
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ternary transition-metal fluoride precursors for the fluorolytic sol–gel route: new insights into speciation and decomposition†
J.
Kohl
a,
D.
Wiedemann
*a,
S. I.
Troyanov
b,
E.
Palamidis
a and
M.
Lerch
a
aTechnische Universität Berlin, Institut für Chemie, Straße des 17. Juni 135, 10623 Berlin, Germany. E-mail: dennis.wiedemann@chem.tu-berlin.de; Fax: +49 30 314-79656; Tel: +49 30 314-26178
bHumboldt-Universität zu Berlin, Institut für Chemie, Brook-Taylor-Straße 2, 12489 Berlin, Germany. E-mail: sergej.troyanov@rz.hu-berlin.de; Fax: +49 30 2093-7468; Tel: +49 30 2093-7303
First published on 24th June 2015
Abstract
The nanoscaled ternary transition-metal fluorides Li3MF6 (M = V, Fe, Mn) and Li2NiF4 are promising candidates for cathode materials in high-voltage lithium-ion batteries. The fluorolytic route to these compounds relies on thermal decomposition of a hitherto uncharacterised precursor mixture produced from acetylacetonates and hydrofluoric acid. By addition of pyridine, different cationic, electroneutral and anionic complexes containing the motifs [MFn](3−n)+ (n = 0–4) have been trapped and characterised by single-crystal X-ray diffraction and IR spectroscopy. Based on the results, a model of successive and incomplete fluorination is proposed for the speciation and formation of the precursor. The decomposition of the latter has been monitored via thermogravimetry (TG) and differential scanning calorimetry (DSC).
Introduction
In search of new cathode materials for lithium-ion batteries during the last decade, ternary fluorides have become candidates of interest, because theoretical calculations had indicated an increase in redox potential when substituting fluorine for oxygen.1 Because of their high lithium content and their variability in oxidation numbers, the ternary fluorides Li3MF6 (M: transition metal) are a focus of research. The preparation of nanosized particles is important with regard to fluorides exhibiting low electrical conductivity. Detailed descriptions of the syntheses and scrutinous electrochemical characterisations of Li3FeF6 and Li3VF6 are available.2–4 In 2012, we reported an approach for the synthesis of ternary fluorides Li3MF6 on the basis of the fluorolytic sol–gel process developed by Kemnitz et al., who described the preparation of a high-surface aluminium fluoride.5,6 The starting materials of our route are lithium tert-butoxide and a transition-metal acetylacetonate [MIII(acac)3], which are reacted with an excess of hydrogen fluoride in an organic solvent (cf.Scheme 1). | ||
| Scheme 1 Preparation of Li3VF6via the fluorolytic sol–gel route (tBu: tert-butyl, Et: ethyl). | ||
The precursor obtained in this first step is then calcined in a dinitrogen atmosphere to form the ternary fluoride. Using this protocol, either modification of Li3VF6 can be prepared in excellent yield: heating of the sample to 300 °C followed by slow cooling to room temperature results in the formation of monoclinic β-Li3VF6, whereas heating the precursor to 700 °C and quenching to room temperature results in the formation of orthorhombic α-Li3VF6. Moreover, β-Li3VF6 can be prepared with domain sizes smaller than 50 nm. While monoclinic β-Li3CrF6 can be synthesised in good yield, the yields of other ternary fluorides are considerably lower—and Li3MnF6 as well as Li3CoF6 cannot at all be synthesised following this route. Monoclinic Li3FeF6 and Li2MnF5 are obtained in moderate to low yields together with the difluorides MF2. In contrast to this, the method can be successfully applied to the system Li–Ni–F: Li2NiF4 is prepared in high yields with domain sizes of 55 nm.7 The precursors contain highly amorphous fractions, making it impossible to get information about their composition by X-ray powder diffraction techniques. Only the existence of LiF and Li2SiF6 in the precursor materials has been confirmed so far. As shown by elemental analyses, the precursors contain high amounts of carbon, hydrogen and oxygen indicating the presence of organic residues. For example, the Li3VF6 precursor additionally contains acetylacetonato ligands bound to vanadium(III) as well as hexafluoridovanadate(III). This has been confirmed by IR spectroscopy and 1H-NMR spectroscopy in solution.6 The material obtained from precursor decomposition contains about 2% residual carbon. However, it was suggested that the precursor be a mixture of several species. Accurate information about its composition is essential for optimising precursor synthesis and decomposition. Bearing this in mind, we have chosen to try and stabilise single species by providing additional donor molecules and crystallise the compounds thus obtained.8 Using this technique, the first of a whole cascade of fluorination steps has been identified and its product, after addition of acetonitrile, [VIII(acac)2(CH3CN)2]BF4 crystallographically characterised.6 In this work, we report on further experiments of the same kind. By addition of pyridine, we stabilised four new species in the system Li–V–F. These reflect the stepwise fluorination process of the starting material [VIII(acac)3], leading to [VF4]−. We have also successfully transferred this concept to Li2MnF5 and Li2NiF4 precursors.
Experimental
Analysis and characterisation
Preparation of single crystals
![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) max/cm−1 3100–2800w (ν[C–H]py), 1605m (ν[C
max/cm−1 3100–2800w (ν[C–H]py), 1605m (ν[C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C]py), 1575m (ν[CO]acac, ν[CC]acac), 1530m, 1524s (ν[CO]acac, ν[CC]acac), 1484w (ν[CC]py), 1448s (ν[CC]py), 1376s (ν[C–O]acac, γ[C–C]acac), 1286w, 1213w, 1067w, 1043m, 766m (γ[C–H]py), 702s (γ[C–H]py), 638m (γ[C–H]py), 578s, 561vs, 537m (ν[V–O], γ[V–acac]), 450m (γ[Npy–V]), 418w and 330s (ν[V–F]). Anal. found: C, 52.3; H, 5.2; N, 8.2. Calc. for C15H17F2N2O2V (364.25): C, 52.0; H, 4.95; N, 8.1%.
max/cm−1 3150–2800w (ν[C–H]py), 1605s (ν[CC]py), 1483m (ν[CC]py), 1448s (ν[CC]py), 1217m, 1101s, 1058vs, 1043s, 950m, 770m (γ[C–H]py), 709m (γ[C–H]py), 697s (γ[C–H]py), 640m (γ[C–H]py), 520w, 499w, 440m (γ[Npy–V]), 309m (ν[V–F]) and 278m (ν[V–N]). Anal. found: C, 48.95; H, 4.2; N, 11.2. Calc. for C20H20BF6N4V (492.14): C, 48.8; H, 4.1; N, 11.4%.
C]py), 1575m (ν[CO]acac, ν[CC]acac), 1530m, 1524s (ν[CO]acac, ν[CC]acac), 1484w (ν[CC]py), 1448s (ν[CC]py), 1376s (ν[C–O]acac, γ[C–C]acac), 1286w, 1213w, 1067w, 1043m, 766m (γ[C–H]py), 702s (γ[C–H]py), 638m (γ[C–H]py), 578s, 561vs, 537m (ν[V–O], γ[V–acac]), 450m (γ[Npy–V]), 418w and 330s (ν[V–F]). Anal. found: C, 52.3; H, 5.2; N, 8.2. Calc. for C15H17F2N2O2V (364.25): C, 52.0; H, 4.95; N, 8.1%.
max/cm−1 3150–2800w (ν[C–H]py), 1605s (ν[CC]py), 1483m (ν[CC]py), 1448s (ν[CC]py), 1217m, 1101s, 1058vs, 1043s, 950m, 770m (γ[C–H]py), 709m (γ[C–H]py), 697s (γ[C–H]py), 640m (γ[C–H]py), 520w, 499w, 440m (γ[Npy–V]), 309m (ν[V–F]) and 278m (ν[V–N]). Anal. found: C, 48.95; H, 4.2; N, 11.2. Calc. for C20H20BF6N4V (492.14): C, 48.8; H, 4.1; N, 11.4%.
Single-crystal X-ray diffraction
Data were collected at 150.00(10) K using an “Oxford Diffraction Xcalibur S” diffractometer equipped with a goniometer in κ geometry, a “Sapphire 3” CCD-detector, a graphite-monochromated “Enhance” Mo-Kα source (λ = 0.71073 Å) and a “Cryojet XL” sample cooler. Diffraction images were integrated with CRYSALISPRO.9 An analytical numeric absorption correction was performed using a multifaceted-crystal model.10 Structures were solved with SHELXT-2014 using a dual-space method and refined with SHELXL-2014 against Fo2 data using the full-matrix least-squares algorithm.11 Non-hydrogen atoms were refined anisotropically; hydrogen atoms were refined isotropically with standard riding-models. Molecular graphics were produced using ORTEP-3 for Windows.12The crystal of [VIII(acac)F2(py)2] (1a) was a merohedral twin of type II by rotation of 180° around [100] in reciprocal space (fraction of second component: 0.291[1]) as discovered by the use of COSET.13
The tetrafluoroborate ion in [VIIIF2(py)4]BF4 (1b) is disordered over a position of higher symmetry and was treated using enhanced rigid-bond restraints.
The crystal of (pyH)[VIIIF2(py)4]SiF6 (1c) was twinned by rotation of −90.5869° around (001) in direct space (fraction of second component: 0.163[2]). Its pyridinium ion is rotationally disordered over a special position and constructed from two symmetry-independent atoms. It was modelled as an occupationally disordered species with C20/N20 occupations of 0.375/0.125 to keep the stoichiometry. The position was chosen because of the possibility of hydrogen bonding. The structure of 1c contains a void of ca. 67 Å3 that was checked for unmodelled electron density by using difference Fourier maps as well as PLATON/SQUEEZE.14 Neither procedure showed a significant residual.
Structure refinement for [VIIIF3(py)3] (1d) did not lead to satisfactory results. Probably because of unresolvable twinning, partial substitutional (solid solution) or rotational disorder of the complex molecules, R factors of the final model were very high, the weighting scheme did not converge to sensible values and large residual electron-density maxima were found at physically unreasonable positions. Therefore, the suggested structure is to be understood as merely modelling connectivity and cell content for the major component.‡
Because the crystal system of (pyH)[VIIIF4(py)2]·⅔EtOH (1e) initially seemed to be orthorhombic, an inappropriate data-collection strategy had been chosen resulting in a somewhat lower completeness. In spite of this shortcoming, the structure is of high quality. One pyridinium ion (N50–C52) is rotationally disordered over a special position and constructed from three symmetry-independent atoms. It was modelled as an occupationally disordered species with C50/N50 occupations of 0.5 each to keep the stoichiometry. The position was chosen because of the possibility of hydrogen bonding and the electron density found. The oxygen-borne hydrogen atom was refined with a restrained 1,2-distance (0.84 Å) and Uiso(H60) = 1.2Ueq(O60). The nitrogen-borne hydrogen atom in the disordered pyridinium ion was fully constrained to the nitrogen atom. The one in the ordered pyridinium ion (N40–C45) was refined with a restrained 1,2-distance (0.88 Å), a same-1,3-distance restraint and Uiso(H40) = 1.2Ueq(N40).
The crystal of [MnIIIF3(py)3] (2b) was twinned by rotation of ca. 180° around (001) in direct space (fraction of second component: 0.356[3]). Some reflections had to be excluded from the refinement because of problems properly separating them.
CCDC 1063076–1063078 and 1063080–1063083 contain the supplementary crystallographic data for this paper.
Results and discussion
Species from the Li3VF6 precursor
The following pyridine adducts of species contained in the Li3VF6 precursor were isolated (py: pyridine): [VIII(acac)F2(py)2] (1a), [VIIIF2(py)4]BF4 (1b), (pyH)[VIIIF2(py)4]SiF6 (1c), [VIIIF3(py)3] (1d) and (pyH)[VIIIF4(py)2]·⅔EtOH (1e). Because hydrogen fluoride reacts with the glassware used during the precursor synthesis,6 tetrafluoroborate and hexafluorosilicate are present in the precursor solutions. The diffusion method, as described in the experimental part, yielded crystals of compounds 1a, 1b and 1d that always co-crystallise and are prone to twinning. Crystals of 1c are formed on slow evaporation of the solvent in air. Cooling crystallisation of 1e occurred when storing reaction mixtures in pyridine at −30 °C. Crystal data and refinement details for 1a–e are summarised in Table 1.| Crystal data | [VIII(acac)F2(py)2] (1a) | [VIIIF2(py)4]BF4 (1b) | (pyH)[VIIIF2(py)4]SiF6 (1c) | (pyH)[VIIIF4(py)2]·⅔EtOH (1e) |
|---|---|---|---|---|
| a [I > 2σ(I)]. b w = 1/[σ2(Fo2) + (uP)2 + vP] where P = (Fo2 + 2Fc2)/3. | ||||
| CCDC no. | 1063076 | 1063077 | 1063078 | 1063080 |
| Chemical formula | C15H17N2F2O2V | C20H20BF6N4V | C25H26F8N5SiV | C49H60F12N9O2V3 |
| M r | 346.24 | 492.15 | 627.54 | 1187.88 |
| Crystal system, space group | Monoclinic, P21/c | Monoclinic, I2/a | Tetragonal, P4/mbm | Monoclinic, P21/n |
| a (Å) | 16.4170(14) | 14.0214(17) | 10.8047(2) | 8.4345(6) |
| b (Å) | 8.0367(4) | 12.8277(10) | 10.8047(2) | 31.4088(12) |
| c (Å) | 13.1548(12) | 13.4718(17) | 12.6865(4) | 10.9799(6) |
| α (°) | 90 | 90 | 90 | 90 |
| β (°) | 113.588(10) | 114.259(14) | 90 | 111.971(7) |
| γ (°) | 90 | 90 | 90 | 90 |
| V (Å3) | 1590.6(2) | 2209.1(5) | 1481.04(7) | 2697.5(3) |
| Z | 4 | 4 | 2 | 2 |
| ρ (g cm−3) | 1.446 | 1.480 | 1.407 | 1.462 |
| μ (mm−1) | 0.65 | 0.51 | 0.45 | 0.60 |
| Crystal size (mm3) | 0.54 × 0.12 × 0.06 | 0.70 × 0.19 × 0.16 | 0.32 × 0.30 × 0.24 | 0.59 × 0.39 × 0.18 |
| Data collection | ||||
| T min, Tmax | 0.831, 0.971 | 0.812, 0.932 | 0.907, 0.937 | 0.800, 0.912 |
| No. of measured, independent and observeda reflections | 12389, 3116, 2613 | 4570, 2162, 1487 | 2574, 2574, 1293 | 11455, 4725, 3538 |
| R int | 0.0654 | 0.0414 | 0.1390 | 0.0448 |
| R σ | 0.0623 | 0.0652 | 0.0509 | 0.0639 |
(sin![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) θ/λ)max (Å−1) θ/λ)max (Å−1) |
0.617 | 0.617 | 0.616 | 0.617 |
| Refinement | ||||
| R 1 (observed/all)a | 0.0403/0.0543 | 0.0492/0.0822 | 0.0508/0.1004 | 0.0471/0.0720 |
| wR2 (observed/all)a,b | 0.0723/0.0778 | 0.1096/0.1295 | 0.1201/0.1325 | 0.0998/0.1114 |
| S/S′ | 1.010/1.010 | 1.101/1.041 | 0.844/0.844 | 1.030/1.030 |
| Data/restraints/parameters | 3116/0/202 | 2162/30/165 | 2574/0/60 | 4725/3/347 |
| Δρmax, Δρmin (e Å−3) | 0.30, −0.34 | 0.31, −0.38 | 0.51, −0.25 | 0.57, −0.33 |
| Weighting parameters u, vb | 0.0284, 0 | 0.0516, 0.5074 | 0.0774, 0 | 0.0362, 2.2766 |
In the crystal, [V(acac)F2(py)2] (1a) forms chains along [010] through dispersive C–H⋯F contacts between fluorido ligands and hydrogen atoms of the acetylacetonato ligand (d[H⋯F] ≈ 2.4 Å). The chains are connected by further C–H⋯F contacts along [10![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ] (d[H⋯F] ≈ 2.5 Å) and dispersive aromatic contacts along [101]. The complex is a slightly distorted coordination octahedron with the pyridine ligands in a trans arrangement (cf.Table 2). In 1b and 1c, the [VIIIF2(py)4]+ units adopt a trans octahedral coordination geometry, which is only slightly distorted. The pyridine rings in 1b are not coplanar to the V–N–F coordination planes, but slightly twisted out of the latter, while pairs of diametrically opposed pyridine rings are coplanar (Fig. 1).
] (d[H⋯F] ≈ 2.5 Å) and dispersive aromatic contacts along [101]. The complex is a slightly distorted coordination octahedron with the pyridine ligands in a trans arrangement (cf.Table 2). In 1b and 1c, the [VIIIF2(py)4]+ units adopt a trans octahedral coordination geometry, which is only slightly distorted. The pyridine rings in 1b are not coplanar to the V–N–F coordination planes, but slightly twisted out of the latter, while pairs of diametrically opposed pyridine rings are coplanar (Fig. 1).
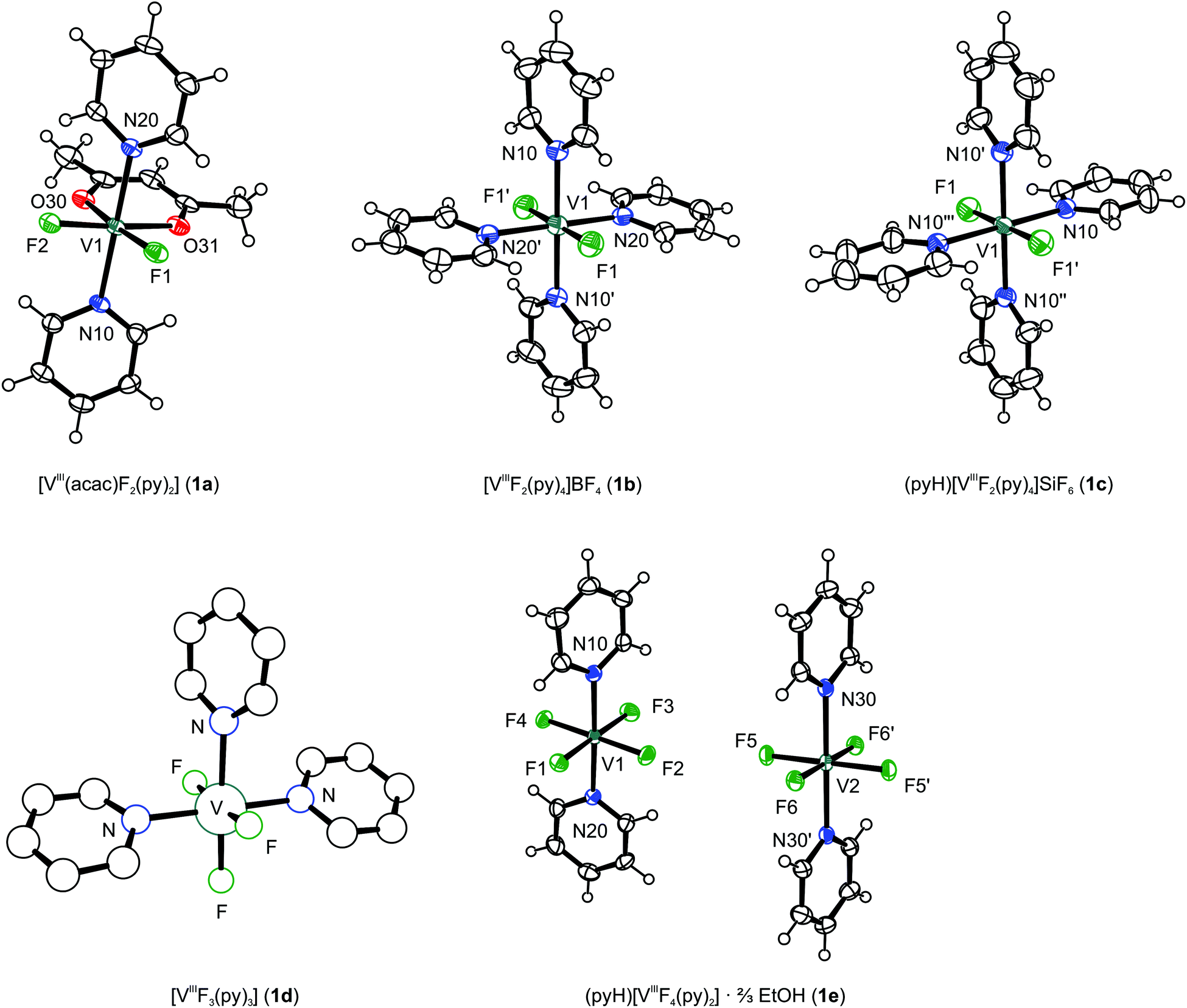 | ||
| Fig. 1 Molecular structures of vanadium(III) complexes 1a–c, 1e (ORTEP representation with ellipsoids of 50% probability; counter ions and solvent molecules omitted for clarity) and connectivity map of 1d (PLUTON representation). | ||
| [VIII(acac)F2(py)2] | [VIIIF2(py)4]BF4 | (pyH)[VIIIF2(py)4]SiF6 | (pyH)[VIIIF4(py)2]·⅔EtOH (1e) | ||
|---|---|---|---|---|---|
| (1a) | (1b) | (1c) | [(V1) IIIF4(py)2]− | [(V2) IIIF4(py)2]− | |
| V–F | 1.8515(17) | 1.7658(16) | 1.762(3) (2×) | 1.8832(19) | 1.8876(17) |
| 1.8603(17) | 1.7659(16) | 1.8837(19) | 1.8876(18) | ||
| 1.9170(18) | 1.9229(18) (2×) | ||||
| 1.919(2) | |||||
| V–N | 2.156(2) | 2.145(2) | 2.134(3) (2×) | 2.161(3) | 2.158(3) (2×) |
| 2.164(2) | 2.150(2) | 2.171(3) | |||
| V–O | 2.011(2) | ||||
| 2.015(2) | |||||
The [VIIIF2(py)4]+ and BF4− ions in 1b form a three-dimensional network. 1c crystallises in a layered structure. In [001] direction, layers of [VIIIF2(py)4]+ units alternate with layers of [SiF6]2− and pyridinium ions, which are associated by hydrogen bonds. Along [001], the [SiF6]2− ions are located in channels built by the cations. The V–F bonds in the cationic complexes 1b and 1c (ca. 1.76 Å) are shorter than in the electroneutral complex 1a (1.85–1.86 Å). The anionic complex 1e has the longest V–F bonds of the vanadium species described in this work with up to 1.9229(18) Å (cf.Table 2). This trend is easily rationalised by assuming an increasingly effective shielding of the central ion's positive charge by the growing number of negatively charged ligands and, thus, a decrease in the electrostatic attraction per fluorido ligand. For comparison, it should be noted that the V–F distance in VF3 is 1.94 Å, whereas it varies from 1.89 to 1.97 Å in β-Li3VF6.15,16 In both cases, the octahedral (bridging) F6 coordination environment effectively shields the charge of the central vanadium(III) ion. Examples with vanadium(III) in a pure F2(py)4 coordination have hitherto been unknown.
However, an example for a compound with a different F2N4 coordination environment is the 5,7,12,14-tetramethyldibenzo[b,i][1,4,7,11]-tetraazacyclodecine complex [VIII(C22H23N4)F2]PF6 described by Schumann,17 in which the configuration of the VF2 unit is not yet clarified. Another known compound is the complex [VIII(bipy)F2]BF4 (bipy: 2,2′-bipyridine), in which a vanadium ion is coordinated octahedrally by two cis-fluorido and the bidentate 2,2′-bipyridine ligands.18
The crystals obtained from 1d were of insufficient quality to allow more than an assessment of connectivity and cell content for the major component, the [VIIIF3(py)3] molecule, that contains an octahedrally coordinated vanadium ion and assumes a mer configuration.‡
(pyH)[VIIIF4(py)2]·⅔EtOH (1e) is the only anionic vanadium species isolated from the precursor. The asymmetric unit contains one and a half complex anions differing in the orientation of the pyridine ligands. While the mean planes of the pyridine ligands N10–C15 and N20–C25 are rotated by 56.3(1)° with respect to each other, the rings are coplanar through symmetry in the complex containing N30–C35. The central ions are coordinated in a distorted octahedral fashion by four fluorido and two trans arranged pyridine ligands. As regards bond lengths and angles, the complex containing V2 is more symmetric than the one containing V1. The V2 unit interacts with two pyridinium ions (containing N40) via hydrogen bonds. Such are also formed between the V1 unit and the ethanol molecule as well as the disordered pyridinium ion containing N50. The packing of the molecules in 1e can be described as a layered structure parallel to (010). Every layer is built of three sublayers with the sequence V1–V2–V1. Within these sublayers, complexes interact via the hydrogen bonds mentioned above.
Species from the Li2MnF5 and Li2NiF4 precursors
As noted in the introduction, the species [MnIIIF2(py)4][SiF5(py)] (2a), [MnIIIF3(py)3] (2b) and [NiIIF2(py)4] (3a) were isolated from the systems Li–Mn–F and Li–Ni–F and crystallographically characterised (see Table 3). The fluorosilicate anion again is a result of the reaction of hydrogen fluoride with the glassware during precursor synthesis. 3a has long been known,19,20 but no crystal structure has been reported so far. The hydrates [NiIIF2(py)4]·xH2O (x = 2 and 7/3) have been known and characterised before.21,22 From the same mother liquor as 3a, purple crystals of [NiII(acac)2(py)2] (3b) formed after four weeks. This compound has already been reported and both, crystallographically and spectroscopically, characterised.23–25| Crystal data | [MnIIIF2(py)4][SiF5(py)] (2a) | [MnIIIF3(py)3] (2b) | [NiIIF2(py)4] (3a) |
|---|---|---|---|
| a [I > 2σ(I)]. b w = 1/[σ2(Fo2) + (uP)2 + vP] where P = (Fo2 + 2Fc2)/3. | |||
| CCDC no. | 1063081 | 1063082 | 1063083 |
| Chemical formula | C25H25F7MnN5Si | C15H15F3MnN3 | C20H20F2N4Ni |
| M r | 611.53 | 349.24 | 413.11 |
| Crystal system, space group | Monoclinic, C2/c | Monoclinic, Pc | Monoclinic, I2/a |
| a (Å) | 21.4549(11) | 15.9240(19) | 15.1861(10) |
| b (Å) | 10.3397(5) | 8.1630(8) | 8.4089(4) |
| c (Å) | 11.9769(7) | 12.4914(13) | 15.7527(9) |
| α (°) | 90 | 90 | 90 |
| β (°) | 98.648(5) | 102.981(12) | 110.717(7) |
| γ (°) | 90 | 90 | 90 |
| V (Å3) | 2626.7(2) | 1582.2(3) | 1881.5(2) |
| Z | 4 | 4 | 4 |
| ρ (g cm−3) | 1.546 | 1.466 | 1.458 |
| μ (mm−1) | 0.62 | 0.87 | 1.06 |
| Crystal size (mm3) | 0.67 × 0.51 × 0.28 | 0.45 × 0.33 × 0.26 | 1.35 × 0.62 × 0.54 |
| Data collection | |||
| T min, Tmax | 0.778, 0.873 | 0.747, 0.842 | 0.480, 0.648 |
| No. of measured, independent and observeda reflections | 5581, 2590, 2331 | 5530, 5530, 4185 | 3859, 1839, 1706 |
| R int | 0.0192 | 0.083 | 0.0149 |
| R σ | 0.0271 | 0.0726 | 0.0202 |
| (sinθ/λ)max (Å−1) |
0.617 | 0.617 | 0.617 |
| Refinement | |||
| R 1 (observed/all)a | 0.0324/0.0385 | 0.0596/0.0787 | 0.0247/0.0277 |
| wR2 (observed/all)a,b | 0.0743/0.0768 | 0.1612/0.1737 | 0.0642/0.0654 |
| S/S′ | 1.078/1.078 | 1.010/1.011 | 1.056/1.056 |
| Data/restraints/parameters | 2590/0/180 | 5530/2/398 | 1839/0/124 |
| Δρmax, Δρmin (e Å−3) | 0.26, −0.32 | 0.49, −0.58 | 0.24, −0.35 |
| Weighting parameters u, vb | 0.0328, 1.9060 | 0.1136, 0 | 0.0302, 0 |
2a forms a structure with alternating layers of [MnIIIF2(py)4]+ and [SiF5(py)]− ions in (100). The trivalent manganese complex has a nearly undistorted octahedral coordination sphere with the fluorido ligands in a trans position (see Fig. 2). The pyridine ligands containing N20 are twisted by 33.6° relative to the N10–V1–F1 plane. At 1.7884(10) Å, the Mn–F bonds (see Table 4) are slightly shorter than those reported for MnF3 (1.7977–2.1057 Å)26,27 and MnF3·3H2O (1.795(3)–1.856(3) Å).28 A [MnIIIF2(py)4]+ species has not previously been described in the literature.
 | ||
| Fig. 2 Molecular structures of manganese(III) complexes 2a–b and nickel(II) complex 3a (ORTEP representation with ellipsoids of 50% probability; counter ions omitted for clarity). | ||
| [MnIIIF2(py)4][SiF5(py)] (2a) | [MnIIIF3(py)3] (2b) | [NiIIF2(py)4] (3a) | ||
|---|---|---|---|---|
| M = Mn | [(Mn1)IIIF3(py)3] | [(Mn2)IIIF3(py)3] | M = Ni | |
| M–F | 1.7884(10) (2×) | 1.811(8) | 1.815(8) | 2.0061(9) (2×) |
| 1.834(8) | 1.816(8) | |||
| 1.815(8) | 1.845(8) | |||
| M–N | 2.1157(15) (2×) | 2.088(11) | 2.094(11) | 2.1102(13) (2×) |
| 2.2397(15) (2×) | 2.304(11) | 2.313(11) | 2.1251(13) (2×) | |
| 2.331(10) | 2.323(11) | |||
| Si–F | 1.6658(11) (2×) | |||
| 1.6703(12) (2×) | ||||
| 1.6623(15) | ||||
| Si–N | 2.016(2) | |||
In the group of divalent manganese compounds [MnIIX2(py)4], species with X = Cl, Br and I (but not F) are known,29,30 having a molecular structure similar to 2a. There are several compounds containing the structure motif [MIIIF2(py)4]+ (M: transition metal), e.g., the chromium compounds [CrF2(py)4]X (X = ClO4, Br, I, NO3, PF6)31,32 and the cobalt compound [CoF2(py)4]ClO4·½H2O.33 Together with 1b and 1c, 2a adds to the hitherto known set of [MIIIF2(py)4]+ species. A special feature of 2a is the [SiF5(py)]− ion. Pyridine fluorosilicates or their polymeric forms have not previously been reported. In contrast, compounds (NR4)[SiF5(NH3)] with R = Me, Et or H exist.34–36 Furthermore, pyridine adducts of tetrafluorosilane, SiF4·py and SiF4·2py, are known.37,38 Si–F and Si–N bond lengths in [SiF5(py)]− are listed in Table 4. For (NH4)[SiF5(NH3)], the Si–N bond distance is reported to be 1.902(4) Å and is therefore shorter than in 2a. At 1.6623(15)–1.6703(12) Å, the Si–F bonds in 2a are somewhat shorter than in (NH4)[SiF5(NH3)] (1.678(1) and 1.680(2) Å). [MnIIIF3(py)3] (2b) and its perdeuterated derivative were described in 1989.39 The authors reported twinning problems. The single crystals obtained in our experiments were also twinned, but the twin law was found, which allowed for a high-quality refinement of the structure. We found similar cell parameters, but assigned a different space group (Pc instead of P21/c as emulated by twinning). The asymmetric unit contains two complexes differing in the orientation of the pyridine ligands with respect to each other and their tilting with respect to the metal–nitrogen bond. In 2b, both crystallographically independent moieties form layers in (100) connecting symmetry equivalent complexes only through dispersive C–H⋯F contacts. The layers interact via π stacks along [010] to form a three-dimensional network.
The molecular structure of [NiIIF2(py)4] (3a) is comparable to the [MIIIF2(py)4]+ species discussed before (see Table 4). A distorted octahedral coordination with trans aligned fluorido ligands is the basic structural motif. At 2.0061(9) Å, the Ni–F bond length is in the same range as for NiF2 (given as 1.98(1)–2.04(2) Å or 1.997(1)–2.011(1) Å),40,41 and for the hydrates [NiIIF2(py)4]·2H2O (2.0424(8) Å) and [NiIIF2(py)4]·7/3H2O (2.015(4) Å). The Ni–N bond lengths and coordination angles are also comparable to those in the latter compounds. The molecules of 3a form chains along a, in which the N10-containing pyridine ligands interact via π stacks.
Successive flourination of the primary metal precursors
The existence of certain LixMFy precursor species can be inferred from the isolation of their solvent adducts. Incorporating [VIII(acac)2(CH3CN)2]BF4,6 the most complete sequence of fluorination products was found for the Li3VF6 precursor. A suitable model for the fluorination of metal acetylacetonates in the precursor-formation process using the example of Li3VF6 is shown in Scheme 2. Starting from [VIII(acac)3], five fluorination steps can be distinguished: first, an acetylacetonato ligand has to be eliminated, leaving behind a cationic {VIII(acac)2}+ core as evidenced by the isolation of [VIII(acac)2(CH3CN)2]BF4 and [VIII(acac)2F].6,42 After elimination of another acetylacetonato ligand and further fluorination, a {VIII(acac)F2} core is formed, which has been trapped in 1a. Neither [VIII(acac)F2] nor [VIII(acac)F2(py)2] have been described in the literature so far. The {VIII(acac)F2} core reacts with two more equivalents of HF, forming {VF3} and finally {VF4}−, which were trapped in 1d and 1e as shown in Scheme 2. | ||
| Scheme 2 Stepwise fluorination of [V(acac)3] and formation of the isolated complexes. | ||
When the starting materials [VIII(acac)3] and LiOtBu are reacted with hydrogen–fluoride solution, increasingly more highly fluorinated species (up to {VF4}−) are formed within a chemical equilibrium. Although an excess of hydrogen fluoride is used, exhaustive fluorination is not accomplished.
The question remains how Li3VF6 is formed during the decomposition of the inhomogeneous precursor material. For precursor synthesis, LiOtBu and [VIII(acac)3] are reacted in the ratio 3:1 with an excess of HF to achieve a stoichiometry of F6Li3V. However, the precursor can neither be Li3VF6 nor a stoichiometric mixture of LiF and VF3, as elemental analysis shows carbon, hydrogen and oxygen to be present in considerable amounts. During precursor decomposition, the following steps occur: while heating, residual solvent evaporates and is carried away by the gas flow. In general, at about 200 °C, acetylacetonates start to decompose;43 pure [VIII(acac)3] was reported to decompose at about 250 °C.44 To avoid oxidation, working under a dinitrogen atmosphere is necessary. Under these conditions, organic residues are carbonised, giving rise to the black colour of the Li3VF6 material and its residual carbon content. Before reaching 300 °C, the decomposition of Li2SiF6 to LiF and SiF4 starts. Gaseous SiF4 is also carried away by the gas flow. Normally, the decomposition of Li2SiF6 should occur at higher temperature as reported in the literature: at 290 °C45 or 350–370 °C46 in dry air. We have not found any Li2SiF6 in Li3VF6 precursor samples prepared under dinitrogen at 300 °C. This indicates that the decomposition of Li2SiF6 occurs at lower temperatures under precursor decomposition conditions as compared to pure samples. To substantiate this, a sample of pure Li2SiF6 prepared from Li2CO3 and aqueous H2SiF6 was calcined for four hours at 240 °C under dinitrogen flow. X-ray powder diffraction showed that the product was a mixture of Li2SiF6 and LiF. At 300 °C, the formation of monoclinic β-Li3VF6 as the main component is observed, so that the decomposition of the fluorinated species and their reaction with LiF can be assumed.
To verify this concept, the precursor decomposition was investigated by thermogravimetry (TG) and differential scanning calorimetry (DSC, see Fig. 3). The sample loses 12.9% of its mass up to 180 °C. This is in the temperature range of a possible solvent loss. Between 180 and 350 °C, the sample loses another 12.5% of its mass, while acetylacetonate and Li2SiF6 decompose. From 233 to 250 °C, a DSC signal for an exothermic process appears with its maximum at about 239 °C. As the precursor contains a plethora of species, which can react with each other during decomposition, we have unfortunately been unable to clarify the origin of this signal. However, we have ruled out the decomposition of Li2SiF6 or acetylacetonate compounds (e.g., [V(acac)3]), because these processes are known to be endothermic.44,47
 | ||
| Fig. 3 TG/DSC during decomposition of the Li3VF6 precursor (synthesised in EtOH) in a dinitrogen atmosphere. Blue curve: TG signal, red curve: DSC signal, black arrows: mass changes. ΔmTGm−1/%: mass loss in per cent. | ||
Upon reaching the final temperature of 600 °C, the sample had lost a total of 30.3% of its mass. This value is in the typical range of losses observed during decomposition (25–40% for Li3VF6 precursors).
It is safe to assume that similar processes occur with Li2MnF5 and Li2NiF4 precursors. In the system Li–Mn–F, {MnF2}+ and {MnF3}+ were captured—in analogy to the system Li–V–F (see Scheme 3). Species with the core {MnIIIFx}(x−3)− have not been found so far for x = 4 and 5. In the system Li–N–F, [Ni(acac)2] partly reacts with hydrogen fluoride to form NiF2, as indicated by the presence of 3a (see Scheme 4). It is as yet unknown if anionic fluoridometal species exist in these systems. Finally, Li2MnF5 and Li2NiF4 will be formed when the mixture of transition-metal species and LiF is heated under dinitrogen flow.
 | ||
| Scheme 3 Stepwise fluorination of [Mn(OAc)3] and formation of the isolated complexes. | ||
 | ||
| Scheme 4 Stepwise fluorination of [Ni(acac)2] and formation of the isolated complexes. | ||
Conclusions
The results presented above give detailed insight into the processes of the fluorolytic sol–gel synthesis of ternary lithium transition-metal fluorides. After stabilising precursor species with pyridine and growing single crystals from the pyridine solutions, it became apparent that the precursor is a mixture of transition-metal compounds with different degrees of fluorination. On the basis of these results, previous analytical data from X-ray diffraction and spectroscopic methods may be better understood. In the case of the synthesis of Li3VF6 starting from [VIII(acac)3], four fluorination steps were identified by isolating and characterising the pyridine adducts 1a–e. Further experiments with Li2MnF5 and Li2NiF4 precursors led to the species 2a–b and 3a. In addition, first attempts to isolate species from the system Li–Fe–F already indicate the presence of [FeIIIF2(py)4]BF4. This demonstrates that the conclusions made for Li3VF6 are transferable to the fluorolytic sol–gel synthesis of other ternary fluorides in general. The elucidation of these processes is necessary for further research on the notoriously low yields of Li2MnF5 and Li3FeF6, as well as the reduction occurring in these systems. As mentioned in the introduction, Li2MnF5 and Li3FeF6 were invariably obtained together with the difluorides MF2. The question, whether divalent species are already formed during precursor synthesis, is important for addressing this issue. Stabilisation with additional solvent ligands such as pyridine is a promising method to help clarify these open questions.Acknowledgements
The authors thank Ms Paula Nixdorf (TU Berlin) for the measurement of X-ray diffractograms and Professor Andreas Grohmann (TU Berlin) for fruitful discussions.Notes and references
- J. Koyama, I. Tanaka and H. Adachi, J. Electrochem. Soc., 2000, 147, 3633 CrossRef PubMed.
- E. Gonzalo, A. Kuhn and F. Garcia-Alvarado, J. Power Sources, 2010, 195, 4990 CrossRef CAS PubMed.
- A. Basa, E. Gonzalo, A. Kuhn and F. Garcia-Alvarado, J. Power Sources, 2012, 207, 160 CrossRef CAS PubMed.
- I. Gocheva, K. Chihara, S. Okada and J. Yamaki, Proceedings of the Lithium Battery Discussions 2011 – Electrode Materials, Arcachon, France, 2011 Search PubMed.
- E. Kemnitz, U. Groß, S. Rüdiger and C. S. Shekar, Angew. Chem., Int. Ed., 2003, 115, 4383 CrossRef PubMed.
- J. Kohl, D. Wiedemann, S. Nakhal, P. Bottke, N. Ferro, T. Bredow, E. Kemnitz, M. Wilkening, P. Heitjans and M. Lerch, J. Mater. Chem., 2012, 22, 15819 RSC.
- J. Kohl, S. Nakhal, N. Ferro, P. Bottke, M. Wilkening, T. Bredow and M. Lerch, Z. Anorg. Allg. Chem., 2013, 639, 326 CrossRef CAS PubMed.
- A. Dimitrov, J. Koch, S. I. Troyanov and E. Kemnitz, Eur. J. Inorg. Chem., 2009, 35, 5299 CrossRef PubMed.
- Agilent Technologies, CrysAlisPro Software System, version 171.37, Agilent Technologies Ltd., Oxford, UK, 2014 Search PubMed.
- R. C. Clark and J. S. Reid, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 1995, 51, 887 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 2012, 45, 849 CrossRef CAS.
- P. Boyle, J. Appl. Crystallogr., 2014, 47, 467–470 CrossRef CAS.
- P. van der Sluis and A. L. Spek, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, 46, 194 CrossRef.
- W. Massa, Z. Kristallogr., 1980, 153, 201 CrossRef CAS PubMed.
- K. H. Jack and V. Gutmann, Acta Crystallogr., 1951, 4, 246 CrossRef CAS.
- H. Schumann, Polyhedron, 1996, 15, 845 CrossRef CAS.
- S. J. Kavitha, K. Panchanatheswaran, J. N. Low and C. Glidewell, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2005, 61, m1965 CAS.
- I. I. Kalinichenko and A. A. Knyazeva, Dokl. Akad. Nauk SSSR, 1966, 169, 887 CAS.
- A. A. Knyazeva, I. I. Kalinichenko and T. A. Degtyareva, Zh. Neorg. Khim., 1967, 12, 1213 CAS.
- J. Holzbock, W. Sawodny and L. Walz, Z. Kristallogr., 1997, 212, 115 CrossRef CAS.
- P. Halasyamani, M. J. Willis, C. L. Stern and K. R. Poeppelmeier, Inorg. Chim. Acta, 1995, 240, 109 CrossRef CAS.
- J. T. Hashagen and J. P. Fackler, J. Am. Chem. Soc., 1965, 87, 2821 CrossRef CAS.
- R. C. Elder, Inorg. Chem., 1968, 7, 2316 CrossRef CAS.
- K. Nakamoto, C. Udovich and J. Takemoto, J. Am. Chem. Soc., 1970, 92, 3973 CrossRef CAS.
- F. Schrötter and B. G. Müller, Z. Anorg. Allg. Chem., 1993, 619, 1426 CrossRef PubMed.
- M. A. Hepworth and K. H. Jack, Acta Crystallogr., 1957, 10, 345 CrossRef CAS.
- M. Molinier and W. Massa, J. Fluorine Chem., 1992, 57, 139 CrossRef CAS.
- M. A. Araya, F. A. Cotton, J. H. Matonic and C. A. Murillo, Inorg. Chem., 1995, 34, 5424 CrossRef CAS.
- C. J. H. Jacobsen, E. Pedersen, J. Villadsen and H. Weihe, Inorg. Chem., 1993, 32, 1216 CrossRef CAS.
- J. Glerup, J. Josephsen, K. Michelsen, E. Pedersen and C. E. Schäfer, Acta Chem. Scand., 1970, 24, 247 CrossRef CAS PubMed.
- G. Fochi, J. Strähle and F. Gingl, Inorg. Chem., 1991, 30, 4669 CrossRef CAS.
- G. Glerup, C. E. Schäffer and J. Springborg, Acta Chem., Scand. Ser. A, 1978, 32, 673 CrossRef PubMed.
- I. Wharf and M. Onyszchuk, Can. J. Chem., 1972, 50, 3450 CrossRef CAS.
- H. C. Clark, K. R. Dixon and J. G. Nicholson, Inorg. Chem., 1969, 8, 450 CrossRef CAS.
- C. Plitzko and G. Meyer, Z. Anorg. Allg. Chem., 1996, 622, 1646 CrossRef CAS PubMed.
- E. L. Muetterties, J. Am. Chem. Soc., 1960, 82, 1082 CrossRef CAS.
- V. A. Bain, R. C. Killean and M. Webster, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1969, 25, 156 CrossRef CAS.
- W. Sawodny, K. N. Rau and W. Bolkart, J. Fluorine Chem., 1989, 43, 119 CrossRef CAS.
- J. W. Stout and S. A. Reed, J. Am. Chem. Soc., 1954, 76, 5279 CrossRef CAS.
- M. M. Costa, J. A. Paixão, M. J. de Almeida and L. C. Andrade, Acta Crystallogr., Sect. B: Struct. Sci., 1993, 49, 591 CrossRef.
- M. K. Chaudhuri and S. K. Ghosh, Inorg. Chem., 1984, 23, 2367 CrossRef CAS.
- J. von Hoene, R. G. Charles and W. M. Hickam, J. Phys. Chem., 1958, 62, 1098 CrossRef CAS.
- G. A. M. Hussein, Thermochim. Acta, 1995, 256, 347 CrossRef CAS.
- Y. N. Moskvich, B. I. Cherkasov and G. I. Dotsenko, Zh. Strukt. Khim., 1979, 20, 348 CAS.
- D. L. Deadmore, J. S. Machin and A. W. Allen, J. Am. Ceram. Soc., 1962, 45, 120 CrossRef CAS PubMed.
- R. R. Patil, M. Rujuta Barve, M. S. Kulkarni, B. C. Bhatt and S. V. Moharil, Phys. B, 2012, 407, 629 CrossRef CAS PubMed.
Footnotes |
| † CCDC 1063076–1063078 and 1063080–1063083. For crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt01748c |
| ‡ Crystal data. C15H15N3F3V, M = 345.24, orthorhombic, a = 8.2723(5) Å, b = 12.6821(13) Å, c = 30.6745(19) Å, T = 150.00(10) K, space group Pbca, Z = 8. |
| This journal is © The Royal Society of Chemistry 2015 |