
DFT-calculation-assisted prediction of the copolymerization between cyclic ketene acetals and traditional vinyl monomers†

Abstract
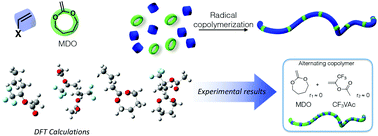
The radical ring-opening polymerization (rROP) of cyclic ketene acetals (CKAs) by free radical or controlled radical mechanisms attracts considerable research interest as it presents an alternative route for the synthesis of aliphatic polyesters. These monomers can undergo radical addition to their C=C double bonds which subsequently leads to propagation by ring opening. CKA/vinyl monomer copolymerization appears to be an elegant method to produce partially or fully degradable copolymers depending on the proportion of the ester functionality incorporated into the copolymer backbone. Although this approach seems promising, some important limitations still remain. Owing to DFT calculations, we are now able to understand the reactivity of CKAs and common vinyl monomers. Indeed, the calculations confirm that cross-addition is not a key parameter for the copolymerization and the reactivity ratios are linked to the homopolymerization rate coefficients of the comonomer pair. In particular, it was demonstrated that trifluoromethyl vinyl acetate (CF3VAc) should provide alternating copolymers. These structures were confirmed experimentally with reactivity ratios close to 0 for the MDO/CF3VAc system (MDO = 2-methylene-1,3-dioxepane). A solid understanding of the reactivity of CKA monomers which allows for the tunable incorporation of main-chain functionalities into copolymers would open up exciting prospects in the field of degradable materials.
- This article is part of the themed collections: Polymer Chemistry Lectureship Winners and Polymer Chemistry Pioneering Investigators 2021
 Please wait while we load your content...
Please wait while we load your content...

