
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Improved photocatalytic carbon dioxide reduction over Bi-doped CeO2 by strain engineering†
Ang
Tian
a,
Ziyu
Mei
b,
Luyuan
Wang
b,
Guangliang
Liu
c,
Zhiguo
Liu
d,
Guangming
Kong
d,
Wenjun
Tang
*e and
Chuangwei
Liu
 *b
*b
aLiaoning Provincial Key Laboratory of Metallurgical Resources Circulation Science, Northeastern University, Shenyang, 110819, P. R. China
bState Key Laboratory of Catalysis, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, China. E-mail: cwliu@dicp.ac.cn
cSichuan Anxinda Rare Earth Technology Co. Ltd, Ya'an, 625400, P. R. China
dNaval Aviation University Qingdao Campus, Qingdao, Shandong 266041, China
eSchool of Materials and Energy, University of Electronic Science and Technology of China, Chengdu 610054, P. R. China. E-mail: wenjuntang9711@gmail.com
First published on 27th February 2024
Abstract
Room-temperature photocatalytic carbon dioxide reduction reaction (CRR) is an essential method for reducing carbon footprint and achieving valuable fuels. The key challenge to accelerating the process is enhancing the catalytic rate and product selectivity. Herein, we investigate the conversion of carbon dioxide to formic acid on Bi-doped CeO2 in the presence of tensile and compressive strain by using density functional theory corrected for on-site coulombic interactions. As demonstrated, the dopant atom not only benefits the oxygen vacancy formed, but also transfers some electrons to the Ti3+ site, which is the main catalytic site for the CRR. The promising model has excellent product selectivity, offering the best catalytic performance for formic acid (ΔGmax = 0.64 eV). Moreover, the catalytic performance is further improved by the compressive strain. The work provides novel insights into designing environment-friendly and low-cost CeO2-based photocatalysts for carbon reduction.
Introduction
The combustion of fossil fuels not only provides power for society's development, but also releases large amounts of greenhouse gas, especially carbon dioxide (CO2).1–3 To mitigate human-induced climate change, conversion of CO2 to valuable carbon-based fuels is deemed as an ideal technology to store renewable energy and alleviate the global warming issue.4,5 Therefore, many catalytic methods were developed for the carbon dioxide reduction reaction (CRR) under ambient conditions, including thermal catalysis, electrocatalysis, and photocatalysis.6–8 In comparison with electrochemical and thermal catalysis all needing external massive energy input, photocatalytic reduction of CO2 on semiconductor materials is driven by solar electricity and water at room temperature.9–11 On account of the inert nature of the CO2 molecule, the CRR is a significantly challenging process.12,13 In addition, owing to the product diversity, it is hard to acquire a pristine valuable chemical during the reduction process.13–15 Therefore, it is urgent to find promising photocatalysts that hold excellent catalytic performance and product selectivity.16,17In the literature, numerous photocatalysts have been extensively reported, including Zn,18,19 noble metals,20–22 heterostructures,23,24 TiO2,14,25 MgO,26 In2O3,27 and CeO2.28,29 However, the low conversion efficiency hinders the commercial application of those photo-materials in the CRR.30,31 Ideally, the photocatalyst should have high light utilization and low electron–hole carrier recombination rate.32,33 Among those candidate materials, CeO2-based photocatalysts take advantage of thermal stability, cheap price, good redox ability, and high oxygen storage capacity.34–36 Methanol (CH3OH) was successfully synthesized on CeO2 nanoparticles by introducing oxygen vacancies under the assistance of sunlight.37 In addition, CO2 was converted to dimethyl carbonate on the pure CeO2 surface with surface vacancy clusters. It is claimed that frustrated Lewis pairs benefit the C–C coupling and stabilize the key intermediates.38 Unfortunately, narrow light absorption and product diversity make pure materials far away from the industrial requirement. To improve the reaction rate of the CRR and light absorption efficiency, a dopant is introduced into CeO2, achieving excellent catalytic performance, because it enhances the ionic conductivity, and manipulates the electronic structure and oxygen vacancy formation. Consequently, many individual dopants were investigated such as Ga, Sm, La, Bi, and Pr.39–42 Importantly, compressive strain engineering also has been widely conducted to tune the electronic structure and decrease the recombination rate of electron–hole pairs.7,40
In this work, we chose the bismuth (Bi) atom as the dopant for improving the CRR catalytic performance on the CeO2 (111). It has two reasons: first, the ionic conductivity and band gap of CeO2 are significantly improved by the addition of Bi3+/Bi5+. Second, the Bi atom has been confirmed as a formate-producing material with high product selectivity. Then, we compare the CRR activity on oxygen vacancies (VO), substituting one lattice Ce (SBi) with an oxygen vacancy (VBi), and substituting two lattice Ce atoms with an oxygen vacancy (VBis), by using density functional theory corrected for on-site coulombic interactions (DFT + U, Uf = 4). More importantly, the CRR activity of promising photocatalysts is improved by introducing the compressive strain. This exploration offers insights into the strain engineering influence on the turnover frequency (TOF) by tuning the intermediates and kinetic barrier.
Computational methods
In this work, all the computations about those structures and energies were based on the standard density functional theory using the Vienna ab initio Simulation Package (6.3.2 VASP).43 The electron-exchange correlation functional was treated through the generalized gradient approximation (GGA) with the revised Perdew–Burke–Ernzerhof (RPBE), due to the functional giving more suitable results for adsorbed intermediates.44,45 The 500 eV energy cutoff was adopted and the Brillouin zone was sampled with 3 × 3 × 1 k-points using the Monkhorst–Pack scheme grid for geometry optimization and self-consistent calculations. We adopted 9 × 9 × 1 k-points for band gap and density of state (DOS) calculations. A Hubbard U term was used to describe the electron localization on the 4f shells of the Ce atom.41 A vacuum space exceeding 20 Å was employed to avoid the interaction between two periodic units. The atomic position was fully relaxed until the maximum force on each atom was less than −0.01 eV Å−1 and 10−5 eV. Thermal and zero-point energy (ZPE) corrections were calculated over Γ points. The van der Waals interaction has been considered using the DFT-D3 scheme.46,47 The free energy of adsorption of A species (ΔGA*) can be calculated by| ΔGA* = ΔEA* + ΔEZPE – TΔSA* |
| ΔEA* = EA* − Ecat − EA |
And the ΔEZPE is the zero-point energy difference between the adsorbed and the free state, and ΔSA* is their entropy difference. The energetic span approximation is applied to calculate the TOF:48
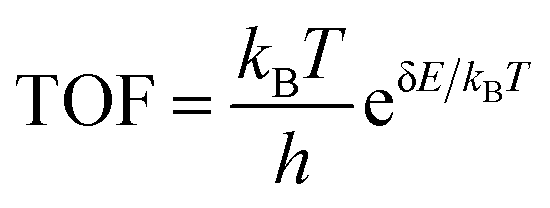
In this approximation, the free energies of the TOF-determining transition state (GTDTS) and TOF-determining intermediate state (GTDI), and the free energy of the reaction (ΔGr) define δE:
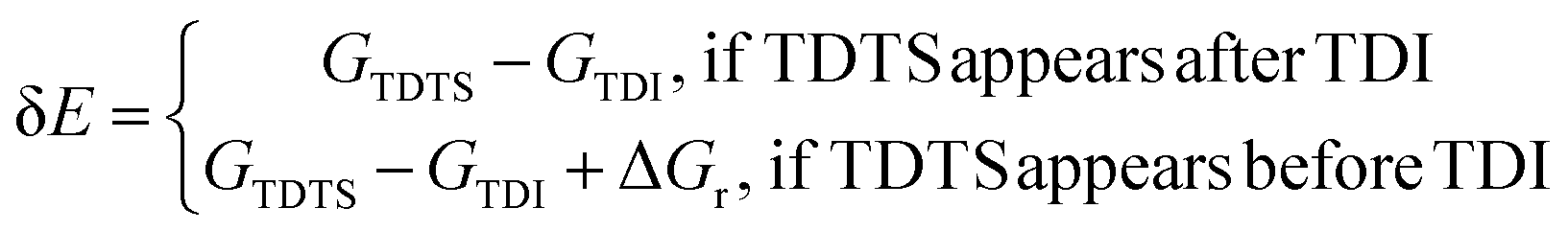
To approximate the energetic span, we calculate the thermodynamic energetic span as δE′ = GHI − GLI + ΔGr, where GHI is the free energy of the reaction intermediate with the highest free energy and GLI is the free energy of the reaction intermediate with the lowest free energy.
Results and discussion
The optimized configurations of the above four potential models are shown in Fig. S1,† corresponding to the side and top views, respectively. As shown in Fig. S1b,† the equilibrium structures of SBi retain perfect CeO2(111) structural characteristics, indicating that the Bi atom has a similar size to the Ce. Fig. 1a shows the different distributions of Bi doping with or without oxygen vacancies in the CeO2(111). Initially, the thermodynamic stability of these concept photocatalysts is evaluated by the formation energy, as shown in Fig. 1b. The formation energy of simple substitution (SBi) and oxygen vacancies (VO) is positive, especially the value of VO is up to 1.85 eV. Therefore, the VO structure is very unstable under mild conditions. Interestingly, the formation energy changes to negative when introducing the Bi dopant into the pure material with one oxygen vacancy. The value decreases with the increase of dopants, and the formation energy of VBis almost closes to zero (−0.03 eV). As described by the energy, those concept-catalysts could be easily synthesized by experimental methods.49,50 Then the CRR performance of SBi, VBi, and VBis is further evaluated in the following part. By comparing the work function (Fig. 1c) and optimized structures of these surfaces, the following results can be deduced: (1) the Bi-dopant improves the ability of electromigration; (2) VBi has the lowest work function. Then the potentially active surface is further analyzed by electron localization function and Bader charge studies (Fig. 1d–f). Every oxygen atom receives about 1.15|e| electron from the Ce atom, losing about 2.30|e|. While the Ce and O atoms still keep the same number of electron exchanges in the SBi structure. In other words, the Bi transfers the same number of electrons (2.30|e|) to the oxygen atom. The Bader charge of the O atom still keeps the same value after introducing the oxygen vacancy, and electron transfer of Bi and Ce is decreased. In addition, some pioneering studies confirmed that oxygen vacancies in the CeO2 surface benefit the frustrated Lewis pair (FLP) catalysts formed.38,51,52 The local electron density and the steric electron transferred are redistributed in the surface defects in CeO2. The Bi–O–Ce structure not only narrows the band gap of CeO2, but also enhances the rapid transfer of electrons between the reaction intermediates and the catalyst.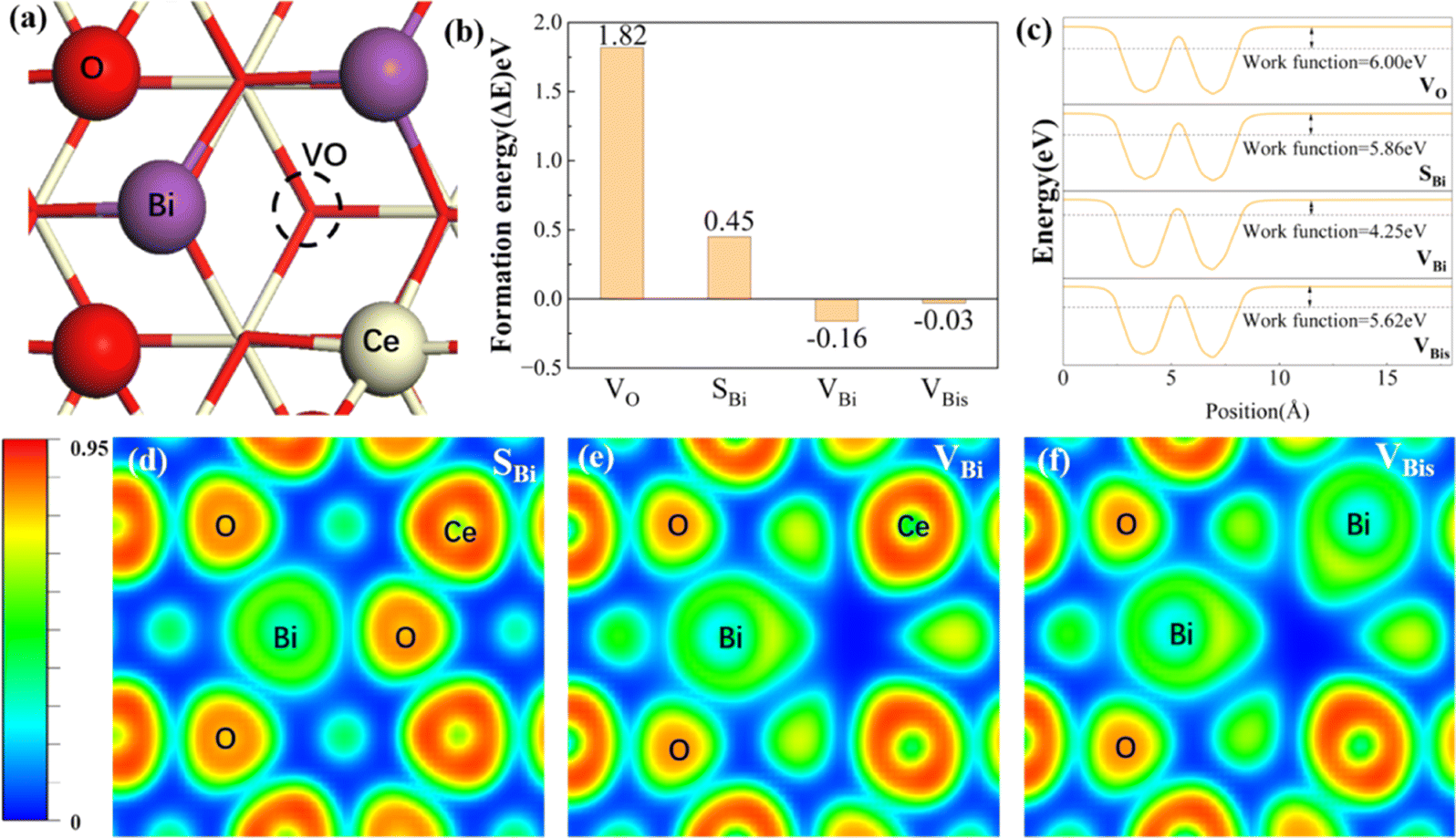 | ||
| Fig. 1 (a) Configuration of four concept structures in CeO2 (111). The formation energy (b), work function (c), and electron localization function (d–f) of the above four materials. White, red, and purple represent Ce, O, and Bi, respectively. | ||
As we know, the CRR is a complicated process, and many products will occur on the different catalysts, such as CO, CH4, HCOOH, and C2+. Various efforts confirm the first reduction intermediate is key for the selectivity of CO2 reduction.53 For example, the *OCHO is considered as a feasible indicator for HCOOH, however, the adsorption energy of *CO has a high relationship with the C2+ products. In the initial adsorption process, the distance between the CO2 and active site is larger than 3.5 Å with a small energy input. In other words, the CO2 molecule is adsorbed on the above three promising surfaces by physical adsorption. For the first reduction step (CO2 → *COOH), the activation energy on the SBi needs about 2.22 eV, and the intermediate (*COOH) still lies in the vacuum space (Fig. S2†). Therefore, the structure is not fit for the CRR under ambient conditions, while two intermediates, including *COOH and *OCHO, are produced on the VBi model in the first reduction step. Interestingly, the catalytically active site is the Ce atom instead of the Bi site. The first reduction step with ΔG values being 0.89 eV and −0.69 eV for *COOH and *OCHO intermediates, respectively, indicates that the *OCHO configuration should dominate the reduction process. Specifically, the bond length of C–O is up to 1.28 Å (1.21 Å for free gas) indicating that the Ce remarkably injects electrons into the intermediate. Bader charge analyses indicate that the *COOH and *OCHO receive 0.68 and 0.76|e| from the substrate, respectively, in which the Ce atom transfers the most electrons to intermediates. In the following hydrogenation step, the reaction energy still needs about 0.31 eV, and CO is provided during the step. Subsequently, the CO will be easily released with an exothermic process. During the *OCHO → *HCOOH step, the reaction is endothermic in the energy profile by 0.64 in Fig. 2b. The formic acid is spontaneously released from the VBi surface with a small energy release (0.43 eV). No matter what, for the two hydrogenation pathways of CO2, *OCHO exhibits a stronger thermodynamic tendency compared to *COOH, and particularly noteworthy is that the further hydrogenation of *OCHO in the VBi structure reaches a ΔG of −0.86 eV, indicating that this pathway is thermodynamically the most favourable.
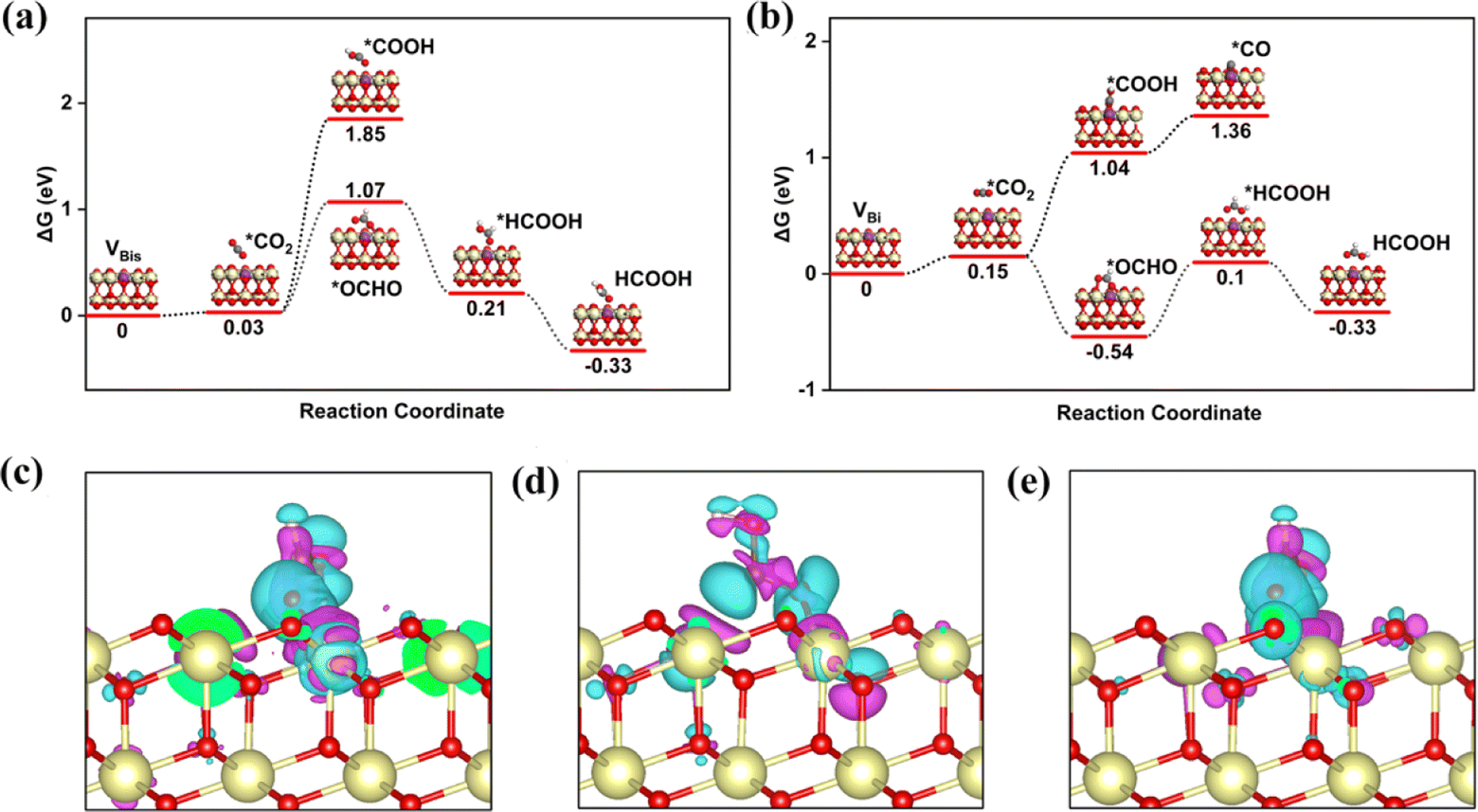 | ||
| Fig. 2 Free energy profile for the CRR on VBi (a) and VBis (b) with various intermediates. Electron density difference plots for *OCHO (c), *COOH (d) on VBi, and *COOH (d) on VBis. The isosurface value was set to 0.003 e Å−3. The charge accumulation and loss are shown in the cyan and pink regions, respectively. Black represents C. | ||
The isosurfaces of the charge density of *OCHO and *COOH configurations for VBi have been investigated in Fig. 2c and d, which indicate that *OCHO is the favorable product from the more charge transfer from the substrate to *OCHO. Therefore, the water-solvation mechanism is the main reaction pathway. Following this route, the CRR performance of the VBis catalyst is estimated. The *COOH intermediate cannot be adsorbed on the VBis surface with an endothermic reaction process. But this reaction pathway is not eligible for CO2 activation due to the requirement of a heavy energy input (1.82 eV). For the *OCHO intermediate, the C–O bond length is up to 1.26 Å from 1.21 Å, indicating that the C–O is activated slightly in this process. And the input activation energy in this way is about 1.04 eV. Then the HCOOH is released by two exothermic processes. And the *OCHO chemisorption on VBis is presented in Fig. 2e, along with the corresponding charge density differences. The charge accumulation and Bader charge studies of *OCHO (0.66|e|) indicate that the intermediate gains some electrons from the substrate. From the above results, four features are achieved: (a) a slight energy requirement for CO2 physical adsorption is observed on the catalyst surface; (b) the catalytic site is the Ce atom, while the Bi atom benefits the oxygen vacancy formed; (c) HCOOH is the preferred product; (d) the VBi structure not only holds the lowest work function, but also presents the best catalytic performance for the CRR (ΔGmax = 0.64 eV).
Researchers found that the oxygen vacancy formation energy and bandgap for the Bi-doped CeO2 are reduced by electronic and structural modification, improving the redox properties.54,55 In addition, the Bi-doped CeO2 with one oxygen vacancy remarkably benefits the photogenerated electron–hole pair separation, indicating that more electrons are produced during light irradiation. In order to deeply study the role of the Bi atom on the CeO2 (111) structure for effective CRR, the electronic band structure is calculated for the bulk-CeO2, VBi, and VBis, as shown in Fig. 3a and b. The bandgaps for the three structures are 1.98, 1.76, and 1.85 eV, respectively, by using RPBE + U. It can be seen that the value of the bulk phase is consistent with the previously reported values. The neutral oxygen vacancy with Bi-doping leaves two electrons to the adjacent Ce atoms, reducing the valency from Ce4+ to Ce3+. Moreover, the dopant presents Bi5+ under this condition, which makes the surrounding Ce atoms receive more electrons. As is known, O2P presents the valence band (VB), and the metal atom donates the conduction band (CB). In addition, the oxygen vacancy states provide the occupied CB states, confirming the presence of Ce3+ ions. Moreover, the electronic band structure of CeO2 is changed by the d orbital of the Bi atom, and a new band between the VB and CB is contributed. The reduced bandgap indicates that the conductive properties of Bi-doped CeO2 are changed, because the Bi improves ionic conductivity. The high ionic conductivity also further decreases the formation energy of oxygen vacancies. Amazingly, the VBi structure has the relatively lowest bandgap, which is consistent with the work function result, overall, the VBi structure is confirmed as an excellent photocatalyst for the CRR.
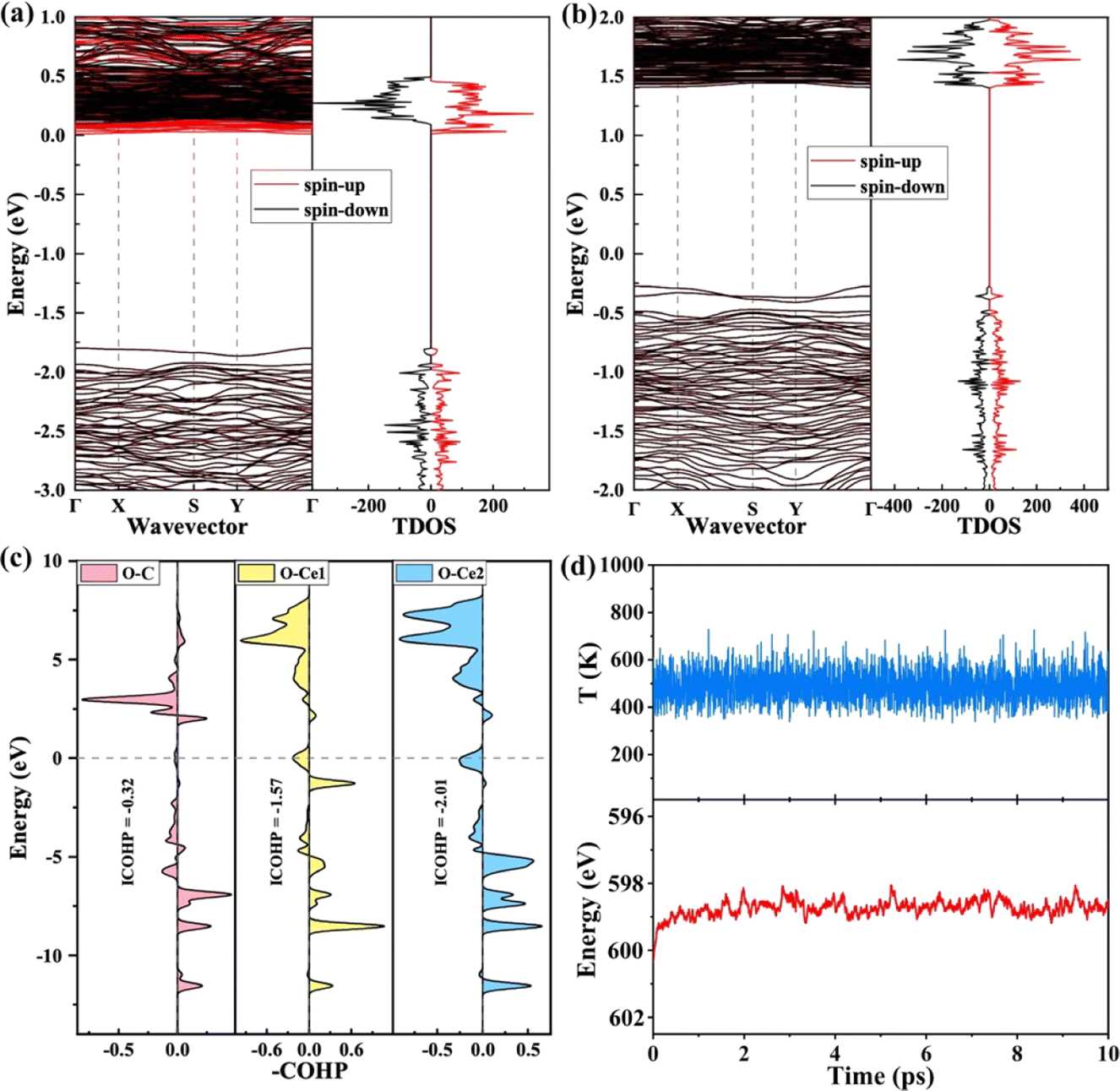 | ||
| Fig. 3 Calculated electronic structures of VBi (a) and VBis (b). The Fermi energy is set as zero. (c) Projected crystal orbital Hamilton population (pCOHP) between O–C, O1–Ce1 and O2–Ce2. (d) Fluctuation of temperature and energy versus time (10 ps). | ||
Generally, the electron transfer between the active site and intermediate is essential for the reduction process. In other words, the electron-accumulated catalytic site can transfer more electrons to the *OCHO on the VBi structure. To support this claim, the projected crystal orbital Hamilton populations (pCOHP) of O–C, O1–Ce1, and O2–Ce2 have been summarized in Fig. 3c. Ce1 and Ce2 (yellow and blue lines) dominate all the valence bond states, which all agree with the Ce–O bonding nature. The bond O2–Ce2 is slightly stronger than the O1–Ce1 below the Fermi level. The conclusion is verified by the Bader charge result, according to that the O1 and O2 gain 1.14 and 1.23|e| from the catalytic sites, respectively. In brief, Bi–O–Ce structure coexistence can tune the electron distribution and build a synergetic effect mechanism for the CRR. To further guide the experimental work, the stability of the VBi structure is studied by Ab Initio Molecular Dynamics simulation (AIMD) under 500 K, as shown in Fig. 3d. The model still keeps the same structure under this temperature, indicating excellent thermodynamic stability under experimental conditions. The fluctuation temperature and energy of the promising material are further investigated about 10 ps. The plot shows that the fluctuation of the two features is close to the equilibrium state. Then, it can be claimed that the potential structure not only possesses excellent photocatalytic activity for the CRR under visible light, but also presents outstanding stability under the experimental reaction conditions.
Many theoretical and experimental studies confirmed that the strain plays an important role in modulating the adsorption energy of the intermediate and tuning the kinetic barriers. The first reduction step (*OCHO → *HCOOH) is the rate-determining step (RDS) in this work. So, the kinetic barrier of this step is investigated by using the Langmuir–Hinshelwood mechanism, as shown in Fig. 4a. The thermodynamic barrier (Ea) for hydrogenation along each reaction pathway enhances with increasing tensile strain, while the adsorption energy of intermediates reduces with increasing compressive strain. The adsorption energies of the two intermediates have a scaling relation with applied compressive strain, indicating that strain-induced stronger adsorption can stabilize transition states and reduce the kinetic barrier, which is critical to achieving large reaction rates. The result shows that the *OCHO and transition state (*OCHO → *HCOOH) have been considered as the TDI (TOF-determining intermediate) and TDTS (TOF-determining transition state), respectively. Finally, the Energetic Span Model is employed to calculate the turnover frequency (TOF) under 300 K, and the calculation results are summarized in Fig. 4b. The profile indicates that the potential catalyst presents the best CRR catalytic performance under comprehensive strain. Our conclusion offers a direct relationship between kinetic parameters and experimentally accessible descriptors.
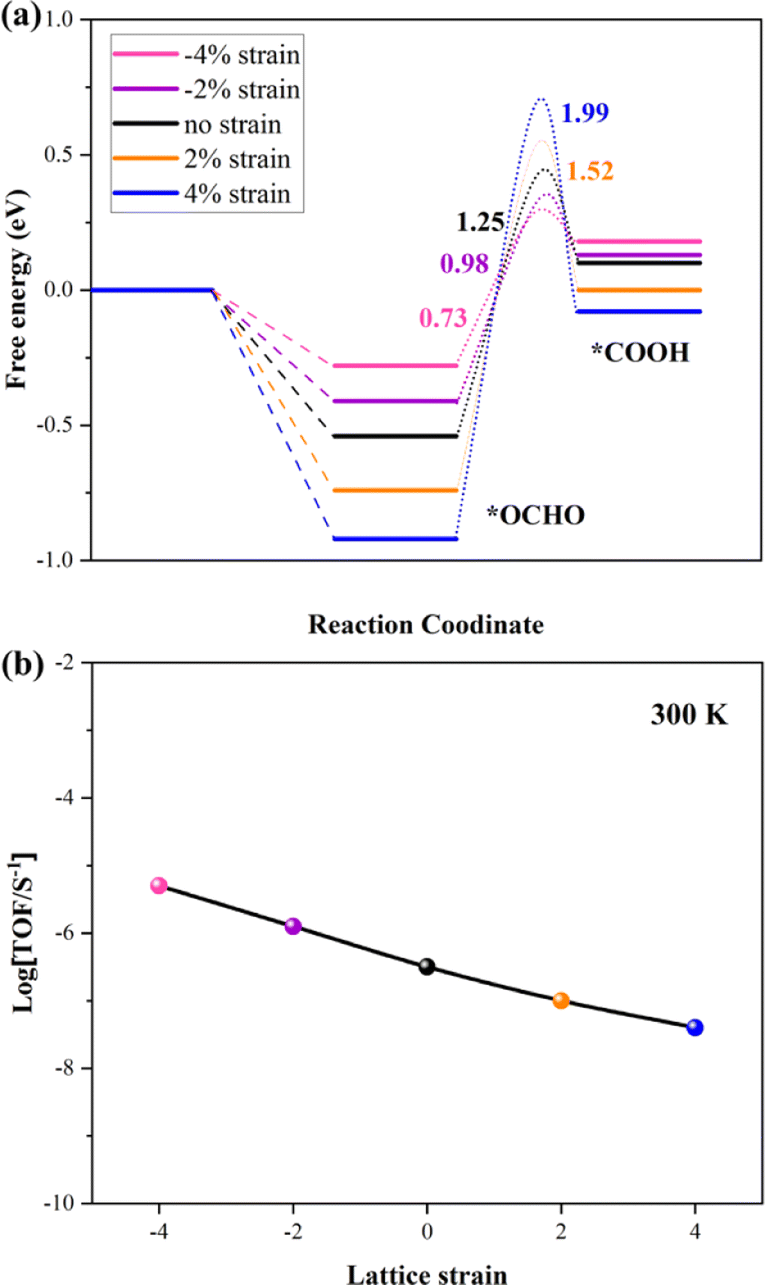 | ||
| Fig. 4 (a) Kinetic barriers for *OCHO to *COOH on the VBi structure with different strains. (b) TOF of the CRR versus different strains under 300 K. | ||
Conclusions
In summary, by performing first-principles calculations, a new single Bi atom doped CeO2 catalyst for the conversion of CO2 to HCOOH is proposed under ambient conditions. By comparing the reaction activity and product selectivity, the VBi structure is considered an outstanding photocatalyst for the CRR under ambient conditions. While the Ti with one oxygen vacancy is the main catalytic site, the Bi-dopant benefits the vacancy formed and rearranges the electrons on the surface. The main product is formic acid, having a relatively low thermodynamics (0.64 eV). Furthermore, the AIMD results show that the model expresses good kinetic stability with experimental methods. With applying compressive strain, the adsorption energy of the intermediate and the kinetic barrier of RDS all decreased. In other words, the catalytic activity increases with an increase in the compressive stain. TOF achieves the maximum value under 4% compressive strain under 300 K. These results provide novel routes for the rational design and development of excellent CeO2-based photocatalysts toward carbon reduction.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the Foundation of China's National Key R&D Programme (grant no. 2023YFB3810600) and Basic Scientific Research Foundation of Central College (grant no. 2125032). The authors also acknowledge funding of Technology and Innovation (grant no. KJ2021-01), which is supported by Ya'an City.References
- C. Liu, A. Tian, Q. Li, T. Wang, G. Qin, S. Li and C. Sun, Adv. Funct. Mater., 2023, 33, 2210759 CrossRef CAS.
- Q. Hao, C. Liu, G. Jia, Y. Wang, H. Arandiyan, W. Wei and B.-J. Ni, Mater. Horiz., 2020, 7, 1014–1029 RSC.
- H. Yuan, J.-H. Mei, Y.-N. Gong, D.-C. Zhong and T.-B. Lu, Tungsten, 2023 DOI:10.1007/s42864-023-00208-2.
- Y. Yu, D. Wang, Y. Hong, T. Zhang, C. Liu, J. Chen, G. Qin and S. Li, Chem. Commun., 2022, 58, 11163–11166 RSC.
- Q. Wu, C. Liu, X. Su, Q. Yang, X. Wu, H. Zou, B. Long, X. Fan, Y. Liao, L. Duan, Z. Quan and S. Luo, ACS Nano, 2023, 17, 402–410 CrossRef CAS PubMed.
- S. Das, J. Pérez-Ramírez, J. Gong, N. Dewangan, K. Hidajat, B. C. Gates and S. Kawi, Chem. Soc. Rev., 2020, 49, 2937–3004 RSC.
- C. Liu, T. Wang, D. Hao, Q. Li, S. Li and C. Sun, J. Mater. Sci. Technol., 2022, 110, 96–102 CrossRef CAS.
- C. L. Hugelshofer, A. Borgschulte, E. Callini, S. K. Matam, J. Gehrig, D. T. Hog and A. Züttel, J. Phys. Chem. C, 2014, 118, 15940–15945 CrossRef CAS.
- E. V. Kondratenko, G. Mul, J. Baltrusaitis, G. O. Larrazábal and J. Pérez-Ramírez, Energy Environ. Sci., 2013, 6, 3112–3135 RSC.
- C. Liu, D. Hao, J. Ye, S. Ye, F. Zhou, H. Xie, G. Qin, J. Xu, J. Liu, S. Li and C. Sun, Adv. Energy Mater., 2023, 13, 2204126 CrossRef CAS.
- Ž. Kovačič, B. Likozar and M. Huš, ACS Catal., 2020, 10, 14984–15007 CrossRef.
- C. Costentin, M. Robert and J.-M. Savéant, Chem. Soc. Rev., 2013, 42, 2423–2436 RSC.
- Y. Jiao, Y. Zheng, P. Chen, M. Jaroniec and S.-Z. Qiao, J. Am. Chem. Soc., 2017, 139, 18093–18100 CrossRef CAS PubMed.
- J.-Y. Liu, X.-Q. Gong and A. N. Alexandrova, J. Phys. Chem. C, 2019, 123, 3505–3511 CrossRef CAS.
- Y. Chen, G. Jia, Y. Hu, G. Fan, Y. H. Tsang, Z. Li and Z. Zou, Sustainable Energy Fuels, 2017, 1, 1875–1898 RSC.
- Y. Miao, Y. Zhao, S. Zhang, R. Shi and T. Zhang, Adv. Mater., 2022, 34, 2200868 CrossRef CAS PubMed.
- B. Su, M. Zheng, W. Lin, X. F. Lu, D. Luan, S. Wang and X. W. Lou, Adv. Energy Mater., 2023, 13, 2203290 CrossRef CAS.
- Y. Zhao, G. Chen, T. Bian, C. Zhou, G. I. N. Waterhouse, L.-Z. Wu, C.-H. Tung, L. J. Smith, D. O'Hare and T. Zhang, Adv. Mater., 2015, 27, 7824–7831 CrossRef CAS PubMed.
- X. Xiong, Y. Zhao, R. Shi, W. Yin, Y. Zhao, G. I. N. Waterhouse and T. Zhang, Sci. Bull., 2020, 65, 987–994 CrossRef CAS PubMed.
- Q. L. Cheng Luo, B. Cheng, B. Zhu and L. Wang, Acta Phys.-Chim. Sin., 2023, 39, 2212026 Search PubMed.
- G. Huang, Q. Niu, J. Zhang, H. Huang, Q. Chen, J. Bi and L. Wu, Chem. Eng. J., 2022, 427, 131018 CrossRef CAS.
- B. Su, Y. Kong, S. Wang, S. Zuo, W. Lin, Y. Fang, Y. Hou, G. Zhang, H. Zhang and X. Wang, J. Am. Chem. Soc., 2023, 145, 27415–27423 CrossRef CAS PubMed.
- M. Hao, D. Wei and Z. Li, Energy Fuels, 2022, 36, 11524–11531 CrossRef CAS.
- L. Wang, X. Zhao, D. Lv, C. Liu, W. Lai, C. Sun, Z. Su, X. Xu, W. Hao, S. X. Dou and Y. Du, Adv. Mater., 2020, 32, 2004311 CrossRef CAS PubMed.
- C. Liu, A. Tian, H. Yang and X. Xue, Mater. Lett., 2013, 106, 1–4 CrossRef CAS.
- Y. Kohno, H. Ishikawa, T. Tanaka, T. Funabiki and S. Yoshida, Phys. Chem. Chem. Phys., 2001, 3, 1108–1113 RSC.
- K. K. Ghuman, T. E. Wood, L. B. Hoch, C. A. Mims, G. A. Ozin and C. V. Singh, Phys. Chem. Chem. Phys., 2015, 17, 14623–14635 RSC.
- K. R. Hahn, M. Iannuzzi, A. P. Seitsonen and J. Hutter, J. Phys. Chem. C, 2013, 117, 1701–1711 CrossRef CAS.
- A. Tian, T. Ma, X. Shi, D. Wang, W. Wu, C. Liu and W. Pei, Coatings, 2021, 11, 1095 CrossRef CAS.
- M. Kanemoto, T. Shiragami, C. Pac and S. Yanagida, J. Phys. Chem., 1992, 96, 3521–3526 CrossRef CAS.
- Y. S. Chaudhary, T. W. Woolerton, C. S. Allen, J. H. Warner, E. Pierce, S. W. Ragsdale and F. A. Armstrong, Chem. Commun., 2012, 48, 58–60 RSC.
- K. Li, B. Peng and T. Peng, ACS Catal., 2016, 6, 7485–7527 CrossRef CAS.
- W. Tu, Y. Zhou and Z. Zou, Adv. Mater., 2014, 26, 4607–4626 CrossRef CAS PubMed.
- Y.-Q. Su, Y.-Y. Qin, T. Wu and D.-Y. Wu, J. Catal., 2022, 407, 353–363 CrossRef CAS.
- J. Cai, D. Li, L. Jiang, J. Yuan, Z. Li and K. Li, Energy Fuels, 2023, 37, 4878–4897 CrossRef CAS.
- T. Wu, T. Vegge and H. Anton Hansen, J. Catal., 2023, 420, 1–8 CrossRef CAS.
- A. Hezam, K. Namratha, Q. A. Drmosh, D. Ponnamma, J. Wang, S. Prasad, M. Ahamed, C. Cheng and K. Byrappa, ACS Appl. Nano Mater., 2020, 3, 138–148 CrossRef CAS.
- L. Li, W. Liu, R. Chen, S. Shang, X. Zhang, H. Wang, H. Zhang, B. Ye and Y. Xie, Angew. Chem., Int. Ed., 2022, 61, e202214490 CrossRef CAS PubMed.
- A. Shahzadi, S. Moeen, A. D. Khan, A. Haider, J. Haider, A. Ul-Hamid, W. Nabgan, I. Shahzadi, M. Ikram and A. Al-Shanini, ACS Omega, 2023, 8, 8605–8616 CrossRef CAS PubMed.
- S. Tang, L. Xu, B. Peng, F. Xiong, T. Chen, X. Luo, X. Huang, H. Li, J. Zeng, Z. Ma and L.-L. Wang, Appl. Surf. Sci., 2022, 575, 151655 CrossRef CAS.
- T. Wu, T. Vegge and H. A. Hansen, J. Catal., 2021, 402, 310–314 CrossRef CAS.
- T. Wu, Q. Deng, H. A. Hansen and T. Vegge, J. Phys. Chem. C, 2019, 123, 5507–5517 CrossRef CAS.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- C. Liu, Q. Li, C. Wu, J. Zhang, Y. Jin, D. R. MacFarlane and C. Sun, J. Am. Chem. Soc., 2019, 141, 2884–2888 CrossRef CAS PubMed.
- L. M. Azofra, N. Li, D. R. MacFarlane and C. Sun, Energy Environ. Sci., 2016, 9, 2545–2549 RSC.
- G. Mills, H. Jónsson and G. K. Schenter, Surf. Sci., 1995, 324, 305–337 CrossRef CAS.
- C. Liu, Q. Wang, J. Guo, T. Vegge, P. Chen and H. A. Hansen, ACS Catal., 2022, 12, 4194–4202 CrossRef CAS.
- C. Santra, A. Auroux and B. Chowdhury, RSC Adv., 2016, 6, 45330–45342 RSC.
- B. Cui, Y. Li, S. Li, Y. Xia, Z. Zheng and Y.-Q. Liu, Energy Fuels, 2020, 34, 9932–9939 CrossRef CAS.
- J. C. M. Pereira, M. Sajid, G. Kehr, A. M. Wright, B. Schirmer, Z.-W. Qu, S. Grimme, G. Erker and P. C. Ford, J. Am. Chem. Soc., 2014, 136, 513–519 CrossRef CAS PubMed.
- D. W. Stephan, Acc. Chem. Res., 2015, 48, 306–316 CrossRef CAS PubMed.
- X. Zhi, Y. Jiao, Y. Zheng, A. Vasileff and S.-Z. Qiao, Nano Energy, 2020, 71, 104601 CrossRef CAS.
- D. A. Andersson, S. I. Simak, B. Johansson, I. A. Abrikosov and N. V. Skorodumova, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 035109 CrossRef.
- Y. Tang, H. Zhang, L. Cui, C. Ouyang, S. Shi, W. Tang, H. Li, J.-S. Lee and L. Chen, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 125104 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3se01680c |
| This journal is © The Royal Society of Chemistry 2024 |