
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Separation of rare earth elements and nickel harnessing electrochemistry and reactive CO2 capture and mineralization†
Prince
Ochonma
 a,
Akanksha
Srivastava
b,
Christopher
Noe
c,
Tianhe
Yin
b,
Prarabdh
Jain
a and
Greeshma
Gadikota
*ab
a,
Akanksha
Srivastava
b,
Christopher
Noe
c,
Tianhe
Yin
b,
Prarabdh
Jain
a and
Greeshma
Gadikota
*ab
aSmith School of Chemical and Biomolecular Engineering, Cornell University, Ithaca, NY 14853, USA. E-mail: gg464@cornell.edu
bSchool of Civil and Environmental Engineering, Cornell University, Ithaca, NY 14853, USA
cDepartment of Chemistry, College of Art and Science, Stony Brook University, Stony Brook, NY 11790, USA
First published on 9th November 2024
Abstract
The aim is to probe the chemical mechanisms underlying the use of multifunctional solvents to simultaneously capture and convert CO2 into insoluble rare earth element (REE)-carbonates, while forming soluble complexes with nickel for separation. Subsequent nickel electrodeposition regenerates the CO2-loaded solvent for reuse.
Advances in technologies that harness CO2 emissions for the recovery of energy relevant metals are crucial for a sustainable and secure energy future.1 The growing demand for critical metals such as REEs and nickel, driven by their limited supply and rapid depletion of conventional high grade ores,2 has sparked interest in recovering these metals from unconventional sources such as industrial residues3 and recycled materials.4 Current methods including pyrometallurgy (heat-based extraction)5 and hydrometallurgy (chemical leaching and liquid–liquid extraction) are used for separation.6–8 Despite significant progress, the greenhouse gas footprint associated with heating, or the use of these reagents is significant,9 and alternative environmentally sustainable pathways are needed. One promising approach involves the capture and use of CO2 for metal separations. Metal chelating amine ligands have been demonstrated for simultaneous CO2 mineralization and recovery of metals such as Ni, Fe, and Cr in the solution phase.10 However, the use of CO2 capture solvents for the separation of REEs and transition metals such as Ni remains less explored.
In this context, it is well-known that REE-carbonates have low solubilities in water and can be preferentially separated via crystallization using CO2 capture solvents that solubilize CO2 and regenerate upon REE – carbonate precipitation. The separation of La, Ni, and Co was reported using diethylenetriamine and carbon dioxide – bearing flue gas.4 After lanthanum carbonate precipitation, ethanol and CO2 were used to separate Ni2+ and Co2+ ions. While effective, this approach relies on multiple solvent-based techniques for sequential separation and requires efficient solvent regeneration for cost-effectiveness. An alternative approach would be to utilize multifunctional solvents that can effectively capture CO2, precipitate REE-carbonates, and serve as an effective medium for Ni precipitation without undergoing degradation. This approach eliminates the need for additional solvents for Ni recovery and instead regenerates the multifunctional solvent which can be looped multiple times for separating REEs (La3+, Pr3+, Nd3+, Eu3+, Dy3+) ions from Ni2+ ions.
These scientific possibilities and challenges motivate the investigation of CO2 capture solvents such as aqueous ammonium hydroxide (NH4OH), aqueous monoethanolamine (MEA), and aqueous diethylenetriamine (DETA). NH4OH is effective in forming soluble complexes with Ni,11 MEA efficiently captures CO2 and converts it to REE-carbonates,12 and DETA has been reported for separating La and Ni in solvent – based extraction4 though its effectiveness in electroplating remains unexplored. Despite these advances, the following specific research questions remain unexplored: (1) what are the chemical mechanisms associated with the use of amine bearing CO2 capture solvents for REE and Ni separations? (2) After the recovery of REE-carbonates, what is the efficacy and associated coulombic efficiencies of Ni electrodeposition? (3) What is the influence of chemical speciation on product yields, purities, and morphologies? Addressing these questions will unlock new insights into the multifunctional role of solvents in capturing CO2, enabling the separation of REEs as water – insoluble carbonates, and mediating Ni electrodeposition (see Fig. 1).
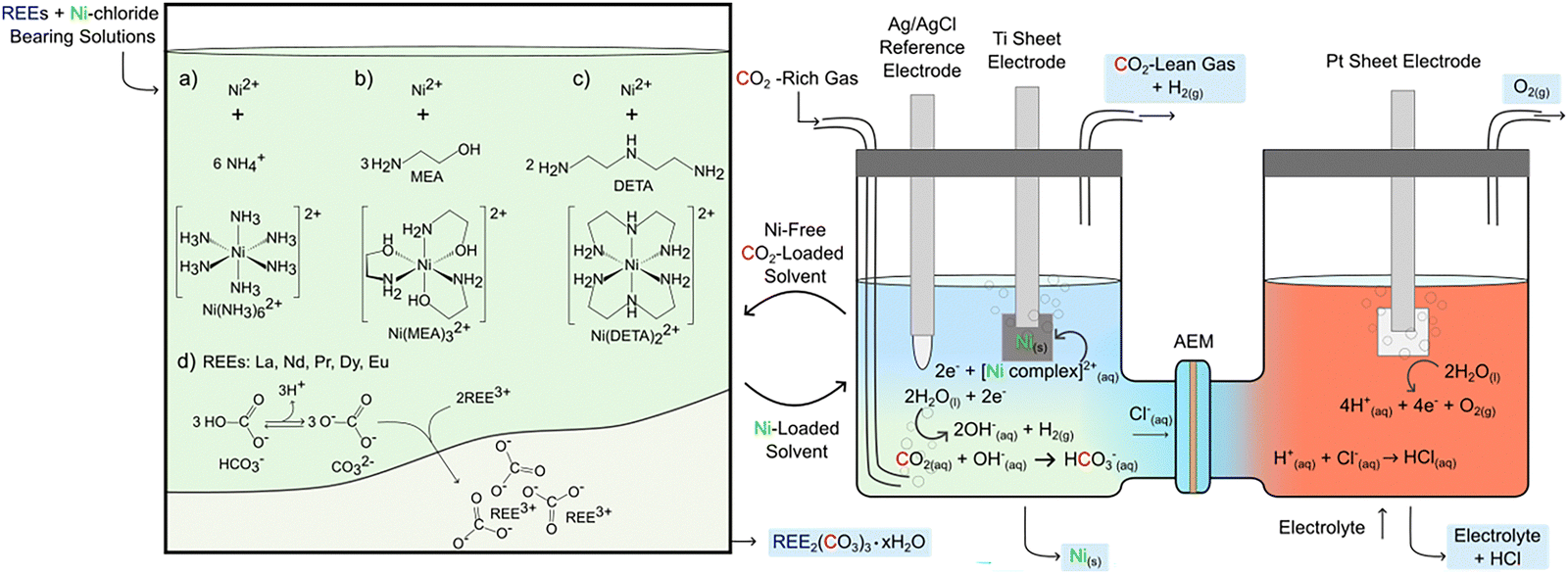 | ||
| Fig. 1 Proposed approach to separate La and Ni using CO2 and multifunctional solvents. Preferred reactions illustrated with (a) ammonium hydroxide (NH4OH), (b) monoethanolamine (MEA) and (c) diethylenetriamine (DETA) are based on the most stable complexes shown in ESI,† Table S2. Products include REE-carbonates, electrodeposited nickel, H2, O2, and regenerated solvent for the next cycles. All measurements are performed at room temperature. | ||
To elucidate the importance of CO2 capture solvents in enhancing CO2 solubility, and facilitating REE separation, two control experiments are conducted with solutions bearing 588 ppm La and 1176 ppm Ni. In the first control experiment, CO2 is bubbled directly through the aqueous La/Ni solution for 12 hours resulting in no precipitation. Consequently, no carbonate formation is observed due to the low solubility of CO2 in pure water, indicating that CO2 capture solvents are needed. In the second control experiment, equal volumes of CO2 loaded aqueous NaOH, and aqueous La/Ni solution are mixed, which resulted in La recovery efficiencies as lanthanum carbonate up to 99.9 (±0.1)%. However, this is also accompanied by 70.7% (±0.5)% of Ni precipitation resulting in La-carbonate purity and separation factor (β) of 46.4 (±0.7)% and 370.5 (±8.2), respectively. These base case separation factors are within a similar range of 314.6–3827.8 for La/Ni separation reported using other separation processes.6–8 Nonetheless, more selective separation of La and Ni can be achieved by co-utilizing CO2 and soluble metal chelating agents, which motivated the investigation of NH4OH, MEA, and DETA.
As shown in Fig. 2 and Table S1 (ESI†), La recovery efficiencies exceeding 99.5% are reported with NH4OH, MEA, and DETA. In addition, suppressed Ni co-extraction is observed (0–17.53% co-recovery efficiencies) at similar conditions with the control experiments (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 for La:Ni) resulting in separation factors of (4524–11630), (2131–4457), and (8784 – no Ni detected in carbonate phase) for NH4OH, MEA, and DETA, respectively. Increasing Ni co-extraction efficiency is reported in the order of DETA < NH4OH < MEA. Notably, this trend in Ni co-extraction efficiency is significantly influenced by the ability for Ni to form stable complexes in solution. Table S2 (ESI†) shows that Ni-DETA complexes have higher stabilities compared to those with NH4OH or MEA. The formation of this soluble complex is also evident from the change in the colour of the solution bearing Ni from green to purple on DETA addition (Fig. S1(d), ESI†). The effectiveness of harnessing NH4OH for separating other REEs such as praseodymium (Pr), neodymium (Nd), europium (Eu), and dysprosium (Dy) is also investigated. From a thermodynamic standpoint, the respective stability constants of REE-carbonates are similar, which is an indicator of similar behaviour as with La while keeping all other factors constant. Fig. 2(A) and Table S1 (ESI†) also show that recoveries exceeding 98% are also observed for all the four REEs of interest. Moreover, the regeneration of the CO2-free solvent can be achieved at higher REE concentrations as demonstrated with MEA in Fig. S2(a)–(d) (ESI†).
:2 for La:Ni) resulting in separation factors of (4524–11630), (2131–4457), and (8784 – no Ni detected in carbonate phase) for NH4OH, MEA, and DETA, respectively. Increasing Ni co-extraction efficiency is reported in the order of DETA < NH4OH < MEA. Notably, this trend in Ni co-extraction efficiency is significantly influenced by the ability for Ni to form stable complexes in solution. Table S2 (ESI†) shows that Ni-DETA complexes have higher stabilities compared to those with NH4OH or MEA. The formation of this soluble complex is also evident from the change in the colour of the solution bearing Ni from green to purple on DETA addition (Fig. S1(d), ESI†). The effectiveness of harnessing NH4OH for separating other REEs such as praseodymium (Pr), neodymium (Nd), europium (Eu), and dysprosium (Dy) is also investigated. From a thermodynamic standpoint, the respective stability constants of REE-carbonates are similar, which is an indicator of similar behaviour as with La while keeping all other factors constant. Fig. 2(A) and Table S1 (ESI†) also show that recoveries exceeding 98% are also observed for all the four REEs of interest. Moreover, the regeneration of the CO2-free solvent can be achieved at higher REE concentrations as demonstrated with MEA in Fig. S2(a)–(d) (ESI†).
 | ||
| Fig. 2 (A) Recovery efficiencies and product purity for REE/Ni separation using NH4OH at REE:Ni of 1:2. (B) The product purity at different concentration ratios for NH4OH, MEA and DETA. | ||
Evidence of REE carbonate formation is determined by investigating the thermal decomposition behaviour using TGA (Fig. S3, ESI†). Weight losses in the range of 80–240 °C accounts for ∼6.7%, 7.2% and 5.7% of the sample weight in NH4OH, MEA and DETA respectively are associated with the loss of H2O molecules implying the presence of hydrated carbonate species.13,14 Higher loss of H2O observed with MEA and NH4OH is likely due to the presence of hydrated Ni-carbonate. This observation is confirmed by the weight loss in the temperature range of 210–430 °C associated with the loss of CO2 in Ni-carbonate.14 Fig. S4(a) (ESI†) shows the TGA, DTG and DSC profile of pure hydrated Ni- carbonate for comparison. It is important to note that Ni-hydroxide and Ni-carbonate decompose at overlapping temperature ranges.14 However, FTIR analyses confirm the absence of the characteristic OH stretching vibrations of Ni(OH)2 typically observed at 3645 (±3) cm−1 (ref. 15) as shown in Fig. S4(b) (ESI†). In the next step, at temperatures in the range of 260–580 °C, lanthanum carbonate La2(CO3)3 decomposes into La2O2(CO3), releasing CO2.13 The fourth weight loss is perhaps the most distinct characteristic weight loss feature of lanthanum carbonate which represents the decomposition of La2O2CO3 to release CO2 and produces La2O3.13 DETA is observed to have the highest weight loss (∼7%) compared with ∼4% in MEA and NH4OH implying a relatively higher purity of La-carbonate. This observation is further confirmed by the distinct green colour of Ni in the final product recovered from experiments performed using NH4OH and MEA (Fig. S5, ESI†) contrasted with a product free of any coloration obtained from DETA post separation. SEM images shown in Fig. S3(d)–(f) (ESI†) reveal the presence of aggregated clusters with rosette and flat morphologies in La-carbonate produced.3,10,16–18
Cathodic reactions E0 vs. SHE, pH = 7
| N(NH3)6Cl2(aq) + 2e− → Ni(s) + 6NH3(aq) + 2Cl(aq)− E0 = −0.48 V | (R1) |
| Ni(H2NC2H4OH)3Cl2(aq) + 2e− → Ni(s) + 3H2NC2H4OH(aq) + 2Cl(aq)− | (R2) |
| Ni(C14H13N3)2Cl2(aq) + 2e− → Ni(s) + 2C14H13N3(aq) + 2Cl(aq)− | (R3) |
| 2H2O(aq) + 2e− → 2OH(aq)− + H2(g) E0 = −0.41 V | (R4) |
| OH(aq)− + CO2(g) → HCO3(aq)− | (R5) |
| OH(aq)− + HCO3(aq)− → CO32−(aq) + H2O | (R6) |
Anodic reactions E0 vs. SHE, pH = 7
| H2O(aq) → 2H(aq)+ + 1/2O2(g) + 2e− E0 = 0.82 V | (R7) |
| H+(aq) + Cl(aq)− → HCl(aq) | (R8) |
The reactions proposed for Ni recovery are shown in (R1)–(R8). Ni deposition at the cathode is facilitated by decomplexation through a gain of electrons to deposit solid Ni species as shown in (R1)–(R3). Thermodynamic plots shown in Fig. S6 (ESI†) show the possibility for Ni to form complexes of varying oxidation states including [Ni(NH3)x]2+, [Ni(H2NC2H4OH)x]2+, and [Ni(C14H13N3)x]2+, where x typically varies from 1–6, 1–3, and 1–2 in NH4OH, MEA, and DETA, respectively. Nonetheless, the most stable Ni complexes in this study are those with x = 6, 3, and 2 for NH4OH, MEA, and DETA, respectively (see Table S2, ESI†). One key side reaction to consider is the hydrogen evolution reaction (HER) at the cathode, which competes for electrons (R4). HER requires a similar number of electrons as Ni electrodeposition which implies that neither has a kinetic advantage. However, HER is favoured at higher potentials, and lower Ni concentrations,19 due to rapidly depleting supersaturation around the electrode surface leading to lower Ni electrodeposition Coulombic efficiencies (CE).16
To evaluate the effect of NH4OH, MEA, and DETA on the electrochemical reduction of Ni, linear sweep voltammetry (LSV) curves are obtained in Fig. S7 (ESI†). Two significant differences were observed among the three solvents. First, the reduction potentials for Ni were more negative for DETA (−0.638 V vs. RHE) compared to MEA (−0.496 V vs. RHE) and NH4OH (−0.454 V vs. RHE). The more negative reduction potential with DETA is attributed to the formation of more stable Ni complexes, requiring greater energy for Ni deposition. Although Ni–MEA complexes are less stable than Ni–NH3, MEA shows a more negative reduction potential, likely due to slower mass transport of the bulkier Ni−MEA complex, resulting in diffusion-limited behavior. Second, the slopes of the LSV curves decrease in the order: NH4OH > MEA > DETA, consistent with decreasing mass transport rates of Ni ions toward the electrode surface.11
Since regeneration of the impurity-free CO2 loaded solvent is of importance, it is essential that these reactions are carried out in a two-chamber cell to facilitate the migration of Cl− ions. It has been reported that the chlorine evolution reactions could occur in a one-chamber cell at the anode leading to the evolution of chlorine gas.16 Moreover the formation of Cl2 gas could result in a homogenous reaction with NH3 to produce N2 leading to ammonia consumption.11 Ammonia consumption through this reaction has been reported in the order of 0.193 kg of NH3/kg of Ni.11 Furthermore, the buildup of Cl− ions could lead to increased acidity and solvent degradation. These side reactions can be avoided by the proposed two chamber cell configuration with an AEM to facilitate the migration of Cl− ions to the anode. At the anode, oxygen evolution reaction (OER) occurs producing protons that stabilize the Cl− ions to generate HCl as shown in (R7)–(R8).
Fig. 3(A) shows the (CE) and the yield rate of Ni per unit area of titanium electrode for each solvent at a galvanostatic hold of 100 mA for 1 hour. The highest yield rates and CE of 32 mg h−1 cm−1 and 29% respectively, are obtained with NH4OH. The CE and rate of deposition of these systems is dependent on the starting concentration of Ni and the electrolyte.19 CE of 45% have been reported for electrodeposition of 1700 ppm Ni from a single chamber cell in an ammoniacal buffer system consisting of (NH4)2SO4, NH3 and H2SO4.16 Moreover the CE is observed to slowly decrease with time (Fig. S8, ESI†), which matches the asymptotic behaviour also observed with Ni recovery efficiencies as a function of time shown in Fig. 3(B). Up to 90% of Ni is electrodeposited after 4 hours of electrolysis from NH4OH and MEA, and up to 4% with DETA. DETA shows early asymptotic behavior, resulting in slower deposition rates, likely due to the transformation of NiDETA into the more stable Ni(DETA)2 complex as the pH rises due to competing hydrogen evolution reaction (HER) (Table S2, ESI†). A pH increase of 3.64 from neutral conditions is observed, suggesting that Ni(DETA)2 complex becomes more prominent in this system according to thermodynamic speciation calculations shown in Fig. S6 (ESI†), implying that a buffer system is required. A similar slow pH increase is observed with NH4OH and MEA which is detrimental for electrodeposition as Ni species undergo hydrolysis at higher pH conditions to precipitate as hydroxides.
 | ||
| Fig. 3 (A) Ni yield rate and coulombic efficiencies in different CO2 capture solvents at galvanostatic hold of 100 mA for 1 h. (B) Ni recovery efficiency vs. time for NH4OH, MEA and DETA. (C) Effect of CO2 loading on Ni recovery. (D) Integrated experiments showing product distribution as carbonates, electrodeposited material and unrecovered metal. | ||
The pH in these systems is typically modulated by adding a buffer, with boric acid commonly used to both regulate pH and reduce overpotential by forming weak Ni borate complex (Ni(H2BO3)2) in solution.20 Alternatively, we propose using CO2, which dissolves and speciates into carbonate and bicarbonate, consuming OH− ions from the HER (R4)–(R6), and controlling pH. CO2 also forms carbamate species with amine which could also aid in Ni extraction. As shown in Fig. 3(C), continuous CO2 supply during electrodeposition improves recovery efficiencies by 1.02, 1.03, and 2.98 times for NH4OH, MEA and DETA respectively after 120 min. Further, the pH change was −0.01, −0.21, and 0.57 for NH4OH, MEA and DETA respectively, compared to 2.37, 3.03, and 3.64 obtained without CO2. The relatively higher ΔpH with DETA compared to MEA and NH4OH is also an indication of the lower Ni extraction CE obtained in Fig. 3(A) due to HER. The effect of CO2 on Ni electrodeposition appears to be primarily through pH modulation, not complexation. Experiments performed using CO2 loaded solvent after REE separation showed no significant enhancement in Ni recovery, despite NMR analysis confirming the presence of similar amine-CO2 species such as carbamates, HCO3/CO32− ions (Fig. S9, ESI†).
The chemical phases, structural and morphological features of the electrodeposited Ni, is discussed using XRD, SEM and XPS analyses. Fig. S10(a)–(c) (ESI†) shows the formation of dark shiny particles on the surface of the electrode indicating a successful electrowinning process. Ni particles of different sizes are observed in Fig. S11(a)–(f) (ESI†). The Ni deposit obtained for NH4OH, MEA, and DETA at galvanostatic hold of 100 mA appeared to be a non-compact, silvery dark powder with spherical morphology which could be easily scraped off the titanium sheet electrode (Fig. S11(a)–(f), ESI†). It is important to also disclose that a different sheet-like morphology is observed when working at lower current densities <5 mA (see Fig. S12c, ESI†). XRD analysis on the scraped powder showed the presence of 111, 200, and 220 phases in Fig. S12 (ESI†) associated with pure metallic Ni (PDF 03-065-2865). This is confirmed by high resolution XPS scans which showed the characteristic binding energy of Ni0 for 2p3/2 at 852.7 eV with ΔE of 17.27 from 2p1/2 indicating the presence of pure metallic Ni. This Ni0 peak decreased in intensity when comparing NH4OH to MEA and could not be detected in the Ni species from DETA. XRD analysis on the Ti electrode bearing Ni for DETA confirms the presence of Ni oxides as opposed to pure metallic Ni observed with NH4OH and MEA.
To illustrate the flexibility of this concept for integrated CO2 capture and the separation of REEs and Ni, we performed a stepwise separation of La and Ni using CO2 loaded aqueous NH4OH, MEA and DETA. Lanthanum carbonate phases with > 99.5% yield were observed in all solvents (Fig. 3(D)), however, 12.5% and 15.3% Ni-carbonates are also co-recovered. Product purity could be improved by controlling CO2 concentration in the solution. The similar stability constants of NiCO3(aq) and Ni-NH4OH(aq)/Ni-MEA(aq) complexes led to partial Ni precipitation as carbonate. However, insignificant quantities of Ni are observed with DETA due to the formation of a significantly stronger complex. This stronger complexation challenges electrochemical Ni recovery in the subsequent step. Ni recovery up to 85% and 81% is obtained using NH4OH and MEA, but only 11% using DETA. To improve Ni electrodeposition, higher temperatures can enhance electrolyte conductivity, ion diffusivity, and charge transfer rates leading to better recovery efficiencies, however, the risk of solvent decomposition exists. Alternatively, solvents that binds Ni preferentially over REE (logK values 10–18) could be explored, ensuring Ni binding is not too strong to hinder electrodeposition. Additionally, solvents must be chemically stable and efficient in CO2 capture.
In summary, using CO2 capture solvents to separate REEs and Ni is highly effective, achieving REE yields up to 99% with product purities of 80%, 79% and 98% for NH4OH, MEA and DETA, respectively. While DETA is more effective for REE–Ni separation, its strong Ni-complex formation challenges nickel recovery during electrodeposition. Also, the supply of CO2 during electrodeposition improves Ni recovery by modulating pH and forming alternative complexes. Scalable deployment can be achieved by optimizing this approach to maximize H2 and O2 recovery and ensure efficient solvent regeneration over multiple cycles.
This work utilized the Cornell Center for Materials Research Facility. This work is supported by Cornell’s Engineering Learning Initiatives and Sprout Awards, and the Link Foundation Energy Fellowship.
Data availability
The data supporting this article has been included as part of the ESI.†Conflicts of interest
G. G. co-founded Carbon To Stone to valorize emissions for producing high value products.Notes and references
- Dolf. Gielen, International Renewable Energy Agency, 2021.
- É. Lèbre, et al. , Environ. Sci. Technol., 2019, 53, 10571–10579 CrossRef PubMed.
- P. Ochonma, et al. , Acc. Chem. Res., 2024, 57, 267–274 CrossRef CAS PubMed.
- J. Septavaux, et al. , Nat. Chem., 2020, 12, 202–212 CrossRef CAS PubMed.
- E. G. Polyakov and A. S. Sibilev, Metallurgist, 2015, 59, 368–373 CrossRef CAS.
- A. Rout and K. Binnemans, Phys. Chem. Chem. Phys., 2016, 18, 16039–16045 RSC.
- A. Rout, et al. , RSC Adv., 2014, 4, 5753–5758 RSC.
- S. Satpathy and S. Mishra, Sep. Purif. Technol., 2017, 179, 513–522 CrossRef CAS.
- T. Norgate and S. Jahanshahi, Miner. Eng., 2011, 24, 1563–1570 CrossRef CAS.
- S. Katre, et al. , Phys. Chem. Chem. Phys., 2024, 26, 9264–9283 RSC.
- R. Cruz-Gaona, et al., Electrochemistry in Mineral and Metal Processing VI: Proceedings of the International Symposium, 2003, 304.
- P. Kim, et al. , J. Mater. Eng. Perform., 2020, 29, 5564–5573 CrossRef CAS.
- X. H. Zhang, et al. , J. Therm. Anal. Calorim., 2015, 119, 1713–1722 CrossRef CAS.
- M. A. Rhamdhani, et al. , Metall. Mater. Trans. B, 2008, 39, 218–233 CrossRef.
- M. S. Vidhya, et al. , RSC Adv., 2020, 10, 19410–19418 RSC.
- C. Lupi, et al. , Miner. Eng., 2006, 19, 1246–1250 CrossRef CAS.
- P. Ochonma, et al. , React Chem. Eng., 2023, 8, 1943–1959 RSC.
- M. Kim, et al. , Energy Fuel, 2024, 38, 15812–15822 CrossRef CAS.
- T. Tawonezvi, et al. , Chem. Eng. J. Adv., 2024, 17, 100579 CrossRef CAS.
- J. Ji, et al. , J. Appl. Electrochem., 1995, 25, 642–650 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cc04632c |
| This journal is © The Royal Society of Chemistry 2025 |