
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Detour to success: photoswitching via indirect excitation
Kim
Kuntze
 a,
Jussi
Isokuortti
b,
Jacob J.
van der Wal
c,
Timo
Laaksonen
ad,
Stefano
Crespi
*c,
Nikita A.
Durandin
*a and
Arri
Priimagi
*a
a,
Jussi
Isokuortti
b,
Jacob J.
van der Wal
c,
Timo
Laaksonen
ad,
Stefano
Crespi
*c,
Nikita A.
Durandin
*a and
Arri
Priimagi
*a
aFaculty of Engineering and Natural Sciences, Tampere University, Tampere, Finland. E-mail: arri.priimagi@tuni.fi; nikita.durandin@tuni.fi
bDepartment of Chemistry, University of Texas at Austin, Austin, TX, USA
cDepartment of Chemistry, Ångström Laboratory, Uppsala University, Uppsala, Sweden. E-mail: stefano.crespi@kemi.uu.se
dFaculty of Pharmacy, University of Helsinki, Helsinki, Finland
First published on 2nd July 2024
Abstract
Photoswitchable molecules that undergo nanoscopic changes upon photoisomerisation can be harnessed to control macroscopic properties such as colour, solubility, shape, and motion of the systems they are incorporated into. These molecules find applications in various fields of chemistry, physics, biology, and materials science. Until recently, research efforts have focused on the design of efficient photoswitches responsive to low-energy (red or near-infrared) irradiation, which however may compromise other molecular properties such as thermal stability and robustness. Indirect isomerisation methods enable photoisomerisation with low-energy photons without altering the photoswitch core, and also open up new avenues in controlling the thermal switching mechanism. In this perspective, we present the state of the art of five indirect excitation methods: two-photon excitation, triplet sensitisation, photon upconversion, photoinduced electron transfer, and indirect thermal methods. Each impacts our understanding of the fundamental physicochemical properties of photochemical switches, and offers unique application prospects in biomedical technologies and beyond.
Introduction
Photoswitches are molecules that convert reversibly between isomers upon excitation with light. A key example of a naturally occurring photoisomerisable molecule is retinal that undergoes Z → E isomerisation after absorbing a photon, which initiates the cellular signalling cascade responsible for vision.1 During the last century, chemists have designed a myriad of artificial photoswitch structures: azobenzenes,2 (stiff-)stilbenes,3 indigoids,4 diarylethenes,5 norbornadienes/quadricyclanes,6 spiropyrans/merocyanines,7 and donor–acceptor Stenhouse adducts (DASAs),8 to name a few (Fig. 1A). The physicochemical properties of a switch change upon isomerisation and give rise to photoresponsive functions. For instance, the E–Z isomerisation of azobenzenes, stilbenes and indigoids can be utilised to control the supramolecular interactions of molecular systems or to induce strain into a macroscopic material. The electrocyclisation of diarylethenes and spiropyrans, on the other hand, drastically changes the conjugation and dipole moment of these molecules, respectively. These phenomena can be exploited in photoresponsive systems operating on the molecular or macroscopic level in medicine,9 biosciences,10,11 catalysis,12 energy storage,13 electronics,14 optics,15 3D printing,16 soft robotics17 and other materials applications.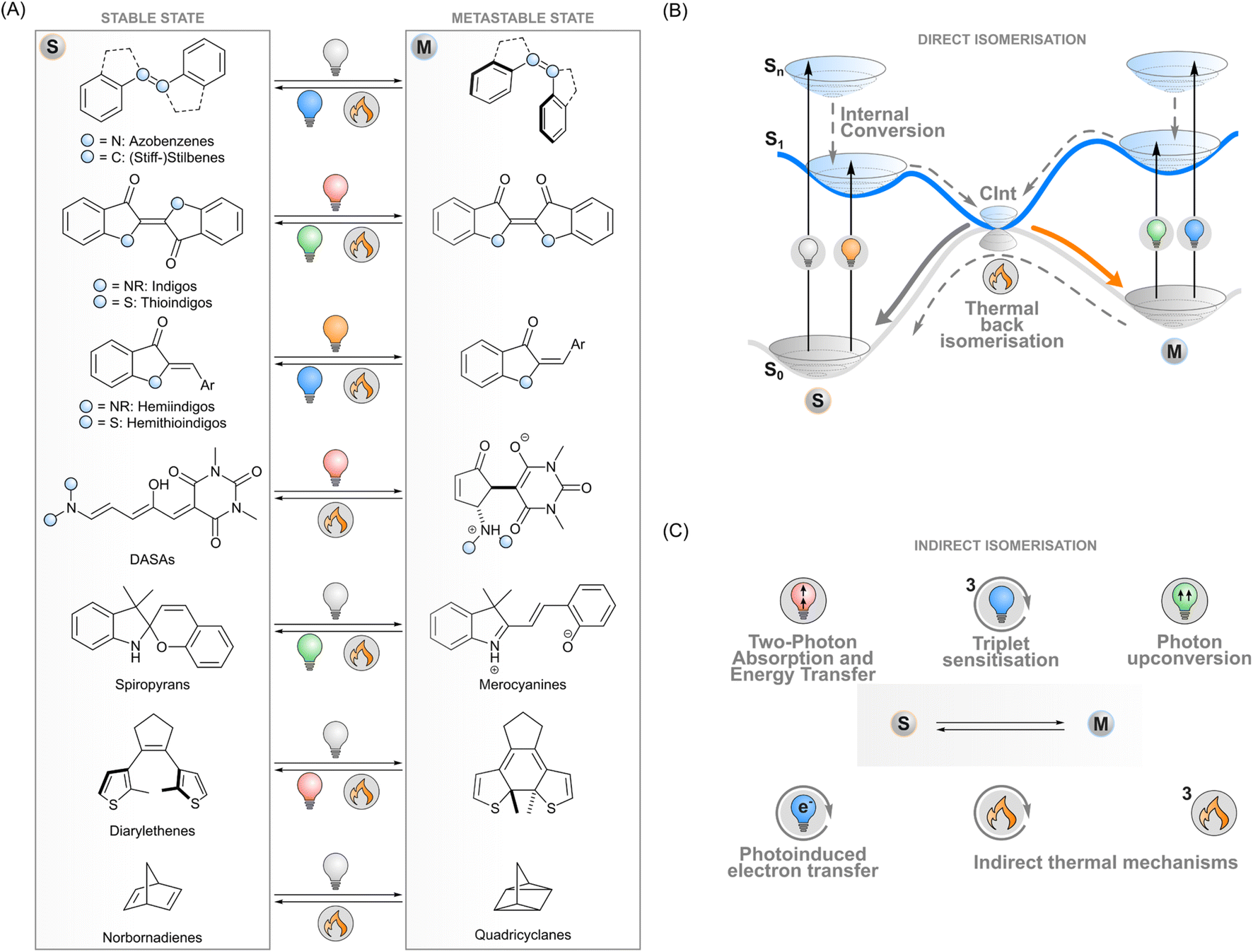 | ||
| Fig. 1 (A) Selected photoswitch families, showing the stable and metastable isomers as well as typical stimuli used to bring about the isomerisation. (B) A schematic view of the processes related to isomerisation with direct excitation. (C) Isomerisation methods utilising indirect excitation, i.e. excitation of a proxy system that induces the isomerisation. | ||
In general terms, photoisomerisation is based on exciting a molecule to an excited singlet state by irradiating one of its absorption bands. Subsequent events on the potential energy surfaces then lead to isomerisation with a certain efficiency i.e. isomerisation quantum yield (Fig. 1B). If the isomers have distinct absorption profiles, it is possible to favour isomerisation in one direction by choosing an appropriate excitation wavelength, leading to photostationary states where one of the isomers is enriched. One isomer typically has a higher ground-state energy and is denoted as the metastable isomer. Thermal back-isomerisation to the stable isomer is typically observed as well; ground-state isomerisation half-lives range from nanoseconds to years and thus partly determine the research directions to which each photoswitch is applicable.5,16
The majority of photoswitches suffer from one critical limitation which hinders their applicability on different research fronts: at least one of the two isomers absorbs only ultraviolet light. This high-energy irradiation often suffers from unwanted absorption or scattering by the matrix surrounding the photoswitches, which causes limited penetration depth and spatial resolution, may have a degrading effect on many materials, and is particularly detrimental to living systems. The ability to control the isomerisation with visible light or with other stimuli would thus be beneficial for a range of applications.18–20 In medicinal and biotechnological applications, the excitation wavelengths should optimally fall inside the bio-optical window located in the red and near-infrared light region (>650 nm) where penetration depths through tissue are the best. To meet these requirements, the absorption bands of conventional photoswitches such as azobenzenes can be red-shifted through structural modifications,18 albeit often at the expense of reduced thermal stability of the metastable isomer,21,22 which limits the scope of these switches. Novel photoswitch scaffolds that inherently absorb red light such as DASAs,23N,N-difunctionalised indigos24 and peri-anthracenethioindigos25 have also been designed, but so far, they have not reached the versatility of more conventional photoswitches in terms of applications.
To overcome these limitations of directly exciting the photoswitch core, another approach is possible. Indirect isomerisation leaves the intrinsic properties (such as the stability of the metastable state) of the photoswitch intact but expands the excitation modality beyond its capabilities by introducing a proxy system responsible for harvesting the excitation energy and transferring it to the photoswitch (Fig. 1C). When designed properly, this enables photoisomerisation with lower-energy photons than absorbed by the photoswitch. Several concepts for indirect excitation of photoswitches have been demonstrated: two-photon excitation of an antenna moiety and subsequent Förster (singlet) energy transfer to the photoswitch,26,27 triplet sensitisation,28,29 photon upconversion,30,31 and photoinduced electron transfer.32 These photochemical pathways are often accompanied by thermal mechanisms that may have undesired effects on the switching system but could also provide an additional way of tuning the isomerisation dynamics. To fully harvest the potential of these strategies, a deep understanding of the underlying mechanisms is paramount.
In this perspective we shall explore these indirect photoisomerisation strategies from theoretical and practical points of view, giving insight into their fundamental mechanisms and demonstrating how they have been exploited to enable photoswitching with low-energy light. We will cover various photoswitch families with a particular focus on azoarenes and stilbenes and guide the reader to recent reviews regarding some other photoswitch families such as diarylethenes and norbornadienes.33,34 We also delve into the often-overlooked thermal isomerisation routes and propose how they can be either suppressed or exploited. Finally, we provide an outlook on the remaining challenges and opportunities in this promising field of research.
Indirect photoexcitation mechanisms
All the indirect photoexcitation mechanisms described hereafter involve a photon absorber (a sensitiser) that may be a molecular fraction covalently linked to the photoswitch or an independent molecule (or nanoparticle). To understand the role of the sensitisers in indirect photoswitching, it is important to first understand the energy transfer pathways involved. After initial excitation to the first excited singlet state (1Sens*), the sensitiser can relax back to the ground state thermally or radiatively (internal conversion or fluorescence, respectively), or it can undergo intersystem crossing (ISC) to the lower-energy first excited triplet state (3Sens*). Since ISC involves a change in the spin state of the sensitiser, it can only compete with the singlet → singlet transitions when the spin–orbit coupling is strong. The most common examples of dyes that exhibit efficient ISC are porphyrin and phthalocyanine complexes that contain transition metals such as palladium or platinum. If crossing to the triplet state occurs, the excited state lifetime is typically prolonged by orders of magnitude due to the low probability of the spin-forbidden 3Sens* → 1Sens transition.Out of the four indirect excitation mechanisms described below, triplet energy transfer and photon upconversion depend on the sensitiser's triplet state, whereas two-photon absorption and subsequent energy transfer is typically a singlet process. Electron transfer reactions are possible with both spin states, although the pathway is more efficient if the longer-living triplet state can be formed, especially in diffusion-controlled systems. In the next sections, we will cover these indirect isomerisation approaches separately, first going through the photochemistry of the sensitiser and then following up with the isomerisation mechanism of the switch.
Two-photon absorption and subsequent energy transfer
One way to avoid the use of high-energy photons for photoswitching is two-photon absorption where two light quanta, each with half the energy required for one-photon excitation, are absorbed (quasi-)simultaneously. While direct two-photon excitation of a photoswitch is in principle possible for any structure and has been used for 3D data storage35 and surface microstructuring,36 pulsed light with extremely high peak intensity (even TW cm−2) is typically required due to low nonlinear absorption cross-sections. Lower intensities may be applied for some switches with push–pull motifs that increase the probability of simultaneous absorption.37 More generally, two-photon excitation of a photoswitch can be achieved indirectly (Fig. 2) by utilising a molecular fragment with a high nonlinear absorption cross-section, and the capability to act as a donor in resonance energy transfer to the photoswitch, i.e. a high fluorescence quantum yield, favourable relative orientation between the donor and photoswitch transition dipole moments, and emission that overlaps with the absorption spectrum of a photoswitch. Commonly, large π systems such as naphthalene, pyrene, or anthracene derivatives are utilised for this purpose, often with a donor–acceptor nature that increases the nonlinear absorption cross-section.38 After two-photon excitation with low-energy light, the singlet-excited absorber transfers its energy to the photoswitch, typically through the aforementioned Förster resonance energy transfer, leading to a singlet-excited photoswitch molecule. Subsequent events on the excited state potential energy surface of the photoswitch lead to isomerisation. The resulting photostationary state is determined by the overlap between the emission spectrum of the two-photon absorber and the absorption spectra of the photoswitch isomers, as well as the quantum yields of relaxation to either isomer from the excited state.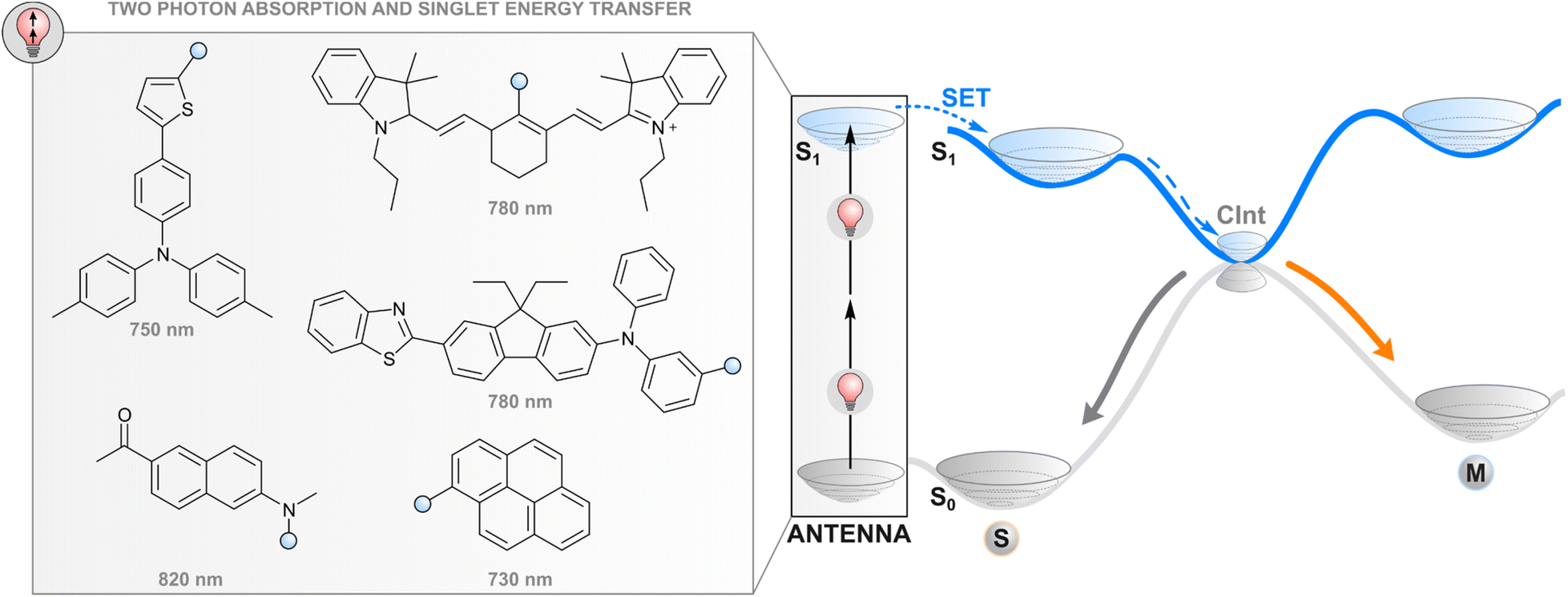 | ||
| Fig. 2 Two-photon excitation of a covalently linked absorber (antenna), subsequent singlet energy transfer to the photoswitch, and the following events in the excited state. | ||
Efficient photoswitching based on indirect two-photon excitation, typically with wavelengths from 750 to 800 nm, has been demonstrated for azobenzenes,39,40 azoheteroarenes,41 overcrowded alkene motors,42 and diarylethenes,26 highlighting the universal applicability of the method. Potential applications have also been shown. Utilising azobenzene nanoimpellers that physically trap anticancer agents inside mesoporous SiO2 nanoparticles as the E isomer, E → Z isomerisation via indirect two-photon excitation led to in vitro cancer cell death with 760 nm light irradiation.27 Also activation of light-gated ion receptors has been accomplished in living cells via indirect two-photon excitation, enabling neuronal and astrocytic stimulation.43 Also a photochromic imidazole dimer has been isomerised with two-photon excitation,44 but in that case the absorption of photons occurs sequentially. The mechanism furthermore proceeds through charge transfer and is hence highlighted in the section covering photoinduced electron transfer.
Since the process does not involve crossing to a triplet state, the difficulties related to the quenching of the excitation by molecular oxygen are avoided (vide infra). On the other hand, efficient energy transfer typically requires covalent linkage to the photoswitch core due to the short lifetime of the excited singlet state of the absorber as compared to triplet sensitisers. Although extreme excitation intensities can be avoided by utilising state-of-the-art nonlinear absorbers, the required intensities are still orders of magnitude larger than those needed by other indirect excitation methods or direct one-photon excitation, and require focused laser beams.37 For this reason, the utility of two-photon processes is limited in, for instance, photoresponsive materials that would require fast and efficient photoswitching simultaneously on a large area. However, the need for a focused photon flux can also be seen as an asset, as it enables a very high (sub-μm-scale) spatial precision in photoisomerisation, which is advantageous for micro- and nanolithography, some drug delivery systems and the manipulation of intra- and extracellular processes in specific parts of the tissue.37
Triplet sensitisation
If the excited sensitiser can cross to its triplet state, a new phenomenon may take place: triplet energy transfer (TET) from the triplet-excited sensitiser to a ground-state photoswitch (Fig. 3A), resulting in a singlet ground-state sensitiser and a triplet-excited photoswitch that would be difficult to obtain through direct excitation due to the forbidden nature of the singlet-to-triplet transition. In the case of triplet sensitisation, this transition is allowed since the reaction proceeds via electron exchange energy transfer where the total spin is conserved and hence, the total spin multiplicity does not change. Since the Dexter mechanism relies on orbital overlap, the sensitiser and photoswitch need to be in close proximity for the energy transfer to take place. This can be accomplished by covalently linking the two together (which enables mechanisms such as superexchange and hopping45,46) or, more commonly, simply relying on collisions of separate molecules, owing to the relatively long lifetime of the triplet state. In this case, the rate of TET depends on diffusion and the electronic coupling between the two molecules. The latter is typically evaluated thermodynamically by comparing the triplet energies of the sensitiser and the photoswitch: larger exoergic gap ΔE from the sensitiser to the photoswitch leads to a higher rate of triplet energy transfer. Traditionally, this has led to the design principle of triplet photosensitised systems where the triplet energy of the sensitiser should be higher than that of the acceptor/photoswitch. However, we will explore the limits of this energetic requirement in this perspective.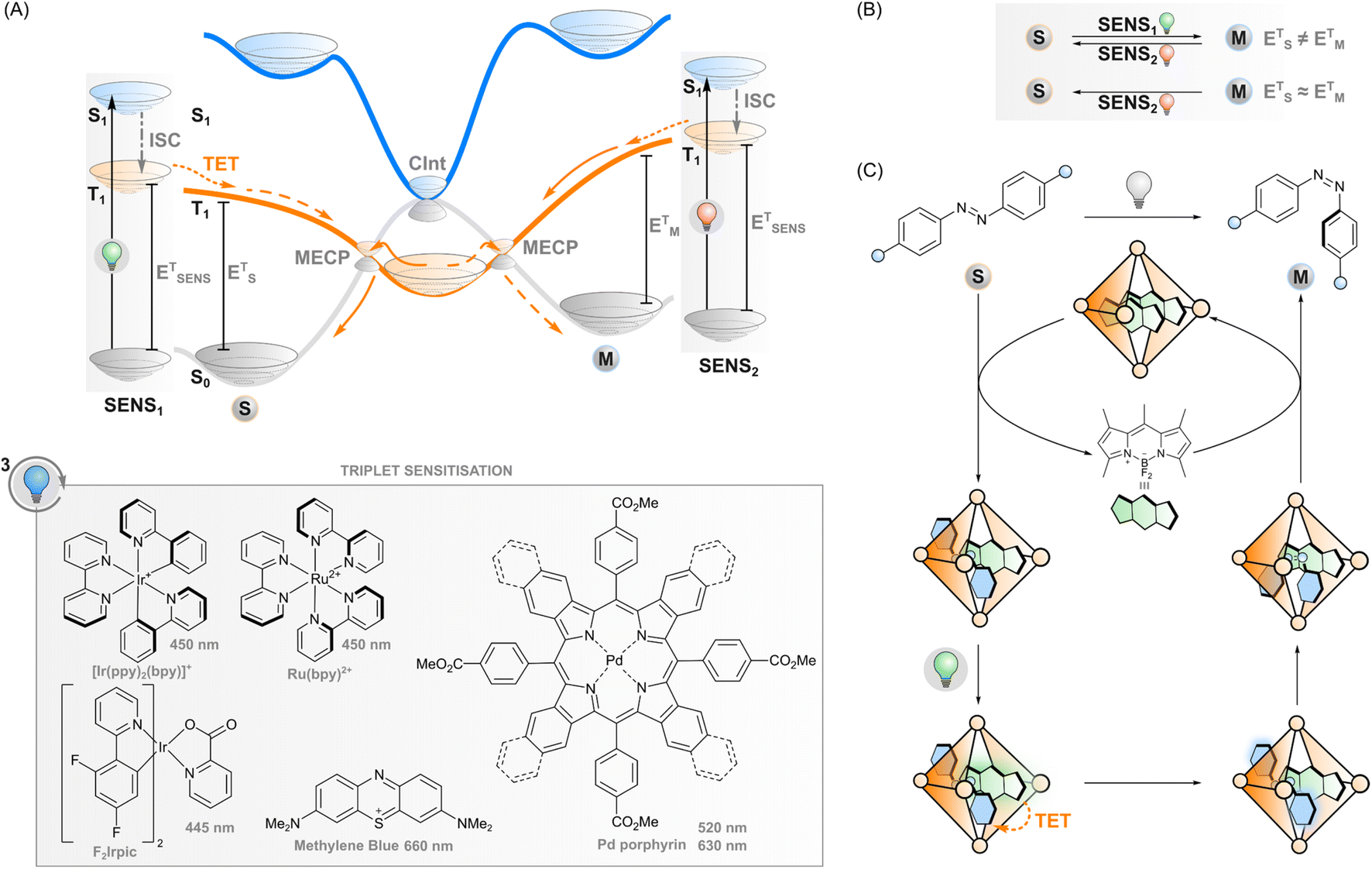 | ||
| Fig. 3 (A) Triplet sensitisation: excitation of the sensitiser (SENS), intersystem crossing (ISC) to its triplet state, triplet energy transfer (TET) to the photoswitch, and ultimately relaxation to the ground state via one of the minimum energy crossing points (MECPs). Selected sensitisers and their absorption maxima are presented. (B) If the triplet energies of the stable and metastable state (EST, EMT) differ, one of them can be selectively sensitised by choosing a sensitiser with matching triplet energy (ESENST), leading to isomerisation from stable to metastable (dashed orange line in (A)) or metastable to stable (solid orange line in (A)). If EST ≈ EMT, sensitisation leads to the same isomer composition regardless of ESENST. (C) DESC utilises flexible coordination cages that can accommodate an E-azoarene and a sensitiser but not a Z-azoarene. Although the quantum yield of Z-azoarene through sensitisation is low, the formed Z-azoarenes are expelled from the cage; thus, over time the sensitisation leads to E → Z isomerisation. | ||
After sensitisation to the triplet-excited state, the events depend on the potential energy surfaces of the photoswitch (Fig. 3A) and, subsequently, the rates of intersystem crossing to the ground state of either isomer. In the case of azobenzenes, the T1 state is barrierless and crosses S0 at two N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N twist angles, approximately 65° and 120°, with the T1 minimum (at 110°) close to the latter. Furthermore, the crossing point at 120° and the T1 minimum are nearly degenerate, while a small energy barrier exists between the 65° crossing and T1 minimum.47,48 This clearly indicates that isomerisation via excitation of the T1 strongly favours the E isomer.28,49,50 As the product of the almost negligible energy barrier and considerable spin–orbit coupling, overall T1–S0 intersystem crossing in azobenzene is very rapid with an estimated rate constant of 1011 s−1.47 Against this backdrop, the triplet sensitisation of azobenzene offers an efficient mode for rapid and selective Z–E isomerisation, yielding a PSS with >95% E. Triplet sensitisation is relatively universal as the triplet energy of azobenzenes is relatively unperturbed by structural modifications.50 However, azoheteroarenes such as arylazopyrazoles51 and the Z isomer of diazocines (bridged azobenzenes)52 exhibit considerably higher triplet energies than conventional azobenzene or the corresponding E isomers. This may be an obstacle to using low-energy light but also an asset when selective sensitisation of either isomer is desired.
N twist angles, approximately 65° and 120°, with the T1 minimum (at 110°) close to the latter. Furthermore, the crossing point at 120° and the T1 minimum are nearly degenerate, while a small energy barrier exists between the 65° crossing and T1 minimum.47,48 This clearly indicates that isomerisation via excitation of the T1 strongly favours the E isomer.28,49,50 As the product of the almost negligible energy barrier and considerable spin–orbit coupling, overall T1–S0 intersystem crossing in azobenzene is very rapid with an estimated rate constant of 1011 s−1.47 Against this backdrop, the triplet sensitisation of azobenzene offers an efficient mode for rapid and selective Z–E isomerisation, yielding a PSS with >95% E. Triplet sensitisation is relatively universal as the triplet energy of azobenzenes is relatively unperturbed by structural modifications.50 However, azoheteroarenes such as arylazopyrazoles51 and the Z isomer of diazocines (bridged azobenzenes)52 exhibit considerably higher triplet energies than conventional azobenzene or the corresponding E isomers. This may be an obstacle to using low-energy light but also an asset when selective sensitisation of either isomer is desired.
Although the phenomenon has been known for decades, only recently have triplet sensitisers such as methylene blue32 and transition metal porphyrin and phthalocyanine complexes29,53 been used to enable red light excitation (via exciting at their Q-bands). The higher-energy S0-to-S2 transition (also known as the Soret band) of the porphyrin photosensitiser in the blue end of the visible spectrum may overlap with the absorption of the azobenzene and cause incomplete E → Z isomerisation. However, by judicious selection of the photoswitch/sensitiser pair with minimised spectral overlap and by tuning their concentration ratios, systems that use visible light for both direct E → Z (525 nm) and triplet-sensitised Z → E isomerisation (770 nm) can be realised.29 Although the triplet energies of porphyrins that absorb red and near-infrared light are rather low and in some cases even lower than that of the photoswitch, sensitisation can be efficient due to entropic contributions and negligible back-energy transfer from the azobenzene, owing to its ultrashort triplet lifetime, to the sensitiser. In addition to different azobenzenes (including ortho-fluorinated ones), also the Z → E isomerisation of arylazopyrazoles was recently induced with red light (630 nm), using a palladium porphyrin sensitiser.51
Furthermore, in an important step forward from purely fundamental solution studies, the use of a red-light-absorbing porphyrin or a triplet-energy-deficient phthalocyanine in TET-catalysed azobenzene isomerisation was recently demonstrated in a bioplastic material.53
Besides azobenzenes, only one other type of photoswitch has seen such extensive characterisation in the triplet manifold. Stilbene has been the subject of triplet sensitisation studies since the 1960s.54,55 The triplet-sensitised isomerisation of olefins in general is an important photochemical transformation in alkene synthesis, and we would like to point interested readers to the recent (2022) review.56 Unlike azobenzene, stilbene undergoes triplet-sensitised isomerisation readily in both directions (Fig. 3B). The triplet state energies of both isomers are considerably higher than in azobenzene, 2.14 and 2.35 eV for E and Z isomers, respectively.54 Thus, relatively selective isomerisation can be achieved by selecting a sensitiser with triplet energy close to the desired acceptor isomer. Sensitisation thus leads to a mixture of isomers whose isomer composition depends on the relative triplet energy of the sensitiser to both isomers.
Delightfully, triplet sensitisation of stilbene isomerisation has recently leapt from fundamental research to applications. Photoactuation of a polymer film doped with an iridium triplet sensitiser where the stilbene moiety was part of the polymer chain was demonstrated.57 Most importantly, this approach afforded a substantial redshift from UV (365 nm) to visible blue (445 nm) in the excitation wavelength. In photopharmacology, a ruthenium photosensitiser was used to isomerise several stilbene-based drug molecules to achieve selective cytotoxicity and disruption of the cellular microtubule network under 450 nm (one-photon) or 916 nm (two-photon) excitation of the sensitiser.58 So far, these works serve as the crucial first steps towards applications of triplet-sensitised photoswitching, more of which are surely to follow, due to the apparent milder, redshifted illumination conditions. In the future, the inherently high spatial resolution of triplet sensitisation (in the order of nanometer) could be of fundamental interest in looking at, e.g., intracellular processes or pathways with a confined triplet sensitiser.
In addition to azobenzene and stilbene, several other types of E–Z photoswitches have been shown to undergo isomerisation via triplet sensitisation. In general, triplet-sensitised isomerisation can be expected for molecules that can rotate about a CC, NN or CN59 bond. As such, triplet-sensitised isomerisation has been demonstrated for overcrowded-alkene-based molecular motors,60 hydrazones61 and thioindigos.62 Compared to E–Z isomerisation, photoswitches that undergo electrocyclisation are underrepresented in the triplet manifold. Triplet-sensitised isomerisation of norbornadienes/quadricyclanes and diarylethenes were recently reviewed,33,34 and we gladly point the interested reader their way. In addition to these two types of electrocyclisation photoswitches, also spiropyrans/merocyanines have been demonstrated to be capable of isomerisation upon triplet energy transfer.63
Regarding azoarenes, we would like to highlight two special cases of triplet isomerisation: bridged azobenzenes (also known as diazocines) and DESC (disequilibration of azoarenes under confinement). A bridged azobenzene, unlike a regular azobenzene, has substantially different triplet energies in E (1.53 eV) and Z (1.89 eV) isomers. This property enables, like in the case of stilbene, selective sensitisation of either isomer, which has recently been utilised to construct a purely triplet-sensitised bidirectional photoswitching system, i.e. without direct excitation of the photoswitch. PSS isomer distributions of 49% E and >99% Z could be acquired upon excitation with 530 and 740 nm, respectively.52 The other special case of triplet-sensitised photoswitching leads to the metastable isomer exclusively, a previously unattained feat. The DESC approach (Fig. 3C) utilises confinement to drive the isomerisation of virtually any azoarene from E to Z – even in aqueous environments. Flexible coordination cages are used to confine the E-azoarene and a triplet sensitiser. Following excitation of the sensitiser with visible (up to 635 nm) light, intersystem crossing and TET to the azoarene, the photoswitch can either relax back to the E isomer or the Z isomer. The confinement may induce some pre-twisting of the azoarene, which increases the quantum yield of the latter possibility, but it is still rather low. However, after isomerisation to the bent Z isomer, the cage cannot accommodate the azoarene anymore, and it is expelled from confinement. Over time, the system will thus efficiently isomerise the switches from E to Z, in many cases more efficiently than via direct excitation.64
The power of triplet sensitisation lies in its universality: it can be used to isomerise several photoswitch families, and since inside a given photoswitch family (e.g., azobenzenes) the triplet energy is apparently little affected by structural changes, the same sensitiser can be used for almost any compound. The biggest drawback in indirect isomerisation via TET is its susceptibility to quenching by molecular oxygen which exists as a triplet in its ground state and is readily excited to the singlet state by visible-light triplet sensitisers,65 which in turn hampers the sensitisation of the photoswitch molecules. In addition, singlet oxygen is extremely reactive and may degrade either the azobenzene or the sensitiser. As the concentration of oxygen in ambient solutions lies in the range of millimoles per litre, any experiments featuring micromolar concentrations of the sensitiser–azobenzene system are seriously affected without precautions. Common measures to tackle the problem include the deaeration of the solvent via freeze–pump–thaw cycles or sparging with an inert gas, and the use of oxygen scavengers.29 In the aforementioned solid-state study, the sensitiser and azobenzene molecules were confined within a gelatine matrix that blocks the diffusion of oxygen, thus removing the need for further deaeration steps.53 Similarly, the confinement in DESC hinders the diffusion of oxygen to the sensitiser–azoarene pair and at the same time ensures the proximity of the triplet energy donor and acceptor. This allows for a fast (on the ns timescale) TET process that successfully competes with oxygen quenching.64
Although the triplet sensitisation of switches like azobenzenes, stilbenes and norbornadienes has been thoroughly studied, several new discoveries have been made in the last few years, indicating that much progress is still to come. Systematic fundamental studies are needed for these well-known molecular families and especially for less explored switches to determine how electronic and conformational changes modify their triplet properties and whether the insight gained into the excited state properties of conventional switches can be generalised for other families. Furthermore, more applied research lines should be ventured to bring the fundamental research into practice. For example, utilising longer wavelengths will be undoubtedly beneficial for biomedical applications, e.g. light-triggered drug release66 and imaging67 or in chemical biology as a sensory tool.68 However, caution must be taken since both at in vivo and in vitro conditions the concentration of oxygen is sufficient to quench the triplet state, leading to the formation of reactive oxygen species. This in turn can result in either degradation of the photoswitch and/or the sensitiser or even the system under study. Therefore, we foresee that this approach will be utilised for, e.g., energy storage and release devices, sensors, and biomedicine applications for which hypoxic/anoxic conditions are relevant. Intriguingly, inherent oxygen sensitivity could be turned into an advantage by using triplet-based photoswitching to target hypoxic disease sites, e.g. solid tumours, which poorly respond to conventional photodynamic therapy.
Photon upconversion
As an alternative to TET described in the previous section, one may employ a more complex phenomenon also dependent on a triplet state: triplet–triplet annihilation upconversion (TTAUC) or triplet fusion upconversion.69 The beauty of this process lies in the fact that it allows the generation of high-energy photons (e.g., blue) upon excitation of a system with low-energy photons (e.g., red light) with a theoretical quantum yield of 50%. To make it happen, two types of molecules (in addition to the photoswitch) are usually needed, a sensitiser and an annihilator. Upon excitation of a sensitiser molecule, it undergoes intersystem crossing to generate a triplet-excited state and subsequent TET to an annihilator molecule. Then two triplet-excited annihilator molecules may collide, leading to triplet fusion (annihilation) where one singlet excited state and one singlet ground state of annihilator will be formed. The singlet excited state of an annihilator will eventually emit a photon of higher energy than initially absorbed by the sensitiser, resulting in anti-Stokes emission that can be absorbed by the switch (Fig. 4). Thanks to a wide range of TTAUC sensitiser/annihilator pairs including commercially available ones,69–71 excitation and emission wavelengths can be tuned to induce the isomerisation of a photoswitch of interest. In contrast to two-photon absorption, the process of TTAUC is efficient even by using non-coherent, low-intensity light, including the sun.72 A potential hurdle to utilising TTAUC for photoswitching is TET from the sensitiser to the photoswitch rather than to the annihilator, leading to triplet sensitisation rather than photon upconversion. This may be avoided by physically separating the TTAUC pair and the photoswitch, or by using upconversion nanoparticles (UCNPs, typically based on NaYF4 doped with lanthanides) instead of organic molecules (vide infra).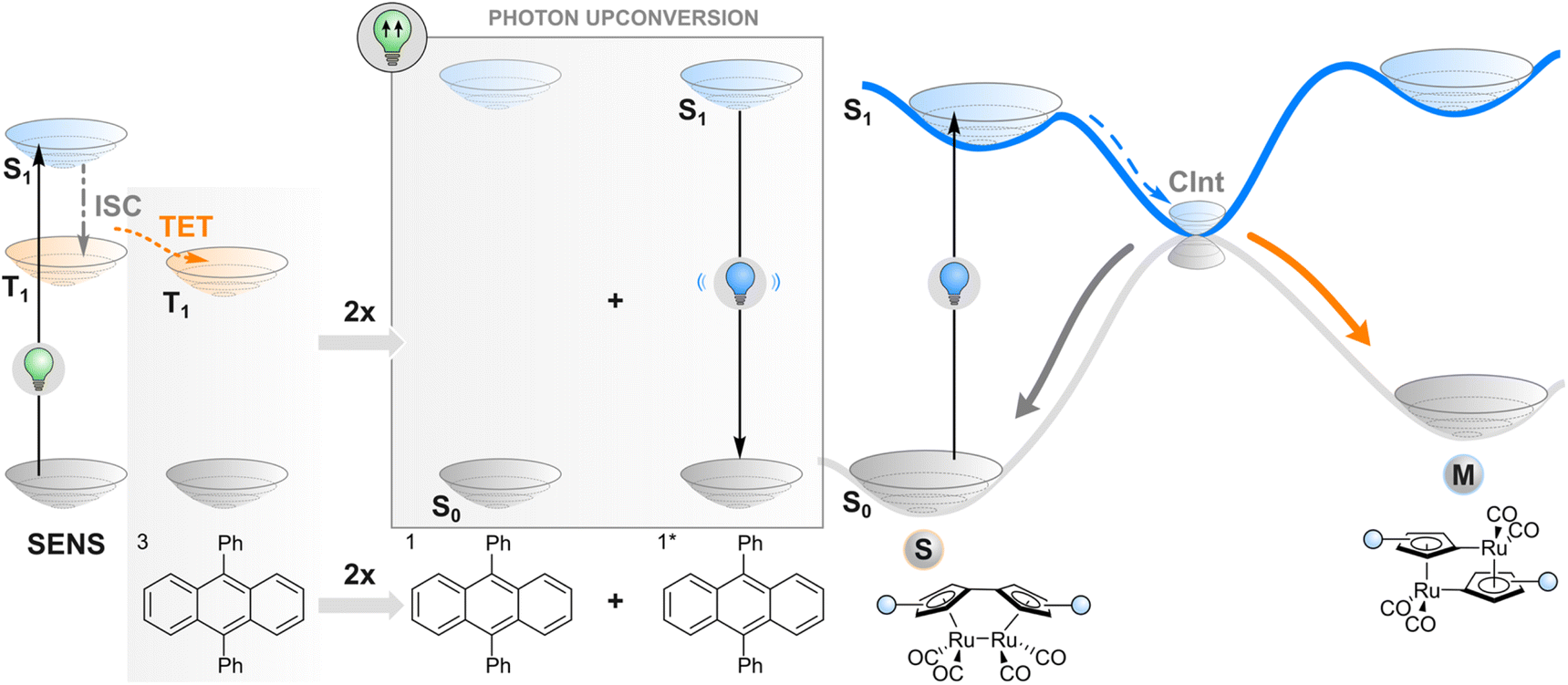 | ||
| Fig. 4 Triplet–triplet annihilation upconversion. Following the excitation of a sensitiser (SENS) and triplet energy transfer (TET) to an annihilator, two triplet-excited (T1) annihilators react to form one singlet-excited (S1) annihilator. It fluoresces light that is absorbed by the photoswitch, which ultimately leads to isomerisation from stable to metastable state. | ||
The first example of utilising TTAUC for photoswitching was published in 2013. A sandwich-like microfluidic device was created for solar energy storage consisting of two chips, one of which contained the TTAUC pair and the other the photoswitch, a fulvalene diruthenium derivative. The upconversion system depended on the absorption of 500–550 nm green light by a palladium(II) porphyrin derivative and consequent upconverted emission of violet light by a diphenylanthracene annihilator, which ultimately transformed the photoswitch into its higher-energy isomer capable of storing the light energy.30 Since then, similar approaches (with slightly different TTAUC pairs in different environments) have been utilised to isomerise a diarylethene,73 a styryl hemicyanine barium(II) complex,74 an azotolane crosslinker for photoactuation,31 and a hemiindigo.75 In the latter case, the authors were able to indirectly induce isomerisation in both directions by using a pH-responsive TTAUC system where the wavelength of the upconverted light could be tuned by adding acid or base.
TTAUC has also been combined with other phenomena covered in this perspective. Two different approaches were utilised to induce the isomerisation of stilbene with red (635 nm) light either from Z to E or from E to Z. For the former direction, an annihilator was used that, in its singlet excited state, could abstract an electron from the stilbene and thus catalyse the isomerisation to the more stable E form (photoinduced electron transfer; vide infra).76 Contrary to most triplet-fusion-based systems, the TTAUC pair and the photoswitch were not separated physically, which enabled the electron transfer. Isomerisation in the reverse direction was induced by first creating upconverted light in a physically separate vessel and then using this to excite a ruthenium(III) triplet sensitiser, followed by TET to the photoswitch and finally E → Z conversion.77 A third combination of TTAUC and another photochemical reaction was enabled by tethering a perylene annihilator into a photochromic phenoxyl–imidazolyl radical complex. After the initial excitation and annihilation steps, a fast and efficient intramolecular charge transfer from perylene to the photoswitch takes place, ultimately resulting in the latter's isomerisation after a sophisticated cascade of photochemical processes.78
In all the aforementioned TTAUC examples, the TTAUC pair and the photoswitch are either physically separated or the desired photoreaction between the singlet-excited annihilator molecule and the photoswitch is something else than simple emission-reabsorption. However, as already mentioned, the trivial emission of upconverted light without competing photochemical reactions is also possible without physical separation if UCNPs are used instead of TTAUC pairs since the upconversion processes are then confined within the nanoparticles (and often do not even produce triplet states) and do not lead to the triplet sensitisation of the photoswitch molecules. The upconversion inside UCNPs can occur either through a mechanism similar to TTAUC, e.g. energy transfer upconversion, or other ones including ground state absorption/excited state absorption, cooperative luminescence, cooperative sensitisation, cross-relaxation, photon avalanche processes, and energy migration-mediated upconversion.79,80 Regardless of the mechanism, it results in the emission of upconverted light that can be used to isomerise virtually any photoswitch system via e.g. Förster Resonance Energy Transfer (FRET) of simple re-absorption employing singlet manifold of a photoswitch. For more details on UCNP-mediated photoswitching, we would like to direct the reader to a recent (2023) review.81 Unfortunately, high power densities (in order of kW cm−2) that are needed to reach reasonably high quantum yields (even 1%) hamper the applicability of UCNPs. The excess of excitation energy may lead to, e.g., heating of the sample, which will promote undesired isomerisation of the photoswitch from its metastable isomer to the thermodynamically stable one. However, FRET or re-absorption usually successfully overcompete with the thermal isomerisation effect.82–84
Photoinduced electron transfer
In addition to the energy transfer processes discussed above, the interaction between an excited photosensitiser and a photoswitch may result in either reductive or oxidative charge transfer (photoinduced electron transfer, PET, Fig. 5); the same effect can also be induced electrochemically without light (vide infra). The utility of this process for catalysing the isomerisation is based on the formation of anionic or cationic photoswitch radicals that both isomerise to the thermodynamically more favourable isomer at an extremely fast rate (Fig. 5). In the case of published data for azobenzenes, the ground-state Z → E isomerisation rates are 1012 to 1017 times higher than for the respective neutral molecules.32,85,86 After isomerisation, the formed cationic or anionic radical E-AB˙(+/−) abstracts an electron from another Z-azobenzene, thus creating a chain reaction that is evident as isomerisation quantum yields above unity. As a consequence of the high rate of isomerisation and the high quantum yield, the overall reaction rate is governed by diffusion and the efficiency of the initial intermolecular electron transfer step. For the latter, the reduction/oxidation potentials of the excited sensitiser and photoswitch must be sufficiently matched; the oxidative pathway is more probable for electron-rich photoswitches, whereas the reductive one may be useful for electron-poor compounds. The mechanism is promoted by a polar medium that can stabilise the pair of radical ions formed.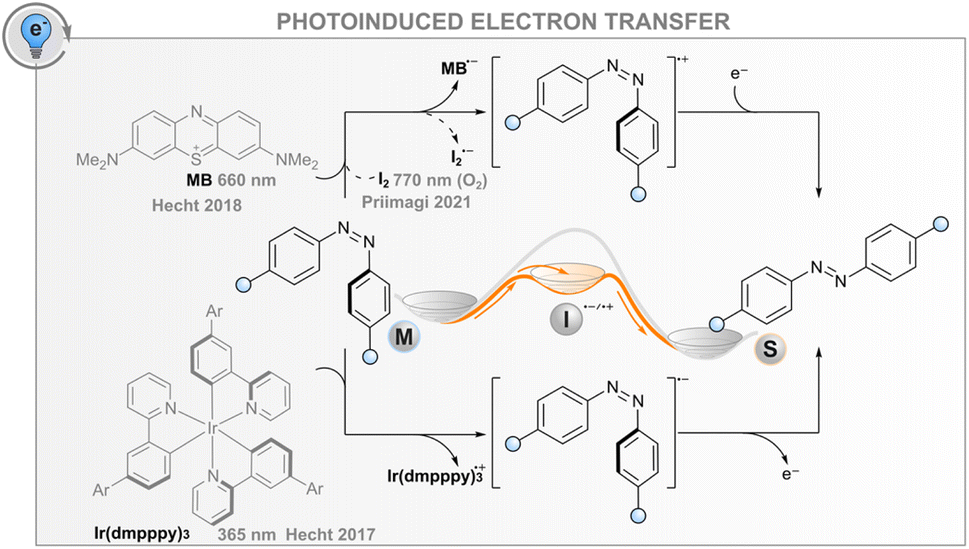 | ||
| Fig. 5 Oxidative (top) and reductive (bottom) photoinduced electron transfer routes. | ||
Catalysing the Z → E isomerisation of azobenzenes via the reductive route has been demonstrated with an iridium(III) photosensitiser but only using high-energy excitation (365 nm).86 In this regard, an oxidative route seems more feasible: the Z → E isomerisation of suitably electron-rich azobenzenes can be catalysed with a red-light photosensitiser methylene blue (MB) via the formation of an azobenzene radical cation.32 For example, 4,4′-dimethoxyazobenzene has an oxidation potential of 1.41 V, matching well the reduction potential of MB in either singlet or triplet excited state (1.56 and 1.60 V, respectively). Thus, MB can be used as a red-light photosensitiser to bring about quantitative and relatively fast isomerisation. It is worth noting that without the sensitiser, complete isomerisation to the E isomer is only possible thermally, as direct irradiation leads at best to a PSS with a Z-fraction of 31% and even this is only possible with blue light (∼440 nm). Since the system does not depend on triplet states, it tolerates oxygen, albeit with slightly diminished efficiency. As another example, it was shown that the Z → E isomerisation of moderately electron-rich azobenzenes could be catalysed with molecular iodine with up to near-infrared (770 nm) light.87 Surprisingly, the reaction was not only tolerated but mediated by molecular oxygen. The catalytic cycle was proposed to include the formation of singlet oxygen that subsequently abstracts an electron from the azobenzene, initiating the radical cation chain reaction. Iodine seems to have another function, perhaps later in the cycle, since the same oxygen-dependent catalytic reaction was observed when a palladium(II) phthalocyanine singlet oxygen sensitiser was used instead of iodine, but the reaction was considerably slower.
For both MB and iodine, two major problems exist. The sensitiser absorbs over most of the visible light region, meaning that E → Z isomerisation of the azobenzene can only be carried out with UV light or the sensitiser must be applied after initial direct photoisomerisation. In addition, hole catalysis is only feasible for suitably electron-rich compounds that match the oxidation potential of the sensitiser (or singlet oxygen): less oxidisable switches do not form the radical cation, whereas too electron-rich compounds over-oxidise irreversibly. Both MB and I2 can also catalyse the reaction for compounds without optimal oxidation properties, but in that case, the MB reaction proceeds via triplet sensitisation and is impeded by oxygen,32 and the I2-catalysed reaction relies on the formation of I˙ radicals that are formed when exciting iodine with green light.88,89 Additionally, iodine is impractical and not suitable for biological applications due to its toxicity. New photosensitisers should therefore be screened for this purpose.
The aforementioned routes depend, at least partly, on the formation of a relatively long-lived triplet species. The use of exclusively singlet-excited electron transfer agents was only recently studied.90 Since the lifetime of a singlet-excited photosensitiser is extremely short, the authors tethered different electron transfer agents to various azobenzenes. They provided the redox potentials and spectroscopic data of 33 azobenzenes and 11 non-phosphorescent (i.e., staying within the singlet manifold) biocompatible chromophores and demonstrated that by combining each azobenzene with a suitable chromophore, it was possible to catalyse the quantitative Z → E isomerisation of each azobenzene through singlet electron transfer. In some cases, even near-infrared light could be used. Because triplet states were avoided altogether, no quenching by molecular oxygen was observed.
As an example besides the E–Z isomerisation of double bonds, photoinduced electron transfer has featured in the isomerisation mechanism of a photochromic imidazole dimer.38 After sequential two-photon absorption of a porphyrin antenna, electron transfer to the imidazole takes place, ultimately leading to cleavage of the imidazole–imidazole bond. However, examples of purposefully leveraging electron transfer have not been reported widely. Since redox-catalysed isomerisation has already been demonstrated for azobenzenes, stilbenes,91 and quadricyclanes,92 catalytic conversion from the metastable to stable isomer via PET might be feasible for some other photoswitches as well. However, the screening of new photosensitisers with suitable spectral and electrochemical properties is required to broaden the scope of this method to photoswitches of different oxidation potentials and, particularly, to minimise the spectral overlap between the sensitiser and the photoswitch. However, already at its current stage, PET catalysis may enable new opportunities for, e.g., the release of stored solar energy or photoactuation, owing to its ability to induce the back-isomerisation fast, efficiently, and quantitatively, which could be of particular importance in optically dense systems such as polymers, liquid crystals and hydrogels where direct excitation of the whole sample is challenging.
Thermal mechanisms to control the isomerisation
Until now, we have focused our attention on the population of excited states of photochemical switches by means of an indirect photochemical stimulus that involves another species, e.g., a sensitiser, to drive the isomerisation to completion. Another means of externally controlling the thermal transformation of a switch with a high isomerisation barrier is by altering the potential energy surface that connects the metastable state to the stable state. By doing so, novel thermal pathways involving reaction intermediates can be opened, fine-tuning the thermal lifetime of back-isomerisation. Achieving this high level of control has a direct impact on the release of energy on-demand, which affects several applications, such as photochemical actuation93 or molecular solar thermal energy storage (MOST).94Azobenzenes offer an efficient platform to showcase the different strategies implemented to this end. Most approaches aim to diminish the double-bond character of the NN group to lower the barrier of isomerisation of the metastable state. Similarly to the photochemical oxidative process presented in the previous section, electron and hole catalysis have been implemented both with chemical oxidants and electrochemistry to regulate the lifetime of the Z form of azobenzenes.32,86 Ablated gold,85,95,96 zirconium oxide97 and silver98 nanoparticles were also found to be excellent oxidants for the thermal Z–E conversion of azobenzenes. Alternatively, the use of Lewis bases and acids99 as well as protic acid sensitisers100 is well known and reported in the literature. Along this line, by modifying the structure of the azobenzene to accommodate groups bearing acidic protons (e.g. NH or OH), it is possible to regulate the rate of thermal back-relaxation by modulating the water content of the solvent mixture, to promote the formation of the corresponding hydrazone tautomers.101,102 Similarly, a more general effect on the water concentration on the thermal back isomerisation of photoswitches was discovered in monoarylated indigo derivatives monosubstituted with an NH.103 In this particular case, the solvent interactions lead to a 300-fold enhancement of the thermal reaction of the metastable form of the switch.
Herein, however, we would like to underline another peculiarity of the thermal isomerisation of azobenzenes which was overlooked until recently in the literature.104 Since the discovery of its isomerism in 1937,105 the nature of the pathway that the molecule follows both at the excited and at the ground states were the source of fierce debates. During the thermal event, two mechanistic pathways were classically considered, inversion around or rotation about the central NN azo bond following a classical adiabatic mechanism on the singlet ground state surface,106–108 with results often influenced by the substituents and the level of constrain in the molecule.109 Nevertheless, the experimental evidence was not consistently backed up by the computational interpretation of the results. Most strikingly, the entropy of activation that is experimentally obtained does not follow the sign (for the inversion) and the magnitude (for the rotation) of the computed ones, affording the so-called “entropy puzzle”.110 An alternative mechanism for the azobenzene thermal isomerisation was first proposed in 2004 in a computational investigation of the excited state decay and thermal isomerisation routes of Z-azobenzene on the S0, S1, and T1 states.47 According to this study, the thermal reaction proceeds following a multistate process involving both the S0 and T1 states which cross in a region close to the transition state associated with the rotation mechanism (Fig. 6). Interestingly, this same multistate mechanism was proposed in 1941, where the authors discussed the potential for rotation about an ethylenic double bond to occur in two fashions. This proposal was later rejected for molecules bearing CC double bonds.111
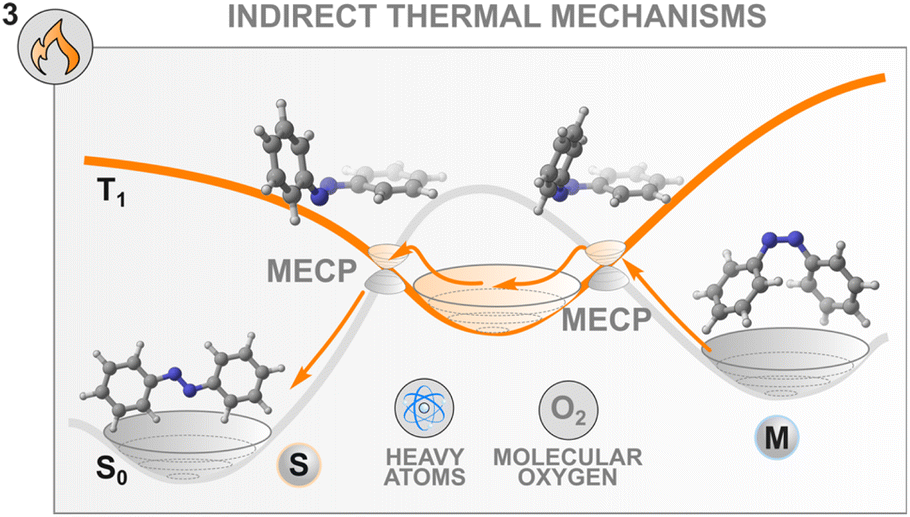 | ||
| Fig. 6 The mechanism of thermal isomerisation of azobenzene involves crossing to the T1 state via two minimum energy crossing points (MECPs). The presence of heavy atoms and molecular oxygen influence the thermal process. | ||
Recently, the thermal population of the lowest triplet state of azobenzenes was probed experimentally.104 This multistate mechanism involves two surface crossings involving the S0 and T1 states and is classified as a type II rotation mechanism (Fig. 6) to differentiate it from the adiabatic one (type I rotation). To describe such a mechanism, Eyring's transition state theory (TST) is no longer valid, therefore non-adiabatic TST needs to be applied. The S0 and T1 surfaces intersect at two minimum energy crossing points which play a similar role to the transition state in conventional Eyring's TST. In this case, the transmission coefficient depends explicitly on the spin–orbit coupling.103 The mechanism seems general for the different azoarenes.112–114
These new insights into the mechanism of azobenzene offer a fresh perspective on the thermal control of the stability of the metastable state of a switch. Indeed, control of the thermal back-isomerisation can be exerted by using additives with heavy atoms in the solvent mixture that can modulate the spin–orbit coupling between the surfaces with different multiplicities and facilitate (or hinder) the thermal back-isomerisation. This was demonstrated in a study where 0.25 M tetrabutylammonium iodide in the solution (of 60 μM azobenzene) slightly accelerated the Z → E isomerisation, especially at elevated temperatures, by increasing the activation entropy.104 The acceleration of thermal isomerisation was earlier observed in the presence of palladium and platinum porphyrins29 as well as molecular iodine87 but these effects were not investigated, making it unsure whether they originate from the heavy atom effect or other interactions such as coordination to the nitrogen lone pairs.
Interestingly, the thermal isomerisation of some switches is apparently dependent on a triplet quencher. BF2-coordinated azo complexes show an unusual influence of oxygen on the thermal relaxation of the metastable state, increasing the switching rate more than twenty-fold.115 Also the thermal isomerisation promoted by molecular iodine seems to be oxygen-dependent.87 The origin and generality of this effect are yet unknown, but it could possibly be leveraged for controlling the thermal back-isomerisation of at least some photoswitches.
These results are the corollary of a scientific story started in 1941 but open more possibilities for the precise control of the thermal isomerisation of molecular switches by multiple surface-crossings. Even though for CC double bonds the mechanism was ruled out, other switches, especially azobenzenes, do manifest peculiar thermal behaviour in the presence of heavy atoms or triplet quenchers. Recent findings demonstrate that (i) this phenomenon can be exploited to control the back-isomerisation rate without a light stimulus, and that (ii) there are still details in the isomerisation that we have not completely unveiled.
Conclusions and outlook
A wide range of photoswitches have been developed starting from the mid-20th century, each showing unique nanoscopic changes upon photoisomerisation that, when incorporated into a larger structure, can bring about macroscopic changes in properties ranging from colour and solubility to shape and motion. Owing to the large variety of different photoswitch structures, these molecules have found use in many fields of chemistry, physics, biology, and materials science. To meet the requirements of such diverse application areas, substitution patterns have been designed that tune the essential photoswitching parameters, including the wavelengths the switches absorb, the spectral separation of the two isomers and consequently the photostationary state isomer distributions, quantum yields of isomerisation, and the thermal stability of the metastable isomer. Particular efforts have been made to shift the absorption wavelengths away from the ultraviolet region and even to the red end of the visible spectrum – or beyond, to the near-infrared region. However, it has proven difficult to decouple the absorption profile from other properties: typically, structural changes that shift absorption towards the red end of the visible spectrum also lead to fast thermal back-isomerisation as well as diminished robustness, which is an obstacle to their wide use. Indirect photoisomerisation, based on exciting a proxy system that induces the isomerisation event, provides a way to using longer wavelengths without making changes to the photoswitch core, thus keeping its favourable properties intact. In this perspective, we have explored four different indirect excitation methods that can be applied to photoswitching: (i) two-photon excitation and subsequent singlet energy transfer, (ii) triplet sensitisation, (iii) photon upconversion, and (iv) photoinduced electron transfer. We have also introduced thermal processes that can be utilised to affect the isomerisation of certain photoswitches. For each indirect isomerisation strategy, we have provided theoretical background, state-of-the-art examples, and practical considerations.Each approach possesses unique prospects for applications. Two-photon excitation of an antenna and subsequent singlet energy transfer can be used to drive the isomerisation in either direction with near-infrared light, but it requires a focused high-power laser beam even with a good absorber. This method enables local isomerisation with extremely high precision, which could be useful for certain applications, but also makes uniform isomerisation in a large sample impossible, narrowing the applicability of this strategy. In addition, the absorber needs to be covalently linked to the photoswitch, which poses additional synthetic strain compared to other strategies that can be exploited by using separate sensitiser molecules with low-power, non-coherent light sources such as LEDs or the sun. Like two-photon excitation, photon upconversion can be leveraged for isomerisation in either direction as the conversion of low-energy photons into high-energy photons is decoupled from the isomerisation events; however, this usually only works if the upconversion system and the photoswitch are physically separated. Otherwise, energy or electron transfer events can take place before upconverted fluorescence, which may be an impediment but also a pathway to eloquent isomerisation mechanisms. The remaining two approaches do not have such a requirement. The function of triplet sensitisation depends on the photoswitch – for instance, stilbenes and diazocines can be isomerised in either direction by choosing suitable sensitisers, while the sensitisation of azoarenes yields mostly the E isomer and thus only catalyses the Z → E isomerisation unless novel strategies such as isomer-specific confinement (the DESC approach) are implemented. The biggest drawback related to working within the triplet manifold is the quenching by molecular oxygen that can pose challenges to many otherwise potential applications, although properly chosen strategies to shield the systems from oxygen or scavenge it may solve this issue. In this sense, isomerisation via photoinduced electron transfer is a more robust method. Moreover, thanks to the chain reaction character of indirect photoswitching induced by PET, a quantum yield of 199% may be reached.32 This is in striking contrast to the QY values obtained employing triplet sensitisation (up to 28%)28,32 and DESC (6.4%).64 However, it can only induce isomerisation from the metastable to the stable state (Z → E in the case of azoarenes and stilbenes) and has, so far, a rather limited scope. The same negatives apply to indirect thermal methods. However, each indirect excitation method can potentially benefit some application areas.
Indirect photoswitching has so far provided intriguing alternative handles for manipulating photoswitches. These efforts have revealed both fundamental (photo)physicochemical properties of different photoswitching families and improved or even introduced new applications for them. As stated above, it is foreseen that the applications that can most benefit from indirect photoswitching would in general meet the following criteria: (i) the absorption band(s) of the molecule are inconvenient in terms of the intended use (e.g. biological samples and UV absorption), and (ii) the photoswitch cannot be chemically altered to fine-tune its photoswitching properties without changing the application–relevant properties. As both of these are true for biological applications of many photoswitches,116 it is expected that many future applications could be found in biomedicine and biotechnology.
The fundamental side of the field presents at least two clear avenues of research: (i) expanding the scope of indirect photoswitching to new photoswitch families and (ii) systematic studies on structure–property relationships on different sensitiser–photoswitch pairs. Under more applied themes, fearless and unprejudiced probing into new application areas as well as finding ways of circumventing such obstacles as oxygen-sensitivity are required to realise the full potential of indirect photoswitching. The field will undoubtedly benefit from a holistic approach in designing the indirect photoswitching system, where both the thermal (ground state) and photophysical (photoexcited state) properties of the switch and the sensitiser and how they influence each other are considered. This approach requires combined computational, synthetic, and spectroscopic efforts, underlining the complex nature yet vast capabilities and tunability of these systems.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Author contributions
K. K. and A. P. conceived the idea for the perspective. K. K., J. I., J. J. W., S. C. and N. A. D. wrote the manuscript, with K. K. being primarily responsible for the work. S. C. created the figures. T. L., N. A. D., S. C. and A. P. acquired funding. All authors provided critical feedback and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the financial support offered by the European Research Council (Consolidator projects MULTIMODAL, No. 101045223, for A. P. and PADRE, No. 101001016, for T. L.). We also thank the Research Council of Finland for their support through the Center of Excellence Programme on Life-Inspired Hybrid Materials Research, LIBER, No. 346107 (A. P.) and Flagship Programme on Photonics Research and Innovation, PREIN, No. 320165 (A. P.; T. L.), and Academy Research Fellowship project, No. 354792 (N. A. D). S. C. thanks the Swedish Vetenskapsrådet for a Starting Grant (2021-05414). J. I. gratefully acknowledges the Finnish Academy of Science and Letters for a Foundations' Post Doc Pool grant.Notes and references
- G. Wald, Molecular Basis of Visual Excitation, Science, 1968, 162, 230–239 Search PubMed.
- F. A. Jerca, V. V. Jerca and R. Hoogenboom, Advances and opportunities in the exciting world of azobenzenes, Nat. Rev. Chem, 2022, 6, 51–69 CrossRef PubMed.
- D. Villarón and S. J. Wezenberg, Stiff-Stilbene Photoswitches: From Fundamental Studies to Emergent Applications, Angew. Chem., Int. Ed., 2020, 59, 13192–13202 Search PubMed.
- C. Petermayer and H. Dube, Indigoid Photoswitches: Visible Light Responsive Molecular Tools, Acc. Chem. Res., 2018, 51, 1153–1163 CrossRef CAS PubMed.
- M. Irie, T. Fukaminato, K. Matsuda and S. Kobatake, Photochromism of diarylethene molecules and crystals: Memories, switches, and actuators, Chem. Rev., 2014, 114, 12174–12277 CrossRef CAS PubMed.
- J. Orrego-Hernández, A. Dreos and K. Moth-Poulsen, Engineering of Norbornadiene/Quadricyclane Photoswitches for Molecular Solar Thermal Energy Storage Applications, Acc. Chem. Res., 2020, 53, 1478–1487 CrossRef PubMed.
- R. Klajn, Spiropyran-based dynamic materials, Chem. Soc. Rev., 2014, 43, 148–184 RSC.
- M. M. Lerch, W. Szymański and B. L. Feringa, The (photo)chemistry of Stenhouse photoswitches: guiding principles and system design, Chem. Soc. Rev., 2018, 47, 1910–1937 RSC.
- M. J. Fuchter, On the Promise of Photopharmacology Using Photoswitches: A Medicinal Chemist’s Perspective, J. Med. Chem., 2020, 63, 11436–11447 CrossRef CAS PubMed.
- W. Szymański, J. M. Beierle, H. A. V Kistemaker, W. A. Velema and B. L. Feringa, Reversible Photocontrol of Biological Systems by the Incorporation of Molecular Photoswitches, Chem. Rev., 2013, 113, 6114–6178 CrossRef PubMed.
- C. Fedele, T.-P. Ruoko, K. Kuntze, M. Virkki and A. Priimagi, New tricks and emerging applications from contemporary azobenzene research, Photochem. Photobiol. Sci., 2022, 21, 1719–1734 CrossRef CAS PubMed.
- S. P. Ihrig, F. Eisenreich and S. Hecht, Photoswitchable polymerization catalysis: state of the art, challenges, and perspectives, Chem. Commun., 2019, 55, 4290–4298 RSC.
- Z. Wang, P. Erhart, T. Li, Z.-Y. Zhang, D. Sampedro, Z. Hu, H. A. Wegner, O. Brummel, J. Libuda, M. B. Nielsen and K. Moth-Poulsen, Storing energy with molecular photoisomers, Joule, 2021, 5, 3116–3136 CrossRef CAS.
- X. Huang and T. Li, Recent progress in the development of molecular-scale electronics based on photoswitchable molecules, J. Mater. Chem. C, 2020, 8, 821–848 RSC.
- S. L. Oscurato, F. Reda, M. Salvatore, F. Borbone, P. Maddalena and A. Ambrosio, Shapeshifting Diffractive Optical Devices, Laser Photonics Rev., 2022, 16, 2100514 CrossRef.
- M. Regehly, Y. Garmshausen, M. Reuter, N. F. König, E. Israel, D. P. Kelly, C.-Y. Chou, K. Koch, B. Asfari and S. Hecht, Xolography for linear volumetric 3D printing, Nature, 2020, 588, 620–624 CrossRef CAS PubMed.
- H. Zeng, P. Wasylczyk, D. S. Wiersma and A. Priimagi, Light Robots: Bridging the Gap between Microrobotics and Photomechanics in Soft Materials, Adv. Mater., 2018, 30, 1703554 CrossRef PubMed.
- Z. Zhang, W. Wang, M. O'Hagan, J. Dai, J. Zhang and H. Tian, Stepping Out of the Blue: From Visible to Near-IR Triggered Photoswitches, Angew. Chem., Int. Ed., 2022, 61, e202205758 CrossRef CAS PubMed.
- A.-L. Leistner and Z. L. Pianowski, Smart Photochromic Materials Triggered with Visible Light, Eur. J. Org. Chem., 2022, 2022, e202101271 CrossRef CAS.
- D. Bléger and S. Hecht, Visible-Light-Activated Molecular Switches, Angew. Chem., Int. Ed., 2015, 54, 11338–11349 CrossRef PubMed.
- J. R. Hemmer, S. O. Poelma, N. Treat, Z. A. Page, N. D. Dolinski, Y. J. Diaz, W. Tomlinson, K. D. Clark, J. P. Hooper, C. Hawker and J. Read de Alaniz, Tunable Visible and Near Infrared Photoswitches, J. Am. Chem. Soc., 2016, 138, 13960–13966 CrossRef CAS PubMed.
- M. Dong, A. Babalhavaeji, S. Samanta, A. A. Beharry and G. A. Woolley, Red-Shifting Azobenzene Photoswitches for in Vivo Use, Acc. Chem. Res., 2015, 48, 2662–2670 CrossRef CAS PubMed.
- S. Helmy, S. Oh, F. A. Leibfarth, C. J. Hawker and J. Read de Alaniz, Design and Synthesis of Donor–Acceptor Stenhouse Adducts: A Visible Light Photoswitch Derived from Furfural, J. Org. Chem., 2014, 79, 11316–11329 CrossRef CAS PubMed.
- C.-Y. Huang, A. Bonasera, L. Hristov, Y. Garmshausen, B. M. Schmidt, D. Jacquemin and S. Hecht, N,N′-Disubstituted Indigos as Readily Available Red-Light Photoswitches with Tunable Thermal Half-Lives, J. Am. Chem. Soc., 2017, 139, 15205–15211 CrossRef CAS PubMed.
- L. Köttner, E. Ciekalski and H. Dube, Peri-Anthracenethioindigo: A Scaffold for Efficient All-Red-Light and Near-Infrared Molecular Photoswitching, Angew. Chem., Int. Ed., 2023, 62, e202312955 CrossRef PubMed.
- K. Mori, Y. Ishibashi, H. Matsuda, S. Ito, Y. Nagasawa, H. Nakagawa, K. Uchida, S. Yokojima, S. Nakamura, M. Irie and H. Miyasaka, One-Color Reversible Control of Photochromic Reactions in a Diarylethene Derivative: Three-Photon Cyclization and Two-Photon Cycloreversion by a Near-Infrared Femtosecond Laser Pulse at 1.28 μm, J. Am. Chem. Soc., 2011, 133, 2621–2625 CrossRef CAS PubMed.
- J. Croissant, M. Maynadier, A. Gallud, H. Peindy N'Dongo, J. L. Nyalosaso, G. Derrien, C. Charnay, J.-O. Durand, L. Raehm, F. Serein-Spirau, N. Cheminet, T. Jarrosson, O. Mongin, M. Blanchard-Desce, M. Gary-Bobo, M. Garcia, J. Lu, F. Tamanoi, D. Tarn, T. M. Guardado-Alvarez and J. I. Zink, Two-Photon-Triggered Drug Delivery in Cancer Cells Using Nanoimpellers, Angew. Chem., Int. Ed., 2013, 52, 13813–13817 CrossRef CAS PubMed.
- L. B. Jones and G. S. Hammond, Mechanisms of Photochemical Reactions in Solution. XXX.1 Photosensitized Isomerization of Azobenzene, J. Am. Chem. Soc., 1965, 87, 4219–4220 CrossRef CAS.
- J. Isokuortti, K. Kuntze, M. Virkki, Z. Ahmed, E. Vuorimaa-Laukkanen, M. A. Filatov, A. Turshatov, T. Laaksonen, A. Priimagi and N. A. Durandin, Expanding excitation wavelengths for azobenzene photoswitching into the near-infrared range via endothermic triplet energy transfer, Chem. Sci., 2021, 12, 7504–7509 RSC.
- K. Börjesson, D. Dzebo, B. Albinsson and K. Moth-Poulsen, Photon upconversion facilitated molecular solar energy storage, J. Mater. Chem. A, 2013, 1, 8521–8524 RSC.
- Z. Jiang, M. Xu, F. Li and Y. Yu, Red-Light-Controllable Liquid-Crystal Soft Actuators via Low-Power Excited Upconversion Based on Triplet–Triplet Annihilation, J. Am. Chem. Soc., 2013, 135, 16446–16453 CrossRef CAS PubMed.
- A. Goulet-Hanssens, C. Rietze, E. Titov, L. Abdullahu, L. Grubert, P. Saalfrank and S. Hecht, Hole Catalysis as a General Mechanism for Efficient and Wavelength-Independent Z → E Azobenzene Isomerization, Chem, 2018, 4, 1740–1755 CAS.
- Z. Li, X. Zeng, C. Gao, J. Song, F. He, T. He, H. Guo and J. Yin, Photoswitchable diarylethenes: From molecular structures to biological applications, Coord. Chem. Rev., 2023, 497, 215451 CrossRef CAS.
- Z. Wang, L. Fernandez, A. S. Aslam, M. Shamsabadi, L. M. Muhammad and K. Moth-Poulsen, Chasing the rainbow: Exploiting photosensitizers to drive photoisomerization reactions, Responsive Mater., 2023, 1, e20230012 CrossRef.
- A. S. Dvornikov, E. P. Walker and P. M. Rentzepis, Two-Photon Three-Dimensional Optical Storage Memory, J. Phys. Chem. A, 2009, 113, 13633–13644 CrossRef CAS PubMed.
- A. Ambrosio, P. Maddalena, A. Carella, F. Borbone, A. Roviello, M. Polo, A. A. R. Neves, A. Camposeo and D. Pisignano, Two-Photon Induced Self-Structuring of Polymeric Films Based on Y-Shape Azobenzene Chromophore, J. Phys. Chem. C, 2011, 115, 13566–13570 CrossRef CAS.
- M. Dudek, N. Tarnowicz-Staniak, M. Deiana, Z. Pokładek, M. Samoć and K. Matczyszyn, Two-photon absorption and two-photon-induced isomerization of azobenzene compounds, RSC Adv., 2020, 10, 40489–40507 RSC.
- M. Albota, D. Beljonne, J.-L. Brédas, J. E. Ehrlich, J.-Y. Fu, A. A. Heikal, S. E. Hess, T. Kogej, M. D. Levin, S. R. Marder, D. McCord-Maughon, J. W. Perry, H. Röckel, M. Rumi, G. Subramaniam, W. W. Webb, X.-L. Wu and C. Xu, Design of Organic Molecules with Large Two-Photon Absorption Cross Sections, Science, 1998, 281, 1653–1656 CrossRef CAS PubMed.
- J. Moreno, M. Gerecke, L. Grubert, S. A. Kovalenko and S. Hecht, Sensitized Two-NIR-Photon Z → E Isomerization of a Visible-Light-Addressable Bistable Azobenzene Derivative, Angew. Chem., Int. Ed., 2016, 55, 1544–1547 CrossRef CAS PubMed.
- Ó. Guzmán-Méndez, E. Villatoro, M. M. Reza, M. E. Sandoval, J. Jara-Cortés, M.-E. García-Aguilera, M. Bravo-Romero, J. G. López-Cortés and J. Peon, Non-linear photo-switching in molecular actuators through intra-molecular energy transfer from an electron donating core, J. Mater. Chem. C, 2023, 11, 10598–10612 RSC.
- E. Villatoro, L. Muñoz-Rugeles, J. Durán-Hernández, B. Salcido-Santacruz, N. Esturau-Escofet, J. G. López-Cortés, M. C. Ortega-Alfaro and J. Peón, Two-photon induced isomerization through a cyaninic molecular antenna in azo compounds, Chem. Commun., 2021, 57, 3123–3126 RSC.
- L. Pfeifer, N. V Hoang, M. Scherübl, M. S. Pshenichnikov and B. L. Feringa, Powering rotary molecular motors with low-intensity near-infrared light, Sci. Adv., 2020, 6, eabb6165 CrossRef CAS PubMed.
- M. Izquierdo-Serra, M. Gascón-Moya, J. J. Hirtz, S. Pittolo, K. E. Poskanzer, È. Ferrer, R. Alibés, F. Busqué, R. Yuste, J. Hernando and P. Gorostiza, Two-Photon Neuronal and Astrocytic Stimulation with Azobenzene-Based Photoswitches, J. Am. Chem. Soc., 2014, 136, 8693–8701 CrossRef CAS PubMed.
- Y. Kobayashi, T. Katayama, T. Yamane, K. Setoura, S. Ito, H. Miyasaka and J. Abe, Stepwise Two-Photon-Induced Fast Photoswitching via Electron Transfer in Higher Excited States of Photochromic Imidazole Dimer, J. Am. Chem. Soc., 2016, 138, 5930–5938 CrossRef CAS PubMed.
- B. Albinsson and J. Mårtensson, Long-range electron and excitation energy transfer in donor–bridge–acceptor systems, J. Photochem. Photobiol., C, 2008, 9, 138–155 CrossRef CAS.
- Z. Huang, Z. Xu, T. Huang, V. Gray, K. Moth-Poulsen, T. Lian and M. L. Tang, Evolution from Tunneling to Hopping Mediated Triplet Energy Transfer from Quantum Dots to Molecules, J. Am. Chem. Soc., 2020, 142, 17581–17588 CrossRef CAS PubMed.
- A. Cembran, F. Bernardi, M. Garavelli, L. Gagliardi and G. Orlandi, On the Mechanism of the cis–trans Isomerization in the Lowest Electronic States of Azobenzene: S0, S1, and T1, J. Am. Chem. Soc., 2004, 126, 3234–3243 CrossRef CAS PubMed.
- L. Gagliardi, G. Orlandi, F. Bernardi, A. Cembran and M. Garavelli, A theoretical study of the lowest electronic states of azobenzene: the role of torsion coordinate in the cis–trans photoisomerization, Theor. Chem. Acc., 2004, 111, 363–372 Search PubMed.
- P. Bortolus and S. Monti, Cis–trans photoisomerization of azobenzene. Solvent and triplet donors effects, J. Phys. Chem., 1979, 83, 648–652 CrossRef CAS.
- S. Monti, S. Dellonte and P. Bortolus, The lowest triplet state of substituted azobenzenes: An energy transfer investigation, J. Photochem., 1983, 23, 249–256 CrossRef CAS.
- M. Morikawa, M. Mizuno, N. Harada and N. Kimizuka, On-demand Triplet-sensitized Photoswitching of Arylazopyrazoles, Chem. Lett., 2023, 52, 727–731 CrossRef CAS.
- J. Isokuortti, T. Griebenow, J.-S. von Glasenapp, T. Raeker, M. A. Filatov, T. Laaksonen, R. Herges and N. A. Durandin, Triplet sensitization enables bidirectional isomerization of diazocine with 130 nm redshift in excitation wavelengths, Chem. Sci., 2023, 14, 9161–9166 RSC.
- P. Bharmoria, S. Ghasemi, F. Edhborg, R. Losantos, Z. Wang, A. Mårtensson, M. Morikawa, N. Kimizuka, Ü. İşci, F. Dumoulin, B. Albinsson and K. Moth-Poulsen, Far-red triplet sensitized Z-to-E photoswitching of azobenzene in bioplastics, Chem. Sci., 2022, 13, 11904–11911 RSC.
- G. S. Hammond and J. Saltiel, Photosensitized Cis–Trans Isomerization of the Stilbenes, J. Am. Chem. Soc., 1962, 84, 4983–4984 CrossRef CAS.
- G. S. Hammond, J. Saltiel, A. A. Lamola, N. J. Turro, J. S. Bradshaw, D. O. Cowan, R. C. Counsell, V. Vogt and C. Dalton, Mechanisms of Photochemical Reactions in Solution. XXII.1 Photochemical cis–trans Isomerization, J. Am. Chem. Soc., 1964, 86, 3197–3217 CrossRef CAS.
- T. Neveselý, M. Wienhold, J. J. Molloy and R. Gilmour, Advances in the E → Z Isomerization of Alkenes Using Small Molecule Photocatalysts, Chem. Rev., 2022, 122, 2650–2694 CrossRef PubMed.
- D. Beery, E. Stanisauskis, G. M. McLeod, A. Das, G. A. Guillory, J. G. Kennemur, W. S. Oates and K. Hanson, Enabling Lower Energy Light Harvesting in Stilbene-Based Photomechanical Polymers via Triplet Sensitization, ACS Appl. Polym. Mater., 2022, 4, 4081–4086 CrossRef CAS.
- E. E. Watson, F. Russo, D. Moreau and N. Winssinger, Optochemical Control of Therapeutic Agents through Photocatalyzed Isomerization, Angew. Chem., Int. Ed., 2022, 61, e202203390 CrossRef CAS PubMed.
- A. Padwa and F. Albrecht, Photochemical syn-anti isomerization about the carbon-nitrogen double bond, J. Am. Chem. Soc., 1974, 96, 4849–4857 CrossRef CAS.
- A. Cnossen, L. Hou, M. M. Pollard, P. V Wesenhagen, W. R. Browne and B. L. Feringa, Driving Unidirectional Molecular Rotary Motors with Visible Light by Intra- And Intermolecular Energy Transfer from Palladium Porphyrin, J. Am. Chem. Soc., 2012, 134, 17613–17619 CrossRef CAS PubMed.
- L. L. Costanzo, U. Chiacchio, S. Giuffrida and G. Condorelli, Sensitized photoisomerization of pyridylhydrazones, J. Photochem., 1980, 14, 125–132 CrossRef CAS.
- G. M. Wyman, B. M. Zarnegar and D. G. Whitten, Excited-state chemistry of indigoid dyes. III. Interaction of indigo and thioindigo with tin(IV) tetraphenyltetrahydroporphyrin triplets. Photosensitized isomerization of thioindigo, J. Phys. Chem., 1973, 77, 2584–2586 CrossRef CAS.
- H. Görner, L. S. Atabekyan and A. K. Chibisov, Photoprocesses in spiropyran-derived merocyanines: singlet versus triplet pathway, Chem. Phys. Lett., 1996, 260, 59–64 CrossRef.
- J. Gemen, J. R. Church, T.-P. Ruoko, N. Durandin, M. J. Białek, M. Weißenfels, M. Feller, M. Kazes, M. Odaybat, V. A. Borin, R. Kalepu, Y. Diskin-Posner, D. Oron, M. J. Fuchter, A. Priimagi, I. Schapiro and R. Klajn, Disequilibrating azobenzenes by visible-light sensitization under confinement, Science, 2023, 381, 1357–1363 CrossRef CAS PubMed.
- C. Schweitzer and R. Schmidt, Physical Mechanisms of Generation and Deactivation of Singlet Oxygen, Chem. Rev., 2003, 103, 1685–1758 CrossRef CAS PubMed.
- H.-B. Cheng, S. Zhang, J. Qi, X.-J. Liang and J. Yoon, Advances in Application of Azobenzene as a Trigger in Biomedicine: Molecular Design and Spontaneous Assembly, Adv. Mater., 2021, 33, 2007290 CrossRef CAS PubMed.
- M. Olesińska-Mönch and C. Deo, Small-molecule photoswitches for fluorescence bioimaging: engineering and applications, Chem. Commun., 2023, 59, 660–669 RSC.
- M. Müller, K. Niemeyer, N. Urban, N. K. Ojha, F. Zufall, T. Leinders-Zufall, M. Schaefer and O. Thorn-Seshold, BTDAzo: A Photoswitchable TRPC5 Channel Activator, Angew. Chem., Int. Ed., 2022, 61, e202201565 CrossRef PubMed.
- T. N. Singh-Rachford and F. N. Castellano, Photon upconversion based on sensitized triplet–triplet annihilation, Coord. Chem. Rev., 2010, 254, 2560–2573 CrossRef CAS.
- N. Yanai and N. Kimizuka, New Triplet Sensitization Routes for Photon Upconversion: Thermally Activated Delayed Fluorescence Molecules, Inorganic Nanocrystals, and Singlet-to-Triplet Absorption, Acc. Chem. Res., 2017, 50, 2487–2495 CrossRef CAS PubMed.
- J. Zhao, S. Ji and H. Guo, Triplet–triplet annihilation based upconversion: from triplet sensitizers and triplet acceptors to upconversion quantum yields, RSC Adv., 2011, 1, 937–950 RSC.
- S. Baluschev, T. Miteva, V. Yakutkin, G. Nelles, A. Yasuda and G. Wegner, Up-Conversion Fluorescence: Noncoherent Excitation by Sunlight, Phys. Rev. Lett., 2006, 97, 143903 CrossRef CAS PubMed.
- W. Larsson, M. Morimoto, M. Irie, J. Andréasson and B. Albinsson, Diarylethene Isomerization by Using Triplet–Triplet Annihilation Photon Upconversion, Chem.–Eur. J., 2023, 29, e202203651 CrossRef CAS PubMed.
- A. Vasilev, R. Dimitrova, M. Kandinska, K. Landfester and S. Baluschev, Accumulation of the photonic energy of the deep-red part of the terrestrial sun irradiation by rare-earth metal-free E–Z photoisomerization, J. Mater. Chem. C, 2021, 9, 7119–7126 RSC.
- Y. Wei, H. Xian, X. Lv, F. Ni, X. Cao and C. Yang, Triplet–triplet annihilation upconversion with reversible emission-tunability induced by chemical-stimuli: a remote modulator for photocontrol isomerization, Mater. Horiz., 2021, 8, 606–611 RSC.
- F. Glaser and O. S. Wenger, Sensitizer-controlled photochemical reactivity via upconversion of red light, Chem. Sci., 2023, 14, 149–161 RSC.
- J. B. Bilger, C. Kerzig, C. B. Larsen and O. S. Wenger, A Photorobust Mo(0) Complex Mimicking [Os(2,2′-bipyridine)3]2+ and Its Application in Red-to-Blue Upconversion, J. Am. Chem. Soc., 2021, 143, 1651–1663 CrossRef CAS PubMed.
- A. Tokunaga, L. M. Uriarte, K. Mutoh, E. Fron, J. Hofkens, M. Sliwa and J. Abe, Photochromic Reaction by Red Light via Triplet Fusion Upconversion, J. Am. Chem. Soc., 2019, 141, 17744–17753 CrossRef CAS PubMed.
- E. M. Chan, E. S. Levy and B. E. Cohen, Rationally Designed Energy Transfer in Upconverting Nanoparticles, Adv. Mater., 2015, 27, 5753–5761 CrossRef CAS PubMed.
- X. Cheng, D. Tu, W. Zheng and X. Chen, Energy transfer designing in lanthanide-doped upconversion nanoparticles, Chem. Commun., 2020, 56, 15118–15132 RSC.
- K. Malhotra, D. Hrovat, B. Kumar, G. Qu, J. Van Houten, R. Ahmed, P. A. E. Piunno, P. T. Gunning and U. J. Krull, Lanthanide-Doped Upconversion Nanoparticles: Exploring A Treasure Trove of NIR-Mediated Emerging Applications, ACS Appl. Mater. Interfaces, 2023, 15, 2499–2528 CrossRef CAS PubMed.
- A. Bednarkiewicz, M. Nyk, M. Samoc and W. Strek, Up-conversion FRET from Er3+/Yb3+:NaYF4 Nanophosphor to CdSe Quantum Dots, J. Phys. Chem. C, 2010, 114(41), 17535–17541 CrossRef CAS.
- A. Das, C. C. Magot, E. Rappeport, T. B. Tis and W. Park, Quantitative modeling and experimental verification of Förster resonant energy transfer in upconversion nanoparticle biosensors, J. Appl. Phys., 2021, 130, 023102 CrossRef CAS.
- S. Schimka, D. T. Klier, A. López de Guereñu, P. Bastian, N. Lomadze, M. U. Kumke and S. Santer, Photo-isomerization of azobenzene containing surfactants induced by near-infrared light using upconversion nanoparticles as mediator, J. Phys.: Condens.Matter, 2019, 31(12), 125201 CrossRef PubMed.
- E. Titov, L. Lysyakova, N. Lomadze, A. V Kabashin, P. Saalfrank and S. Santer, Thermal Cis-to-Trans Isomerization of Azobenzene-Containing Molecules Enhanced by Gold Nanoparticles: An Experimental and Theoretical Study, J. Phys. Chem. C, 2015, 119, 17369–17377 CrossRef CAS.
- A. Goulet-Hanssens, M. Utecht, D. Mutruc, E. Titov, J. Schwarz, L. Grubert, D. Bléger, P. Saalfrank and S. Hecht, Electrocatalytic Z → E Isomerization of Azobenzenes, J. Am. Chem. Soc., 2017, 139, 335–341 CrossRef CAS PubMed.
- K. Kuntze, J. Isokuortti, A. Siiskonen, N. Durandin, T. Laaksonen and A. Priimagi, Azobenzene Photoswitching with Near-Infrared Light Mediated by Molecular Oxygen, J. Phys. Chem. B, 2021, 125, 12568–12573 CrossRef CAS PubMed.
- S. Yamashita, Iodine-photocatalyzed cis–trans Isomerization of Stilbene, Bull. Chem. Soc. Jpn., 1961, 34, 972–976 CrossRef CAS.
- S. Yamashita, D. P. Cosgrave, H. Ono and O. Toyama, The Photoisomerization of cis-Azobenzene in the Presence of Iodine, Bull. Chem. Soc. Jpn., 1963, 36, 688–692 CrossRef CAS.
- B. Baumgartner, V. Glembockyte, R. Mayer, A. Gonzalez-Hernandez, R. Kindler, A. Valavalkar, A. Wiegand, A. Müller-Deku, L. Grubert, F. Steiner, C. Gross, M. Reynders, V. Grenier, J. Broichhagen, S. Hecht, P. Tinnefeld, A. Ofial, B. Dietzek-Ivanšic, J. Levitz and O. Thorn-Seshold, Azobenzenes can achieve near-infrared photocontrol in biological systems, with quantitative Z → E photoisomerization, via singlet manifold photoredox, ChemRxiv, 2023, preprint, DOI:10.26434/chemrxiv-2023-37sv4.
- F. D. Lewis, J. R. Petisce, J. D. Oxman and M. J. Nepras, One-way photoisomerization of cis-stilbene via a cation radical chain mechanism, J. Am. Chem. Soc., 1985, 107, 203–207 CrossRef CAS.
- F. Waidhas, M. Jevric, M. Bosch, T. Yang, E. Franz, Z. Liu, J. Bachmann, K. Moth-Poulsen, O. Brummel and J. Libuda, Electrochemically controlled energy release from a norbornadiene-based solar thermal fuel: increasing the reversibility to 99.8% using HOPG as the electrode material, J. Mater. Chem. A, 2020, 8, 15658–15664 RSC.
- A. Ryabchun, Q. Li, F. Lancia, I. Aprahamian and N. Katsonis, Shape-Persistent Actuators from Hydrazone Photoswitches, J. Am. Chem. Soc., 2019, 141, 1196–1200 CrossRef CAS PubMed.
- Z. Wang, H. Hölzel and K. Moth-Poulsen, Status and challenges for molecular solar thermal energy storage system based devices, Chem. Soc. Rev., 2022, 51, 7313–7326 RSC.
- G. L. Hallett-Tapley, C. D'Alfonso, N. L. Pacioni, C. D. McTiernan, M. González-Béjar, O. Lanzalunga, E. I. Alarcon and J. C. Scaiano, Gold nanoparticle catalysis of the cis–trans isomerization of azobenzene, Chem. Commun., 2013, 49, 10073–10075 RSC.
- S. Simoncelli and P. F. Aramendía, Mechanistic insight into the Z–E isomerization catalysis of azobenzenes mediated by bare and core–shell gold nanoparticles, Catal. Sci. Technol., 2015, 5, 2110–2116 RSC.
- J. L. Kanyak, C. A. Pointer and D. C. Achey, Control of Intramolecular Isomerization Reactivity of an Azobenzene Derivative Anchored to ZrO2 Nanoparticle Thin Films, J. Phys. Chem. C, 2018, 122, 1589–1594 CrossRef CAS.
- G. Angelini, L. Scotti, A. Aceto and C. Gasbarri, Silver nanoparticles as interactive media for the azobenzenes isomerization in aqueous solution: From linear to stretched kinetics, J. Mol. Liq., 2019, 284, 592–598 CrossRef.
- C. D. Hall and P. D. Beer, Trico-ordinate phosphorus compounds as catalysts for the isomerization of (Z)-to (E)-azobenzene, J. Chem. Soc., Perkin Trans. 2, 1991, 1947–1950 RSC.
- S. Ciccone and J. Halpern, Catalysis of the cis–trans isomerization of azobenzene by acids and cupric salts, Can. J. Chem., 1959, 37, 1903–1910 CrossRef CAS.
- N. A. Simeth, S. Crespi, M. Fagnoni and B. König, Tuning the Thermal Isomerization of Phenylazoindole Photoswitches from Days to Nanoseconds, J. Am. Chem. Soc., 2018, 140, 2940–2946 CrossRef CAS PubMed.
- M. Poutanen, Z. Ahmed, L. Rautkari, O. Ikkala and A. Priimagi, Thermal Isomerization of Hydroxyazobenzenes as a Platform for Vapor Sensing, ACS Macro Lett., 2018, 7, 381–386 CrossRef CAS PubMed.
- L. A. Huber, P. Mayer and H. Dube, Photoisomerization of Mono-Arylated Indigo and Water-Induced Acceleration of Thermal cis-to-trans Isomerization, ChemPhotoChem, 2018, 2, 458–464 CrossRef CAS.
- M. Reimann, E. Teichmann, S. Hecht and M. Kaupp, Solving the Azobenzene Entropy Puzzle: Direct Evidence for Multi-State Reactivity, J. Phys. Chem. Lett., 2022, 13, 10882–10888 CrossRef CAS PubMed.
- G. S. Hartley, 113. The cis-form of azobenzene and the velocity of the thermal cis → trans-conversion of azobenzene and some derivatives, J. Chem. Soc., 1938, 633–642 RSC.
- J. Dokić, M. Gothe, J. Wirth, M. V Peters, J. Schwarz, S. Hecht and P. Saalfrank, Quantum Chemical Investigation of Thermal Cis-to-Trans Isomerization of Azobenzene Derivatives: Substituent Effects, Solvent Effects, and Comparison to Experimental Data, J. Phys. Chem. A, 2009, 113, 6763–6773 CrossRef PubMed.
- A. Muždalo, P. Saalfrank, J. Vreede and M. Santer, Cis-to-Trans Isomerization of Azobenzene Derivatives Studied with Transition Path Sampling and Quantum Mechanical/Molecular Mechanical Molecular Dynamics, J. Chem. Theory Comput., 2018, 14, 2042–2051 CrossRef PubMed.
- S. Crespi, N. A. Simeth, A. Bellisario, M. Fagnoni and B. König, Unraveling the Thermal Isomerization Mechanisms of Heteroaryl Azoswitches: Phenylazoindoles as Case Study, J. Phys. Chem. A, 2019, 123, 1814–1823 CrossRef CAS PubMed.
- T. Asano, Pressure effects on the thermal cis–trans isomerization of 4-dimethylamino-4’-nitroazobenzene. Evidence for a change of mechanism with solvent, J. Am. Chem. Soc., 1980, 102, 1205–1206 CrossRef CAS.
- C. Rietze, E. Titov, S. Lindner and P. Saalfrank, Thermal isomerization of azobenzenes: on the performance of Eyring transition state theory, J. Phys.: Condens.Matter, 2017, 29, 314002 CrossRef PubMed.
- M. C. Lin and K. J. Laidler, Some aspects of cis–trans-isomerization mechanisms, Can. J. Chem., 1968, 46, 973–978 CrossRef CAS.
- S. Axelrod, E. Shakhnovich and R. Gómez-Bombarelli, Thermal Half-Lives of Azobenzene Derivatives: Virtual Screening Based on Intersystem Crossing Using a Machine Learning Potential, ACS Cent. Sci., 2023, 9, 166–176 CrossRef CAS PubMed.
- N. K. Singer, K. Schlögl, J. P. Zobel, M. D. Mihovilovic and L. González, Singlet and Triplet Pathways Determine the Thermal Z/E Isomerization of an Arylazopyrazole-Based Photoswitch, J. Phys. Chem. Lett., 2023, 14, 8956–8961 CrossRef CAS PubMed.
- A. H. Heindl and H. A. Wegner, Rational Design of Azothiophenes—Substitution Effects on the Switching Properties, Chem.–Eur. J., 2020, 26, 13730–13737 CrossRef CAS PubMed.
- Y. Yang, R. P. Hughes and I. Aprahamian, Visible light switching of a BF 2-coordinated azo compound, J. Am. Chem. Soc., 2012, 134, 15221–15224 CrossRef CAS PubMed.
- B. Baumgartner, V. Glembockyte, A. J. Gonzalez-Hernandez, A. Valavalkar, R. J. Mayer, L. L. Fillbrook, et al., A general method for near-infrared photoswitching in biology, demonstrated by the >700 nm photocontrol of GPCR activity in brain slices, ChemRxiv, 2024, preprint, DOI:10.26434/chemrxiv-2024-vm4n3.
| This journal is © The Royal Society of Chemistry 2024 |