Stabilization of optically inactive α-helices of peptidic foldamers through sequence control and i, i + 4 stapling†
Received
11th February 2025
, Accepted 4th March 2025
First published on 5th March 2025
Abstract
We report the effect of a minimal (i, i + 4) staple on the dynamic interconversion between right-handed (P) and left-handed (M) forms of an optically inactive α-helical peptide composed only of helicogenic achiral amino acids, such as 1-amino-cyclohexanecarboxylic acid (Ac6c) and 4-aminopiperidine-4-carboxylic acid (Api) residues. The P/M interconversion rate of the peptide with a flexible hydrocarbon-based staple was estimated to be 0.41 s−1 at 298 K through variable temperature 1H NMR measurements in 1,1,2,2-tetrachloroethane-d2. A combined analysis using 1H NMR spectroscopy, single-crystal X-ray crystallography, and density functional theory (DFT) calculations revealed that the present flexible stapling does not effectively constrain the conformational freedom of the helical peptide. DFT calculations revealed that Ac6c residues exhibit a stronger propensity for α-helical conformation over the 310-helix than α-aminoisobutyric acid (Aib) residues, with their influence being highly dependent on position and sequence within the oligopeptides.
Introduction
The stabilization of the α-helical conformation of short oligopeptides has attracted much attention because of its potential therapeutic application.1–5 Among the methods for such stabilization, intramolecular cross-linking between peptide side chains, i.e. stapling, is one of the powerful strategies to stabilize the helical conformation not only of α-helical peptides1–8 but also of helical polymers9–11 and oligomers (foldamers) composed of abiotic backbones.12–16 It is also known that the incorporation of strongly helicogenic chiral or achiral Cα-tetrasubstituted amino acid residues into oligopeptides instead of naturally occurring Cα-trisubstituted ones stabilizes 310- or α-helical conformations of peptides.17–21 When only achiral Cα-tetrasubstituted amino acids, such as α-aminoisobutyric acid (Aib),17,22,23 1-amino-cyclohexanecarboxylic acid (Ac6c),24–26 and 4-aminopiperidine-4-carboxylic acid (Api),27,28 are used to build oligopeptides, the achiral peptides fold into racemic mixtures of both right-handed (P) and left-handed (M) helices in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio. For example, homo-Aib oligomers are known to adopt a dynamically racemic 310-helical conformation rather than the corresponding α-helical one, most likely due to a strong 310-helical propensity of the Aib residues,18,22,29–31 and to undergo rapid interconversion between enantiomeric (P)- and (M)-forms on the millisecond time scale.32–36 This rapid interconversion rate of Aib-based 310-helical peptides can be reduced by a factor of approximately 106 through a single staple between the side chains of Api residues introduced at the ith and i + 3th positions, which are located on the same side of the 310-helix.37,38 Similar to these 310-helices, we have recently reported stapled α-helical peptides that exhibit a slow interconversion between (P)- and (M)-forms on a time scale of minutes.39 These peptides are composed mainly of Ac6c and its piperidine analog Api residues, with intramolecular cross-linking between the side chains of the Api residues introduced at the ith and i + 7th positions using a biphenyl-based cross-linker spanning two α-helical turns. Their P/M interconversion can be almost completely suppressed by introducing double staples, effectively transforming dynamic α-helices into static ones. Thanks to this static nature, doubly stapled α-helical peptides synthesized exclusively from achiral components can be separated into enantiomeric (P)- and (M)-α-helices using chiral HPLC. Moreover, the P/M interconversion rate is highly dependent on the stiffness of the staple moiety;40 for instance, replacing the biphenyl-based staple with a more flexible hydrocarbon staple significantly accelerates the P/M interconversion of the corresponding α-helical peptides. While (i, i + 7) staples spanning two α-helix turns have been employed in Ac6c-based peptides to construct both dynamic and static α-helical peptides,39,40 the effect of a minimal stapling – i.e., an (i, i + 4) staple spanning one α-helical turn-on the P/M interconversion of optically inactive α-helical peptides remains unknown. In addition, the helix type propensity (310vs. α-helix) of Ac6c residues compared to that of Aib residues is still poorly understood.24,25 Herein, we report the synthesis and structural characterization of an Ac6c/Api-based nonapeptide with an (i, i + 4) staple and investigate the effect of minimal stapling on its P/M interconversion (Fig. 1). In addition, we also report a density functional theory (DFT)-based theoretical study that examines the effect of peptide chain length in homo-Ac6c and alternative Aib-Ac6c sequences on the 310-/α-helix propensities, as well as the effects of sequence and position of Ac6c residues within Aib-based helical oligopeptides.
:1 molar ratio. For example, homo-Aib oligomers are known to adopt a dynamically racemic 310-helical conformation rather than the corresponding α-helical one, most likely due to a strong 310-helical propensity of the Aib residues,18,22,29–31 and to undergo rapid interconversion between enantiomeric (P)- and (M)-forms on the millisecond time scale.32–36 This rapid interconversion rate of Aib-based 310-helical peptides can be reduced by a factor of approximately 106 through a single staple between the side chains of Api residues introduced at the ith and i + 3th positions, which are located on the same side of the 310-helix.37,38 Similar to these 310-helices, we have recently reported stapled α-helical peptides that exhibit a slow interconversion between (P)- and (M)-forms on a time scale of minutes.39 These peptides are composed mainly of Ac6c and its piperidine analog Api residues, with intramolecular cross-linking between the side chains of the Api residues introduced at the ith and i + 7th positions using a biphenyl-based cross-linker spanning two α-helical turns. Their P/M interconversion can be almost completely suppressed by introducing double staples, effectively transforming dynamic α-helices into static ones. Thanks to this static nature, doubly stapled α-helical peptides synthesized exclusively from achiral components can be separated into enantiomeric (P)- and (M)-α-helices using chiral HPLC. Moreover, the P/M interconversion rate is highly dependent on the stiffness of the staple moiety;40 for instance, replacing the biphenyl-based staple with a more flexible hydrocarbon staple significantly accelerates the P/M interconversion of the corresponding α-helical peptides. While (i, i + 7) staples spanning two α-helix turns have been employed in Ac6c-based peptides to construct both dynamic and static α-helical peptides,39,40 the effect of a minimal stapling – i.e., an (i, i + 4) staple spanning one α-helical turn-on the P/M interconversion of optically inactive α-helical peptides remains unknown. In addition, the helix type propensity (310vs. α-helix) of Ac6c residues compared to that of Aib residues is still poorly understood.24,25 Herein, we report the synthesis and structural characterization of an Ac6c/Api-based nonapeptide with an (i, i + 4) staple and investigate the effect of minimal stapling on its P/M interconversion (Fig. 1). In addition, we also report a density functional theory (DFT)-based theoretical study that examines the effect of peptide chain length in homo-Ac6c and alternative Aib-Ac6c sequences on the 310-/α-helix propensities, as well as the effects of sequence and position of Ac6c residues within Aib-based helical oligopeptides.
 |
| | Fig. 1 (a) Synthesis of the stapled optically inactive α-helical peptide 1C7 with an (i, i + 4) hydrocarbon-based staple. (b) Schematic representation of the deceleration of P/M interconversion in an optically inactive α-helical peptide upon minimal (i, i + 4) stapling. | |
Results and discussion
Theoretical study of 310-/α-helix type propensities of the Ac6c and Aib residues
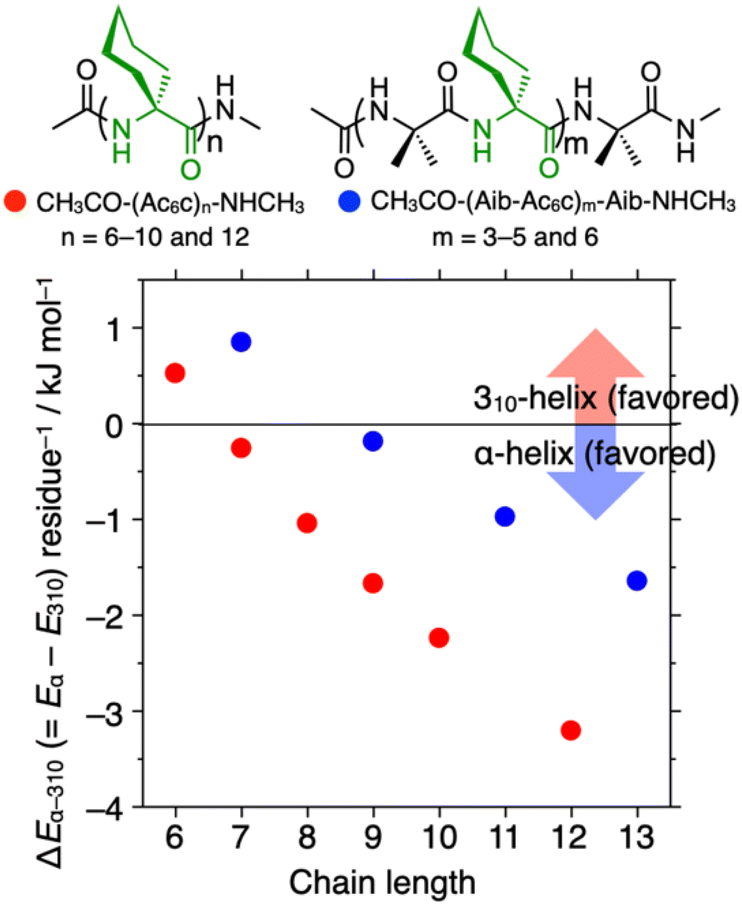
In the previous study, oligopeptides containing a tetrameric or hexameric homo-Ac6c segment were found to undergo 310/α-helix transitions in response to solvent changes, with 310- and α-helical conformations induced in less polar CHCl3 and polar 2,2,2-trifluoroethanol (TFE), respectively.41 In sharp contrast, oligopeptides composed mainly of alternative (Aib-Ac6c)n sequences (n = 4 and 8) did not adopt an α-helical conformation, even in TFE.42 To gain further insight into the 310/α-helix type propensity of Ac6c residues in optically inactive helical peptides, we performed DFT calculations to estimate the energy differences (ΔEα-310 per residue, where ΔEα-310 = Eα − E310, in kJ mol−1) between α- and 310-helical conformations of -(Ac6c)n- and -(Aib-Ac6c)m-based helical peptides with varying repeat numbers (Fig. 2). The ΔEα-310 per residue values for acetyl-(Ac6c)n-NHCH3 (n = 6) and acetyl-(Aib-Ac6c)m-Aib-NHCH3 (m = 3) were positive, indicating their preferred formation of the 310-helix. In sharp contrast, when the peptide chain length was sufficiently long (n ≥ 7 and m ≥ 4), the α-helical conformation became energetically favored over the 310-helix. Moreover, the α-helix formation propensity of homo-Ac6c peptides was significantly stronger than that of alternative Aib-Ac6c peptides, consistent with the experimental observations described above (Fig. 2).41,42
 |
| | Fig. 2 Plots of the energy difference (ΔEα-310 per residue, where ΔEα-310 = Eα − E310, in kJ mol−1) between the α- and 310-helical conformations of acetyl-(Ac6c)n-NHCH3 (n = 6, 7, 8, 9, and 12) (red circles) and acetyl-(Aib-Ac6c)m-Aib-NHCH3 (m = 3, 4, 5, and 6) (blue circles) versus peptide chain length. | |
Next, we estimated the ΔEα-310 and the ΔEα-310/n (where n is number of Ac6c residues, in kJ mol−1) values of the optically inactive helical nonapeptides I–IX, acetyl-(AA)9-NHCH3 (AA = Ac6c and/or Aib), with different amino acid sequences listed in Table 1, to clarify the effects of the number and position of Ac6c residues in Aib-based nonapeptides on α-helix formation. Interestingly, replacement of the Aib(7) residue (the number in parentheses represents the residue number from the N-terminus) of acetyl-(Aib-Ac6c)4-Aib-NHCH3 (I) with Ac6c resulted in a significant increase in α-helical preference (peptides I and II in Table 1). This effect was further enhanced when the Aib(5) residue in peptide II was also substituted with Ac6c (peptides II and III in Table 1). However, the further replacement of the Aib(3) residue in peptide III with Ac6c, as well as its combination with Ac6c substitutions at both termini produced only a relatively modest enhancement of α-helical preference compared to the previous substitutions. These results suggest that segments composed of more than three consecutive Ac6c residues strongly promote α-helical conformation in the corresponding nonapeptides. Additionally, peptide VI which contains two Ac6c residues at the ith and i + 3th positions, destabilizes the 310-helical conformation, likely due to steric repulsion between the Ac6c side chains. Similarly, Ac6c substitutions at the ith and i + 4th positions (peptide VII) destabilize the α-helical conformation, making these Ac6c residues less effective in stabilizing the α-helix compared to those in other peptides listed in Table 1. Interestingly, the introduction of two consecutive Ac6c residues in the middle of the Aib-based peptide (peptide VIII) enhanced α-helical preference more effectively than the corresponding peptide containing a single Ac6c residue in the middle (peptide IX).
Table 1 Effect of the number and position of Ac6c residue in the optically inactive helical nonapeptides I–IX (acetyl-(AA)9-NHCH3, AA = Ac6c and/or Aib) on the energy difference (ΔEα-310 and ΔEα-310/n, where ΔEα-310 = Eα − E310 and n = number of Ac6c residues, in kJ mol−1) between α- and 310-helical conformationsa
| Peptide |
Amino acid sequence from N-terminus (1) to C-terminus (9) |
ΔEα-310 (ΔEα-310/n) (kJ mol−1) |
| 1 |
2 |
3 |
4 |
5 |
6 |
7 |
8 |
9 |
|
The averaged dihedral angles (|ϕ|/|ψ|) of all energy-minimized α- and 310-helical structures were varied within |56|/|45–48| and |54–56|/|30–32|, respectively.
|
|
I
|
Aib |
Ac6c |
Aib |
Ac6c |
Aib |
Ac6c |
Aib |
Ac6c |
Aib |
–1.72 (−0.41) |
|
II
|
Aib |
Ac6c |
Aib |
Ac6c |
Aib |
Ac6c |
Ac6c |
Ac6c |
Aib |
–6.51 (−1.31) |
|
III
|
Aib |
Ac6c |
Aib |
Ac6c |
Ac6c |
Ac6c |
Ac6c |
Ac6c |
Aib |
–10.3 (−1.71) |
|
IV
|
Aib |
Ac6c |
Ac6c |
Ac6c |
Ac6c |
Ac6c |
Ac6c |
Ac6c |
Aib |
–11.3 (−1.61) |
|
V
|
Ac6c |
Ac6c |
Ac6c |
Ac6c |
Ac6c |
Ac6c |
Ac6c |
Ac6c |
Ac6c |
–15.1 (−1.67) |
|
VI
|
Aib |
Aib |
Ac6c |
Aib |
Aib |
Ac6c |
Aib |
Aib |
Aib |
–1.59 (−0.80) |
|
VII
|
Aib |
Aib |
Ac6c |
Aib |
Aib |
Aib |
Ac6c |
Aib |
Aib |
–0.51 (−0.25) |
|
VIII
|
Aib |
Aib |
Aib |
Aib |
Ac6c |
Ac6c |
Aib |
Aib |
Aib |
–2.42 (−1.21) |
|
IX
|
Aib |
Aib |
Aib |
Aib |
Ac6c |
Aib |
Aib |
Aib |
Aib |
–0.56 (−0.56) |
Synthesis and conformational analysis of the stapled peptide 1C7
Based on the previous study39,40 and the DFT study discussed above, we chose Ac6c and its piperidine analog, Api, to construct an optically inactive stapled α-helical nonapeptide. In this design, two Api residues were incorporated at the ith and i + 4th positions, namely the 3rd and 7th from the N-terminus, to be tethered by a minimal staple. Our modeling study suggested that a flexible cross-linker based on heptanedioic acid would be a minimal i, i + 4 staple suitable for cross-linking between the side chains of the Api residues incorporated at the i and i + 4 positions of the Ac6c-based achiral peptide chain. The linear peptide, Z-(Ac6c)2-Api(Boc)-(Ac6c)3-Api(Boc)-Ac6c-Aib-OMe (Z = benzyloxycarbonyl, Boc = tert-butoxycarbonyl, and OMe = methoxy), was prepared via stepwise liquid phase synthesis (Fig. 1a and Scheme S1†). After deprotection of the Boc groups at the Api side chains, the resulting peptide was reacted with the activated diester stapling reagent (2), containing a C7 linker, in the presence of base under dilute conditions, affording the stapled peptide 1C7 in moderate yield (Fig. 1a, see ESI† for more details). The stapled peptide 1C7 was identified by electrospray ionization time-of-flight (ESI-TOF) mass spectrometry, and its purity was confirmed by high-performance liquid chromatography (HPLC) analysis (Fig. S1†).43
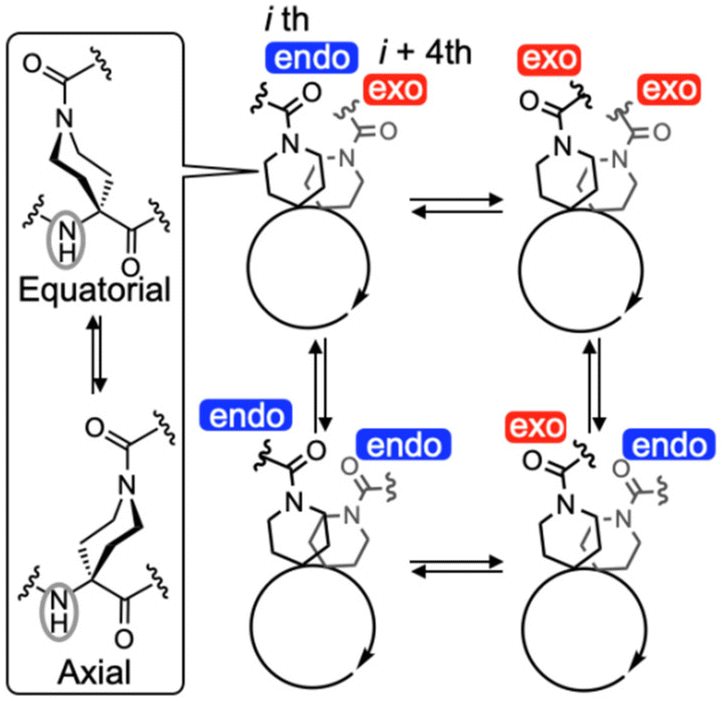
Interestingly, the 1H NMR spectral pattern of 1C7 in 1,1,2,2-tetrachloroethane-d2 (TCE-d2) at 298 K was complicated (Fig. 3), due to the presence of four possible side-chain amide carbonyl (–CO-N(CH2)2–) orientations: endo/exo, exo/exo, exo/endo, and endo/endo (Fig. 4). These orientations interconvert slowly on the NMR time scale, with endo and exo referring to their orientations relative to a plane passing through the midpoint of the two amide side chains and the center of the helix. This complexity is evident in the N-terminal urethane N(1)H proton signal, observed around 5.6 ppm, which appears as a dominant peak accompanied by three minor peaks. In contrast, previously reported α-helical peptides composed mainly of Ac6c and Api residues with (i, i + 7) flexible hydrocarbon stapling exhibit only a single orientation (exo/exo).40 DFT calculations of 1C7 with these four amide carbonyl orientations suggest that the major conformation could be assigned to an α-helical conformation with the endo/exo amide carbonyl orientation (conf-1 in Fig. S2a†), which is the lowest energy conformation among them (Fig. S2†). This structural assignment is further supported by single-crystal X-ray diffraction, which reveals that 1C7 adopts an α-helical conformation with the endo/exo orientation. The analyzed crystal contains two crystallographically independent but conformationally similar molecules, A and B, both of which adopt a typical α-helical conformation (average dihedral angles (|ϕ|/|ψ|) excluding the C-terminal Aib residue: 57°/44° for A and 56°/46° for B) (Fig. 5). Interestingly, the amide NH groups of the Api(3) residues of these peptides adopt an axial orientation, whereas those of the Api(7) residues are found in an equatorial orientation (Fig. 4, left panel and 5). In addition, the energy difference (ΔE) between the energy-minimized structure obtained by DFT calculations using molecule B as an initial structure and the corresponding structure with all equatorial positions is only 2.2 kJ mol−1 (Fig. S2a and f†). This axial–equatorial conformational fluctuation is most likely due to the high flexibility of the stapling moiety.
 |
| | Fig. 3 Temperature-dependent 1H NMR (600 MHz, C2D2Cl4, 5.0 mM) spectral changes of 1C7. The expanded spectra of the highlighted region (indicated by the red dotted rectangle) is shown in the right panel, illustrating the temperature-dependent behavior of the N-terminal methylene protons and the amide N(1)H signals. | |
 |
| | Fig. 4 Schematic representation of the interconversions between four possible orientations (endo or exo) of the side-chain amide carbonyl (–CO-N(CH2)–) groups on the Api residues in the (P)-α-helical 1C7. Here, endo and exo represent their orientations relative to a plane passing through the midpoint of the two amide side chains and the center of the helix. Additionally, the interconversion between the equatorial and axial positions of the amide NH group on the piperidine ring of the Api residue is also show. | |
 |
| | Fig. 5 The X-ray crystal structures of (P)-α-helical 1C7. Crystallographically independent molecules A (a) and B (b) are shown. Only the (P)-helix is shown. All of the hydrogen atoms, disordered atoms and solvent molecules are omitted for clarity. Dotted lines represent intramolecular hydrogen bonds. The dihedral angles (ϕ, ψ and ω) and hydrogen-bonding parameters of 1C7 are summarized in Tables S13 and S16.† The number in parentheses represents the residue number from the N-terminus. | |
The 1H NMR spectrum of 1C7 in TCE-d2 at 298 K displayed diastereotopic splitting of the methylene protons of the N-terminal Z group (Δν = 103 Hz), indicating that the interconversion between the (P) and (M)-helices is slow on the NMR time scale (Fig. 3). To estimate the free energy of activation (ΔG‡, kJ mol−1) for the P/M interconversion of 1C7 at the coalescent temperature (Tc) of the signal splitting of the methylene protons, we performed variable-temperature 1H NMR measurements. As the temperature increased from 298 K to 348 K, the four N(1)H signals, corresponding to the different side chain amide carbonyl orientations, gradually broadened and coalesced. However, the N-terminal methylene proton signals remained split throughout this temperature range, indicating that the P/M interconversion is much slower than the interconversion between these four conformers. At 373 K, the methylene proton signals finally coalesced, giving the ΔG‡ value of 75.2 kJ mol−1. The rate constants (k, sec−1) for the P/M interconversion were estimated to be 229 s−1 at Tc and 0.41 s−1 at 298 K using the equation k = (kBT/h)exp(−ΔG‡/RT). Surprisingly, the k298 value of 1C7 is approximately 102 times faster than that of a previously reported α-helical peptide with a rigid (i, i + 7) biphenyl-based staple, which has only a single set of the amide carbonyl orientations on the Api side chains.39 This suggests that the rigidity of the stapling moiety plays a crucial role in restricting the overall conformational flexibility of the peptide including intermediate structures during the helix reversal process.
Conclusions
We have successfully designed and synthesized a dynamically optically inactive α-helical peptide foldamer composed mainly of achiral Ac6c and Api residues with an (i, i + 4) hydrocarbon-based staple. While this single staple effectively slowed the P/M interconversion rate of the peptide, it did not significantly restrict the conformational freedom of the Api side-chain amide carbonyl linked by the staple. Furthermore, in helical oligopeptides composed mainly of Aib and Ac6c residues, we found that Ac6c promotes α-helical formation more strongly than Aib, favoring the α-helix over the 310-helix in a highly position-dependent manner. This α-helical propensity is further enhanced when a segment contains more than three consecutive Ac6c residues within the oligopeptides. Our findings may provide not only a deeper understanding of how staple length and rigidity influence α-helical stability, but also a rational design strategy for unique helical peptides capable of stimuli-responsive 310/α-helix transitions and tunable P/M interconversion rates.
Author contributions
N.O. conceived the project, designed and performed the experiments, analysed the data, and wrote the paper. S.A. performed X-ray crystallographic analysis. All authors discussed the results and commented on the manuscript.
Data availability
The data supporting this article have been included as part of the ESI.† Additional data are available upon request from the authors. Crystallographic data for compound 1C7 have been deposited at the Cambridge Crystallographic Data Centre [CCDC 2421064].†
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported in part by JSPS KAKENHI (JP22K05220 (N. O.)) and the World Premier International Research Center Initiative (WPI), MEXT, Japan (N. O., S. A., and M. J. M.).
References
- G. L. Verdine and G. J. Hilinski, Methods Enzymol., 2012, 503, 3–33 CAS.
- A. M. Ali, J. Atmaj, N. Van Oosterwijk, M. R. Groves and A. Dömling, Comput. Struct. Biotechnol. J., 2019, 17, 263–281 CrossRef CAS PubMed.
- R. Mourtada, H. D. Herce, D. J. Yin, J. A. Moroco, T. E. Wales, J. R. Engen and L. D. Walensky, Nat. Biotechnol., 2019, 37, 1186–1197 CrossRef CAS PubMed.
- H. Yokoo, M. Hirano, T. Misawa and Y. Demizu, ChemMedChem, 2021, 16, 1226–1233 CrossRef CAS PubMed.
- Y. Li, M. Wu, Y. Fu, J. Xue, F. Yuan, T. Qu, A. N. Rissanou, Y. Wang, X. Li and H. Hu, Pharmacol. Res., 2024, 203, 107137 CrossRef CAS PubMed.
- L. D. Walensky, A. L. Kung, I. Escher, T. J. Malia, S. Barbuto, R. D. Wright, G. Wagner, G. L. Verdine and S. J. Korsmeyer, Science, 2004, 305, 1466–1470 CrossRef CAS PubMed.
- Y. H. Lau, P. De Andrade, Y. T. Wu and D. R. Spring, Chem. Soc. Rev., 2015, 44, 91–102 RSC.
- X. Li, S. Chen, W. D. Zhang and H. G. Hu, Chem. Rev., 2020, 120, 10079–10144 CrossRef CAS PubMed.
- S. Hecht and A. Khan, Angew. Chem., Int. Ed., 2003, 42, 6021–6024 CrossRef CAS PubMed.
- K. Maeda, H. Mochizuki, M. Watanabe and E. Yashima, J. Am. Chem. Soc., 2006, 128, 7639–7650 CrossRef CAS PubMed.
- A. Hashimoto, H. Sogawa, M. Shiotsuki and F. Sanda, Polymer, 2012, 53, 2559–2566 CrossRef CAS.
- N. Fuentes, A. Martin-Lasanta, L. Alvarez de Cienfuegos, R. Robles, D. Choquesillo-Lazarte, J. M. García-Ruiz, L. Martínez-Fernández, I. Corral, M. Ribagorda, A. J. Mota, D. J. Cárdenas, M. C. Carreño and J. M. Cuerva, Angew. Chem., Int. Ed., 2012, 51, 13036–13040 CrossRef CAS PubMed.
- C. Tsiamantas, X. de Hatten, C. Douat, B. Kauffmann, V. Maurizot, H. Ihara, M. Takafuji, N. Metzler-Nolte and I. Huc, Angew. Chem., Int. Ed., 2016, 55, 6848–6852 CrossRef CAS PubMed.
- L. Zheng, C. Yu, Y. Zhan, X. Deng, Y. Wang and H. Jiang, Chem. – Eur. J., 2017, 23, 5361–5367 CrossRef CAS PubMed.
- H. Abe, C. Sato, Y. Ohishi and M. Inouye, Eur. J. Org. Chem., 2018, 3131–3138 CrossRef CAS.
- N. Miki, R. Inoue and Y. Morisaki, Bull. Chem. Soc. Jpn., 2021, 95, 110–115 CrossRef.
- I. L. Karle and P. Balaram, Biochemistry, 1990, 29, 6747–6756 CrossRef CAS PubMed.
- C. Toniolo and E. Benedetti, Trends Biochem. Sci., 1991, 16, 350–353 CrossRef CAS PubMed.
- C. Toniolo, M. Crisma, F. Formaggio and C. Peggion, Biopolymers, 2001, 60, 396–419 CrossRef CAS PubMed.
- M. Tanaka, Chem. Pharm. Bull., 2007, 55, 349–358 CrossRef CAS PubMed.
- M. Crisma and C. Toniolo, Biopolymers, 2015, 104, 46–64 CrossRef CAS PubMed.
- N. Shamala, R. Nagaraj and P. Balaram, J. Chem. Soc., Chem. Commun., 1978, 996–997 RSC.
- V. Moretto, M. Crisma, G. M. Bonora, C. Toniolo, H. Balaram and P. Balaram, Macromolecules, 1989, 22, 2939–2944 CrossRef CAS.
- P. K. C. Paul, M. Sukumar, R. Bardi, A. M. Piazzesi, G. Valle, C. Toniolo and P. Balaram, J. Am. Chem. Soc., 1986, 108, 6363–6370 CrossRef CAS.
- M. Crisma, G. M. Bonora, C. Toniolo, A. Bavoso, E. Benedetti, B. Di Blasio, V. Pavone and C. Pedone, Macromolecules, 1988, 21, 2071–2074 CrossRef CAS.
- V. Pavone, E. Benedetti, V. Barone, B. Di Blasio, F. Lelj, C. Pedone, A. Santini, M. Crisma, G. M. Bonora and C. Toniolo, Macromolecules, 1988, 21, 2064–2070 CrossRef CAS.
- C. L. Wysong, T. S. Yokum, G. A. Morales, R. L. Gundry, M. L. McLaughlin and R. P. Hammer, J. Org. Chem., 1996, 61, 7650–7651 CrossRef CAS PubMed.
- T. S. Yokum, T. J. Gauthier, R. P. Hammer and M. L. McLaughlin, J. Am. Chem. Soc., 1997, 119, 1167–1168 CrossRef CAS.
- Y. Paterson, S. M. Rumsey, E. Benedetti, G. Nemethy and H. A. Scheraga, J. Am. Chem. Soc., 1981, 103, 2947–2955 CrossRef CAS.
- C. Toniolo, M. Crisma, G. M. Bonora, E. Benedetti, B. Dl Blasio, V. Pavone, C. Pedone and A. Santini, Biopolymers, 1991, 31, 129–138 CrossRef CAS.
- P. Kumar, N. G. Paterson, J. Clayden and D. N. Woolfson, Nature, 2022, 607, 387–392 CrossRef CAS PubMed.
- R.-P. Hummel, C. Toniolo and G. Jung, Angew. Chem., Int. Ed. Engl., 1987, 26, 1150–1152 CrossRef.
- M. Kubasik and A. Blom, ChemBioChem, 2005, 6, 1187–1190 CrossRef CAS PubMed.
- M. Kubasik, J. Kotz, C. Szabo, T. Furlong and J. Stace, Biopolymers, 2005, 78, 87–95 CrossRef CAS PubMed.
- J. Clayden, A. Castellanos, J. Solà and G. A. Morris, Angew. Chem., Int. Ed., 2009, 48, 5962–5965 CrossRef CAS PubMed.
- B. A. F. Le Bailly and J. Clayden, Chem. Commun., 2016, 52, 4852–4863 RSC.
- N. Ousaka, T. Sato and R. Kuroda, J. Am. Chem. Soc., 2008, 130, 463–465 CrossRef CAS PubMed.
- N. Ousaka, Y. Inai and R. Kuroda, J. Am. Chem. Soc., 2008, 130, 12266–12267 CrossRef CAS PubMed.
- N. Ousaka, M. J. MacLachlan and S. Akine, Nat. Commun., 2023, 14, 6834 CrossRef CAS PubMed.
- N. Ousaka, M. J. MacLachlan and S. Akine, Chem. – Eur. J., 2024, 30, e202402704 CrossRef CAS PubMed.
- F. Mamiya, N. Ousaka and E. Yashima, Angew. Chem., Int. Ed., 2015, 54, 14442–14446 CrossRef CAS PubMed.
- N. Ousaka, Y. Takeyama and E. Yashima, Chem. – Eur. J., 2013, 19, 4680–4685 CrossRef CAS PubMed.
- We did not perform CD measurements of the stapled peptide 1C7 to distinguish between α- and 310-helical conformations, as 1C7 is composed solely of achiral components. Consequently, CD spectroscopy would not provide meaningful information in this case. Instead, the distinction between α- and 310-helices was determined using a combination of DFT calculations, X-ray crystallographic analysis, and temperature-dependent 1H NMR data (for a detailed discussion, see the main text).
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *ab,
Mark J.
MacLachlan
*ab,
Mark J.
MacLachlan