
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Reactivities of phosphaalkynes towards diverse bis-silylenes†
Xiaofei
Sun
 a,
Da
Jin
a,
Stefanie
Maier
a,
Alexander
Hinz
a and
Peter W.
Roesky
*ab
a,
Da
Jin
a,
Stefanie
Maier
a,
Alexander
Hinz
a and
Peter W.
Roesky
*ab
aInstitute for Inorganic Chemistry, Karlsruhe Institute of Technology (KIT), Engesserstr. 15, 76131, Karlsruhe, Germany. E-mail: roesky@kit.edu
bInstitute for Nanotechnology, Karlsruhe Institute of Technology (KIT), Kaiserstr. 12, 76131, Karlsruhe, Germany
First published on 3rd June 2024
Abstract
Bis-silylenes do not only act as strong chelating σ-donor ligands, but also exhibit cooperative behaviour in the activation of small molecules. Three different P–Si containing molecules were prepared from the reaction between tBuC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) P and different bis-silylenes, which are bridged by ferrocenediyl, diaminobenzene, or o-carborane.
P and different bis-silylenes, which are bridged by ferrocenediyl, diaminobenzene, or o-carborane.
Introduction
The chemistry of silylenes has witnessed significant advancements since Jutzi's seminal report on the first divalent silicon compound1 and West's ground-breaking discovery of the first stable N-heterocyclic silylene.2 Over recent decades, several examples of bis-silylenes have emerged,3–11 primarily utilising the amidinate-based Si(II) fragment, owing to the straightforward functionalisation starting from the parent chlorosilylene [LSiCl]12,13 (L = PhC(tBuN)2). Altering the spacer moiety allows for adjusting the electronic properties as well as the Si⋯Si distance, which has led to varied reactivities.10,14 Apart from serving as strong σ-donating chelate ligands, bis-silylenes exhibit the capability to cooperatively activate small molecules owing to the two Si(II) centres in close proximity. This distinctive feature cannot be achieved using mono-silylenes. In addition to different small molecules such as P4, CO2, N2O, whose reactivity has been studied using bis-silylenes, attention has recently shifted towards organic substrates with triple bonds.15–17As a heavier congener of nitriles, phosphaalkynes feature a polarised Cδ−Pδ+ triple bond. Since the isolation of the first stable phosphaalkyne tBuCP by Becker in 1981,18 this major breakthrough has paved the way for scientists to achieve a series of kinetically stable phosphaalkynes.19,20 Their versatility as building block in synthetic chemistry has been demonstrated. In the area of low-valent silicon chemistry, earlier reports showcased the diverse reactivities with phosphaalkynes to form Si–P heterocycles via cycloaddition21–23 and insertion reactions.16,24–27 Intrigued by this, we started our investigation of reactions between different bis-silylenes towards tBuCP.
Results and discussion
Synthesis and characterisation of 1
The reaction between the ferrocenediyl-bridged bis-silylene [LSiFcSiL]6 (Fc = ferrocenediyl) and tBuCP yielded the double [1 + 2] cycloaddition product 1 in 83% yield (Scheme 1). The molecular structure was unequivocally determined by single crystal X-ray diffraction (SCXRD) analysis and is depicted in Fig. 1. Compound 1 crystallises in the orthorhombic space group Pbca. It consists of a four-membered “butterfly-shaped” 1-phospha-2,4-disilabicyclo[1.1.0]butane as central structural motif, in which the SiPCSi ring is folded about the P–C axis by 64.49°. The strongly bent core structure might arise from the steric constraints from the ferrocene-based linker. Structurally, the interatomic Si⋯Si separation of 2.97 Å clearly rules out any bonding interaction. The P–C bond (2.056(2) Å) is slightly longer than classical P–C single bond lengths, but the P–Si bonds (2.2716(9) and 2.2310(9) Å) and the Si–C1 bonds (1.867(2) and 1.875(2) Å) fall within the range of the respective single bond lengths.28 The two Si atoms are pentacoordinated by two C atoms, one P atom and two N atoms and the P atom is in a trigonal pyramidal coordination environment. At first glance, the structure motif of 1 resembles that of the zwitterionic SiPSiC ring26 formed from the reaction between the mono-silylene [LSiCl]12,13 and 1-AdCP.
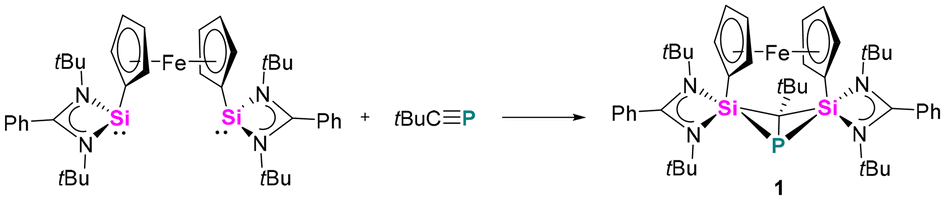 | ||
| Scheme 1 Reaction of [LSiFcSiL] and tBuCP. | ||
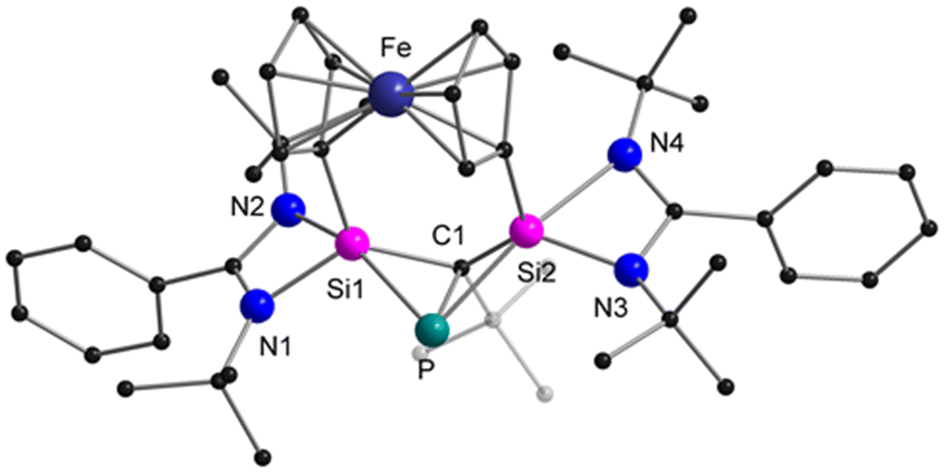 | ||
| Fig. 1 Molecular structure of compound 1 in the solid state. Hydrogen atoms and non-coordinating solvent molecules are omitted for clarity. Selected bond distances [Å] and angles [°]: P–C1 2.056(2), P–Si1 2.2716(9), P–Si2 2.2310(9), Si1–C1 1.867(2), Si2–C1 1.875(2), Si1–N1 1.858(2), Si1–N2 2.080(2), Si2–N3 1.817(2), Si2–N4 2.221(2); N1–Si1–N2 66.49(8), N3–Si2–N4 65.22(8), Si1–C1–Si2 105.07(11), Si1–P–Si2 82.54(3), Si1–C1–P 70.58(8), Si2–C1–P 68.97(8), Si1–P–C1 50.81(7), Si2–P–C1 51.68(7). | ||
However, the predominant difference lies in the geometry of the SiPSiC cycle which is in this case perfectly planar (sum of inner angles 360°) and the phosphorus atom is only dicoordinated by two Si atoms, due to the cleavage of the P–C bond. In contrast, the P–C bond in compound 1 is intact.
In the 1H NMR spectrum of 1, the tBu protons appear as three singlets at 1.53, 1.34, and 1.31 ppm. The singlet signal at −294.2 ppm in the 31P{1H} NMR spectrum is assigned to the tricoordinated P atom, which is shifted to lower frequencies compared to tBuCP (−69 ppm).29 In the 29Si{1H} NMR spectrum, a doublet signal is detected at −76.4 ppm with 1JSiP of 44.8 Hz. The addition of the bis-silylene moiety to the phosphaalkyne induces a significant highfield shift of the resonance.6
Synthesis and characterisation of 2
Next, we investigated the reaction between the diaminobenzene-functionalised bis-silylene [LSiCSiL]9 (C = 2,6-{(EtN)2C6H3}) and tBuCP29 (Scheme 2). After stirring both precursors at room temperature for 30 min, all the starting materials were consumed. Concentrating the solution and storing in a −40 °C freezer resulted in the formation of single crystals of the cyclic reaction product 2, which significantly differs from 1. The molecular structure of compound 2 was determined via SCXRD analysis and is depicted in Fig. 2. Compound 2 crystallises in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) and the core structure comprises a seven-membered SiC2NSiPC heterocycle. Notably, the Si1–P bond distance of 2.1322(8) Å is significantly shorter than the reported Si–P single bonds (∼2.25 Å)30 but only marginally longer than the Si–P double bond (∼2.09 Å), indicating its double bond character. The Si1–P bond length found in compound 2 is comparable with other literature-reported Si–P heterocycles26,31 or chain-type species.32,33
and the core structure comprises a seven-membered SiC2NSiPC heterocycle. Notably, the Si1–P bond distance of 2.1322(8) Å is significantly shorter than the reported Si–P single bonds (∼2.25 Å)30 but only marginally longer than the Si–P double bond (∼2.09 Å), indicating its double bond character. The Si1–P bond length found in compound 2 is comparable with other literature-reported Si–P heterocycles26,31 or chain-type species.32,33
 | ||
| Scheme 2 Reaction of [LSiCSiL] and tBuCP. | ||
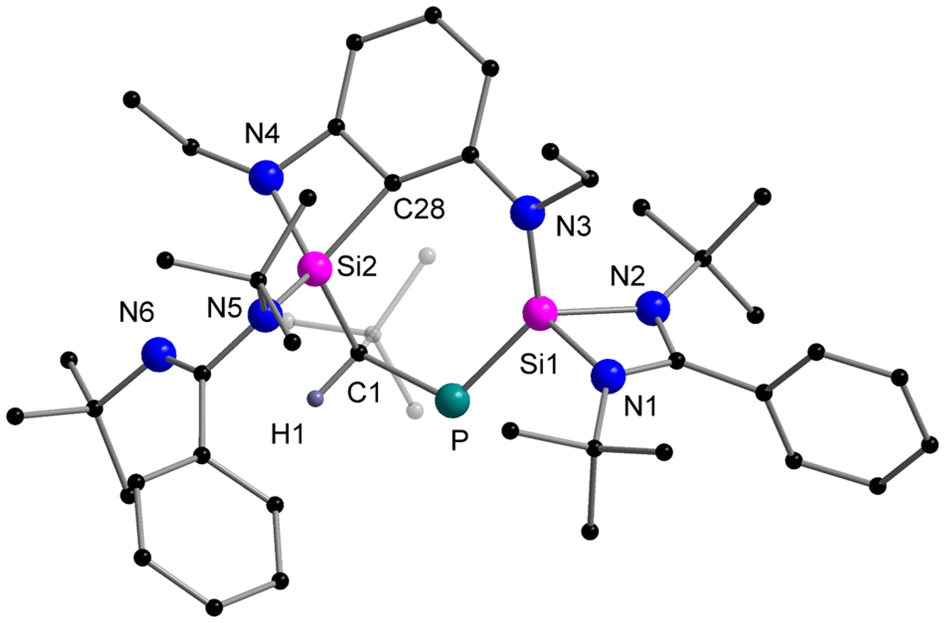 | ||
| Fig. 2 Molecular structure of compound 2 in the solid state. Hydrogen atoms are omitted for clarity. Selected bond distances [Å] and angles [°]: Si1–P 2.1322(8), Si1–N1 1.835(2), Si1–N2 1.889(2), Si1–N3 1.728(2), P–C1 1.897(2), Si2–C1 1.881(2), Si2–C28 1.855(2), Si2–N4 1.777(2), Si2–N5 1.748(2); N1–Si1–N2 70.42(8), Si1–P–C1 116.28(7), P–C1–Si2 106.15(10), N4–Si2–C28 75.40(9). | ||
In contrast, the P–C1 bond distance (1.897(2) Å) falls within the range of P–C single bonds.28 The formation of the seven-membered ring is facilitated by an intramolecular C–H bond activation. Si2 in compound 2 is coordinated by two amido groups and two C-substituents C1 and C28 in a distorted tetrahedral environment. The presence of the proton at C1 was detected at 2.21 ppm as a doublet signal due to the 2JHP (23.2 Hz) coupling with the 31P nucleus. Correspondingly, the 31P NMR resonance was observed at −173.0 ppm as a doublet (2JHP = 23.2 Hz) and the 29Si{1H} NMR resonances were found at 2.5 ppm and −28.1 ppm as two doublets with coupling constants of 209.3 Hz (1JSiP) and 10.8 Hz (2JSiP), respectively.
Critically, there is no suitable C–H proton available in the ferrocenediyl-bridged bis-silylene [LSiFcSiL], which leads to the formation of 1. Thus, the C–H bond activation in [LSiCSiL] is a key factor for the formation of the two different products 1 and 2. Therefore, we aimed to investigate the reactivity of the diaminopyridine-bridged bis-silylene [LSiNSiL] (N = 2,6-{(EtN)2C5H3N}),4 which is analogous to [LSiCSiL] but lacks a proton at 1-position of the aromatic ring. However, the NMR-scale reaction in C6D6 revealed the formation of a mixture of several reaction products, and unfortunately, no single product could be crystallised and identified.
DFT calculations for the reaction pathway of the formation of 2
The reaction pathway for the formation of 2 was investigated by DFT calculations (Scheme 3). The initial step is most likely to be well-established the [2 + 1] cycloaddition of the PC triple bond on one of the silylenes. This is exergonic by −69.0 kJ mol−1 and leads to Int1. Subsequently, the strained phosphasilirene21 is opened to give the nine-membered cyclic species Int2 in which both a P–C bond order is reduced to 1 and both a Si–C and Si–P double bond exist. This step can proceed by opening either the Si–C or a Si–P bond of Int1. Both are feasible, but the activation barrier for the latter path is considerably lower (ΔG = 39.5 kJ mol−1). From Int2 there is a path with a very low activation barrier that directly leads to Int3 (=2) where simultaneously, the former phosphaalkyne C atom abstracts a hydrogen atom from the central phenyl ring and the nine-membered heterocycle contracts to the seven-membered cyclic Int3. Alternatively, the H atom could be transferred to the phosphorus atom (Int6), but the subsequent H atom migration has a rather high activation barrier, which makes the direct process the most probable one.
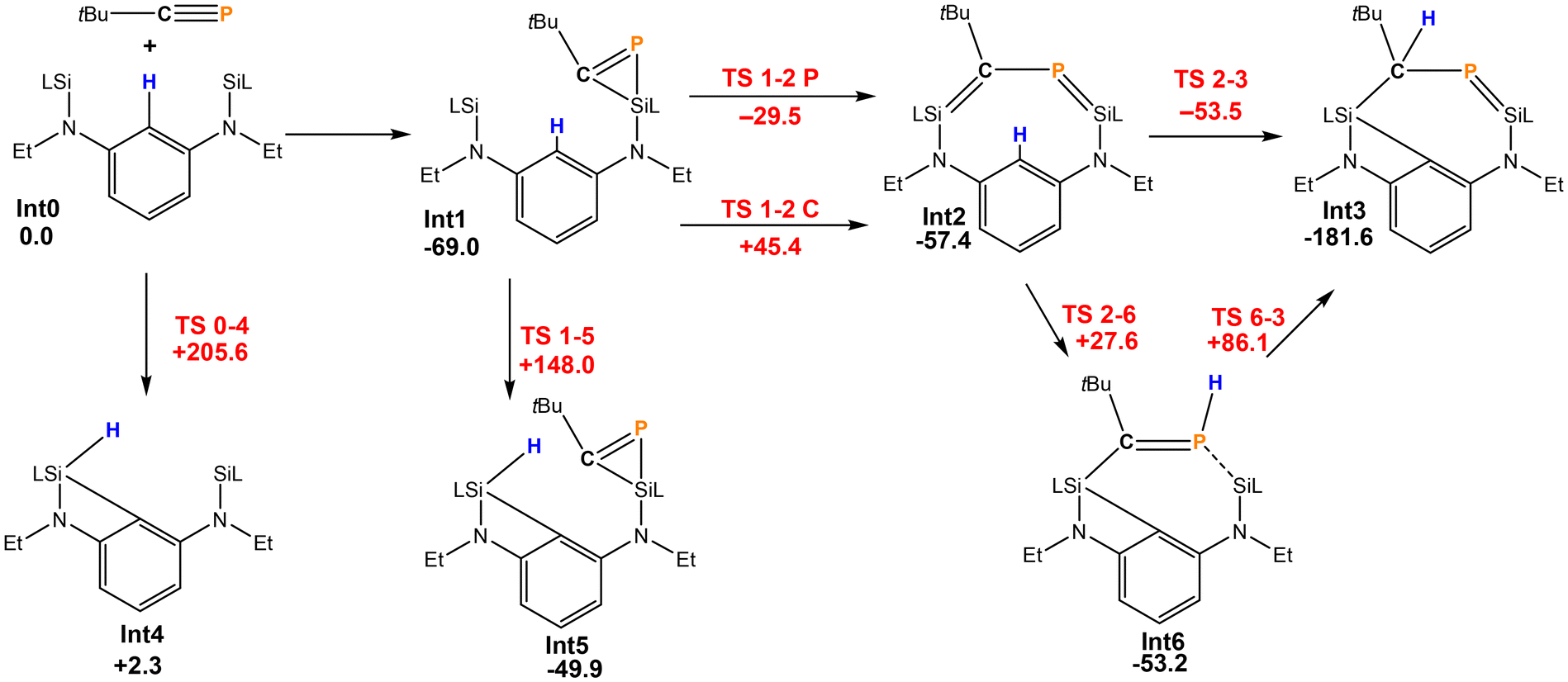 | ||
| Scheme 3 Reaction of [LSiCSiL] and tBuCP (energies in kJ mol−1, intermediates in black, transition states in red). | ||
It should be noted that without the C–P unit bridging both silylenes, the C–H activation barrier by the silylene of 148.0 or 205.6 kJ mol−1 is much too large to be feasible. Therefore, the rigid linker of the bis-silylene first enables the formation of the unusual intermediate Int2. The additional strain enforced closer proximity of the silylene moiety and the Ph group that allows then C–H activation. The more flexible ferrocenediyl backbone can facilitate closer Si⋯Si distances so that the bicyclic 1 can form. This is inhibited in the formation of 2.
Synthesis and characterisation of 3
When the o-carborane-substituted bis(silylene) [LSiBSiL]11 (B = C2B10H10) was treated with tBuCP, a single reaction product 3 was formed and isolated in 51% yield as yellow crystals (Scheme 4). Compound 3 crystallises in the monoclinic space group P21/n and consists of two fused four-membered heterocycles Si2PC and SiC2P (Fig. 3). In this transformation, one of the Si–C bonds is cleaved and a Si–Si single bond (2.352(2) Å) is newly formed. Notably, the C–P bond of the phosphaalkyne remains intact and the P–C1 bond of 1.780(5) Å is slightly shorter compared to classical P–C single bonds (∼1.87 Å). In contrast, the P–C6 bond distance of 1.964(5) Å is elongated than typical P–C single bonds.
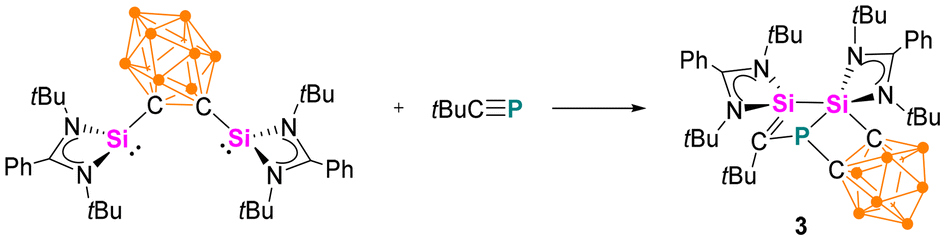 | ||
| Scheme 4 Reaction of [LSiBSiL] and tBuCP. | ||
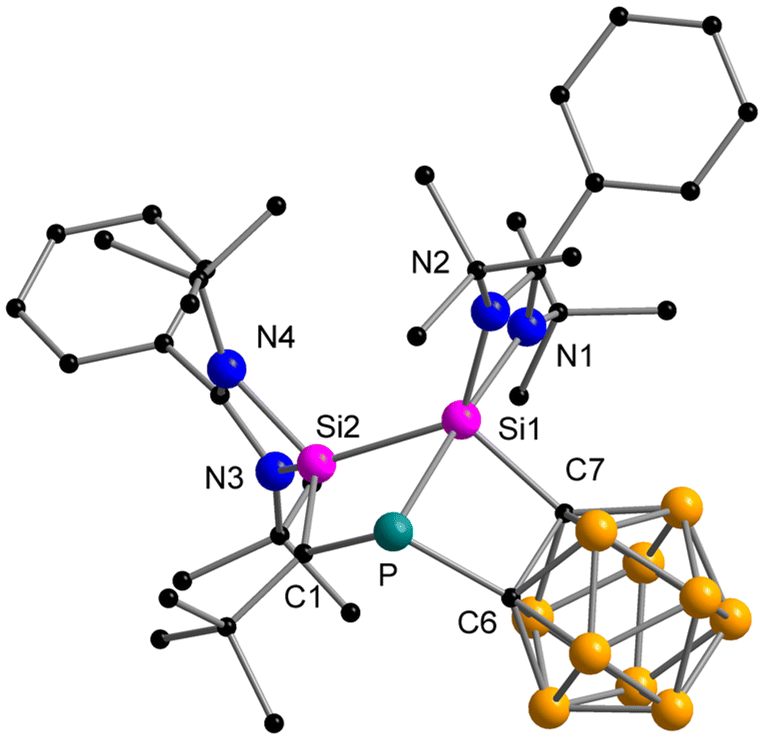 | ||
| Fig. 3 Molecular structure of compound 3 in the solid state. Hydrogen atoms and non-coordinating solvent molecules are omitted for clarity. Selected bond distances [Å] and angles [°]: Si1–Si2 2.352(2), Si2–C1 1.761(5), C1–P 1.780(5), P–Si1 2.343(2), P–C6 1.964(5), Si1–C7 1.969(5), Si1–N1 1.956(4), Si1–N2 1.857(4), Si2–N3 1.894(4), Si2–N4 1.884(4); N1–Si1–N2 68.7(2), N3–Si2–N4 69.2(2), Si1–P–C1 89.5(2), Si1–P–C6 79.93(14), Si1–Si2–C1 89.7(2), Si2–Si1–P 73.84(6), Si2–Si1–C7 118.53(14), Si2–C1–P 105.6(3), C1–P–C6 110.9(2), C7–Si1–P 80.64(14). | ||
Moreover, the Si2–C1 bond distance of 1.761(5) Å is much shorter than literature-known Si–C single bonds (1.86–1.93 Å)34 but well-comparable to that of Brook's silene [(Me3Si)2Si![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C(OSiMe3)Ad] (1.764(3) Å)35 and other cyclic silenes.25,36 During the preparation of this work, the group of Li and Su reported a similar reaction between the bis-silylene [LSiBSiL] and 1-AdCP, which gave a comparable product.16 The bonding metrics of the two compounds are very similar. In their work, DFT calculations were performed to elucidate the mechanistic pathway, which is initiated by the [2 + 2] cycloaddition between the PC bond and one Si–C bond. Following this, one C–C bond is formed and one Si–C bond is cleaved to rearrange to the reaction product.
C(OSiMe3)Ad] (1.764(3) Å)35 and other cyclic silenes.25,36 During the preparation of this work, the group of Li and Su reported a similar reaction between the bis-silylene [LSiBSiL] and 1-AdCP, which gave a comparable product.16 The bonding metrics of the two compounds are very similar. In their work, DFT calculations were performed to elucidate the mechanistic pathway, which is initiated by the [2 + 2] cycloaddition between the PC bond and one Si–C bond. Following this, one C–C bond is formed and one Si–C bond is cleaved to rearrange to the reaction product.
In the 1H NMR spectrum of 3, five singlet signals (0.93, 1.08, 1.18, 1.26, 1.54 ppm) were found for the five tBu groups, and at 3.15 ppm a broad signal was detected for the ten remaining protons of the o-carborane. In the 31P{1H} NMR spectrum, a singlet signal appeared at 96.9 ppm and in the 29Si{1H} NMR spectrum, two doublet signals were detected at −21.0 and −59.1 ppm, with coupling constants of 31.8 and 7.3 Hz due to the 1JSiP and 2JSiP coupling with the 31P atom. These resonances are similar to that found in the Ad-functionalised species.16 A generalisation of reactivity patterns is difficult, because the characteristics of the ligand scaffolds are quite distinct: in [LSiFcSiL], a high degree of flexibility is enabled by rotation of the Fc moiety, allowing for both large and small Si⋯Si distances without reactive sites in close proximity. In contrast, with the more rigid [LSiCSiL], a longer Si⋯Si distance is enforced, but the reactive C–H opens up a pathway independent of the Si⋯Si distance. Lastly, with the [LSiBSiL] scaffold a short Si⋯Si contact is enforced, but the anchoring bond between backbone and silylene is broken upon reaction with the phosphaalkyne. This is not a frequently observed mode of reaction and is likely a property owing to the carborane scaffold than the bis-silylene.
Conclusions
In summary, we have prepared three P–Si containing molecules starting from the reaction between the different bis-silylenes [LSiFcSiL], [LSiCSiL] and [LSiBSiL] with tBuCP. When the ferrocenediyl-functionalised bis-silylene was employed, a double cycloaddition took place, giving rise to compound 1 featuring a butterfly-shaped SiPCSi molecule. When changing from the ferrocene-linker to a diaminobenzene unit, C–H activation at the benzene ring occurred and compound 2 was isolated. With the o-carborane-bridged bis-silylene, compound 3 with two fused four-membered heterocycles formed. Thus, subtle changes in ligand structure and properties can significantly impact the behaviour in small molecule activation by changing Si⋯Si distances and Si–linker bond stability. This study provides valuable insights into the reactivity of bis-silylenes with phosphaalkynes, expanding the synthetic possibilities of silicon–phosphorus heterocycles and contributing to the field of low-valent silicon chemistry.
Experimental section
General procedures
All air- and moisture-sensitive manipulations were performed under dry N2 or Ar atmosphere using standard Schlenk techniques or in an argon-filled MBraun glovebox, unless otherwise stated. n-Pentane and toluene were dried using an MBraun solvent purification system (SPS-800) and degassed. n-Hexane was distilled under nitrogen from potassium benzophenone ketyl. C6D6 and THF-d8 were dried over Na-K alloy and degassed by freeze–pump–thaw cycles. tBuCP29 (synthesised as a hexamethyldisiloxane solution, ca. 1130 mg solution per 1 mmol tBuCP, the concentration (mol g−1) of the silylether solution of tBuCP was determined using 31P NMR spectroscopy: 46 mg of the tBuCP solution and 17.2 mg of bis(diphenylphosphino)methan (DPPM) were mixed in an NMR tube and the relaxation time (D1) was set to 120 s, the concentration was calculated by comparing the integrations), [LSiFcSiL] (L = PhC(tBuN)2, Fc = ferrocenediyl),6 [LSiCSiL] (C = 2,6-{(EtN)2C6H3})9 and [LSiBSiL] (B = C2B10H10)11 were prepared according to the literature procedures. All other chemicals were obtained from commercial sources and used without further purification. Elemental analyses were carried out with an Elementar vario MICRO cube. NMR spectra were recorded on Bruker spectrometers (Avance Neo 300 MHz, Avance Neo 400 MHz or Avance III 400 MHz). Chemical shifts are referenced internally using signals of the residual protio solvent (1H) or the solvent (13C{1H}) and are reported relative to tetramethylsilane (1H, 13C{1H}), or externally relative to BF3·Et2O (11B), tetramethylsilane (29Si) or 85% phosphoric acid (31P). All NMR spectra were measured at 298 K, unless otherwise specified. The multiplicity of the signals is indicated as s = singlet, d = doublet, dd = doublet of doublets, dq = doublet of quartets, t = triplet, q = quartet, m = multiplet and br = broad. Assignments were determined based on unambiguous chemical shifts, coupling patterns and 13C-DEPT experiments or 2D correlations (1H–1H COSY, 1H–13C HMQC and 1H–13C HMBC). Infrared (IR) spectra were recorded in the region 4000–400 cm−1 on a Bruker Tensor 37 FTIR spectrometer equipped with a room temperature DLaTGS detector, a diamond attenuated total reflection (ATR) unit. In terms of their intensity, the signals were classified into different categories (vs = very strong, s = strong, m = medium, w = weak, and sh = shoulder). Details on crystallography and quantum chemical calculations are provided in the ESI.†Synthesis of compound 1
To a J. Young NMR tube containing a C6D6 (ca. 0.4 mL) solution of [LSiFcSiL] (50.0 mg, 0.07 mmol) was added tBuCP (8 mg (375 mg solution), 0.08 mmol). The NMR tube was carefully shaken and immediately, single crystals suitable for X-ray diffraction analysis precipitated. The solution was carefully decanted and the remaining crystals were washed twice with n-hexane (ca. 1 mL) and dried under vacuum for 30 min. Yield (based on crystals): 83% (49.8 mg, 0.06 mmol). Anal. calcd for C45H61FeN4PSi2 (0.5 C6D6) (843.08 g mol−1): C 68.38; H 8.01; N 6.65. Found: C 68.87; H 8.52; N 6.91. 1H NMR (400.3 MHz, THF-d8): δ (ppm) = 7.39–7.26 (m, 10H, CHPh), 4.63–4.53 (m, 4H, CHFc), 4.19–4.15 (m, 4H, CHFc), 1.53 (s, 9H, PC(CH3)3), 1.34 and 1.31 (two br s, 36H, NC(CH3)3). 13C{1H} NMR (101.67 MHz, THF-d8): δ (ppm) = 171.0 (NCN), 138.1 (Ph–Cq), 130.8 (CHPh), 130.01 (CHPh), 129.95 (CHPh), 128.4 (CHPh), 128.0 (CHPh), 80.2 (Fc–CH), 76.3 (Fc–CH), 75.6 (Fc–Cq), 72.0 (Fc–CH), 70.4 (Fc–CH), 56.2 (NC(CH3)3), 54.7 (NC(CH3)3), 34.3 (NC(CH3)3), 33.7 (CC(CH3)3), 33.5 (NC(CH3)3). The two quaternary carbon atoms PCCtBu and PCCtBu could not be detected. 29Si{1H} NMR (79.49 MHz, C6D6): δ (ppm) = −76.4 (d, 1JSiP = 44.8 Hz). 31P{1H} NMR (162.04 MHz, THF-d8): δ (ppm) = −294.2 (s). IR (ATR):![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) (cm−1) = 3084 (vw), 2959 (vs), 2933 (s), 2867 (m), 1642 (w), 1614 (w), 1538 (vw), 1477 (m), 1446 (m), 1393 (s), 1360 (s), 1245 (m), 1192 (s), 1161 (m), 1100 (vw), 1073 (w), 1036 (m), 1018 (m), 1003 (m), 975 (s), 922 (w), 799 (w), 768 (m), 705 (m), 637 (w), 616 (w), 580 (w), 573 (w), 496 (w), 475 (w).
(cm−1) = 3084 (vw), 2959 (vs), 2933 (s), 2867 (m), 1642 (w), 1614 (w), 1538 (vw), 1477 (m), 1446 (m), 1393 (s), 1360 (s), 1245 (m), 1192 (s), 1161 (m), 1100 (vw), 1073 (w), 1036 (m), 1018 (m), 1003 (m), 975 (s), 922 (w), 799 (w), 768 (m), 705 (m), 637 (w), 616 (w), 580 (w), 573 (w), 496 (w), 475 (w).
Synthesis of compound 2
To a Schlenk flask containing a toluene (ca. 5 mL) solution of [LSiCSiL] (81.2 mg, 0.119 mmol) was added tBuCP (13 mg (605 mg solution), 0.130 mmol) at room temperature and the solution became immediately dark red. After stirring at room temperature for 30 min, the solution was concentrated to ca. one third of the original volume. Crystals of the title compound could be obtained after storing the solution in a −40 °C freezer for 3 days. The mother liquid was carefully removed via syringe and the obtained crystals were dried, washed with a small amount of n-hexane and the remaining crystals were dried under vacuum for 1 h. Yield (based on crystals): 57% (53.1 mg, 0.068 mmol). Anal. calcd for C45H69N6PSi2 (781.23 g mol−1): C 69.18; H 8.90; N 10.76. Found: C 69.55; H 9.27; N 10.44. 1H NMR (400.3 MHz, C6D6): δ (ppm) = 8.20 (d, 3JHH = 7.2 Hz, 2H, HAr), 7.31 (dd, 3JHH = 8.3, 7.3 Hz, 1H, Hbenz), 7.19–7.17 (m, 2H, HAr), 7.10–7.08 (m, 2H, HAr), 6.93–6.90 (m, 1H, HAr), 6.85–6.81 (m, 2H, HAr), 6.77–6.75 (m, 1H, HAr), 6.32 (d, 3JHH = 8.3 Hz, 1H, Hbenz), 6.20 (d, 3JHH = 7.3 Hz, 1H, Hbenz), 3.90–3.70 (overlapped dq, 2JHH = 13.2 Hz, 3JHH = 6.7 Hz, 2H, CH2CH3), 3.63 (dq, 2JHH = 13.2 Hz, 3JHH = 6.7 Hz, 1H, CH2CH3), 3.35 (dq, 2JHH = 14.3 Hz, 3JHH = 7.2 Hz, 1H, CH2CH3), 2.21 (d, 2JHP = 23.2 Hz, 1H, PCH), 1.56 (t, 3JHH = 7.1 Hz, 3H, CH2CH3), 1.51 (s, 9H, C–C(CH3)3), 1.38 (s, 9H, N–C(CH3)3), 1.35–1.34 (overlapped s and t, 21H, 18H of N–C(CH3)3 + 3H of CH2CH3), 1.05 (s, 9H, N–C(CH3)3). 13C{1H} NMR (100.67 MHz, C6D6): δ (ppm) = 174.3 (NCN–Cq), 165.8 (NCN–Cq), 162.8 (benz–Cq), 145.2 (benz–Cq), 142.1 (Ph–Cq), 132.7 (CHbenz), 131.83 (Ph–Cq), 131.78 (CHPh), 131.7 (CHPh), 130.1 (CHPh), 128.9 (CHPh), 128.4 (CHPh), 128.3 (CHPh), 128.2 (CHPh), 127.5 (CHPh), 127.1 (two CHPh), 126.7 (benz–Cq), 104.9 (CHbenz), 97.3 (CHbenz), 55.5 (N–C(CH3)3), 54.6 (N–C(CH3)3), 54.3 (N–C(CH3)3), 39.2 (CH2CH3), 38.8 (CH2CH3), 34.6 (PCC(CH3)3), 34.1 (d, 1JCP = 60.6 Hz, PCtBu), 33.1 (2 N–C(CH3)3), 32.3 (d, 3JCP = 16.1 Hz, C–C(CH3)3), 32.2 (d, 3JCP = 4.2 Hz, N–C(CH3)3),37 31.4 (N–C(CH3)3), 16.0 (CH2CH3), 14.6 (CH2CH3). One carbon signal CAr could not be detected. 29Si{1H} NMR (79.52 MHz, C6D6): δ (ppm) = 2.5 (d, 1JSiP = 209.3 Hz), −28.1 (d, 2JSiP = 10.8 Hz). 31P NMR (162.04 MHz, C6D6): δ (ppm) = −173.0 (d, 2JHP = 23.2 Hz). 31P{1H} NMR (162.04 MHz, C6D6): δ (ppm) = −173.0 (s, 1JSiP = 209.0 Hz, determined from the 29Si satellites). IR (ATR):(cm−1) = 3059 (vw), 2963 (vs), 2928 (s), 2865 (m), 1646 (vw), 1589 (m), 1571 (s), 1530 (w), 1474 (m), 1443 (m), 1394 (s), 1362 (s), 1313 (w), 1286 (w), 1266 (m), 1243 (s), 1198 (s), 1188 (s), 1159 (m), 1092 (m), 1071 (m), 1024 (w), 1008 (m), 975 (w), 934 (w), 905 (w), 840 (m), 790 (w), 766 (s), 731 (w), 709 (s), 633 (m), 590 (w), 575 (w), 534 (w), 469 (w), 436 (vw).
Synthesis of compound 3
To a J. Young NMR tube containing a C6D6 solution (ca. 0.4 mL) of [LSiBSiL] (43.0 mg, 0.075 mmol) was added tBuCP (8 mg (375 mg solution), 0.08 mmol). The NMR tube was carefully shaken for monitoring the reaction. A 31P{1H} NMR spectrum was recorded, which confirmed the formation of a single reaction product. The solution was carefully concentrated to ca. 0.1 mL and kept at room temperature. After three days, yellow block-shaped crystals have formed. Yield (based on crystals): 51% (32.4 mg, 0.038 mmol). Anal. calcd for C37H65B10N6Psi2 0.15 (C6H18Si2O) (785.56 g mol−1): C 57.95; H 8.85; N 7.13. Found: C 57.93; H 7.85; N 6.54. 1H NMR (400.3 MHz, C6D6): δ (ppm) = 7.20–7.18 (m, 1H, CHPh), 6.94–6.76 (m, 9H, CHPh), 3.15 (br, 10H, BH), 1.54 (s, 9H, C–C(CH3)3), 1.26 (s, 9H, N–C(CH3)3), 1.18 (s, 9H, N–C(CH3)3), 1.08 (s, 9H, N–C(CH3)3), 0.93 (s, 9H, N–C(CH3)3), 0.00 (s, ca. 2.6H, O{Si(CH3)3}2, co-crystallised solvent). 11B{1H} NMR (128.38 MHz, C6D6): δ (ppm) = 26.7, 2.1, −2.2, −4.5, −5.4, −7.9, −13.5, −14.9. 13C{1H} NMR (100.62 MHz, C6D6): δ (ppm) = 174.6 (NCN), 174.2 (NCN), 133.0 (Ph–Cq), 131.7 (Ph–Cq), 130.32 (CHPh), 130.28 (CHPh), 129.7 (CHPh), 129.4 (CHPh), 128.4 (CHPh), 128.1 (CHPh), 127.8 (CHPh), 127.72 (CHPh), 127.68 (CHPh), 126.6 (CHPh), 98.1 (d, 1JCP = 48.3 Hz, SiC), 91.5 (d, 1JCP = 133.7 Hz, cage-C), 89.4 (d, 2JCP = 13.1 Hz, cage-C), 55.7 (N–C(CH3)3), 55.1 (N–C(CH3)3), 54.4 (N–C(CH3)3), 54.1 (N–C(CH3)3), 37.4 (d, 2JCP = 10.7 Hz, C–C(CH3)3), 35.3 (d, JCP = 7.7 Hz, N–C(CH3)3),37 33.4 (d, JCP = 13.8 Hz, C–C(CH3)3), 32.7 (two N–C(CH3)3), 32.4 (N–C(CH3)3), 2.1 (O{Si(CH3)3)2}, co-crystallised solvent). 29Si{1H} NMR (79.52 MHz, C6D6): δ (ppm) = −21.0 (d, 1JSiP = 31.8 Hz), −59.1 (d, 2JSiP = 7.3 Hz). 31P{1H} NMR (162.04 MHz, C6D6): δ (ppm) = 96.9 (s). IR (ATR): (cm−1) = 3059 (vw), 2971 (vs), 2953 (vs), 2930 (vs), 2856 (m), 2565 (vs), 1605 (vw), 1529 (w), 1474 (m), 1456 (m), 1447 (m), 1406 (vs), 1394 (vs), 1380 (vs), 1363 (vs), 1251 (m), 1236 (m), 1222 (m), 1195 (vs), 1071 (m), 1042 (w), 1021 (m), 966 (vw), 925 (w), 894 (m), 841 (w), 786 (m), 759 (m), 747 (m), 729 (m), 706 (s), 688 (vw), 634 (w), 610 (m), 581 (vw), 493 (w), 479 (w), 440 (w).
Author contributions
X. S. – investigation, synthesis, crystallography, manuscript writing (original draft); D. J. – investigation, manuscript writing (editing); S. M. – investigation, manuscript writing (editing); A. H. – theoretical calculation, manuscript writing (editing); P. W. R. – conceptualisation, supervision, manuscript writing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Deutsche Forschungsgemeinschaft (RO 2008/25-1, 546228048).References
- P. Jutzi, D. Kanne and C. Krüger, Angew. Chem., Int. Ed., 1986, 25, 164 CrossRef
.
- M. Denk, R. Lennon, R. Hayashi, R. West, A. V. Belyakov, H. P. Verne, A. Haaland, M. Wagner and N. Metzler, J. Am. Chem. Soc., 1994, 116, 2691 CrossRef CAS
- X. Chen, H. Wang, S. Du, M. Driess and Z. Mo, Angew. Chem., Int. Ed., 2021, 61, e2021145 Search PubMed
- D. Gallego, S. Inoue, B. Blom and M. Driess, Organometallics, 2014, 33, 6885 CrossRef CAS
- S. Li, Y. Wang, W. Yang, K. Li, H. Sun, X. Li, O. Fuhr and D. Fenske, Organometallics, 2020, 39, 757 CrossRef CAS
- W. Wang, S. Inoue, S. Enthaler and M. Driess, Angew. Chem., Int. Ed., 2012, 51, 6167 CrossRef CAS PubMed
- X. Wang, B. Lei, Z. Zhang, M. Chen, H. Rong, H. Song, L. Zhao and Z. Mo, Nat. Commun., 2023, 14, 2968 CrossRef CAS PubMed
- R. Yadav, X. Sun, R. Köppe, M. T. Gamer, F. Weigend and P. W. Roesky, Angew. Chem., Int. Ed., 2022, 61, e202211115 CrossRef CAS PubMed
- H. Yang, A. Hinz, Q. Fan, S. Xie, X. Qi, W. Huang, Q. Li, H. Sun and X. Li, Inorg. Chem., 2022, 61, 19710 CrossRef CAS PubMed
- S. Yao, A. Saddington, Y. Xiong and M. Driess, Acc. Chem. Res., 2023, 56, 475 CrossRef CAS PubMed
- Y.-P. Zhou, S. Raoufmoghaddam, T. Szilvási and M. Driess, Angew. Chem., Int. Ed., 2016, 55, 12868 CrossRef CAS PubMed
- S. S. Sen, H. W. Roesky, D. Stern, J. Henn and D. Stalke, J. Am. Chem. Soc., 2010, 132, 1123 CrossRef CAS PubMed
- C.-W. So, H. W. Roesky, J. Magull and R. B. Oswald, Angew. Chem., Int. Ed., 2006, 45, 3948 CrossRef CAS PubMed
- C. Shan, S. Yao and M. Driess, Chem. Soc. Rev., 2020, 49, 6733 RSC
- Y. Chen, J. Li, Y. Zhao, L. Zhang, G. Tan, H. Zhu and H. W. Roesky, J. Am. Chem. Soc., 2021, 143, 2212 CrossRef CAS PubMed
- R. Liu, Y. Tang, C. Wang, Z.-F. Zhang, M.-D. Su and Y. Li, Inorg. Chem., 2023, 62, 1095 CrossRef CAS PubMed
- J. Schoening, A. Gehlhaar, C. Wölper and S. Schulz, Chem. – Eur. J., 2022, 28, e202201031 CrossRef CAS PubMed
- G. Becker, G. Gresser and W. Uhl, Z. Naturforsch., B: J. Chem. Sci., 1981, 36b, 16 CrossRef CAS
- A. Chirila, R. Wolf, J. Chris Slootweg and K. Lammertsma, Coord. Chem. Rev., 2014, 270–271, 57 CrossRef CAS
- M. Regitz, Chem. Rev., 1990, 90, 191 CrossRef CAS
- M. P. Müller and A. Hinz, Chem. – Eur. J., 2023, 29, e202302311 CrossRef PubMed
- A. Schäfer, M. Weidenbruch, W. Saak and S. Pohl, Angew. Chem., Int. Ed. Engl., 1987, 26, 776 CrossRef
- M. Weidenbruch, S. Olthoff, K. Peters and H. Georg von Schnering, Chem. Commun., 1997, 1433, 10.1039/A703054A
- S. S. Sen, J. Hey, M. Eckhardt, R. Herbst-Irmer, E. Maedl, R. A. Mata, H. W. Roesky, M. Scheer and D. Stalke, Angew. Chem., Int. Ed., 2011, 50, 12510 CrossRef CAS PubMed
- S. S. Sen, J. Hey, R. Herbst-Irmer, H. W. Roesky and D. Stalke, J. Am. Chem. Soc., 2011, 133, 12311 CrossRef CAS PubMed
- S. S. Sen, S. Khan, H. W. Roesky, D. Kratzert, K. Meindl, J. Henn, D. Stalke, J.-P. Demers and A. Lange, Angew. Chem., Int. Ed., 2011, 50, 2322 CrossRef CAS PubMed
- C. Wang, M.-D. Su, Z. Fang, J. Zhou, H. Zhang, X. Li, D. Zuo, Z.-F. Zhang and Y. Li, Chem. Commun., 2023, 59, 10275 RSC
-
E. Wiberg, Lehrbuch der anorganischen Chemie, Walter de Gruyter GmbH & Co KG, 2019 Search PubMed
-
C. A. Russell and N. S. Townsend, in Phosphorus(III) Ligands in Homogeneous Catalysis: Design and Synthesis, ed. P. W. N. M. van Leeuwen and P. C. J. Kamer, John Wiley & Sons, West Sussex, United Kingdom, 2012, ch. 11, p. 343 Search PubMed
- P. Pyykkö and M. Atsumi, Chem. – Eur. J., 2009, 15, 12770 CrossRef PubMed
- Y. Xiong, S. Dong, S. Yao, C. Dai, J. Zhu, S. Kemper and M. Driess, Angew. Chem., Int. Ed., 2022, 61, e202209250 CrossRef CAS PubMed
- S. Khan, R. Michel, S. S. Sen, H. W. Roesky and D. Stalke, Angew. Chem., Int. Ed., 2011, 50, 11786 CrossRef CAS PubMed
- X. Sun, A. Hinz and P. W. Roesky, CCS Chem., 2022, 4, 1843 CrossRef CAS
-
M. Kaftory, M. Kapon and M. Botoshansky, in The Chemistry of Organic Silicon Compounds, ed. Z. Rappoport and Y. Apeloig, John Wiley & Sons, West Sussex, United Kingdom, 1998, ch. 5, p. 181 Search PubMed
- A. G. Brook, F. Abdesaken, B. Gutekunst, G. Gutekunst and R. K. Kallury, J. Chem. Soc., Chem. Commun., 1981, 191 RSC
- X. Sun, A. Hinz, H. Kucher, M. T. Gamer and P. W. Roesky, Chem. – Eur. J., 2022, 28, e202201963 CrossRef CAS PubMed
- J.-C. Hierso, Chem. Rev., 2014, 114, 4838 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2345640–2345643. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt01367k |
| This journal is © The Royal Society of Chemistry 2024 |