
Structures, electronic properties and charge carrier mobility of graphdiyne-like BN nanoribbons
Yanli Suna,
Hongcun Baib and
Yuanhe Huang*a
aCollege of Chemistry, Beijing Normal University, Beijing, 100875, China. E-mail: yuanhe@bnu.edu.cn; Fax: +86-010-58802075; Tel: +86-010-58805425
bKey Laboratory of Energy Sources and Chemical Engineering, State Key Laboratory Cultivation Base of Natural Gas Conversion, Ningxia University, Yinchuan, Ningxia 750021, China
First published on 22nd December 2014
Abstract
In this study, one-dimensional (1D) graphdiyne-like BN (BN-diBN) nanoribbons (NRs) with armchair and zigzag edges are studied using the self-consistent field crystal orbital method based on the density functional theory. The structures, stabilities, electronic properties and charge carrier mobility of these NRs with different widths are investigated and compared to their isoelectronic equivalents, namely, the corresponding graphdiyne NRs. The formation of most of the BN-diBN NRs is energetically favorable according to the calculated Gibbs free energies. The stabilities of these BN-diBN NRs increase as their widths increase. The calculations show that the BN-diBN strips are all semiconductors with wide band gaps. The variation of the band gaps with respect to the NR widths is different for the two patterns of BN-diBN NRs. The mobilities of charge carriers for these BN-diBN NRs are calculated based on the deformation potential theory and effective mass approach. The mobilities are not the monotonic function of the NR widths and have different dependencies on the NR widths for BN-diBN NRs with different edge structures. It is found that the armchair and zigzag BN-diBN NRs are more favorable for the transportation of holes and electrons, respectively.
1. Introduction
As the structural analogues of carbon-based materials, boron nitride nanotubes and nanosheets have attracted considerable interest.1–11 A two-dimensional (2D) graphene-like BN sheet has already been successfully synthesized and it shows a large optical band gap and high transparency in the UV-visible region.12 A theoretical study shows that the BN sheet is a semiconductor with a wide band gap.13 These new discoveries stimulate great efforts to explore new low-dimensional boron nitride nanostructures based on the BN sheet.14–17 It is well known that a graphdiyne sheet composed of sp2 and sp hybridized C atoms has been synthesized,18 in which the hexagonal rings are connected through diacetylenic linkages. Moreover, the 1D NRs obtained by cutting graphdiyne and BN sheets have also been explored,19–22 revealing that their electronic and transport properties are tunable through the variation of NR widths and edge structures. The structure analogues and isoelectronic equivalents of graphdiyne, i.e. graphdiyne-like BN sheets and BN-diyne sheets, which are composed of hexagonal rings with diacetylenic linkages have also been proposed and investigated.14,23,24 With novel electronic properties, the BN analogues of graphdiyne have optimistic prospects and potential applications in nanoelectronics.Inspired by the experimental and theoretical discoveries, we are interested in the properties that the new material would have if a graphdiyne-like BN sheet is cut into strips, i.e., graphdiyne-like BN nanoribbons (denoted as BN-diBN NR). The comparison of the two isoelectronic analogues of carbon- and BN-based NRs would be significant.
In this paper, we perform a theoretical investigation on 1D graphdiyne-like BN NRs with the self-consistent field crystal orbital (SCF-CO) method under the periodical boundary condition. The structures, electronic property and mobility of these BN-diBN NRs are calculated and compared with those of BN and graphdiyne NRs. According to our knowledge, there are no reports of SCF-CO calculations on the BN-diBN NRs to date.
2. Models and computational methods
The two patterns of 1D BN-diBN NRs with armchair and zigzag edges (denoted as A-BN-diBN-M and Z-BN-diBN-M) are shown in Fig. 1. The number M indexes the size of the unit cells for these BN-diBN NR models. Taking M = 1–12, we can study the effect of the quantum confinement for 1D BN-diBN NRs with widths from 10–100 Å. The dangling bonds at both edges in both the zigzag and armchair BN-diBN NRs are terminated by hydrogen atoms, which is similar to the treatment for graphene and graphdiyne NRs.19,25,26 A unit cell contains (9M + 5) BN units and four hydrogen atoms for A-BN-diBN-M, but contains (9M + 10) BN units and eight hydrogen atoms for Z-BN-diBN-M. | ||
| Fig. 1 Models of 1D BN-diBN NRs with (a): armchair edges; (b): zigzag edges. a is the lattice constant of 1D BN-diBN NRs. | ||
The band structures and electronic properties are calculated by the SCF-CO method based on DFT with full structural optimization and CRYSTAL09 program27 for all the models studied. In the DFT SCF-CO calculations, the exchange–correlation functional PBEsol density functional28 and 6–21G(d,p)29 basis set implemented in the program for solid-state calculations are used. Moreover, 41 k-point samplings in the first Brillouin zone are adopted for these 1D BN-diBN NRs and the default values of convergence criteria in CRYSTAL09 are used (total energy change less than 10−6 hartree per cell and geometry optimization with maximum force less than 0.00045 hartree per bohr).
3. Results and discussions
3.1 Structures and stabilities
The widths of A-BN-diBN-M and Z-BN-diBN-M (M = 1–12) are in the range of 11–102 Å and 13–66 Å, respectively. The optimized lattice lengths are from 9.658 Å to 9.687 Å and from 16.736 Å to 16.748 Å for the armchair and zigzag NRs, respectively, which are 0.164–0.298 Å longer than those of the corresponding graphdiyne NRs.19For comparison, the 2D BN-diBN sheet is also investigated with 20 × 20 k-point samplings in the first Brillouin zone and the same calculation method. The calculated B–N bond lengths for the BN-diBN sheet are 1.482 Å in the hexagonal rings, 1.392 Å between the hexagonal rings and diBN linkages, and 1.286 Å and 1.346 Å in the diBN linkages, respectively. Here, the optimized lattice length of the 2D BN-diBN sheet is 9.66 Å. The values of these structure parameters are close to the corresponding values obtained by the projector augmented wave method (PAW),14 and the differences are less than 0.01 Å and 0.03 Å respectively for B–N bond lengths and lattice length. The B–N bond lengths in the hexagonal rings of the graphyne-like BN sheet is 1.423 Å (ref. 30), and is calculated by means of the density-functional based tight-binding method (DF-TB), which is close to that (1.472 Å) obtained by PAW.14 It appears that the influence of these BN-based sheets on the geometric structure is not considerable for different calculation methods. The order of bond lengths for the BN-diBN sheet is the same as that of the corresponding bond lengths for its isoelectronic graphdiyne.31 As for the B–N bond in the 1D BN-diBN NRs, the bond lengths in the middle part of the ribbons gradually reach the corresponding values of 2D BN-diBN with the increase of the ribbon width. The difference in the B–N bond lengths between the 2D sheet and 1D NRs is less than 0.01 Å except for the B–N bonds in the hexagonal rings at the edges of the armchair BN-diBN NRs. The largest difference in the B–N bond lengths between the edges and middle is 0.061 Å for the BN-diBN NRs. Therefore, the edge effect is small on the bond lengths. These results are not unusual because the B and N atoms in the hexagonal rings are all bonded with sp2 hybridization whether they are in middle or at the edges for these 1D BN-diBN NRs. The structural features of the BN-diBN NRs studied are considerably similar to those of the corresponding graphdiyne NRs,19 indicating that the two isoelectronic equivalents are indeed structural analogues.
As the 1D BN-diBN NRs have different chemical compositions, we adopt the approach customarily used in binary phase thermodynamics to account for the chemical composition, which was utilized previously to analyze the relative stability of graphene NRs,32 graphdiyne NRs,19 endohedral silicon nanowires33 and BN NRs.34 The Gibbs free energy δG is defined as follows:
| δG = −E(coh) + xHμH + (1 − xH)μBN |
 | ||
| Fig. 2 δG–W relationship of the BN-diBN NRs. | ||
The Fig. 2 shows that the values of the Gibbs free energies are all negative except for Z-BN-diBN-1 and 2, indicating that the formation for most of the BN-diBN NRs is energetically favorable with respect to the constituents. However, the calculated Gibbs free energies are all positive for the corresponding graphdiyne NRs by the same method.19 The δG of a zigzag BN NR is also positive by the HSE/6-31G** calculation.34 The BN-diBN NRs appear to be the structures that are more stable than the BN and graphdiyne NRs in terms of Gibbs free energy. For a better understanding of the thermodynamic stability of the BN-diBN NRs, the phonon dispersions of the first two smallest NRs for the two different edge types are calculated based on the Hessian matrix at the same computing level.35 Fig. 3 shows the 10 lowest phonon bands for these BN-diBN NRs. The armchair BN-diBN NRs have positive frequencies in the Brillouin zone, except for a few of them that have imaginary frequencies in the small region of k → 0, which is similar to the case in the BN chain and 2D BN sheet.36 The region where imaginary frequencies occur is large in the zigzag BN-diBN NRs, even throughout the Brillouin zone for Z-BN-diBN-1. The occurrence of imaginary frequencies would indicate the structural instabilities. However, the artifacts of the numerical calculations may also result in imaginary frequencies.15 The absolute values of the imaginary frequencies are small and less than 19 cm−1 for the BN-diBN NRs. If the numerical calculation accuracy is not high enough, it may lead to imaginary frequencies. We also performed the calculations of vibration frequencies only at the Γ(k = 0) point for the NRs, which showed that the imaginary frequency did not exist. To further explore the stability of the BN-diBN NRs, the molecular dynamics (MD) simulations on the smallest NRs, A-BN-diBN-1 and Z-BN-diBN-1, were carried out using the DF-TB method37 under the condition of constant-temperature constant-volume at T = 300 K for 5 ps with the time step size as 1 fs. The structures of MD simulations with time evolution for A-BN-diBN-1 and Z-BN-diBN-1 are shown in Fig. 4. It can be seen that the structures of the NRs are stable and change little in the MD simulation. The differences for the B–N bond lengths before and after the MD simulations for 2 ps are less than 0.067 Å and 0.072 Å, respectively, for A-BN-diBN-1 and Z-BN-diBN-1. The corresponding values before and after the MD simulations for 5 ps are even less and are smaller than 0.057 Å and 0.063 Å for A-BN-diBN-1 and Z-BN-diBN-1, respectively. Thus, the BN-diBN NRs should be stable in terms of the MD simulations. Fig. 2 shows that the values of δG decrease monotonically for both the armchair and zigzag 1D BN-diBN NRs with the increase of NR widths. Therefore, the stabilities of these 1D BN-diBN NRs increase as their widths increase. Because the smallest NRs with the largest δG are stable, the larger 1D BN-diBN structures should be stable due to smaller δG. This is considerably different from the case in the corresponding 1D graphdiyne NRs, whose stability decreases with the increase of the NR widths.19 Here, the armchair BN-diBN NRs are more stable than the zigzag BN-diBN NRs, which is also contrary to the case in the graphdiyne NRs.19 Therefore, the two isoelectronic equivalents have similar structures, but have different structure–stability relationships, which reflects the effect of the different components on the stability. The first-principle investigations also showed that the armchair BN NRs were more stable than the zigzag NRs,21,38 which implies that the insertion of the diBN linkages in the BN NRs does not alter the relative stabilities for the two different types of BN-based NRs. In addition, the δG for the armchair BN-diBN NRs changes more smoothly than that for the zigzag BN-diBN NRs with the increase of the widths. The NR width thus has a smaller effect on the stability of the armchair BN-diBN NRs. These results indicate that the edge structure has an important influence on the relationship of stability–NR widths for the 1D BN-diBN NRs.
 | ||
| Fig. 3 Calculated phonon bands of the armchair and zigzag BN-diBN NRs. | ||
 | ||
| Fig. 4 The structures of MD simulations with time evolution for 5 ps: (a) A-BN-diBN-1 and (b) Z-BN-diBN-1. | ||
3.2 Band structures and electronic properties
The calculated band structures of 1D BN-diBN NRs are presented in Fig. 5. From the band structures, it can be seen that all the 1D BN-diBN NRs have a direct band gap (Eg) at the Γ point, regardless of the edge structures. Therefore, the 1D BN-diBN NRs are all semiconductors. Thus, the semiconducting property of these BN-diBN NRs is edge-independent, which is similar to the case in the corresponding graphdiyne NRs.19,39 The calculated band gaps are in the range of 3.76–3.99 eV and 3.38–3.53 eV with M = 1–12 for the armchair and zigzag BN-diBN NRs, respectively. Thus, these BN-diBN NRs are semiconducting materials with a wide band gap. | ||
| Fig. 5 Band structures of (a) A-BN-diBN-M and (b) Z-BN-diBN-M with M = 1–12. | ||
The band gap variations of the BN-diBN NRs with respect to NR width (Eg–W relationship) are shown in Fig. 6. We can see that the band gaps of the armchair BN-diBN NRs decrease as their widths increase, whereas those of the zigzag BN-diBN NRs increase with the increase of the NR widths. However, the band gaps of both the armchair and zigzag graphdiyne NRs decrease monotonically as the widths increase.19,39 Although the BN-diBN and graphdiyne NRs are structural analogues and isoelectronically equivalent, the dependence of the band gaps on the NR widths is different due to the different compositions. The band gaps of both the types of BN-diBN NRs are close to that of the 2D sheet. The calculated Eg of the 2D BN-diBN sheet here is 3.77 eV, which is close to that (3.88 eV) obtained by the PAW method,14 but smaller than those of the BN sheets by the PBE functional (4.7 eV)40 and by DF-TB (4.17 eV).30 Moreover, the band gaps of these BN-diBN NRs are smaller than those of the BN NRs.41 Like the diacetylenic linkages in the BN-diyne,23 the diBN linkages can also decrease the band gap from BN to BN-diBN NRs.
 | ||
| Fig. 6 Eg–W relationship of BN-diBN NRs. | ||
Compared to the graphdiyne NRs, the BN-diBN NRs have a smaller change range of Eg with the variation of the NR widths. The differences of the band gaps are less than 0.23 and 0.15 eV for the armchair and zigzag BN-diBN NRs with M = 1–12, respectively, which is larger than that by metal adatoms for the armchair graphdiyne NRs.42 However, the band gaps change from 0.97 eV to 0.48 eV for the armchair graphdiyne NRs and from 1.65 eV to 0.56 eV for the zigzag graphdiyne NRs with the change of the NR widths.19,39 As a matter of fact, the changes of the band gap for both the armchair and zigzag BN-diBN NRs are less than 0.1 eV with M ≥ 3. Therefore, different from the graphdiyne NRs, the BN-diBN NRs have a quite limited adjustment of the band gaps with the change of the NR widths. Perhaps, this character may be suitable for the subtle adjustment of the band gaps. Moreover, the Eg–W relationship of the BN-diBN NRs is also different from that of the BN NRs. The PAW calculations show that the band gaps oscillate or fluctuate for the armchair BN NRs but decrease for the zigzag BN NRs with the increase of the NR widths.36,41 This indicates that inserting diBN units between the hexagonal rings in the BN NRs alters the dependence of the band gaps on the NR widths.
From Fig. 5, it can also be seen that the frontier bands are very narrow for the zigzag BN-diBN NRs. Here, the frontier bands refer to the highest occupied band (HOB) and the lowest unoccupied band (LUB). The HOB and LUB bandwidths of the zigzag BN-diBN NRs are about 0.057–0.063 and 0.082–0.099 eV, respectively. It is known that the bandwidths are concerned with the degree of orbital overlap between the neighboring unit cells. As a representation, the highest occupied and the lowest unoccupied crystal orbitals (HOCO and LUCO) in a unit cell are shown in Fig. 7 for Z-BN-diBN-6 and A-BN-diBN-6. It can be seen that both the HOCO and LUCO mainly come from the contribution of the π atomic orbitals at only one edge for Z-BN-diBN-6. As a result, the orbital overlap between the neighboring unit cells is very small, leading to a narrow bandwidth. Moreover, the bond lengths are slightly changed as the NR width increases. The distances between the bonding orbital interaction in HOCO of Z-BN-diBN-6 are slightly shorter (<0.003 Å) than those of Z-BN-diBN-1, but the case is just contrary to the distance of the antibonding interaction (0.002–0.004 Å longer), which leads to the lower HOCO of Z-BN-diBN-6. As for the LUCOs in the two NRs, the orbitals in the edge hexagonal rings have the largest contribution to antibonding interaction. Compared with Z-BN-diBN-1, Z-BN-diBN-6 has slightly longer bond lengths of antibonding interaction in the hexagonal rings (about 0.001 Å longer). However, the bonding and antibonding lengths in Z-BN-diBN-6 are slightly longer and shorter than those in Z-BN-diBN-1 along the BN chains connecting the two edge hexagonal rings, respectively, which partially compensates the reduction of antibonding interaction in the hexagonal rings. Therefore, the LUCO falls less than the HOCO for Z-BN-diBN-6. The HOCO and LUCO of Z-BN-diBN-6 are 0.16 eV and 0.05 eV lower than those of Z-BN-diBN-1, respectively. The band gap of Z-BN-diBN-6 thus is 0.11 eV larger than that of Z-BN-diBN-1. In addition, because the changes in the structure are considerably small, the energy variation of the frontier orbitals is small, resulting in the small change of the band gaps with the change of the zigzag NR widths (less than 0.15 eV). Compared with Z-BN-diBN-6, the π atomic orbitals at most parts of A-BN-diBN-6 have contributions to both the HOCO and LUCO except for those at the edges, resulting in a larger orbital overlap between the adjacent unit cells. The bandwidths of the HOBs and LUBs are 0.24–0.30 and 0.17–0.20 eV for the armchair BN-diBN NRs, respectively, except for the LUBs of A-BN-diBN-1 (0.36 eV) and 2 (0.26 eV). Usually, a wider bandwidth would be favorable to the movement of the charge carriers.
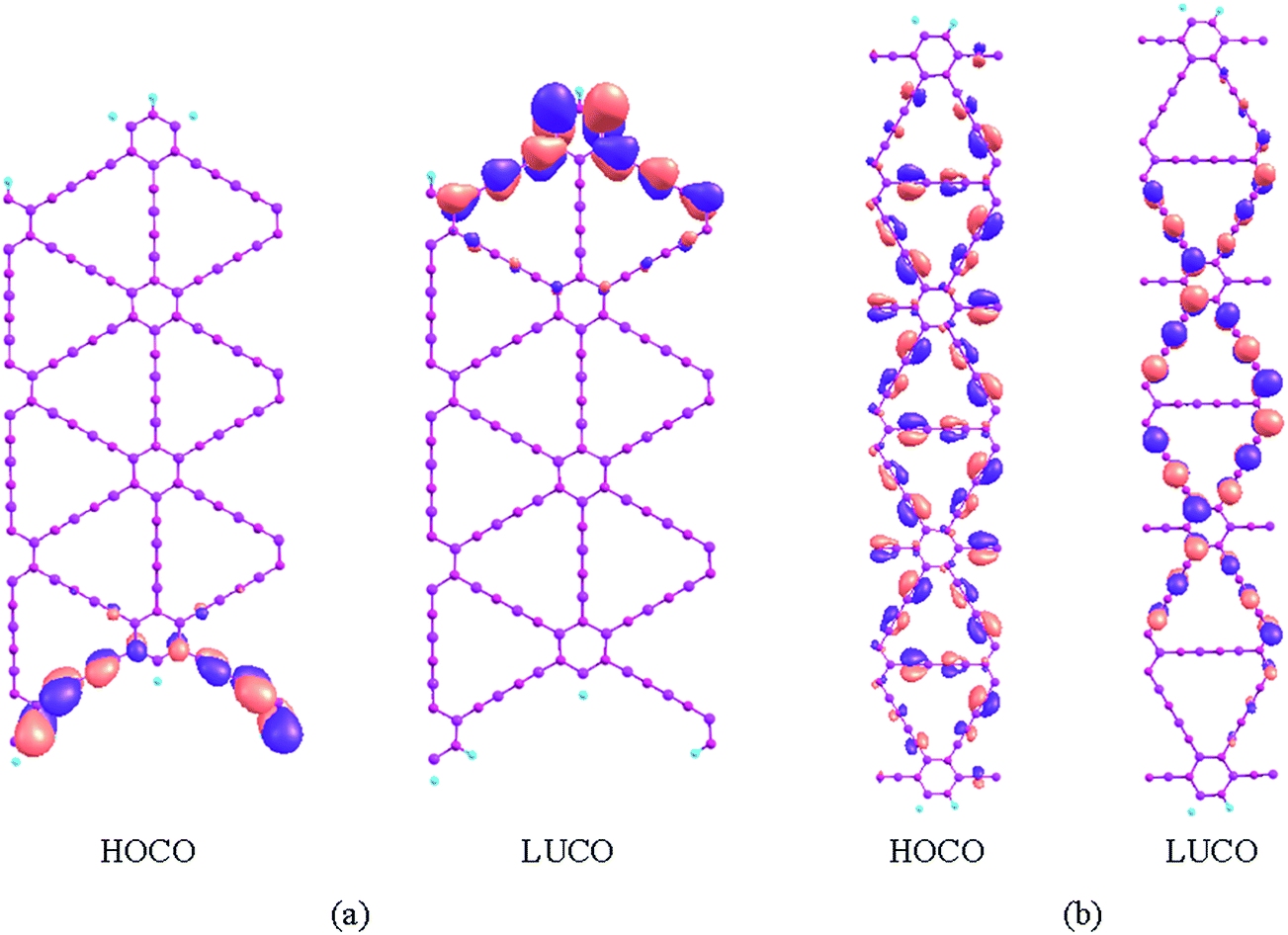 | ||
| Fig. 7 Frontier crystal orbitals of (a) Z-BN-diBN-6 and (b) A-BN-diBN-6. | ||
3.3 Carrier mobilities
The mobility of the charge carrier for these 1D BN-diBN NRs is calculated using the equation , which was derived with the deformation potential (DP) theory and effective mass approximation43,44 and was successfully applied to 1D conjugated polymers,45 graphdiyne,19,46 graphene NRs,47–49 and carbon nanotubes.50,51 In this equation, C is the stretching modulus of a 1D crystal, m* is the effective mass of the charge carriers, and E1c and E1v are the DP constants for electrons and holes, respectively. These parameters are obtained using the procedure described in ref. 19.
, which was derived with the deformation potential (DP) theory and effective mass approximation43,44 and was successfully applied to 1D conjugated polymers,45 graphdiyne,19,46 graphene NRs,47–49 and carbon nanotubes.50,51 In this equation, C is the stretching modulus of a 1D crystal, m* is the effective mass of the charge carriers, and E1c and E1v are the DP constants for electrons and holes, respectively. These parameters are obtained using the procedure described in ref. 19.
The calculated mobilities of the charge carriers for these BN-diBN NRs at temperature T = 298 K are shown in Table 1. For the armchair type, the mobilities of electrons (μe) and holes (μh) are in the range of 102–103 and 103–104 cm2 V−1 s−1, respectively. The μe is one or two orders of magnitude smaller than that of the armchair graphdiyne NRs, but the μh is considerably larger than that of the corresponding graphdiyne NRs.19 The μe and μh change smoothly and have an increasing trend with the increase of the NR widths. The difference between the largest and the smallest mobility values for the same charge carriers (μe or μh) is less than one order of magnitude, indicating the small effect of the NR width on the mobilities. Moreover, the μh is always larger than the μe for any one of the armchair BN-diBN NRs. Therefore, armchair BN-diBN NRs are more favorable to the movement of holes, which is contrary to the case in the corresponding graphdiyne NRs.19,46 Here, the calculated bandwidth of the HOB is wider than that of the LUB for A-BN-diBN-M (≥3). A wider bandwidth means better delocalization, which is favorable for moving the charge carriers due to a smaller effective mass. However, the holes in A-BN-diBN-1 with a wider LUB still have larger mobility than the electrons. Moreover, the difference of the effective mass between the electrons and holes is not significant, which cannot make the hole mobility one order of magnitude larger than the electron mobility. Here, the effective masses of the charge carriers in the BN-diBN NRs are considerably larger than those in the corresponding graphdiyne NRs19,46 and 2D graphdiyne sheet.52 From Table 1, it can be seen that the DP constants of the electrons (E1c) are all more than three times larger than the corresponding DP constants of the holes (E1v) for all the armchair BN-diBN NRs. The larger difference between E1c and E1v indicates very different strengths of scattering by the longitudinal acoustic wave for the two different charge carriers. Therefore, it is the stronger electron scattering in the bottom of the LUB that results in the smaller electron mobility. Moreover, it can be seen that the BN chains parallel to the ribbon translational direction (denoted as p-BN chains) do not contribute to the LUCO of A-BN-diBN-6 in Fig. 7(b). Therefore, it is hard for the electrons to move along the p-BN chains. The atoms with and without contribution to the LUCO are arranged alternately along the BN chains nonparallel to the ribbon axis, and the orbital overlap among the atoms in these chains is very less, resulting in the poor delocalization of the LUCO and difficulty for the electrons to move. It has also been pointed out that the charge carriers are more easily scattered by acoustic phonons in the orbital with more nodes in the dilation direction.46 The LUCOs in Fig. 7 exhibit that the BN chains along the extended direction in the LUCO of A-BN-diBN-6 has more nodes than the edge BN chain in the LUCO of Z-BN-diBN-6. From Table 1, it can be seen that the DP constants (E1c) of the armchair NRs are considerably larger than those of the zigzag NRs, which indicates stronger scattering in the armchair NRs than in the zigzag NRs. Therefore, the armchair NRs have lower electron mobility than the zigzag NRs. We would like to point out that some of the atoms in the p-BN chains also contribute π orbitals to the LUCOs for the smaller A-BN-diBN-M (M < 5), but the atoms with and without the contribution to the LUCO are also arranged alternately. The delocalization of the LUCOs is still poor for the smaller armchair BN-diBN NRs.
| NR | C | m*e | m*h | E1v | E1c | μe | μh |
|---|---|---|---|---|---|---|---|
| A-BN-diBN-1 | 166 | 0.339 | 0.406 | 0.966 | 5.653 | 2.105 × 102 | 5.501 × 103 |
| A-BN-diBN-2 | 241 | 0.384 | 0.438 | 1.022 | 5.400 | 2.779 × 102 | 6.368 × 103 |
| A-BN-diBN-3 | 339 | 0.483 | 0.433 | 0.956 | 4.148 | 4.696 × 102 | 1.041 × 104 |
| A-BN-diBN-4 | 425 | 0.777 | 0.726 | 0.964 | 3.830 | 3.384 × 102 | 5.914 × 103 |
| A-BN-diBN-5 | 512 | 0.751 | 0.706 | 0.965 | 3.706 | 4.582 × 102 | 7.414 × 103 |
| A-BN-diBN-6 | 600 | 0.706 | 0.654 | 0.964 | 3.638 | 6.114 × 102 | 9.766 × 103 |
| A-BN-diBN-7 | 687 | 0.725 | 0.670 | 0.959 | 3.611 | 6.828 × 102 | 1.090 × 104 |
| A-BN-diBN-8 | 773 | 0.712 | 0.665 | 0.957 | 3.601 | 7.938 × 102 | 1.245 × 104 |
| A-BN-diBN-9 | 861 | 0.687 | 0.637 | 0.955 | 3.607 | 9.297 × 102 | 1.486 × 104 |
| A-BN-diBN-10 | 949 | 0.699 | 0.651 | 0.949 | 3.594 | 1.006 × 103 | 1.605 × 104 |
| A-BN-diBN-11 | 1036 | 0.689 | 0.634 | 0.945 | 3.576 | 1.133 × 103 | 1.838 × 104 |
| A-BN-diBN-12 | 1122 | 0.693 | 0.643 | 0.942 | 3.577 | 1.216 × 103 | 1.962 × 104 |
| Z-BN-diBN-1 | 113 | 0.876 | 1.214 | 2.696 | 0.749 | 1.965 × 103 | 9.298 × 101 |
| Z-BN-diBN-2 | 163 | 0.797 | 1.223 | 3.419 | 0.171 | 6.268 × 104 | 8.248 × 101 |
| Z-BN-diBN-3 | 213 | 0.813 | 1.123 | 3.867 | 0.254 | 3.603 × 104 | 9.575 × 101 |
| Z-BN-diBN-4 | 262 | 0.726 | 1.134 | 4.241 | 0.507 | 1.318 × 104 | 9.650 × 101 |
| Z-BN-diBN-5 | 311 | 0.726 | 1.124 | 4.556 | 0.917 | 4.783 × 103 | 1.006 × 102 |
| Z-BN-diBN-6 | 359 | 0.739 | 1.197 | 4.825 | 0.718 | 8.769 × 103 | 9.420 × 101 |
| Z-BN-diBN-7 | 409 | 0.726 | 1.095 | 5.055 | 1.416 | 2.638 × 103 | 1.118 × 102 |
| Z-BN-diBN-8 | 456 | 0.774 | 1.194 | 5.277 | 1.409 | 2.698 × 103 | 1.004 × 102 |
| Z-BN-diBN-9 | 506 | 0.861 | 1.217 | 5.443 | 0.412 | 2.985 × 104 | 1.018 × 102 |
| Z-BN-diBN-10 | 556 | 0.878 | 1.241 | 5.617 | 0.253 | 8.447 × 104 | 1.020 × 102 |
| Z-BN-diBN-11 | 606 | 0.732 | 1.166 | 5.762 | 0.842 | 1.092 × 104 | 1.160 × 102 |
| Z-BN-diBN-12 | 653 | 0.754 | 1.151 | 5.901 | 1.290 | 4.795 × 103 | 1.215 × 102 |
As for the zigzag BN-diBN NRs, the electron mobility μe displays fluctuating changes with the increase of NR widths. The two peak values of μe occur in Z-BN-diBN-2 and Z-BN-diBN-10. Contrary to the situation in the armchair BN-diBN NRs, the zigzag BN-diBN NRs have higher electron mobilities, which are in the range of 103–104 cm2 V−1 s−1 and 1–2 orders of magnitude larger than the corresponding μh, mainly due to the smaller E1c. Similar to the corresponding graphdiyne NRs,19,46 the zigzag BN-diBN NRs are more favorable for the transport of electrons. The LUCOs of the zigzag BN-diBN NRs are all similar to that shown in Fig. 7(a) for Z-BN-diBN-6. The electron path thus is mainly constructed by one edge of the zigzag ribbons. All the atoms at the edge contribute π orbitals to the LUCO except for the atoms bonding with H atoms. Therefore, the orbital delocalization along the electron path of the zigzag BN-diBN NRs is higher than that of the armchair BN-diBN NRs, leading to higher electron mobility. The edge structures thus have great influence on the behaviors of the different charge carriers for these BN-diBN NRs. The charge carriers in the BN-diBN NRs with different edge structures would have different interactions with the acoustic phonon, i.e. different scatterings by the acoustic wave.
It should be noted that the relationship of the mobility with the change of NR widths for the BN-diBN NRs is considerably different from that for the corresponding isoelectronic species graphdiyne NRs in a term of the calculations. The mobilities of both the electrons and holes all increased monotonously with the increase of the NR widths for the graphdiyne NRs.19 In addition, both the armchair and zigzag graphdiyne NRs have the same relationship of electron or hole mobility–NR width (W). As for the BN-diBN NRs, however, the mobilities of the charge carriers do not always change monotonously but have at least a fluctuation within the range of the NR width studied. The mobility–W relationship is edge-dependent and very different for the two different types of BN-diBN NRs. As discussed in the above section, the BN-diBN and graphdiyne NRs also have different dependencies of band gaps on the NR widths. These indicate that both the structure and composition have a significant influence on the electronic and transport properties for the two isoelectronic equivalents with similar structures.
3.4 Young's moduli
The Young's modulus (Y) calculated as .
.  is the second derivative of the energy (E) of a unit cell with respect to the uniaxial linear strain ε along the 1D direction. V0 is the volume of a unit cell and ε is a small deformation of the lattice constants. Here, the thickness of the ribbons is taken as 3.4 Å to evaluate the volume, which is the sum of the van der Waals radii of B (1.8 Å) and N (1.6 Å).53 The calculated Young's moduli of these NRs are in the range of 518–719 GPa for A-BN-diBN-M and a little larger than those of 428–470 GPa for Z-BN-diBN-M. Moreover, the Young's moduli of the BN-diBN NRs are slightly smaller than those of the corresponding graphdiyne NRs.19 Therefore, the BN-diBN NRs should have a weaker resistance to the strain along the axis direction than the graphdiyne NRs. For comparison, the Young's moduli of two armchair graphene NRs with widths of about 2.7 and 4.0 nm are also calculated to be 1184 and 1035 GPa at the same computational level, respectively. Compared to the BN-diBN and graphdiyne NRs, the larger Young's moduli of the graphene NRs are resulted from the sp2 net structure of the denser carbon atom distribution. Similar to the situation in the graphdiyne NRs, A-BN-diBN-1 with the narrowest width has the largest Young's modulus among these 1D NRs due to the largest atom distribution density among these 1D BN-diBN NRs.
is the second derivative of the energy (E) of a unit cell with respect to the uniaxial linear strain ε along the 1D direction. V0 is the volume of a unit cell and ε is a small deformation of the lattice constants. Here, the thickness of the ribbons is taken as 3.4 Å to evaluate the volume, which is the sum of the van der Waals radii of B (1.8 Å) and N (1.6 Å).53 The calculated Young's moduli of these NRs are in the range of 518–719 GPa for A-BN-diBN-M and a little larger than those of 428–470 GPa for Z-BN-diBN-M. Moreover, the Young's moduli of the BN-diBN NRs are slightly smaller than those of the corresponding graphdiyne NRs.19 Therefore, the BN-diBN NRs should have a weaker resistance to the strain along the axis direction than the graphdiyne NRs. For comparison, the Young's moduli of two armchair graphene NRs with widths of about 2.7 and 4.0 nm are also calculated to be 1184 and 1035 GPa at the same computational level, respectively. Compared to the BN-diBN and graphdiyne NRs, the larger Young's moduli of the graphene NRs are resulted from the sp2 net structure of the denser carbon atom distribution. Similar to the situation in the graphdiyne NRs, A-BN-diBN-1 with the narrowest width has the largest Young's modulus among these 1D NRs due to the largest atom distribution density among these 1D BN-diBN NRs.
4. Conclusions
The two patterns of 1D BN-diBN NRs with different widths have been investigated using the SCF-CO method based on DFT calculations.The calculated Gibbs free energies are negative for most of the BN-diBN NRs studied, which indicates that the formation of these NRs is energetically favorable with respect to the constituents. Different from the graphdiyne NRs, the stabilities of these BN-diBN NRs increase as their widths increase, and the armchair BN-diBN NRs are more stable than the zigzag ones. Both the armchair and zigzag BN-diBN NRs are wide band gap semiconductors. The band gaps decrease and increase with the increase of NR widths for the armchair and zigzag BN-diBN NRs, respectively, showing that the Eg–W relationship is different from that for the graphdiyne NRs.
The mobilities of the charge carriers are calculated using the DP theory and effective mass approach. Different from the case for the graphdiyne NRs, the mobilities of both the electrons and holes are not monotonic functions of the NR widths for the BN-diBN NRs. The mobility of the electrons is about 1–2 orders of magnitude smaller than that of holes in the armchair NRs, but just opposite in the zigzag NRs. The electron mobility in the zigzag BN-diBN NRs and hole mobility in the armchair BN-diBN NRs are about 103–104 cm2 V−1 s−1. Therefore, the edge structures of the BN-diBN NRs greatly affect the behavior of the charge carriers. These BN-diBN NRs can be considered as possible candidates for 1D transport materials with high mobility. Moreover, we can select the different patterns of BN-diBN NRs as transport materials for different charge carriers. The calculated values of the Young's moduli show that the BN-diBN NRs have smaller Young's moduli than the corresponding graphdiyne NRs.
The BN-diBN and graphdiyne NRs have similar geometrical structures, but the transport properties are very different for the two isoelectronic equivalents. They also have different dependencies of band gaps on the NR widths. These manifest that the different compositions play important roles in the electronic and transport properties of the two isoelectronic equivalents.
Acknowledgements
This work is supported by the National Natural Science Foundation of China (Grant no. 20873009 and 21363017).References
- D. Golberg, Y. Bando, Y. Huang, T. Terao, M. Mitome, C. C. Tang and C. Y. Zhi, ACS Nano, 2010, 4, 2979–2993 CrossRef CAS PubMed.
- S. Tang and Z. Cao, Comput. Mater. Sci., 2010, 48, 648 CrossRef CAS PubMed.
- A. Rubio, J. L. Corkill and M. L. Cohen, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 5081–5084 CrossRef CAS.
- X. Blase, A. Rubio, S. G. Louie and M. L. Cohen, Europhys. Lett., 1994, 28, 335–340 CrossRef CAS.
- S. Alexandre, M. S. C. Mazzoni and H. Chacham, Appl. Phys. Lett., 1999, 75, 61–63 CrossRef CAS PubMed.
- N. Chopra, R. J. Luyken and K. Cherrey, Science, 1995, 269, 966–967 CrossRef CAS PubMed.
- J. Cumings and A. Zettl, Chem. Phys. Lett., 2000, 316, 211–216 CrossRef CAS.
- V. Barone and J. E. Peralta, Nano Lett., 2008, 8, 2210–2214 CrossRef CAS PubMed.
- C. Park and S. G. Louie, Nano Lett., 2008, 8, 2200–2203 CrossRef CAS PubMed.
- M. S. Si and D. S. Xue, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 193409 CrossRef.
- Y. Zhang, K. Zhou, X. Gou, K. Xie, H. Zhang and Y. Peng, Chem. Phys. Lett., 2010, 484, 266–270 CrossRef CAS PubMed.
- L. Song, L. Ci, H. Lu, P. B. Sorokin, C. Jin, J. Ni, A. G. Kvashnin, D. G. Kvashnin, J. Lou, B. I. Yakobson and P. M. Ajayan, Nano Lett., 2010, 10, 3209–3215 CrossRef CAS PubMed.
- X. Blase, A. Rubio, S. G. Louie and M. L. Cohen, Phys. Rev. B: Condens. Matter Mater. Phys., 1995, 51, 6868–6875 CrossRef CAS.
- X. Cao, Y. Li, X. Cheng and Y. Zhang, Chem. Phys. Lett., 2011, 502, 217–221 CrossRef CAS PubMed.
- V. O. Özçelik and S. Ciraci, J. Phys. Chem. C, 2013, 117, 2175–2182 Search PubMed.
- J. Gong, Y. Tang and P. Yang, J. Mol. Struct., 2014, 1064, 32–36 CrossRef CAS PubMed.
- D. Ghosh, P. Parida and S. K. Pati, J. Mater. Chem. C, 2014, 2, 392–398 RSC.
- G. Li, Y. Li, H. Liu, Y. Guo, Y. Li and D. Zhu, Chem. Commun., 2010, 46, 3256–3258 RSC.
- H. Bai, Y. Zhu, W. Qiao and Y. Huang, RSC Adv., 2011, 1, 768–775 RSC.
- J. Kang, F. Wu and J. Li, J. Phys.: Condens. Matter, 2012, 24, 165301 CrossRef PubMed.
- R. Mukherjee and S. Bhowmick, J. Chem. Theory Comput., 2011, 7, 720–724 CrossRef CAS.
- Y. Ding, Y. Wang and J. Ni, Appl. Phys. Lett., 2009, 94, 233107 CrossRef PubMed.
- J. Zhou, K. Lv, Q. Wang, X. S. Chen, Q. Sun and P. Jena, J. Chem. Phys., 2011, 134, 174701 CrossRef PubMed.
- H. Bu, M. Zhao, H. Zhang, X. Wang, Y. Xi and Z. Wang, J. Phys. Chem. A, 2012, 116, 3934–3939 CrossRef CAS PubMed.
- K. Nakada, M. Fujita, G. Dresselhaus and M. S. Dresselhaus, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 17954–17961 CrossRef CAS.
- R. H. Baughman and H. Eckhardt, J. Chem. Phys., 1987, 87, 6687–6699 CrossRef CAS PubMed.
- R. Dovesi, V. R. Saunders, C. Roetti, R. Orlando, C. M. Zicovich-Wilson, F. Pascale, K. Doll, N. M. Harrison, B. Civalleri, I. J. Bush, Ph. D'Arco and M. Llunell, CRYSTAL09 User's Manual, University of Torino, Torino, 2010 Search PubMed.
- J. Perdew, A. Ruzsinsky, G. I. Csonka, O. A. Vydrov, G. E. Scuseria, L. A. Constantin, X. Zhou and K. Burke, Phys. Rev. Lett., 2008, 100, 136406 CrossRef.
- R. Dovesi, M. Causa, R. Orlando, C. Roetti and V. R. Saunders, J. Chem. Phys., 1990, 92, 7402–7411 CrossRef CAS PubMed.
- A. N. Enyashin and A. L. Ivanovskii, Chem. Phys. Lett., 2011, 509, 143–147 CrossRef CAS PubMed.
- N. Narita, S. Nagai, S. Suzuki and K. Nakao, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 58, 11009–11014 CrossRef CAS.
- V. Barone, O. Hod and G. E. Scuseria, Nano Lett., 2006, 6, 2748–2754 CrossRef CAS PubMed.
- T. Dumitric, M. Hua and B. I. Yakobson, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 70, 241303 CrossRef.
- D. Krepel and O. Hod, J. Chem. Theory Comput., 2014, 10, 373–380 CrossRef CAS.
- F. Pascale, C. M. Zicovich-Wilson, F. Lopez Gejo, B. Civalleri, R. Orlando and R. Dovesi, J. Comput. Chem., 2004, 25, 888–897 CrossRef CAS PubMed.
- M. Topsakal, E. Aktürk and S. Ciraci, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 115442 CrossRef.
- M. Elstner, D. Porezag, G. Jungnickel, J. Elsner, M. Haug, T. Frauenheim, S. Suhai and G. Seifert, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 58, 07260 CrossRef CAS.
- B. Huang, H. Lee, B. Gu, F. Liu and W. Duan, Nano Res., 2012, 5, 62–72 CrossRef CAS.
- L. D. Pan, L. Z. Zhang, B. Q. Song, S. X. Du and H. J. Gao, Appl. Phys. Lett., 2011, 98, 173102 CrossRef PubMed.
- J. Zhou, Q. Wang, Q. Sun and P. Jena, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 085442 CrossRef.
- A. J. Du, S. C. Smith and G. Q. Lu, Chem. Phys. Lett., 2007, 447, 181–186 CrossRef CAS PubMed.
- Z. Z. Lin, Q. Wei and X. Zhu, Carbon, 2014, 66, 504–510 CrossRef CAS PubMed.
- Y. Huang and R. Liu, Chem. Res. Chin. Univ., 1991, 7, 107–113 CAS.
- F. B. Beleznay, F. Bogár and J. Ladik, J. Chem. Phys., 2003, 119, 5690–5695 CrossRef CAS PubMed.
- G. Wang and Y. Huang, J. Phys. Chem. Solids, 2007, 68, 2003–2007 CrossRef CAS PubMed.
- M. Long, L. Tang, D. Wang, Y. Li and Z. Shuai, ACS Nano, 2011, 5, 2593–2600 CrossRef CAS PubMed.
- G. Wang, Phys. Chem. Chem. Phys., 2011, 13, 11939–11945 RSC.
- M. Long, L. Tang, D. Wang, L. Wang and Z. Shuai, J. Am. Chem. Soc., 2009, 131, 17728–17729 CrossRef CAS PubMed.
- L. Chen, L. Wang and D. Beljonne, Carbon, 2014, 77, 868–879 CrossRef CAS PubMed.
- G. Wang and Y. Huang, J. Phys. Chem. Solids, 2008, 69, 2531–2534 CrossRef CAS PubMed.
- W. Qiao, H. Bai, Y. Zhu and Y. Huang, J. Phys.: Condens. Matter, 2012, 24, 185302 CrossRef PubMed.
- G. Luo, X. Qian, H. Liu, R. Qin, J. Zhou, L. Li, Z. Gao, E. Wang, W. Mei, J. Lu, Y. Li and S. Nagase, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 075439 CrossRef.
- S. S. Batsanov, Inorg. Mater., 2001, 37, 871–885 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2015 |