
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnantioselective C–H functionalization: logic and applications in the total synthesis of natural products
Pu-Fan
Qian
 a and
Bing-Feng
Shi
*abcd
a and
Bing-Feng
Shi
*abcd
aState Key Laboratory of Soil Pollution Control and Safety, Department of Chemistry, Zhejiang University, Hangzhou 310058, China. E-mail: bfshi@zju.edu.cn
bState Key Laboratory of Coordination Chemistry, Nanjing University, Nanjing 210023, China
cSchool of Chemistry and Chemical Engineering, Henan Normal University, Xinxiang 453007, China
dCollege of Material Chemistry and Chemical Engineering, Key Laboratory of Organosilicon Chemistry and Material Technology, Ministry of Education, Hangzhou Normal University, Hangzhou 311121, China
First published on 16th January 2026
Abstract
Natural products, with their profound structural diversity and potent bioactivities, represent an invaluable source of inspiration for drug discovery and biomaterial development. The integration of enantioselective C–H functionalization into synthetic planning has emerged as a transformative strategy, offering streamlined and atom-economical routes to complex molecular architectures. This review comprehensively summarizes the pivotal role of these methodologies in the asymmetric total synthesis of natural products. We have organized recent breakthroughs according to three distinct C–H functionalization pathways: (1) radical hydrogen atom abstraction (HAA), (2) metallocarbene C–H insertion, and (3) C–H metalation. Through illustrative case studies, we dissect how these strategies function as the key steps to construct stereodefined frameworks, thereby enabling the concise synthesis of target molecules, confirming absolute configurations, and facilitating biological evaluation. We anticipate that the continued maturation of enantioselective C–H activation will further democratize and accelerate the synthesis of complex natural products and their analogues.
 Pu-Fan Qian | Pu-Fan Qian was born in Zhejiang, China. He joined the research group of Prof. Bing-Feng Shi in 2019 and then received a BSc degree from Zhejiang University in 2022. Now he is a PhD candidate in Zhejiang University under the guidance of Prof. Bing-Feng Shi, Dr Tao Zhou, Prof. Qi-Jun Yao and Prof. Dawei Di. His research interests focus on the development of transition metal-catalyzed site- and enantioselective C–H functionalization and its application in the asymmetric synthesis of chiral functional molecules. |
 Bing-Feng Shi | Bing-Feng Shi was born in Shandong, China. He received his BS degree from Nankai University in 2001 and a PhD degree from the Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences under the guidance of Professor Biao Yu in 2006. Following time as a postdoctoral fellow at the University of California, San Diego (2006–2007), he moved to The Scripps Research Institute working with Professor Jin-Quan Yu as a research associate. In 2010, he joined the Department of Chemistry at Zhejiang University as a professor. His research focus is transition metal-catalyzed C–H functionalization and the application in the synthesis of biologically important small molecules. |
1 Introduction
Natural products, isolated from living organisms, are a significant class of chemical compounds known for their diverse and unique bioactivities. Consequently, these natural products and their analogues have found extensive applications across pharmaceuticals, agrochemicals, cosmetics, and biomaterials science.1 Since Wöhler accomplished the first total synthesis of urea 3 from inorganic salts—silver cyanate 1 and ammonium chloride 2—in 1828 (Scheme 1a),2 chemists have recognized the potential for artificially synthesizing bioactive organic compounds in abiotic systems. In 1917, the tropinone 7 was established by Robinson via a twofold Michael addition (Scheme 1b).3 Compared with the first 21-step approach reported by Willstätter,4 this landmark one-pot synthesis5 further highlighted the power of innovative methodologies for enhancing efficiency in total synthesis.6 | ||
| Scheme 1 Advancements in the efficiency of total synthesis driven by innovations in synthetic methodology and strategy. | ||
Today, the advancements in organic chemistry have opened up new avenues for imaginative disconnections in the retrosynthesis of complex structures. It appears that nearly all natural products, regardless of their complexity, can be synthesized in the laboratory or industry,7 providing an alternative to biosynthesis for compounds that are insufficiently available from natural sources. However, challenges remain, as unsatisfactory overall yields and tedious synthetic procedures continue to limit the practical applications of many natural products.1,8 For example, Taxol (paclitaxel) 9, a well-known and effective natural-source antitumor drug, still primarily relies on semi-synthesis from the isolated intermediate 10-deacetylbaccatin III (10-DAB) 8 (Scheme 1c).9 Thus, there is an urgent need to develop new synthetic protocols for the total syntheses of complex molecules, which may achieve higher atom economy, yield, and stereoselectivity within shorter and scalable synthetic routes.10
The ubiquitous C–H bond in organic molecules presents an attractive target for direct and precise transformation without pre-functionalization. This approach offers a more convenient, cost-effective and sustainable pathway for target-oriented organic synthesis, and has a long-standing history in the total synthesis of natural products.11 The pioneering work of Löffler and colleagues in 1909 applied a site-selective radical C–H functionalization in the synthesis of nicotine 11 through intramolecular 1,5-hydrogen atom transfer (HAT, Scheme 1d).12,13 In the following decades, significant contributions were made. In 1977, the Barton group utilized diastereoselective C–H oxidation as a key step in synthesizing aldosterone acetate 13 from a photochemically unstable nitrite ester by taking advantage of the rigid conformation of a steroid (Scheme 1e).14 Cory and colleagues employed intramolecular C–H insertion for the construction of ishwarane 16 with an in situ generated carbene (Scheme 1f),15 trimming the route to 6 steps from the original 16-step synthesis.16 Additionally, Trost demonstrated a Pd-catalyzed C–H activation/reductive Heck coupling in the total synthesis of ibogamine (Scheme 1g).17 Despite these advances, the generation of new chiral elements in these early examples often depended on pre-existing stereocenters within the molecules, necessitating the use of chiral precursors that are challenging to prepare in many cases, thereby restricting the enantioselective synthesis of chiral natural products.
Recently, transition metal-catalyzed enantioselective C–H functionalization—including radical hydrogen atom abstraction (HAA), metallocarbene/nitrene insertion, and C–H metalation strategies—has garnered significant attention.18 This approach offers an innovative avenue for the streamlined synthesis of chiral complex compounds from prochiral substrates and enables the late-stage decoration of highly functionalized intermediates under mild conditions, balancing reactivity and selectivity among numerous similar inert C–H bonds.11 Over the past three decades, enantioselective C–H functionalization has emerged as a promising strategy for constructing both molecular skeletons and stereocenters in the total synthesis of natural products with minimized synthetic steps and waste (Scheme 1h). To summarize recent progress and offer new perspectives on this burgeoning field, this review will comprehensively discuss the contributions of total syntheses that benefit from transition metal-catalyzed enantioselective C–H functionalization. We will emphasize the interconnections between synthetic advancements and the corresponding innovations in methodology. To provide a clearer focus, this review will primarily cover the construction of chirality during the C–H activation step. Processes involving functionalization of unsaturated bonds after C–H activation to generate chirality are beyond the scope of this discussion.
2 Syntheses via HAA/radical relay
In the metabolism of organisms, C-centered radicals serve as prevalent intermediates for selective C–H functionalization via a radical rebound pathway. This process can be facilitated by reactive metal–oxo species under mild conditions through enzyme catalysis.19 The precise manipulation of free radicals generated through hydrogen atom abstraction (HAA) poses a significant challenge due to their instability and high reactivity.20 To address this issue, a second species—often a transition metal catalyst—is introduced to trap the diffusible C-centered radical intermediate, enabling excellent regio- and stereoselectivity. This approach is known as a radical relay process and is typically achieved using cobalt, copper, or nickel catalysts.18k,m,212.1 Cu-catalyzed enantioselective C(sp3)–H oxidation
Polyacetylenic alcohols virol A 19, virol B 20 and virol C 21 (Scheme 2a) are three minor toxic components isolated from water hemlock,22Cicuta virosa (Umbelliferae), a plant widespread throughout temperate areas. These compounds are extremely poisonous for tonic and clonic convulsions and respiratory paralysis to both livestock and humans through their impact on the central nervous system. Compared with their analogue cicutoxin 22, the major toxin of the plant,23 virols A, B and C are more chemically stable compounds, which are suitable for pharmacological action investigations. In 1999, Oshima and coworkers disclosed a total synthesis of virols A and C from (S)-oct-1-yn-3-ol 24 and (S,S)-methylbenzenesulfonate 28 respectively,24 which could be prepared from L-(+)-dimethyl tartrate 23via several transformations in moderate yield (Scheme 2b and d).25 Alternatively, the enantioselective Kharasch–Sosnovsky reaction, a copper-catalyzed direct oxidation of allyl, propargylic or benzylic C–H bonds,26 is obviously a more ideal approach of these chiral propargyl alcohols, while the enantiocontrol was poor and an excessive amount of hydrocarbon substrate was required.26c Very recently, building on their sustained endeavors in enantioselective C–H functionalization via Cu-catalyzed radical relay,21d,27 Liu, Lin and coworkers showcased a biomimetic propargylic C(sp3)–H acyloxylation with excellent site- and enantioselectivity assisted by chiral bisoxazoline (BOX) ligand L1.28 Leveraging their radical C(sp3)–H oxidation methodology, the nearly 10-step syntheses of chiral fragments 25 and 29 could be simplified to 3 steps from dimethylphenylsilyl alkynes 26 and 31 in the absence of stoichiometric chiral synthons (Scheme 2c and e). Similarly, the formal synthesis of alfaprostol 35 was also performed with a chiral side chain constructed via this Cu-catalyzed radical relay (Scheme 2f).29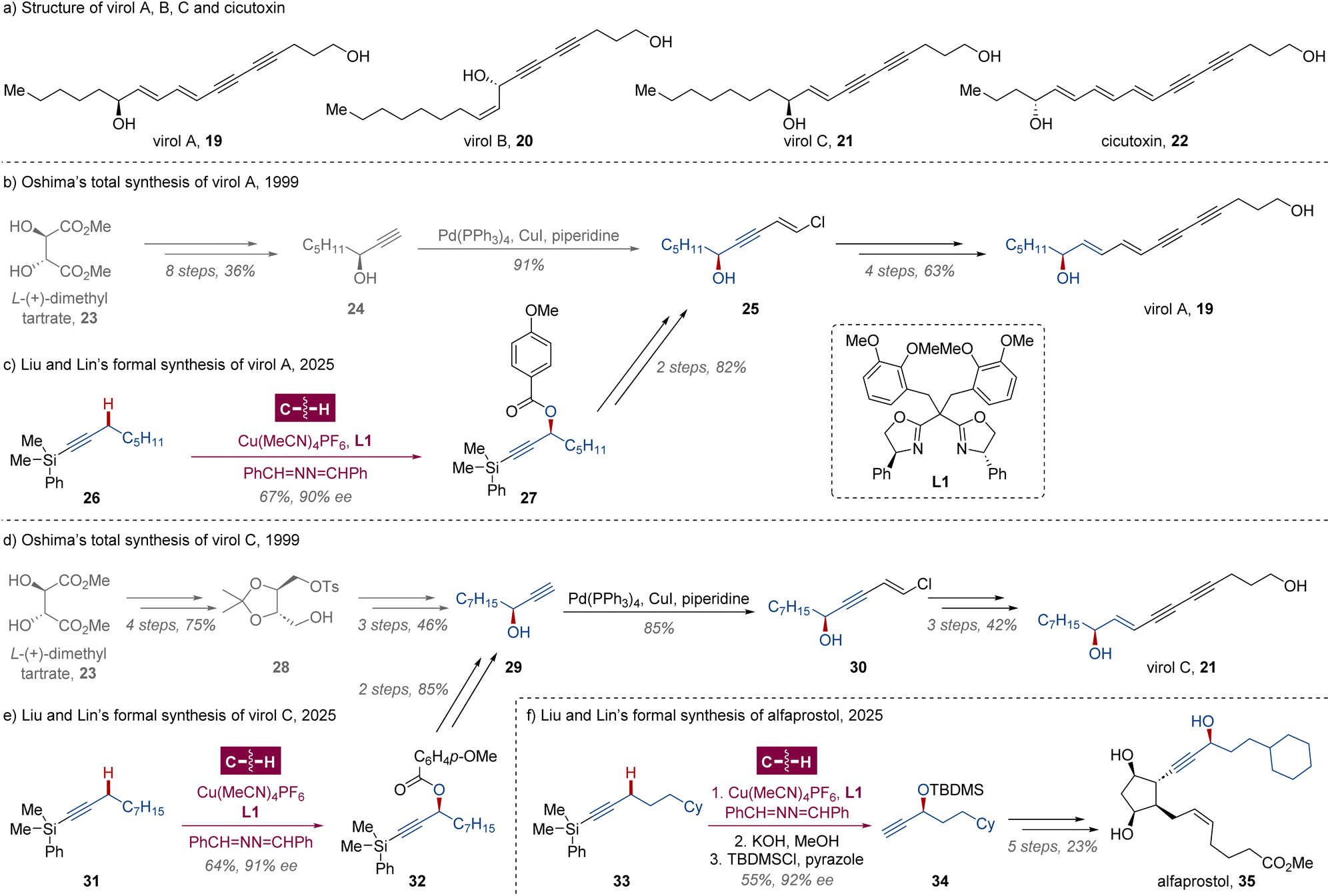 | ||
| Scheme 2 Liu and Lin's formal syntheses of virol A, virol C and alfaprostol via Cu-catalyzed radical relay. | ||
2.2 Ni-catalyzed enantioselective C(sp3)–H alkylation and amination
Indolizidine and pyrrolizidine alkaloids, featuring a 5/6- or 5/5-membered fused azacycle system as the core skeleton, respectively (Scheme 3a), are widely present among a multitude of marine and terrestrial living beings, such as trees, fungi, frogs and so on, illustrating numerous distinctive biological activities.30 For these natural products, a major synthetic challenge is the construction of an α-tertiary or quaternary chiral center on the fused N-heterocycle framework. | ||
| Scheme 3 Huo's total synthesis of (−)-indolizidine 201 and Kong and Ping's total synthesis of (−)-coniceine. | ||
In 2022, Yu, Yuan and colleagues presented a total synthesis of (−)-indolizidine 201 36 from N-Boc-2-formylpyrrolidine 44 in 27% yield over 8 steps by means of a photoexcited Cu/BOX-catalyzed kinetic resolution of alkenes as the key stereo-introducing step (Scheme 3b).31 Drawing from previous studies,32 in 2024, the Huo group demonstrated a photoredox nickel-catalyzed enantioselective α-C(sp3)–H alkylation of N-Bz-pyrrolidine 45 with in situ prepared N-hydrophthalimide (NHPI) ester utilizing chiral BOX ligand L2, and the (−)-indolizidine 201 36 was obtained in 59% yield over 3 steps with additional acidic deprotection and diastereoselective reductive amination of the corresponding compound 46 to construct the 5/6-membered fused ring (Scheme 3c).33
Very recently, Kong, Ping and collaborators revealed a similar enantioselective α-C(sp3)–H alkylation of saturated heterocycles with olefins enabled by Ni/L3 catalysis.34 Applying this protocol, the 3-step total synthesis of the simplest indolizidine alkaloid (−)-coniceine 37 was performed (Scheme 3d), which was similar to Huo's route of (−)-indolizidine 201 36 and more concise than the previous synthetic approach.35 Meanwhile, the authors also achieved the enantio- and diastereoselective formation of secondary–secondary C(sp3)–C(sp3) bonds using alkenyl boronates as alkylation agents to restrain potential chain walking by stabilizing the branched organonickel intermediate,36 which streamlined the construction of more complicated indolizidine and pyrrolizidine alkaloids (Scheme 4). The alkylated products 48 and 49 were oxidized to alcohol and benzylated with stereochemical retention in 66% and 77% yields, respectively. Cp2ZrHCl-mediated debenzoylation was then performed at 0 °C, followed by desilylation for 50 with tetrabutylammonium fluoride (TBAF) or debenzylation for 51 with trifluoroacetic acid (TFA). The alcohol 53 was oxidized to carboxylic acid by (1-hydroxycyclohexyl)phenylmethanone (1-HCPK) via a base-promoted hydrogen atom transfer,37 and 8-hydroxy-5-indolizidinone 54 was afforded after amidation and debenzylation as the key synthon for the formal synthesis of pumiliotoxin 251D 38,38 a major component in the secretion of Dendrobates tricolor (Ecuador) with myotonic and cardiotonic properties.39 The natural alkaloid (−)-1-hydroxypyrrolizidine 39 was generated in an overall yield of 12% after further Mitsunobu-type nucleophilic cyclization and debenzylation of free alcohol 52, as well as (−)-perhydro-8-indolizinol 40 in 17% total yield via compound 53. With a stereoconserving Matteson boronate rearrangement process of 48 and 49 for homologation followed by the same benzylation–deprotection–cyclization–debenzylation sequence described above, pyrrolizidine (−)-trachelanthamidine 41 and indolizidine (−)-tashiromine 42 were formed in 7 steps with overall yields of 11% and 8%, respectively. Moreover, the azabicyclo[3.3.0]octane ring of (−)-trachelanthamidine 41 was a core framework of selective serotonin 5-HT4 receptor agonists and antagonists, e.g. SC-53606 43.40
 | ||
| Scheme 4 Kong and Ping's total (or formal) syntheses of chiral indolizidine and pyrrolizidine alkaloids. | ||
Chiral pyrrolidines, usually bearing an α-C-stereocenter, are found as a common structural motif in numerous biologically active natural products, mainly prepared from enantiopure proline.41 As prevalent in approximately 20% of saturated N-heterocycle drugs, pyrrolidines are recognized as a treasure trove for pharmaceutical design.42 Evidently, the enantioselective α-C(sp3)–H alkylation developed by the Huo and Kong groups would be applicable in asymmetric pyrrolidine synthesis, especially for the rapid construction of an analogue library.33,34 Employing these methodologies, the synthesis of (−)-norbgugaine 61 and Ca-sensing receptor antagonist precursor 62 was refined to 2 steps from N-Bz-pyrrolidine 45via nickel-catalyzed enantioselective α-C(sp3)–H alkylation and Cp2ZrHCl-mediated debenzoylation (Scheme 5a), compared with the original approach from L-proline.43a,b (−)-Bgugaine 63 and (−)-irnidine 64, two antibacterial and antimycotic natural alkaloids isolated from tubers of Arisarum vulgare,44 were produced through a similar enantioselective α-C(sp3)–H alkylation and debenzoylation sequence with an additional methylation in improved overall yields of 47% and 39%, respectively (Scheme 5b).43c
 | ||
| Scheme 5 Huo's and Kong and Ping's total syntheses of chiral pyrrolidines. | ||
(+)-α-Lipoic acid 67 is identified as a coenzyme participating in a wide range of biological processes among animals, plants and microorganisms.45 Especially, it is both an intracellular and an extracellular metabolic antioxidant for reactive oxygen species (ROS) scavenging, exhibiting potent neuroprotection, anti-inflammation, anti-cancer and anti-human immunodeficiency virus (HIV) activities and so on.46 In both the enzymatic and transition metal-catalyzed asymmetric syntheses of (+)-α-lipoic acid 67, methyl (S)-6,8-dihydroxyoctanoate 66 was always involved as the key intermediate, which entailed multiple steps to build the stereocenter and the 1,3-diol structure.47 To address this synthetic challenge, guided by their previous accomplishment in the asymmetric syntheses of chiral alcohol-derived drugs via photoredox C(sp3)–H arylation,48 Kong, Ping and coworkers explored whether their enantioselective α-C(sp3)–H alkylation protocol would serve as a superior solution with an extended heterocycle scope to saturated oxacycles.34 After replacement of the oxidant from dicumyl peroxide (DCP) to di-tert-butyl peroxide (DTBP) for the HAT process, the chiral segment 66 was obtained in 54% yield and 97% ee with BOX ligand ent-L3 (Scheme 6). In a similar manner, harnessing this convenient one-pot synthesis, key intermediate (S)-tetradecane-1,3-diol 68 was constructed with excellent enantioselectivity for the formal synthesis of (−)-tetrahydrolipstatin 69,49 a saturated counterpart of lipstatin with strong and irreversible inhibition to lipase isolated from Streptomyces toxytricini, which is a medication against obesity marketed as Xenical.50
 | ||
| Scheme 6 Kong and Ping's formal syntheses of (+)-α-lipoic acid and (−)-tetrahydrolipstatin. | ||
Beyond enantioselective C–C formation, related transformations such as C–N bond formation have also been developed for complex molecule synthesis. For instance, Guo and coworkers reported a nickel-catalyzed electrochemical enantioselective dehydrogenative C–H amination.51 This method was successfully applied to the concise synthesis of bioactive agents, including the (+)-γ-secretase inhibitor and the agrochemicals (+)-flamprop-methyl and (+)-flamprop-isopropyl, illustrating the potential of these reactions in target-oriented synthesis.
3 Syntheses via metallocarbene C–H insertion
Direct carbene C–H insertion offers another valuable route to the formation of C(sp3)–C(sp3) bonds. The enantioselective version of this transformation was pioneered by chiral dirhodium(II) catalysts bearing carboxylate or carboxamidate ligands.18a,g,52 Subsequently, these same catalysts were successfully extended to asymmetric nitrene insertions into C(sp3)–H bonds.52a,53 More recently, the catalytic manifold has expanded to include other transition metals—such as Fe, Co, Cu, Ru and Ir—for enantioselective alkylation and amination via carbene and nitrene transfer, respectively.53,54 Despite the rapid progress, the application of these methods in natural product total synthesis remains limited. To date, only asymmetric dirhodium-carbenoid C(sp3)–H insertion has been employed in a handful of cases to forge C(sp3)–C(sp3) bonds.11c,55 Furthermore, these applications often rely on diastereoselective control, achieved by combining a chiral dirhodium catalyst with a pre-installed chiral auxiliary;56 they primarily establish stereogenicity on the diazo-derived fragment rather than on the functionalized hydrocarbon backbone.57 As a result, enantioselective C–H functionalization via carbene or nitrene insertion has not yet been widely adopted to enable concise total syntheses, representing a significant area for future development.3.1 Rh-catalyzed intramolecular C(sp3)–H carbene insertion
(−)-Enterolactone 77 is the most commonly synthesized lactone with anticarcinogenic and antiestrogenic properties in the lignan library widely distributed in plants,58 which was fortuitously observed and characterized from female urine with a periodic excretion during the menstrual cycle as well as pregnancy59 and then synthesized by Groen.60 In a previous study, the fabrication of chiral γ-lactone ring was quite tricky and troublesome.61 As an efficient approach for the asymmetric γ-lactone construction, Doyle, Simonsen, Lynch and coworkers disclosed a refined synthesis of (−)-enterolactone 77 employing the dirhodium-catalyzed enantioselective intramolecular C(sp3)–H carbene insertion in 1995 (Scheme 7a).62 The C–C double bond in commercially available substrate meta-methoxycinnamic acid 70 was initially hydrogenated in the presence of 5% Pd/C, followed by the reduction of carboxyl group with LiAlH4 to form 3-(meta-methoxyphenyl)propan-1-ol 71 in 98% yield over two steps. The diazoacetate reactant 72 was then delivered through esterification with diketene, diazotization with methanesulfonyl azide and deacetylation with LiOH·H2O. After optimization of conditions for the key step in the entire route, the intramolecular C(sp3)–H carbene insertion, the authors uncovered that the carboxamidate-coordinated dirhodium catalyst Rh2(R-MPPIM)4Rh1 was preferable for this transformation, with the (R)-γ-lactone 76 afforded in 63% yield and 93% ee. The 2-substituent was introduced with outstanding diastereoselectivity by lithium diisopropylamide (LDA)-mediated deprotonation and nucleophilic substitution with meta-benzyl bromide. With methyl groups removed by BBr3, the desired lignan (−)-enterolactone 77 was eventually produced in 26% yield over 8 steps. Meanwhile, with the enantiomer of dirhodium catalyst Rh1 provided from L-asparagine, (+)-enterolactone 78 was also synthesized smoothly via the same procedure with Rh2(S-MPPIM)4ent-Rh1 in an overall yield of 27% with 91% ee. | ||
| Scheme 7 Doyle, Simonsen and Lynch's total (or formal) syntheses of chiral lignan and phenolic natural products. | ||
Taking advantage of this efficient intramolecular C(sp3)–H carbene insertion for enantioselective γ-lactone construction catalyzed by newly designed dirhodium Rh2(R-MPPIM)4Rh1 and Rh2(S-MPPIM)4ent-Rh1, Doyle and collaborators also studied the streamlined synthesis of other lignan natural products.62 (−)-Hinokinin 82,63 (+)-hinokinin 83 and (+)-arctigenin 88 (ref. 64) were analogues of enterolactone with subtle differences in O-containing moieties on arenes, which were unobstructedly synthesized with 94% ee in overall yields of 27%, 25% and 19% applying their enantioselective C(sp3)–H insertion to form the chiral γ-lactone cycle respectively (Scheme 7b and c). Similar to the total synthesis of (+)-hinokinin 83, the key chiral building block (S)-γ-lactone ent-81 was delivered with Rh2(S-MPPIM)4ent-Rh1 in 67% yield and 94% ee, and the target lignan (+)-isodeoxypodophyllotoxin 87 (ref. 65) was provided through nucleophilic addition to 3,4,5-trimethoxybenzaldehyde and TFA-promoted Friedel–Crafts alkylation in 68% yield and >99% ee after crystallization (Scheme 7d). Additionally, the chiral lactone 90, obtained from the corresponding diazoacetate 89 in 59% yield and 97% ee via Rh2(R-MPPIM)4Rh1-catalyzed enantioselective C(sp3)–H insertion, is also a key synthon in syntheses of lignan compounds,66 including (−)-matairesinol 91,61b,67 (−)-anhydrosecoisolariciresinol 92,68 (−)-secoisolariciresinol 93 (ref. 69) and (+)-isolariciresinol 94 (ref. 70) (Scheme 7e). Notably, this enantioselective C(sp3)–H insertion methodology was also viable for complex chiral lactam structures, such as bioactive (−)-rolipram.71
Furthermore, since the chiral γ-lactone scaffold could be easily opened through hydrolysis, nucleophilic substitution or other potential process, the rhodium catalysis mentioned above was also feasible for the asymmetric synthesis of linear natural products. Isolated from a diuretic and anti-inflammatory Chinese traditional medicine—Imperata cylindrica, (+)-imperanene 98 is a homoallylic alcohol with a tertiary C-stereocenter proximal to the C–C double bond, illustrating platelet aggregation inhibitory activity.72 In 2002, the Doyle group realized the first enantioselective total synthesis of (+)-imperanene 98 (Scheme 7f).73 The tert-butyldiphenylsilyl (TBDPS)-protected diazoacetate 95 was prepared in 51% yield from the inexpensive starting material 4-hydroxyl-3-methoxycinnamic acid over 6 steps, and then the dirhodium Rh2(S-MPPIM)4ent-Rh1-catalyzed enantioselective C(sp3)–H carbene insertion/lactonization occurred, generating γ-lactone 96 with 68% yield and 93% ee in the S configuration. Compound 96 was reduced to a hemiacetal with diisobutylaluminium hydride (DIBAL-H), followed by stereospecific ring-opening nucleophilic addition with aryllithium to yield acyclic 1,4-diol 97 as an approximately 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of diastereomers. The primary hydroxy group was selectively silylated, and the 12-step total synthesis of homoallylic (+)-imperanene 98 was accomplished in 12% overall yield after the ultimate elimination and desilylation sequence.
:1 mixture of diastereomers. The primary hydroxy group was selectively silylated, and the 12-step total synthesis of homoallylic (+)-imperanene 98 was accomplished in 12% overall yield after the ultimate elimination and desilylation sequence.
3.2 Rh-catalyzed C(sp3)–H intermolecular carbene insertion
Pseudopterogorgia elisabethae (Octocorallia) is a Caribbean gorgonian coral found in the West Indies, where the gorgonian octocorals flourish more than anywhere else in the world.74 Since the pioneering discovery of diterpene-glycoside seco-pseodopteosins by Fenical in 1987,75 plenty of marine secondary metabolites in Pseudopterogorgia elisabethae have been isolated and characterized as a family of diterpenes. These compounds usually possess distinctive structural architectures which are rarely found in terrestrial organisms,74 exhibiting diverse bioactivities including antibacterial, anti-inflammatory, antitubercular and anticancer properties.74,76 Originating from the common precursor (+)-elisabethatriene 99 in biological synthesis,77 these natural products typically possess three stereocenters (Scheme 8a), which are daunting to be constructed simultaneously in the correct configuration with excellent enantioselectivity and diastereoselectivity.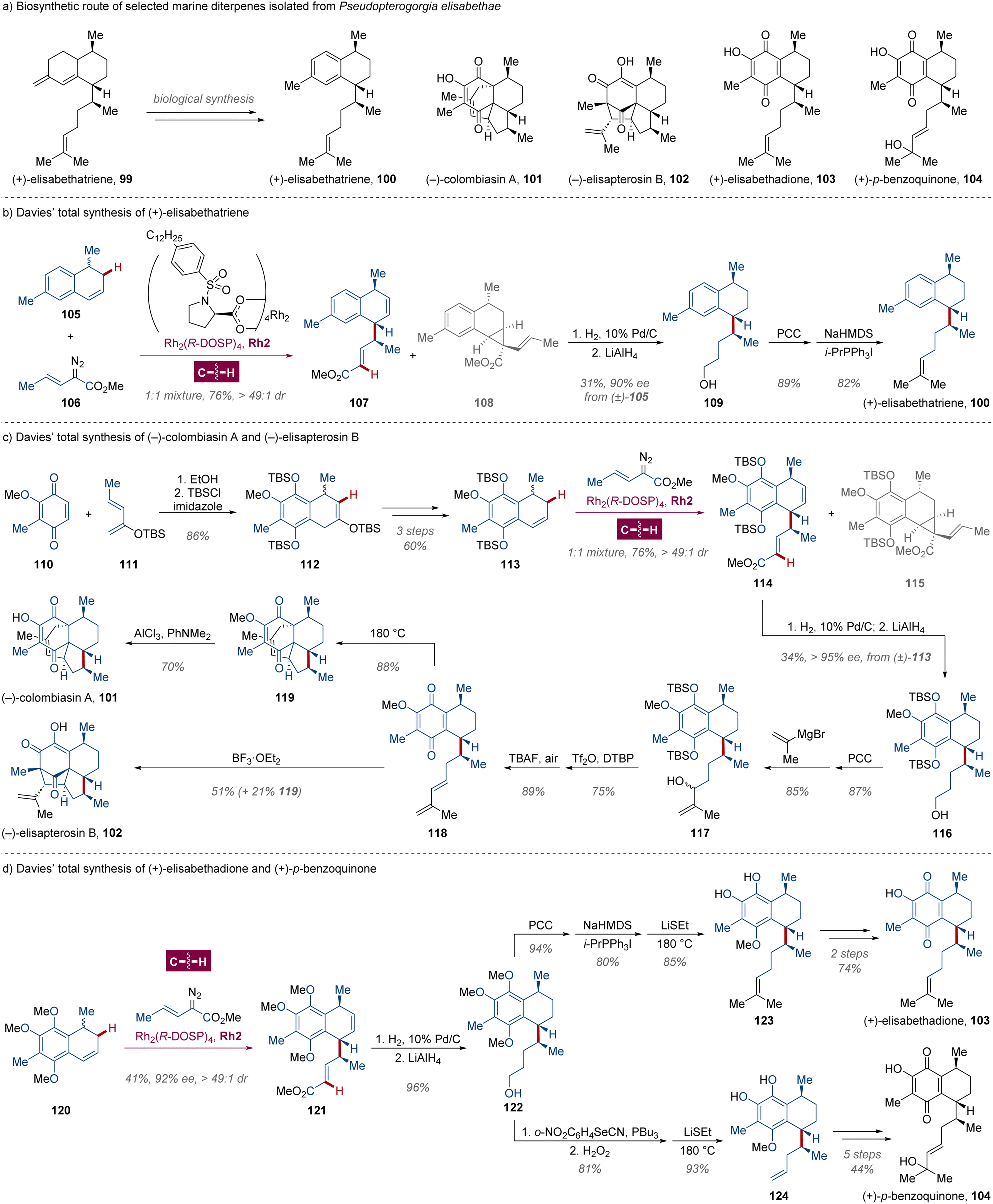 | ||
| Scheme 8 Davies' total syntheses of marine secondary metabolite diterpenes. | ||
Leveraging their previous achievements in dirhodium-catalyzed intermolecular enantiodifferentiating C(sp3)–H carbene insertion/Cope rearrangement,78 Davies and colleagues materialized the total synthesis of antitubercular marine diterpene (+)-erogorgiaene 100 (ref. 79) via a kinetic enantiodivergent functionalization in 2005 (Scheme 8b).80 Using commercially available 4-oxo-4-p-tolylbutanoic acid as the starting reagent, racemic dihydronaphthalene 105 was obtained in 28% yield through a methylation–vinylation–hydrogenation–hydrolysis–cyclization–reduction–elimination sequence. Controlled by the steric effect of the chiral dirhodium catalyst Rh2(R-DOSP)4Rh2, the asymmetric C(sp3)–H carbene insertion proceeded at the 2-position for (S)-105, followed by an enantiospecific Cope arrangement to afford compound 107 bearing three aspired tertiary C-stereocenters in >49:1 dr. In contrast, under the same conditions, only a C–C double bond cyclopropanated product was detected for its enantiomer (R)-105, attaining the efficient resolution of enantiomers and establishing the fundamental architecture of the (+)-erogorgiaene 100. The C–C double bond and carbomethoxy group were subsequently reduced to avoid the propensity of retro-Cope arrangement, and alcohol 109 was isolated from the corresponding 1:1 mixture of compounds 107 and 108 by chromatography in 34% yield over 3 steps from (±)-105, which could be transformed to (+)-erogorgiaene 100 by oxidation and Wittig olefination.
Undoubtedly, polycyclic diterpenes (−)-colombiasin A 101 (ref. 81) and (−)-elisapterosin B 102 (ref. 82) are more sophisticated and highly functionalized, featuring two quaternary and three tertiary C-stereocenters. Stimulated by previous approaches,83 the authors envisioned that the late-stage intermediate 118 for [4 + 2] or [5 + 2] cycloaddition could be settled through the cascade asymmetric C(sp3)–H carbene insertion/Cope rearrangement with ease (Scheme 8c).84 The core framework of dihydronaphthalene (±)-113 was generated from diene 110 and dienophile 111via Diels–Alder reaction after the cleavage of the TBS-enol group and migration of the C–C double bond.83a,b Then the combined enantiodifferentiating C(sp3)–H allylation/Cope rearrangement was investigated with dirhodium Rh2(R-DOSP)4Rh2 catalysis, and the TBS group was proven to be vital for excellent enantiocontrol with sufficient steric hindrance provided, since the highest enantiomeric excess (>95% ee) was measured for further reduced alcohol 116 from TBS-protected p-hydroquinone (±)-113 compared to those from dimethyl 133 or diacetyl polyhydroxy dihydronaphthalene derivatives in proper stereochemistry. Within 4 standard steps, the late-stage p-quinone intermediate bearing a diene group was constructed, which could be smoothly transformed to (−)-colombiasin A 101 through Diels–Alder reaction/demethylation and (−)-elisapterosin B 102 undergoing a BF3·OEt2-mediated [5 + 2] cycloaddition as single diastereomers.83 In a similar manner, assisted by a selective demethylation of adjacent methoxy groups with LiSEt developed by Schmalz,85 gorgonian octocoral bicyclic metabolites (+)-elisabethadione 103 (ref. 86) and (+)-p-benzoquinone 104 (ref. 87) were synthesized from trimethoxy-1,4-dihydronaphthalene 121, which was obtained through Rh2(R-DOSP)4Rh2 catalyzed enantiodivergent C–H functionalization of 120 respectively (Scheme 8d).88
In addition to the chemodivergent kinetic resolution via C(sp3)–H carbene insertion/Cope rearrangement, the dirhodium(II)-catalyzed direct diastereoselective construction of two adjacent tertiary C-stereocenters located on both the C–H fragment and diazo skeleton has also been unveiled. Using dirhodium-catalyzed diastereo- and enantioselective C–H functionalization, the Sorensen group uncovered the rapid access toward several benzo-fused analogues of the indoxamycin natural product family in 2016.89 Subsequently, Shaw and collaborators employed this method in the construction of a multi-substituted isochroman skeleton,90 which is a vital scaffold existing in a lot of natural products and biologically active compounds. Initially, the authors found that the structure of panowamycin A 131 was misassigned by Ōmura,91 and the actual structure was predicted by computational NMR studies with Mahmud's updated spectra.92 Starting from benzyl alcohol 125, the precursor of donor/donor carbene, phenylethylidene hydrazine 127, was prepared with (S)-Roche ester in 7 steps (Scheme 9). With hydrazone 127 treated with MnO2 in MeCN, the generated diazo intermediate was added into the dilute solution of Rh2(S-TCPTTL)4Rh3 dropwise, and the isochroman-cored 128 and epi-128 were produced in 79% yield and 4:1 dr. Fortunately, nearly complete cis-selectivity (>19:1 cis/trans) was measured at C10 and C11 position in both compounds 128 and epi-128, which is consistent with the configuration in the target molecules. Although the starting material 127 contains a chiral side chain, the stereoselectivity at C10 and C11 was predominantly governed by the chiral dirhodium catalyst Rh3 during the C–H functionalization step. Lactone 129 was then obtained via oxidation with pyridinium chlorochromate (PCC) from 128. Further elaboration of 129 yielded panowamycin A 131, TM-135 132, and panowamycin B 133. Meanwhile, epi-128 was converted into veramycin F 134, a compound isolated from Streptomyces sp. ST157608,93 through sequential PCC oxidation and Wacker oxidation.
 | ||
| Scheme 9 Shaw's total syntheses of panowamycins A, B, TM-135 and veramycin F. | ||
(−)-Cylindrocyclophane A 146 is a [7,7]paracyclophane natural product isolated from cytotoxic Nostocaceae blue-green algae in 1990,94 and various synthetic attempts of this unique all-carbon macrocyclic structure have been reported with stereocenters settled by enzyme catalysis or chiral auxiliary ever since.95 Building on the model synthesis of simplified [7,7]paracyclophane with previously developed dirhodium species Rh5,96 Davies, Stoltz and coworkers demonstrated the asymmetric total synthesis of (−)-cylindrocyclophane A 146via multifold C–H functionalizations (Scheme 10).97 Controlled by sterically hindered catalyst Rh2(R-p-PhTPCP)4Rh4, a site- and enantioselective primary C(sp3)–H alkylation of trans-2-hexene 136 was carried out with diazocompound 135 at the less crowded allylic position.96,98 The olefin 137 could be easily obtained in 25 g scale with 73% yield, >20:1 rr and 96% ee, and the aryl diazoacetate 138 was prepared after hydronation and a formal C–H arylation of α-diazo ester. Strikingly, the [7,7]-paracyclophane structure in 141 could be established within a one-pot direct cyclodimerization of 138via Rh2(R-2-Cl-5-BrTPCP)4Rh5-catalyzed twofold regio- and diastereoselective C(sp3)–H carbenoid insertion in 19% yield and 6:1 dr, while a superior result as 36% overall yield and 8:1 dr would be gained through a three-step macrocyclization process. Selective sequential deesterification and derivatization of the trichloroethyl and trifluoroethyl esters in 141 were conducted respectively, followed by the palladium-catalyzed acetoxylation of four C(sp2)–H bonds in macrocycle 143 with 5-(trifluoromethyl)pyridine-3-sulfonic acid used as a ligand.99 Eventually, the asymmetric total synthesis of (−)-cylindrocyclophane A 146 was finished by installation of two alkyl side chains and final deprotection.
 | ||
| Scheme 10 Davies and Stoltz's total synthesis of (−)-cylindrocyclophane A. | ||
Very recently, the Davies group also illustrated an elegant total synthesis of the 2,3-dihydroxypyrrolidine natural product (−)-dragocin D, a cytotoxic metabolite of marine cyanobacteria,100 with their newly developed dirhodium-catalyzed diastereoselective C(sp3)–H alkylation of N-Boc-2,5-dihydro-1H-pyrrole (Scheme 11).101 Modulated by Rh2(S-PTAD)4Rh6 at a mere catalyst loading of 0.05 mol%, the C–C bond in compound 149 was formed from Boc-protected 2,5-dihydropyrrole 147via an 100 mmol scale diastereoselective intermolecular C–H insertion with aryl diazoacetate 148 in 87% yield, 96% ee and >20:1 dr. The C–C double bond was epoxidized by m-CPBA, and the epoxide ring was attacked from the opposite face in situ during the zinc-induced deprotection of the trichloroethyl group. Through a reduction–silylation–selective hydrolysis102–reoxidation sequence, γ-butyrolactone 150 was converted into carboxylic acid 152 smoothly, followed by a photopromoted intramolecular decarboxylative carbamylation in 68% yield and 20:1 dr.103 Subsequently, the bicyclic carbamate 153 was reduced by DIBAL-H, and the absolute configuration of the benzylic hydroxyl group of alcohol 154 was inverted, providing the corresponding hydrofluoride salt of (−)-dragocin D 156 in 15:1 dr.
 | ||
| Scheme 11 Davies' total synthesis of (−)-dragocin D. | ||
3.3 V-catalyzed C(sp3)–H oxidative kinetic resolution
As an isoelectronic species of metal–carbenoid complex, metal–oxos are widely involved in the single-electron C–H functionalization process via HAA/radical rebound.19,104 Intriguingly, a unique two-electron HAA mechanism was observed in the vanadium-catalyzed enantioselective C–H oxidation,105 which could be regarded as a special form of the two-electron C–H insertion with a metal–oxo intermediate. In 2008, the Toste group applied this C–H oxidative kinetic resolution in the de novo synthesis of (−)-octalactin A 164,106 which is a medium-ring metabolite isolated from the marine microorganism, Streptomyces sp.107 As drawn in Scheme 12, the reactant rac-158 for C–H oxidation was prepared within 4 steps from benzyl chloroacetate 157. Catalyzed by VO(Oi-Pr)3 and (R)-2-(3,5-di-tert-butylsalicylideneamino)-tert-butyl-1-ethanol L4, all three stereocenters in the retained (2S,3S)-158 and generated (R)-159 were constructed upon the pivotal asymmetric C–H oxidation of rac-158 using 1 atm of molecular oxygen as the stoichiometric oxidant. Both (2S,3S)-158 and (R)-159 were utilized in subsequent transformation, and two modified resolution fragments, aldehyde 160 and ketophosphonate 161, were recombined through olefination, affording the linear alkene 162 in 82% yield with complete E-selectivity. By employing Arndt–Eistert homologation/Wolff rearrangement for lactonization, the lactone 163 was obtained in 26% yield over 9 steps, which could be converted into (−)-octalactin A 164 with the installation of a side chain through Nozaki–Hiyama–Kishi coupling with vinyl iodide. | ||
| Scheme 12 Toste's total synthesis of (−)-octalactin A. | ||
4 Syntheses via C–H metalation/functionalization
Opposite to the HAA/radical relay or metallocarbene C–H insertion processes discussed above, enantioselective C–H metalation/functionalization involves an inner-sphere mechanism C–H cleavage with the formation of a new carbon–metal bond, which is identified as the strictly speaking C–H activation.18e,f,108 Since the pioneering palladium catalysis developed by the Yu group with N-monoprotected amino acid (MPAA) as chiral ligands,109 a variety of enantioselective C–H activation methodologies have been developed ranging from noble metal Pd, Rh, and Ir to earth abundant metal Co, Ni, and Cu catalysis and so on.18h–j,l,110 To date, these transformations have become pivotal tools in the efficient synthesis of complex molecules, including the total synthesis of natural products.11n,o4.1 Pd-catalyzed enantioselective C(sp2)–H activation
The cyclopenta[de]isoquinoline alkaloid delavatine A 172 is a potential anticancer agent with considerable cytotoxicity isolated from Incarvillea delavayi, a mountain flowering plant in Chinese herbal medicine blooming at high altitudes in Southwest China.111 In 2017, Zhang and Li hypothesized that the enantioenriched precursor 168 for the construction of an isoquinoline ring could be produced from kinetic resolution via triflamide-directed palladium-catalyzed enantioselective C(sp2)–H olefination followed by an ozonolysis quenched by NaBH4 (Scheme 13).112 After optimization of conditions, the intended alkenylated product 167 was obtained in 46% yield and 92% ee on 5-gram scale from racemic indane 166 employing N-Boc-protected D-tert-leucine (N-Boc-D-Tle-OH) as a chiral ligand. With hydroxymethyl triflamide 168 in hand, the tetrahydroisoquinoline moiety was cyclized through Mitsunobu substitution in 90% yield. Subsequently, the triflyl group was removed by E1cb-type reaction with KHMDS, and an oxidative aromatization took place in situ under O2 atmosphere to afford isoquinoline 170. A trifloxy group was installed in 81% yield from methoxy for the final Stille–Migita cross-coupling with stannane synthon arisen from (+)-pulegone. Notably, within this route, each of the steps could be amplified to gram-scale preparation with delavatine A 172 obtained over 1 g, illustrating the robustness and convenience of enantioselective C–H activation in total synthesis.10c | ||
| Scheme 13 Zhang and Li's total synthesis of delavatine A. | ||
Dibenzocyclooctadiene represents another significant class of natural lignan skeletons, comprising an additional unique chiral C–C axis within the biaryl framework which is arduous to be settled.58,113 Guided by their persistent efforts in asymmetric synthesis of axially chiral compounds via atroposelective C–H activation,114 Shi and coworkers assumed that the Pd-catalyzed enantioselective C–H alkynylation facilitated by a chiral transient directing group (cTDG) would be a mature strategy115 for these cyclic axially chiral lignans,116 such as (+)-isoschizandrin 177 from Schisandra chinensis,117 (−)-steganone ent-182 from Steganotaenia araliacea118 and their analogues. With an imine formed in situ as the cTDG group, the transannular C(sp2)–H activation was conducted, and the atroposelectivity was modulated by the residue of amino acid L-tert-leucine (L-Tle-OH), generating axially chiral biaryls 174 and 179 in 2-gram and 3-gram scale respectively (Scheme 14a and b). Aldehyde 174 was transformed to dimethyl acetal for desilylation and homologation, and the Z-olefin 176, a key intermediate for (+)-isoschizandrin 177,119 was created in 1.35 g with 98% ee through stereoselective reduction by Ti(Oi-Pr)4 and i-PrMgCl.120 Similarly, the precursor to (+)-steganone 182 (ref. 121) was then yielded in 1.11 g with 96% ee from C(sp2)–H alkynylated product 179, avoiding the usage of stoichiometric sulfoxide as a chiral auxiliary in previous work via asymmetric C(sp2)–H alkenylation.122
 | ||
| Scheme 14 Shi's total (or formal) syntheses of dibenzocyclooctadiene and dihydrophenanthrene-9,10-diol natural products. | ||
Capitalizing on a negligibly adjusted Pd/cTDG strategy for the establishment of axially chiral biaryls, in 2019, Shi and colleagues realized the enantioselective total synthesis of TAN-1085 190 through axial-to-central chirality transfer (Scheme 14c),123 which is a trans-9,10-dihydrophenanthrene-9,10-diol-cored antibiotic produced by Streptomyces sp. S-11106.124 Prepared by Suzuki–Miyaura cross-coupling of aryl bromide 183 and pinacol boronate 184, the reactant 185 was atroposelectively alkenylated by n-butyl acrylate catalyzed by Pd(OAc)2 and L-Tle-OH, providing enantiopure biaryl 186 in 1.08 g with 99% ee. The C–C double bond was cleaved to form dialdehyde 186, followed by a SmI2-mediated radical cyclization with an in situ site-selective monobenzoylation of trans-9,10-diol to fabricate the fundamental scaffold of TAN-1085 190, which was eventually afforded after further glycosylation, oxidation and deprotection.
4.2 Co-catalyzed enantioselective C(sp2)–H annulation
Owing to its low toxicity, abundant availability, cost-effectiveness and unique reactivity, cobalt-catalyzed enantioselective C–H activation has attracted widespread attention in recent years.110a,b,125 With long-term interests in cobalt catalysis,126 the Shi group reported an elegant protocol for asymmetric total syntheses of chiral 1-substituted tetrahydroisoquinoline (THIQ) natural alkaloids in 2023 (Scheme 15).127 Combined with salicyloxazoline (Salox) L5, 1-(3,4-dimethylbenzyl)-1,2-dihydroisoquinoline 192 was obtained in 1.42 g with 98% ee via cobalt-catalyzed kinetic resolution (Scheme 15a). Enantiopure natural product (−)-norlaudanosine 195 was then synthesized from C–H annulated intermediate 192 undergoing a desilylation–hydrogenation–deacylation sequence seamlessly, which could be transformed to opium alkaloids (+)-laudanosine 196 and (−)-xylopinine 198via reductive amination and Mannich aminomethylation in yields of 76% and 81% respectively,128 as well as the morphinandienone alkaloid (+)-sebiferine 197 with further oxidative dearomatization/cyclization treated with the mixture of [bis(trifluoroacetoxy)iodo]benzene (PIFA), phosphotungstic acid (HPA) and BF3·OEt2 from (+)-laudanosine 196.129 For tetrahydroisoquinoline alkaloid (+)-cryptostyline II 202, a probe for dopamine receptor D1 isolated from Cryptostylis fulva,130 the authors carried out the desymmetrization of benzhydrylamine derivative 199via Co-catalyzed enantioselective C–H annulation with Salox L6, resulting in the enantioenriched product 200 in 1.52 g with 96% ee (Scheme 15b). Within 4 standard reactions similar to (+)-laudanosine 196, the desired natural product (+)-cryptostyline II 202 was generated in 99% ee. Meanwhile, this strategy was also manifested as a fruitful method for the construction of N-α-chiral bioactive molecules, involving (+)-solifenacin,131 (+)-FR115427 (ref. 132) and (+)-NPS R-568.133 | ||
| Scheme 15 Shi's total synthesis of chiral 1-substituted tetrahydroisoquinoline alkaloids. | ||
4.3 Pd-catalyzed enantioselective C(sp3)–H arylation
In view of the inert molecular orbital profile and lower acidity than C(sp2)–H bond for metalation, the asymmetric C(sp3)–H activation has been relatively less investigated and mainly limited to palladium catalysis,18f,134 while the majority of applications in total synthesis are diastereoselective transformations.135 Based on their intensive efforts on Pd(0) catalyzed enantioselective C(sp3)–H activation,136 Baudoin and collaborators accomplished the asymmetric total synthesis and structural revision of (+)-indidene C 212 (Scheme 16),137 a prenylated polyketide with anticancer properties isolated from Streblus indicus grown in South Asia.138 With the substrate 207 prepared through an SN2 reaction between substituted benzyl bromide 204 and dimethyl malonate derivative 206, the enantioselective C(sp3)–H intramolecular arylation was actualized by the cooperation with palladium and a bulky IBiox-type chiral N-heterocyclic carbene (NHC) ligand L7 revealed in their previous study,139 yielding cyclopentane compound (R)-208 with 98% ee. Then the corresponding diol 209 was obtained through a cascade of diastereoselective Krapcho decarboxylation, double Grignard addition and Pd/C-catalyzed hydrogenolysis. After a mild oxidation of the primary hydroxyl group with 2,2,6,6-tetramethylpiperidin-1-oxy (TEMPO) and (diacetoxyiodo)benzene (PIA), the lactone 210 was created in 70% yield, of which the absolute configuration was determined as (5S,9R) by vibrational circular dichroism (VCD) analysis.140 Followed by site-selective bromination, Suzuki–Miyaura cross-coupling and deprotection, (+)-indidene C 212 was ultimately generated within 13 steps. | ||
| Scheme 16 Baudoin's total synthesis of (+)-indidene C. | ||
5 Conclusion and outlook
In summary, with the flourishing methodologies of transition metal-catalyzed enantioselective C–H functionalization, a range of sophisticated natural products were synthesized from prochiral starting materials through streamlined and scaled-up procedures, facilitating the exploration of their potential bioactivities and applications as new pharmaceutical candidates and others. The HAA/radical relay strategy serves as an efficient approach for chiral alcohols and amines, especially for pyrrolidines bearing an α-C-stereocenter next to nitrogen atom, while the enantioselective metal–carbenoid insertion represents an alternative protocol for methylene C(sp3)–H functionalization within the aliphatic strain particularly in the absence of any heteroatoms. On the other side, those traditional asymmetric C–H activation methodologies undergoing an inner-sphere C–H metalation mechanism, significantly promote the construction of multifarious compounds incorporating chirality elements other than C-centrally chiral molecules. Remarkably, considering the unnecessity of pre-functionalization, these highly stereoselective transformations could be expanded to gram scale or even decagram scale with minimum waste, accelerating their future adoption in industrial manufacturing.Despite considerable progress, several challenges remain in applying stereoselective C–H functionalization to target-oriented synthesis. First, current methods are predominantly limited to C–C bond formations using a narrow set of well-established catalytic systems. The development of novel catalysis to directly forge C–N and C–O bonds—ubiquitous linkages in natural products—would therefore significantly broaden the scope and utility of this approach. Furthermore, these synthetic methodologies are frequently employed for chiral induction at an early stage rather than the establishment of key molecular scaffolds in complex natural products. There is a pressing need to apply these methods to access sophisticated biogenic molecules with multiple chiral elements, such as the glycopeptide antibiotic vancomycin, which features central, axial, and planar chirality.141 Finally, while asymmetric C–H functionalization offers clear benefits in step- and atom-economy, the complexity and cost of catalysts and ligands can hinder broader application. Simplifying catalytic systems and developing inexpensive, readily accessible chiral ligands are crucial for enabling more extensive and streamlined synthetic campaigns. In conclusion, we hope this review will inspire further innovations towards more efficient, sustainable, and simplified total syntheses of natural metabolites via enantioselective C–H functionalization, ultimately contributing to their practical application for human benefit.
Author contributions
B.-F. S. conceived the topic of this review. P.-F. Q. developed the conceptual framework and summarized the relevent literature. P.-F. Q. and B.-F. S. wrote the manuscript.Conflicts of interest
The authors declare no conflict of interest.Data availability
No primary research results, software or code have been included and no new data were generated or analyzed as part of this review.Acknowledgements
Financial support from the National Natural Science Foundation of China (9225630, 222525011, U25A20547, and U22A20388 for B.-F. S. and 223B2118 for P.-F. Q.) is gratefully acknowledged.Notes and references
- (a) K. C. Nicolaou and T. Montagnon, Molecules That Changed the World: A Brief History of the Art and Science of Synthesis and Its Impact on Society, John Wiley & Sons, Inc., New York, USA, 2008 Search PubMed; (b) S. Danishefsky, Nat. Prod. Rep., 2010, 27, 1114–1116 RSC; (c) L. Katz and R. H. Baltz, J. Ind. Microbiol. Biotechnol., 2016, 43, 155–176 CrossRef CAS PubMed; (d) X. Zhang, M. Jiang, N. Niu, Z. Chen, S. Li, S. Liu and J. Li, ChemSusChem, 2018, 11, 11–24 CrossRef PubMed; (e) D. J. Newman and G. M. Cragg, J. Nat. Prod., 2020, 83, 770–803 CrossRef CAS PubMed; (f) N. Echegaraya, N. Guzel, M. Kumar, M. Guzel, A. Hassoun and J. M. Lorenzo, Food Chem., 2023, 404, 134453 CrossRef PubMed; (g) P. Chunarkar-Patil, M. Kaleem, R. Mishra, S. Ray, A. Ahmad, D. Verma, S. Bhayye, R. Dubey, H. N. Singh and S. Kumar, Biomedicines, 2024, 12, 201 CrossRef CAS PubMed; (h) K. Lewis, R. E. Lee, H. Brötz-Oesterhelt, S. Hiller, M. V. Rodnina, T. Schneider, M. Weingarth and I. Wohlgemuth, Nature, 2024, 632, 39–49 CrossRef CAS PubMed.
- F. Wöhler, Ann. Phys. Chem., 1828, 88, 253–256 CrossRef.
- R. Robinson, J. Chem. Soc., Trans., 1917, 111, 762–768 RSC.
- R. Willstätter, Adv. Cycloaddit., 1901, 317, 204–265 Search PubMed.
- J. W. Medleya and M. Movassaghi, Chem. Commun., 2013, 49, 10775–10777 RSC.
- E. J. Corey and X.-M. Cheng, The Logic of Chemical Synthesis, John Wiley & Sons, Inc., New York, USA, 1989 Search PubMed.
- (a) W.-j. Chung and C. D. Vanderwal, Angew. Chem., Int. Ed., 2016, 55, 4396–4434 Search PubMed; (b) M. D. Kärkäs, J. A. Porco and C. R. J. Stephenson, Chem. Rev., 2016, 116, 9683–9747 CrossRef PubMed; (c) L. Li, Z. Chen, X. Zhang and Y. Jia, Chem. Rev., 2018, 118, 3752–3832 CrossRef CAS PubMed; (d) M. M. Heravi, V. Zadsirjan, P. Saedia and T. Momeni, RSC Adv., 2018, 8, 40061–40163 RSC; (e) S. Woo and R. A. Shenvi, Acc. Chem. Res., 2021, 54, 1157–1167 Search PubMed; (f) A. J. Frontier and P. P. Sinclair, Acc. Chem. Res., 2021, 54, 1817–1829 Search PubMed; (g) R. Parella, S. Jakkampudi and J. C.-G. Zhao, ChemistrySelect, 2021, 6, 2252–2280 CrossRef CAS; (h) S. P. Pitre and L. E. Overman, Chem. Rev., 2022, 122, 1717–1751 CrossRef CAS PubMed; (i) B. A. Wright and R. Sarpong, Nat. Rev. Chem., 2024, 8, 776–792 CrossRef CAS PubMed; (j) Y. Wang and J. Gui, Acc. Chem. Res., 2024, 57, 568–579 CrossRef CAS PubMed.
- (a) K. C. Nicolaou, J. S. Chen and S. M. Dalby, Bioorg. Med. Chem., 2009, 17, 2290–2303 Search PubMed; (b) P. A. Wender and B. L. Miller, Nature, 2009, 460, 197–201 Search PubMed.
- (a) M. C. Wani, H. L. Taylor, M. E. Wall, P. Coggon and A. T. McPhail, J. Am. Chem. Soc., 1971, 93, 2325–2327 CrossRef CAS PubMed; (b) P. B. Schiff, J. Fant and S. B. Horwitz, Nature, 1979, 277, 665–667 CrossRef CAS PubMed; (c) J. N. Denis, A. E. Greene, D. Guenard, F. Gueritte-Voegelein, L. Mangatal and P. Potier, J. Am. Chem. Soc., 1988, 110, 5917–5919 CrossRef CAS; (d) J. Yared and K. Tkaczuk, Drug Des., Dev. Ther., 2012, 6, 371–384 CAS; (e) L. Min, J.-C. Han, W. Zhang, C.-C. Gu, Y.-P. Zou and C.-C. Li, Chem. Rev., 2023, 123, 4934–4971 CrossRef CAS PubMed; (f) S. Zhang, T. Ye, Y. Liu, G. Hou, Q. Wang, F. Zhao, F. Li and Q. Meng, Molecules, 2023, 28, 7517 CrossRef CAS PubMed.
- (a) B. M. Trost, Science, 1991, 254, 1471–1477 Search PubMed; (b) T. Newhouse, P. S. Baran and R. W. Hoffmann, Chem. Soc. Rev., 2009, 38, 3010–3021 Search PubMed; (c) C. A. Kuttruff, M. D. Eastgate and P. S. Baran, Nat. Prod. Rep., 2014, 31, 419–432 RSC; (d) P.-Q. Huang, Z.-J. Yao and R. P. Hsung, Efficiency in Natural Product Total Synthesis, John Wiley & Sons, Inc., New York, USA, 2018 CrossRef; (e) D. S. Peters, C. R. Pitts, K. S. McClymont, T. P. Stratton, C. Bi and P. S. Baran, Acc. Chem. Res., 2021, 54, 605–617 Search PubMed; (f) Y. Hayashi, Acc. Chem. Res., 2021, 54, 1385–1398 Search PubMed.
- (a) K. Godula and D. Sames, Science, 2006, 312, 67–72 CrossRef CAS PubMed; (b) L. McMurray, F. O'Hara and M. J. Gaunt, Chem. Soc. Rev., 2011, 40, 1885–1898 RSC; (c) W. R. Gutekunst and P. S. Baran, Chem. Soc. Rev., 2011, 40, 1976–1991 RSC; (d) M. P. Doyle, M. Ratnikov and Y. Liu, Org. Biomol. Chem., 2011, 9, 4007–4016 RSC; (e) J. Yamaguchi, A. D. Yamaguchi and K. Itami, Angew. Chem., Int. Ed., 2012, 51, 8960–9009 Search PubMed; (f) D. Y.-K. Chen and S. W. Youn, Chem.–Eur. J., 2012, 18, 9452–9474 Search PubMed; (g) Y. Qiu and S. Gao, Nat. Prod. Rep., 2016, 33, 562–581 Search PubMed; (h) P. B. Brady and V. Bhat, Eur. J. Org Chem., 2017, 5179–5190 Search PubMed; (i) D. J. Abrams, P. A. Provencher and E. J. Sorensen, Chem. Soc. Rev., 2018, 47, 8925–8967 RSC; (j) S. K. Sinha, G. Zanoni and D. Maiti, Asian J. Org. Chem., 2018, 7, 1178–1192 CrossRef CAS; (k) F. Li, X. Zhang and H. Renata, Curr. Opin. Chem. Biol., 2019, 49, 25–32 CrossRef CAS PubMed; (l) O. Baudoin, Angew. Chem., Int. Ed., 2020, 59, 17798–17809 CrossRef CAS PubMed; (m) T. Dalton, T. Faber and F. Glorius, ACS Cent. Sci., 2021, 7, 245–261 CrossRef CAS PubMed; (n) N. Y. S. Lam, K. Wu and J.-Q. Yu, Angew. Chem., Int. Ed., 2021, 60, 15767–15790 CrossRef CAS PubMed; (o) S. K. Sinha, P. Ghosh, S. Jain, S. Maiti, S. A. Al-Thabati, A. A. Alshehri, M. Mokhtar and D. Maiti, Chem. Soc. Rev., 2023, 52, 7461–7503 RSC; (p) I. Bakanas, R. F. Lusi, S. Wiesler, J. H. Cooke and R. Sarpong, Nat. Rev. Chem., 2023, 7, 783–799 CrossRef CAS PubMed; (q) D. Chandra, T. Sharma, Sarthi, Sachin and U. Sharma, J. Catal., 2024, 439, 115756 CrossRef CAS.
- (a) A. W. Hofmann, Ber. Dtsch. Chem. Ges., 1883, 16, 558–560 CrossRef; (b) K. Löffler and C. Freytag, Ber. Dtsch. Chem. Ges., 1909, 42, 3427–3431 Search PubMed.
- K. Löffler and S. Kober, Ber. Dtsch. Chem. Ges., 1909, 42, 3431–3438 Search PubMed.
- (a) D. H. R. Barton and J. M. Beaton, J. Am. Chem. Soc., 1960, 82, 2641 CrossRef CAS; (b) D. H. R. Barton, J. M. Beaton, L. E. Geller and M. M. Pechet, J. Am. Chem. Soc., 1960, 82, 2640–2641 CrossRef CAS.
- R. M. Cory and F. R. McLaren, J. Chem. Soc., Chem. Commun., 1977, 587–588 RSC.
- R. B. Kelly, J. Zamecnik and B. A. Beckett, J. Chem. Soc. D, 1971, 479 RSC.
- B. M. Trost, S. A. Godleski and J. P. Genet, J. Am. Chem. Soc., 1978, 100, 3930–3931 CrossRef CAS.
- (a) H. M. L. Davies and R. E. J. Beckwith, Chem. Rev., 2003, 103, 2861–2903 CrossRef CAS PubMed; (b) R. Giri, B.-F. Shi, K. M. Engle, N. Maugel and J.-Q. Yu, Chem. Soc. Rev., 2009, 38, 3242–3272 RSC; (c) C. Zheng and S.-L. You, RSC Adv., 2014, 4, 6173–6214 RSC; (d) J. F. Hartwig, J. Am. Chem. Soc., 2016, 138, 2–24 CrossRef CAS PubMed; (e) C. G. Newton, S.-G. Wang, C. C. Oliveira and N. Cramer, Chem. Rev., 2017, 117, 8908–8976 CrossRef CAS PubMed; (f) T. G. Saint-Denis, R.-Y. Zhu, G. Chen, Q.-F. Wu and J.-Q. Yu, Science, 2018, 359, eaao4798 Search PubMed; (g) H. M. L. Davies and K. Liao, Nat. Rev. Chem., 2019, 3, 347–360 CrossRef CAS PubMed; (h) T. Yoshino, S. Satake and S. Matsunaga, Chem.–Eur. J., 2020, 26, 7346–7357 CrossRef CAS PubMed; (i) B.-B. Zhan, L. Jin and B.-F. Shi, Trends Chem., 2022, 4, 220–235 Search PubMed; (j) Q. Zhang, L.-S. Wu and B.-F. Shi, Chem, 2022, 8, 384–413 CrossRef CAS; (k) Z. Zhang, P. Chen and G. Liu, Chem. Soc. Rev., 2022, 51, 1640–1658 Search PubMed; (l) P.-F. Qian, J.-Y. Li, Y.-B. Zhou, T. Zhou and B.-F. Shi, SynOpen, 2023, 7, 466–485 CrossRef CAS; (m) W.-C. C. Lee and X. P. Zhang, Angew. Chem., Int. Ed., 2024, 63, e202320243 CrossRef CAS PubMed.
- (a) B. Meunier, S. P. de Visser and S. Shaik, Chem. Rev., 2004, 104, 3947–3980 CrossRef CAS PubMed; (b) P. R. Ortiz de Montellano, Chem. Rev., 2010, 110, 932–948 CrossRef CAS PubMed; (c) J. C. Lewis, P. S. Coelhob and F. H. Arnold, Chem. Soc. Rev., 2011, 40, 2003–2021 RSC.
- D. P. Curran, N. A. Porter and B. Giese, Stereochemistry of Radical Reactions: Concepts, Guidelines, and Synthetic Applications, Wiley-VCH, Weinheim, Germany, 1996 Search PubMed.
- (a) H. Yi, G. Zhang, H. Wang, Z. Huang, J. Wang, A. K. Singh and A. Lei, Chem. Rev., 2017, 117, 9016–9085 CrossRef CAS PubMed; (b) H.-M. Huang, M. H. Garduño-Castro, C. Morrill and D. J. Procter, Chem. Soc. Rev., 2019, 48, 4626–4638 RSC; (c) C. Zhang, Z.-L. Li, Q.-S. Gu and X.-Y. Liu, Nat. Commun., 2021, 12, 475 CrossRef CAS PubMed; (d) Y. Zhang, T. Zhang and S. Das, Chem, 2022, 8, 3175–3201 CrossRef CAS.
- T. Ohta, K. Uwai, R. Kikuchi, S. Nozoe, Y. Oshima, K. Sasaki and F. Yoshizaki, Tetrahedron, 1999, 55, 12087–12098 CrossRef CAS.
- E. F. L. J. Anet, M. H. Silk and S. Trippett, J. Chem. Soc., 1953, 309–322 RSC.
- K. Uwai and Y. Oshima, Tetrahedron, 1999, 55, 9469–9480 CrossRef CAS.
- (a) S. Takano, T. Sugihara and K. Ogasawara, Heterocycles, 1990, 31, 1721–1725 Search PubMed; (b) H. Suemune, T. Harabe and K. Sakai, Chem. Pharm. Bull., 1988, 36, 3632–3637 CrossRef CAS; (c) E. Hungerbühler and D. Seebach, Helv. Chim. Acta, 1981, 64, 687–702 Search PubMed.
- (a) M. S. Kharasch and G. Sosnovsky, J. Am. Chem. Soc., 1958, 80, 756 CrossRef CAS; (b) M. S. Kharasch, G. Sosnovsky and N. C. Yang, J. Am. Chem. Soc., 1959, 81, 5819–5824 CrossRef CAS; (c) S. Tang, H. Song and S. Yu, Org. Lett., 2024, 26, 10036–10040 CrossRef CAS PubMed.
- (a) W. Zhang, F. Wang, S. D. McCann, D. Wang, P. Chen, S. S. Stahl and G. Liu, Science, 2016, 353, 1014–1018 Search PubMed; (b) J. Li, Z. Zhang, L. Wu, W. Zhang, P. Chen, Z. Lin and G. Liu, Nature, 2019, 574, 516–521 CrossRef CAS PubMed; (c) F. Wang, P. Chen and G. Liu, Acc. Chem. Res., 2018, 51, 2036–2046 CrossRef CAS PubMed.
- H. Zhang, Y. Zhou, T. Yang, J. Wu, P. Chen, Z. Lin and G. Liu, Nat. Catal., 2025, 8, 58–66 CrossRef CAS.
- H. Baars, M. J. Classen and V. K. Aggarwal, Org. Lett., 2017, 19, 6008–6011 CrossRef CAS PubMed.
- (a) J. Zhang, S. L. Morris-Natschke, D. Ma, X.-F. Shang, C.-J. Yang, Y.-Q. Liu and K.-H. Lee, Med. Res. Rev., 2021, 41, 928–960 CrossRef CAS PubMed; (b) J. W. Daly and C. W. Myers, Science, 1967, 156, 970–973 CrossRef CAS PubMed; (c) J. R. Liddell, Nat. Prod. Rep., 1999, 16, 499–507 RSC.
- H. Zhang, C. Huang, X.-A. Yuan and S. Yu, J. Am. Chem. Soc., 2022, 144, 10958–10967 CrossRef CAS PubMed.
- (a) X. Cheng, H. Lu and Z. Lu, Nat. Commun., 2019, 10, 3549 CrossRef PubMed; (b) X. Shu, L. Huan, Q. Huang and H. Huo, J. Am. Chem. Soc., 2020, 142, 19058–19064 CrossRef CAS PubMed.
- J. Li, B. Cheng, X. Shu, Z. Xu, C. Li and H. Huo, Nat. Catal., 2024, 7, 889–899 Search PubMed.
- Z. Zhou, Y. Ke, R. Miao, F. Hu, X. Wang, Y. Ping, S. Xu and W. Kong, Nat. Chem., 2025, 17, 344–355 CrossRef CAS PubMed.
- (a) H. Yoda, H. Katoh, Y. Ujihara and K. Takabe, Tetrahedron Lett., 2001, 42, 2509–2512 CrossRef CAS; (b) S. H. Park, H. J. Kang, S. Ko, S. Park and S. Chang, Tetrahedron: Asymmetry, 2001, 12, 2621–2624 CrossRef CAS.
- (a) S. Bera and X. Hu, Angew. Chem., Int. Ed., 2019, 58, 13854–13859 CrossRef CAS PubMed; (b) Y. Zhang, B. Han and S. Zhu, Angew. Chem., Int. Ed., 2019, 58, 13860–13864 CrossRef CAS PubMed; (c) S. Bera, R. Mao and X. Hu, Nat. Chem., 2021, 13, 270–277 CrossRef CAS PubMed.
- W.-Y. Tan, Y. Lu, J.-F. Zhao, W. Chen and H. Zhang, Org. Lett., 2021, 23, 6648–6653 CrossRef CAS PubMed.
- (a) A. Sudau, W. Munch, J.-W. Bats and U. Nubbemyer, Eur. J. Org Chem., 2002, 3315–3325 CrossRef CAS; (b) L. E. Overman and A. S. Franklin, Chem. Rev., 1996, 96, 505–522 Search PubMed.
- (a) J. W. Daly, T. Tokuyama, T. Fujiwara, R. J. Highet and I. L. Karle, J. Am. Chem. Soc., 1980, 102, 830–836 CrossRef CAS; (b) J. W. Daly, S. I. Secunda, H. M. Garraffo, T. F. Spande, A. Wisnieski and J. F. Cover, Toxicon, 1994, 32, 657–663 CrossRef CAS PubMed.
- D. P. Becker, D. L. Flynn, A. E. Moormann, R. Nosal, C. I. Villamil, R. Loeffler, G. W. Gullikson, C. Moummi and D.-C. Yang, J. Med. Chem., 2006, 49, 1125–1139 CrossRef CAS PubMed.
- C. Bhat and S. G. Tilve, RSC Adv., 2014, 4, 5405–5452 RSC.
- E. Vitaku, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257–10274 CrossRef CAS PubMed.
- (a) M. S. Majik, D. Naik, C. Bhat, S. Tilve, S. Tilvi and L. D'Souza, Bioorg. Med. Chem. Lett., 2013, 23, 2353–2356 CrossRef CAS PubMed; (b) W. Yang, Y. Wang, J. Y. Roberge, Z. Ma, Y. Liu, R. M. Lawrence, D. P. Rotella, R. Seethala, J. H. M. Feyen and J. K. Dickson, Bioorg. Med. Chem. Lett., 2005, 15, 1225–1228 CrossRef CAS PubMed; (c) C. J. Maddocks, K. Ermanis and P. A. Clarke, Org. Lett., 2020, 22, 8116–8121 CrossRef CAS PubMed.
- A. Melhaoui, M. Mallea, A. Jossang and B. Bodo, Nat. Prod. Lett., 1993, 2, 237–242 CrossRef CAS.
- (a) U. Schmidt, P. Grafen, K. Altland and H. W. Goedde, Adv. Enzymol. Relat. Areas Mol. Biol., 1969, 32, 423–469 CAS; (b) H. Sigel, Angew. Chem., Int. Ed. Engl., 1982, 21, 389–400 CrossRef; (c) M. H. Brookes, B. T. Golding, D. A. Howes and A. T. Hudsonb, J. Chem. Soc., Chem. Commun., 1983, 1051–1053 RSC.
- (a) A. Baur, T. Harrer, M. Peukert, G. Jahn, J. R. Kalden and B. Fleckenstein, Klin. Wochenschr., 1991, 69, 722–724 CrossRef CAS PubMed; (b) L. Packer, H. J. Tritschler and K. Wessel, Free Radical Biol. Med., 1997, 22, 359–378 CrossRef CAS PubMed.
- (a) P. C. B. Page, C. M. Rayner and I. O. Sutherland, J. Chem. Soc., Chem. Commun., 1986, 1408–1409 RSC; (b) L. Dasaradhi, N. W. Fadnavis and U. T. Bhalerao, J. Chem. Soc., Chem. Commun., 1990, 729–730 RSC; (c) S. Zhang, X. Chen, J. Zhang, W. Wang and W. Duan, Synthesis, 2008, 3, 383–386 Search PubMed; (d) J.-Q. Wang, X. Ling, H.-J. Wang and F.-E. Chen, RSC Adv., 2023, 13, 36346–36363 RSC.
- S. Xu, Y. Ping, W. Li, H. Guo, Y. Su, Z. Li, M. Wang and W. Kong, J. Am. Chem. Soc., 2023, 145, 5231–5241 CrossRef CAS PubMed.
- J. S. Yadav, M. S. Reddy and A. R. Prasad, Tetrahedron Lett., 2006, 47, 4995–4998 CrossRef CAS.
- (a) E. K. Weibel, P. Hadvary, E. Hochuli, E. Kupfer and H. Lengsfeld, J. Antibiot., 1987, 40, 1081–1085 CrossRef CAS PubMed; (b) E. Hochuli, E. Kupfer, R. Maurer, W. Meister, Y. Mercadal and K. Schmidt, J. Antibiot., 1987, 40, 1086–1091 CrossRef CAS PubMed; (c) P. Barbier, F. Schneider and U. Widmer, Helv. Chim. Acta, 1987, 70, 196–202 CrossRef CAS.
- K. Liang, Q. Zhang and C. Guo, Nat. Synth., 2023, 2, 1184–1193 CrossRef CAS.
- (a) H. M. L. Davies and J. R. Manning, Nature, 2008, 451, 417–424 CrossRef CAS PubMed; (b) M. P. Doyle, R. Duffy, M. Ratnikov and L. Zhou, Chem. Rev., 2010, 110, 704–724 CrossRef CAS PubMed; (c) H. M. L. Davies and D. Morton, Chem. Soc. Rev., 2011, 40, 1857–1869 Search PubMed; (d) Y. He, Z. Huang, K. Wu, J. Ma, Y.-G. Zhou and Z. Yu, Chem. Soc. Rev., 2022, 51, 2759–2852 Search PubMed.
- (a) F. Collet, C. Lescot and P. Dauban, Chem. Soc. Rev., 2011, 40, 1926–1936 Search PubMed; (b) H. Hayashi and T. Uchida, Eur. J. Org Chem., 2020, 909–916 Search PubMed; (c) M. Ju and J. M. Schomaker, Nat. Rev. Chem., 2021, 5, 580–594 Search PubMed; (d) H.-H. Li, X. Chen and S. Kramer, Chem. Sci., 2023, 14, 13278–13289 RSC.
- (a) M.-Y. Teng, T. Han, E.-H. Huang and L.-W. Ye, Chin. J. Org. Chem., 2022, 42, 3295–3301 CrossRef CAS; (b) W.-C. C. Lee and X. P. Zhang, Angew. Chem., Int. Ed., 2024, 63, e202320243 CrossRef CAS PubMed.
- J. Egger and E. M. Carreira, Nat. Prod. Rep., 2014, 31, 449–455 RSC.
- (a) W. Kurosawa, T. Kan and T. Fukuyama, J. Am. Chem. Soc., 2003, 125, 8112–8113 CrossRef CAS PubMed; (b) Y. Koizumi, H. Kobayashi, T. Wakimoto, T. Furuta, T. Fukuyama and T. Kan, J. Am. Chem. Soc., 2008, 130, 16854–16855 CrossRef CAS PubMed; (c) D.-H. Wang and J.-Q. Yu, J. Am. Chem. Soc., 2011, 133, 5767–5769 CrossRef CAS PubMed; (d) P. Lu, A. Mailyan, Z. Gu, D. M. Guptill, H. Wang, H. M. L. Davies and A. Zakarian, J. Am. Chem. Soc., 2014, 136, 17738–17749 CrossRef CAS PubMed; (e) B. Hong, C. Li, Z. Wang, J. Chen, H. Li and X. Lei, J. Am. Chem. Soc., 2015, 137, 11946–11949 CrossRef CAS PubMed.
- (a) H. M. L. Davies and Q. Jin, Tetrahedron: Asymmetry, 2003, 14, 941–949 CrossRef CAS; (b) N. A. Falcone, A. T. Bosse, H. Park, J.-Q. Yu, H. M. L. Davies and E. J. Sorensen, Org. Lett., 2021, 23, 9393–9397 CrossRef CAS PubMed.
- (a) D. A. Whiting, Nat. Prod. Rep., 1985, 2, 191–211 Search PubMed; (b) D. A. Whiting, Nat. Prod. Rep., 1987, 4, 499–525 RSC; (c) K. D. R. Setchell and H. Adlercreutz, in Role of the Gut Flora Toxicity and Cancer Mammalian Lignans and Phytoestrogens: Recent Study on the Formation Metabolism and Biological Role in Health and Disease, ed. I. R. Rowland, Academic Press, London, UK, 1988, pp. 315–345 Search PubMed.
- (a) S. R. Stitch, J. K. Toumba, M. B. Groen, C. W. Funke, J. Leemhuis, J. Vink and G. F. Woods, Nature, 1980, 287, 738–740 CrossRef CAS PubMed; (b) M. Axelson, J. Sjövall, B. E. Gustafsson and K. D. R. Setchell, Nature, 1982, 298, 659–660 CrossRef CAS PubMed.
- M. B. Groen and J. Leemhuis, Tetrahedron Lett., 1980, 21, 5043–5046 CrossRef CAS.
- (a) M. Asaoka, N. Fujii, K. Shima and H. Takei, Chem. Lett., 1988, 17, 805–808 CrossRef; (b) P. Eklund, A. Lindholm, J.-P. Mikkola, A. Smeds, R. Lehtilä and R. Sjöholm, Org. Lett., 2003, 5, 491–493 CrossRef CAS PubMed; (c) M. Ghosh, Tetrahedron, 2007, 63, 11710–11715 CrossRef CAS.
- (a) M. P. Doyle, M. N. Protopopova, Q.-L. Zhou, J. W. Bode, S. H. Simonsen and V. Lynch, J. Org. Chem., 1995, 60, 6654–6655 CrossRef CAS; (b) J. W. Bode, M. P. Doyle, M. N. Protopopova and Q.-L. Zhou, J. Org. Chem., 1996, 61, 9146–9155 Search PubMed.
- (a) Y. Yoshiki und and T. Ishiguro, J. Pharm. Soc. Jpn., 1933, 53, 73–151 Search PubMed; (b) B. R. Prabhu and N. B. Mulchandani, Phytochemistry, 1985, 24, 329–331 CrossRef CAS; (c) S. H. Cavalcante, M. Yoshida and O. R. Gottlieb, Phytochemistry, 1985, 24, 1051–1055 CrossRef CAS.
- H. Suzuki, K. H. Lee, M. Haruna, T. Iida, K. Ito and H.-C. Huang, Phytochemistry, 1982, 21, 1824–1825 Search PubMed.
- (a) M. Kuhn and A. von Wartburg, Helv. Chim. Acta, 1967, 50, 1546–1565 Search PubMed; (b) F. Zavala, D. Guenard, J.-P. Robin and E. Brown, J. Med. Chem., 1980, 23, 546–549 Search PubMed.
- (a) E. Brown and A. Daugan, Heterocycles, 1987, 26, 1169–1172 CrossRef CAS; (b) E. Brown and A. Daugan, J. Nat. Prod., 1991, 54, 110–118 CrossRef.
- (a) T. H. Easterfield and J. Bee, J. Chem. Soc., Trans., 1910, 97, 1028–1032 RSC; (b) R. D. Haworth and T. Richardson, J. Chem. Soc., 1935, 633–636 Search PubMed.
- (a) R. D. Haworth and D. Woodcock, J. Chem. Soc., 1939, 1054–1057 RSC; (b) K. I. Lapteva, N. Y. Tyukavkina and L. I. Ryzhova, Chem. Nat. Compd., 1971, 7, 802–803 CrossRef; (c) J. D. Ford, K.-S. Huang, H.-B. Wang, L. B. Davin and N. G. Lewis, J. Nat. Prod., 2001, 64, 1388–1397 CrossRef CAS PubMed.
- (a) L. H. Briggs, R. C. Cambie and J. L. Hoare, Tetrehedron, 1959, 7, 262–269 CrossRef CAS; (b) R. G. Powell and R. D. Plattner, Phytochemistry, 1976, 15, 1963–1965 CrossRef CAS; (c) R. D. Plattner and R. G. Powell, Phytochemistry, 1978, 17, 149–150 CrossRef CAS.
- K. Weinges, Tetrahedron Lett., 1960, 1, 1–2 CrossRef.
- (a) M. Anada, O. Mita, H. Watanabe, S. Kitagaki and S. Hashimoto, Synlett, 1999, 11, 1775–1777 CrossRef; (b) W.-J. Liu, Z.-L. Chen, Z.-Y. Chen and W.-H. Hu, Tetrahedron: Asymmetry, 2005, 16, 1693–1698 CrossRef CAS.
- K. Matsumaga, M. Shibuya and Y. Ohizumim, J. Nat. Prod., 1995, 58, 138–139 CrossRef PubMed.
- M. P. Doyle, W. Hu and M. V. Valenzuela, J. Org. Chem., 2002, 67, 2954–2959 CrossRef CAS PubMed.
- (a) A. D. Rodríguez, Tetrahedron, 1995, 51, 4571–4618 CrossRef PubMed; (b) T. J. Heckrodt and J. Mulzer, Top. Curr. Chem., 2005, 244, 1–41 CrossRef CAS.
- S. A. Look and W. Fenical, Tetrahedron, 1987, 43, 3363–3370 CrossRef CAS.
- (a) W. Fenical, J. Nat. Prod., 1987, 50, 1001–1008 CrossRef CAS PubMed; (b) I. I. Rodríguez, Y.-P. Shi, O. J. García, A. D. Rodríguez, A. M. S. Mayer, J. A. Sánchez, E. Ortega-Barria and J. González, J. Nat. Prod., 2004, 67, 1672–1680 CrossRef PubMed; (c) A. M. S. Mayer, K. B. Glaser, C. Cuevas, R. S. Jacobs, W. Kem, R. D. Little, J. M. McIntosh, D. J. Newman, B. C. Potts and D. E. Shuster, Trends Pharmacol. Sci., 2010, 31, 255–265 CrossRef CAS PubMed; (d) W.-Y. Lu, H.-J. Li, Q.-Y. Li and Y.-C. Wu, Bioorg. Med. Chem., 2021, 35, 116058 CrossRef CAS PubMed.
- A. C. Coleman and R. G. Kerr, Tetrahedron, 2000, 56, 9569–9574 CrossRef CAS.
- (a) H. M. L. Davies and Q. Jin, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 5472–5475 CrossRef CAS PubMed; (b) H. M. L. Davies and Q. Jin, J. Am. Chem. Soc., 2004, 126, 10862–10863 CrossRef CAS PubMed.
- A. D. Rodríguez and C. Ramírez, J. Nat. Prod., 2001, 64, 100–102 CrossRef PubMed.
- H. M. L. Davies and A. M. Walji, Angew. Chem., Int. Ed., 2005, 44, 1733–1735 CrossRef CAS PubMed.
- A. D. Rodríguez and C. Ramírez, Org. Lett., 2000, 2, 507–510 CrossRef PubMed.
- A. D. Rodríguez, C. Ramírez, I. I. Rodríguez and C. L. Barnes, J. Org. Chem., 2000, 65, 1390–1398 CrossRef PubMed.
- (a) K. C. Nicolaou, G. Vassilikogiannakis, W. Mägerlein and R. Kranich, Angew. Chem., Int. Ed., 2001, 40, 2482–2486 CrossRef CAS; (b) K. C. Nicolaou, G. Vassilikogiannakis, W. Mägerlein and R. Kranich, Chem.–Eur. J., 2001, 7, 5359–5371 CrossRef CAS PubMed; (c) A. I. Kim and S. D. Rychnovsky, Angew. Chem., Int. Ed., 2003, 42, 1267–1270 CrossRef CAS PubMed; (d) D. C. Harrowven, D. D. Pascoe, D. Demurtas and H. O. Bourne, Angew. Chem., Int. Ed., 2005, 44, 1221–1222 CrossRef CAS PubMed; (e) A. A. Boezio, E. R. Jarvo, E. R. Lawrence and E. N. Jacobsen, Angew. Chem., Int. Ed., 2005, 44, 6046–6050 Search PubMed.
- H. M. L. Davies, X. Dai and M. S. Long, J. Am. Chem. Soc., 2006, 128, 2485–2490 CrossRef CAS PubMed.
- F. Dehmel and H.-G. Schmalz, Org. Lett., 2001, 3, 3579–3582 CrossRef CAS PubMed.
- A. Ata, R. G. Kerr, C. E. Moya and R. S. Jacobs, Tetrahedron, 2003, 59, 4215–4222 CrossRef CAS.
- A. D. Rodríguez and Y.-P. Shi, Tetrahedron, 2000, 56, 9015–9023 CrossRef.
- H. M. L. Davies and X. Dai, Tetrahedron, 2006, 62, 10477–10484 Search PubMed.
- T. A. Bedell, G. A. B. Hone, D. Valette, J.-Q. Yu, H. M. L. Davies and E. J. Sorensen, Angew. Chem., Int. Ed., 2016, 55, 8270–8274 CrossRef CAS PubMed.
- B. D. Bergstrom, A. T. Merrill, J. C. Fettinger, D. J. Tantillo and J. T. Shaw, Angew. Chem., Int. Ed., 2022, 61, e202203072 CrossRef CAS PubMed.
- J. Hashida, M. Niitsuma, M. Iwatsuki, M. Mori, A. Ishiyama, M. Namatame, A. Nishihara-Tsukashima, A. Matsumoto, I. Ara, Y. Takahashi, H. Yamada, K. Otoguro, K. Shiomi and S. Ōmura, J. Antibiot., 2012, 65, 197–202 Search PubMed.
- (a) W. Zhou, P. Posri, M. E. Abugrain, A. J. Weisberg, J. H. Chang and T. Mahmud, ACS Chem. Biol., 2020, 15, 3217–3226 CrossRef CAS PubMed; (b) W. Zhou, P. Posri, X.-J. Liu, Z. Ju, W.-J. Lan and T. Mahmud, J. Nat. Prod., 2021, 84, 2411–2419 CrossRef CAS PubMed.
- D. Dardić, N. Böhringer, A. Plaza, F. Zubeil, J. Pohl, S. Sommer, L. Padva, J. Becker, M. A. Patras, M.-K. Bill, M. Kurz, L. Toti, S. W. Görgens, S. M. M. Schuler, A. Billion, O. Schwengers, P. Wohlfart, A. Goesmann, N. Tennagels, A. Vilcinskas, P. E. Hammann, T. F. Schäberle and A. Bauer, Org. Chem. Front., 2022, 9, 1604–1615 RSC.
- B. S. Moore, J. L. Chen, G. M. L. Patterson, R. E. Moore, L. S. Brinen, Y. Kato and J. Clardy, J. Am. Chem. Soc., 1990, 112, 4061–4063 CrossRef CAS.
- D. Berthold and B. Breit, Synlett, 2021, 32, 436–446 CrossRef CAS.
- (a) L. Fu, J. D. Mighion, E. A. Voight and H. M. L. Davies, Chemistry, 2017, 23, 3272–3275 CrossRef CAS PubMed; (b) W. Liu, Z. Ren, A. T. Bosse, K. Liao, E. L. Goldstein, J. Bacsa, D. G. Musaev, B. M. Stoltz and H. M. L. Davies, J. Am. Chem. Soc., 2018, 140, 12247–12255 CrossRef CAS PubMed.
- A. T. Bosse, L. R. Hunt, C. A. Suarez, T. D. Casselman, E. L. Goldstein, A. C. Wright, H. Park, S. C. Virgil, J.-Q. Yu, B. M. Stoltz and H. M. L. Davies, Science, 2024, 386, 641–646 CrossRef CAS PubMed.
- C. Qin and H. M. L. Davies, J. Am. Chem. Soc., 2014, 136, 9792–9796 CrossRef CAS PubMed.
- (a) G. Li, L. Wan, G. Zhang, D. Leow, J. Spangler and J.-Q. Yu, J. Am. Chem. Soc., 2015, 137, 4391–4397 CrossRef CAS PubMed; (b) H. Park, N. Chekshin, P.-X. Shen and J.-Q. Yu, ACS Catal., 2018, 8, 9292–9297 CrossRef CAS PubMed.
- H. Choi, N. Engene, T. Byrum, S. Hwang, D.-C. Oh and W. H. Gerwick, Org. Lett., 2019, 21, 266–270 CrossRef CAS PubMed.
- T.-T. H. Nguyen, K. Shimabukuro, D. G. Musaev, J. Bacsa, A. Navarro and H. M. L. Davies, J. Am. Chem. Soc., 2025, 147, 28098–28106 CrossRef CAS PubMed.
- K. Sapkota and F. Huang, Synlett, 2019, 30, 1895–1898 CrossRef CAS.
- S. Senaweera, K. C. Cartwright and J. A. Tunge, J. Org. Chem., 2019, 84, 12553–12561 CrossRef CAS PubMed.
- (a) A. Gunay and K. H. Theopold, Chem. Rev., 2010, 110, 1060–1081 Search PubMed; (b) A. S. Borovik, Chem. Soc. Rev., 2011, 40, 1870–1874 Search PubMed; (c) K. P. Bryliakov, Coord. Chem. Rev., 2024, 508, 215793 CrossRef CAS; (d) A. Palone, M. Bietti and M. Costas, Synthesis, 2025, 57, 1280–1294 CrossRef CAS.
- A. T. Radosevich, C. Musich and F. D. Toste, J. Am. Chem. Soc., 2005, 127, 1090–1091 Search PubMed.
- A. T. Radosevich, V. S. Chan, H.-W. Shih and F. D. Toste, Angew. Chem., Int. Ed., 2008, 47, 3755–3758 CrossRef CAS PubMed.
- D. M. Tapiolas, M. Roman, W. Fenical, T. J. Stout and J. Clardy, J. Am. Chem. Soc., 1991, 113, 4682–4683 CrossRef.
- J. F. Hartwig, Organotransition Metal Chemistry: From Bonding to Catalysis, University Science Books, Sausalito, USA, 2010 Search PubMed.
- B.-F. Shi, N. Maugel, Y.-H. Zhang and J.-Q. Yu, Angew. Chem., Int. Ed., 2008, 47, 4882–4886 Search PubMed.
- (a) Ł. Wozniak and N. Cramer, Trends Chem., 2019, 1, 471–484 Search PubMed; (b) J. Loup, U. Dhawa, F. Pesciaioli, J. Wencel-Delord and L. Ackermann, Angew. Chem., Int. Ed., 2019, 58, 12803–12818 Search PubMed; (c) Q. Shao, K. Wu, Z. Zhuang, S. Qian and J.-Q. Yu, Acc. Chem. Res., 2020, 53, 833–851 Search PubMed; (d) T. K. Achar, S. Maiti, S. Jana and D. Maiti, ACS Catal., 2020, 10, 13748–13793 Search PubMed; (e) X. Yu, Z.-Z. Zhang, J.-L. Niu and B.-F. Shi, Org. Chem. Front., 2022, 9, 1458–1484 RSC; (f) Q. Yue, B. Liu, G. Liao and B.-F. Shi, ACS Catal., 2022, 12, 9359–9396 CrossRef CAS; (g) C.-X. Liu, S.-Y. Yin, F. Zhao, H. Yang, Z. Feng, Q. Gu and S.-L. You, Chem. Rev., 2023, 123, 10079–10134 Search PubMed; (h) K. Wu, N. Lam, D. A. Strassfeld, Z. Fan, J. X. Qiao, T. Liu, D. Stamos and J.-Q. Yu, Angew. Chem., Int. Ed., 2024, 63, e202400509 Search PubMed.
- Z. Zhang, F. Yang, J.-J. Fu, Y.-H. Shen, W. He and W.-D. Zhang, RSC Adv., 2016, 6, 65885–65888 Search PubMed.
- Z. Zhang, J. Wang, J. Li, F. Yang, G. Liu, W. Tang, W. He, J.-J. Fu, Y.-H. Shen, A. Li and W.-D. Zhang, J. Am. Chem. Soc., 2017, 139, 5558–5567 Search PubMed.
- J. Chang, J. Reiner and J. Xie, Chem. Rev., 2005, 105, 4581–4609 Search PubMed.
- (a) G. Liao, T. Zhou, Q.-J. Yao and B.-F. Shi, Chem. Commun., 2019, 55, 8514–8523 Search PubMed; (b) Y.-J. Wu, G. Liao and B.-F. Shi, Green Synth. Catal., 2022, 3, 117–136 Search PubMed; (c) P.-F. Qian, T. Zhou and B.-F. Shi, Chem. Commun., 2023, 59, 12669–12684 Search PubMed; (d) G. Liao and B.-F. Shi, Acc. Chem. Res., 2025, 58, 1562–1579 CrossRef CAS PubMed.
- (a) Q.-J. Yao, S. Zhang, B.-B. Zhan and B.-F. Shi, Angew. Chem., Int. Ed., 2017, 56, 6617–6621 Search PubMed; (b) G. Liao, T. Zhang, Z.-K. Lin and B.-F. Shi, Angew. Chem., Int. Ed., 2020, 59, 19773–19786 CrossRef CAS PubMed.
- G. Liao, Q.-J. Yao, Z.-Z. Zhang, Y.-J. Wu, D.-Y. Huang and B.-F. Shi, Angew. Chem., Int. Ed., 2018, 57, 3661–3665 CrossRef CAS PubMed.
- Y. Ikeya, H. Taguchi, H. Mitsuhashi, S. Takeda, Y. Kase and M. Aburada, Phytochemistry, 1988, 27, 569–573 Search PubMed.
- S. M. Kupchan, R. W. Britton, M. F. Ziegler, C. J. Gilmore, R. J. Restivo and R. F. Bryan, J. Am. Chem. Soc., 1973, 95, 1335–1336 Search PubMed.
- G. A. Molander, K. M. George and L. G. Monovich, J. Org. Chem., 2003, 68, 9533–9540 CrossRef CAS PubMed.
- (a) K. Harada, H. Urabe and F. Sato, Tetrahedron Lett., 1995, 36, 3203–3206 Search PubMed; (b) N. L. Hungerford and W. Kitching, Chem. Commun., 1996, 1697–1698 RSC.
- A. I. Meyers, J. R. Flisak and R. A. Aitken, J. Am. Chem. Soc., 1987, 109, 5446–5452 CrossRef CAS.
- Q. Dherbassy, J. Wencel-Delord and F. Colobert, Tetrahedron, 2016, 72, 5238–5245 CrossRef CAS.
- J. Fan, Q.-J. Yao, Y.-H. Liu, G. Liao, S. Zhang and B.-F. Shi, Org. Lett., 2019, 21, 3352–3356 CrossRef CAS PubMed.
- (a) T. Kanamaru, Y. Nozaki and M. Muroi, Kokai Tokkyo Koho, JP 02-289-532/1990, 1991Chem. Abstr. 1991, 115, 47759n Search PubMed; (b) K. Ohmori, K. Mori, Y. Ishikawa, H. Tsuruta and S. Kuwahara, Angew. Chem., Int. Ed., 2004, 43, 3167–3171 CrossRef CAS PubMed.
- (a) W. Xua and M. Ye, Synthesis, 2022, 54, 4773–4783 CrossRef; (b) Y. Zheng, C. Zheng, Q. Gu and S.-L. You, Chem Catal., 2022, 2, 2965–2985 CAS; (c) B. Garai, A. Das, D. V. Kumar and B. Sundararaju, Chem. Commun., 2024, 60, 3354–3369 RSC.
- (a) W.-K. Yuan and B.-F. Shi, Angew. Chem., Int. Ed., 2021, 60, 23187–23192 CrossRef CAS PubMed; (b) Y.-H. Liu, P.-P. Xie, L. Liu, J. Fan, Z.-Z. Zhang, X. Hong and B.-F. Shi, J. Am. Chem. Soc., 2021, 143, 19112–19120 CrossRef CAS PubMed; (c) Q.-J. Yao, J.-H. Chen, H. Song, F.-R. Huang and B.-F. Shi, Angew. Chem., Int. Ed., 2022, 61, e202202892 CrossRef CAS PubMed; (d) Q.-J. Yao and B.-F. Shi, Acc. Chem. Res., 2025, 58, 971–990 CrossRef CAS PubMed.
- Y.-J. Wu, J.-H. Chen, M.-Y. Teng, X. Li, T.-Y. Jiang, F.-R. Huang, Q.-J. Yao and B.-F. Shi, J. Am. Chem. Soc., 2023, 145, 24499–24505 CrossRef CAS PubMed.
- D. Mujahidin and S. Doye, Eur. J. Org Chem., 2005, 2689–2693 Search PubMed.
- H. Hamamoto, Y. Shiozaki, H. Nambu, K. Hata, H. Tohma and Y. Kita, Chem.–Eur. J., 2004, 10, 4977–4982 Search PubMed.
- (a) K. Leander, B. Luning and E. Ruusa, Acta Chem. Scand., 1969, 23, 244 CrossRef CAS; (b) A. Brossi and S. Teitel, Helv. Chim. Acta, 1971, 54, 1564–1571 Search PubMed; (c) D. L. Minor, S. D. Wyrick, P. S. Charifson, V. J. Watts, D. E. Nichols and D. E. Mailman, J. Med. Chem., 1994, 37, 4317–4328 CrossRef CAS PubMed.
- (a) R. Naito, Y. Yonetoku, Y. Okamoto, A. Toyoshima, K. Ikeda and M. Takeuchi, J. Med. Chem., 2005, 48, 6597–6606 Search PubMed; (b) S. Wang, M. B. Onaran and C. T. Seto, Org. Lett., 2010, 12, 2690–2693 Search PubMed; (c) Z.-S. Ye, R.-N. Guo, X.-F. Cai, M.-W. Chen, L. Shi and Y.-G. Zhou, Angew. Chem., Int. Ed., 2013, 52, 3685–3689 CrossRef CAS PubMed.
- (a) M. Ludwig, C. E. Hoesl, G. Höfner and K. T. Wanner, Eur. J. Med. Chem., 2006, 41, 1003–1010 Search PubMed; (b) X. Li and I. Coldham, J. Am. Chem. Soc., 2014, 136, 5551–5554 CrossRef CAS PubMed.
- (a) E. F. Nemeth, M. E. Steffey, L. G. Hammerland, B. C. P. Hung, B. C. Van Wagenen, E. G. DelMar and M. F. Balandrin, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 4040–4045 Search PubMed; (b) R. M. Barmore, S. R. Logan and B. C. Van Wagenen, Tetrahedron Lett., 1998, 39, 3451–3454 CrossRef CAS; (c) N. Yamazaki, M. Atobe and C. Kibayashi, Tetrahedron Lett., 2001, 42, 5029–5032 CrossRef CAS.
- (a) J. He, M. Wasa, K. S. L. Chan, Q. Shao and J.-Q. Yu, Chem. Rev., 2017, 117, 8754–8786 Search PubMed; (b) Q. Zhang and B.-F. Shi, Chin. J. Chem., 2019, 37, 647–656 CrossRef CAS; (c) P.-S. Wang and L.-Z. Gong, Acc. Chem. Res., 2020, 53, 2841–2854 CrossRef CAS PubMed; (d) Q. Zhang and B.-F. Shi, Acc. Chem. Res., 2021, 54, 2750–2763 CrossRef CAS PubMed; (e) B. Liu, A. M. Romine, C. Z. Rubel, K. M. Engle and B.-F. Shi, Chem. Rev., 2021, 121, 14957–15074 CrossRef CAS PubMed; (f) R. Kaur and N. Jain, Chem.–Asian J., 2022, 17, e202200944 CrossRef CAS PubMed; (g) P.-S. Wang and L.-Z. Gong, Chin. J. Chem., 2023, 41, 1841–1848 CrossRef CAS; (h) Y.-Q. Han and B.-F. Shi, Acta Chim. Sin., 2023, 81, 1522–1540 CrossRef CAS.
- (a) E. M. Stang and M. C. White, Nat. Chem., 2009, 1, 547–551 CrossRef CAS PubMed; (b) Y. Feng and G. Chen, Angew. Chem., Int. Ed., 2010, 49, 958–961 Search PubMed; (c) W. R. Gutekunst and P. S. Baran, J. Am. Chem. Soc., 2011, 133, 19076–19079 Search PubMed; (d) F. Frébault and N. Maulide, Angew. Chem., Int. Ed., 2012, 51, 2815–2817 CrossRef PubMed; (e) W. R. Gutekunst, R. Gianatassio and P. S. Baran, Angew. Chem., Int. Ed., 2012, 51, 7507–7510 Search PubMed; (f) C. P. Ting and T. J. Maimone, Angew. Chem., Int. Ed., 2014, 53, 3115–3119 CrossRef CAS PubMed; (g) D. H. O’ Donovan, P. Aillard, M. Berger, A. de la Torre, D. Petkova, C. Knittl-Frank, D. Geerdink, M. Kaiser and N. Maulide, Angew. Chem., Int. Ed., 2018, 57, 10737–10741 CrossRef; (h) R. Melot, M. V. Craveiro, T. Bürgi and O. Baudoin, Org. Lett., 2019, 21, 812–815 Search PubMed; (i) C. P. Ting, E. Tschanen, E. Jang and T. J. Maimone, Tetrahedron, 2019, 75, 3299–3308 Search PubMed; (j) S.-L. Fang, M.-X. Jiang, S. Zhang, Y.-J. Wu and B.-F. Shi, Org. Lett., 2019, 21, 4609–4613 Search PubMed; (k) W.-Y. Qi, S.-L. Fang, X.-T. Xu, K. Zhang and B.-F. Shi, Org. Chem. Front., 2021, 8, 1802–1807 Search PubMed; (l) S.-Q. Wang, W.-Y. Qi, X.-S. Yin and B.-F. Shi, Org. Chem. Front., 2021, 8, 3360–3365 RSC; (m) X.-S. Yin, W.-Y. Qi and B.-F. Shi, Chem. Sci., 2021, 12, 13137–13143 Search PubMed; (n) Q.-Z. Li, S.-H. Hou, J.-C. Kang, P.-F. Lian, Y. Hao, C. Chen, J. Zhou, T.-M. Ding and S.-Y. Zhang, Angew. Chem., Int. Ed., 2022, 61, e202207088 CrossRef CAS PubMed; (o) Y.-C. Lin, F. Schneider, K. J. Eberle, D. Chiodi, H. Nakamura, S. H. Reisberg, J. Chen, M. Saito and P. S. Baran, J. Am. Chem. Soc., 2022, 144, 14458–14462 Search PubMed.
- (a) O. Baudoin, Acc. Chem. Res., 2017, 50, 1114–1123 CrossRef CAS PubMed; (b) O. Vyhivskyi, A. Kudashev, T. Miyakoshi and O. Baudoin, Chem.–Eur. J., 2021, 27, 1231–1257 Search PubMed.
- A. Kudashev, S. Vergura, M. Zuccarello, T. Bürgi and O. Baudoin, Angew. Chem., Int. Ed., 2024, 63, e202316103 CrossRef CAS PubMed.
- R. He, X. Huang, Y. Zhang, L. Wu, H. Nie, D. Zhou, B. Liu, S. Deng, R. Yang, S. Huang, Z. Nong, J. Li and Y. Huang, J. Nat. Prod., 2016, 79, 2472–2478 Search PubMed.
- R. Melot, M. Zuccarello, D. Cavalli, N. Niggli, M. Devereux, T. Bürgi and O. Baudoin, Angew. Chem., Int. Ed., 2021, 60, 7245–7250 CrossRef CAS PubMed.
- C. Merten, T. P. Golub and N. M. Kreienborg, J. Org. Chem., 2019, 84, 8797–8814 Search PubMed.
- (a) C. M. Harris, H. Kopecka and T. M. Harris, J. Am. Chem. Soc., 1983, 105, 6915–6922 CrossRef CAS; (b) J. F. Levine, Med. Clin. North Am., 1987, 71, 1135–1145 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2026 |