dimethylformamide in some cases), glycomonomers 15, 16 and 17 were formed in yields ranging between 60 and 99%. The particular success of the reaction to form compound 17 (99% yield) was attributed to the absence of N,N-dimethylformamide in the reaction vessel due to the high solubility of 10 in toluene. This result is in accord with the observation that copper-catalyzed Huisgen 1,3-dipolar cycloaddition reactions work very well in non-metal coordinating solvent systems, such as toluene.35 | ||
| Scheme 3 Attachment of oligosaccharides 8, 9 and 10 to either pre-monomer 1 or 2 was achieved via a Huisgen 1,3-dipolar cycloaddition reaction, generating glycomonomers 15, 16 and 17. The * indicates DMF was added to the reaction to aid with solubility. | ||
Following workup of each reaction, the glycomonomers 15, 16 and 17 were purified via silica column chromatography. The carbohydrate-containing monomers were fully characterized, with 1D-TOCSY and 2D NMR techniques particularly useful in the assignment of the complex proton and carbon NMR spectra. In addition, a simple comparison between the infrared spectra of the starting materials versus the products provided additional evidence toward the successful formation of the triazole moiety in each cycloaddition product. An example of this is shown in Fig. 3, which shows the infrared spectra of the propargylated pre-monomer 1 (top), disaccharide 8 (middle) and the corresponding product 15 (bottom). Key absorptions corresponding to the alkyne and azide moieties in the starting materials are clearly absent in the infrared spectrum of compound 15 indicating that these functionalities are not present in the cycloaddition product.
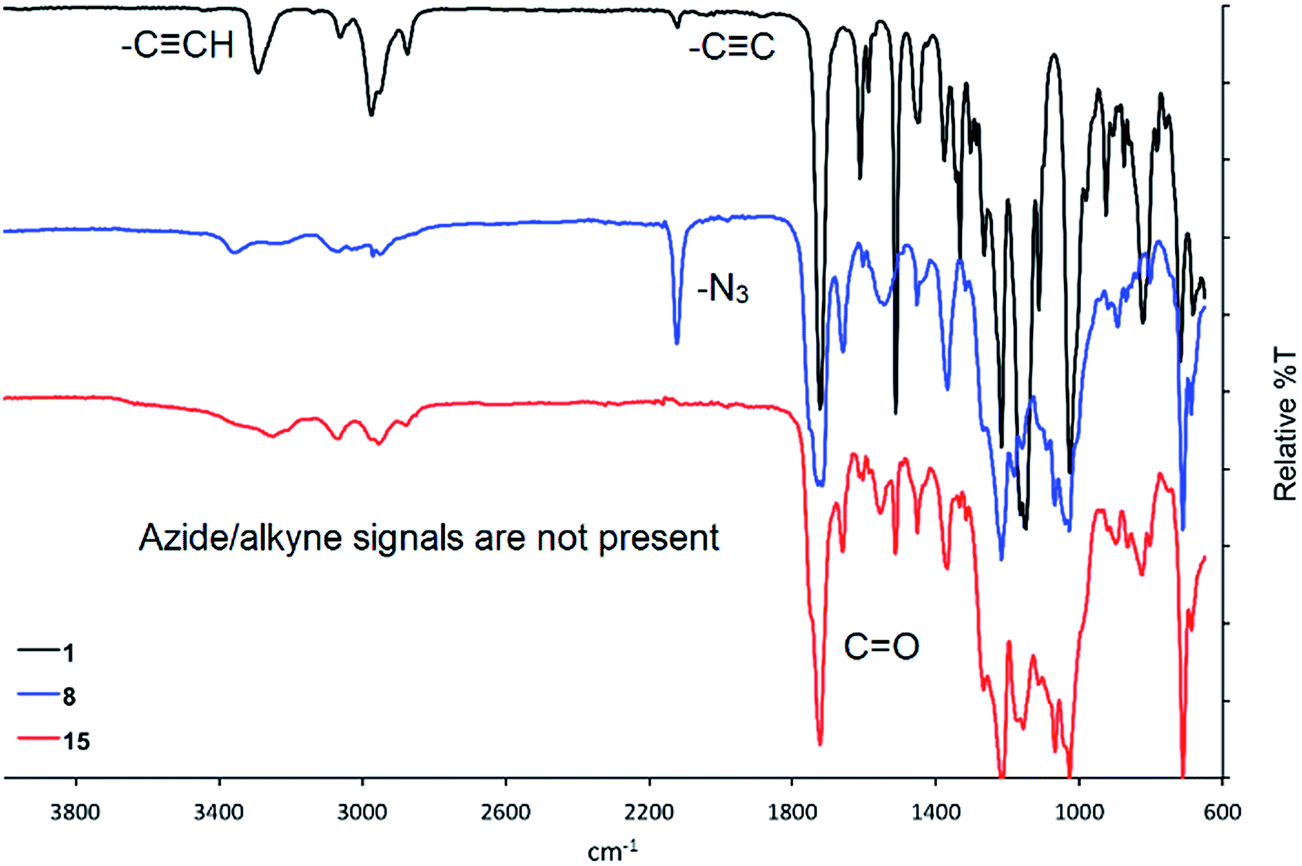 | ||
| Fig. 3 Comparison of the infrared spectra of pre-monomer 1 (black), disaccharide 8 (blue) and the cycloaddition product 15 (red). | ||
Synthesis and characterization of the glycopolymers
Each of the three glycomonomers (15, 16 and 17) was tested for their ability to polymerize under various reaction conditions (Scheme 4) with the results summarized in Table 1. Monomer 15 was found to undergo facile polymerization over 24 hour periods at room temperature in THF. Treatment with two different ratios of Grubbs' 3rd generation catalyst (G3; method 1a) gave, as expected, polymers (18 and 19) that had different degrees of polymerization and relatively low polydispersity indexes (Đ < 1.5; Table 1). In contrast, the amide-linked monomer 17 with the same per-benzoylated carbohydrate adjacent to the norbornenyl group (R′ = Bz) could only be polymerized in a mixture of benzene and THF at 60 °C. The reaction was sluggish, requiring lengthy reaction periods to generate the desired product 20, which had a polydispersity index of 1.36. Amide-linked monomer 16 containing the per-acetylated carbohydrate moiety in the same position (R′ = Ac) was found to be too unreactive to be polymerized under any conditions trialed. | ||
| Scheme 4 Glycomonomers 15 and 17 were polymerized under optimized ROMP conditions using Grubbs' 3rd generation catalyst (G3) to give glycopolymers 18, 19 and 20, respectively. Following polymerization, ethylvinyl ether was added to quench the reactions.36–38 | ||
| Monomer | X | R; R′; R′′ | Conc. (M) | [M]/[I] | Polymera | Yieldb | ![[M with combining macron]](https://www.rsc.org/images/entities/i_char_004d_0304.gif) n (calc)c n (calc)c |
n (GPC)d |
|
Đd |
|---|---|---|---|---|---|---|---|---|---|---|
| a Conditions: (a) G3, THF, 25 °C, 24 h; (b) G3, C6H6, THF, 60 °C, 96 h; all were quenched with ethylvinylether.b Isolated yield.c Calculated from: ([M] × [MWmonomer] × conversion) + end groups.d THF vs. polystyrene standards. | ||||||||||
| 15 | O | Bz; Ac; TFA | 0.033 | 25:1 |
18a | 80% | 2.8 × 104 | 1.9 × 104 | 17 | 1.22 |
| 15 | O | Bz; Ac; TFA | 0.039 | 100:1 |
19a | 37% | 1.1 × 105 | 4.3 × 104 | 39 | 1.47 |
| 16 | NH | Ac; Ac; Ac | 0.035 | 100:1 |
— | — | — | — | — | — |
| 17 | NH | Bz; Bz; TFA | 0.025 | 25:1 |
20b | 65% | 3.3 × 105 | 9.8 × 103 | 8 | 1.36 |

Polymers 18, 19 and 20 were each purified via column chromatography over silica and triplicate precipitation from dichloromethane into diethyl ether to remove the catalyst and residual monomer, as NMR and GPC analyses of the crude reactions showed incomplete monomer conversions. The silica used for glycopolymer purification by chromatography had a larger pore size than silica that is conventionally used and, as a result the polymers were not irreversibly adsorbed onto the column. This purification method was used in preference to size exclusion chromatography, as it was more time efficient. Unreacted glycomonomer was adequately separated from the desired polymer due to the large differences in polarity.
The GPC traces of the purified polymers are shown in the ESI† and there are several points to note. First, for each polymer there is a main fraction with a relatively low dispersity corresponding to the polymer products 18, 19 and 20, respectively (Table 1). The Mn of polymer 19 was the highest and that of compound 20 the lowest, which is consistent with the overall monomer conversion for each polymer (from the 1H NMR). It is clear from Table 1 that there are discrepancies between the Mn values calculated from the 1H NMR and experimental Mn values from GPC. Given the distinctly different structural elements between the new polymers and the polystyrene standards this is not surprising as each polymer will adopt a different shape in solution due to the differences in polarity, and the measured Mn is dependent on the hydrodynamic volume. In addition to the main fraction, the GPC trace of polymer 18 showed an apparent “higher” molecular component (‘A’ in GPC trace – see ESI†). However, given the low degree of polymerization and the fact that the monomer was returned during the purification step, we believe that the higher molecular weight material corresponds to aggregation. In the case of 19 and 20 there was some material that had a longer retention time (‘B’ in GPC traces – see ESI†). It is not clear what the origin of this material is because the 1H NMR did not have any sharp peaks that would correspond to monomers or dimers. It is interesting to note that the proportion of this “lower” molecular weight material appears to be highest for 20, which has the lowest degree of polymerization.
The 1H NMR was also found to be consistent with the structure of the polymers. The spectra all had broad signals, consistent with macromolecule formation, and the signals corresponding to the olefinic backbone protons were not easily distinguishable due to the high number of carbohydrate resonances (see ESI†). 2D 1H NMR analysis enabled assignment of the CH and CH2 signals of the five-membered rings of the polymeric backbone to be identified, and were found in the general range of ∼0.70–2.50 ppm. This indicates that the backbone units are not adopting a uniform conformation in the polymers, which is not surprising given the size of the pendant side chains and the potential for the formation of numerous intra- and inter-molecular hydrogen bonds between the carbohydrates. In contrast, the majority of the carbohydrate proton signals, although broad, appear at similar chemical shifts to that of the corresponding monomeric units.
Overall, it is immediately clear from the above results that the ester-linked glycomonomer 15 was polymerized more efficiently than the corresponding amide-linked monomers 16 and 17. For example, the polymerization reactions using monomer 15 (R′ = Bz) proceeded at room temperature, in tetrahydrofuran, and had a relatively low polydispersity index. On the other hand, glycomonomer 17 (despite being structurally similar and with R′ = Bz) required heat, a mixture of solvents to solubilize the starting materials, intermediates and products, and longer reaction times to generate polymer products with a significantly lower degree of polymerization and with a higher polydispersity index than those generated by the polymerization of 15. Given that glycomonomers 15 and 17 have the same protecting groups on the glycoside closest to the monomer unit, the most likely explanation for this difference is the linkage at the norbornenyl–benzyl junction. By changing the ester linkage to an amide, the overall polarity of the glycomonomer and polymers increased, and the reaction efficiency decreased. This is most likely due to formation of additional inter- and intra-molecular hydrogen bonds between the amide-linked glycomonomers, the catalyst and the solvent mixture.
It is important to note our observation that the type of linkage close to the site of reactivity has influence over the polymerization outcome is only preliminary in nature – although it appears general for the small number of groups, solvents and conditions that we have tested. However, further work is required to determine whether it is a universal effect. A similar effect was previously noticed by Liaw et al. during the photo-initiated free radical polymerization of other norbornene-based monomers, whereby the presence of an amide close to the reaction center resulted in the formation of a dormant propagating radical and thus a low monomer conversion.39 To the authors' knowledge however, this is the first report of this kind in relation to the reactivity of norbornene derivatives polymerized under ROMP conditions.
This claim is further supported by evidence outlined previously, which showed that carbohydrate protecting groups nearest the site of polymerization played a role in the rate of polymerization. As previously highlighted, by simply replacing the benzoyl protecting groups with acetates on the glycoside nearest the norbornenyl moiety, the polymerization was effectively retarded – as evidenced by the unreactivity of glycomonomer 16 under many different conditions, such as various solvent mixtures, reaction temperatures and reaction lengths, and using either Grubbs 2nd or 3rd generation catalysts.
Conclusions
Through the preparation of two different alkynyl-functionalized pre-monomer units 1 and 2, three different glycomonomer building blocks were synthesized via copper-catalyzed Huisgen 1,3-dipolar cycloaddition reactions. The monomers with benzoyl protecting groups on the glycoside nearest the norbornenyl moiety were found to polymerize with Grubbs 3rd generation catalyst. Each of the polymers were analyzed by NMR and GPC, and were found to consist of a low dispersity entity containing a backbone with pendent carbohydrate groups. The ester-linked monomer 15 proved to polymerize much more efficiently than its corresponding amide-linked counterparts (16 and 17) – an effect attributed to the greater polarity of the amide linkage over the ester linkage. Thus, we have developed an approach for the creation of biofilm-inspired polymers and investigations are continuing into the application of these unique biomacromolecules.Acknowledgements
Lucy Weaver was supported by an Australian Postgraduate Award (APA). This work was performed in part at the Queensland node of the Australia National Fabrication Facility (ANFF) – a company established under the National Collaborative Research Infrastructure Strategy to provide nano and microfabrication facilities for Australia's researchers. Paul Burn is a University of Queensland Research Focused Fellow.References
- R. Sunasee and R. Narain, Macromol. Biosci., 2013, 13, 9 CrossRef CAS PubMed.
- N. C. Reichardt, M. Martin-Lomas and S. Penades, Chem. Soc. Rev., 2013, 42, 4358 RSC.
- V. Ladmiral, E. Melia and D. Haddleton, Eur. Polym. J., 2004, 40, 431 CrossRef CAS.
- S. M. Muthana, C. T. Campbell and J. C. Gildersleeve, ACS Chem. Biol., 2011, 7, 31 CrossRef PubMed.
- M. Rawat, C. I. Gama, J. B. Matson and L. C. Hsieh-Wilson, J. Am. Chem. Soc., 2008, 130, 2959 CrossRef CAS PubMed.
- S. R. S. Ting, G. Chen and M. H. Stenzel, Polym. Chem., 2010, 1, 1392 RSC.
- G. Yilmaz and C. R. Becer, Eur. Polym. J., 2013, 49, 3046 CrossRef CAS.
- C. R. Becer, Macromol. Rapid Commun., 2012, 33, 742 CrossRef CAS PubMed.
- K. Godula and C. R. Bertozzi, J. Am. Chem. Soc., 2012, 134, 15732 CrossRef CAS PubMed.
- B. Lepenies, J. Yin and P. H. Seeberger, Curr. Opin. Chem. Biol., 2010, 14, 404 CrossRef CAS PubMed.
- H. Park, R. R. Rosencrantz, L. Elling and A. Böker, Macromol. Rapid Commun., 2015, 36, 45 CrossRef CAS PubMed.
- B. Voit and D. Appelhans, Macromol. Chem. Phys., 2010, 211, 727 CrossRef CAS.
- R. Okoth and A. Basu, Beilstein J. Org. Chem., 2013, 9, 608 CrossRef CAS PubMed.
- C. Fraser and R. H. Grubbs, Macromolecules, 1995, 28, 7248 CrossRef CAS.
- K. H. Mortell, R. V. Weatherman and L. L. Kiessling, J. Am. Chem. Soc., 1996, 118, 2297 CrossRef CAS.
- M. Okada, Prog. Polym. Sci., 2001, 26, 67 CrossRef CAS.
- M. Semsarilar, V. Ladmiral and S. Perrier, Macromolecules, 2010, 43, 1438 CrossRef CAS.
- V. Ladmiral, G. Mantovani, G. J. Clarkson, S. Cauet, J. L. Irwin and D. M. Haddleton, J. Am. Chem. Soc., 2006, 128, 4823 CrossRef CAS PubMed.
- B. S. Sumerlin and A. P. Vogt, Macromolecules, 2010, 43, 1 CrossRef CAS.
- S. Jabbouri and I. Sadovskaya, FEMS Immunol. Med. Microbiol., 2010, 59, 280 CAS.
- K. K. Jefferson, FEMS Microbiol. Lett., 2004, 236, 163 CrossRef CAS PubMed.
- A. Kropec, T. Maira-Litrán, K. K. Jefferson, M. Grout, S. E. Cramton, F. Götz, D. A. Goldmann and G. B. Pier, Infect. Immun., 2005, 73, 6868 CrossRef CAS PubMed.
- A. S. Anderson, A. A. Miller, R. G. K. Donald, I. L. Scully, J. S. Nanra, D. Cooper and K. U. Jansen, Hum. Vaccines Immunother., 2012, 8, 1585 CrossRef CAS PubMed.
- J. Broughan, R. Anderson and A. S. Anderson, Expert Rev. Vaccines, 2011, 10, 695 CrossRef CAS PubMed.
- T. Maira-Litrán, A. Kropec, C. Abeygunawardana, J. Joyce, G. Mark, D. Goldmann and G. Pier, Infect. Immun., 2002, 70, 4433 CrossRef.
- D. Skurnik, M. R. Davis, D. Benedetti, K. L. Moravec, C. Cywes-Bentley, D. Roux, D. C. Traficante, R. L. Walsh, T. Maira-Litrán, S. K. Cassidy, C. R. Hermos, T. R. Martin, E. L. Thakkallapalli, S. O. Vargas, A. J. Mcadam, T. D. Lieberman, R. Kishony, J. J. Lipuma, G. B. Pier, J. B. Goldberg and G. P. Priebe, J. Infect. Dis., 2012, 205, 1709 CrossRef CAS PubMed.
- B. Spellberg and R. Daum, Semin. Immunopathol., 2012, 34, 335 CrossRef CAS PubMed.
- E. Yamashita, K. Okubo, K. Negishi and T. Hasegawa, Chem. Lett., 2009, 38, 122 CrossRef CAS.
- S. Kanamathareddy and C. Gutsche, J. Org. Chem., 1996, 61, 2511 CrossRef CAS.
- S. Binauld, D. Damiron, T. Hamaide, J.-P. Pascault, E. Fleury and E. Drockenmuller, Chem. Commun., 2008, 4138 RSC.
- J.-Y. Wang, J.-M. Han, J. Yan, Y. Ma and J. Pei, Chem.–Eur. J., 2009, 15, 3585 CrossRef CAS PubMed.
- W. Steglich and G. Höfle, Angew. Chem., Int. Ed., 1969, 8, 981 CrossRef CAS.
- L. G. Weaver, Y. Singh, J. T. Blanchfield and P. L. Burn, Carbohydr. Res., 2013, 371, 68 CrossRef CAS PubMed.
- E. Walker, Carbohydr. Res., 1974, 32, 145 CrossRef CAS.
- C. A. Bell, Z. Jia, S. Perrier and M. J. Monteiro, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 4539 CrossRef CAS.
- C. W. Bielawski and R. H. Grubbs, Prog. Polym. Sci., 2007, 32, 1 CrossRef CAS.
- R. H. Grubbs, Tetrahedron, 2004, 60, 7117 CrossRef CAS.
- J. Murphy, K. Nomura and R. Paton, Macromolecules, 2006, 39, 3147 CrossRef CAS.
- D.-J. Liaw, C.-C. Huang, S.-M. Hong, W.-H. Chen, K.-R. Lee and J.-Y. Lai, Polymer, 2006, 47(13), 4613–4621 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Containing full experimental procedures and spectral data. See DOI: 10.1039/c5ra25732h |
| This journal is © The Royal Society of Chemistry 2016 |