
tert-Butyl hypochlorite mediated diastereoselective oxidative coupling: access to 1-functionalized tetrahydrocarbazoles†
Long Jianga,
Xiaoni Xiea and
Liansuo Zu*abc
aDepartment of Pharmacology and Pharmaceutical Sciences, School of Medicine, Tsinghua University, Beijing, 100084 China. E-mail: zuliansuo@biomed.tsinghua.edu.cn
bCollaborative Innovation Center for Biotherapy, Tsinghua University, Beijing, China
cCollaborative Innovation Center for Biotherapy, State Key Laboratory of Biotherapy and Cancer Center, West China Hospital, West China Medical School, Sichuan University, Chengdu, China
First published on 23rd December 2014
Abstract
A mild and operationally simple protocol for the direct C–H functionalization of the 1-position of tetrahydrocarbazoles is reported. The diastereoselective oxidative coupling process is mediated by tert-butyl hypochlorite, and allows the successful generation of 1-functionalized tetrahydrocarbazoles with good to excellent yields and good functional group tolerance.
1-Functionalized tetrahydrocarbazoles have attracted attention due to their potent and diverse biological activities. For example, a variety of 1-substituted tetrahydrocarbazoles have been reported for their therapeutic potential as anti-HPV agents, NPY-1 antagonists, androgen receptor modulators, and DP1 antagonists (Fig. 1).1 1-Functionalized (heteroatom substituted) tetrahydrocarbazoles are also key structural features of some complex indole alkaloids, such as alstoscholarine and gilbertine (Fig. 1).2 Thus, the direct C–H functionalization of the 1-position of tetrahydrocarbazoles via an oxidative coupling approach,3,4 particularly in complex settings, could provide a shortcut to these medicinally important structures and offer distinct bond disconnection strategy in natural product total synthesis.
 | ||
| Fig. 1 1-Functionalized tetrahydrocarbazoles. | ||
Recently, the direct C–H functionalization of the 1-position of tetrahydrocarbazoles via an oxidation process has been documented using different oxidants.5 Despite the significance of these approaches, there are still some limitations that remain to be addressed: (1) as the majority of the developed approaches utilize simple tetrahydrocarbazoles (with variations only on the phenyl ring) as substrates, reactivity in terms of functional group tolerance and diastereoselectivity, which are key factors in complex molecule synthesis, remains unexplored. (2) Most oxidative species that promote the direct C–H functionalization are generated in situ through the combined use of other reagents. Mild and operational simple protocols using single stable oxidant are highly desirable. (3) In most cases, protecting groups on the nitrogen are essential to govern the reactivity, and thus the overall synthetic efficiency is diminished.
tert-Butyl hypochlorite is a stable, commercial available and inexpensive oxidizing reagent. Its ability to promote the 2α-functionalization of 2,3-disubstituted indoles has been reported, as exemplified by the work of Gassman and co-workers on 2,3-dimethyl indole6 and the chloroindolenine-based coupling strategy utilized in the total synthesis of vinblastine and Iboga alkaloids.7 However, attempts to direct functionalize the 1-position of tetrahydrocarbazole using this reagent under various reaction conditions failed,8 indicating that the highly reactive chloroindolenine intermediate formed could undergo several competing reaction pathways. To provide a solution to this unsolved synthetic problem, address some limitations associated with the existing approaches and develop a mild, operational simple and diastereoselective protocol for the preparation of 1-functionalized tetrahydrocarbazoles, we set out to systematically investigate this C–H functionalization via an oxidative coupling process using tert-butyl hypochlorite with the hypothesis that suitable reaction conditions in terms of solvent, additives and temperature might play important roles in governing the reactivity.
A model oxidative coupling reaction between 1a and benzyl amine (2a) was carried out first to identify the optimized reaction conditions (Table 1). 1a, readily prepared by the thermal Diels–Alder reaction between 2-vinyl indole and the corresponding α,β-unsaturated ketone,9,10 represents a suitable substrate to examine the diastereoselectivity and functional group tolerance of this oxidative coupling reaction. Indeed, as we envisioned, the outcome of the reaction was highly depended on the solvent used and the reaction temperature. The chloroindolenine 4 (X = Cl) was formed in good yields when DMF, DCM, and acetonitrile were utilized as the reaction media; however, the reactivity was completely switched and the desired product 3a was isolated in good yields when ethyl acetate and THF was used as the solvent at −20 °C. It should be noted that the reaction was promoted by tert-butyl hypochlorite as the single activating reagent, without the addition of any additive(s), such as acid or base. Moreover, the oxidative coupling reaction tolerated both the ketone and ester groups and proceeded with excellent diastereoselectivity (>20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). The reaction yield was further improved by using only 1.0 equiv. of tert-butyl hypochlorite (entry 6). Other halogenating reagents, such as NBS, iodine, and NIS were also screened, but none of them promoted the formation of 3a.‡
:1). The reaction yield was further improved by using only 1.0 equiv. of tert-butyl hypochlorite (entry 6). Other halogenating reagents, such as NBS, iodine, and NIS were also screened, but none of them promoted the formation of 3a.‡
|
||||
|---|---|---|---|---|
| Entry | Oxidant | Solvent | Yield of 3aa (%) | Yield of 4a (%) |
| a Isolated yield.b Using 1.0 equiv. of tert-butyl hypochlorite and 3.0 equiv. of benzyl amine.c Using 1.0 equiv. of tert-butyl hypochlorite and 1.5 equiv. of benzyl amine. | ||||
| 1 | t-BuOCl | DMF | 0 | 73 |
| 2 | t-BuOCl | DCM | 0 | 68 |
| 3 | t-BuOCl | EtOAc | 71 | 0 |
| 4 | t-BuOCl | CH3CN | 0 | 75 |
| 5 | t-BuOCl | THF | 60 | 0 |
| 6 | t-BuOCl | THF | 81b | 0 |
| 7 | t-BuOCl | THF | 65c | 0 |
| 8 | NBS | THF | 0 | 82 |
| 9 | I2 | THF | 0 | 0 |
| 10 | NIS | THF | 0 | 0 |

The generality and substrate scope of the oxidative coupling reaction was next investigated (Scheme 1). Under optimized reaction conditions, a variety of different tetrahydrocarbazoles underwent effective coupling with benzyl amine, generating the directly 1-fuctionalized products in good to excellent yields. Substituents on the phenyl rings could be varied, and the electronic properties did not seem to affect the reaction yields significantly (3b–3e). Substituents at the 2,3,4-positions of the tetrahydrocarbazoles were also well tolerated (3a, 3f–3l), providing a direct entry to multisubstituted 1-fuctionalized tetrahydrocarbazoles, which are difficult to be synthesized by conventional methods. Notably, a variety of functional groups, including ketone, ester, ketal, lactone, and heterocyclic pyrrole (3i), remained intact during the oxidative coupling reactions. In addition, good to excellent diastereoselectivities were observed with substituents at 3 and/or 4 positions, albeit a 1:1 mixture of two isomers was obtained using 2-methyl tetrahydrocarbazole (3l).
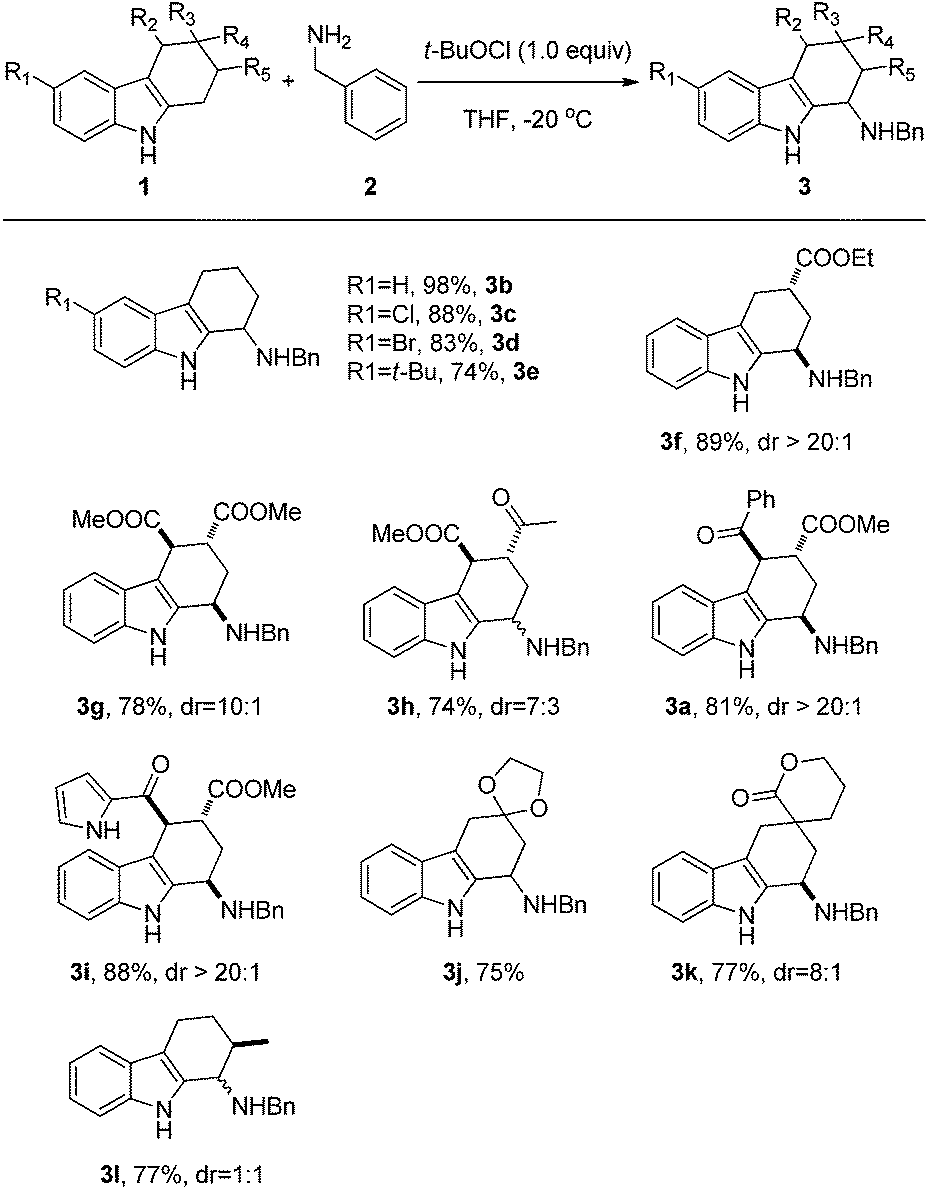 | ||
| Scheme 1 The generality and scope of the oxidative coupling reaction: different tetrahydrocarbazoles. | ||
We next examined the usage of other nucleophilic reagents in the tert-butyl hypochlorite mediated oxidative coupling reaction (Scheme 2). In addition to benzyl amine, cyclic and acyclic secondary amines (3m, 3n, 3o), and aniline (3p) were also effective coupling partners. The characterization data of 3p is identical with that reported.5f Attempts to induce diastereoselectivity using chiral benzylic amine only resulted in a mixture of two isomers (3q, 2:3). Oxygen based nucleophiles (3r, 3s, 3t) could also participate in the oxidative coupling reaction with good yields. The surprisingly high diastereoselectivity observed with 3r (compared to that of 3s, 3t) might be attributed to the recemizable nature of the newly generated stereogenic center and the related dynamic resolution process. It should be mentioned that 1-oxygenated tetrahydrocarbazoles have been demonstrated to serve as effective precursors for the subsequent acid-promoted coupling reactions with a variety of different nucleophilic reagents.5b,11 In addition, C–C bond formation was also successfully achieved using sodium salt of malonate diester as the coupling partner, further expanding the chemical diversity of the products generated. Similarly, the reaction of cyclohepta [1,2-b] indole afforded 3v in good yield.
 | ||
| Scheme 2 The generality and scope of the oxidative coupling reaction: different nucleophiles. | ||
To probe whether the chloroindolenine 4 was the first formed intermediate leading to the desired coupling product 3a, control experiments were carried out (Scheme 3). The reaction between 1a and tert-butyl hypochlorite in dichloromethane led exclusively to the formation of chloroindolenine 4. When 4 was subjected to the standard reaction condition for the oxidative coupling with benzyl amine in THF, 3a was isolated in good yield, indicating that the chloroindolenine 4 was indeed an intermediate en route to 3a. Presumably both of A and B can be possible intermediates, and nucleophilic attack through either SN2′ or conjugate addition are possible pathways. In either mechanism, the observed diastereoselectivity might be attributed to the axial attack of the nucleophilic reagents from the opposite side of the most hindered substituted group.
 | ||
| Scheme 3 Control experiments and proposed mechanism. | ||
Conclusions
In conclusion, we have developed a mild and operational simple protocol for the direct C–H functionalization of the 1-position of tetrahydrocarbazoles. The diastereoselective oxidative coupling process is mediated by tert-butyl hypochlorite as the single oxidant, and allows the successful generation of a diverse array of 1-functionalized tetrahydrocarbazoles with good to excellent yields and good functional group tolerance. Our method provides a solution to an unsolved synthetic problem and address some limitations of the existing approaches. With the significance of 1-functionalized tetrahydrocarbazoles as structural elements in medicinally important compounds and complex natural products, we expect this strategy will find applications in chemical synthesis and drug discovery.Acknowledgements
The authors are grateful to Tsinghua University, “1000 Talents Recruitment Program”, Collaborative Innovation Center for Biotherapy (Tsinghua University and Sichuan University) and National Natural Science Foundation of China (Grant no. 21302107) for financial support.Notes and references
-
(a) R. DiFabio, R. Giovannini, B. Bertani, M. Borriello, A. Bozzoli, D. Donati, A. Falchi, D. Ghirlanda, C. P. Leslie, A. Pecunioso, G. Rumboldt and S. Spada, Bioorg. Med. Chem. Lett., 2006, 16, 1749 CrossRef CAS PubMed
; (b) K. S. Gudmundsson, P. R. Sebahar, L. D. Richardson, J. G. Catalano, S. D. Boggs, A. Spaltenstein, P. B. Sethna, K. W. Brown, R. Harvey and K. R. Romines, Bioorg. Med. Chem. Lett., 2009, 19, 3489 CrossRef CAS PubMed
-
(a) X. H. Cai, Z. Z. Du and X. D. Luo, Org. Lett., 2007, 9, 1817 CrossRef CAS PubMed
- For selected reviews, see:
(a) C. S. Yeung and V. M. Dong, Chem. Rev., 2011, 111, 1215 CrossRef CAS PubMed
- For selected examples of oxidative coupling reactions of indoles, see:
(a) P. S. Baran and J. M. Richter, J. Am. Chem. Soc., 2004, 126, 7450 CrossRef CAS PubMed
-
(a) Y. Nakano and D. W. Lupton, Chem. Commun., 2014, 50, 1757 RSC
- P. G. Gassman, G. A. Campbell and G. Mehta, Tetrahedron, 1972, 28, 2749 CrossRef CAS
-
(a) N. Neuss, M. Gorman, N. J. Cone and L. L. Huckstep, Tetrahedron Lett., 1968, 783 CrossRef CAS
-
(a) R. J. Owellen, J. Org. Chem., 1974, 39, 69 CrossRef CAS
- For selected examples of the Diels–Alder reactions of 2-vinyl indoles, see:
(a) Y. A.-M. Mohamed, F. Inagaki, R. Takahashi and C. Mukai, Tetrahedron, 2011, 67, 5133 CrossRef CAS PubMed
- The unexpected regioselectivity might be directed by the π–π interaction between the diene and dienophile. See ESI.†.
- 1-OAc substituted tetrahydrocarbazole was obtained by treatment of 3r with acetic acid.
Footnotes |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures, characterization data, and copies of 1H and 13C spectra of all products. See DOI: 10.1039/c4ra14092c |
| ‡ Representative procedure: to a solution of 1a (50.0 mg, 0.15 mmol) in THF (1.0 mL) at −20 °C was added t-BuOCl (16.2 mg, 0.15 mmol) dropwisely. After being stirred at −20 °C for 10 min, benzyl amine (2a) (48.0 mg, 0.45 mmol) was added. The resulting mixture was stirred at −20 °C for additional 2 hours and then quenched by the addition of aqueous sodium bicarbonate (5 mL). After warmed to room temperature, the reaction mixture was extracted with dichloromethane (3 × 10 mL). The combined organic layers were dried over anhydrous sodium sulfate, and concentrated under reduced pressure. The crude residue was purified by silica gel chromatography (dichloromethane/methanol = 50:1) to yield the desired product 3a (53.3 mg, 81% yield) as a light yellow solid. |
| This journal is © The Royal Society of Chemistry 2015 |