
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSilicon nanocrystal hybrid photocatalysts as models to understand solar fuels producing assemblies†
Simran S.
Saund
 a,
Abha
Dabak-Wakankar
a,
Melissa K.
Gish
a and
Nathan R.
Neale
*ab
a,
Abha
Dabak-Wakankar
a,
Melissa K.
Gish
a and
Nathan R.
Neale
*ab
aChemistry and Nanoscience Center, National Renewable Energy Laboratory, 15013 Denver West Parkway, Golden, Colorado 80401, USA. E-mail: Nathan.neale@nrel.gov
bRenewable and Sustainable Energy Institute, University of Colorado Boulder, Boulder, Colorado 80309, USA
First published on 12th December 2023
Abstract
Direct coupling of light-harvesting semiconductors and molecular catalysts is an attractive approach in designing new systems for artificial photosynthesis. Understanding the energetic requirements to enable photogenerated charge transfer from the semiconductor to the molecular catalyst is an essential design component of photoelectrochemical schemes. Here, we explore a model system to study these requirements by tethering a rhenium carbonyl coordination complex to a dodecyl-terminated silicon nanocrystal using a two-step surface functionalization process. Diffuse reflectance infrared Fourier transform spectroscopy and cyclic voltammetry characterized successful surface attachment and redox properties of the hybrid structure. UV-visible spectroelectrochemical measurements confirmed the formation of a known Re(0) reduction product in thin films of the hybrid assembly under cathodic potentials. During reduction, only spectroscopic features associated with the monomer were observed, not the Re–Re dimer, suggesting that the hybrid structure prevents a common deactivation pathway for Lehn-type CO2 reduction catalysts. Initial photocatalytic data showed that the NC-catalyst hybrid structure retained CO2 reduction activity to CO. Finally, transient absorption spectroscopy was used to examine the ultrafast dynamics of the system and shed light on the energetic alignment between the silicon semiconductor and the rhenium carbonyl complex.
Introduction
Photocatalytic conversion of small molecules such as carbon dioxide (CO2) and water (H2O) to thermodynamically uphill products, such as carbon monoxide (CO), hydrogen (H2) and oxygen (O2), has gained interest as one method for storing energy from sunlight in fuels and chemicals.1 To meet the exigencies of an evolving energy economy that requires both short- and long-term energy storage, new, more efficient methods for solar photoconversion are required that can mimic the natural photosynthetic process.Silicon's high natural abundance and well-controlled optoelectronic properties have made it the dominant semiconductor absorber material for photovoltaic devices2 and photoelectrochemical (PEC) solar fuels platforms.3 In Si PEC systems, the inherent catalytic inactivity of Si requires coupling with a suitable electrocatalyst. Most Si surface functionalization schemes tether catalysts to oxide surface layers,4–10 and examples of direct covalent attachment to Si are rare.11–14 Understanding the effects of surface immobilization on the catalyst's electrochemical activity as well as its ability to accept photogenerated charges from Si is an essential step in developing a practical PEC system.
In addition to bulk silicon, quantum-confined silicon nanocrystals (Si NCs) have arisen as a highly tuneable form of the popular semiconductor. With advances in synthesis and processing, high purity Si NCs with widely tunable band gaps are now accessible.15–20 Together, the excellent photophysical properties and high surface areas of Si NCs make them ideal candidates as model systems for fuel-forming solar photoconversion using bulk Si photoelectrodes.21,22 While metal chalcogenide NCs/quantum dots as the light harvesting semiconductor in a photocatalytic hybrid assembly with a surface-adsorbed catalyst has been explored widely,23–32 there are no reports of similar Si NC-catalyst hybrid assemblies. The only examples of Si NCs evolving H2 or reducing CO2 utilize the native Si NC as a stoichiometric hydride donor requiring elevated temperatures to operate.21,22,33–37
In this work we develop a Si NC-catalyst hybrid structure to enable fundamental studies on catalyst activity and photogenerated charge dynamics. We choose a Lehn-type Re complex given the well understood CO2 reduction reaction (CO2RR) mechanisms of this class of electrocatalysts. Briefly, Lehn-type [Re(bpy)(CO)3X] catalysts (bpy = 2,2′-bipyridine, X = halide) undergo two sequential electron transfer (ET) reactions where the first reduction occurs at the bpy ligand and the second reduction occurs at the metal center to yield the catalytically active, five-coordinate, doubly reduced complex (Scheme S1†). The first reduction is followed by halide ion dissociation, where the open coordination site at the Re metal center is filled by a Lewis basic solvent molecule such as acetonitrile. This charge neutral, singly reduced [Re(bpy)(CO)3L] (L = neutral donor ligand) undergoes L group loss and dimerization that deactivates the catalyst toward CO2RR (Scheme S1†). Thus, in any covalently tethered hybrid system with a Lehn-type CO2 reduction catalyst, a key goal is preventing catalyst deactivation via dimerization.
Here, we demonstrate tethering a molecular catalyst to a Si NC surface maintains catalyst activity toward CO2RR and arrests deleterious dimer formation using steady state spectroelectrochemical and CO product quantification measurements. However, we find from transient absorption spectroscopy that the energetics between the Si NC and CO2RR catalyst do not permit photoexcited electron transfer from the semiconductor to the electrocatalyst, highlighting the need for careful design of semiconductor-molecular catalyst assemblies.
Results and discussion
In this work, Si NCs are prepared by nonthermal plasma-enhanced chemical vapor deposition as previously reported.17,18 Stabilization of as-synthesized, 3.9 nm diameter, hydride-terminated Si NCs is carried out by the radical initiated reaction between 1-dodecene and silicon hydride surface sites to give dodecyl-terminated Si NCs (Si-C12). Dodecyl ligands provide excellent colloidal stability in non-polar solvents such as toluene, passivate surface defects and slow oxidation. Previously, our group has shown that surface saturation by long chain hydrophobic ligands requires at least 3 h reaction times at the operant temperature.18 The main goal in this work is to add functionality to the Si NC by decorating the surface with a Lehn-type CO2RR catalyst [Re(bpy)(CO)3Br],38,39 a process precluded by complete surface saturation with a hydrophobic ligand. Thus, we adopt a two-step synthetic approach (Scheme 1). First, we generate Si NCs with sparse dodecyl group coverage by limiting the reaction time to under 12 min instead of 3 h. This short reaction time provides a surface containing sufficient dodecyl groups to readily disperse the Si NCs in toluene but leaves available surface area for further functionalization. The second step involves tethering the catalyst via aldehyde insertion across a surface silicon hydride. We chose the Re precursor [Re(mcabpy)(CO)3Br] (where mcabpy is 4-methyl-2,2′-bipyridyl-4′-carboxaldehyde), the bromo analog of a previously reported complex,40 to generate the [Re(mcabpy)(CO)3Br]-functionalized Si NC product, termed Si-[Re].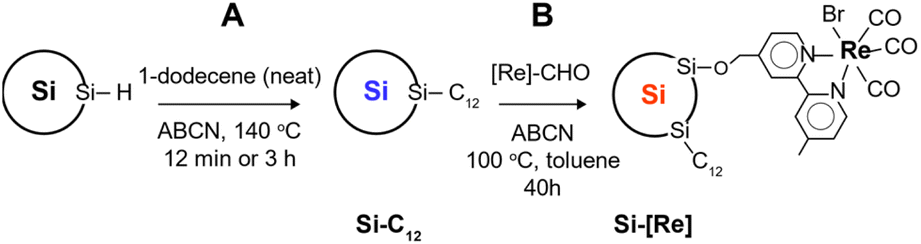 | ||
| Scheme 1 Synthetic scheme for Si-C12 ((A), 3 h reaction) and Si-[Re] ((A), 12 min reaction time, then (B), 40 h reaction time). | ||
Diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) of Si-[Re] thin films (Fig. 1 and S1†) confirm the facial Re(CO)3 motif is retained when tethered to the Si NC surface as identified by modes from 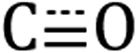 in-phase symmetric (A′(1), 2023 cm−1), equatorial asymmetric (A′′, 1924 cm−1), and out of phase symmetric (A′(2), 1904 cm−1) stretches.41,42 Conveniently, these
in-phase symmetric (A′(1), 2023 cm−1), equatorial asymmetric (A′′, 1924 cm−1), and out of phase symmetric (A′(2), 1904 cm−1) stretches.41,42 Conveniently, these 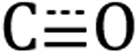 stretching frequencies are close to those of Si NC *Si–Hx (∼2090 cm−1) but are offset far enough towards lower energies to resolve all features independently. Due to the similar relative magnitude of the high oscillator strength
stretching frequencies are close to those of Si NC *Si–Hx (∼2090 cm−1) but are offset far enough towards lower energies to resolve all features independently. Due to the similar relative magnitude of the high oscillator strength 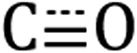 stretches and the *Si–Hx stretching region, it is reasonable to assume that the total surface [Re] coverage of individual Si NCs is quite low. Experiments to control and quantify the amount of [Re(mcabpy)(CO)3Br] are found to successfully lower the surface-bound [Re] loading (Fig. S2†), but quantification was not possible likely due to the variable dodecyl group coverage from step A in Scheme 1. Based on these and other experiments, we estimate the bounds of catalyst loading to be 1–10 [Re]/Si NC.
stretches and the *Si–Hx stretching region, it is reasonable to assume that the total surface [Re] coverage of individual Si NCs is quite low. Experiments to control and quantify the amount of [Re(mcabpy)(CO)3Br] are found to successfully lower the surface-bound [Re] loading (Fig. S2†), but quantification was not possible likely due to the variable dodecyl group coverage from step A in Scheme 1. Based on these and other experiments, we estimate the bounds of catalyst loading to be 1–10 [Re]/Si NC.
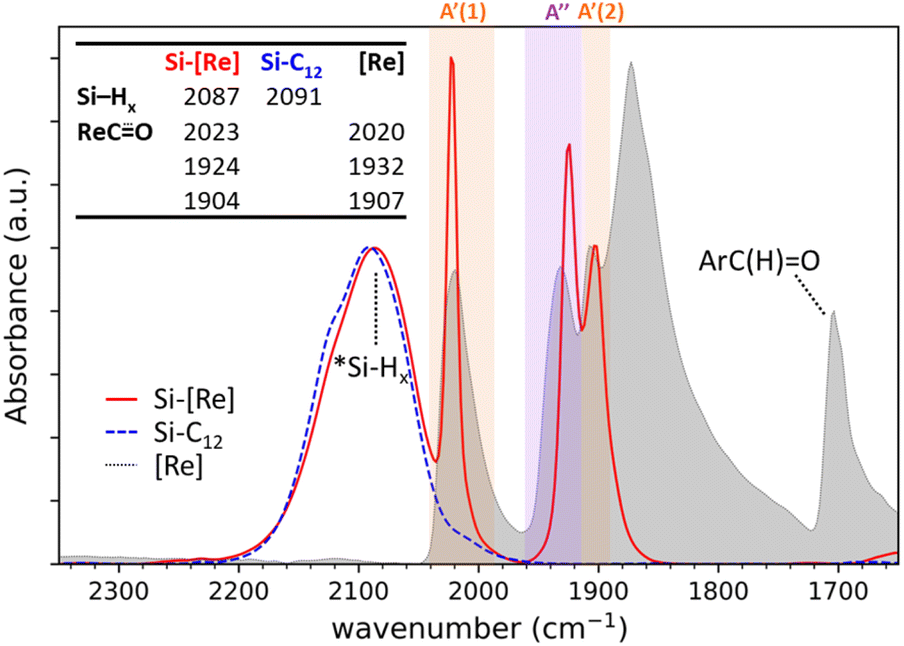 | ||
Fig. 1 DRIFTS spectra of the Si–Hx (unshaded), 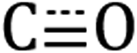 symmetric (orange shading) and asymmetric (purple shading) stretching regions of Si-[Re] (red solid line), Si-C12 (blue dashed line), and [Re] (grey fill). The Si NC spectra are normalized to the Si–Hx stretching region intensity and [Re] is scaled to the lowest energy Si-[Re] Re symmetric (orange shading) and asymmetric (purple shading) stretching regions of Si-[Re] (red solid line), Si-C12 (blue dashed line), and [Re] (grey fill). The Si NC spectra are normalized to the Si–Hx stretching region intensity and [Re] is scaled to the lowest energy Si-[Re] Re 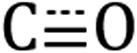 stretch. Specific peak values (in cm−1) for Si–Hx and Re stretch. Specific peak values (in cm−1) for Si–Hx and Re 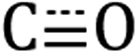 are listed in the inset table. are listed in the inset table. | ||
Next, we compare the features observed for Si-[Re] to DRIFTS spectra of the [Re] precursor and dodecyl-saturated Si-C12. The *Si–Hx stretching region for Si-C12 NCs (centered at 2091 cm−1) and Si-[Re] (2087 cm−1) are nearly identical, indicating that the Si-[Re] NC surface is dominated by dodecyl ligands. However, a slight shift in the *Si–Hx stretching region to lower energy for the Si-[Re] complex suggests that the Si-[Re] NCs are slightly more electron rich than Si-C12 NCs, consistent with binding of an electron-rich transition-metal complex. Similarly, the 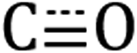 stretches for Si-[Re] NC and the Re precursor (2020, 1932, and 1907 cm−1) are closely matched, with the symmetric stretches at nearly indistinguishable frequencies and the Si-[Re] asymmetric
stretches for Si-[Re] NC and the Re precursor (2020, 1932, and 1907 cm−1) are closely matched, with the symmetric stretches at nearly indistinguishable frequencies and the Si-[Re] asymmetric 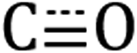 stretch shifted towards a slightly lower energy, a change characteristic of a more electron rich Re center. Combined with the disappearance of the intense aldehyde C
stretch shifted towards a slightly lower energy, a change characteristic of a more electron rich Re center. Combined with the disappearance of the intense aldehyde C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretch at 1703 cm−1 in the spectra of Si-[Re] NCs, these data provide strong evidence that the reaction chemistry in Scheme 1 is successful and occurs via reduction of the carboxaldehyde into an alkoxy anchoring group. After Si NC functionalization with [Re], a broad peak at 1062 cm−1 and a small shoulder at ∼2250 cm−1 appears for in the spectrum of Si-[Re] (Fig. S3†), characteristic of a surface Si–O stretch and a so-called ‘back-bonded’ (O)*Si–Hx stretch, respectively, providing additional evidence of the atomic surface structure as a methoxysilyl ether functionality.
O stretch at 1703 cm−1 in the spectra of Si-[Re] NCs, these data provide strong evidence that the reaction chemistry in Scheme 1 is successful and occurs via reduction of the carboxaldehyde into an alkoxy anchoring group. After Si NC functionalization with [Re], a broad peak at 1062 cm−1 and a small shoulder at ∼2250 cm−1 appears for in the spectrum of Si-[Re] (Fig. S3†), characteristic of a surface Si–O stretch and a so-called ‘back-bonded’ (O)*Si–Hx stretch, respectively, providing additional evidence of the atomic surface structure as a methoxysilyl ether functionality.
To further confirm that the reaction in Scheme 1 generates a surface-tethered Re complex and not residual precursor trapped in a Si-C12 matrix, we conduct a control experiment in which [Re(mcabpy)(CO)3Br] is replaced by [Re(bpy)(CO)3Br] (where bpy is 2,2′-bipyridyl). The lack of an aldehyde moiety on the bpy ligand prevents surface tethering through the aldehyde insertion reaction, which should leave free [Re(bpy)(CO)3Br] in solution with the colloidal Si-C12 NCs. We term this sample Si + [Re] to distinguish that this is a physical mixture. As shown in Fig. S4,† no trace of carbonyl 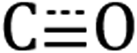 stretches are observed in DRIFTS spectra of the Si + [Re] sample, suggesting that the washing methodology completely removes free, unbound [Re(bpy)(CO)3Br] and further corroborates that the Si-[Re] NC sample contains surface-bound [Re(mcabpy)(CO)3Br].
stretches are observed in DRIFTS spectra of the Si + [Re] sample, suggesting that the washing methodology completely removes free, unbound [Re(bpy)(CO)3Br] and further corroborates that the Si-[Re] NC sample contains surface-bound [Re(mcabpy)(CO)3Br].
We further characterize the optical properties of the Si-[Re] NCs and find that the addition of a Re complex on the NC surface has little to no effect on the Si NC absorptive or emissive properties. First, the absorption spectra (normalized at 380 nm) of Si-C12 and Si-[Re] NC samples are nearly indistinguishable with Si-[Re] displaying only a slight increase in relative optical density between 280–350 nm that could be attributed to additional absorption from the surface-bound [Re] complex (Fig. S5†). Second, we observe no noticeable differences between the photoluminescence (PL) of Si-[Re] and Si-C12 (Fig. S6†).
Redox properties of the Si-[Re] NCs with surface-bound Re complexes are next characterized by a series of electrochemical studies on Si-[Re] NC thin films. Cyclic voltammograms are acquired in acetonitrile (CH3CN) with 100 mM tetrabutylammonium hexafluorophosphate (TBAPF6) as supporting electrolyte, a silver pseudo reference electrode, and a platinum wire counter electrode. All potentials are calibrated to the ferrocene (Fc+/0) redox couple. Working electrodes are prepared by dipping a freshly polished glassy carbon working electrode (GCE) into a toluene colloid of Si-[Re] NCs for approximately one second, removing the electrode and allowing the film to dry evaporatively. Upon scanning the Si-[Re] NC-coated working electrode cathodically, a reductive feature is observed with a peak potential (Ep) at −2.18 V vs. Fc+/0 (Fig. 2, red trace). On the reverse scan, three small anodic features are observed at −2.06, −1.79, and −0.57 V vs. Fc+/0. As controls, we electrochemically characterize films of the Si-C12 and Si + [Re] samples in the same manner (Fig. 2, blue dotted trace, and Fig. S7,† dashed orange trace, respectively). Neither of these experiments yield redox behavior in the same scan window, confirming that the features observed for thin films of Si-[Re] NCs originate from the surface bound Re complex. Blank CVs are acquired on the electrolyte with a clean GCE before and after each Si NC sample to check for degradative desorption of any redox active species from the Si NC thin films during cycling, for which no changes are observed in the baseline (Fig. S8†), suggesting that the catalyst does not desorb during the experiments.
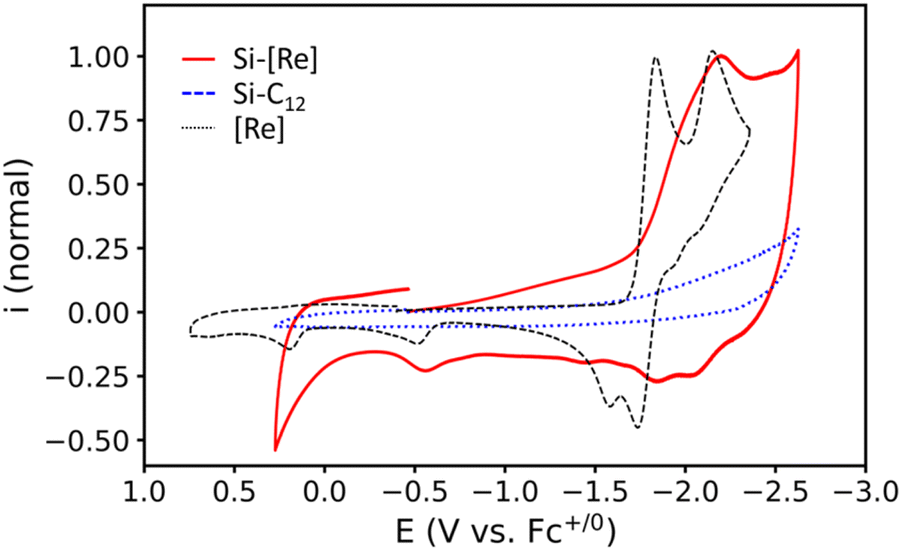 | ||
| Fig. 2 CVs of Si-[Re] (red solid line) and Si-C12 (blue dotted line) thin films on GCE, and a 1 mM [Re] solution (black dashed line) taken with an unaltered GCE. The red and black traces are normalized to their respective peaks near −2.15 V vs. Fc+/0. The blue trace is scaled to the same current magnitude as the red trace. All voltammograms were taken at 50 mV s−1 in CH3CN under an Ar atmosphere with 100 mM TBAPF6 as supporting electrolyte. | ||
To explore the nature of the reduced state of Si-[Re] we employ UV-visible spectroelectrochemistry to thin films on Si-[Re] deposited on ITO coated glass slides by drop casting. The electrode is placed in a 1.7 mm pathlength cuvette filled with CH3CN and 100 mM TBAPF6 as supporting electrolyte. A Ag pseudo reference electrode and Pt wire counter electrode are employed and the reference electrode is calibrated to a Fc+/0 standard solution before the experiment. A potential of −2.63 V vs. Fc+/0 is applied to the film and electronic difference spectra are acquired after 30 s. During electrolysis, a new peak at 521 nm grows in over the course of 10 min (Fig. 3). This absorption energy is consistent with formation of the singly reduced Re(0) bpy radical anion [Re(bpy˙−)(CO)3L]n where L is either a bromide ligand (n = −1) or a solvent ligand (n = 0).12,14,19–23 This reduced species is a key intermediate in catalytic CO2RR activity by Lehn type catalysts and suggests that the surface-bound catalysts exhibit similar behavior to homogeneous catalysts. Notably, the ∼600 and ∼800 nm peaks characteristic of the catalytically inactive Re–Re dimer43–46 are not observed (grey trace, Fig. 3). As stated above, dimerization of Re complexes in the Re(0) state represents a catalytically parasitic side reaction and the most successful catalysts of this class avoid Re–Re bond formation.47,48 Notably, we also do not observe the large ∼580 nm peak associated with the two electron reduced, five coordinate Re(−1) species expected at such negative potentials. We consider two possibilities for this discrepancy. First, the highly non-polar environment may discourage formation of such an unstable charged species by hindering ionic mobility to [Re] sites in the film (vide infra). Second, based on the PL data and the Si-[Re] hybrid ultrafast dynamics (vide infra), the HOMO of the highly reducing Re(−1) complex lies higher in energy than the conduction band of the Si NC. Thus, reduction of a surface [Re] by two electrons may result in a surface Re(0) complex and an electrochemically n-doped Si NC. But based on electrochemical analysis (vide infra), we favor the first explanation.
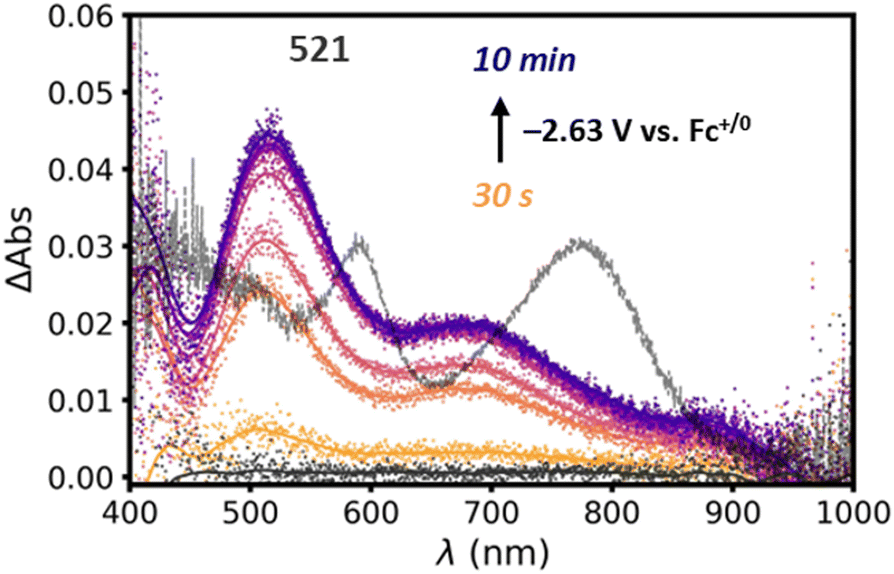 | ||
| Fig. 3 Electronic difference spectra of a Si-[Re] NC film on an ITO-coated glass slide submerged in CH3CN with 100 mM TBAPF6 as supporting electrolyte taken before (black symbols) and during (orange to purple symbols). A spectrum of the unbound [Re]–[Re] dimer is overlayed (grey line) and scaled to fit in the window. For clarity Savitzky–Golay smoothed representations of the raw data are included (solid lines). | ||
Finally, we note the trend in charging currents between the three samples Si-[Re], Si-C12, and Si + [Re] (Fig. S7†) in the pre-reduction region (−0.50 to −1.6 V vs. Fc+/0) and compare each to the baseline voltametric curve acquired with a clean GCE (pre-blank CV, Fig. S8†). From these data we conclude that the sparse dodecyl coverage on Si-[Re] and Si + [Re] leads to lower insulating properties for the film compared with the full dodecyl coverage for saturated Si-C12 (Fig. S7† blue and orange traces) and that the inclusion of [Re] tethered to the surface of Si-[Re] further enhances the dark conductive characteristics of the film presumably by allowing a pathway for electron hopping between discrete [Re] sites and/or Si-[Re] hybrids in the adsorbed matrix (Fig. S7† red and orange traces).
We next compare the CV data acquired for Si-[Re] NCs with that of dissolved [Re(bpy)(CO)3Br] (Fig. 2, black dashed trace). The electrochemical behavior of [Re(bpy)(CO)3Br] is well described in the literature and these data are consistent with those prior reports.43,49–51 In brief, the multiple redox waves in the CV for the [Re(bpy)(CO)3Br] complex are attributed to formal reduction of Re(I) to Re(0) (E0′ = −1.79 V vs. Fc+/0) and further reduction of Re(0) to Re(−1) (Ep,c = −2.15 V vs. Fc+/0) with concomitant dissociation of the labile Br− ligand (Scheme S1†). More sophisticated analysis has shown that the electron from the first reduction resides on the bpy ligand, such that the Re only is reduced upon the 2nd reduction,46,52 but we use the formal Re oxidation states for simplicity. It is also known that the Re(0) bromide complex undergoes dimerization following a relatively slow halide dissociation,45,46 and the resulting Re–Re bond is inert toward reaction with CO2; consequently, its formation is a parasitic side reaction during CO2RR catalysis.49,53 Specifically, on the return scan, anodic features are expected for the oxidation of the Re(0) bromide complex (Ep,a = −1.74 V vs. Fc+/0),49,54 Re(0) solvent complex (Ep,a = −1.57 V vs. Fc+/0)54,55 and the Re(0) dimer (Ep,a = −0.51 V vs. Fc+/0).53 The anodic wave with Ep,a = +0.20 V vs. Fc+/0 corresponds to irreversible oxidation to Re(II).54 CVs of Si-[Re] NC thin films display the same features observed for [Re(bpy)(CO)3Br], however, the waves are broadened and do not obey the canonical waveform expected for freely diffusing analytes. Interestingly, an anodic feature is observed at Ep,a = −0.58 V vs. Fc+/0 that is at a similar oxidation potential as the Re(0) dimer, but we discount the possibility of a dimer for two main reasons: (1) dimer formation would require inter-NC Re–Re linkages that are unlikely in the solid-state especially given the low [Re] complex surface coverage, and (2) spectroelectrochemical data suggests that dimerization does not occur (Fig. 3). Additionally, the presence of a small oxidative wave in this region has been observed under conditions where no dimer is detected spectroelectrochemically.56 The presence of observable Re-centered reductions in thin films of the hybrid assembly suggests a lack of coupling between the semiconductor and tethered [Re] complexes.57
To monitor the integrity of the hybrid assembly, we perform a set of multi-cycle CVs on the Si-[Re] thin films (Fig. S9†) and observe an ∼50% loss of current compared to the first reductive wave in subsequent scans. The broad cathodic feature observed on the initial cycle also resolves into two quasi-reversible waves (E0′ = −1.83 and −2.03 V vs. Fc+/0, ΔEp = +140 and +124 mV, respectively). We attribute these observations to release of the bromide ligand during the first scan, which provides a convenient first order charge balancing reaction. However, diffusion of the released bromide away from the electrode hampers charge neutralizing ion diffusion on subsequent scans, negatively impacting electron transfer kinetics and resulting in significant charging current. Lastly, we find that applying anodic potentials (>0 V vs. Fc+/0) does not change the intensity of the species with the prominent 521 nm peak in spectroelectrochemical measurements shown in Fig. 3, which is consistent with the irreversible formation of this presumed Re(0) monomer ([Re(bpy)(CO)3NCCH3], Scheme S1†) complex in thin films.
To test the photocatalytic activity of Si-[Re], we irradiate a CO2 saturated tetrahydrofuran (THF) solution containing Si-[Re], 100 mM 1,3-dimethyl-2-phenyl-2,3-dihydro-1H-benzo[d]imidazole (BIH) as a sacrificial electron donor, and 103 mM 2,2,2-trifluoroethanol (TFE) as a proton source with a 405 nm LED light source. Products are characterized by gas chromatography (GC) and compared to a calibration curve to determine total CO produced. Over the first 4 h, CO production occurs at a rate of 2.1 mmol g−1 h−1, which reduces to 0.2 mmol g−1 h−1 over the next 4–24 h (Fig. 4). We attribute this time dependent loss in activity to consumption of CO2, BIH, TFE, and/or photocatalyst degradation. As a control, we repeat the experiment with Si-C12 having no [Re] present. No photolysis activity for CO2 reduction to CO is observed over the same 24 h period.
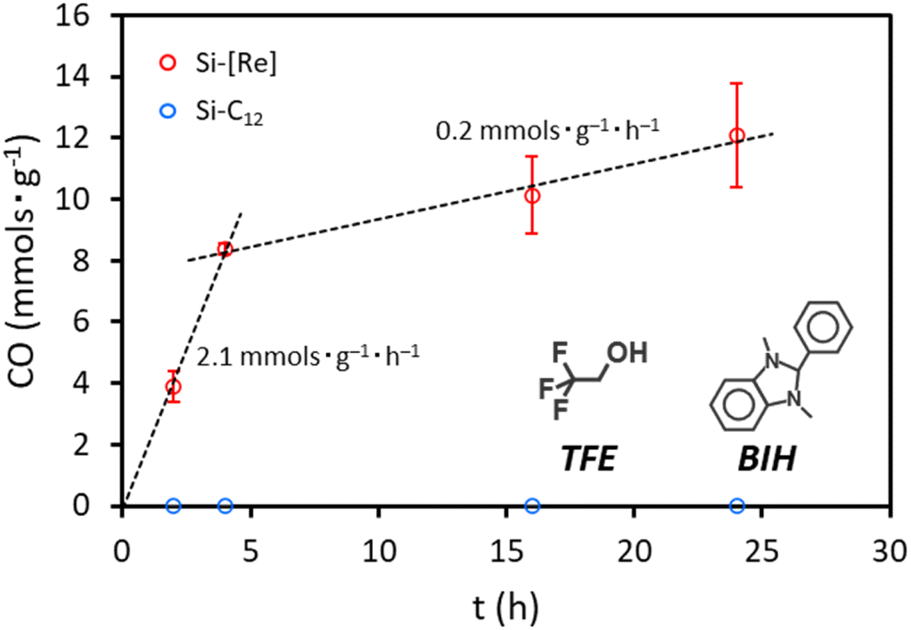 | ||
| Fig. 4 Photocatalytic activity of Si-[Re] (red) and Si-C12 (blue) under 405 nm irradiation. Results were acquired in CO2 saturated THF with 100 mM BIH and 103 mM TFE. | ||
Additional control experiments are performed on [Re] catalyst only given that the broad emission feature of the 405 nm LED overlaps the MLCT band for the [Re] complex (Fig. S10†). That Si-[Re] shows catalytic activity even though we are unable to observe the Re(−1) signal by spectroelectrochemistry could be due to several causes. First, the environment experienced by surface tethered Si-[Re] in thin films is a highly nonpolar dodecyl matrix. Under photocatalytic conditions, the Si-[Re] are freely dispersed in THF and each [Re] is exposed to the more polar THF solvent sphere. This may allow formation of Re(−1) in the THF solvent sphere that does not occur in the nonpolar film environment. Alternately, an equilibrium may exist between valence states of Si-[Re] such that a small population of Si-[Re(−1)] exists but is not observed spectroscopically. A third possibility is that the reduction of CO2 could occur by a surface bound Si-[Re(0)] species since it has been shown that the [Re(0)] species can be catalytically active for CO2 reduction.58 Photolysis of [Re] shows similar absolute CO production during irradiation under identical conditions and excluding any Si NC material (Fig. S11†). We also calculate turnover number (TON) as mol(CO) mol(catalyst)−1, wherein mol(catalyst) for Si-[Re] is calculated on a per nanocrystal basis. We note that the apparent enhanced performance of Si-[Re] relative to [Re] is likely due to multiple discrete [Re] complexes on each Si-[Re] nanocrystal; though attempts to quantify the amount of [Re] on each Si-[Re] hybrid were unsuccessful, we estimate that a normalized TON per [Re] would be similar for Si-[Re] and [Re]. Thus, the [Re] CO2RR catalyst activity appears to be unperturbed by Si NC surface tethering, which suggests that there may be little photogenerated charge transfer between the Si NC and the surface-bound [Re].
To probe this latter question, we explore the photodynamics using a series of ultrafast transient absorption (TA) experiments. The excited state charge dynamics of colloidal Si-[Re] in toluene are compared to colloidal Si-C12 in toluene or [Re] dissolved in CH3CN (Fig. 5). These TA data show that the Si-[Re] hybrid structure exhibits a small change in the response at ultrafast time scales (<5 ps) relative to Si-C12; however, between 5–5000 ps, the broad visible absorbance of the Si NC exciton dominates the spectrum for both samples, suggesting that there is no significant contribution of any [Re] species to the TA spectra. The kinetics of Si-C12 and Si-[Re] monitored at 545 nm (Fig. 5b) show minimal difference in behaviour after 10 ps (Table S1†). The initial change in the Si-[Re] kinetics may indicate two [Re] excited state quenching pathways. The first is fast excited state electron transfer from the catalyst into the Si NC and the second is rapid energy transfer from [Re] to the Si NC. Regardless of which mechanism is operative, these TA data are clear that there is no evidence of Si NC excited state interaction with ground state [Re]. These time-resolved data as well as those from electrochemical and steady-state optical measurements are consistent with an energetic arrangement where the [Re(I)] LUMO lies above the Si NC conduction band minimum.
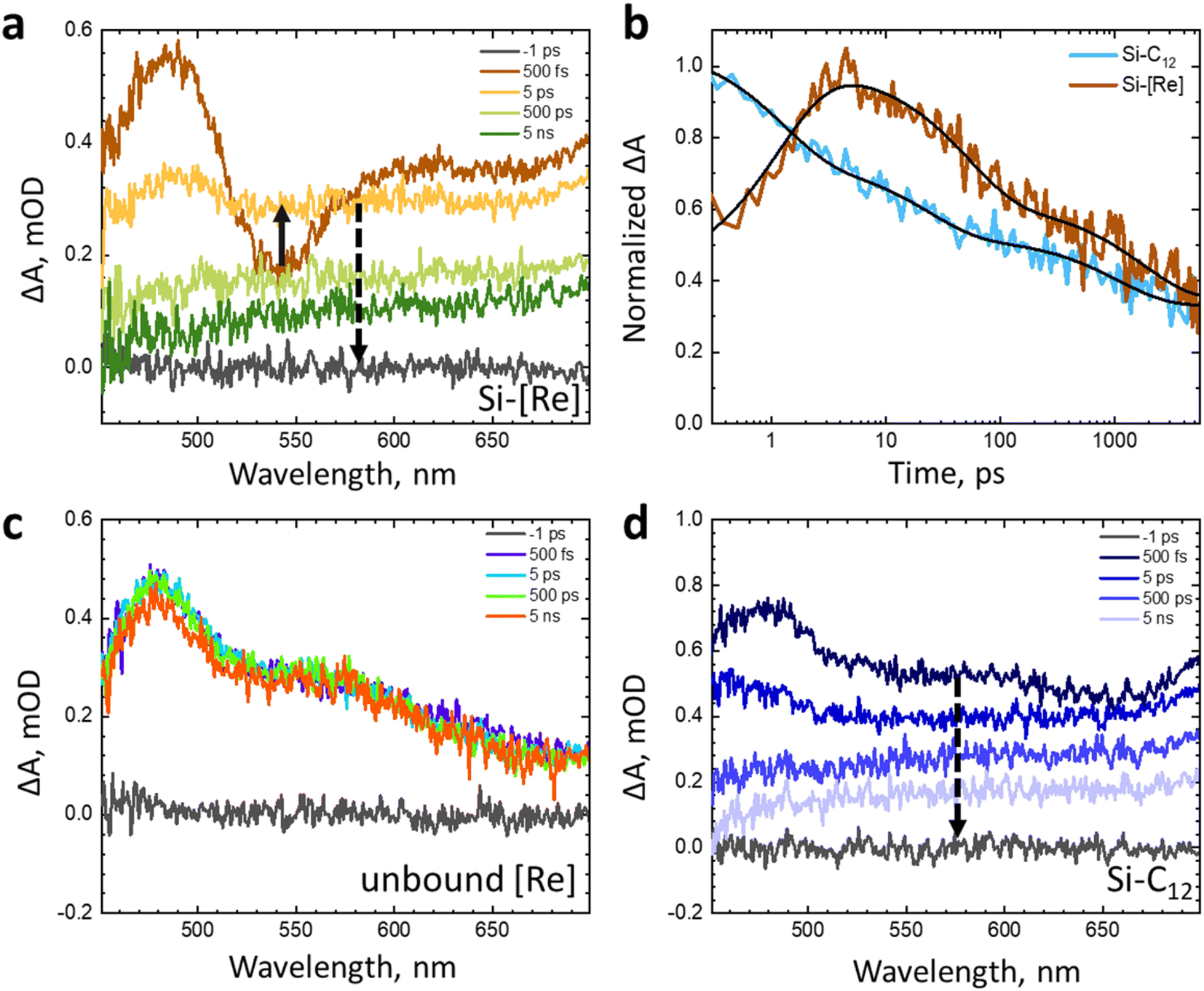 | ||
| Fig. 5 (a) Transient absorption spectra of colloidal Si-[Re] in toluene monitored at 500 fs (dark orange) through 5 ns (dark green). (b) Normalized transient absorption kinetics of colloidal Si-C12 (blue) and Si-[Re] (red) in toluene monitored at 545 nm. Fits to the data are shown as black lines. Results of the fits can be found in the ESI (Table S1).† (c) Transient absorption spectra of unbound [Re] in acetonitrile monitored at 500 fs (purple) through 5 ns (orange). (d) Transient absorption spectra of colloidal Si-C12 in toluene monitored at 500 fs (dark blue) through 5 ns (light blue). All samples were prepared in a glovebox and excited at 400 nm (180 nJ per pulse). | ||
Conclusions
In summary, we prepared and characterized partially dodecyl-terminated Si NCs decorated with a Lehn-type rhenium tricarbonyl bpy complex. The successful inclusion of the Re complex on the NC surface was confirmed by FTIR, CV, and comparison to control species. The hybrid structure displayed redox characteristics consistent with unbound Re complexes of the same class as demonstrated by electrochemical and spectroelectrochemical analysis, including observation of a monomeric Re(0) complex with no propensity towards dimerization. Photocatalysis results indicated that the Si-[Re] hybrid assembly exhibits CO2RR activity to CO under 405 nm irradiation comparable to [Re] only. One key conclusion from these data is that surface tethering does not impact the catalyst activity and can have a beneficial impact on longer-term activity by preventing catalyst dimerization. Transient absorption spectroscopic data was used to rationalize this observation and confirmed that there is minimal electronic coupling between the Si NC and [Re] electrocatalyst. Future work to tune the energetics of the Si NC or the electrocatalyst, or both, to enable electronic coupling is under active investigation.Author contributions
The project was conceived by N. R. N. with help from S. S. S. Experiments were performed by S. S. S., A. D. W., and M. K. G. All authors analysed the data and S. S. S., M. K. G., and N. R. N. co-wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to acknowledge Chris Urban for his help with GC experiments and Taylor Thomas for growth of Si NCs. This work was authored by the National Renewable Energy Laboratory, operated by the Alliance for Sustainable Energy, LLC, for the U.S. Department of Energy DOE under Contract No. DE-AC36-08GO28308. Funding provided by U.S. Department of Energy, Office of Science, Basic Energy Sciences, Division of Chemical Sciences, Geosciences, and Biosciences, Solar Photochemistry Program. The views expressed in the article do not necessarily represent the views of the DOE or the U.S. Government. The U.S. Government retains and the publisher, by accepting the article for publication, acknowledges that the U.S. Government retains a nonexclusive, paid-up, irrevocable, worldwide license to publish or reproduce the published form of this work, or allow others to do so, for U.S. Government purposes.Notes and references
- Q. Wang, C. Pornrungroj, S. Linley and E. Reisner, Nat. Energy, 2022, 7, 13–24 CrossRef.
- C. Ballif, F. J. Haug, M. Boccard, P. J. Verlinden and G. Hahn, Nat. Rev. Mater., 2022, 7, 597–616 CrossRef.
- N. R. Neale and R. T. Pekarek, Molecular Functionalization of Semiconductor Surfaces, in Spring Handbook of Inorganic Photochemistry, ed. D. Bahnemann and A. O. T. Patrocinio, Springer International Publishing, 2022, pp. 923–964 Search PubMed.
- B. Seger, K. Herbst, T. Pedersen, B. Abrams, P. C. K. Vesborg, O. Hansen and I. Chorkendorff, J. Electrochem. Soc., 2014, 161, H722–H724 CrossRef.
- H. J. Kim, J. Seo and M. J. Rose, ACS Appl. Mater. Interfaces, 2016, 8, 1061–1066 CrossRef CAS PubMed.
- J. J. Leung, J. Warnan, D. H. Nam, J. Z. Zhang, J. Willkomm and E. Reisner, Chem. Sci., 2017, 8, 5172–5180 RSC.
- B. Shan, M. K. Brennaman, L. Troian-Gautier, Y. Liu, A. Nayak, C. M. Klug, T. T. Li, R. M. Bullock and T. J. Meyer, J. Am. Chem. Soc., 2019, 141, 10390–10398 CrossRef CAS PubMed.
- L. Gong, H. Yin, C. Nie, X. Sun, X. Wang and M. Wang, ACS Appl. Mater. Interfaces, 2019, 11, 34010–34019 CrossRef CAS PubMed.
- S. Chandrasekaran, N. Kaeffer, L. Cagnon, D. Aldakov, J. Fize, G. Nonglaton, F. Baleras, P. Mailley and V. Artero, Chem. Sci., 2019, 10, 4469–4475 RSC.
- C. Nie, C. Liu, L. Gong and M. Wang, J. Mater. Chem. A, 2021, 9, 234–238 RSC.
- G. F. Moore and I. D. Sharp, J. Phys. Chem. Lett., 2013, 4, 568–572 CrossRef CAS PubMed.
- J. R. C. Lattimer, J. D. Blakemore, W. Sattler, S. Gul, R. Chatterjee, V. K. Yachandra, J. Yano, B. S. Brunschwig, N. S. Lewis and H. B. Gray, J. Chem. Soc., Dalton Trans., 2014, 43, 15004–15012 RSC.
- J. Seo, R. T. Pekarek and M. J. Rose, Chem. Commun., 2015, 51, 13264–13267 RSC.
- B. L. Huffman, G. P. Bein, H. Atallah, C. L. Donley, R. T. Alameh, J. P. Wheeler, N. Durand, A. K. Harvey, M. C. Kessinger, C. Y. Chen, Z. Fakhraai, J. M. Atkin, F. N. Castellano and J. L. Dempsey, ACS Appl. Mater. Interfaces, 2023, 15, 984–996 CrossRef CAS PubMed.
- D. Jurbergs, E. Rogojina, L. Mangolini and U. Kortshagen, Appl. Phys. Lett., 2006, 88, 233116 CrossRef.
- R. J. Anthony, D. J. Rowe, M. Stein, J. Yang and U. Kortshagen, Adv. Funct. Mater., 2011, 21, 4042–4046 CrossRef CAS.
- L. M. Wheeler, N. C. Anderson, P. K. B. Palomaki, J. L. Blackburn, J. C. Johnson and N. R. Neale, Chem. Mater., 2015, 27, 6869–6878 CrossRef CAS.
- G. M. Carroll, R. Limpens and N. R. Neale, Nano Lett., 2018, 18, 3118–3124 CrossRef CAS PubMed.
- R. Limpens, G. F. Pach and N. R. Neale, Chem. Mater., 2019, 31, 4426–4435 CrossRef CAS.
- G. F. Pach, G. M. Carroll, H. Zhang and N. R. Neale, Faraday Discuss., 2020, 222, 201–216 RSC.
- W. Sun, C. Qian, L. He, K. K. Ghuman, A. P. Y. Wong, J. Jia, A. A. Jelle, P. G. O'Brien, L. M. Reyes, T. E. Wood, A. S. Helmy, C. A. Mims, C. V. Singh and G. A. Ozin, Nat. Commun., 2016, 7, 12553 CrossRef CAS PubMed.
- S. A. Martell, Y. Lai, E. Traver, J. MacInnis, D. D. Richards, S. MacQuarrie and M. Dasog, ACS Appl. Nano Mater., 2019, 2, 5713–5719 CrossRef CAS.
- Z. Chai, Q. Li and D. Xu, RSC Adv., 2014, 4, 44991–44995 RSC.
- S. Lian, M. S. Kodaimati, D. S. Dolzhnikov, R. Calzada and E. A. Weiss, J. Am. Chem. Soc., 2017, 139, 8931–8938 CrossRef CAS PubMed.
- M. F. Kuehnel, K. L. Orchard, K. E. Dalle and E. Reisner, J. Am. Chem. Soc., 2017, 139, 7217–7223 CrossRef CAS PubMed.
- S. Lian, M. S. Kodaimati and E. A. Weiss, ACS Nano, 2018, 12, 568–575 CrossRef CAS PubMed.
- M. F. Kuehnel, C. D. Sahm, G. Neri, J. R. Lee, K. L. Orchard, A. J. Cowan and E. Reisner, Chem. Sci., 2018, 9, 2501–2509 RSC.
- H. Lu, Z. Huang, M. S. Martinez, J. C. Johnson, J. M. Luther and M. C. Beard, Energy Environ. Sci., 2020, 13, 1347–1376 RSC.
- F. Arcudi, L. Dordević, B. Nagasing, S. I. Stupp and E. A. Weiss, J. Am. Chem. Soc., 2021, 143, 18131–18138 CrossRef CAS PubMed.
- C. Gimbert-Suriñach, J. Albero, T. Stoll, J. Fortage, M. N. Collomb, A. Deronzier, E. Palomares and A. Llobet, J. Am. Chem. Soc., 2014, 136, 7655–7661 CrossRef PubMed.
- M. Sandroni, R. Gueret, K. D. Wegner, P. Reiss, J. Fortage, D. Aldakov and M. N. Collomb, Energy Environ. Sci., 2018, 11, 1752–1761 RSC.
- J. L. Dempsey, C. M. Heyer and G. J. Meyer, Electrochem. Soc. Interface, 2021, 30, 65–68 CrossRef CAS.
- D. Neiner and S. M. Kauzlarich, Chem. Mater., 2010, 22, 487–493 CrossRef CAS.
- F. Peng, J. Wang, G. Ge, T. He, L. Cao, Y. He, H. Ma and S. Sun, Mater. Lett., 2013, 92, 65–67 CrossRef CAS.
- M. Dasog, G. B. de Los Reyes, L. v. Titova, F. A. Hegmann and J. G. C. Veinot, ACS Nano, 2014, 8, 9636–9648 CrossRef CAS PubMed.
- M. Dasog, S. Kraus, R. Sinelnikov, J. G. C. Veinot and B. Rieger, Chem. Commun., 2017, 53, 3114–3117 RSC.
- A. P. Y. Wong, W. Sun, C. Qian, A. A. Jelle, J. Jia, Z. Zheng, Y. Dong and G. A. Ozin, Adv. Sustainable Syst., 2017, 1, 1700118 CrossRef.
- J. Hawecker, J.-M. Lehn and R. Ziessel, J. Chem. Soc., Chem. Commun., 1983, 536–538 RSC.
- J. Hawecker, J.-M. Lehn and R. Ziessel, J. Chem. Soc., Chem. Commun., 1984, 328–330 RSC.
- B. Gholamkhass, H. Mametsuka, K. Koike, T. Tanabe, M. Furue and O. Ishitani, Inorg. Chem., 2005, 44, 2326–2336 CrossRef CAS PubMed.
- A. el Nahhas, C. Consani, A. M. Blanco-Rodríguez, K. M. Lancaster, O. Braem, A. Cannizzo, M. Towrie, I. P. Clark, S. Záliš, M. Chergui and A. Vlček, Inorg. Chem., 2011, 50, 2932–2943 CrossRef CAS PubMed.
- A. Ge, B. Rudshteyn, P. E. Videla, C. J. Miller, C. P. Kubiak, V. S. Batista and T. Lian, Acc. Chem. Res., 2019, 52, 1289–1300 CrossRef CAS PubMed.
- A. I. Breikss and H. D. Abruña, J. Electroanal. Chem., 1986, 201, 347–358 CrossRef CAS.
- G. J. Stor, F. Hartl, J. W. M. van Outersterp and D. J. Stufkens, Organometallics, 1995, 14, 1115–1131 CrossRef CAS.
- Y. Hayashi, S. Kita, B. S. Brunschwig and E. Fujita, J. Am. Chem. Soc., 2003, 125, 11976–11987 CrossRef CAS PubMed.
- E. Fujita and J. T. Muckerman, Inorg. Chem., 2004, 43, 7636–7647 CrossRef CAS PubMed.
- J. M. Smieja and C. P. Kubiak, Inorg. Chem., 2010, 49, 9283–9289 CrossRef CAS PubMed.
- M. D. Sampson, A. D. Nguyen, K. A. Grice, C. E. Moore, A. L. Rheingold and C. P. Kubiak, J. Am. Chem. Soc., 2014, 136, 5460–5471 CrossRef CAS PubMed.
- B. Patrick Sullivan, C. Mark Bolinger, D. Conrad, W. J. Vining and T. J. Meyer, J. Chem. Soc., Chem. Commun., 1985, 1414–1416 RSC.
- F. P. A. Johnson, M. W. George, F. Hartl and J. J. Turner, Organometallics, 1996, 15, 3374–3387 CrossRef CAS.
- C. W. Machan, M. D. Sampson, S. A. Chabolla, T. Dang and C. P. Kubiak, Organometallics, 2014, 33, 4550–4559 CrossRef CAS.
- E. E. Benson, M. D. Sampson, K. A. Grice, J. M. Smieja, J. D. Froehlich, D. Friebel, J. A. Keith, E. A. Carter, A. Nilsson and C. P. Kubiak, Angew. Chem., Int. Ed., 2013, 52, 4841–4844 CrossRef CAS PubMed.
- E. E. Benson and C. P. Kubiak, Chem. Commun., 2012, 48, 7374–7376 RSC.
- J. V. Caspar and T. J. Meyer, J. Phys. Chem., 1983, 87, 952–957 CrossRef CAS.
- M. L. Clark, P. L. Cheung, M. Lessio, E. A. Carter and C. P. Kubiak, ACS Catal., 2018, 8, 2021–2029 CrossRef CAS.
- S. S. Saund, M. A. Siegler and V. S. Thoi, Inorg. Chem., 2021, 60, 13011–13020 CrossRef CAS PubMed.
- M. N. Jackson, S. Oh, C. J. Kaminsky, S. B. Chu, G. Zhang, J. T. Miller and Y. Surendranath, J. Am. Chem. Soc., 2018, 140, 1004–1010 CrossRef CAS PubMed.
- G. Sahara and O. Ishitani, Inorg. Chem., 2015, 54, 5096–5104 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3se01512b |
| This journal is © The Royal Society of Chemistry 2024 |