
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Solvatomorphism of a 2,6-pyridyldicarboxamide-based foldamer†
Sena
Ozturk
a,
Alexander R.
Davis
a,
Colin C.
Seaton
 b,
Louise
Male
a and
Sarah J.
Pike
*a
b,
Louise
Male
a and
Sarah J.
Pike
*a
aSchool of Chemistry, University of Birmingham, Edgbaston, Birmingham, B15 2TT, UK. E-mail: s.j.pike@bham.ac.uk
bSchool of Chemistry and Biosciences, Faculty of Life Sciences, University of Bradford, Bradford, West Yorkshire, BD7 1DP, UK
First published on 21st March 2025
Abstract
A detailed solvatomorphism study conducted on a diamine-terminated 2,6-pyridyldicarboxamide-based foldamer 1 is reported. This investigation establishes the influence of a diverse range of polar and non-polar solvents including chloroform (1A), a trifluorotoluene/dichloromethane mixture (1A), dimethylformamide/diethyl ether (1B), tetrahydrofuran (1·THF), butanone (1·butanone), dichloromethane (1·DCM), a methanol/dichloromethane mixture (1·MeOH) and dimethylsulfoxide (1·DMSO) on the solid-state conformation and crystal packing behaviour of this supramolecular scaffold. Single-crystal X-ray diffraction analysis of the seven solvatomorphs of the studied foldamer (1A, 1B, 1·DCM, 1·THF, 1·butanone, 1·MeOH and 1·DMSO) identified that 1·DCM, 1·THF, 1·butanone, 1·MeOH and 1·DMSO form supramolecular aggregates (e.g., channels/cavities) which incorporate solvent molecules within the voids of the system, leading them to adopt channels of differing dimensions between 3.5 and 9.0 Å. Solid-state analysis identified that a diverse array of intermolecular non-covalent interactions form between the foldamer and the solvent molecule, including N–H⋯O, N–H⋯Cl, O–H⋯O, N–H⋯Cl and C–H⋯O hydrogen-bonding interactions, stabilising the formation of these solvent-mediated channel aggregates within the different solvatomorphs of the studied foldamer. We envisage that these solvatomorphism studies will facilitate the future design of foldamers, particularly given the emerging solid-state applications of foldamers which could hold relevance in the field of crystal engineering or for the uptake of small molecules for long-term use in energy storage and materials chemistry.
Introduction
Foldamers are synthetic oligomers that adopt stable conformations inspired by those found in natural systems (e.g., helices).1–3 Foldamers have garnered much attention due to their widespread applications in a diverse range of areas including synthesis as catalysts,4,5 supramolecular chemistry as sensors and probes6–8 and biological chemistry due to their potent anti-cancer, anti-fungal and anti-bacterial activities.9,10 Moreover, in recent years, foldamers have found an increasing number of applications in the solid state, including crystal engineering11 and materials science,12,13 for example, with reports of their molecular transport properties.14Solvatomorphism refers to the ability of a compound to exist in different solid-state forms. Solvatomorphs exhibit a number of crystalline states wherein their unit cells show distinct elemental compositions due to the incorporation of one or more solvent molecules.15 The propensity of different solvatomorphs of a compound to exhibit distinct physical properties (e.g., stability, bioavailability and solubility) can affect their activity.15 Consequently, this has led to them attracting great interest, particularly in pharmaceuticals,15 and more recently, in supramolecular chemistry.16–18
The role of solvent in controlling the behaviour of foldamers has been studied for a range of foldamer classes including aromatic oligoamides,19,20mPE21 and aminoisobutyric acid foldamers.22 However, many of these solvent-based studies conducted on foldamers focus on their behaviour in solution. Conversely, solid-state studies undertaken on foldamers are generally conducted to elucidate the nature of the intramolecular non-covalent interactions that govern the conformational behaviour23–25 and self-assembly26 of these supramolecular scaffolds without extensively exploring solvent effects in the solid state. The influence of solvent on the self-assembly of foldamers has been used to control the formation of vesicles and organogels.27 Solvent effects have also been used to control the adoption of topologically distinct structures within foldamers including regulating the interplay between linear structures and dimers28 and controlling the preference for dimerization between helices with opposite or identical handedness in stable head-to-tail dimers in different chlorinated solvents.29 Solvent effects can also play an important role in the formation of solvent channels and pores in the solid state in a diverse array of structures including metal–organic frameworks,30 carbon–organic frameworks,31 proteins32 and other materials.33 However, despite the importance of crystallographic studies into the solvatomorphism of foldamers, studies which systematically consider the role of solvent in the solid state are limited.34,35 This lack of data leaves this important area largely unexplored for these supramolecular scaffolds which, in turn, restricts the development of the field.
In 2016, Nissinen and co-workers highlighted the importance of solvatomorphism in controlling the conformational behaviour of foldamers, when they used single-crystal X-ray diffraction analysis to assess the influence of a range of solvents including polar protic (e.g., methanol and ethanol), polar non-protic (e.g., dimethylsulfoxide, tetrahydrofuran, dioxane, acetonitrile, dimethylacetamide, and ethyl acetate) and non-polar aprotic solvents (e.g., toluene) on the solid-state behaviour of a series of aromatic oligoamide foldamers.36 In this solvatomorphism study, it was identified that two general conformations of a protohelical @-conformation and a sigmoidal S-conformation were adopted for the studied aromatic oligoamide foldamers. In the protohelical @-conformation, the packing coefficients were generally slightly larger than in the sigmoidal S-conformation as it was established that the @-conformation lacked void spaces. Conversely, the sigmoidal S-conformation for these scaffolds exhibits a less compact crystal packing arrangement wherein for a selection of the studied aromatic oligoamide foldamers, solvent molecules (e.g., dimethylacetamide) occupy the void spaces within the structure.37 However, despite these reports and the growing solid-state applications of foldamers (e.g., in crystal engineering and materials science), studies into the solvatomorphism of these supramolecular scaffolds remain rare, and this lack of information limits the progression of the field.
Herein, we report on a solvatomorphism study of a diamine terminated 2,6-pyridyldicarboxamide-based foldamer 1 (see Fig. 1).38 The incorporation of amine functionalities at the termini of the foldamer offers the opportunity for these supramolecular scaffolds to interact with a diverse range of solvents (e.g., by forming hydrogen bonds with an appropriate solvent molecule). This detailed investigation of seven solvatomorphs of 1A, 1B, 1·DCM, 1·THF, 1·butanone, 1·MeOH and 1·DMSO identifies the influence of solvent on governing the conformational preferences and crystal packing arrangements of these supramolecular scaffolds. Furthermore, crystallographic analysis has identified the presence of a range of supramolecular aggregates in these solvatomorphs including channels and one-dimensional hydrogen-bonding chains. In-depth solid-state analysis has also established that the dimensions of the supramolecular channel aggregates within the solvatomorphs of the studied foldamer vary significantly (between 3.5 and 9.0 Å) as a function of the solvent used under the crystallisation conditions.
 | ||
| Fig. 1 Structure of the amine-terminated 2,6-pyridyldicarboxamide foldamer 1 whose solvatomorphism is investigated in this work using single-crystal X-ray diffraction analysis. | ||
Results and discussion
Synthesis and crystallographic conditions for foldamer 1
The amine-terminated foldamer 1 was synthesised using a linear five-step procedure in accordance with known literature procedures (see the ESI†).39,40 To determine the influence of solvent on the solid-state behaviour of 1, single crystals suitable for X-ray diffraction analysis were grown in a diverse array of solvents including chloroform, trifluorotoluene/dichloromethane, dimethylformamide, butanone, tetrahydrofuran, dichloromethane, methanol and dimethylsulfoxide. From this solvent screening, seven distinct solvatomorphs of the foldamer including 1A (from chloroform and trifluorotoluene/dichloromethane respectively), 1B (from dimethylformamide), 1·DCM (from dichloromethane), 1·THF (from tetrahydrofuran), 1·butanone (from butanone), 1·MeOH (from methanol) and 1·DMSO (from dimethylsulfoxide) were successfully accessed. Unfortunately, the additional tested solvents of acetone, ethanol, pyridine, 1,4-dioxane, dimethylformamide/diethyl ether, ethyl acetate, toluene, xylene, 2-decanone, 2-hexanone, 3-hexanone and 2-ethoxyethanol did not lead to crystals of sufficiently good quality for single-crystal X-ray diffraction analysis (see the ESI† for further details).Crystallographic analysis of foldamer solvatomorphs
Crystallographic analysis carried out on 1A, 1B, 1·DCM, 1·THF, 1·butanone, 1·MeOH and 1·DMSO allowed for the determination of the conformational preferences of the scaffolds in the solid state (i.e., the presence of a stable helical conformation) and the quantification of the helical pitch values of the foldamer scaffolds.41 Furthermore, solid-state analysis of these solvatomorphs has established the presence or absence of supramolecular aggregates, including channels/cavities and one-dimensional hydrogen-bonded chains, in the solid state. The influence of the solvent used under the crystallisation conditions on the nature and size of the supramolecular channel aggregates formed in the solid state has been determined through quantification of the channel cavity dimensions.42 Additionally, X-ray crystallographic analysis has identified the presence of a range of intramolecular and intermolecular non-covalent interactions including π–π stacking and the presence of N–H⋯O, N–H⋯Cl, O–H⋯O and C–H⋯O hydrogen-bonding interactions43 that support the crystal packing arrangements observed in these structures. Selected crystallographic data and details for 1A, 1B, 1·DCM, 1·THF, 1·butanone, 1·MeOH and 1·DMSO, including crystallisation solvent, compound name, whether the solvatomorph is a racemate or a conglomerate, and the crystal packing arrangement, are presented in Table 1.| Crystallisation | Compound | Racemate/conglomerate | Packing arrangementb |
|---|---|---|---|
| Solvent | |||
| a The following abbreviations have been used; DCM = dichloromethane, DMSO = dimethylsulfoxide, MeOH = methanol and THF = tetrahydrofuran. b MoloVol42 was used for crystallographic analysis of the solid state structures and to quantify the dimensions of the supramolecular aggregates (e.g., channels). c Disorder within the crystal structure led to two different channel dimensions being calculated, so an average of the structures was determined. d Diameters of the cavities were estimated using the van der Waals radii of atoms surrounding the depicted structure to calculate the distance as a cross section through the cavity in Olex2. | |||
| Chloroform | 1A | Racemate | No channel |
| Trifluorotoluene/dichloromethane | 1A | Racemate | No channel |
| Dimethylformamide/diethyl ether | 1B | Racemate | No channel |
| Tetrahydrofuran | 1·THF | Conglomerate | 7.8 Å channel |
| Butanone | 1·butanone | Conglomerate | 9.0 Å channelc |
| Dichloromethane | 1·DCM | Racemate | 4.3 Å channeld |
| Methanol | 1·MeOH | Racemate | 3.5 Å channeld |
| Dimethylsulfoxide | 1·DMSO | Conglomerate | 8.7 Å channelc |
General similarities in the solid-state behaviour of the foldamer solvatomorphs
In all of the crystal structures obtained for the foldamer (i.e., 1A, 1B, 1·DCM, 1·THF, 1·butanone, 1·MeOH and 1·DMSO), the scaffold adopts a helical conformation that is stabilised by the presence of hydrogen-bonding interactions formed between the N atom of the central pyridyl unit and the NH atoms of the adjacent amide bonds (see Fig. 2a and the ESI†).44 This intramolecular hydrogen-bonding motif enables the adoption of a syn–syn conformation that introduces curvature into the scaffold.45 Additionally, the incorporation of the azobenzene units, both of which adopt E-geometry, on either side of the central pyridyl unit further extends the helical conformation of 1 in all the studied foldamers (see Fig. 2a and the ESI†).46 | ||
| Fig. 2 X-ray structure of 1: (a) viewed from above showing the adoption of a helical conformation in the solid state stabilised by the presence of the intramolecular hydrogen-bonding interactions between the NH groups of the carboxamide units and the N atom of the central pyridine; (b) viewed side-on showing the presence of the columnar crystal packing arrangement composed of stacks of helical molecules of alternating screw-sense preferences. C atoms are shown in grey, O in red, N in light blue and H in white. H-bonding interactions are shown as dashed red lines. H atoms have been removed from (b) for clarity. | ||
For all the studied structures of 1A, 1B, 1·DCM, 1·THF, 1·butanone, 1·MeOH and 1·DMSO, comparable dimensions for the central 2,6-pyridyldicarboxamide cavity are observed (see the ESI†). As a consequence of the restricted size and shape of the central 2,6-pyridyldicarboxamide cavities in the studied foldamers, no solvent molecules are encapsulated into this central 2,6-pyridyldicarboxamide cavity under the studied crystallisation conditions (see Fig. 2 and the ESI†). Presumably, the encapsulation of solvent molecules within this central 2,6-pyridyldicarboxamide cavity of the foldamer is not favourable as it could disrupt the intramolecular hydrogen-bonding N–H⋯N⋯H–N motif that stabilises the syn–syn conformation of the scaffold. Further inspection of the crystal packing behaviour of 1A, 1B, 1·DCM, 1·THF, 1·butanone, 1·MeOH and 1·DMSO shows that for all the studied systems, the same columnar packing arrangement composed of stacks of foldamer molecules is observed (see Fig. 2b and the ESI†). This crystal packing arrangement is supported by a range of intermolecular non-covalent interactions including intermolecular offset, face-to-face π–π stacking interactions,43 and intermolecular N–H⋯O, N–H⋯Cl, O–H⋯O and C–H⋯O hydrogen-bonding interactions (see the ESI†).43
Detailed solid-state analysis of the foldamer solvatomorphs
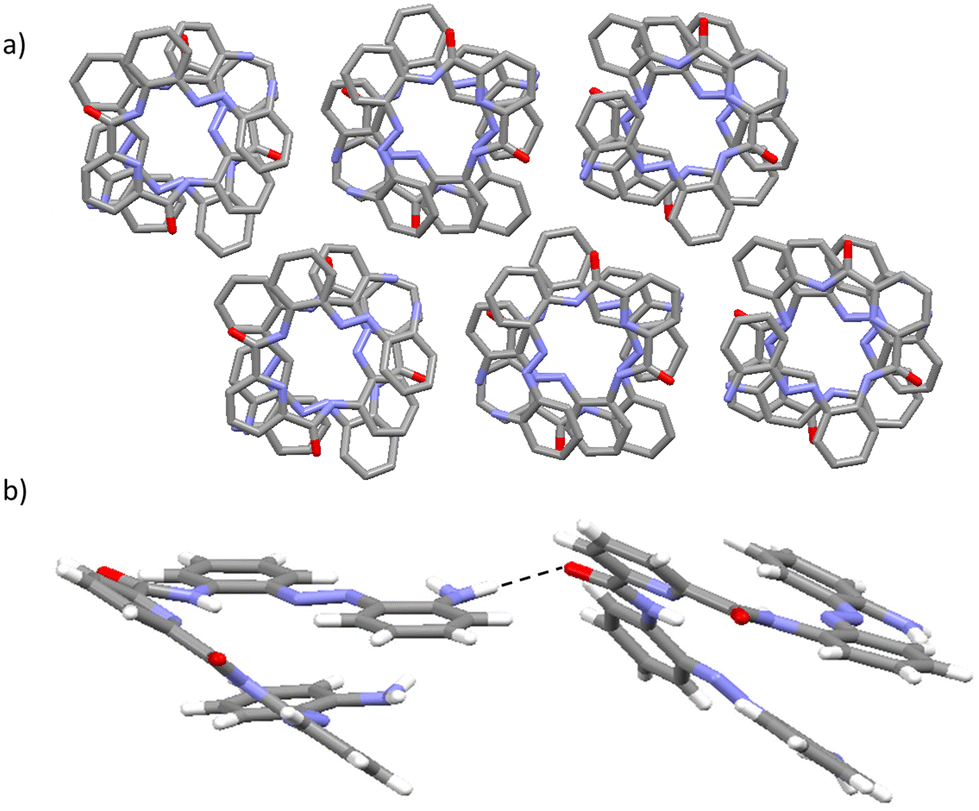 | ||
| Fig. 3 X-ray structure of 1A: (a) viewed along the a-axis highlighting the presence of supramolecular aggregates in the form of channels within the void spaces in the solid state; (b) highlighting the presence of an intermolecular N–H⋯O hydrogen-bonding interaction between foldamer molecules of opposite handedness in adjacent columnar stacks. C atoms are shown in grey, O in red, N in light blue and H in white. H-bonding interactions are shown as dashed red lines. H atoms have been removed from (a) for clarity. | ||
 | ||
| Fig. 4 X-ray structure of 1·THF: (a) viewed along the b-axis highlighting the presence of the tetrahydrofuran solvent molecules within the void spaces of the crystal packing arrangement of the structure; (b) showing the presence of intermolecular N–H⋯O and hydrogen-bonding interactions formed between the foldamer and a tetrahydrofuran solvent molecule which help stabilise the incorporation of tetrahydrofuran solvent molecules into the channel cavities within the structure; (c) showing the calculation of the channel cavity dimensions using MoloVol.42 C atoms are shown in grey, O in red, N in light blue and H in white. H-bonding interactions are shown as dashed red lines. H atoms have been removed from (a) for clarity. The disorder about the THF solvent molecules has been removed for clarity. | ||
In the crystal packing arrangement of 1·THF, columnar stacks composed of molecules of only P-helicity are observed. Hence, the crystal packing of 1·THF differs from that observed in 1A and 1B which exhibit a columnar arrangement composed of alternating molecules of opposite helical handedness within the same stack due to the differing adoption of conglomerates (in 1·THF) and racemates (in 1A and 1B). Within the crystal packing arrangement of 1·THF, the presence of channels is observed wherein disordered THF molecules reside within the void spaces of these supramolecular aggregates. Due to the disorder about the THF solvent molecules, it is not possible to comment on their relative arrangement within the channel's cavity (see the ESI†). The channels in 1·THF exhibit a diameter of 7.8 Å (see Table 1 and Fig. 4c). The encapsulation of the THF molecules within the void spaces of the channels in 1·THF is stabilised by the formation of intermolecular hydrogen-bonding interactions between the foldamer and solvent molecules. Specifically, there is an intermolecular N–H⋯O hydrogen-bonding interaction44 between the NH atom of one of the terminal amine functionalities and the O atom of the THF solvent molecule (see Fig. 4b).
 | ||
| Fig. 5 X-ray structure of 1·butanone: (a) viewed along the b-axis highlighting the presence of the butanone solvent molecules occupying spaces within the channel crystal packing arrangement of the structure; (b) showing the presence of intermolecular N–H⋯O and hydrogen-bonding interactions formed between the foldamer and a butanone solvent molecule which help stabilise the incorporation of butanone solvent molecules into the channel cavities within the structure. C atoms are shown in grey, O in red, N in light blue and H in white. H-bonding interactions are shown as dashed red lines. H atoms have been removed from (a) for clarity. | ||
In an analogous manner to 1·THF, the butanone-containing solvatomorph of 1 exhibits a crystal packing arrangement wherein columnar stacks are composed of foldamer molecules adopting only one helical screw sense (e.g., only P-helicity). Furthermore, as with 1·THF, butanone solvent molecules reside within the void spaces of the channel aggregates that are formed within this structure. These channels have a diameter of 9.0 Å and the observed increase in dimensions compared to 1·THF (cf. 7.8 Å for 1·THF vs. 9.0 Å for 1·butanone, see Table 1) is likely a consequence of the differing sizes, shapes and/or orientations of the THF and butanone solvent molecules within the channels. Unlike in 1·THF, there is no disorder about the solvent molecules in 1·butanone so it is possible to identify the relative head-to-tail orientation of the solvent molecules within the channel cavities (see Fig. 5a). The presence of the butanone solvent molecules within the channels of 1·butanone is stabilised by the formation of an intermolecular hydrogen-bonding interaction between the foldamer and solvent molecules for the majority component. Hence, as in 1·THF, there is an intermolecular N–H⋯O hydrogen-bonding interaction involving the NH atom of one of the terminal amine functionalities of the foldamer and the O atom of the solvent molecule which stabilises the observed crystal packing arrangement (see Fig. 5b).49
 | ||
| Fig. 6 X-ray structure of 1·DCM: (a) viewed along the c-axis highlighting the presence of the dichloromethane solvent molecules within the void spaces of the crystal packing arrangement of the structure; (b) showing the presence of intermolecular N–H⋯Cl and aliphatic C–H⋯O hydrogen-bonding interactions between the foldamer and the dichloromethane solvent molecules which help stabilise their incorporation into the channel cavities within the structure. C atoms are shown in grey, O in red, N in light blue, Cl in light green and H in white. H-bonding interactions are shown as dashed red lines. H atoms have been removed from (a) for clarity. | ||
The crystal packing arrangement of 1·DCM is stabilised by the presence of two types of intermolecular hydrogen bonding interactions formed between one dichloromethane solvent molecule and two foldamers of opposite helical screw-sense preferences in adjacent columnar stacks (see Fig. 6b). First, an intermolecular N–H⋯Cl hydrogen-bonding interaction50 is observed between an NH on a terminal amine group of a foldamer molecule and the Cl atom of a dichloromethane solvent molecule. Second, an aliphatic C–H⋯O interaction51 is present between an H atom on the same dichloromethane solvent molecule and an O atom of the carbonyl bond in a second foldamer molecule in a neighbouring columnar stack.
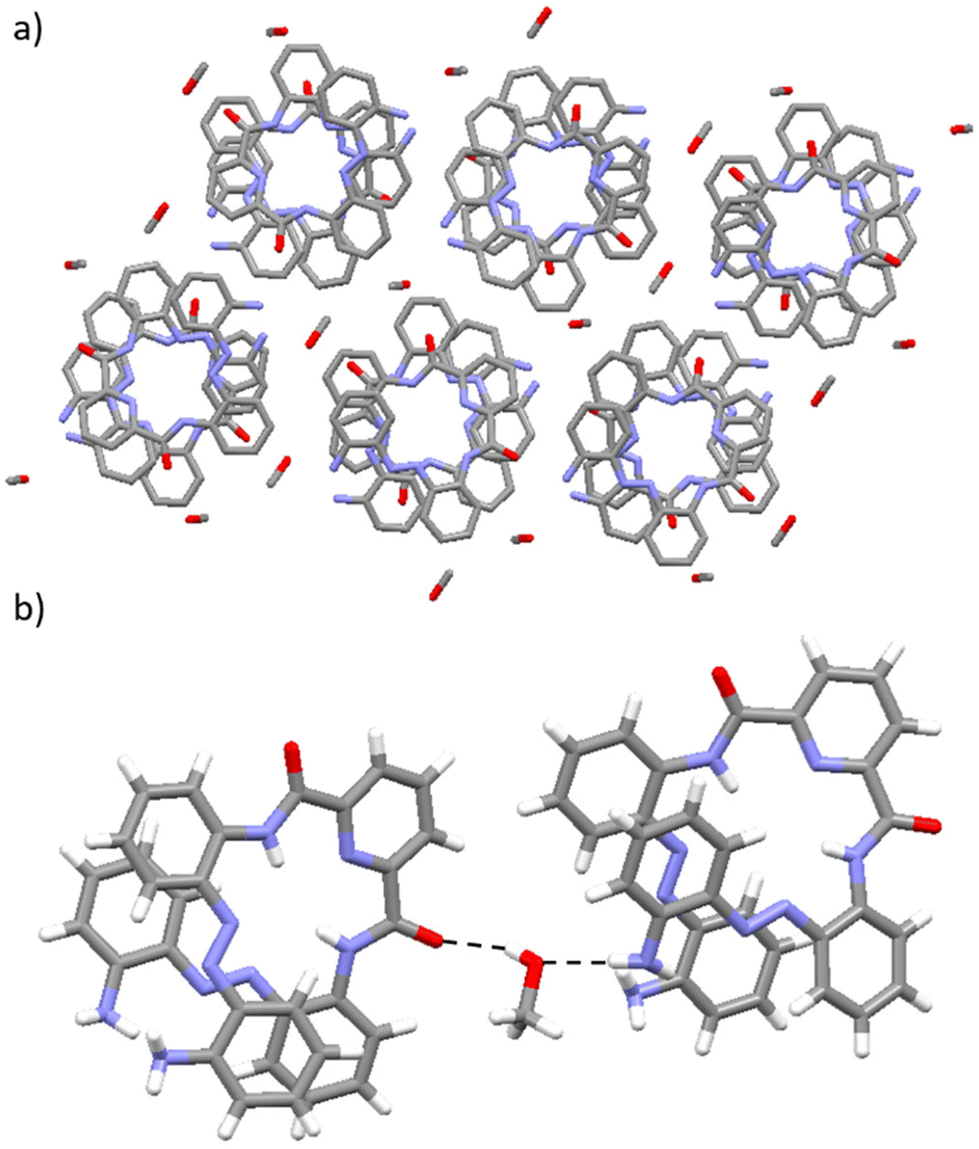 | ||
| Fig. 7 X-ray structure of 1·MeOH: (a) highlighting the presence of the methanol solvent molecules within the void spaces of the crystal packing arrangement of the structure; (b) showing the presence of intermolecular N–H⋯O and O–H⋯O hydrogen-bonding interactions between the foldamer and the methanol solvent molecules which stabilise their incorporation into the channel cavities within the structure. C atoms are shown in grey, O in red, N in light blue and H in white. H-bonding interactions are shown as dashed red lines. H atoms have been removed from (a) for clarity. | ||
In the crystal packing arrangement of 1·MeOH, columnar stacks composed of M- and P-helical foldamers are observed, which is in line with the structures exhibited by the other identified racemates of 1A, 1B and 1·DCM. 1·MeOH forms cavities within the solid state with a diameter of 3.5 Å (see Table 1). These cavities are considerably smaller than those observed for 1·THF and 1·butanone of 7.8 Å and 9.0 Å respectively and slightly smaller than those observed in 1·DCM of 4.3 Å. In an analogous manner to 1·THF, 1·butanone, 1·CH2Cl2 and 1·DMSO, the solvent molecules in 1·MeOH are encapsulated within the channel cavities of the structure. However, it is likely that the differing sizes, shapes and/or orientations of the distinct solvent molecules within these solvatomorphs (of 1·THF, 1·butanone, 1·CH2Cl2 and 1·MeOH) and their varying ability to form a range of non-covalent interactions with the foldamer molecule, to stabilise their encapsulation, strongly influence the dimensions of the channels and cavities adopted within the solid state.
As with 1·DCM, the methanol-containing solvatomorph exhibits a crystal packing arrangement where each foldamer molecule is oriented closely to six solvent molecules (see Fig. 7a). The crystal packing arrangement in 1·MeOH is stabilised through two distinct types of hydrogen-bonding interactions formed between the methanol solvent molecule and the foldamer. First, intermolecular N–H⋯O hydrogen-bonding interactions are present between an amine functionality at the termini of the foldamer and the O atom within the methanol solvent molecule (see Fig. 7b). Second, intermolecular O–H⋯O hydrogen-bonding interactions are formed between the hydroxyl group of a methanol solvent molecule and the O atom of the carbonyl group in the foldamer (see Fig. 7b).49
 | ||
| Fig. 8 X-ray structure of 1·DMSO: (a) viewed along the b-axis highlighting the presence of the DMSO solvent molecules within the void spaces of the crystal packing arrangement of the structure; (b) showing the presence of intermolecular C–H⋯O hydrogen-bonding interactions between DMSO solvent molecules, which form the basis of a one-dimensional hydrogen-bonding chain. C atoms are shown in grey, O in red, N in light blue, S atoms in yellow and H in white. H-bonding interactions are shown as dashed red lines. H atoms have been removed from (a) for clarity. The disorder about the DMSO solvent molecules and the aromatic ring and amine functionality of the foldamer have been removed for clarity. | ||
The crystal packing arrangement observed in 1·DMSO involves the adoption of columnar stacks of foldamer molecules exhibiting only M-helicity (see Fig. 8a). This is in contrast to the other studied solvatomorphs of the foldamer which exhibit columnar stacks of alternating M- and P-helical foldamers as they are racemates (e.g., 1A, 1B and 1·DCM). In an analogous manner to 1·THF and 1·butanone, solvent molecules are incorporated into the cavities of the channels formed within the columnar stack crystal packing arrangement of 1·DMSO. Furthermore, MoloVol42 analysis of 1·DMSO shows that this structure exhibits a channel packing arrangement with a diameter of 8.7 Å (see Table 1). This is considerably larger than those observed in 1·DCM and 1·MeOH but slightly larger than those seen in 1·THF (cf. 7.8 Å for 1·THF vs. 8.7 Å for 1·DMSO, see Table 1). The varying diameters of the identified channels in the different solvatomorphs are likely due to varying sizes and shapes of the solvent molecules that are incorporated within the channels and/or their relative orientations within the supramolecular channel aggregates.
The presence of the DMSO solvent molecules within 1·DMSO is stabilised by intermolecular C–H⋯O hydrogen-bonding interactions that are formed between the aliphatic protons of the DMSO solvent molecule and the O atom of an adjacent DMSO solvent molecule (see Fig. 8b). These intermolecular non-covalent interactions formed between DMSO solvent molecules lead to the formation of a one-dimensional C–H⋯O hydrogen-bonded chain in the solid state wherein the DMSO solvent molecules adopt a head-to-tail geometry within the structure. No intermolecular hydrogen-bonding interactions are observed which involve the amine functionality in the foldamer and solvent molecule. Instead, the NH atoms in the terminal amine functionalities of the foldamers of 1·DMSO form an intermolecular N–H⋯O hydrogen-bonding interaction with the O atom of the amide bond in the central carboxamide moiety of an adjacent foldamer molecule (see the ESI†).
Conclusions
The solvatomorphism of a diamine-terminated 2,6-pyridyldicarboxamide-based foldamer 1 has been investigated in a range of solvents through the solid-state analysis of a range of solvatomorphs including 1A, 1B, 1·DCM, 1·THF, 1·butanone, 1·MeOH and 1·DMSO. Single-crystal X-ray diffraction analysis has been used to establish the influence of the nature of the solvent on the conformational behaviour and crystal packing arrangements adopted in these supramolecular systems. 1A, 1B, 1·DCM and 1·MeOH have been shown to exist as racemates in the solid state whilst 1·THF, 1·butanone and 1·DMSO form conglomerates. These conglomerates could be a useful source of enantio-enriched solid foldamers in the future.47All of the studied solvatomorphs, of 1A, 1B, 1·DCM, 1·THF, 1·butanone, 1·MeOH and 1·DMSO, adopt a stable helical conformation, which establishes that the nature of the solvent does not perturb the secondary structure of these supramolecular scaffolds in the solid state. Notably, the solvent-free solvatomorphs of 1 (i.e., 1A, 1B) do not exhibit channels whilst those incorporating solvents into the solid-state structure (i.e., 1·DCM, 1·THF, 1·butanone, 1·MeOH and 1·DMSO) adopt supramolecular aggregates of channels. Additionally, in the case of 1·DMSO, a solvent-based one-dimensional hydrogen-bonded chain is observed.
Crystallographic analysis shows that the dimensions of the channels found in 1·DCM, 1·THF, 1·butanone, 1·MeOH and 1·DMSO vary significantly in diameter between 3.5 and 9.0 Å. These observed variations in the cavity sizes within the crystal packing arrangements of the studied solvatomorphs of 1, 1·DCM, 1·THF, 1·butanone, 1·MeOH and 1·DMSO are likely due to their ability to encapsulate the appropriate solvents, which have differing sizes, shapes and orientations, within the channel cavities (i.e., dichloromethane for 1·DCM, tetrahydrofuran for 1·THF, butanone for 1·butanone, methanol for 1·MeOH and dimethylsulfoxide for 1·DMSO). Finally, it has been established that the incorporation of the different solvents into the channel cavities of the distinct solvatomorphs of the studied foldamers is stabilised by the presence of a diverse array of intermolecular hydrogen-bonding interactions, including N–H⋯O, N–H⋯Cl, O–H⋯O, N–H⋯Cl and C–H⋯O, formed between the foldamer and/or solvent molecule(s).
Given the relevance of systematic solid-state studies in improving our understanding of the fundamental features that govern the conformational behaviour of foldamers and the underexplored role of solvatomorphism in these supramolecular scaffolds, we anticipate that this work will help enable the future design of new foldamers and optimise the performance of existing ones. This is particularly relevant to the emerging solid-state applications of foldamers (e.g., in crystal engineering) as well as their potential long-term applications (e.g., in biological chemistry). Additionally, supramolecular scaffolds that can form solvent-mediated 1D networks are the subject of great interest due to their potential applications in the uptake and storage of small molecules for potential uses in energy and materials chemistry.
Data availability
The data supporting this article including crystallisation screening conditions and crystallographic analysis have been included as part of the ESI.† Crystallographic data for 1A, 1B, 1·THF, 1·butanone, 1·MeOH and 1·DMSO have been deposited at the CCDC and can be found in CIF and other electronic formats with the following CCDC numbering; 1A: 2426430, 1A′: 2426870, 1B: 2426871, 1·THF: 2426432, 1·butanone: 2426872, 1·DCM: 2426428, 1·MeOH: 2426873, 1·DMSO: 2426874.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the UKRI Future Leaders Fellowship [MR/S035486/2], the Royal Society of Chemistry Research Grant [RF19-4013], the Royal Society Research Grant [RGS/R1/231242] and the Leverhulme Trust Research Project Grant [RPG-2022-118] for financial support. SJP also acknowledges the University of Birmingham for financial support through the University of Birmingham Fellowship Scheme and the Talent Stabilisation Fund. We acknowledge Diamond Light Source for providing time on Beamline I19 under Proposal CY28766 and CY36069. We acknowledge Katherine Deverson for undertaking preliminary studies. SJP is a Birmingham Fellow and a UKRI Future Leaders Fellow.References
- (a) H. Juwarker, J.-M. Suk and K.-S. Jeong, Chem. Soc. Rev., 2009, 38, 3316–3325 RSC; (b) D.-W. Zhang, X. Zhao, J.-L. Hou and Z. T. Li, Chem. Rev., 2012, 112, 5271–5316 CrossRef CAS PubMed.
- (a) D. J. Hill, M. J. Mio, R. B. Prince, T. S. Hughes and J. S. Moore, Chem. Rev., 2001, 101, 3893 CrossRef CAS PubMed; (b) I. Saraogi and A. D. Hamilton, Chem. Soc. Rev., 2009, 38, 1726 RSC; (c) T. A. Martinek and F. Fülöp, Chem. Soc. Rev., 2012, 41, 687 RSC.
- (a) G. Guichard and I. Huc, Chem. Commun., 2011, 47, 5933 RSC; (b) B. A. F. Le Bailley and J. Clayden, Chem. Commun., 2016, 52, 4852 Search PubMed.
- (a) Z. C. Girvin and S. H. Gellmann, J. Am. Chem. Soc., 2020, 142, 17211 CAS; (b) D. Bécart, V. Diemer, A. Salaün, M. Oiarbide, Y. R. Nelli, B. Kauffmann, L. Fishcer, C. Palomo and G. Guichard, J. Am. Chem. Soc., 2017, 139, 12524 Search PubMed; (c) C. Ma, J. Tang, L. Yu, K. Wen and Q. Gan, Chem. – Eur. J., 2022, 28, e202200834 CAS.
- Q. Lin, C. Ma, R. T. Stendall, K. Shankland, R. A. Musgrave, P. N. Horton, C. Baldauf, H.-J. Hofmann, C. P. Butts, M. M. Müller and A. J. A. Cobb, Angew. Chem., Int. Ed., 2023, 62, e202305326 CAS.
- H. Juwarker and K.-S. Jeong, Chem. Soc. Rev., 2010, 39, 3664 CAS.
- (a) D.-W. Zhang, X. Zhao and Z.-T. Li, Acc. Chem. Res., 2014, 47, 1961 CAS; (b) J. Shen, C. Ren and H. Zeng, J. Am. Chem. Soc., 2017, 139, 3587 Search PubMed.
- (a) Y. Zhao and Z. Zeng, J. Am. Chem. Soc., 2006, 128, 9988 CAS; (b) H. Li, L. Liang, B. Ku, W. Zhao, X.-J. Yang and B. Wu, Chem. Sci., 2022, 13, 4915 CAS.
- (a) M. Pasco, C. Dolain and G. Guichard, Comprehensive Supramolecular Chemistry II, Elsevier, 2017, p. 89 Search PubMed; (b) D. Deepak, J. Wu, V. Corvaglia, L. Allmendinger, M. Schechenbach, P. Tinnefeld and I. Huc, Angew. Chem., Int. Ed., 2024, e202422958 Search PubMed.
- (a) A. D. Peters, S. Borsley, F. della Sala, D. F. Cairns-Gibson, M. Leonidou, J. Clayden, G. F. S. Whitehead, I. J. Vitórica-Yrezábal, E. Takano, J. Burthem, S. L. Cockroft and S. J. Webb, Chem. Sci., 2020, 11, 7023 CAS; (b) R. Gopalakrishnan, A. I. Frolov, L. Knerr, W. J. Drury III and E. Valeur, J. Med. Chem., 2016, 59, 9599 CAS.
- (a) G. W. Collie, K. Pulka-Ziach and G. Guichard, Chem. Sci., 2016, 7, 3377 CAS; (b) V. Pavone, S.-Q. Zhang, A. Merlino, A. Lombardi, Y. Wu and W. F. DeGrado, Nat. Commun., 2014, 5, 3581 CrossRef PubMed; (c) L. A. Estroff, C. D. Incarvito and A. D. Hamilton, J. Am. Chem. Soc., 2003, 126, 2 CrossRef PubMed; (d) X. Hu, S. J. Dawson, P. K. Madal, X. de Hatten, B. Baptiste and I. Huc, Chem. Sci., 2017, 8, 3741 RSC.
- (a) J. Solà, M. Bolte and I. Alfonso, CrystEngComm, 2016, 18, 3793 RSC; (b) C. Tomasini, N. Castellucci, V. C. Caputo, L. Milli, G. Battistelli, S. Fermani and G. Falini, CrystEngComm, 2014, 17, 116 RSC; (c) C. Dutta, P. Krishnamurthy, D. Su, S. H. Yoo, G. W. Collie, M. Pasco, J. K. Marzinek, P. J. Bond, C. Verma, A. Gréland, A. Loquet, J. Li, M. Luo, M. Barboiu, G. Guichard, R. M. Kini and P. P. Kumar, Chem, 2023, 9, 2237 CAS.
- C. Dutta, P. Krushnamurthy, D. Su, S. H. Yoo, G. W. Collie, M. Pasco, J. K. Marzinek, P. J. Bond, C. Verma, A. Grélard, A. Loquet, J. Li, M. Luo, M. Barboiu, G. Guichard, R. M. Kini and P. P. Kumar, Chem, 2023, 9, 2237 CAS.
- F. Qin, M. Sheng, R. Li, D. Liu, M. Ren, R. Tian, L. Douadji, D. Wang, Q. Gan and L. Liang, Chem. Eng. J., 2024, 489, 151006 CAS.
- H. G. Brittain, J. Pharm. Sci., 2012, 101, 464 CAS.
- M.-I. Delegkou, N. Panagiotou, C. Papatriantafyllopoulou, A. Tasipoulos, D. Papaioannou, S. P. Perlepes and V. Nastopoulos, CrystEngComm, 2024, 26, 3574 CAS.
- (a) S. J. Pike, A. D. Bond and C. A. Hunter, CrystEngComm, 2018, 20, 2912 CAS; (b) P. Panini, E. Boel, L. Van Meerpikvelt and G. Van den Mooter, Cryst. Growth Des., 2022, 22, 2703 CAS; (c) H. Lemmer, N. Steiger, W. Liebenberg and M. R. Caira, Cryst. Growth Des., 2012, 12, 1683 CAS.
- M. Jurić, B. Perić, N. Brničević, P. Planinić, D. Pajić, K. Zadro, G. Giester and B. Kaitner, Dalton Trans., 2008, 842 Search PubMed.
- A. Suhonen, E. Nauha, K. Salorinne, K. Helttunen and M. Nissinen, CrystEngComm, 2012, 14, 7398 CAS.
- T. Qi, V. Maurizot, H. Noguchi, T. Charoenraks, B. Kauffmann, M. Takafuji, H. Ihara and I. Huc, Chem. Commun., 2012, 48, 6337 CAS.
- M. T. Stone, J. M. Fox and J. S. Moore, Org. Lett., 2004, 6, 3317 CAS.
- B. A. F. Le Bailly, L. Byrne, V. Diemer, M. Foroozandeh, G. A. Morris and J. Clayden, Chem. Sci., 2015, 6, 2313 CAS.
- (a) F. Meng, X. Zhang, T. Sarma, N. Yuan, Y. Yin, Z. Duan, C. Lei, Q. Qian, L. Ding and Z. Zhang, CrystEngComm, 2019, 21, 3906 CAS; (b) A. A. Nalawade, N. G. Kumar, K. R. P. Kumar, M. Singh, S. Dey and H. N. Gopi, CrystEngComm, 2024, 26, 913 Search PubMed.
- (a) F. S. Menke, B. Wicher, L. Allmendinger, V. Maurizot and I. Huc, Chem. Sci., 2023, 14, 3742 RSC; (b) A. Meunier, M. L. Singleton, B. Kauffmann, T. Granier, G. Lautrette, Y. Ferrand and I. Huc, Chem. Sci., 2020, 11, 12178 RSC; (c) K. Urushibara, Y. Ferrand, Z. Liu, K. Katagirim, M. Kawahata, E. Morvan, R. D'Elia, V. Pophristic, A. Tanatani and I. Huc, Chem. – Eur. J., 2021, 27, 11205 CrossRef CAS PubMed.
- (a) S. J. Pike, J. E. Jones, J. Raftery, J. Clayden and S. J. Webb, Org. Biomol. Chem., 2015, 13, 9580 RSC; (b) S. J. Pike, T. Boddaert, J. Raftery, S. J. Webb and J. Clayden, New J. Chem., 2015, 39, 3288 RSC; (c) S. J. Pike, J. Raftery, S. J. Webb and J. Clayden, Org. Biomol. Chem., 2014, 12, 4124 RSC.
- (a) F. S. Menke, B. Wicher, V. Maurizot and I. Huc, Angew. Chem., Int. Ed., 2023, 62, e202217325 CAS; (b) A. Borissov, I. Marques, J. Y. C. Lim, V. Félix, M. D. Smith and P. D. Beer, J. Am. Chem. Soc., 2019, 141, 4119 CAS; (c) S. J. Pike, V. Diemer, J. Raftery, S. J. Webb and J. Clayden, Chem. – Eur. J., 2014, 20, 15981 CrossRef CAS PubMed; (d) C.-Z. Liu, S. Koppireddi, H. Wang, D.-W. Zhang and Z.-T. Li, Angew. Chem., Int. Ed., 2018, 58, 226 Search PubMed.
- (a) W. Cai, G.-T. Wang, Y.-X. Xu, X.-K. Jiang and Z.-T. Li, J. Am. Chem. Soc., 2008, 130, 6936 CrossRef CAS PubMed; (b) L.-Y. You, G. T. Wang, X.-K. Jiang and Z. T. Li, Tetrahedron, 2009, 65, 9494 CAS.
- C.-Z. Liu, C. Zhang, C.-G. Li, H. B. Chen, W. Yang, Z.-Y. Li, Z.-Y. Hu, L. Xu, B. Zhai and Z.-T. Li, Chem. – Eur. J., 2024, 30, e202401150 CrossRef CAS PubMed.
- F. S. Menke, B. Wicher, V. Maurixot and I. Huc, Angew. Chem., Int. Ed., 2023, 62, e202217325 CrossRef CAS PubMed.
- R. A. Dodson, A. P. Kalenak and A. J. Matzger, J. Am. Chem. Soc., 2020, 142, 20806 CrossRef CAS PubMed.
- H. Yang, H. Zhang, C. J. Kang, C. Ji, D. Shi and D. Zhao, Sci. Adv., 2024, 10, eads0260 CrossRef CAS PubMed.
- A. A. Jones and C. D. Snow, Chem. Commun., 2024, 60, 5790 RSC.
- D. R. Brown, M. J. Hurlock, T. M. Nenoff and J. M. Rimsza, ACS Appl. Mater. Interfaces, 2025, 17, 5496 CrossRef PubMed.
- A. Suhonen, M. Kortelainen, E. Nauha, S. Yliniemelä-Sipari, P. M. Pihko and M. Nissinen, CrystEngComm, 2016, 18, 2005 RSC.
- R. Annala, A. Suhonen, H. Laakkonen, P. Permi and M. Nissinen, Chem. – Eur. J., 2017, 23, 16671 CrossRef CAS PubMed.
- A. Suhonen, M. Kortelainen, E. Nauha, S. Yliniemelä-Sipari, P. M. Pihko and M. Nissinen, CrystEngComm, 2016, 18, 2005 CAS.
- R. Annala, A. Suhonen, H. Laakkonen, P. Permi and M. Nissinen, Chem. – Eur. J., 2017, 23, 16671 CrossRef CAS PubMed.
- (a) A. R. Davis, S. Ozturk, C. C. Seaton, L. Male and S. J. Pike, Chem. – Eur. J., 2024, 30, e202402892 CrossRef CAS PubMed; (b) A. R. Davis, J. O. Dismorr, J. H. R. Tucker and S. J. Pike, Chem. – Eur. J., 2024, 30, e202402423 CrossRef CAS PubMed.
- S. J. Pike, R. Telford and L. Male, Org. Biomol. Chem., 2023, 21, 7717 RSC.
- S. J. Pike, A. Heliot and C. C. Seaton, CrystEngComm, 2020, 40, 4030 Search PubMed.
- In this foldamer class, we have previously defined the helical pitch value as the distance between the C atoms present in terminal amide functionalities, see: A. R. Davis, S. Ozturk, C. C. Seaton, L. Male and S. J. Pike, Chem. – Eur. J., 2024, 30, e202402892 CrossRef CAS PubMed for more detailed examples. However, the lack of terminal amine groups in 1 means that we have calculated the helical pitch values as the distance between the N atoms of the terminal amine functionalities in this study. See ESI† for further details.
- J. B. Maglic and R. Lavendomme, J. Appl. Crystallogr., 2022, 55, 1033 CrossRef CAS PubMed.
- (a) C. A. Hunter, Angew. Chem., Int. Ed., 2004, 43, 5310 CrossRef CAS PubMed; (b) C. A. Hunter, J. Singh and J. M. Thornton, J. Mol. Biol., 1991, 218, 837 CrossRef CAS PubMed.
- I. Rozas, I. Alkorta and J. Elguero, J. Phys. Chem. A, 1998, 102, 9925–9932 CrossRef CAS.
- Y. Hamuro, S. J. Geib and A. D. Hamilton, J. Am. Chem. Soc., 1997, 119, 10587 CrossRef CAS.
- Short contacts between the N atoms of the azo bonds and the H atom of the adjacent amine group are observed in all solid state structures of 1 and these could serve to further stabilise the helical structure of the scaffold. Similar short contacts have been reported by Parquette and co-workers in their related pyridinedicarboxamide/m-(phenylazo)azobenzene foldamers, see: C. Tie, J. C. Gallucci and J. R. Parquette, J. Am. Chem. Soc., 2006, 128, 1162 CAS.
- M. P. Walsh, J. A. Barclay, C. S. Begg, J. Xuan and M. O. Kitching, Cryst. Growth Des., 2023, 23, 2837 CAS.
- H. J. Shen, R. Ye, A. Romanies, A. Roy, F. Chen, C. Ren, Z. Liu and H. Zeng, J. Am. Chem. Soc., 2020, 142, 10050 CrossRef PubMed.
- J. Shen, R. Ye, A. Romanies, A. Roy, F. Chen, C. Ren, Z. Liu and H. Zeng, J. Am. Chem. Soc., 2020, 142, 10050 CrossRef CAS PubMed.
- E. P. Aldrich, K. A. Bussey, J. R. Connell, E. F. Reinhart, K. D. Oshin, B. Q. Mercado and A. G. Oliver, Acta Crystallogr., Sect. E: Crystallogr. Commun., 2016, 72, 40–43 CrossRef CAS PubMed.
- S. Horowitz and R. C. Trievel, J. Biol. Chem., 2012, 287, 41576 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Crystallographic data and conditions for 1A, 1B, 1·DCM, 1·THF, 1·butanone, 1·MeOH and 1·DMSO. CCDC 2426430 (1A), 2426870, 2426871, 2426432 (1·THF), 2426872, 2426428 (1·DCM), 2426873 and 2426874. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5ob00342c |
| This journal is © The Royal Society of Chemistry 2025 |