
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The effect of thiadiazole out-backbone displacement in indacenodithiophene semiconductor polymers†
Miquel
Planells
*a,
Mark
Nikolka
b,
Michael
Hurhangee
a,
Pabitra S.
Tuladhar
a,
Andrew J. P.
White
a,
James R.
Durrant
a,
Henning
Sirringhaus
b and
Iain
McCulloch
a
aDepartment of Chemistry and Centre for Plastic Electronics, Imperial College London, London SW7 2AZ, UK. E-mail: m.planells@imperial.ac.uk
bCavendish Laboratory, University of Cambridge, Cambridge CB3 OHE, UK
First published on 27th August 2014
Abstract
We describe the synthesis and characterisation of two new polymers consisting of an electron-rich backbone containing indacenodithiophene (IDT) and dithiophene (DT) with the electron-poor units benzothiadiazole (BT) and benzopyrazolothiadiazole (BPT) fused on top of DT. The effect of this substitution has been studied and discussed by optical, electrochemical and computational means. Despite having very similar molecular distribution as well as thermal and electrochemical properties, the addition of the stronger electron-withdrawing BPT unit leads to a substantial change on the absorption properties by promoting the intramolecular charge transfer (ICT) band alongside the π–π*. Furthermore, we also report organic field effect transistor and solar cells device results, giving hole mobilities of 0.07 cm2 V−1 s−1 with low threshold voltage (<10 V) and power conversion efficiencies of up to 2.2%.
Introduction
Semiconductor polymers have attracted much attention from scientific community due to their potential low cost production of organic electronic devices, roll-to-roll solution processing as well as their use in lightweight and flexible applications.1–3The indacenodithiophene (IDT) unit has been successfully employed as an electron rich, conformationally rigid repeat unit in a range of high performing polymers for both transistor and solar cell applications.4 There are several features at the molecular level that make this unit attractive. With the aromatic units fixed co-planar due to the bridging groups, a low energetic disorder close packed intramolecular conformation is feasible. It is believed that IDT–BT copolymers can tolerate significant intermolecular disorder, and still maintain good charge transport, in part due to the excellent backbone rigidity, which allows charges to be transported along the polymer chain and π-stacking (usually present in crystalline domains) is only occasionally required.5
The electron poor benzothiadiazole (BT) unit has been used extensively as a co-monomer in many light absorbing donor–acceptor polymers in bulk heterojunction OPV devices, as it promotes efficient molecular orbital hybridisation, which allows fine band gap tuning.2 In addition, the off-axis dipole moment is believed to have a contribution on the charge transport properties of IDT–BT polymers as the sterically free BT units from adjacent polymer backbones can adopt an antiparallel dipole alignment to each other, thus facilitating close intermolecular contacts.6
Therefore, fused systems, which reduce the conformational disorder, containing BT units, which enhance the off axis dipole moment, seem to be promising molecular design guidelines to obtain high charge carrier mobility polymers for both OFET and OPV applications.
In this paper, we describe the synthesis and characterisation of two monomers prepared by fusing dithiophene units with both a new benzopyrazolothiadiazole BPT unit and its BT analogue (Fig. 1).7 We co-polymerize both these monomers with the C16IDT monomer and study the optical, electrochemical and thermal properties of the polymers as well as their performance in OFET and OPV devices.
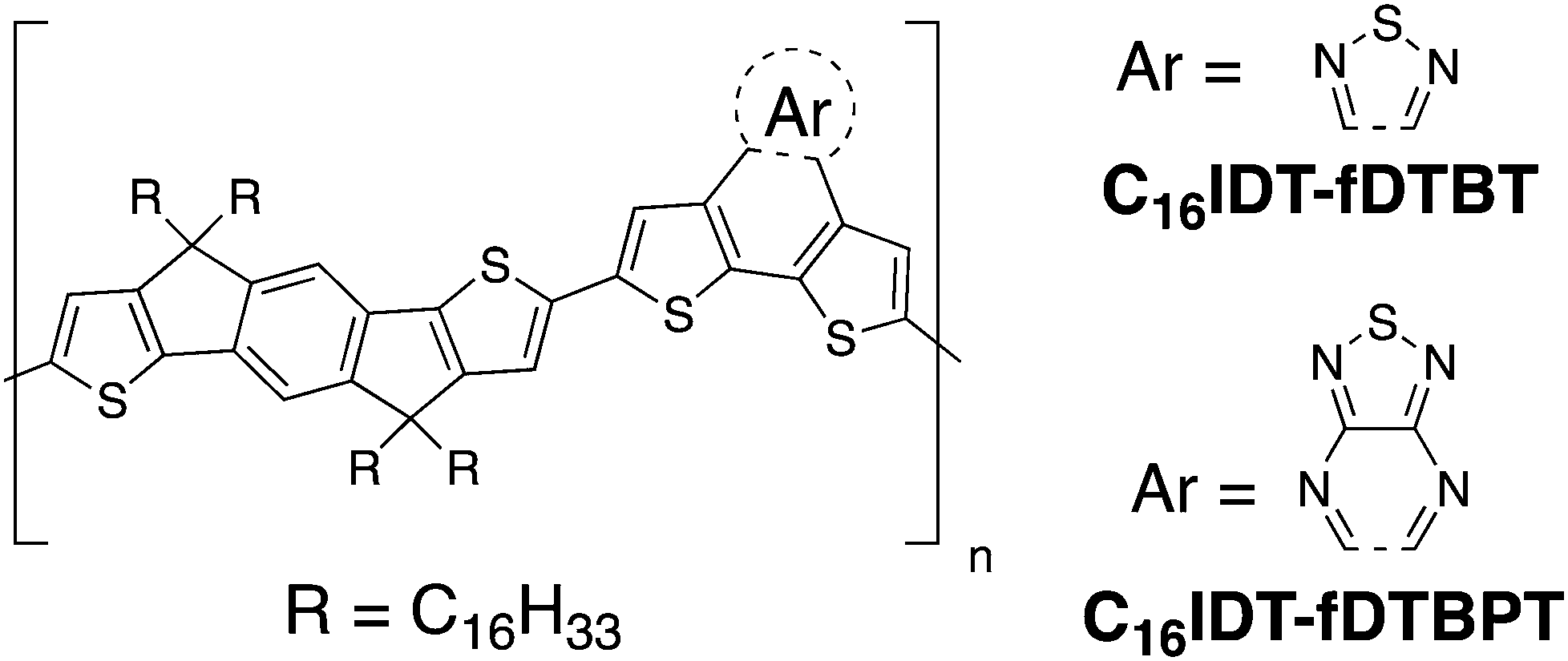 | ||
| Fig. 1 Molecular structures of C16IDT based polymers used in this study. | ||
Experimental section
Synthetic procedure
Synthesis of [1,2,5]thiadiazolo[3,4-b]dithieno[3,2-f:2′,3′-h]quinoxaline (2). 1 (110 mg, 0.5 mmol) and 1,2,5-thiadiazole-3,4-diamine (64 mg, 0.55 mmol) were added to a microwave vial. 10 mL of previously degassed acetic acid was added via syringe and the mixture was stirred overnight at reflux. After cooling down, the product was obtained by filtration and washed with 10 mL of AcOH, MeOH and CHCl3, giving a black solid (73 mg, 50% yield). 1H NMR (400 MHz, TCE-d2) δH: 8.45 (d, J = 5.5 Hz, 2H); 7.66 (d, J = 5.3 Hz, 2H). 13C NMR (100 MHz, TCE-d2) δC: 152.4; 143.7; 138.6; 134.1; 125.8; 125.7.
Synthesis of 2,5-dibromo-[1,2,5]thiadiazolo[3,4-b]dithieno[3,2-f:2′,3′-h]quinoxaline (3). 2 (73 mg, 0.25 mmol) was added to a round bottom flask with 75 mL of CHCl3. Bromine (38 μL, 0.75 mmol) was added and the mixture was stirred at reflux overnight. After cooling down, the product was obtained by filtration and washed with copious amounts of CHCl3 and CH2Cl2. 1H NMR (400 MHz, TCE-d2 @ 120 °C) δH: 8.50 (s, 2H). 13C NMR (100 MHz, TCE-d2 @ 120 °C) δC: 152.8; 142.1; 134.6; 128.4; 120.2; 114.2. MS EI (m/z): [M]+ calcd for C12H2N4S3Br2: 455.7808; found: 455.7810.
Synthesis of benzo[1,2-b:6,5-b′]dithiophene-4,5-diamine (4). 1 (364 mg, 1 mmol) and NH2OH·HCl (173 mg, 2.5 mmol) were added to a round bottom flask with 10 mL of EtOH. The mixture was heated at reflux overnight. Then, the reaction mixture was allowed to cool down to room temperature and Pd/C 10% (20 mg) was added. The reaction mixture was warmed up to 60 °C and N2H4·H2O (1.5 mL) in 2.5 mL EtOH was added via the addition funnel from the top of the condenser. After that, the reaction mixture was heated at reflux overnight. After cooling to room temperature, the crude product was plugged in EtOAc to remove Pd/C and further purified by column chromatography (SiO2, CH2Cl2 up to CH2Cl2–EtOAc 1
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) to afford the product as a yellow solid (190 mg, 86% yield). 1H NMR (400 MHz, DMSO-d6) δH: 7.60 (d, J = 5.4 Hz, 2H); 7.45 (d, J = 5.4 Hz, 2H). 13C NMR (100 MHz, CDCl3) δC: 129.3; 124.7; 122.4; 122.3; 121.8.
:1) to afford the product as a yellow solid (190 mg, 86% yield). 1H NMR (400 MHz, DMSO-d6) δH: 7.60 (d, J = 5.4 Hz, 2H); 7.45 (d, J = 5.4 Hz, 2H). 13C NMR (100 MHz, CDCl3) δC: 129.3; 124.7; 122.4; 122.3; 121.8.
Synthesis of dithieno[3′,2′:3,4;2′,3′:5,6]benzo[1,2-c][1,2,5]thiadiazole (5). S2Cl2 (256 μL, 3.2 mmol) was added to a round bottom flask with 1.6 mL of DMF and stirred at 0 °C under Ar. Then, a previously made solution of 4 (176 mg, 0.80 mmol) in 1.6 mL of DMF was added dropwise. After addition, the mixture was allowed to warm at room temperature and stirred for further 2 hours. Ice-water was added to quench the reaction and extraction was carried out using CH2Cl2. The organic layer was washed with brine, dried over MgSO4, filtered and the solvent was removed. The crude was further purified by column chromatography (SiO2, hexane–CH2Cl2 2
:1) to afford the product as a yellow solid (110 mg, 55% yield). 1H NMR (400 MHz, CDCl3) δH: 8.03 (d, J = 5.2 Hz, 2H); 7.54 (d, J = 5.2 Hz, 2H). 13C NMR (100 MHz, CDCl3) δC: 150.8; 135.8; 129.1; 125.5; 124.4.
Synthesis of 5,8-dibromodithieno[3′,2′:3,4;2′,3′:5,6]benzo[1,2-c][1,2,5]thiadiazole (6). 5 (110 mg, 0.45 mmol) was added to a round bottom flask equipped with a reflux condenser and 36 mL of CHCl3. Bromine (0.05 mL, 0.99 mmol) was added dropwise and the mixture was stirred at reflux for 6 hours, during which a yellow precipitate appeared. After that, the temperature was switched off and the mixture was stirred overnight. The resulting solid was filtered off and washed with copious amounts of CHCl3 and CH2Cl2, giving a bright yellow solid (105 mg, 63% yield). 1H NMR (400 MHz, TCE-d2 @ 120 °C) δH: 8.09 (s, 2H). 13C NMR (100 MHz, TCE-d2 @ 120 °C) δC: 149.1; 129.1; 126.9; 120.2; 114.1.
General polymer synthesis and purification. An oven-dried microwave vial was charged with distannylated C16IDT (149 mg, 0.10 mmol), and 1 eq. of 3 (46 mg, 0.10 mmol) or 6 (41 mg, 0.10 mmol) together with Pd(PPh3)4 (4.7 mg, 4 μmol, 4 mol%). The vial was sealed, and dry o-xylene (1 mL) was added. The reaction mixture was degassed with argon for 30 min before being placed into the microwave reactor and subjected to the following heating conditions: 120 °C for 2 min, 140 °C for 2 min, 160 °C for 2 min, and 180 °C for 40 min. Once the reaction mixture had cooled, polymer crude solution was precipitated by adding it dropwise into an acidic MeOH solution (containing 1% HCl) and stirred for 1–3 h until fine powder was obtained. The precipitate was filtered off into a cellulose thimble and soxhlet extraction in acetone (16 h) and hexane (16 h) was carried out. The remaining solid was soxhlet extracted in CHCl3 for 2 hours in order to extract the polymer. The organic layer was washed with sodium diethyldithiocarbamate aqueous solution three times, dried over Na2SO4, filtered off and solvent was removed. Preparative GPC in chlorobenzene at 80 °C was carried out and the polymer was fractionated by molecular weight (MW). Low MW fractions were discarded and high MW fractions were combined, solvent removed and re-precipitated by adding into a stirring MeOH solution to afford C16IDT–fDTBT (71 mg, 50%) and C16IDT–fDTBPT (66 mg, 45%) as dark solids. The collected polymer was dried under high vacuum for 24 hours before characterisation.
Chemical characterisation. 1H and 13C NMR spectra were recorded on a Bruker Advance 400 spectrometer (400 MHz for 1H and 100 MHz for 13C). The deuterated solvents are indicated; chemical shifts, δ, are given in ppm, referenced to TMS, standardized by the solvent residual signal (1H, 13C). Number-average (Mn) and weight-average (Mw) molecular weights were determined with an Agilent Technologies 1200 series GPC in chlorobenzene at 80 °C, using two PL mixed B columns in series, and calibrated against narrow polydispersity polystyrene standards. DSC experiments were carried out with a TA Instrument DSC Q20 and TGA plots were obtained with a Perkin-Elmer Pyris 1 TGA.
Electrochemical characterisation. All cyclic voltammetry measurements were carried out in dry acetonitrile using a 0.1 M [TBA][PF6] electrolyte in a three-electrode system, with each solution being purged with N2 prior to measurement. The working electrode was ITO treated glass, the reference electrode was Ag/AgCl and the counter electrode was a Pt rod. All cyclic voltammetry (CV) measurements were carried out at room temperature using an AUTOLAB PGSTAT101 potentiostat at a 50 mV s−1 scan rate and referenced to ferrocene.
Optical characterisation. Solution and solid state UV-Visible absorption spectra were recorded using a UV-1601 Shimadzu UV-Vis spectrophotometer. Photoluminescence (PL) spectra were recorded with a Fluoromax-3 fluorimeter. All samples were measured in either a 1 cm cell at room temperature or a spin-coated film.
Computational details. The molecular structures were optimized in a vacuum without any symmetry constraints. All calculations were carried out using the Gaussian 09 program9 with the Becke three parameter hybrid exchange and Lee Yang–Parr correlation functional (B3LYP) level of theory. All atoms were described by the 6-31G(d) basis set. All structures were input and processed using Avogadro software package.10 Time-dependent calculations (TD-DFT) were performed using the same level of theory B3LYP/6-31G(d).11,12 The 10 lowest singlet electronic transitions were calculated and processed using GaussSum software package.13
OFET fabrication. For the fabrication of bottom-contact top-gate OFETs, Ti/Au (10 nm/30 nm) bottom electrodes were patterned by photolithography on clean glass substrates. The patterning was done using a double layer lift-off process in N-methyl-2-pyrrolidone (NMP). Polymers were deposited by spin coating on solvent cleaned electrodes (Sonicated in Acetone and IPA), followed by an annealing step at 100 °C for 1 h. Subsequently, a 500 nm layer of Cytop (Asahi Glass) was spin coated and devices were finished off by evaporating a 20 nm thick gold top gate through a shadow mask. Transistor transfer characteristics were measured with an Agilent 4155B Semiconductor Parameter Analyser with all charge carrier mobility values being determined from the square root of the saturation transfer curve. To guarantee reproducibility, all fabrication steps were performed in an N2 glove box.
OPV fabrication. ITO-coated glass substrates were cleaned with acetone and isopropyl alcohol followed by nitrogen blow-drying and oxygen plasma treatment. A 30 nm layer of PEDOT:PSS (AI4083, Clevios) was spin-coated onto the plasma-treated ITO substrate and annealed at 150 °C for 15 min. An active layer consisting of 1
:3.5 blend of the polymer (12 mg mL−1) and PC71BM (Solenne, BV) dissolved in o-dichlorobenzene (o-DCB) was spin-coated on the PEDOT:PSS layer in air and then the Ca (20 nm)/Al (100 nm) cathode was finally deposited by thermal evaporation under high vacuum (10−6 mbar) through a shadow mask. The pixel size, defined by the spatial overlap of the ITO anode and the Ca/Al cathode, was 0.045 cm2. The device characteristics were measured using a xenon lamp at AM1.5 solar illumination (Oriel Instruments).
Results and discussion
Synthesis and chemical properties
The synthesis of monomers and C16IDT based polymers is shown in Scheme 1. The key intermediate diketone 1 was obtained using a modified reported literature procedure.8 Condensation of 1 with diaminothiadiazole14 in the presence of acetic acid gave 2 in good yields. Diketone 1 was successfully reduced to yield diamine 4 and fused dithienobenzothiadiazole 5 was obtained after ring closure.7 Dibrominated co-monomers 3 and 6 were successfully achieved by refluxing with bromine.7 It is worth mentioning that both co-monomers possessed low solubility.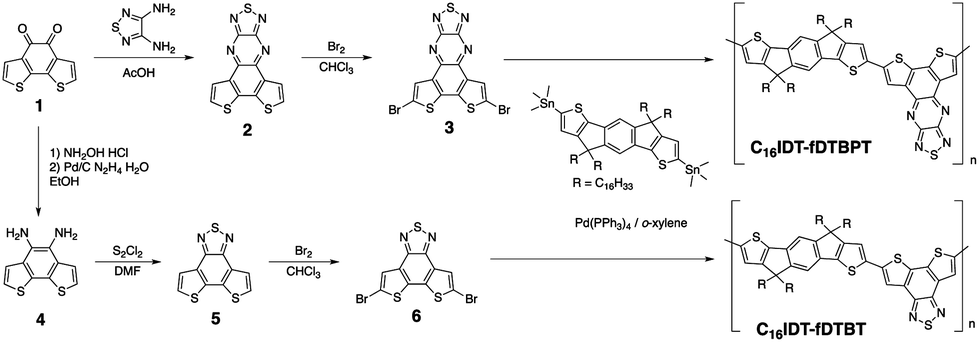 | ||
| Scheme 1 Synthetic pathway to C16IDT based polymers. | ||
Although the packing between monomeric units might not accurately describe the polymer intermolecular interactions between benzothiadiazole moieties, it could definitely provide valuable information. Thus, single crystals suitable for XRD analysis of the fused DTBPT unit 2 were obtained by slow evaporation in chloroform.‡ The crystal structure of 2 (Fig. 2a) shows the molecule to be almost completely flat, all of the non-hydrogen atoms being coplanar to within ca. 0.05 Å. Glide related molecules pack in a canted fashion along the crystallographic a-axis direction such that the major molecular axis (defined as the vector between the centroids of rings A and C) of adjacent molecules is inclined by ca. 135° (see Fig. 2b). The closest approaches are between ring B in one molecule and ring C in the “above” counterpart (centroid⋯centroid and mean interplanar distances of ca. 3.55 and 3.40 Å, rings inclined by ca. 1°), and between ring B in one molecule and ring E in the “below” counterpart (centroid⋯centroid and mean interplanar distances of ca. 3.57 and 3.37 Å, rings inclined by ca. 1°). This quasi head-to-tail packing motif is characteristic for non-symmetrical donor–acceptor molecules, as their molecular dipole moments predispose to cancel each other.15,16
 | ||
| Fig. 2 (a) Crystal structure of fused DTBPT unit 2, (b) its packing on the π–π stacking direction along the a-axis and (c) S–N short contact interactions between adjacent thiadiazole units along the c-axis. | ||
More interestingly, adjacent screw-related molecules are linked by an S⋯N short contact of 3.302(3) Å between S(1) in one molecule and N(2) in the next, forming a chain along the crystallographic c-axis direction (Fig. 2c). This short contact confirms that the intermolecular interactions between fDTBPT units are strong and achievable and they might potentially help inter-backbone polymer charge transport by attracting polymer chains together.
Both monomers were co-polymerised via microwave assisted Stille coupling with stannylated C16IDT.17 Purification of the crude polymers was carried out by Soxhlet extraction with acetone, hexane and then with chloroform to extract the polymer product. Both polymers exhibited good solubility in common organic solvents despite the rigidity of the electron-accepting unit. Preparative GPC in chlorobenzene was also carried out in order to further purify and remove low molecular weight polymer fractions.18
Polymer molecular weights and polydispersity were determined by GPC analysis and referenced to polystyrene standards and are shown in Table 1. Although the PDIs are similar (1.5) for both polymers, lower molecular weights were obtained for the C16IDT–fDTBPT polymer, presumably due to the lower solubility of co-monomer 6.
| Polymer | M n/g mol−1 | M w/g mol−1 | PDI | DPnc | T d/°C |
|---|---|---|---|---|---|
| a Average molecular weight in number (Mn) and in weight (Mw) and weight-average polydispersity PDI (Mw/Mn) as determined by GPC in chlorobenzene at 80 °C and calibrated on polystyrene standards. b Decomposition temperature determined by TGA under N2 and based on 5% weight loss. c The degree of polymerization (DPn) is defined in this case as the number of repeating units. | |||||
| fDTBT | 29000 |
43000 |
1.52 | 20 | 427 |
| fDTBPT | 20000 |
29000 |
1.45 | 13 | 400 |
Thermal stability, which is a very important factor in organic electronic devices, was evaluated by TGA carried out under a N2 atmosphere (Fig. 3). Both polymers showed high temperature decomposition temperatures (5% loss on weight), particularly over 400 °C (Table 1). Furthermore, DSC scans were performed showing no obvious transitions and therefore supporting the evidence that C16IDT based polymers have an amorphous character (Fig. S1†).19
 | ||
| Fig. 3 Thermogravimetric analysis (TGA) of C16IDT–fDTBT (black line) and C16IDT–fDTBPT (red line) measured under N2 atmosphere. Dashed line indicates 5% mass loss. | ||
Energy levels
Most low band gap semiconducting co-polymers consist of alternating electron rich (e.g. IDT) and electron deficient (e.g. BT) units.4 In this case, the electron rich unit is delocalized along the whole polymer backbone while the electron poor unit is localised on one co-monomer (Fig. 4). This configuration is crucial to understand the electrochemical and optical properties and therefore the energy levels of both polymers.20 | ||
| Fig. 4 Schematic representation of C16IDT based co-polymers used in this study, where red and blue colours indicate electron-rich and electron-poor units, respectively. | ||
The HOMO energy levels were determined by cyclic voltammetry (CV) and compared to photoelectron spectroscopy in air (PESA), and they are shown in Table 2. Oxidation potentials of +0.59 V and +0.55 V for C16IDT–fDTBT and C16IDT–fDTBPT were obtained from the oxidation onset after calibrating externally with ferrocene E1/2 (Fig. S2†). The oxidation potentials (V) were converted to EHOMO (eV) by following a procedure which employs an empirical linear translation equal to EHOMO (eV) = −4.88 − EOx (V).21 Not unexpectedly, the oxidation potential was very similar for both polymers. This is because the HOMO is predominantly located on the electron rich IDT and DT units (Fig. 4), common to both polymers and confirmed by DFT calculations (see below). EHOMO values are in good agreement with previous IDT based polymers.17,19 The LUMO energy levels were obtained by addition of the optical gaps (see below) to the HOMO and they differed substantially, being C16IDT–fDTBPT 0.6 eV deeper than C16IDT–fDTBT. This can be attributed to the stronger electron-withdrawing ability of the fDTBPT unit.
| Polymer | λ max/nm | λ onset/nm | E gap /eV | E Ox /V | E HOMO /eV | E LUMO /eV |
|---|---|---|---|---|---|---|
| a Optical gap from the onset of absorption spectrum. b CV measured from 0.1 M [TBA][PF6] in CH3CN and referenced to ferrocene. c E HOMO (eV) = −4.88 − EOx (V). PESA results are shown in brackets. d E LUMO = EHOMO + Egap. | ||||||
| fDTBT | 577 | 614 | 2.02 | +0.59 | −5.47 | −3.45 |
| (−5.46) | ||||||
| fDTBPT | 730 | 900 | 1.38 | +0.55 | −5.43 | −4.05 |
| (−5.42) | ||||||
UV-Visible absorption profiles were acquired in both solution and solid state (Fig. 5). The main absorption bands in chloroform solution were located at 566 nm and 534 nm for C16IDT–fDTBT and –fDTBPT, respectively, with a well-defined vibronic structure. UV-Visible absorption in the solid-state was only 5 nm red-shifted compared to solution experiments, suggesting the absence of strong aggregation effects. Interestingly, the C16IDT–fDTBPT polymer showed a red-shifted and broad band located at 760 nm, with much lower intensity than the main absorption band.
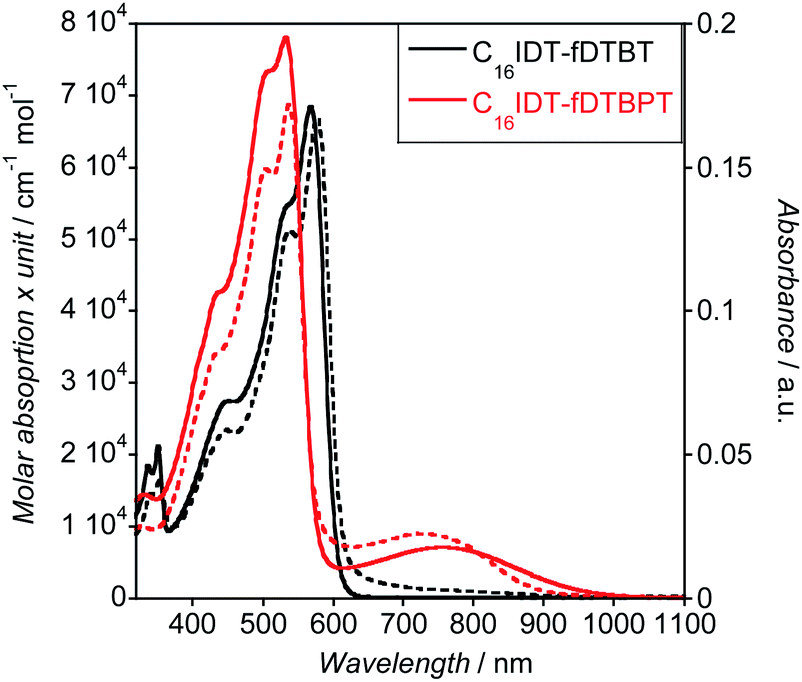 | ||
| Fig. 5 UV-Visible absorption of C16IDT–fDTBT (black line) and C16IDT–fDTBPT (red line) measured at 0.02 mg mL−1 in chloroform (solid line) and in solid state (dashed line). | ||
The molecular (chemical) origin of those absorption bands was not straightforward to establish. Thus, in order to shed some light on the optical results, we performed hybrid DFT calculations at the B3LYP/6-31G(d) level of theory on a trimer system which was chosen as an approximation to our polymers.22 As shown in Fig. 6, the HOMO for both polymers is fully delocalized into the π orbitals and therefore the electron-deficient unit BT or BPT have minimal influence on it. This justifies that both polymers have approximately the same HOMO energy level as shown previously on CV and PESA results. On the other hand, the LUMO is mainly π* based for the C16IDT–fDTBT trimer, with little contribution from the extended BT unit and therefore the main electronic transition is π–π* based. However, for C16IDT–fDTBPT, the LUMO has 3-fold degeneracy (as no symmetry is included on the calculation) and it is exclusively based on the electron-withdrawing BPT unit.
 | ||
| Fig. 6 Molecular orbital distribution of the C16IDT–fDTBT trimer (left) and C16IDT–fDTBPT (right) at the B3LYP/6-31G(d) level of theory (isodensity = 0.02). | ||
Hence, for C16IDT–fDTBPT the first electronic transition is an intramolecular charge transfer (ICT) band from the thiophene bridged system (IDT–DT) to the electron-deficient BPT unit and it relates to the low-energy broad band observed in the UV-Visible spectrum (Fig. 5). Furthermore, the LUMO+1 is π* based and the second electronic transition (HOMO to LUMO+1) corresponds to the sharp and high-energy absorption peak observed in the experimental spectrum and has π–π* character. TD-DFT calculations confirm that both ICT and π–π* take place in C16IDT–fDTBPT with higher oscillator strength for the π–π* electronic transition (Fig. 7 and Table S1†). We believe that the ICT band is not observed for the C16IDT–fDTBT polymer due to the weaker electron-withdrawing ability of the fDTBT moiety, and it is confirmed by TD-DFT as well.
 | ||
| Fig. 7 Simulated UV-Visible absorption (solid line) and its oscillator strength (dashed line) by TD-DFT means at the B3LYP/6-31G(d) level of theory for C16IDT–fDTBT (black line) and C16IDT–fDTBPT (red line) trimer systems. | ||
Interestingly, the double band feature, one low-energy low-intensity transition together with one high-energy high-intensity band, was also observed for the isolated fDTBPT unit 3 and confirmed by TD-DFT as well (Fig. S3†). This suggested that the fDTBPT unit was ultimately responsible for the final polymer UV-Visible trace.
Photoluminescence (PL) spectra were also acquired for both polymers in solution and solid state (Fig. S4 and S5†) by exciting at the main absorption band (i.e. π–π* transition). C16IDT–fDTBT was highly emissive in solution and even visible to the naked eye. However, its PL was quenched dramatically in the solid state. On the other hand, emission from the C16IDT–fDTBPT polymer was very weak in solution and not detectable in the solid state. We could attribute this phenomenon to the presence of low-lying excited states that could promote non-radiative decay pathways as in this particular case, the emission overlaps with the ICT absorption band.
Field-effect transistor performance
Representative transfer and output characteristics of bottom-contact top-gate field effect transistors are shown in Fig. 8. For both polymers, the transfer characteristics show excellent Ion/Ioff ratios of ∼106 with average hole mobilities of 0.07 cm2 V−1 s−1 and 0.03 cm2 V−1 s−1 extracted for C16IDT–fDTBT and C16IDT–fDTBPT, respectively. The polymers show low threshold voltages of −9 V (C16IDT–fDTBT) and −10 V (C16IDT–fDTBPT), which suggests that FET operation is not limited by charge injection. The linear output characteristics recorded for both polymers, as well as the turn-on at ∼0 V furthermore verifies the absence of major injection barriers. The higher mobility obtained for C16IDT–fDTBT could be attributed to higher defect tolerance for this polymer. In this case, the fDTBT unit has lower conformational energy change by rotation compared to fDTBPT.23 It is worth noting that mobilities obtained are 1000-fold higher than the reported polymers containing fDTBT units.24 | ||
| Fig. 8 Transfer (top) and output (bottom) characteristic curves of C16IDT polymers (L = 20 μm, W = 1 mm) measured at VD = −5 V (dashed lines) and VD = −60 V (solid lines). | ||
Solar cell properties
Photovoltaic performance of the polymers was evaluated by preparing bulk-heterojuction solar cells with a conventional device structure containing ITO/PEDOT:PSS/polymer:PC71BM/Ca/Al and tested under simulated 100 mW cm−2 AM 1.5G sun light. The J–V curves are shown in Fig. 9 and the device parameters are summarized in Table 3. The high VOC obtained for both polymers, exceeding +0.80 V, is similar for both polymers and is due to the deep HOMO energy level, confirmed by CV and PESA results. On the other hand, the JSC obtained for C16IDT–fDTBT was about twice the value for C16IDT–fDTBPT. We could partially attribute this to the lower molecular weight for the C16IDT–fDTBPT polymer as well as a very low-lying LUMO energy level of −4.05 eV, which might limit the efficient charge separation in the polymer:PC71BM blend.25,26 In fact, the EQE spectra (Fig. 9 – inset) confirmed that the ICT absorption band (∼750 nm) for the C16IDT–fDTBPT polymer did not contribute to the photocurrent generation, yielding only immobile excitons. Overall, efficiencies of 2.18% and 1.34% were obtained for C16IDT–fDTBT and –fDTBPT, respectively. A major reason for the low PCE values is likely to be due to the presence of long linear, C16, alkyl chains on the IDT unit. In previous studies of IDT polymers, this particular side chain exhibited sub-optimal phase separation and the lowest PCEs.17 However, we were limited to C16 alkyl chains to ensure good polymer solubility, as co-monomers fDTBT and fDTBPT showed limited solubility.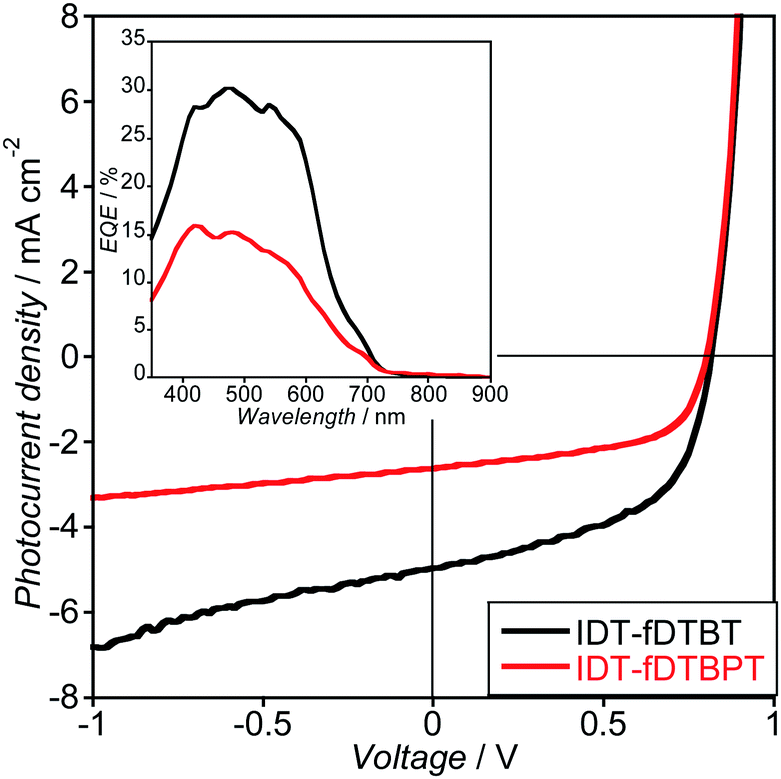 | ||
| Fig. 9
J–V characteristics of C16IDT–fDTBT (black line) and C16IDT–fDTBPT (red line) polymer:PC71BM (ratio 1:3.5) solar cell under AM 1.5G solar illumination. Inset: external quantum efficiency for both C16IDT based polymers. | ||
| Polymer | J SC/mA cm−2 | PCE/% | V OC/V | FF |
|---|---|---|---|---|
| fDTBT | 4.95 | 2.18 | 0.82 | 0.54 |
| fDTBPT | 2.82 | 1.34 | 0.81 | 0.58 |
Conclusions
We have synthesised and characterised two polymers composed of an electron-rich backbone, comprised of alternating IDT and DT units, with electron-deficient BT and BPT units fused on the DT unit. Both polymers showed a decent molecular weight with narrow polydispersity and good thermal stability. Increasing the electron-withdrawing strength of this BT unit by inserting a pyrazine ring led to a significant difference in the optical properties, turning not only the first electronic transition (HOMO to LUMO) from a π–π* based to ICT character, but also red-shifting the absorption by 100 nm. Polymers showed promising OFET performance close to 0.1 cm2 V−1 s−1 with very small turn on and threshold voltages (less than −10 V). Nevertheless, the strong and sharp absorption of the C16IDT–fDTBT polymer makes it potentially suitable as a wide band gap polymer for tandem solar cells. Replacement of the IDT linear C16 alkyl chains for branched ethylhexyl (C2C6) should improve the bulk heterojunction morphology.Acknowledgements
This work was supported by EU grants 604397 (ArtESun) and 287818 (X10D) as well as Imperial College Doctoral Training Centre (DTC) grant EP/G037515/1. M.N. and M.H. thank gratefully the Technology Strategy Board (TSB) PORSCHED project 101238. We are grateful to Scott E. Watkins (CSIRO Materials Science and Engineering, Victoria, Australia) for his contribution to the polymer energy levels determination by UV-PESA measurements.Notes and references
- A. C. Arias, J. D. MacKenzie, I. McCulloch, J. Rivnay and A. Salleo, Chem. Rev., 2010, 110, 3–24 CrossRef CAS PubMed.
- X. Guo, M. Baumgarten and K. Müllen, Prog. Polym. Sci., 2013, 38, 1832–1908 CrossRef CAS PubMed.
- C. B. Nielsen, M. Turbiez and I. McCulloch, Adv. Mater., 2013, 25, 1859–1880 CrossRef CAS PubMed.
- I. McCulloch, R. S. Ashraf, L. Biniek, H. Bronstein, C. Combe, J. E. Donaghey, D. I. James, C. B. Nielsen, B. C. Schroeder and W. Zhang, Acc. Chem. Res., 2012, 45, 714–722 CrossRef CAS PubMed.
- X. Zhang, H. Bronstein, A. J. Kronemeijer, J. Smith, Y. Kim, R. J. Kline, L. J. Richter, T. D. Anthopoulos, H. Sirringhaus, K. Song, M. Heeney, W. Zhang, I. McCulloch and D. M. DeLongchamp, Nat. Commun., 2013, 4, 3238-1–3238-9 Search PubMed.
- H. N. Tsao, D. M. Cho, I. Park, M. R. Hansen, A. Mavrinskiy, D. Y. Yoon, R. Graf, W. Pisula, H. W. Spiess and K. Müllen, J. Am. Chem. Soc., 2011, 133, 2605–2612 CrossRef CAS PubMed.
- F. A. Arroyave, C. A. Richard and J. R. Reynolds, Org. Lett., 2012, 14, 6138–6141 CrossRef CAS PubMed.
- Y. Xie, T. Fujimoto, S. Dalgleish, Y. Shuku, M. M. Matsushita and K. Awaga, J. Mater. Chem. C, 2013, 1, 3467–3481 RSC.
- M. J. Frisch, et al., Gaussian 09, Revision B.01, Gaussian Inc, Wallingford CT, 2009 Search PubMed.
- M. Hanwell, D. Curtis, D. Lonie, T. Vandermeersch, E. Zurek and G. Hutchison, J. Cheminf., 2012, 4, 17 CAS.
- R. Bauernschmitt and R. Ahlrichs, Chem. Phys. Lett., 1996, 256, 454–464 CrossRef CAS.
- D. J. Tozer and N. C. Handy, J. Chem. Phys., 1998, 109, 10180–10189 CrossRef CAS PubMed.
- N. M. O'Boyle, A. L. Tenderholt and K. M. Langner, J. Comput. Chem., 2008, 29, 839–845 CrossRef PubMed.
- H. Bastian and E. Breitmaier, Chem. Ber., 1985, 118, 1278–1281 CrossRef CAS.
- M. Planells and N. Robertson, Eur. J. Org. Chem., 2012, 2012, 4947–4953 CrossRef CAS.
- F. Würthner, S. Yao, T. Debaerdemaeker and R. Wortmann, J. Am. Chem. Soc., 2002, 124, 9431–9447 CrossRef PubMed.
- H. Bronstein, D. S. Leem, R. Hamilton, P. Woebkenberg, S. King, W. Zhang, R. S. Ashraf, M. Heeney, T. D. Anthopoulos, J. d. Mello and I. McCulloch, Macromolecules, 2011, 44, 6649–6652 CrossRef CAS.
- R. S. Ashraf, B. C. Schroeder, H. A. Bronstein, Z. Huang, S. Thomas, R. J. Kline, C. J. Brabec, P. Rannou, T. D. Anthopoulos, J. R. Durrant and I. McCulloch, Adv. Mater., 2013, 25, 2029–2034 CrossRef CAS PubMed.
- W. Zhang, J. Smith, S. E. Watkins, R. Gysel, M. McGehee, A. Salleo, J. Kirkpatrick, S. Ashraf, T. Anthopoulos, M. Heeney and I. McCulloch, J. Am. Chem. Soc., 2010, 132, 11437–11439 CrossRef CAS PubMed.
- P. M. Beaujuge, C. M. Amb and J. R. Reynolds, Acc. Chem. Res., 2010, 43, 1396–1407 CrossRef CAS PubMed.
- B. A. Jones, A. Facchetti, M. R. Wasielewski and T. J. Marks, J. Am. Chem. Soc., 2007, 129, 15259–15278 CrossRef CAS PubMed.
- T. M. McCormick, C. R. Bridges, E. I. Carrera, P. M. DiCarmine, G. L. Gibson, J. Hollinger, L. M. Kozycz and D. S. Seferos, Macromolecules, 2013, 46, 3879–3886 CrossRef CAS.
- H. Sirringhaus, Adv. Mater., 2014, 26, 1319–1335 CrossRef CAS PubMed.
- C.-Y. Mei, L. Liang, F.-G. Zhao, J.-T. Wang, L.-F. Yu, Y.-X. Li and W.-S. Li, Macromolecules, 2013, 46, 7920–7931 CrossRef CAS.
- S. D. Dimitrov, A. A. Bakulin, C. B. Nielsen, B. C. Schroeder, J. Du, H. Bronstein, I. McCulloch, R. H. Friend and J. R. Durrant, J. Am. Chem. Soc., 2012, 134, 18189–18192 CrossRef CAS PubMed.
- S. D. Dimitrov and J. R. Durrant, Chem. Mater., 2013, 26, 616–630 CrossRef.
- SHELXTL, Bruker AXS, Madison, WI CrossRef CAS PubMed; SHELX-97, G. M. Sheldrick, Acta Crystallogr., Sect. A, 2008, 64, 112–122 CrossRef CAS PubMed; SHELX-2013, http://shelx.uni-ac.gwdg.de/SHELX/index.php Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: DSC, CV, 1H-NMR and PL experiments of C16IDT based polymers. UV-Visible absorption of 3 and TD-DFT tables. CCDC 992593. For ESI and crystallographic data in CIF or other electronic format: see DOI: 10.1039/c4tc01500b |
| ‡ Crystal data for 2: C12H4N4S3, M = 300.37, orthorhombic, Pna21 (no. 33), a = 7.22656(17), b = 18.9016(4), c = 8.35979(18) Å, V = 1141.89(4) Å3, Z = 4, Dc = 1.747 g cm−3, μ(Cu-Kα) = 5.844 mm−1, T = 173 K, dark red blocky needles, Agilent Xcalibur PX Ultra A diffractometer; 1453 independent measured reflections (Rint = 0.0209), F2 refinement, R1(obs) = 0.0249, wR2(all) = 0.0637, 1404 independent observed absorption-corrected reflections [|Fo| > 4σ(|Fo|), 2θmax = 147°], and 173 parameters.27 The absolute structure of 2 was determined by a combination of R-factor tests [R1+ = 0.0249 and R1− = 0.0367] and by use of the Flack parameter [x+ = 0.05(2) and x− = 0.94(2)]. CCDC 992593. |
| This journal is © The Royal Society of Chemistry 2014 |