
Behaviors of a proton exchange membrane electrolyzer under water starvation
Shucheng Sunab,
Yu Xiaoa,
Dong Liangc,
Zhigang Shao*a,
Hongmei Yua,
Ming Houa and
Baolian Yia
aFuel Cell System and Engineering Laboratory, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, 457 Zhongshan Road, Dalian 116023, PR China. E-mail: zhgshao@dicp.ac.cn; Fax: +86-411-84379185; Tel: +86-411-84379153
bGraduate University of the Chinese Academy of Sciences, Beijing 100039, PR China
cSunrise Power Co., Ltd., Dalian 116025, China
First published on 8th January 2015
Abstract
Water starvation could be one of the reasons for proton exchange membrane (PEM) water electrolyzer degradation. In this paper, the water starvation phenomena of a unit cell in a PEM electrolyzer stack are investigated. The voltage, current density and temperature distribution are investigated in situ with a segmented electrolyzer. The results show that the voltage of the middle and outlet regions is higher than the inlet voltage, which illustrates that water starvation could occur simultaneously in different regions of the electrolyzer. The water stoichiometries have an important effect on the voltage distribution, current density distribution and temperature distribution at 0.5 A cm−2 and 60 °C. The electrochemical impedance spectra of different segments show that the cell resistance and charge transfer resistance gradually increase along the water flow direction when the water stoichiometry is 3. According to the flow regime map, the critical water stoichiometry for electrolysis is further discussed.
1. Introduction
Hydrogen as a clean and environmentally acceptable fuel is expected be one of the most promising energy carriers in the near future. The proton exchange membrane (PEM) water electrolyzer as a hydrogen production device has demonstrated a number of advantages for the production of electrolytic grade hydrogen, such as higher efficiency, higher current density capability, reliability, higher gas purity (above 99.99%), and the possibility of producing compressed gases (up to 150 bar and more) for direct pressurized storage without any external mechanical compression.1,2 Proton exchange membrane (PEM) water electrolysis is potentially interesting for the decentralized production of hydrogen from renewable energy sources,3,4 which offers a convenient and efficient way to produce hydrogen.Improving water management in PEM is one of the most interesting topics for improving the efficiency and extending the operation of electrolyzer.
It is well known that the membrane humidification in electrolyzer is provided by the high flow of pumped water. When the flow rate of the circulating water (Q) is too high, the pressure drop increases and thus damages the membrane electrode assembly (MEA) by high hydraulic pressure.5,6 However, when the total water supplied to electrolyzer MEA is lower than the total water consumed in the single water electrolyzer, the water starvation would occur, namely the overall starvation.7 On the contrary, the local starvation would occur,8 which often results from uneven water distribution, inappropriate porous current collectors,9 high current density and high pressure operation.10
Water starvation often causes the inhomogeneous water distribution, which would lead to the inhomogeneous distribution of the voltage and current. Moreover, water starvation can cause the local drying of the membrane and “hot spots” in the electrolyzer, which would lead to the electrolyzer failure. Also, it has been shown that the conductivity of PEMs is directly related to the hydration state.10,11 As the water content in the membrane decreases, the higher ohmic losses can happen.
There are many studies on the subject focused on the novel oxygen evolution reaction (OER) electrocatalyst12,13 and metal cations poisoning.14,15 However, no publication is available concerning the characters of PEM electrolyzer under the water starvation. By means of investigating the behaviors under the water starvation, this work will help to identify the cause of the performance degradation and optimize the water amount for a water electrolyzer operation.
In this paper, the water stoichiometry (ξ) indicates the ratio of the circulated water amount to the consumed water amount for electrolysis at a definite current density, and the critical water stoichiometry (ξ*) is namely the ratio, above which a PEM water electrolyzer just operate stably. The behaviors of a PEM electrolyzer under water starvation were in situ investigated. Potential sensors, mini-size thermocouples and segmented electrolyzer were adopted to identify the voltage, temperature and current density distribution under different water stoichiometry. At last, the superficial velocities of gas–liquid two-phase flow were calculated, and the critical water stoichiometry was further explained.
2. Experimental
2.1 Experimental system
To study the behaviors of PEM electrolyzer water starvation, a homemade test station was used, which could be found in our previous paper.16,17Fig. 1 showed the schematic of test station. The DC power source (KIKUSUI PAT60-133T) provided an voltage on the water electrolyzer. The value of the DC voltage was in the range of 0–2.4 V for the paper.
 | ||
| Fig. 1 Schematic diagram of test station for PEM water electrolysis. (1) water tank; (2) power supply; (3) water electrolyzer; (4) micropump; (5) circulating water pump; (6) heat exchanger. | ||
The water electrolyzer was kept at 60 °C and atmospheric pressure during operation. The electrolyzer temperature was controlled by adjusting the temperature of the circulating water through the cathode bipolar plate. The deionized water (Milli-Q, 18.2 MΩ cm) was fed into the anode side of the electrolyzer by a micropump (SYLTECH 510 Prep Hd, USA). Water starvation was made intentionally by adjusting the water flow rate, so that the behaviors of the single electrolyzer under water starvation could be investigated. The produced oxygen was evacuated from the anode upside of electrolyzer, whereas the produced hydrogen and water were evacuated from the cathode upside of electrolyzer.
2.2 Installation of voltage sensors
To study the voltage characteristic of PEM electrolyzer under water starvation, the normal electrolyzer was adopted and the active area was 160 cm2. Three voltage sensors were placed on the surface of anode catalyst layer, and two on the surface of cathode diffusion layer. Static reference hydrogen electrode (RHE) was designed at the hydrogen outlet of cathode, the detailed structure as shown in Fig. 2.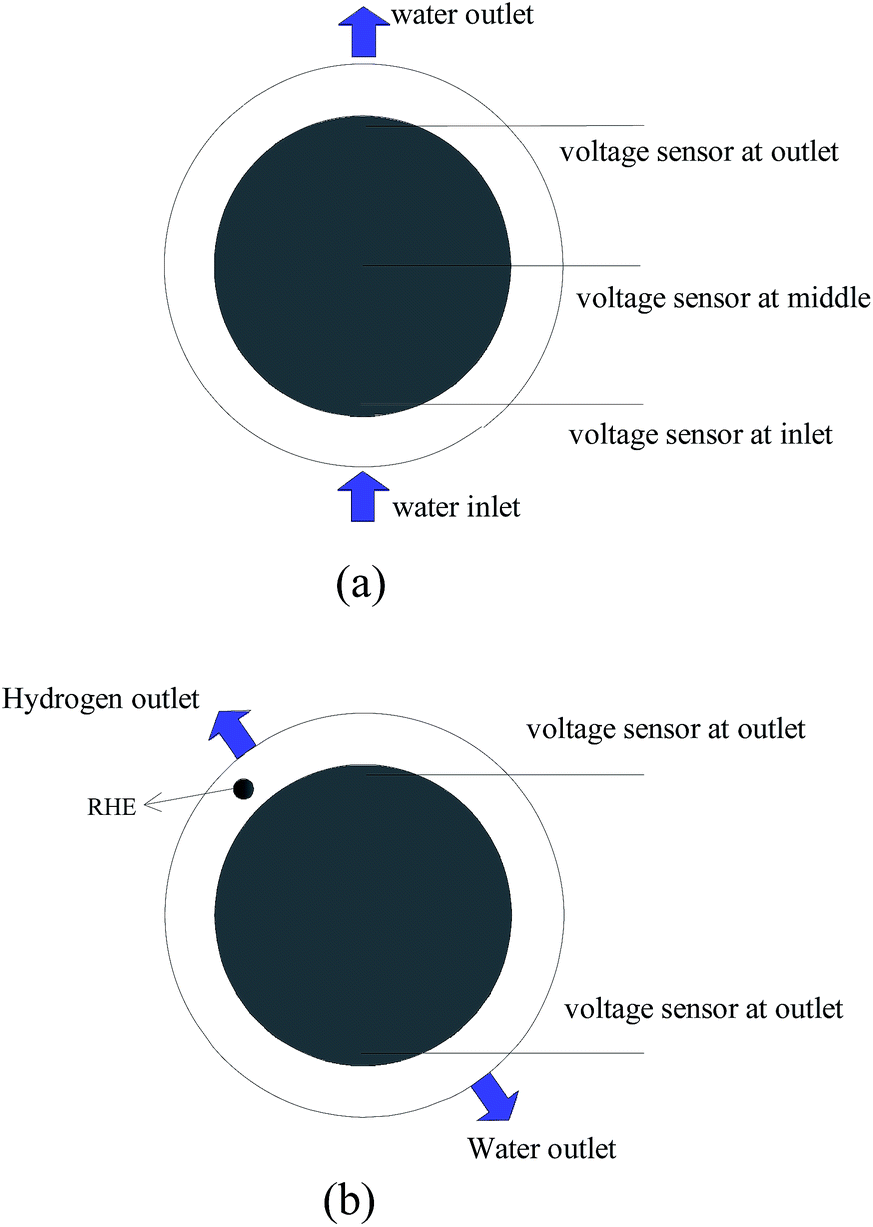 | ||
| Fig. 2 Schematic sketch of the detailed places of voltage sensors: (a) three voltage sensors on the surface of anode catalyst layer; (b) two voltage sensors on the surface of cathode diffusion layer. | ||
The RHE was connected with normal reaction region through proton exchange membrane. The anode and cathode voltage sensors used in this study was made of platinum and copper wires with insulating paint outside, respectively. The diameter of the wires was 0.08 mm. At the end of each voltage sensor being placed on the measured regions, the insulating paint was removed for about 2 mm. Thus, the local voltages could be indicated by the voltage sensors. Because the high purified hydrogen (99.99%) was fed into the anode and cathode side at a flow rate of 50 ml min−1, respectively, the potential of RHE and cathode was assumed to be stable and zero. However, the potential difference between the cathode and the RHE existed, which was used as a modified coefficient of the cathode reference. The mean value of the inlet and outlet potential of the cathode side could be taken as a reference potential. The local voltages of inlet region, middle region and outlet region on anode (hereinafter referred as inlet, middle and outlet, respectively) against the reference were measured and recorded automatically.
2.3 Single electrolyzer structure
Due to our water electrolyzer stack is circular, each cell with 160 cm2 active area. To investigate actually the behaviors of water electrolyzer under starvation, a segmented electrolyzer was constructed to measure the current density distribution, which included a segmented membrane electrode assembly (MEA) and a circular anode end plate.The MEA was specially designed as displayed in Fig. 3. The anode electrode was segmented, the active area of each segment was 4.5 cm2, and the total active area was 49.5 cm2. The segments were numbered from 1 to 5 along the direction of water flow. Segment 1 and 5 were individual segment, whereas each of segment 2, 3 and 4 included three segments, respectively, as shown in Fig. 3. The cathode effective area was 160 cm2 with normal structure. The preparation process of MEA was the same as our previous paper.16 A carbon paper (Toray, Φ 145) and eleven porous titanium disks (Φ 24) electroplated Pt were used as the current collector for cathode and anode of MEA, respectively. Fig. 4 showed the specially made anode current collecting end plate, in which eleven titanium blocks electroplated Pt were embedded corresponding to the segmented MEA. Eleven copper sticks were connected with eleven titanium blocks. The organic glass end plate was carved into parallel channels, the cross-section of the channel was rectangle (0.4 mm × 0.8 mm) and 0.0032 cm2 in area, and the number of the flow channels was 80. In the electrolyzer set-up, the water flow was vertical upward in the parallel flow field. When current distribution was investigated, the electrolysis current was kept at 24.75 A, and the current density was 0.5 A cm−2 with regard to the total effective area of anode.
 | ||
| Fig. 3 Structure of the segmented MEA ((a) segmented anode; (b) proton exchange membrane; (c) cathode; (d) plastic frame). | ||
 | ||
| Fig. 4 Structure of the anode end plate collecting current. | ||
2.4 Segmented performance measurement
First, the electrolyzer was operated normally at 0.5 A cm−2 and 60 °C for 15 min. Then the current density distribution was made a chart with different water stoichiometries after the segmented electrolyzer had run for 10 min. The voltage and temperature distributions were the same as that of current density.2.5 Electrochemical impedance spectroscopy (EIS) measurement
Prior to EIS measurements, the segmented electrolyzer was operated at 0.5 A cm−2 and 60 °C for 15 min. EIS of the segments were conducted at 1.55 V using a Solartron (1287A + 1260) in a range of frequency (0.1 Hz to 10 kHz), and a 5 mV amplitude of voltage perturbation was employed.3. Results and discussion
3.1 Behaviors of electrolyzer voltage
Fig. 5 shows the voltage distributions of different water stoichiometries at fixed current density of 0.5 A cm−2 (with regard to the water stoichiometry 1.5, 2.5, 3, 5 and 8, respectively), and the water flow rate corresponds to 0.21, 0.35, 0.42, 0.7 and 1.12 ml min−1, respectively. As illustrated in Fig. 5, when the water stoichiometry (ξ) decreases from 8 to 3 at 0.5 A cm−2, the average voltage just increases about 10 mV. The voltage distribution is slightly non-homogeneous, the outlet voltages are 10 mV lower than that of inlet and middle, which is attributed to the relatively lower temperature at water inlet. However, when the water stoichiometry is as low as 3 at 0.5 A cm−2, the average voltage distinctly increases from 1.67 V to 1.9 V, and the voltages of middle and outlet are especially higher than that of inlet. | ||
| Fig. 5 Voltage distributions of different water stoichiometries at 0.5 A cm−2. | ||
Fig. 6 shows the voltage difference between outlet and inlet corresponding to Fig. 5. When the water stoichiometry decreases from 8 to 3, the voltage difference is almost constant (ca. −12 mV). However, when the water stoichiometry varies from 3 to 1.5, the voltage difference between outlet and inlet dramatically increases, which approximately increases from −10 mV to 22 mV. The phenomena display that at low water stoichiometry (ξ < 3), the water supply could not meet the demand for the overall MEA, and the water amount decreases down the water flow direction. The water content in the membrane has a more significant effect on ohmic resistance.18,19 Therefore, Fig. 5 and 6 show the membrane water content near the water outlet is quite insufficient for operation at 0.5 A cm−2 when ξ < 3, which increases the outlet voltage to a large extent, and thus it causes the increase of the total voltage.
 | ||
| Fig. 6 The voltage difference between outlet and inlet relating to Fig. 7. | ||
In order to observe obviously the voltage distribution in electrolyzer, the water stoichiometry (ξ = 2.5) was selected.
Fig. 7 displays the voltage distribution inside PEM water electrolyzer under different current densities at fixed water stoichiometry of 2.5 (with regard to the current density 0.1, 0.3, 0.5, 0.6 and 0.8 A cm−2, respectively), and the water flow rate corresponds to 0.23, 0.68, 1.13, 1.35 and 1.8 ml min−1, respectively. It can be seen that the voltage distribution becomes more non-homogeneous with the increase of current density. For the current densities from 0.1 to 0.5 A cm−2, the voltages of middle and outlet are only slightly lower than that of inlet, which is attributed to the slightly lower temperature at water inlet. However, when the current density is above 0.5 A cm−2, the voltages of middle and outlet are obviously higher than that of inlet, and that of outlet is the highest. We can obviously see the phenomenon in Fig. 8, which is a plot of the voltage difference between outlet and inlet corresponding to Fig. 7. As shown in Fig. 8, when the current density is above 0.5 A cm−2, the voltage difference between outlet and inlet increases dramatically. The outlet voltage is even 33 mV higher than that of inlet at 0.8 A cm−2.
 | ||
| Fig. 7 Voltage distribution under different current densities at fixed water stoichiometry of 2.5 (with regard to the current densities 0.1, 0.3, 0.5, 0.6 and 0.8 A cm−2, respectively). | ||
 | ||
| Fig. 8 Voltage difference between outlet and inlet relating to Fig. 7. | ||
This may be attributed to the insufficient amounts of water supplied to the anode. Water amount at anode inlet is the most highest in membrane and decreases along the water flow direction when water stoichiometry is 2.5. Therefore, the drying of membrane near the water outlet occurs at high current density (ca. above 0.5 A cm−2), which will lead to the increase in the local ohmic resistance.
3.2 Behaviors of current density distribution
The process of water starvation can also be illustrated through the current density distribution in the electrolyzer. Fig. 9 shows the dependence of current density distribution on the variation of water stoichiometry (ξ = 1.5, 2.5, 3, 5, 8 and 10) at 0.5 A cm−2, the water flow rate corresponds to 0.21, 0.35, 0.42, 0.7, 1.11 and 1.37 ml min−1, respectively. When the water stoichiometry changes from 10 to 5, the current density of segment 1–5 slightly becomes uneven. While the water stoichiometry approximates 3, the current density distribution is extremely sensitive to the water amount. The current density of segment 1 and 5 deviates obviously from 0.5 A cm−2 at ξ = 3. As the water stoichiometry is less than 3, the current density distribution tends to be more heterogeneous. At segment 1 the local current density is the greatest, whereas at segment 5 the local current density becomes the least. The local current density distribution along the channels is mainly dominated by the gradient of water content in membrane. Because the gradient of water content can cause the difference of membrane ionic conductivity along the channel,20 and ohmic resistance can be determined by the membrane ionic conductivity. Therefore, ohmic loss and concentration loss may increase seriously when ξ < 3, which can not maintain the stability and efficiency of PEM electrolyzer operation. This could be also proved by the result of Fig. 6. | ||
| Fig. 9 Current density distribution varying with water stoichiometries at 0.5 A cm−2. | ||
3.3 Behaviors of temperature distribution
Fig. 10 shows the temperature distribution along the anode channels inside the eletrolyzer under different water stoichiometries. As shown in Fig. 10, when the water stoichiometry changes from 10 to 5, the temperature distribution of electrolyzer is almost homogeneous. | ||
| Fig. 10 Temperature distributions varying with water stoichiometries at 0.5 A cm−2. | ||
As the water stoichiometry is equal to or less than 3, the temperature distribution becomes more heterogeneous. It indicates that the water amount is insufficient to remove the produced heat, the local hot spot in the electrolyzer would be produced, which is likely to cause the local degradation of the MEA.
3.4 Electrochemical impedance spectra (EIS)
Besides, in situ EIS measurement is applied to investigate the water distribution along the water flow direction under the water stoichiometries of 10 and 3. Fig. 11 shows Nyquist plots of different segments at 1.55 V and 60 °C. In general, the cathode polarization is very small and can be neglected. Thus, the obtained impedance spectrum mainly reflects the anode polarization. The impedance spectrum mainly includes three parts: a highest frequency intercept with the real axis denoting the cell resistance (RΩ), a higher frequency arc which reflects the combination of a charge transfer resistance (Rct) and a double layer capacitance (Cdl) within the catalyst layer, and the lower frequency arc reflects the mass-transport process within the current collector.21 The double layer capacitance is considered to be a constant phase element (CPE). The RΩ and Rct can be obtained through simulation with RΩ(RctQdl)(RdQd) equivalent circuit.22 Only the RΩ and Rct fitting data are listed in Table 1. For ξ = 10, RΩ fluctuates from 0.49 to 0.509 mΩ cm2, and Rct fluctuates from 0.234 to 0.246 Ω cm2. However, for ξ = 3, RΩ increases from 0.492 to 0.72 Ω cm2, and Rct increases from 0.243 to 0.36 Ω cm2. It indicates that the water distribution along the water flow direction is almost uniform for ξ = 10, but water starvation takes obviously place for ξ = 3. When ξ = 3, RΩ and Rct gradually increase along the water flow direction, and Rct is particularly higher than that of ξ = 10. In addition, the impedance spectra have the second arc at the lower frequency, which shows that the diffusion impedance exists in the mass-transport limitation for ξ = 3 and 10. | ||
| Fig. 11 Electrochemical impedance spectra of different segments under water stoichiometries: a = 10; b = 3. | ||
| Segments | RΩ (Ω cm2) | Rct (Ω cm2) | ||
|---|---|---|---|---|
| ξ = 3 | ξ = 10 | ξ = 3 | ξ = 10 | |
| 1 | 0.492 | 0.490 | 0.243 | 0.244 |
| 2 | 0.504 | 0.490 | 0.252 | 0.244 |
| 3 | 0.531 | 0.509 | 0.239 | 0.234 |
| 4 | 0.581 | 0.491 | 0.319 | 0.242 |
| 5 | 0.720 | 0.500 | 0.360 | 0.246 |
So we can deduce that the water amount for ξ = 10 has a little influence on RΩ and Rct, whereas for ξ = 3, the performance near the exit of electrolyzer has deteriorated at 0.5 A cm−2 and 60 °C. This coincides with the results in Fig. 6 and 9.
3.5 Flow regime of two-phase flow
In general, the mass transfer of liquid from a channel to an electrode is significantly hindered by the residence of gas bubbles in the flow channel. The flow pattern of the two-phase flow in the flow channel of the anode can indicate the size of gas bubbles. To illuminate above phenomena, the flow regime is investigated as shown in Fig. 12. Our results are compared and analyzed using the flow regime map (Fig. 12a and b) presented by Cubaud and Ho.23 The map can almost describe the flow pattern of the two-phase flow and reflects the wet state of membrane electrode assembly (MEA). | ||
| Fig. 12 Flow regime map expressed by superficial velocity of gas (jG) and liquid (jL) presented by Cubaud and Ho.23 Symbols indicate our calculated velocities in the channel of PEM electrolyzer at 0.5 A cm−2 and 60 °C for different segments. | ||
The flow regime changes depending on the mass flux, quality, or superficial velocity of each phase.5,24 Quality fraction (x) inside the two-phase flow is defined at an arbitrary location along the channel as follows:
 | (1) |
The superficial velocities of the gas and liquid (jG and jL) can be calculated as follows:
 | (2) |
 | (3) |
The x can be calculated by the total mass balance through the anode channel in eqn (3). Table 2 summarizes the detailed definitions of Gcirc, GO2, Gcons and Gdrag. The jG and jL at the channel of different segments can be calculated using eqn (2) and (3).
| Parameter | Equation |
|---|---|
| Gcirc |  |
| GO2 |  |
| Gcons |  |
| Gdrag |  |
| ndrag | 0.016T − 2.89556 (ref. 25) |
When the water stoichiometry (ξ) is equal to 10 at 0.5 A cm−2 and 60 °C, segment 3–5 have located in annular area, as shown in Fig. 12b. Although the diffusion impedance exists for ξ = 10 (Fig. 11a), the MEA should be wet under annular flow so that the electrolyzer can operate having no loss in performance (Fig. 9).
However, when ξ = 3, segment 3, 4 and 5 of electrolyzer have obviously located in dry area (Fig. 12a). However, this conflicts with the critical water stoichiometry (ξ*) to some extent. The difference may result from two causes. Firstly, the flow regime map is based on the assumption that the membrane is in contact and in equilibrium with liquid water on both sides of the electrolyzer. Secondly, the electro-osmotic drag coefficient varies within a wide range, depending on the degree of membrane hydration in contact with liquid water.26 For ξ = 3, the water content of membrane is in nonequilibrium with liquid water, and the electro-osmosis drag coefficient (ndrag) should be less than 2.43 (ref. 23) at 0.5 A cm−2 and 60 °C. Moreover, the back-diffusion water from the cathode may compensate for the electro-osmotic dragged water, which would lead to the net water transport from the anode to cathode decrease.27 As a result, Fig. 12 differs from described above results to some extent.
Consequently, the critical water stoichiometry (ξ*) is obtained for a PEM electrolyzer at 60 °C and 0.5 A cm−2, ξ* is about 3. A PEM water electrolyzer could operate normally on condition that the water stoichiometry is more than ξ*. In addition, heat dissipation in a PEM water electrolyzer also becomes a limiting factor under water starvation. This point also proves that the circulating water does not only act as reactant, but functions as cooling water. Therefore, a heat balance between the heat generation capacity and the ability of the heat dissipation must be considered in some cases of a large cell/stack. In addition, low humidity and high temperature may lead to the membrane degradation such as thermal degradation, mechanical stress, and chemical attack28 during the water starvation.
4. Conclusions
In this work, the behaviors of a unit cell in a PEM electrolyzer stack under water starvation conditions have been investigated. It is found that current density and water stoichiometry have significant effects on behaviors of electrolyzer. With the increase of current density or the decrease of water stoichiometry, the voltage distribution is heterogeneous. The temperature and current density distribution varies more heterogeneous with the decrease of water stoichiometry. The electrochemical impedance spectra of different segments display that RΩ and Rct gradually increase along the water flow direction for ξ = 3. The flow regime map shows that the electro-osmosis drag coefficient (ndrag) should not be constant for ξ*. The critical water stoichiometry (ξ*) is obtained at 60 °C and 0.5 A cm−2. When the water stoichiometry is equal to or less than ξ*, the local hot spots have been observed at the anode water outlet. That would be a possible reason for the degradation of the MEA. Future efforts should focus on the degradation mechanism investigation of electrolyzer under water starvation.Symbols
| ξ | Water stoichiometry |
| ξ* | Critical water stoichiometry |
| Q | Flow rate of circulating water, ml min−1 |
| a | Cross-sectional area of flow channel, cm2 |
| F | Faraday constant = 96![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 485, C mol−1 485, C mol−1 |
| G | Mass flow flux in channel, kg m−2 s−1 |
| nchan | Number of channels in flow field |
| ndrag | Electro-osmosis drag coefficient |
| Rct | Charge transfer resistance |
| Rd | Mass-transport resistance |
| GO2 | Mass flux of produced oxygen gas |
| Gcons | Mass flux consumed by the anode reaction |
| Gdrag | Mass flux of the water dragged by electro-osmosis |
| A | Electrode surface area, cm2 |
| T | Cell temperature, K |
| x | Quality fraction |
| ρL | Liquid density, kg m−3 |
| ρG | Gas density, kg m−3 |
| j | Superficial velocity, m s−1 |
| i | Current density, A cm−2 |
| M | Molar weight, kg mol−1 |
| RΩ | Cell resistance |
| Cdl | Double layer capacitance |
| Gcirc | Mass flux of circulating water |
Acknowledgements
We thank the National High Technology Research and Development Program of China (863 Program, 2013AA110201-1), the Natural Science Foundations of China (no. 21203191) and the National Basic Research Program of China (973 program no. 2012CB215500) for financial support.Notes and references
- http://www.prnewswire.com/news-releases/proton-energy-systems-achieves-breakthrough-in-high-pressure-hydrogen-technology-61978327.html.
- http://www.plantservices.com/vendors/products/2009/080.html.
- C. S. Song, Top. Catal., 2008, 49, 1–3 CrossRef CAS.
- A. H. K. Abdel and M. A. Shalabi, Int. J. Hydrogen Energy, 1992, 17, 359–367 CrossRef.
- H. Ito, T. Maeda, A. Nakano, Y. Hasegawa, N. Yokoi and C. M. Hwang, Int. J. Hydrogen Energy, 2010, 35, 9550–9560 CrossRef CAS PubMed.
- T. Oi and Y. Sakaki, J. Power Sources, 2004, 129, 229–237 CrossRef CAS PubMed.
- A. Taniguchi, T. Akita and K. Yasuda, et al., J. Power Sources, 2004, 130, 42–49 CrossRef CAS PubMed.
- H. Tang, Z. Qi and M. Ramani, et al., J. Power Sources, 2006, 158, 1306–1312 CrossRef CAS PubMed.
- S. A. Grigoriev, P. Millet, S. A. Volobuev and V. N. Fateev, Int. J. Hydrogen Energy, 2009, 34, 4968–4973 CrossRef CAS PubMed.
- T. D. Myles, G. J. Nelson, A. A. Peracchio, R. J. Roy, B. L. Murach, G. A. Adamson and W. K. S. Chiu, Int. J. Hydrogen Energy, 2012, 37, 12451–12463 CrossRef CAS PubMed.
- Y. Sone, P. Ekdunge and D. Simonsson, J. Electrochem. Soc., 1996, 143, 1254–1259 CrossRef CAS PubMed.
- G. F. Li, H. M. Yu, X. Y. Wang, D. L. Yang, Y. K. Li, Z. G. Shao and B. L. Yi, J. Power Sources, 2014, 249, 175–184 CrossRef CAS PubMed.
- G. F. Li, H. M. Yu, W. Song, X. Y. Wang, Y. K. Li, Z. G. Shao and B. L. Yi, Int. J. Hydrogen Energy, 2012, 37, 16786–16794 CrossRef CAS PubMed.
- L. S. Zhang, X. Jie, Z. G. Shao, X. Y. Wang and B. L. Yi, J. Power Sources, 2013, 241, 341–348 CrossRef CAS PubMed.
- L. S. Zhang, X. Jie, Z. G. Shao, Z. M. Zhou, G. Xiao and B. L. Yi, Int. J. Hydrogen Energy, 2012, 37, 1321–1325 CrossRef CAS PubMed.
- S. Sun, Z. Shao, H. Yu, G. Li and B. Yi, J. Power Sources, 2014, 267, 515–520 CrossRef CAS PubMed.
- D. Liang, Q. Shen, M. Hou, Z. Shao and B. Yi, J. Power Sources, 2009, 194, 847–853 CrossRef CAS PubMed.
- F. N. Buchi and G. G. Scherer, J. Electrochem. Soc., 2001, 148, A183–A188 CrossRef CAS PubMed.
- M. Erni, S. M. H. Nik, R. W. D. Wan, H. M. Edy and I. R. Masli, Int. J. Hydrogen Energy, 2013, 38, 9401–9408 CrossRef PubMed.
- D. Lee and J. Bae, J. Power Sources, 2009, 191, 390–399 CrossRef CAS PubMed.
- T. E. Springer, T. A. Zawodzinski, M. S. Wilson and S. Gottesfeld, J. Electrochem. Soc., 1996, 143, 587–599 CrossRef CAS PubMed.
- J. Hou, H. Yu, S. Zhang, S. Sun, H. Wang, B. Yi and P. W. Ming, J. Power Sources, 2006, 162, 513–520 CrossRef CAS PubMed.
- T. Cubaud and C. M. Ho, Phys. Fluids, 2004, 16, 4575–4585 CrossRef CAS PubMed.
- H. Ito, T. Maeda, A. Nakano, C. M. Hwang, M. Ishida, A. Kato and T. Yoshida, Int. J. Hydrogen Energy, 2012, 37, 7418–7428 CrossRef CAS PubMed.
- X. J. Li, S. G. Qu, H. M. Yu, M. Hou, Z. G. Shao and B. L. Yi, J. Power Sources, 2009, 190, 534–537 CrossRef CAS PubMed.
- F. Liu, G. Lu and C.-Y. Wang, J. Membr. Sci., 2007, 287, 126–131 CrossRef CAS PubMed.
- Q. Yan, H. Toghiani and J. Wu, J. Power Sources, 2006, 158, 316–325 CrossRef CAS PubMed.
- C. Chen and T. F. Fuller, Polym. Degrad. Stab., 2009, 94, 1436–1447 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |