
Rapid determination of V isotopes with MC-ICP-MS: new developments in sample purification
Zhen
Zeng
ab and
Fei
Wu
 *a
*a
aSchool of Earth Sciences, State Key Laboratory of Geological Processes and Mineral Resources, China University of Geosciences, Wuhan, 430074, China. E-mail: wufei@cug.edu.cn
bState Key Laboratory of Soil and Sustainable Agriculture, Institute of Soil Science, Chinese Academy of Sciences, Nanjing, 210008, China
First published on 9th November 2023
Abstract
Vanadium (V) isotopes are a powerful tool for fingerprinting the redox-state variations in geological processes, but their widespread application is hindered by the complicated processes of separation of V from matrix elements. In this study, we established a two-stage chromatography technique using cation and anion exchange resins for V purification from natural samples. This method can quantitatively and efficiently separate V from matrix elements, especially Ti and Cr, which introduce spectral interferences. The V isotopic composition was analysed using multi-collector inductively coupled plasma-mass spectrometry. The instrumental mass fractionation of V isotopic ratios was corrected by the sample-standard-bracketing method. The δ51V values of two in-house standard solutions, CUG-V and BDH-V, were 0.04 ± 0.08‰ (2 SD, n = 121) and −1.23 ± 0.08‰ (2 SD, n = 91), respectively. The V isotope compositions of 13 reference materials, including igneous rock, manganese nodules, carbonaceous siliceous shale, sediment and soil, were measured in this study. Based on the repeated measurements of pure solution and reference materials, the long-term reproducibility was better than ± 0.10‰ (2 SD) for δ51V values. The new approach to V isotope analysis presented in this work is both economical and time-efficient, improving the ability for further investigation of V isotope geochemistry in various geological and environmental systems.
1. Introduction
Vanadium (V) is a refractory lithophilic element which also shows weak siderophile affinity. Vanadium exhibits multiple oxidation states (V2+, V3+, V4+ and V5+) in silicates and oxides, and as V0 in the metallic core.1 As a redox-sensitive element, V is widely used to constrain variations in redox conditions in various geological processes, such as terrestrial accretion and core-mantle differentiation,2,3 the oxidation state of the mantle and magmas throughout Earth's history,4–6 as well as changes in marine palaeo-redox states.7,8Vanadium has two long-lived isotopes (50V and 51V) with abundances of 0.24% and 99.76%,9 respectively. With the development of analytical methods using multi-collector inductively coupled plasma-mass spectrometry (MC-ICP-MS), high-precision measurements of V isotopes have recently been developed.10–21 Vanadium isotope compositions are generally reported as δ51V values, which are the deviations of V isotope ratios relative to AA-V solution:10 δ51V = [(51V/50Vsample)/(51V/50VAA) − 1] × 1000‰. Large V isotope fractionations have been predicted by theoretical and experimental studies,22–24 and observed in natural samples, which implies their wide application to study geological processes. The distinct δ51V in chondritic meteorite,25,26 Martian,27 lunar,28 and terrestrial materials29,30 were proposed to reflect V isotope fractionation during planetary differentiation in the Solar System and the formation of the Moon. Vanadium isotope variations observed in mantle-derived magmas are thought to indicate variable isotope fractionation during mantle partial melting, which are likely controlled by the degree of partial melting and fO2 variations in the mantle.29,31–33 The δ51V values of igneous rocks show apparently increasing with the degree of magma differentiation, indicating remarkable V isotope fractionation during magmatic evolution.31,34,35 In addition, different magmatic differentiation trends of V isotopes are observed in different magmatic systems, which is thought to be controlled by variable crystallized minerals with different isotope fractionation factors. Thus, V isotopes could also be used to distinguish between different fractionating assemblages and thus contribute to our understanding of differentiation processes during magma evolution.35
Limited isotope fractionation was observed during rock weathering,36 which is consistent with the observation that the δ51V of average river waters was similar to that of average upper continental crust.20 For comparison, the δ51V of the seawater were significantly heavier than those of river waters due to the fact that the main sinks of V in the ocean, i.e., oxic sediment, anoxic sediment, euxinic sediment and hydrothermal sediment, preferentially captured light V isotopes from seawater.21,37–39 The strong correlation between δ51V in marine authigenic sediments and the overlying redox conditions suggests that vanadium isotopes in sediments have the potential to trace variations in the marine palaeo-redox state, particularly to trace subtle redox variations in local oxygen-deficient to low oxygen environments.40–44 Moreover, the variations in V isotopes in terrestrial materials could be used to trace the sources of vanadium in crude oils, soils, lake sediments and aquatic food chains.15,18,19,45
Despite the promising application of V isotopes, the dataset and investigation of V isotopes are still limited due to the complicated and inefficient chemical purification procedure of V isotopes. There are several reasons for this problem. First, the sample-standard-bracketing (SSB) method is the only applicable method for V isotope measurement, which requires a nearly 100% recovery rate of V to avoid isotopic fractionation during chemical purification. Second, V is a trace element, and the low tolerance of the matrix effect during instrumental measurement requires the quantitative removal of matrices during chemical purification. Last, both 50Ti and 50Cr exhibit isobaric interferences on the minor 50V, which significantly affects the determination of V isotopes. Therefore, the quantitative separation of V from matrix elements, especially Cr and Ti, is crucial to obtain high-precision and accurate V isotope ratios. To overcome the above difficulties, chemical purification of V from matrix elements typically involved multiple sequential ion exchange chromatography columns in previous works. For instance, Nielsen, et al.10 used anion and TRU SPEC resin to separate the majority of matrix elements, followed by quantitative removal of the remaining matrix elements, especially Cr and Ti, via two or more anion exchange columns. In an effort to improve time-efficiency, Wu, et al.13 utilized cation exchange resin to remove the majority of matrix elements. In summary, previous methods required at least four stage ion exchange columns, consuming considerable time and reagents. In this study, we established a simplified purification procedure, consisting of a two-step ion exchange process using one cation exchange column and one anion exchange column to effectively separate V from matrices.
2. Analytical methods
2.1 Reagents, materials, and sample preparation
Electron grade HF, HNO3 and HCl from LIAN SHI NEW MATERIAL CORP., LTD (LSNM, China) were double purified using a DTS-1000 acid purification system (Savillex, USA). The Guaranteed Reagent (GR) HClO4 and H2O2 were purchased from LSNM. The cation-exchange resins (AG50W-X12) and anion-exchange resins (AG 1-X8) required for the purification process were purchased from Bio-Rad. Ultrapure water with a resistivity of 18.2 MΩ cm, obtained from a Milli-Q Element system (Millipore Corporation, USA), was used for reagent dilution and sample preparation.The reference materials used in this study included four igneous rock standards from the United States Geological Survey (USGS), one manganese nodule standard from the USGS, one carbonaceous siliceous shale standard from the Institute of Geophysical and Geochemical Exploration (IGGE) of the Chinese Academy of Geological Sciences and seven soil and sediment standards from the National Institute of Standards and Technology (NIST) and IGGE.
The sample digestion and chemical purification procedures were carried out in an ultraclean laboratory of the State Key Laboratory of Geological Processes and Mineral Resources, China University of Geosciences, Wuhan. Samples (10–100 mg) containing 2–20 μg V were weighed into 7 mL Teflon PFA screw cap vials (Savillex) and digested with concentrated HF and HNO3 (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) at 120–140 °C for 24 h to decompose silicates. The soil and sediment samples were pretreated with HClO4–HNO3–HF mixture acids (1:1:2, v/v/v) at 160 °C for 8 h to digest organic matter before decomposing the silicates. Then, the samples were dried and treated with aqua regia first at room temperature for 12 h and then heated to 100 °C for 4 h to ensure that all V in the solution were oxidized to pentavalent V (V5+). After subsequent evaporation to dryness, samples were treated with 2 mL of 3 mol L−1 (M) HCl to convert all anions to chloride. Finally, the sample was dried and re-dissolved in 1 mL of 1 M HCl at 120 °C for 4 h, and then 1 mL ultrapure H2O was used to dilute the sample solution which was ready for ion-exchange purification in the first column.
:1, v/v) at 120–140 °C for 24 h to decompose silicates. The soil and sediment samples were pretreated with HClO4–HNO3–HF mixture acids (1:1:2, v/v/v) at 160 °C for 8 h to digest organic matter before decomposing the silicates. Then, the samples were dried and treated with aqua regia first at room temperature for 12 h and then heated to 100 °C for 4 h to ensure that all V in the solution were oxidized to pentavalent V (V5+). After subsequent evaporation to dryness, samples were treated with 2 mL of 3 mol L−1 (M) HCl to convert all anions to chloride. Finally, the sample was dried and re-dissolved in 1 mL of 1 M HCl at 120 °C for 4 h, and then 1 mL ultrapure H2O was used to dilute the sample solution which was ready for ion-exchange purification in the first column.
2.2 Chemical purification procedure
A two-step ion exchange scheme for the separation of V was developed in this study. The chemical purification procedures are listed in Table 1. In the first step, 2 mL of cation exchange resin (AG50W-X12, 200–400 mesh, Bio-Rad, USA) was loaded into the Bio-Rad PP chromatography column (2 mL bed volume). A cation exchange resin column was used to remove the majority of all matrix elements. The loaded cation exchange resin was cleaned with 20 mL 4 M HNO3 + 0.5 M HF and 5 mL H2O sequentially, and then conditioned with 5 mL 0.5 M HCl. Fig. 1a illustrates the typical elution curves for V purification procedures in the first column. The sample solutions containing 2–20 μg V in 2 mL 0.5 M HCl were loaded on the column. Five milliliters of 0.1 M HCl + 0.05 M HF mixture followed by 13 mL 0.1 M HCl were used to elute Ti and part of Al and Cr. Vanadium was then collected with 6 mL 0.1 M HNO3 + 2% H2O2. After this step, most matrix elements (e.g., Na, Fe, Mg, Ca, K and Cr) were still held on the cation exchange resin while V was completely eluted. The collected V solution was dried and re-dissolved in 1 mL 0.1 M HCl. Thirty-three microliters of H2O2 (30%, v/v) was added to the sample solution 1–2 h before loading sample solution into the second column.| Resin | Eluent | Vol. (mL) | Comment |
|---|---|---|---|
| Column 1 2 mL AG50W-X12 (200–400 mesh) | 4 M HNO3 + 0.5 M HF | 20 | Clean resin |
| H2O | 5 | Clean resin | |
| 0.5 M HCl | 5 | Condition resin | |
| Load sample in 0.5 M HCl | 2 | — | |
| 0.1 M HCl + 0.05 M HF | 5 | Elute Ti, Al and Cr | |
| 0.1 M HCl | 13 | Elute Ti, Al and Cr | |
| 0.1 M HNO3 + 2% H2O2 | 6 | Collect V | |
| 4 M HNO3 | 20 | Elute matrix elements | |
| Column 2 1 mL AG1-X8 (200–400 mesh) | 1 M HNO3 | 10 | Clean resin |
| H2O | 5 | Clean resin | |
| 0.1 M HCl + 1% H2O2 | 5 | Condition resin | |
| Load sample in 0.1 M HCl + 1% H2O2 | 1 | — | |
| 0.1 M HCl + 1% H2O2 | 15 | Elute matrix elements | |
| 1 M HNO3 | 10 | Collect V |
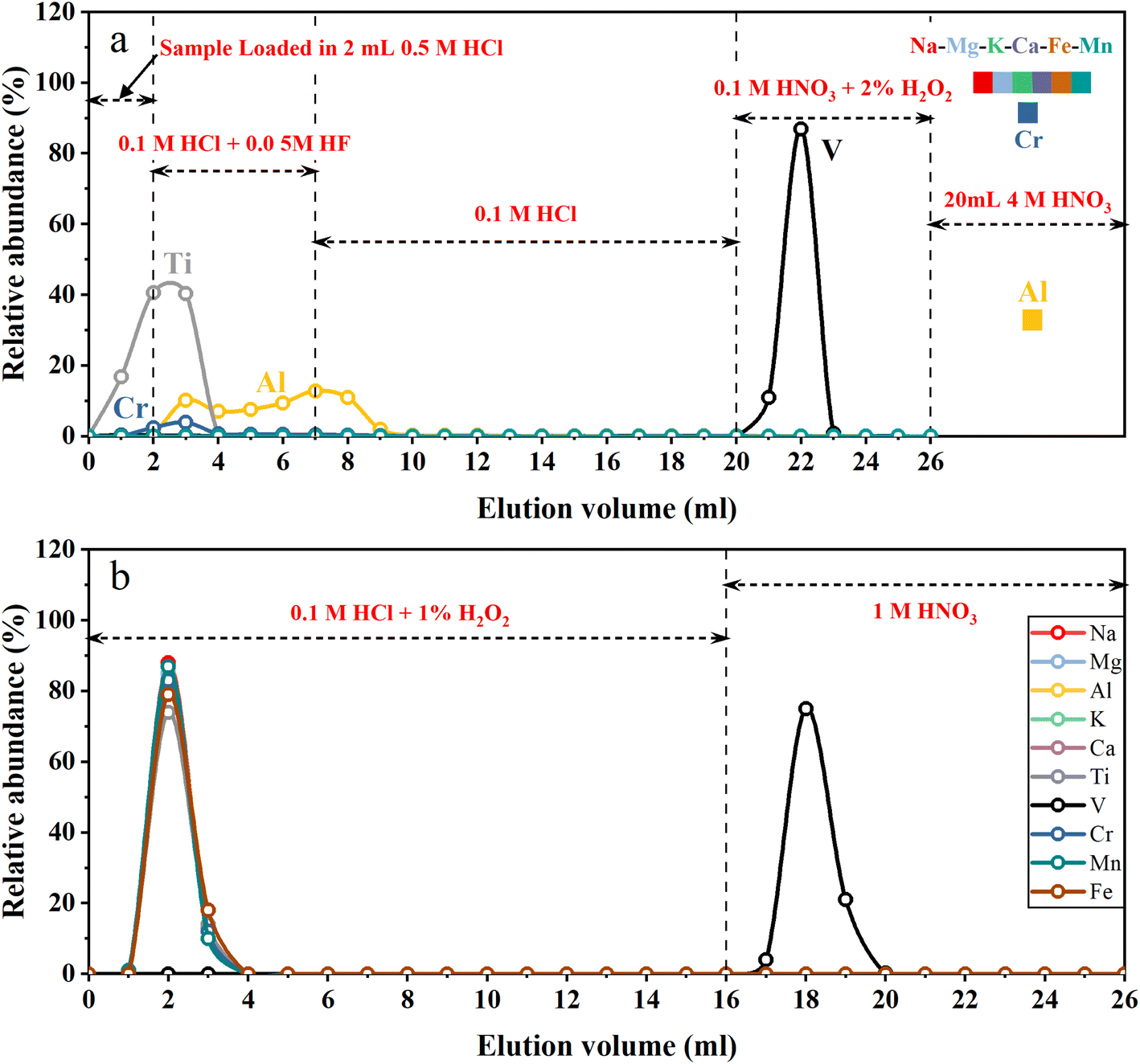 | ||
| Fig. 1 (a) Elution curve for V purification in the first column. The igneous reference material BCR-2 was used to obtain the elution curve. (b) Elution curve for V purification in the second column. The synthetic solution (Na:Mg:Al:K:Ca:Ti:V:Cr:Mn:Fe = 5:5:5:5:5:1:1:1:1:5) was used to obtain the elution curve. | ||
Since V is a trace element, even a small proportion of matrix elements (especially Ti and Cr) remaining in the solution after the first step of purification would significantly influence the V isotope measurement.10,13 With the aim of quantitatively removing the remaining matrix elements, a second purification step was carried out using an anion exchange resin column. One milliliter of anion exchange resins (AG1-X8, 200–400 mesh, Bio-Rad, USA) was loaded into the pipette tip with quartz wool inserted as a porous barrier to retain the resins. Fig. 1b illustrates the typical elution curves for V purification procedures in the second column. After cleaning with 10 mL 1 M HNO3 and 3 mL H2O, the anion exchange resins were conditioned with 3 mL 0.1 M HCl + 1% H2O2. Sample dissolved in 1 mL 0.1 M HCl + 1% H2O2 was loaded onto the resin. Matrix elements were eluted with 15 mL of 0.1 M HCl + 1% H2O2, and V was collected using 10 mL 1 M HNO3 afterwards. After evaporation of the collected V cut, 1 mL of 1:1 (v/v) concentrated HNO3 and 30% H2O2 were added to the beaker, which was closed and heated on a hot plate at 80 °C for four hours to decompose organic matter eluted from resins. Finally, the sample was dried and dissolved in 2% HNO3 (m/m) for instrumental analysis. The total procedure blank was below 2 ng, which is negligible compared to a total of 2–20 μg V in the samples.
2.3 Mass spectrometry
Vanadium isotopes were measured using multi-collector inductively coupled plasma-mass spectrometry (Neptune Plus, Thermo-Fisher Scientific, Bremen, Germany) in the same laboratory. The cup configuration and instrumental parameters are summarized in Table 2. Normal Ni Jet and Ni X-skimmer cones (Thermo-Fisher Scientific, Bremen, Germany) were used for V isotope measurements. High-resolution mode with a resolution greater than 6000 was used to avoid molecular interference (such as 36Ar14N+, 36Ar16O+, and 38Ar14N+). In order to improve the transmission, the sample solutions were introduced into the instrument using an Aridus II desolvator (CETAC Technologies). The sweep Ar gas used had a flow rate of ∼3 L min−1. The typical sensitivity of 51V was approximately 120 V ppm−1. To correct the isobaric interferences of 50Ti and 50Cr on the 50V signal, 49Ti and 52Cr were collected simultaneously during measurement. The ratios of 49Ti/50Ti and 52Cr/50Ti during isobaric interference correction were obtained by determining the pure standard Ti and Cr solution before each measurement sequence began. Data were gathered for one block comprised of 40 cycles, each with an integration time of 4.194 seconds. To prevent any possible cross-contamination, the sample introduction system was cleaned with 5% HNO3 (m/m) for two minutes followed by 2% HNO3 (m/m) for two minutes before each sample or standard measurement. After the cleaning interval, the signal of 51V was reduced to ∼50 mV. To eliminate the influence of background, the signals in 2% HNO3 (m/m) at the beginning of each measurement sequence were subtracted from the analysed signals of each sample. During measurement, the instrumental mass fractionation of V isotope ratios was corrected using the sample-standard-bracketing method.| MC-ICP-MS | Thermo Fisher Scientific, Neptune Plus | |||||
| Cooling Ar | ∼16 L min−1 | |||||
| Auxiliary Ar | ∼0.70 L min−1 | |||||
| Nebulizer Ar | ∼1.05 L min−1 | |||||
| Mass resolution | High resolution | |||||
| Typical sensitivity | ∼120 V per μg mL−1 for 51V | |||||
| Cones | Ni jet cone, X-skimmer cone | |||||
| Desolvator | Aridus II | |||||
| Ar sweep | ∼3 L min−1 | |||||
| Solution uptake rate | ∼50 μL min−1 | |||||
| Faraday cup | L4 | L2 | C | H1 | H3 | H4 |
| Resistor | 1011 Ω | 1011 Ω | 1011 Ω | 1010 Ω | 1011 Ω | 1011 Ω |
| Mass | 47Ti | 49Ti | 50V | 51V | 52Cr | 53Cr |
3. Results and discussion
3.1 Separation scheme of V in the first column
To quantitatively remove matrix elements, especially Ti and Cr from V, previous purification schemes primarily relied on peroxide–V complexes and anion exchange resin. Under weakly acidic conditions (pH 1–3), pentavalent V can form anionic complexes with H2O2.46 Such peroxide–V anionic complexes can firmly bind to anion exchange resin, while other matrix elements can be quantitatively eluted from the resin under such conditions.47,48 However, two difficulties should be addressed to achieve this aim. First, the existence of complicated matrix elements, especially high field-strength elements like Ti, made it unstable for most geological samples with the required mass in weak acid, preventing them from being totally dissolved. Second, H2O2 is easily catalyzed to dissociate into oxygen and water by transition metals such as Fe, which is a major element in most geological samples. This could lead to the destruction of peroxide–V complexes, resulting in a large loss of V from the anion exchange resin. Therefore, multiple separation procedures were designed to remove these critical matrix elements (Ti and Fe) prior to the purification procedure by applying peroxide–V complexes with anion exchange resin. For example, Nielsen, et al.10 used anion exchange resin and TRU SPEC resin to remove Fe and Ti. Wu, et al.13 applied cation exchange resin to remove most of the Ti, Al, Fe and some other matrix elements. Qi, et al.30 combined multiple anion and cation exchange resins to separate V from the majority of matrix elements including Fe and Ti. The anion exchange procedure with peroxide–V complexes was further applied at least twice to quantitatively remove the remaining matrix elements, especially Ti and Cr, by these methods. In conclusion, previous methods required at least four column purifications to achieve thorough separation for reliable instrumental measurement, requiring at least one week to complete the purification process.To overcome the above obstacles, we proposed a novel chromatography strategy with a cation exchange resin (Table 1). As shown in the elution curve (Fig. 1a), after loading sample solution, Ti and Al were first eluted using 0.1 M HCl + 0.05 M HF and 0.1 M HCl, as they form anionic fluorides that have low affinity with cation resin, in the presence of trace HF.49,50 Then, V was eluted from the cation resin with 0.1 M HNO3 + 2% H2O2 (v/v) in the form of a V-peroxide anionic complex, which does not bind with the cation resin. For comparison, other cationic matrix elements were still retained on the cation exchange resin,30 because they have high equilibrium distribution coefficients under low acidity conditions.51 Thus, V could be separated from almost all the matrix elements with this column procedure. After this step of purification, all sample solutions exhibited 49Ti/51V and 52Cr/51V ratios lower than 10−4 and 10−3, respectively, representing a decrease of three to five orders of magnitude (Table 3).
| Isotopic ratio | Sample | Before separation | After first column | After second column |
|---|---|---|---|---|
| 49Ti/51V | GSP-2 | 4.3 | 6.5 × 10−5 | 0.4 × 10−5 |
| SRM2711a | 6.7 | 3.8 × 10−5 | 0.4 × 10−5 | |
| BHVO-2 | 2.8 | 3.2 × 10−5 | 0.3 × 10−5 | |
| BCR-2 | 1.8 | 1.1 × 10−5 | 0.3 × 10−5 | |
| 52Cr/51V | GSP-2 | 0.32 | 2.0 × 10−4 | 0.4 × 10−4 |
| SRM2711a | 0.54 | 1.5 × 10−4 | 0.5 × 10−4 | |
| BHVO-2 | 0.76 | 1.3 × 10−4 | 0.4 × 10−4 | |
| BCR-2 | 0.04 | 0.5 × 10−4 | 0.3 × 10−4 |
The elution curves of cations, such as Mg,52 V,13 and Rb,53 might drift with different amounts of these elements loaded into the cation resin columns. Therefore, to ensure quantitative collection of V during the purification procedure, we conducted experiments to determine the elution curves of different types of samples containing variable amounts of V, ranging from 2 μg to 20 μg. The samples used in this experiment included GSP-2, SRM2711a, BHVO-2 and BCR-2, which represent granodiorite, soil and basalt, respectively. The V concentration of these reference materials ranged from 52 to 420 μg g−1. The results showed that regardless of the variable loading amount of V and matrix elements onto the column, the elution curves of V overlapped perfectly with each other (Fig. 2). Thus, the recovery rates of V with this purification procedure were consistently higher than 99%. This is because previous methods generally used diluted HNO3 (0.2–1.2 M) to gradually elute V from the column,13,15 depending on the distribution coefficient of V in the cation exchange resins. Instead, hydrogen peroxide was used in this procedure to form V-peroxide anion complexes that could not bind to cation exchange resins, allowing V to be directly eluted. It is notable that there were a few small bubbles generated in the column when using 0.1 M HNO3 + 2% H2O2 to elute V, as hydrogen peroxide was partly catalyzed to decomposition by Fe retained in the resin. Despite this issue, the consistently high recovery rates of V for different types of samples with variable Fe/V ratios demonstrate that H2O2 was sufficient to completely form the V-peroxide anion complex. Furthermore, we conducted three repetitions of the purification using the same cation exchange resin and found that the elution curve of V could be well replicated, regardless of the number of usages.
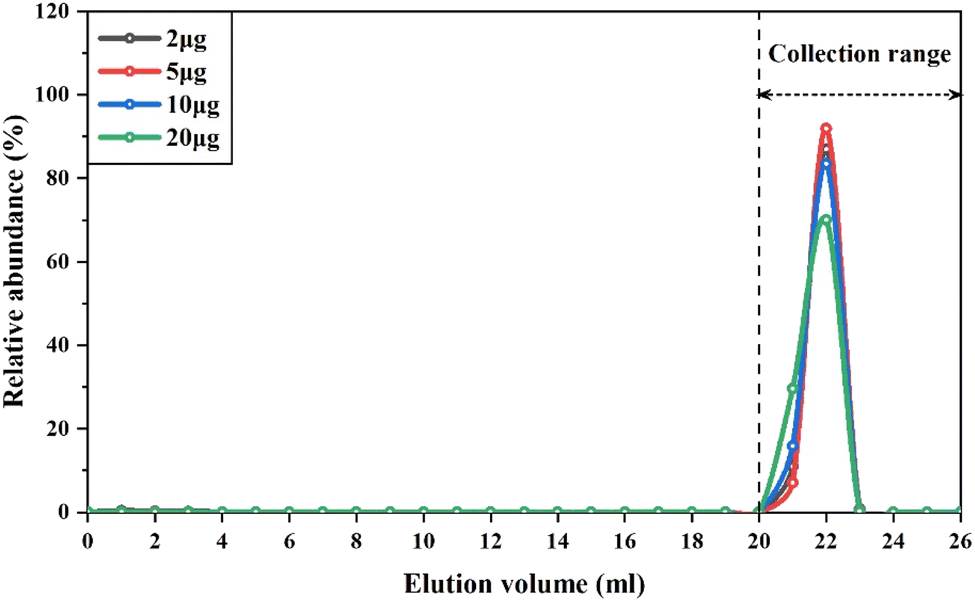 | ||
| Fig. 2 Elution curves for V with different masses of V loaded to the first columns. The reference materials GSP-2, SRM2710a, BHVO-2 and BCR-2 were used to obtain the elution curves with V masses of 2 μg, 5 μg, 10 μg and 20 μg, respectively. | ||
In order to quantitatively separate the remaining Ti, Cr and other matrix elements to meet the requirements of instrumental measurement, an anion resin column procedure using peroxide and weak acid was necessary.10,13 Since our method has already separated the majority of matrix elements after the first column, and with 49Ti/51V and 52Cr/51V below 10−4 and 10−3 respectively (Table 3), we only need to perform one round of anion exchange resin purification (Table 1).
In summary, our purification scheme offers several advantages over previous studies: (1) the purification process requires only two separation steps, greatly reducing the time and reagents required for the whole procedure; (2) the method relies on the desorption of V-peroxide complex anions from cation exchange resin under dilute acid conditions, demonstrating excellent reproducibility and operational feasibility; (3) the elution curves of V in the first column will not shift when increasing the amount of V from 2 μg to 20 μg, obtaining a broader range of applicability; (4) the cation or anion exchange resin used in the method can be repeatedly used, effectively reducing costs. Therefore, our V isotope analysis method is both economical and time-efficient.
3.2 Correction for Ti and Cr interference
To obtain an accurate measurement of the V isotope composition, it is crucial to perform a rigorous correction for isobaric interferences, i.e., 50Ti and 50Cr. The correction of the interference of 50Ti and 50Cr is followed by eqn (1):| 50Vcorrected = 50Vmeasured − 49Timeasured × (50Ti/49Ti)IMF − 52Crmeasured × (50Cr/52Cr)IMF | (1) |
| RT = RIMF × (m1/m2)β | (2) |
Nielsen, et al.10 observed significant V isotope drift in the correction of isobaric interferences from 50Ti or 50Cr when the instrumental mass fractionation factor varied from −1.8 to −2.2. To address this issue, Wu, et al.13 obtained the real-time instrument mass fractionation factor of Ti and Cr isotopes by measuring the pure standard Ti and Cr solution with natural abundance before each measurement sequence began. For comparison, Schuth, et al.16 introduced Fe solution as external standard, and used the instrumental mass fractionation factor of Fe to correct Cr–Ti interferences. Both approaches assumed that the Ti and Cr isotope ratios of residual Cr and Ti in the samples were consistent with those in the Ti and Cr standard solutions, respectively.13,16 However, the Ti and Cr isotopic ratios of natural samples exhibited significant variation.54,55 In addition, isotope fractionation could occur during the removal of Cr and Ti in the column. Thus, the trace Cr and Ti remaining after purification probably have different isotopic compositions from standard Ti and Cr solutions. This could cause V isotopic offsets of the measured δ51V value from the true δ51V value (Δ51V = δ51Vmeasured − δ51Vtrue) after isobaric calibration of Cr and Ti.
In this study, we conducted doping experiments to assess the effect of Cr and Ti isotope differences on the accuracy of V isotope measurements. Variable amounts of GSB-Ti (GSB 04-1757-2004, 1000 μg mL−1) were added to a 1 μg mL−1 AA-V solution to evaluate the extent to which the Ti remaining in the solution could be effectively corrected. First, we used the correction method from Wu, et al.13 to assess the V isotopic offset generated by correcting the isobaric interference when using Ti isotopic ratios of GSB-Ti. We determined the 50Ti/49Ti ratio of 50 ppb GSB-Ti after instrumental fractionation and used this ratio as (50Ti/49Ti)IMF for isobaric correction. The results demonstrated that Δ51V values were close to 0 within uncertainty when the sample had 49Ti/51V < 4 × 10−4 (Fig. 3a). However, when we assumed a ±5‰ difference in Ti isotopes between the remaining Ti in the solution and GSB-Ti, i.e., (50Ti/49Ti)remain/(50Ti/49Ti)GSB-Ti = 0.995 and (50Ti/49Ti)remain/(50Ti/49Ti)GSB-Ti = 1.005, the results showed that Δ51V values deviated obviously from 0 within uncertainty when 49Ti/51V > 4 × 10−5 (Fig. 3a). A similar correction was also applied for the interference of 50Cr. First, we measured the 50Cr/52Cr ratio of 50 ppb GSB-Cr (GSB 04-1723-2004, 1000 μg mL−1) after instrumental fractionation and used this ratio as (50Cr/52Cr)IMF to correct the interference of 50Cr. The results revealed that when the sample had 52Cr/51V < 4 × 10−3, the Δ51V values were close to 0 within uncertainty (Fig. 3b). We also assumed a ±5‰ difference in Cr isotopes between Cr remaining in the solution and GSB-Cr, i.e., (50Cr/52Cr)remain/(50Cr/52Cr)GSB-Cr = 0.995 and (50Cr/52Cr)remain/(50Cr/52Cr)GSB-Cr = 1.005. The Δ51V values shifted distinctly from 0 within uncertainty with 52Cr/51V > 8 × 10−4, when using Cr isotopic ratios different from GSB-Cr for correction (Fig. 3b). Due to the unknown Ti or Cr isotopic composition of natural samples, Δ51V values may also shift significantly from 0 when the sample solution has 49Ti/51V > 4 × 10−5 or 52Cr/51V > 8 × 10−4. In this study, all sample solutions from different types of reference materials after two-step purification procedures had 49Ti/51V < 1 × 10−5 and 52Cr/51V < 1 × 10−4 (Table 3), thereby not affecting our measurement accuracy. This also demonstrates that our chemical purification process is reliable and universally applicable.
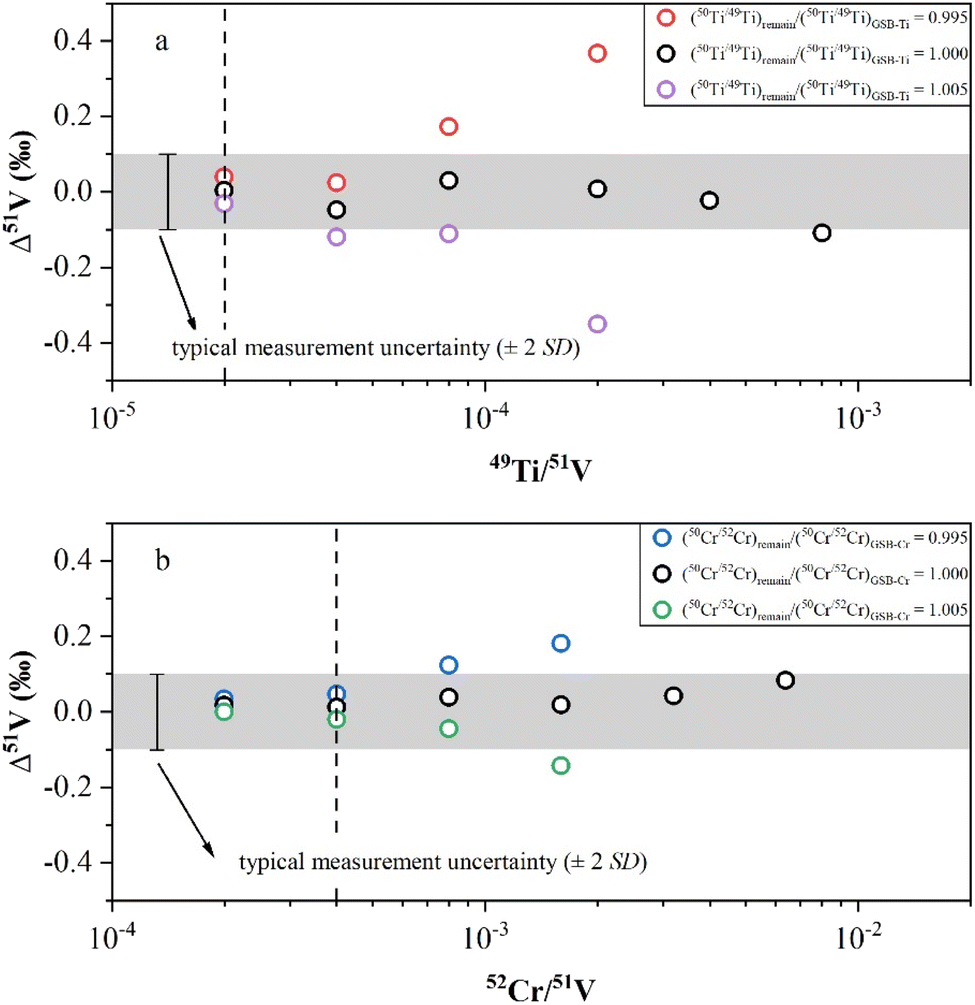 | ||
| Fig. 3 (a) The V isotopic offset (Δ51V = δ51Vmeasured − δ51Vtrue) generated by 50Ti correction, using three different 50Ti/49Ti isotopic ratios (with (50Ti/49Ti)remian/(50Ti/49Ti)GSB-Ti ranging from 0.995 to 1.005) versus the 49Ti/51V ratio of standard solutions containing different amounts of Ti. The (50Ti/49Ti)GSB-Ti was the measured value determined before each measurement sequence began. When 49Ti/51V ratios are 4 × 10−4 and 8 × 10−4, beside of (50Ti/49Ti)remian/(50Ti/49Ti)GSB-Ti = 1.000, the corrections are not shown here because of the large isotopic offset created in these solutions. The dotted line represents the acceptable maximum value of the 49Ti/51V ratio for sample measurements. (b) The V isotopic offset generated by 50Cr correction, using three different 50Ti/49Ti isotopic ratios (with (50Cr/52Cr)remian/(50Cr/52Cr)GSB-Cr ranging from 0.995 to 1.005) versus the 52Cr/51V ratio of standard solutions containing different amounts of Cr. The (50Cr/52Cr)GSB-Cr was the measured value determined before each measurement sequence began. When 52Cr/51V ratios are 3.2 × 10−3 and 6.4 × 10−3, beside of (50Cr/52Cr)remian/(50Cr/52Cr)GSB-Cr = 1.000, the corrections are not shown here because of the large isotopic offset created in these solutions. The dotted line represents the acceptable maximum value of the 52Cr/51V ratio for sample measurements. | ||
3.3 Effect of acid molarity and concentration mismatch
The effects of HNO3 molarity mismatch on V isotope measurements were evaluated in this study. The concentrated AA-V solution (1000 μg mL−1) was diluted to 1 μg mL−1 in HNO3 solution with a range from 1% (m/m) to 3% (m/m). The results showed that the measured δ51V values were consistent with zero within the long-term reproducibility of this study (Fig. 4a). The investigation of the effect of the V concentration mismatch between the sample and standard on the instrumental measurement was also performed in this study. AA-V solutions with concentrations ranging from 0.7 μg mL−1 to 1.4 μg mL−1 were measured by bracketing with a 1 μg mL−1 AA-V solution. The results reveal that δ51V values display no obvious offset with Csample/Cstandard ratios ranging from 0.7 to 1.4 (Fig. 4b). Nevertheless, the Csample/Cstandard ratios were typically adjusted to 0.9–1.1 during instrumental measurements to ensure accurate V isotopic determinations.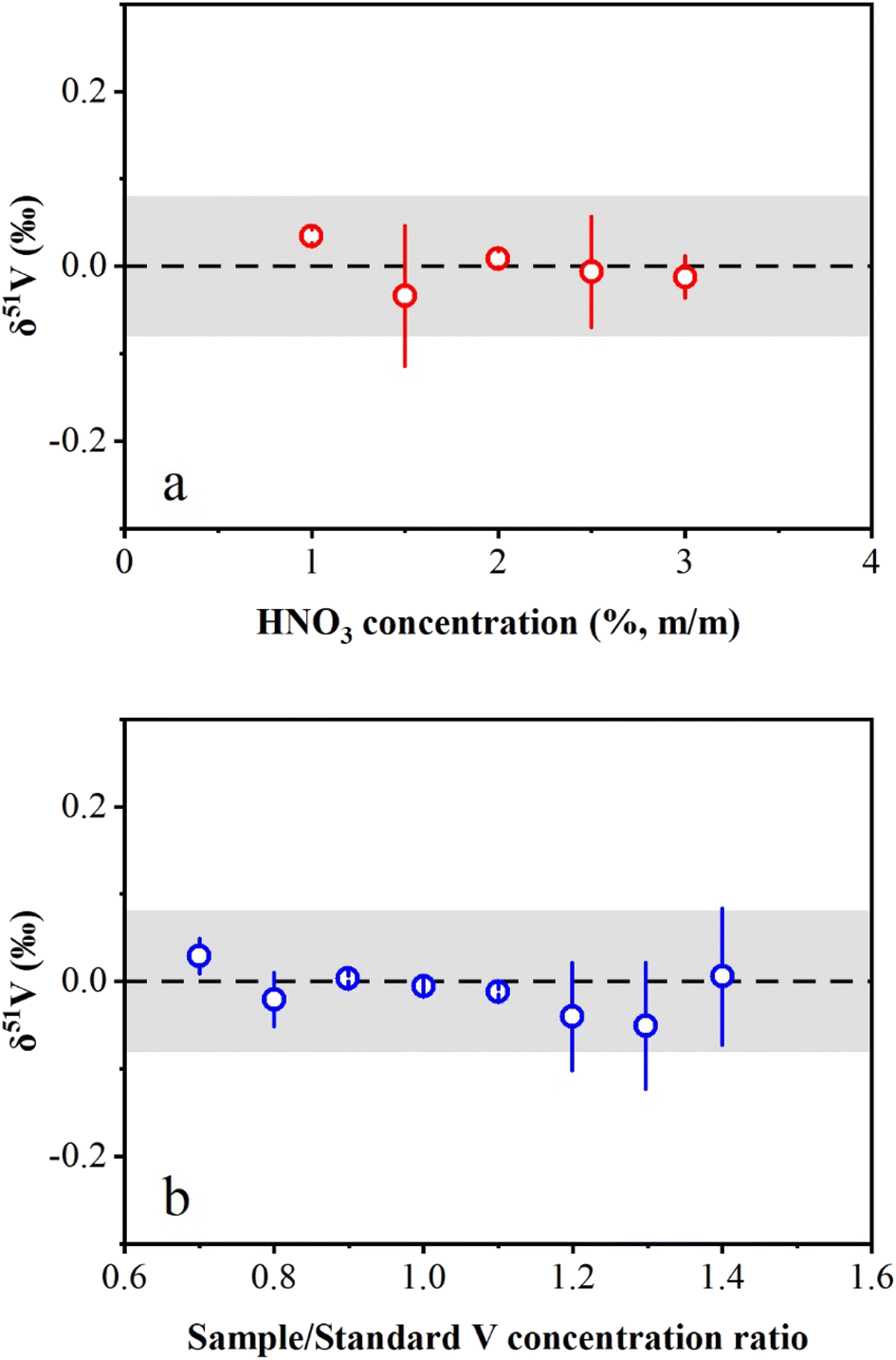 | ||
| Fig. 4 Effects of (a) HNO3 concentration and (b) sample/standard V concentration ratio on the determination of δ51V values using the SSB correction method. The HNO3 concentration of the bracketing standard is 2% (m/m). The error bars represent two standard deviations. | ||
3.4 Precision and accuracy
Two approaches were used in this study to evaluate the precision and accuracy of V isotope measurements. First, we repeatedly determined the pure V solutions, BDH-V and CUG-V (in-house standard purchased from Inorganic Ventures, Lot: M2-V655406, 1000 μg mL−1) relative to AA standard solution to assess the long-term external reproducibility of pure V solution measurement in this instrument. Second, vanadium isotopes of four igneous rock RMs (BHVO-2, BCR-2, AGV-2 and GSP-2) and one manganese nodule RM (NOD-A), which have been reported by previous studies, were measured in the laboratory.The long-term analysis of the BDH-V solution produced an average δ51V value of −1.23 ± 0.08‰ (2 SD, n = 91), which was consistent with the literature values.10,12,13 The repeated determination of the CUG-V solution gave an average δ51V value of 0.04 ± 0.08‰ (2 SD, n = 121). For the reference materials, V isotope analyses were repeatedly conducted with independent digestion and purification through different columns. The δ51V values of these reference materials (BHVO-2, BCR-2, AGV-2, GSP-2 and NOD-A) were −0.87 ± 0.08‰, −0.81 ± 0.09‰, −0.72 ± 0.05‰, −0.61 ± 0.10‰ and −1.02 ± 0.07‰, respectively. These δ51V values are consistent with the literature values.11,13,26 In summary, our V isotopic analytical method has been shown to accurately and precisely measure V isotopes, with a long-term reproducibility of δ51V values better than ±0.10‰ (2 SD) as demonstrated through the analysis of these reference materials.
3.5 Vanadium isotope compositions of reference materials of sediments and soils
The δ51V values of the soil and sediment reference materials are shown in Table 4 and Fig. 5. The carbonaceous siliceous shale standard material (GSR-20) was collected from Yutangba, Enshi, Hubei Province, with a high V content (614 ppm) as well as V/Ti (0.47) and V/Al (0.022). Its average δ51V value is −0.30 ± 0.06‰, heavier than modern oxic or suboxic marine sediments, indicating deposition in a relatively reduced environment.37 GSD-28 and GSD-32 are river sediments from the Lanping lead-zinc mining area in Yunnan and the Rucheng granite area in Hunan, respectively. GSS-24 and GSS-25 are South China Sea tidal flat sediment from Guangdong and Luochuan loess from Shanxi. The δ51V values of these sediments or deposits, ranging from −0.77‰ to −0.73‰, are similar to those of the upper continental crust (UCC, −0.72‰ to −0.60‰),56 supporting that continental weathering plays an insignificant role in V isotopic fractionation. The V isotope compositions in three agricultural soils from San Joaquin (SRM2709a) and Montana (SRM2710a and SRM2711a) are within the range of the average upper continental crust, indicating that human cultivation activities may not significantly alter the V isotopes.| Name | δ 51V (‰) | 2 SD (‰) | n |
|---|---|---|---|
| BHVO-2 | −0.88 | 0.08 | 3 |
| BHVO-2 | −0.85 | 0.07 | 3 |
| BHVO-2 | −0.85 | 0.03 | 3 |
| BHVO-2 | −0.91 | 0.06 | 3 |
| Average | −0.87 | 0.08 | 12 |
| Prytulak, et al.11 | −0.89 | 0.08 | 9 |
| Wu, et al.13 | −0.83 | 0.09 | 22 |
| BCR-2 | −0.83 | 0.05 | 3 |
| BCR-2 | −0.75 | 0.10 | 3 |
| BCR-2 | −0.82 | 0.05 | 3 |
| BCR-2 | −0.82 | 0.10 | 3 |
| BCR-2 | −0.83 | 0.05 | 3 |
| Average | −0.81 | 0.09 | 15 |
| Prytulak, et al.11 | −0.95 | 0.16 | 27 |
| Wu, et al.13 | −0.78 | 0.08 | 36 |
| Nielsen, et al.26 (WHOI) | −0.79 | 0.15 | 24 |
| Nielsen, et al.26 (ICL) | −0.80 | 0.14 | 3 |
| AGV-2 | −0.71 | 0.04 | 3 |
| AGV-2 | −0.74 | 0.05 | 3 |
| AGV-2 | −0.70 | 0.03 | 3 |
| Average | −0.72 | 0.05 | 9 |
| Prytulak, et al.11 | −0.50 | 0.19 | 4 |
| Wu, et al.13 | −0.70 | 0.10 | 37 |
| Nielsen, et al.26 | −0.73 | 0.17 | 16 |
| GSP-2 | −0.57 | 0.10 | 3 |
| GSP-2 | −0.63 | 0.07 | 3 |
| GSP-2 | −0.64 | 0.08 | 3 |
| Average | −0.61 | 0.10 | 9 |
| Prytulak, et al.11 | −0.63 | 0.10 | 6 |
| Wu, et al.13 | −0.62 | 0.07 | 26 |
| Nielsen, et al.26 | −0.76 | 0.15 | 4 |
| NOD-A | −0.97 | 0.10 | 3 |
| NOD-A | −1.05 | 0.06 | 3 |
| NOD-A | −1.06 | 0.04 | 3 |
| NOD-A | −1.02 | 0.02 | 3 |
| NOD-A | −1.02 | 0.05 | 3 |
| NOD-A | −1.02 | 0.03 | 3 |
| Average | −1.02 | 0.07 | 18 |
| Wu, et al.13 | −0.99 | 0.10 | 19 |
| GSR-20 | −0.27 | 0.06 | 3 |
| GSR-20 | −0.32 | 0.06 | 3 |
| GSR-20 | −0.30 | 0.05 | 3 |
| GSR-20 | −0.32 | 0.02 | 3 |
| Average | −0.30 | 0.06 | 12 |
| GSD-28 | −0.79 | 0.10 | 3 |
| GSD-28 | −0.73 | 0.01 | 3 |
| GSD-28 | −0.76 | 0.07 | 3 |
| Average | −0.76 | 0.08 | 9 |
| GSD-32 | −0.76 | 0.06 | 3 |
| GSD-32 | −0.77 | 0.04 | 3 |
| GSD-32 | −0.77 | 0.05 | 3 |
| Average | −0.76 | 0.04 | 9 |
| GSS-24 | −0.74 | 0.03 | 3 |
| GSS-24 | −0.77 | 0.04 | 3 |
| GSS-24 | −0.78 | 0.01 | 3 |
| GSS-24 | −0.81 | 0.04 | 3 |
| GSS-24 | −0.76 | 0.06 | 3 |
| Average | −0.77 | 0.06 | 15 |
| GSS-25 | −0.79 | 0.02 | 3 |
| GSS-25 | −0.75 | 0.02 | 3 |
| GSS-25 | −0.70 | 0.08 | 3 |
| GSS-25 | −0.72 | 0.03 | 3 |
| Average | −0.74 | 0.08 | 12 |
| SRM2709a | −0.76 | 0.03 | 3 |
| SRM2709a | −0.69 | 0.05 | 3 |
| SRM2709a | −0.81 | 0.05 | 3 |
| SRM2709a | −0.77 | 0.06 | 3 |
| Average | −0.76 | 0.10 | 12 |
| SRM2710a | −0.75 | 0.06 | 3 |
| SRM2710a | −0.78 | 0.05 | 3 |
| SRM2710a | −0.69 | 0.10 | 3 |
| Average | −0.74 | 0.10 | 9 |
| SRM2711a | −0.75 | 0.03 | 3 |
| SRM2711a | −0.70 | 0.05 | 3 |
| SRM2711a | −0.75 | 0.08 | 3 |
| Average | −0.73 | 0.07 | 9 |
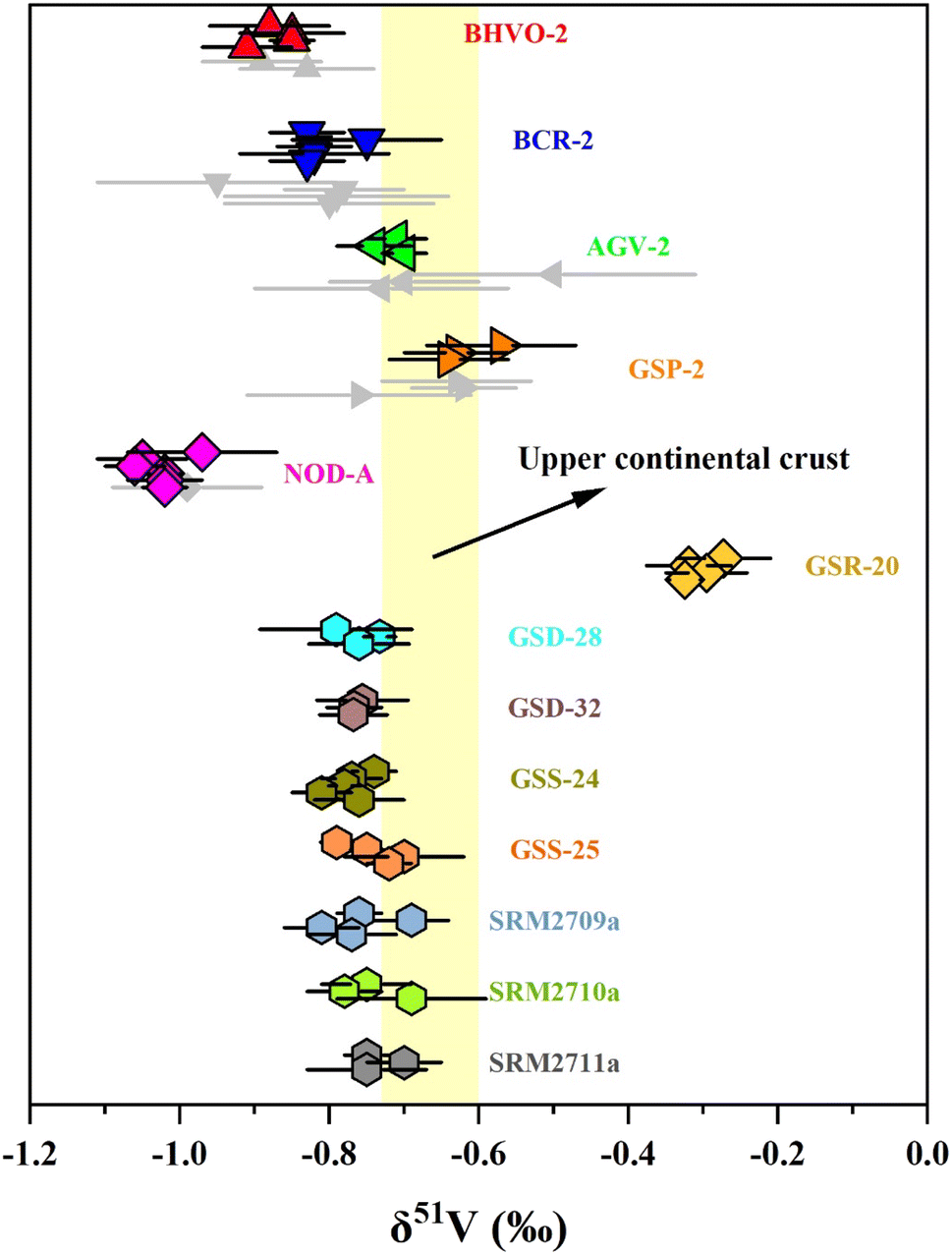 | ||
| Fig. 5 The δ51V values of reference materials. Error bars reflect two standard deviations. Data are from Table 3. The range of δ51V values of the upper continental crust (UCC) is from Tian, et al.56 | ||
4. Conclusions
We have employed a rapid method to purify V from matrix elements through a two-column chromatography technique, significantly improving time efficiency and reducing reagent consumption. The first step of the purification procedure utilized the peroxide–V complexes and cation exchange resin, effectively separating the majority of all matrix elements, particularly Ti and Cr. The recovery rate of V consistently remained >99% for a variety of sample types and V masses, and the total procedure blank was lower than 2 ng. Vanadium isotopic compositions were analysed using MC-ICP-MS combined with the sample-standard-bracketing method. To ensure the accuracy of our measurement, high-resolution mode was employed to avoid the interference of polyatomic interferences. Additionally, we carefully corrected the isobaric interferences from 50Ti and 50Cr during measurement. The vanadium isotopic compositions of 13 reference materials, including igneous rock, manganese nodules, soil, sediment and carbonaceous siliceous shale, were analysed in this study. Based on repeated measurements of pure V solutions and reference materials, the long-term measurement precision was better than ±0.10‰ in this study. Our newly developed method provides an efficient and accurate approach for measuring V isotopic ratios, which will be beneficial for future studies exploring V geochemistry.Author contributions
Zhen Zeng: conceptualization, methodology, investigation, writing – original draft, writing – review & editing. Fei Wu: conceptualization, resources, supervision, methodology, funding acquisition, writing – review & editing.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We are grateful to Hai-Hong Chen, Jie Lin, Wen Zhang for their support on the clean-lab and instrumentation operations. This research was financially supported by the National Science Foundation of China (4222530), and startup grant (101-162301212602) from China University of Geosciences (Wuhan). This manuscript has benefited from reviews by Ashley N. Martin and Stephan Schuth and the editorial handling from Gabriel Clarke.References
- J.-H. Huang, F. Huang, L. Evans and S. Glasauer, Chem. Geol., 2015, 417, 68–89 CrossRef CAS.
- J. Siebert, J. Badro, D. Antonangeli and F. J. Ryerson, Science, 2013, 339, 1194–1197 CrossRef CAS PubMed.
- J. Wade and B. J. Wood, Earth Planet. Sci. Lett., 2005, 236, 78–95 CrossRef CAS.
- Z.-X. A. Li and C.-T. A. Lee, Earth Planet. Sci. Lett., 2004, 228, 483–493 CrossRef CAS.
- M. Laubier, T. L. Grove and C. H. Langmuir, Earth Planet. Sci. Lett., 2014, 392, 265–278 CrossRef CAS.
- R. W. Nicklas, I. S. Puchtel, R. D. Ash, P. M. Piccoli, E. Hanski, E. G. Nisbet, P. Waterton, D. G. Pearson and A. D. Anbar, Geochim. Cosmochim. Acta, 2019, 250, 49–75 CrossRef CAS.
- W. W. Bennett and D. E. Canfield, Earth-Sci. Rev., 2020, 204 Search PubMed.
- T. J. Algeo and J. B. Maynard, Geosphere, 2008, 4, 872–887 CrossRef.
- A. Shore, A. Fritsch, M. Heim, A. Schuh and M. Thoennessen, At. Data Nucl. Data Tables, 2010, 96, 351–357 CrossRef CAS.
- S. G. Nielsen, J. Prytulak and A. N. Halliday, Geostand. Geoanal. Res., 2011, 35, 293–306 CrossRef CAS.
- J. Prytulak, S. G. Nielsen and A. N. Halliday, Geostand. Geoanal. Res., 2011, 35, 307–318 CrossRef CAS.
- S. G. Nielsen, J. D. Owens and T. J. Horner, J. Anal. At. Spectrom., 2016, 31, 531–536 RSC.
- F. Wu, Y. Qi, H. Yu, S. Tian, Z. Hou and F. Huang, Chem. Geol., 2016, 421, 17–25 CrossRef CAS.
- L. H. Dong, W. Wei, C. L. Yu, Z. H. Hou, Z. Zeng, T. Chen and F. Huang, Anal. Chem., 2021, 93, 7172–7179 CrossRef CAS PubMed.
- Y. Gao, J. F. Casey, L. M. Bernardo, W. Yang and K. K. Bissada, Geol. Soc., 2018, 468, 83–103 Search PubMed.
- S. Schuth, I. Horn, A. Bruske, P. E. Wolff and S. Weyer, Ore Geol. Rev., 2017, 81, 1271–1286 CrossRef.
- D. Malinovsky and N. A. Kashulin, Anal. Methods, 2016, 8, 5921–5929 RSC.
- J. Chetelat, S. G. Nielsen, M. Auro, D. Carpenter, L. Mundy and P. J. Thomas, Environ. Sci. Technol., 2021, 55, 4813–4821 CrossRef CAS PubMed.
- G. T. Ventura, L. Gall, C. Siebert, J. Prytulak, P. Szatmari, M. Hurlimann and A. N. Halliday, Appl. Geochem., 2015, 59, 104–117 CrossRef CAS.
- S. Schuth, A. Bruske, S. V. Hohl, S. Y. Jiang, A. K. Meinhardt, D. D. Gregory, S. Viehmann and S. Weyer, Chem. Geol., 2019, 528 Search PubMed.
- F. Wu, J. D. Owens, T. Y. Huang, A. Sarafian, K. F. Huang, I. S. Sen, T. J. Horner, J. Blusztajn, P. Morton and S. G. Nielsen, Geochim. Cosmochim. Acta, 2019, 244, 403–415 CrossRef CAS.
- F. Wu, T. Qin, X. F. Li, Y. Liu, J. H. Huang, Z. Q. Wu and F. Huang, Earth Planet. Sci. Lett., 2015, 426, 216–224 CrossRef CAS.
- P. A. Sossi, J. P. Bullet and H. S. C. O'Neill, Contrib. Mineral. Petrol., 2018, 173 Search PubMed.
- T. Fujii, C. Kato, N. Wada, A. Uehara, P. Sossi and F. Moynier, ACS Earth Space Chem., 2023, 7, 912–925 CrossRef CAS.
- Y. Xue, C.-h. Li, Y. Qi, C. Zhang, B. Miao and F. Huang, Acta Geochim., 2018, 37, 501–508 CrossRef CAS.
- S. G. Nielsen, M. Auro, K. Righter, D. Davis, J. Prytulak, F. Wu and J. D. Owens, Earth Planet. Sci. Lett., 2019, 505, 131–140 CrossRef CAS.
- S. G. Nielsen, D. V. Bekaert, T. Magna, K. Mezger and M. Auro, Geochem. Perspect. Lett., 2020, 15, 35–39 CrossRef.
- S. G. Nielsen, D. V. Bekaert and M. Auro, Nat. Commun., 2021, 12, 1817 CrossRef CAS PubMed.
- J. Prytulak, S. G. Nielsen, D. A. Ionov, A. N. Halliday, J. Harvey, K. A. Kelley, Y. L. Niu, D. W. Peate, K. Shimizu and K. W. W. Sims, Earth Planet. Sci. Lett., 2013, 365, 177–189 CrossRef CAS.
- Y. H. Qi, F. Wu, D. A. Ionov, I. S. Puchtel, R. W. Carlson, R. W. Nicklas, H. M. Yu, J. T. Kang, C. H. Li and F. Huang, Geochim. Cosmochim. Acta, 2019, 259, 288–301 CrossRef CAS.
- F. Wu, Y. H. Qi, M. R. Perfit, Y. J. Gao, C. H. Langmuir, V. D. Wanless, H. M. Yu and F. Huang, Earth Planet. Sci. Lett., 2018, 493, 128–139 CrossRef CAS.
- Z. Chen, X. Ding, E. S. Kiseeva, X. Lin, J. Huang and F. Huang, Lithos, 2023, 442–443 Search PubMed.
- D. Novella, J. Maclennan, O. Shorttle, J. Prytulak and B. J. Murton, Earth Planet. Sci. Lett., 2020, 531 Search PubMed.
- J. Prytulak, P. A. Sossi, A. N. Halliday, T. Plank, P. S. Savage and J. D. Woodhead, Geochem. Perspect. Lett., 2017, 3, 75–84 CrossRef.
- X. Ding, R. T. Helz, Y. H. Qi and F. Huang, Geochim. Cosmochim. Acta, 2020, 289, 114–129 CrossRef CAS.
- Y.-H. Qi, Y.-Z. Gong, F. Wu, Y. Lu, W. Cheng, F. Huang and H.-M. Yu, Geochim. Cosmochim. Acta, 2022, 320, 26–40 CrossRef CAS.
- F. Wu, J. D. Owens, F. Scholz, L. Q. Huang, S. Q. Li, N. Riedinger, L. C. Peterson, C. R. German and S. G. Nielsen, Geochim. Cosmochim. Acta, 2020, 284, 134–155 CrossRef CAS.
- F. Wu, J. D. Owens, L. Tang, Y. Dong and F. Huang, Geochim. Cosmochim. Acta, 2019, 265, 371–385 CrossRef CAS.
- F. Wu, J. D. Owens, C. R. German, R. A. Mills and S. G. Nielsen, Geochim. Cosmochim. Acta, 2022, 328, 168–184 CrossRef CAS.
- A. W. Heard, Y. Wang, C. M. Ostrander, M. Auro, D. E. Canfield, S. Zhang, H. Wang, X. Wang and S. G. Nielsen, Earth Planet. Sci. Lett., 2023, 610, 118127 CrossRef CAS.
- H. Fan, C. M. Ostrander, M. Auro, H. Wen and S. G. Nielsen, Earth Planet. Sci. Lett., 2021, 567 Search PubMed.
- S. Li, O. Friedrich, S. G. Nielsen, F. Wu and J. D. Owens, Earth Planet. Sci. Lett., 2023, 617 Search PubMed.
- W. Wei, X. Chen, H.-F. Ling, F. Wu, L.-H. Dong, S. Pan, Z. Jing and F. Huang, Earth Planet. Sci. Lett., 2023, 602 Search PubMed.
- S. G. Nielsen, Vanadium Isotopes: A Proxy for Ocean Oxygen Variations, in Elements in Geochemical Tracers in Earth System Science, Cambridge University Press, 2020 Search PubMed.
- Y. Huang, Z. Long, D. Zhou, L. Wang, P. He, G. Zhang, S. S. Hughes, H. Yu and F. Huang, Sci. Total Environ., 2021, 791, 148240 CrossRef CAS PubMed.
- G. Kakabadse and H. J. Wilson, Nature, 1957, 180, 861 CrossRef CAS.
- T. Fukasawa and T. Yamane, Anal. Chim. Acta, 1977, 88, 147–153 CrossRef CAS.
- T. Kiriyama and R. Kuroda, Talanta, 1983, 30, 261–264 CrossRef CAS PubMed.
- Y. Fovet, J.-Y. Gal and F. Toumelin-Chemla, Talanta, 2001, 53, 1053–1063 CrossRef CAS.
- M. Frankowski and A. Ziola-Frankowska, Talanta, 2010, 82, 1763–1769 CrossRef CAS.
- F. W. E. Strelow, Anal. Chem., 1960, 32, 1185–1188 CrossRef CAS.
- Y. J. An, F. Wu, Y. X. Xiang, X. Y. Nan, X. Yu, J. H. Yang, H. M. Yu, L. W. Xie and F. Huang, Chem. Geol., 2014, 390, 9–21 CrossRef CAS.
- X. Hu, X.-Y. Nan, H.-M. Yu and F. Huang, J. Anal. At. Spectrom., 2021, 36, 2744–2755 RSC.
- Y. Li, Y. Huang, Z. Li, X. Tang, X. Liu and S. S. Hughes, Ecotoxicol. Environ. Saf., 2022, 242, 113948 CrossRef CAS PubMed.
- Z. B. Deng, M. Chaussidon, P. Savage, F. Robert, R. Pik and F. Moynier, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 1132–1135 CrossRef CAS PubMed.
- S. Tian, X. Ding, Y. Qi, F. Wu, Y. Cai, R. M. Gaschnig, Z. Xiao, W. Lv, R. L. Rudnick and F. Huang, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2220563120 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |