
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInteraction of low-energy electrons with radiosensitizers
Barbora
Sedmidubská
 abc and
Jaroslav
Kočišek
*a
abc and
Jaroslav
Kočišek
*a
aJ. Heyrovský Institute of Physical Chemistry of the CAS, Dolejškova 3, 182223 Prague, Czech Republic. E-mail: kocisek@jh-inst.cas.cz
bDepartment of Nuclear Chemistry, Faculty of Nuclear Sciences and Physical Engineering, Břehová 7, 11519 Prague, Czech Republic
cInstitut de Chimie Physique, UMR 8000 CNRS and Faculté des sciences d'Orsay, Université Paris Saclay, F-91405 Orsay Cedex, France
First published on 15th February 2024
Abstract
We provide an experimentalist's perspective on the present state-of-the-art in the studies of low-energy electron interactions with common radiosensitizers, including compounds used in combined chemo-radiation therapy and their model systems. Low-energy electrons are important secondary species formed during the interaction of ionizing radiation with matter. Their role in the radiation chemistry of living organisms has become an important topic for more than 20 years. With the increasing number of works and reviews in the field, we would like to focus here on a very narrow area of compounds that have been shown to have radio-sensitizing properties on the one hand, and high reactivity towards low-energy electrons on the other hand. Gas phase experiments studying electron attachment to isolated molecules and environmental effects on reaction dynamics are reviewed for modified DNA components, nitroimidazoles, and organometallics. In the end, we provide a perspective on the future directions that may be important for transferring the fundamental knowledge about the processes induced by low-energy electrons into practice in the field of rational design of agents for concomitant chemo-radiation therapy.
1 Introduction
The motivation for studying interactions of low-energy electrons (LEEs) with radiosensitizers results from the possible role of such interactions in chemoradiation synergism. Identification of major processes contributing to the synergism may open the direct way for the efficient design of chemoradiation drugs. The hypothesis that LEEs are actively involved in the synergism during concomitant chemo-radiation therapy is rationalized by two main facts. The first is a high amount of secondary low-energy electrons produced during the passage of high-energy ionizing radiation particles through biological matter. The second fact is that many chemo-radiotherapeutics exhibit strong reactivity with LEE due to the high electron affinity of the molecule or its components. In the introduction, we will zoom in on these two facts as well as better define the scope of the present perspective article.Ionization is a process of removing an electron from the molecular orbital. Depending on the mechanism, electrons with different energies are ejected into the environment. These initial electrons can induce further ionization in the e–2e avalanche process or slow down by electronic excitations. The histogram of the processes contributing to the electron slow-down in the medium is known as the electron degradation spectrum. A good introduction to the initial processes during the physicochemical stage of radiation interaction with living tissue can be found in the book of Bednar1 or reviews on the topic.2,3 Determination of electron degradation spectrum in living tissue is an extremely complicated issue due to the large number of molecules present at different densities and polymeric forms. The number of available experimental cross-sections for electron scattering on the constituents of living tissue is rather low (e.g. ref. 4 and 5), and therefore the electron transport is typically approximated based on theoretical models.6,7 Important in the present content is that the LEEs, in contrast to other secondary species, can be distributed far, several tens of nm from the ionization track of the primary particle.8 Since electron ionization is the lowest energetically accessible process resulting in the multiplication of the number of free electrons in the medium, it is clear that the main component of the electron energy degradation spectrum is formed by electrons with energies below the ionization threshold of the medium.9 Processes induced by such LEEs with sub-ionization energies are of the primary focus in the present perspective.
1.1 LEE-induced processes in isolated molecules
In Fig. 1 we show the main interactions of LEEs with isolated molecules depending on the energy of the incident electron and examples of the relevant potential energy curves. Scattering processes, including elastic and inelastic scattering of electrons (I), electron-induced dissociation (II) and electron-induced fluorescence(III), can occur by simple collision of the incident electron with a molecular system, without the need for the formation of a transient negative ion (TNI). Even though, these processes may be strongly enhanced when LEE interaction time is prolonged by the formation of TNI. Examples are resonant features in vibrational excitation curves10 or LEE-catalysed dissociation.11,12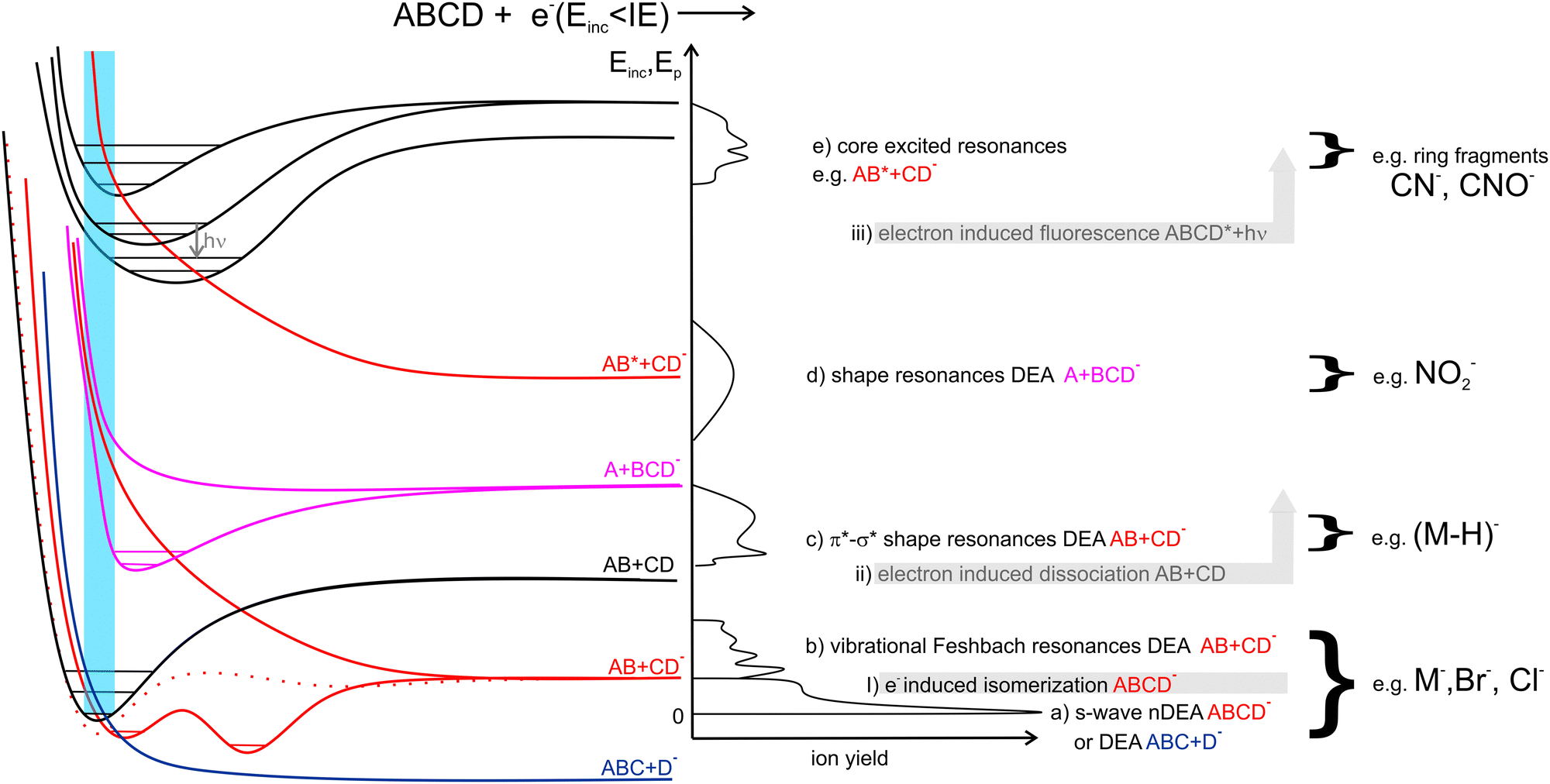 | ||
| Fig. 1 Common LEE interactions with radiosensitizers discussed in the present work. Gas phase experiments focus mainly on the reactions leading to stable anion formation (a)–(e) via electron attachment EA. Scattering processes leading to neutral products I–III are less explored. The left side of the image provides an example of potential energy curves that can be associated with the processes listed on the right side together with some typical products of LEE interactions with radiosensitizers after the parentheses. The central line provides an example of the corresponding anion yield as a function of the energy of the incident electron that can be expected in a typical electron attachment spectroscopy experiment. | ||
Electron attachment processes in Fig. 1(a)–(e), require the formation of TNI, which means that the electron is attached to the molecule on the timescales longer than the typical period for vibrational motion. TNI can be stabilized via intramolecular vibrational energy redistribution (IVR)13,14 or anion isomerization15,16 to form parent molecular anion. Another means of anion stabilization represents the emission of photons, which is closely related to IVR and recently explored mainly for anions of astrophysical relevance.17,18 Alternatively, the TNI can fragment via dissociative electron attachment (DEA), when an anion and a neutral co-products are formed. DEA is interesting from the point of view of reaction enthalpy. Formation of an anion fragment is accompanied by energy surplus due to the fragment electron affinity, significantly lowering the dissociation energy in comparison to the dissociation into neutral fragments.
EA can occur via different scenarios. If the anion's potential energy surface crosses that of the ground state of the neutral and electron attachment is exothermic (Fig. 1a), the cross-section at energies of the incident electron close to 0 eV diverges, theoretically exceeding the values of the elastic scattering cross-section.19
In the range of energies corresponding to the vibrational excitation of the ground state neutral, attachment via (dipole supported) vibrational Feshbach resonances (VFRs) occur (Fig. 1b). VFRs are initiated by the overlap of the neutral and anion vibrational energy levels caused by nuclear displacement, a consequence of the long-range interaction of the incident electron with the molecular dipole.20 As shown by Fabrikant, Hotop, and Allan,21 as well as the more recent experiments,22–24 not only the permanent dipole moment,25 but molecular polarizability as well plays an important role in the formation of dipole bound states. In systems with low-lying electronic excited states such as pyrazolide, the VFR may occur also via a core-excited (two-particle one-hole) process.26 The structure of the dipole-bound anions is very close to that of the neutral27 and therefore, VFRs can be understood only as a doorway mechanism for the formation of energetically lower-lying valence-bound anions.28 However, this is not always the case. In molecules with low or slightly negative electron affinities, the dipole-bound state can lay energetically under that of the valence-bound state.29,30 Moreover, the anion rearrangement from dipole to valence state may require additional energy input forming energetic barriers for the transformation.31,32
In the energy range between vibrational and electronic excitation levels shape resonances (Fig. 1d) are usually observed. These are formed by electron attachment to the unoccupied molecular orbitals LUMO+n of the molecule (virtual orbitals). The energies of these virtual states are directly related to the energies of molecular orbitals of the neutral precursor molecule (see e.g. ref. 33). Theoretically, electrons cannot be attached to the orbitals having zero angular momentum (σ) as they do not provide a barrier against autodetachment. In molecules, however, short-lived Sigma resonances can be detected in the vibrational excitation spectra, usually not resulting in DEA (short lifetime) and having only limited contribution to the total elastic cross section (large width).34–36 Even though the direct DEA mechanism of these resonances has a low probability, the σ and π molecular orbitals in complex biomolecules are often mixed allowing for rich fragmentation dynamics of TNIs. Examples are biomolecules in which hydrogens are displaced off the symmetry axis in CN–H and possibly also CO–H subunits. There, the low-lying shape resonances can have mixed σ*–H* character (Fig. 1c), e.g. ref. 37–39.
Finally, at energies of electronically excited states (Fig. 1(e)), core excited resonances can be detected. These are naturally of Feshbach's character (formation of TNI is stabilizing the system). Core-excited shape resonances are typically observed at significantly higher energies than the ones corresponding to the excitation of the neutral molecule.34 More details on the individual LEE processes can be found in specialized reviews40,41 or books on the topic.42–44
1.2 LEE interactions in solvent
The wavelength of LEE is already larger than molecular dimensions, implying that the cross-section for the molecular interaction can be larger than the geometrical cross-section of a molecule. This is reflected in the high LEE reaction cross-sections measured in the gas phase experiments, discussed in the previous section. On the other hand, in solvent media, the same long-range electron–dipole interaction results in the fast orientation of the neighboring molecules forming so-called solvated electrons. Water, with its large dipole moment, is one of the fastest traps for electrons with the transition to the aqueous state represented by a potential well of ∼3.5 eV45 at the timescale well below a picosecond.46–48In contrast to LEEs, the reactivity of solvated electrons is low. The fact that LEEs in biological media are only transient species limits their studies in realistic environments to state-of-the-art time-resolved experiments.49,50 On the other side, electrons of controlled energy can be also produced artificially in a vacuum, where molecular dynamics approaches can be applied to explore their interactions with model systems of various complexity.51 The present perspective focuses, but is not limited to these gas-phase experimental studies.
The advantage of the gas phase studies with model systems is that they can separate various effects of the environment on the dynamics of TNIs. Reviewing the present state-of-the-art, the four main effects on the reaction dynamics of LEEs in the environment can be identified, which are sketched in Fig. 2 The primary effect of the environment is an energy sink Fig. 2(a). As we discussed previously, electron affinity makes many of the LEE-induced processes exothermic. This energy gain, which induces fragmentation via DEA in the isolated molecules, is often dissipated to the environment. If the solvent molecules are different from the interacting molecule, the most probable mechanism is intermolecular vibrational energy redistribution via momentum transfer of the moving nuclei.52–54 In the case of overlapping energetic levels, such as in the case of molecules of the same type, this process may become more effective via resonant energy transfer.14
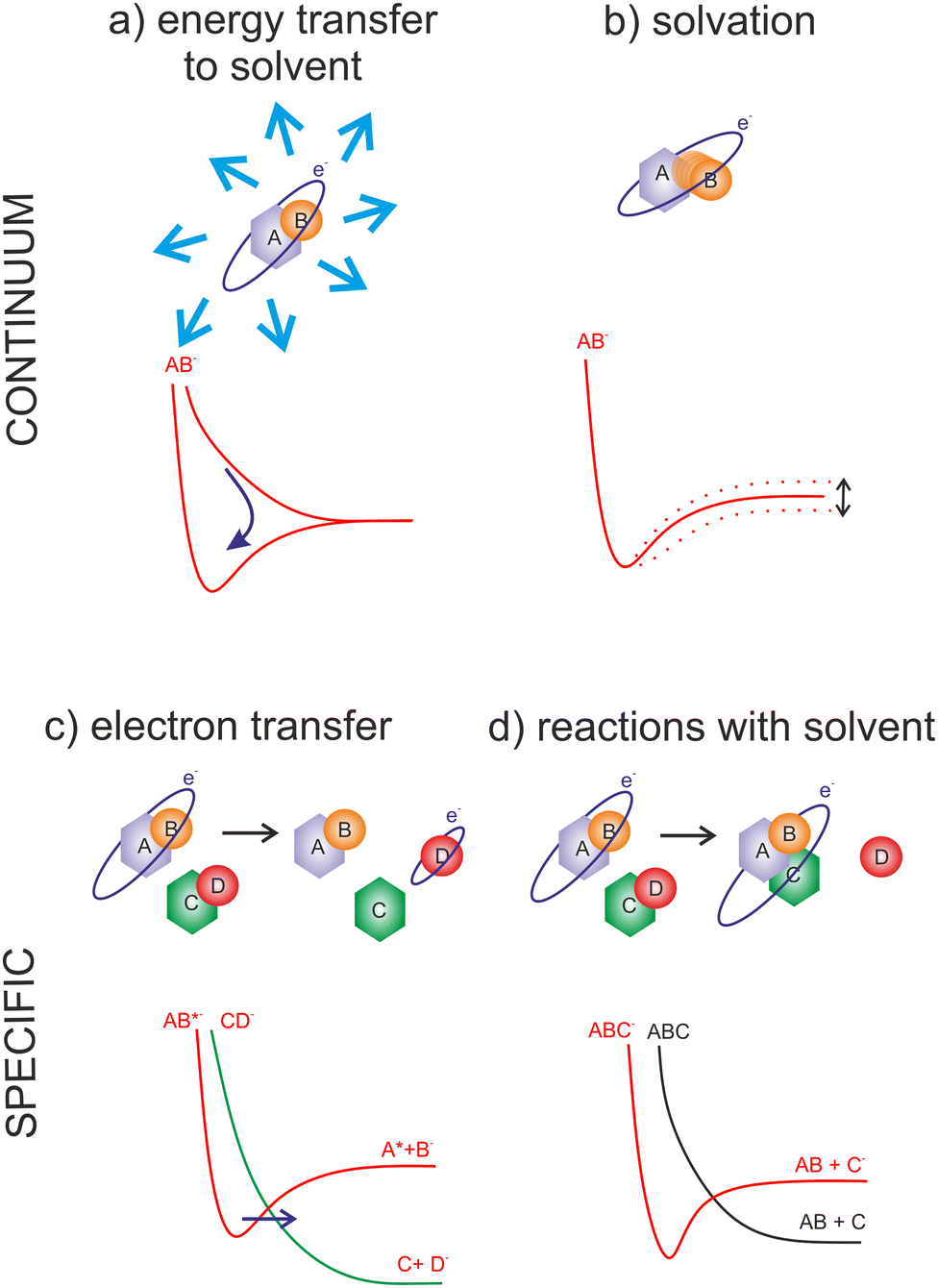 | ||
| Fig. 2 Commonly observed solvent effects on the dynamics of transient negative ions. (a) Energy transfer to the solvent can prevent molecular fragmentation as well as lead to significant heating of the environment; (b) solvation can prevent as well as promote dissociation or isomerization of TNI; (c) the process studied as self-scavenging in small clusters or electron transfer in gas phase experiments results in the formation of TNIs of one molecule via resonant electron attachment to another, in systems with many types of molecules such as in tissue this practically transfers very narrow resonant electron attachment processes into threshold processes operative over a wide range of electron energies; (d) the reactions TNI with solvent molecules can change the final product of electron attachment reaction. Important is also the catalytic role of an electron, in promoting the reactions that could not be possible in between neutrals. | ||
The second effect is the effect of solvation Fig. 2(b) when the neutral or anion geometry is distorted with respect to the gas phase geometry. A good example is the transformation of valence-bound anions of nucleobases with negative electron affinities into positive electron affinities in the solvent (stabilization of valence-bound anions).55,56 Another example is anion predissociation, typical e.g. for halogen-substituted molecules.57
The third effect is electron transfer Fig. 2(c).58,59 In contrast to isolated molecules in the gas phase, in bulk, the molecules are in permanent interaction allowing for the fast electron transfer to the energetically favorable sites. There are many electron transfer sequences known at the cellular level from electron transfer through DNA, through protein-mediated electron transfer chains to cell respiration and metabolism. Free LEEs can enter these sequences at any stage and influence their function. Identification of LEE reactions involved in these cellular mechanisms is one of the important tasks that will require close collaboration of molecular physicists and biophysicists.60 Considering the radiosensitization, the electron attractive centers in DNA are thymine bases61 that may act as an electron transfer terminus in nonsensitized strands. Follow-up electron transfer processes within nucleotides and nucleotide pairs can result in DNA strand breaks.62,63 The radiosensitizers may act as additional electron gain centers allowing for the enhancement of these processes.
The second important fact associated with electron transfer is the energy range under which a particular reaction occurs. As we discussed, the attachment of an electron is always possible only at a specific resonant energy. Therefore, from the wide energy distribution of LEEs, only a small part can interact via a particular DEA channel. This “bond and site selectivity” is an important characteristic of DEA.64–67 In the environment, one molecule can act as an electron scavenger and another can be dissociated upon electron transfer. This way the region of the reaction energies is significantly widened by all possible attachement-transfer sequences. This process is well known in cluster physics as a self- or auto-scavenging.68–71
Finally, reactions with solvent often occur. A good example is the reactivity of nucleotide TNI. McAllister et al. suggested that within the nucleotide, electron attachment to the DNA base will result in fast protonation of the base followed by C–N glycosidic bond cleavage.72 Such description is consistent with experimentally observed fragmentation of microhydrated 2-deoxycytidine-5-monophosphate upon interaction with LEEs.73 It is worth mentioning that electron-induced glycosidic bond cleavage in nucleotide was observed also using transient absorption spectroscopy in the bulk.74
Another good example related to the radiosensitizing molecules is the 5-nitro-2,4-dichloropyrimidine,75 where the DEA channel resulting in the release of the Cl neutral is accompanied by hydroxylation of the formed anion.
1.3 Why radiosensitizers?
Many molecules used in the concomitant chemo-radiation therapy have high electron affinities or bare electron affine groups. Typical examples are halogens or nitro groups. The electron affinity represents a negative (exothermic) contribution to the reaction enthalpy, often in the range of electron-volts. The presence of electron affine groups therefore significantly increases reactivity with LEEs as well as their ability to induce molecular transformations in the medium. This fact has attracted attention, since the fundamental mechanisms of chemo-radiation synergism are not fully understood. LEEs react during the physico-chemical stage of the radiation interaction with tissue and therefore only a tiny enhancement of reactivity towards LEEs can have significant consequences in further stages of radiation interaction with living tissue.3Several LEE-based mechanisms have been suggested so far to contribute to radio-sensitization and synergism. Simple attachment of an electron by a molecule can lead to the release of the energy equivalent to electron affinity into the environment. Considering the high number of LEEs forming around the ionization track, the addition to the linear energy transfer to the tissue can be significant.52 Another contribution of the non-dissociative electron attachment can be enhanced transport through the cellular membranes or accumulation in the cells.76 Except for the enhanced transport, in realistic environments the electron attachment is an important prerequisite of multiple electron reduction, which should be better explored, as will be discussed in the perspectives section. Electron affinity is also reflected in the dissociative processes induced by LEEs. LEEs can induce bond breaking at energies significantly below that required by sole energy input into the systems, such as photoexcitation. Already a single LEE can induce the formation of double-strand breaks in cisplatin-sensitized DNA strands.77,78 Many more DNA intercalating molecules have been suggested as possible radiosensitizers based on their DEA (e.g. ref. 79). However, the DEA to unbound molecules in the cellular medium can produce reactive radicals leading to cell stress and death.80,81 An interesting suggestion is also that the presence of heavy metal atoms, such as Pt can locally enhance the production of reactive LEEs.82
All these processes will be discussed on examples of particular molecules from three main groups of radiosensitizers studied during the last years: modified DNA components, nitroimidazoles and organometallics. In the end, we provide a perspective section focused on future directions that should be addressed to enable some real impact in the field of rational design of radiosensitizers and chemo-radiotherapeutics.
2 DNA components
The most studied derivatives of DNA components concerning the LEE-induced chemistry are halogenated pyrimidine bases, nucleosides and nucleotides (Fig. 3). The primary mechanism of action of these compounds in cancer chemotherapy is pyrimidine antagonism. Their presence inhibits the repair and formation of the DNA via replacing the pyrimidine bases in the DNA or via saturating the nucleotide synthesis proteins. Reduction in the DNA damage site repairs is an important source of combined and synergistic effects of these molecules with radiation.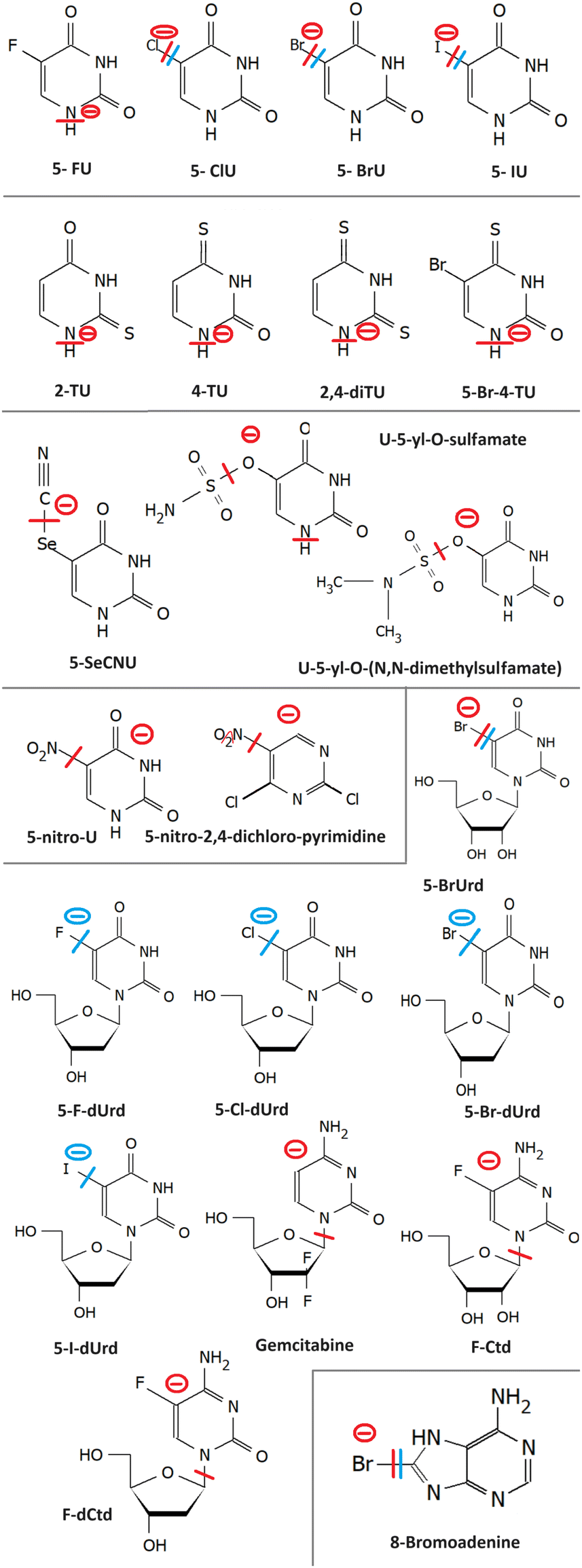 | ||
| Fig. 3 Radiosensitizers from the group of modified DNA components and their model compounds, whose reactivity with LEES is discussed in the text. From top to bottom halouracils (2.1); thiouracils (2.2); 5-selenocyanatouracil (2.3), uracil-5-yl-O-sulfamate and uracil-5-yl-O-(N,N-dimethylsulfamate) (2.4); 5-nitrouracil (2.5) and 5-nitro-2,4,-dichloropyrimidine (2.6); modified nucleosides (2.8) and 8-bromoadenine (2.7). The red line marks the bond broken in the most intense DEA reaction of the molecule identified for the isolated molecule, and the blue line the dissociation for a solvated molecule. The minus sign marks the anionic DEA fragment. | ||
Additionally to the suppression of repair, the molecules can also act as DNA radiosensitizers, increasing the susceptibility of DNA to damage. When incorporated into DNA, mispairings and DNA mutations are formed influencing the DNA secondary structure as well as increasing the number of accessible sites for radicals and LEEs.83 Indeed, the works with oligonucleotides84 as well as short DNA segments85,86 demonstrate enhancement of DNA strand breaks by low-energy electrons due to the presence of halogen-modified nucleosides. The enhancement can be direct, when more single-strand breaks are produced or due to the formation of reactive radicals, enhancing the clustered and double-strand damages.87 The direct and electron transfer mechanisms induced by LEEs are well-reviewed in the works of Von Sonntag,88 Rak et al.89 or Kumar.63 Poorly explored are the effects of mutations on the DNA secondary structure that can lead to better access of reactive species to the DNA components and enhanced damages during irradiation.86,90 Another unexplored field is the effect on hydrogen bonding of DNA bases important in protein interactions and during the DNA repair process.91 In this direction, there is only a handful of studies directed at non-modified DNA bases (e.g. ref. 92–95). An important benchmark to the computational models can be acquired in precisely designed experiments on DNA origami platforms96 but also using various clustering techniques.51,97,98
Generally, pyrimidines are considered better radiosensitizers than purines, which can be related to their higher electron affinities.99 Except for electron affinity, also the DEA was considered an important prerequisite of radio-sensitizing properties of pyrimidine substitutes. Systematic studies on the topic were performed mainly in the J. Rak group. Fig. 5 shows a set of substituted uracils with the main DEA reaction channels identified in the work of Makurat.89 The DFT-based study sorted the studied uracils into three groups according to their DEA fragmentation pattern resulting in the stabilization of parent anions in group A, fragmentation of substituent group B, and formation of reactive oxygen radical anion in group C. Based on the high stability of the parent anion, group A was excluded as not having radiosensitizing potential. However, based on recent studies with different radiosensitizers, DEA is not a necessary condition for radiosensitizing properties. Single electron reduction can result in several sensitizing mechanisms already discussed, such as enhanced linear energy transfer, better transport of the radiosensitizer within the biological system, or an increase in electron transfer and multiple electron reduction rates. In vitro, experimental studies with complexity similar to the DFT study of Makurat would give us much better insight into the undergoing mechanisms. Systematic in vitro studies with model molecular systems are, however, complicated due to a large number of parameters that have to be considered when moving from gas phase experiments to biological buffers.75,100,101
2.1 Halouracils
We will start with probably the most studied systems, 5-halo uracils. Three energy regions for the interaction with LEEs can be identified.In the first region, at energies of the incident electron below 1 eV the electron is attached by the molecular dipole moment into VFRs.102,103 Resulting anion in the vibrationally excited state allows for very different dissociation dynamics of that of ground or electronically excited anion.104–106 The cross section at these low energies has a shape of discrete vibrational bands, whose threshold and intensity depend on the reaction enthalpy for the particular DEA channel. DEA at these energies proceeds via carbon-to-halogen bond breakage resulting in halogen or U-yl anions products. The enthalpy depends on the electron affinity of the fragment anion and bond dissociation energy. Carbon-to-halogen bond dissociation energies in organic molecules decrease from around 5 eV for C–F to around 2.5 eV for C–I.107 Electron affinities of all F to I halogens are higher than 3 eV108,109 while that of 5-U-yl radical is 2.34 eV.110 Simply subtracting the bond dissociation energy and electron affinity can give us energy surplus for some halogen uracils while shortage for others. In gas phase experiments, however, all but FU efficiently fragment via DEA at the energy of incident electron 0 eV, while the calculated reaction enthalpies are typically positive in the 0–1 eV range. This behavior was assigned to the high sensitivity of the vibrational Feshbach resonances to temperature.111 The experiments with isolated molecules are performed via sublimation of the molecules into the gas phase, which requires high temperatures in the order of hundreds of degrees Celsius. Even though only the tail of the Boltzmann distribution may allow for dissociation, the electron interaction probability at near 0 eV diverges, as we already mentioned, and therefore the molecules from this very tail of the thermal distribution can be enough to produce significant DEA ion signals. This is also in agreement with different distributions of the most intense bands in the cross sections from different experiments.104–106
On the other hand, TNI allows also for hydrogen and proton mobility112 or complex rearrangements.113 By way of example, simple hydrogen migration upon electron attachment can result in the formation of N1 U-yl radical with an electron affinity of 3.4810 ± 0.0006 eV.114 instead of simple bond cleavages and 5-U-yl radical formation. Such change can explain several previously puzzling 0 eV DEA peaks. However, the rearrangement barriers will need to be explored in detail, recently done e.g. for 5-bromo-4-thio-uracil.115 An additional explanation for the observed dissociation near 0 eV can be the creation of stable neutral HX molecules during the TNI lifetime, where X is halogen, which was suggested in the mentioned study of 5-bromo-4-thio-uracil as well as several other TNIs.116–118
In the second region of incident electron energies between 1 eV and 4 eV, electrons are attached via shape resonances into the three lowest-lying unoccupied π orbitals. Even though their coupling to the dissociative σ* state still plays an important role in the dissociation process.102,119 The fragmentation spectra at low energies below 4 eV are dominated by halogen or U-yl anions being highly reactive. Formation of these species was observed in early photolysis experiments.120,121 They were also suggested to play an important role in the evolution of other reactive species such as OH radicals.122
In the third energy region, core excited resonances result in complex multiple bond cleavages including the ring breakage and formation of anions such as OCN−, H2C3NO−,123 HC2N−, C3NO− and others.117
When halouracils are submerged in solvent, their dissociation efficiency at low energies is reduced. Studies with microhydrated molecules in clusters demonstrate that while FU doesn't dissociate, BrU and ClU anions can dissociate even when fully surrounded by water molecules.52 In contrast to e.g. thiophenols57 no halogen-water clusters were observed in the fragmentation spectrum of microhydrated uracils. This indicates either a fast dissociation, without the formation of intermediate products, or neutral HBr formation, similar to recently reported HF from fluorouracil.117 Such neutral products, which cannot be detected using mass spectrometry, can be one of the reasons for a significant decrease in the fragmentation signal upon solvation. However, a similar trend of decreasing fragmentation from intense IdU decay to non-decaying FdU was observed also in the pulsed radiolysis study in bulk.124 Further suppression of LEE-induced fragmentation can be expected in biological buffers, as demonstrated by Beach, Fuciarelli, and Zimbrick.125
The suppression of fragmentation in the environment does not necessarily mean the inactivation of LEE-induced radiosensitization. These studies actually demonstrate that even in the solvent conditions, LEE will effectively attach to halogenated DNA bases. When incorporated in the DNA, electrons can be transferred into the backbone,126,127 or negative ions may become attractive for proton transfer from the backbone,128,129 which are both efficient ways for DNA strand breaks. Except for chemical ways of radiosensitization, there are also physical consequences of high electron attachment rate in the solution. The energy gained by the system due to the electron affinity of halogen-modified nucleobases can efficiently dissipate into the solvent as demonstrated in Fig. 4 from the work of Poštulka et al. In other words, electron affinity represents a positive contribution to the linear energy transfer (LET).
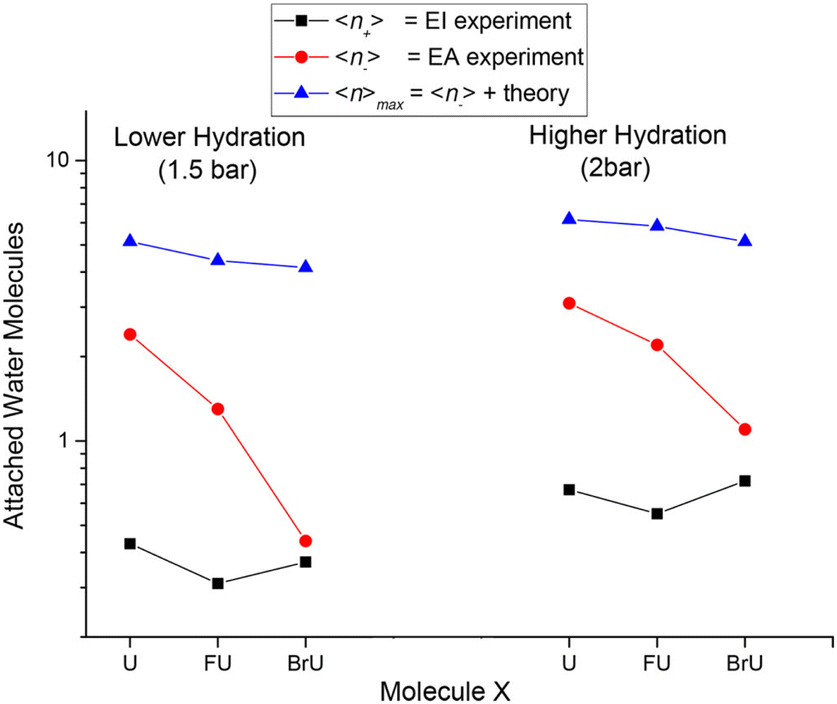 | ||
| Fig. 4 The image from the work of Poštulka et al. showing that theory predicted increase in the energy transfer to solvent due to the electron affinity of the microhydrated halo uracils can reproduce well the fragmentation of their TNIs. For uracil (U), bromouracil (BrU) and fluorouracil (FU) shown on the x-axis the y-axis shows the detected mean size of ionic clusters upon electron attachment (red) and electron ionization (black). The blue points are mean values of neutral cluster sizes estimated based on electron affinities and under the assumption that all energy transferred to solvent leads to the evaporation of water molecules from the cluster. Reproduced from ref. 52 with permission from American Chemical Society, copyright 2017. | ||
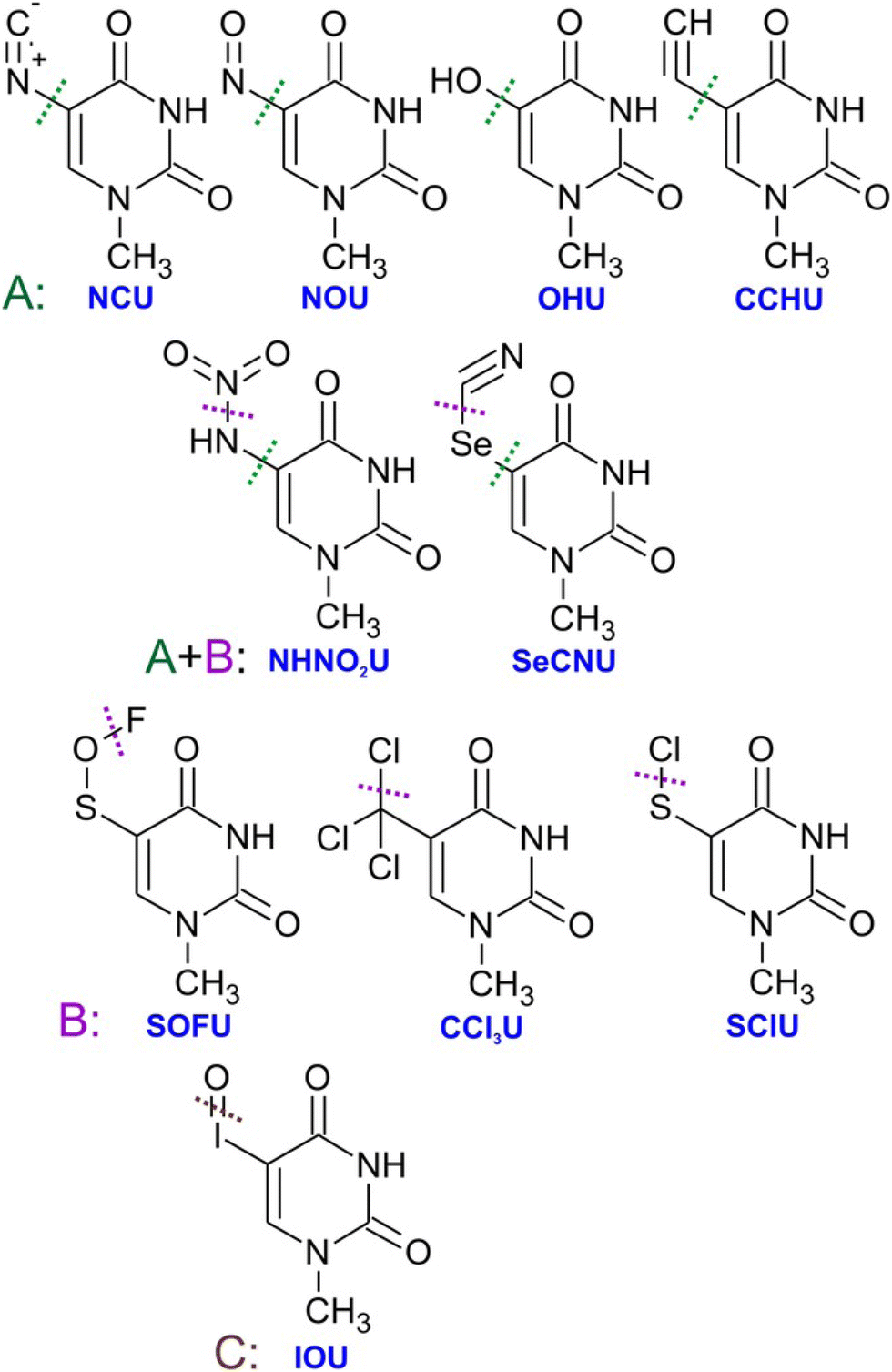 | ||
| Fig. 5 Substituted uracils from the systematic DFT study of Makurat et al. with dotted lines showing main sites of fragmentation via dissociative electron attachment induced by LEEs in solution modelled by polarizable continuum model. Electron attachment to the group A molecules was predicted to result in stable negative ions, while B type ions will fragment by dissocaition of the substituent group and IOU by the formation of reactive oxygen radical anion. Reproduced from ref. 89 with permission from John Wiley and Sons, copyright 2016. | ||
Except for the water solvent, other environmental effects have been studied for halogenated bases, particularly the effect of base pairing.130 Pairing of halogenated bases results in a decrease in their adiabatic electron affinity. Therefore, the mutations in the double strands may be less sensitive to LEEs in comparison to the single strand form, e.g. during repair and replication processes.
2.2 Thiouracils
Electron attachment to 2-TU in the gas phase was studied using electron attachment spectroscopy and ab initio calculations. Low-energy VFRs lead to efficient hydrogen abstraction from nitrogen, with a possible contribution from low-lying π* resonances at the high energy tail of the observed signal. At energies above 4 eV, DEA is proceeding via core excited resonances of the CS bond in a mechanism similar to CS2.131 The most intense DEA channels opening at this energy are leading to S− and (TU-S)−. However, the ion yield of the second most intense decomposition product of the molecule SCN− is slightly shifted towards lower energy, which can be a result of the contribution from the high π* shape resonance to this DEA channel.132 Dependency of the anion signal on temperature has shown only a little effect in comparison to the usual behavior of VFRs.133TU is a nice example demonstrating how reactivity with the solvent (Fig. 2) can change the reaction output of LEE interaction. Prasanthkumar et al.134 studied TU and TC using pulsed radiolysis in solution. Changing buffer solution composition, two modes of interaction were reached dominated by reactions with solvated electrons or CO2− ions. In the first case, the electron attachment and formation of TNI is accompanied by a fast proton transfer from the solvent resulting in the formation of protonated radical. While EA in the gas phase discussed in the previous paragraph, results mainly in hydrogen abstraction, in solution it is the opposite. When CO2− is introduced in solution, it can abstract hydrogens from the molecules, but the formed radicals interact with each other to form sulfur-to-sulfur bound dimers.
More recently, 5-bromo-4-thio-uracil was studied showing strong effects of intramolecular proton transfer on reaction dynamics, partially stabilizing the parent anion and allowing for HBr formation and release during the TNI decay.115 It is worth repeating that the formation of hydrohalic acids seems to be an important process upon LEE interaction with many halogenated molecules116,118,135 however, the biological consequences of this process are not well explored.
Other thiouracils were explored theoretically. The electron affinities were calculated to rise from 2-TU with EA of 0.26 eV to 4-TU (0.61 eV) and 2,4-diTU (0.87 eV). Therefore, observation of stable parent anions should be possible, particularly for di-substituted thiophenols136
2.3 5-Selenocyanatouracil
Based on the already mentioned systematic work of Makurat,89 5-SeCNU was selected for further experimental studies. Isolated 5-SeCNU is more sensitive to EA than BrU and efficiently releases CN−. However, this mechanism does not transfer into a solution, where the U–Se radical stabilizes via the formation of two stable products: dimer U–Se–Se–U and an adduct of radical OtBu to radical U–Se (![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) U–Se–OtBu) due to the used tert-butanol [tBuOH] scavenger in the buffer solution.137 It is worth mentioning that dimer formation was observed also upon 2-TU and 2-TC radiolysis in solution, but their occurrence was buffer dependent.134 Actually, the composition of buffer solutions to separate LEE-induced processes is one of the major issues complicating the experiments as will be mentioned on several places in the present perspective.
U–Se–OtBu) due to the used tert-butanol [tBuOH] scavenger in the buffer solution.137 It is worth mentioning that dimer formation was observed also upon 2-TU and 2-TC radiolysis in solution, but their occurrence was buffer dependent.134 Actually, the composition of buffer solutions to separate LEE-induced processes is one of the major issues complicating the experiments as will be mentioned on several places in the present perspective.
2.4 Uracil-5-yl-O-sulfamate and uracil-5-yl-O-(N,N-dimethylsulfamate)
LEE interaction with isolated uracil-5-yl-O-sulfamate results in complex fragmentation already at near 0 eV energies of the electrons. The main product results from the S–O bond cleavage and neutral NH2SO2 radical release. The other intense fragmentation channels observed at low energies are associated with sulfamate dissociation, particularly the strong SNO−. This is understandable based on the large electron affinity of this fragment138 in comparison to other anions that are commonly observed from sulfoxy compounds, such as SO2− and SO3−, detected with only minor intensity. Upon solvation, DEA channels seem to be closed as demonstrated by radiolysis of the solutions with t-BuOH as a radical scavenger.139 For uracil-5-yl-O-(N,N-dimethylsulfamate), the LEE-induced fragmentation of the isolated molecule is analogous to uracil-5-yl O-sulfamate, only intensities differ because of methylation. e.g. the intensities of 5-U-yl fragments were under the detection limits.1352.5 5-Nitro-uracil
The electron affinity of the nitrogen dioxide is 2.273 ± 0.005 eV,108 close to that of the U-yl radical. While the C–N bond is one of the strongest, in the nitro compounds it has a single bond character with bond dissociation energy values ranging from 2.5 eV to 3 eV.112,140 As a result, DEA via release of neutral NO2, which is the most intense channel, is endothermic. The occurrence of intense 0 eV VFRs in the DEA cross section for this channel is therefore surprising.140 As discussed in the case of halouracils, the exothermicity can be enabled by hydrogen migration over the radical, internal vibrational energy of the neutral, as well as more complex rearrangements during the TNI lifetime. The similar electron affinities of the co-products allowing for charge migration between NO2 and U-yl and suggested rearrangement before the dissociation probably contribute to the long lifetime of the parent anion of 5-nitro-uracil that was also experimentally observed with high intensities.1412.6 5-Nitro-2,4-dichloropyrimidine
5-Nitro-2,4-dichloropyrimidine is a model compound containing two halogen atoms and a nitro group connected to the pyrimidine ring. The molecule has exceptionally high electron affinity and anion lifetime. DEA proceeds via the release of neutral NO and NO2 radicals at near 0 eV energies and Cl− at energies above 2 eV. The reaction channels are suppressed by hydration but NO2 release channel remains open in the solvent, which is not common e.g. for nitroimidazoles that will be discussed further in the perspective. Additionally, reactivity with solvent results in the halogen replacement reaction, where Cl atom is replaced by OH forming a C4H2Cl2N3O3 anion.752.7 8-Bromoadenine
8-Bromoadenosine incorporation into DNA enhances its susceptibility for single and consequently double-strand breaks upon irradiation.87 It has been shown in the experiments on hot electron transfer through DNA vires that strong dehalogenation occurs on the 8BrA-modified sequences, presumably via the DEA mechanism.142 Similar strong dehalogenation was observed also for 8-bromoadenine143 and 8-bromoguanine, directly bound to plasmonic nanoparticles.144Dehalogenation is also a primary channel for DEA to the isolated 8-bromoadenine in the gas phase resulting in both Br− and (M-Br)− anions in approximately 6 to 1 ratio. DEA seems to proceed via VFRs at energies close to 0 eV and a shape resonance peaking around 1.3 eV. The minor (M–H)− channel demonstrates typical behavior of σ*–π* mixing with the sharp onset of the signal, which may be temperature dependent. An intense signal upon electron attachment was observed also for the parent anion. The anion has a structure of pre-dissociated Br− relatively far from the polarized adenine core. This type of non-covalent anion structure was recently identified for several cyclic halocarbons (e.g. ref. 145–147) and may be crucial for understanding also the evolution of cyclic hydrocarbon TNIs in the solution.57,99,148
2.8 Halogenated nucleosides
Halogenated nucleosides are generally more soluble in aqueous solutions than bare DNA bases. As discussed by Falkiewicz et al.149 it is the OH group at the 2nd position in ribose and its electronic and steric effects determining the solubility. Nucleosides are, however also more thermally labile. Therefore, the number of their studies in the gas phase is significantly lower than that for the DNA bases, while there are more studies in bulk.The most studied nucleoside is 5-bromouridine. DEA to isolated molecule leads to the release of Br− anion and 5-BrU-yl anion with high cross sections of 2 × 10−14 cm2 and (9 × 10−16 cm2), respectively.150 Other dissociation channels, including uracil fragmentation are much less intense.151 In water, pulsed radiolysis was employed to demonstrate Br− release by LEEs.152 The mechanism was further confirmed by a more recent study in diethylene glycol.153 A pulsed radiolysis study with F to I deoxyuridines in water then demonstrated a decreasing trend from strongly decaying IdU to practically non-decaying FdU in LEE-induced halogen release reactions.124
Even more effective dissociation upon electron attachment than in 5 substituted species can occur in 6-substituted uridines. However, as demonstrated for 6-iodo-2-deoxyuridine,149 the bond dissociation energies may allow dissociation in solvents before electron attachment. The environmental effects such as solubility and buffer reactivity will be critical for the transfer of further halogen-based radiosensitizers into applications such as 5-iodo-4-thio-2-deoxyuridine154 or 5-bromo-4-thio--deoxyuridine.115
An important difference in comparison to DNA bases is that nucleosides can be modified also on the sugar moiety. A well-known sugar-modified molecule already in use in clinical practice is gemcitabine.155 Comparative experimental study of gemcitabine and fluorocytidine demonstrated that the binding site of the electron affine atom changes the outcome of DEA significantly. The π system of the DNA base is highly attractive for the electron and halogenation of the ring enhances the cross-section for hydrogen loss (Cyt-H)− channel 5.5 times. Fluorine on the sugar moiety still enhances the (Cyt-H)− formation, but only 2.8 times, indicating that overall the gemcitabine is probably a weaker electron scavenger.156
The recent study of 2′-deoxy-2′-fluorocytidine, as a model compound, and 8-bromoadenosine modified DNA studied on a model DNA nanostructure supports87 demonstrated that the DEA intensity may not be the sole difference between sugar and base halogenated radiosensitizers. While 8-bromoadenosine modified strands show an increase in the total number of lesions and consequently in double-strand breaks with respect to the random sequence, the 2′-deoxy-2′-fluorocytidine modified strands show only an increase in the number of double-strand breaks but no enhancement in the total number of lesions. 2′-Deoxy-2′-fluorocytidine is therefore enhancing the probability for double-strand break in the case that single-strand break occurs. Such enhancement was tentatively assigned to forming reactive sugar-based radicals in the modified strands inducing clustered damages.157
The role of LEEs in the formation of sugar-based radicals in radiosensitizer-modified DNA is not sufficiently explored. Tentatively, the mechanism can copy that explored for unmodified DNA using ESR,63,158 where dehydrogenated base radicals can be formed by hydrogen abstraction followed by proton extraction from the sugar moiety. The scenario may be, however, influenced by the environment. While phosphate counter ions have only minor effects on electron affinities159 or electron transfer,94 proton transfer from the solvent may play an important role in the dissociation dynamics (see e.g. ref. 160) and lead to the formation of sugar moiety anions as demonstrated for microhydrated deoxycytidine monophosphate.72,73
Another mechanism is the direct electron attachment to the sugar moiety. For unmodified DNA it was demonstrated already in the seminal work of Boudaiffa.161 Ptasinska and co-workers then showed high electron attachment cross sections for isolated ribose, a model compound of more complex sugars.162 Sugar-based radicals and anions were reported also upon electron attachment to softly desorbed D-ribose-5-phosphate.163 We therefore believe that there should be more focus on the formation of sugar-based radicals and anions upon the interaction of modified DNA with LEEs.
3 Nitroimidazoles
Most azole-based cancer chemotherapeutics (Fig. 6) are antimetabolites, mainly involved in the folate cycle. Folic acid is required to build and repair DNA, and in many types of cancer, the folate receptors are overexpressed. Azoles are also biomimetic to amide bonds and therefore they are known to interact with proteins164 strongly. This fact is only poorly explored concerning the DNA repair mechanisms upon irradiation. Research of the LEE community is mainly focused on nitroimidazoles due to their radiosensitizing potential dependence on electron affinity, which was realized in the 1970s already.165,166 The first works focused on the direct incorporation of nitroazoles into DNA and electron scavenging. Later, many other mechanisms appeared ranging from NOx effects,167 particularly the DNA repair enzyme inhibition,168,169 vasodilation effects, fixation of organic radicals, enzyme-catalyzed depletion of nucleophiles, formation of toxic products, and interference with recombination reactions.170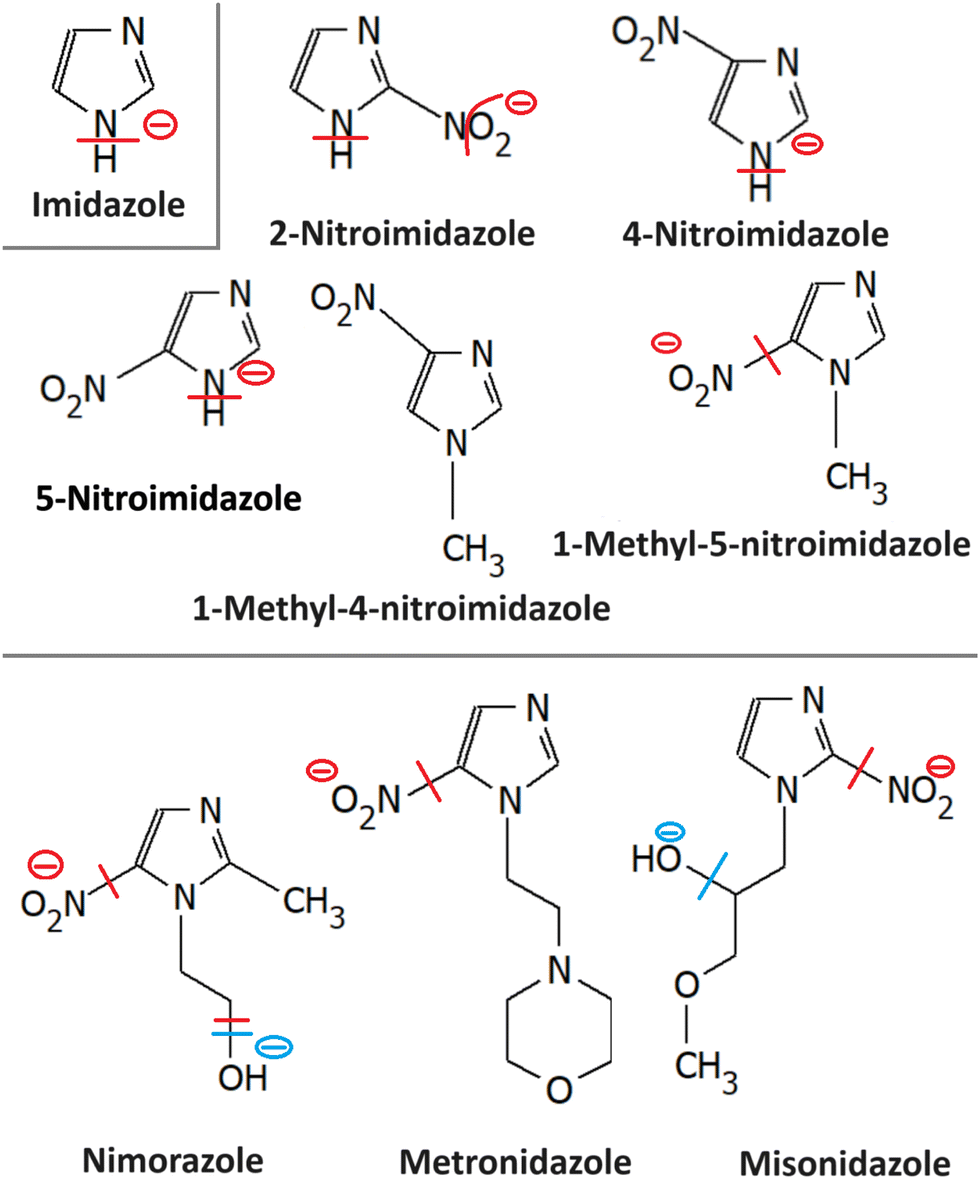 | ||
| Fig. 6 Radiosensitizers from the group of nitroimidazoles and their model compounds, whose reactivity with LEEs is discussed in the text. From top to bottom imidazole (3.1) and nitroimidazoles (3.2); nimorazole (3.4); metronidazole (3.3) and misonidazole (3.5). The red line marks the bond broken in the most intense DEA reaction of the molecule identified for the isolated molecule, and the blue line the dissociation for a solvated molecule. The minus sign marks the anionic DEA fragment. | ||
Nitroimidazoles are efficient under hypoxic conditions, which further support their bioactivation mechanisms by single electron reduction, a process in strong competition with oxygen reduction under normoxic conditions (see Fig. 7).171 The single electron reduction may be induced by electron transfer172 in applications such as antibiotics or antiparasitics, while direct attachment of LEEs may be responsible for their activation during radiotherapy.76 The genotoxicity can be then induced by the nitro anions, reactive oxygen species or redox reaction intermediates. The remaining problem is that the most important mechanisms of genotoxicity have not been identified yet, despite very systematic studies on a range of compounds.173,174 Only a full understanding of the nature and reactivity of nitro anions formed after LEE interaction will enable rational design of new radiosensitizers and possibly also more efficient antibiotics.
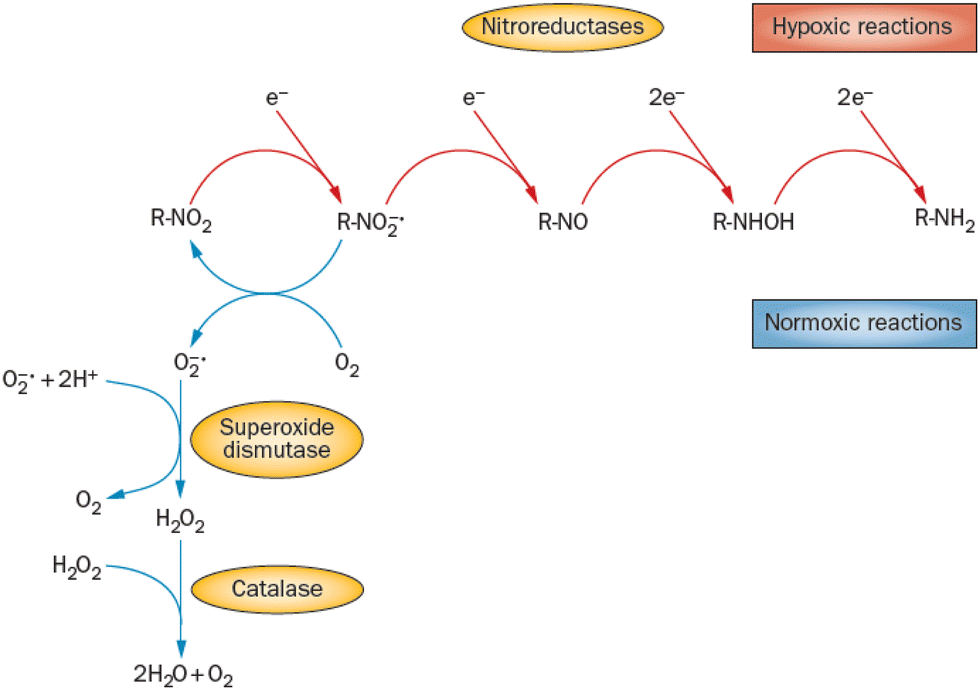 | ||
| Fig. 7 Different radiation chemistry of nitro group under hypoxia and normoxia. While under normoxic conditions single electron reduction is in strong competition with the reduction of oxygen, under hypoxia multiple electron reduction occurs leading to a variety of reactive radical species. Reproduced from ref. 171 with permission from Springer Nature, copyright 2012. | ||
3.1 Imidazole
Similar to pyrimidine bases, imidazole contains N–H bonds prone to dissociation upon low-energy electron attachment. In the unsubstituted imidazole, the N–H bond strength is high and therefore DEA can occur only at energies above 1 eV via π* resonances. Electron attachment, however, can occur also via low-lying VFRs due to the high dipole moments of azoles (e.g. 3.67(5)D for imidazole).175 Such attachment is also discernible from the electron transmission spectroscopy.176 Competition between electron auto detachment, DEA, and valence anion stabilization for these VFRs is then controlled by the functional groups attached to the ring as will be discussed for particular derivatives. Another important similarity to pyrimidines is in the strong coupling of σ* and π* states,177 which is an important characteristic of the CN–H moiety.38Imidazole loses hydrogen via shape resonances in 1.5–4 eV range,176,178 presumably upon electron attachment to the LUMO and LUMO+2 orbitals.177,179,180 In isoxazole additionally, O–N bond can be cleaved at energies as low as 1.5 eV leading to the ring opening.181,182 At higher energies, core excited resonances can be identified in the 6–11 eV range also leading to the N–O bond cleavages and ring opening in imidazole176 or isoxazole.181 An important process observed is multiple hydrogen cleavage at low energies that must be accompanied by hydrogen molecule formation.176,178 The fundamental electron attachment properties of the imidazole ring, its propensity to be opened or fast hydrogen migration across the ring all transform to the LEE-induced chemistry of more complex models of imidazole-based radiosensitizers.
3.2 Nitroimidazole
A range of nitroimidazoles has been studied as model compounds for electron-affinic radiosensitizers. In the systematic DFT exploration and electrospray experiments of Feketeová et al.183 eight derivatives were studied. For all the studied compounds, the most stable anion structure has the additional electron localized on the nitroimidazole with only negligible effect from the other functional groups. Fig. 8. The SOMO energies are influenced only by the nitro group position on the ring. As a result, the adiabatic electron affinity of 4- and 5-nitro imidazole have values around 1.3 eV, while that of 2-nitroimidazole is more than 0.6 eV lower. Despite that, fragmentation induced by LEEs to 2-nitroimidazoles is richer than that of the 4- and 5-nitroimidazoles.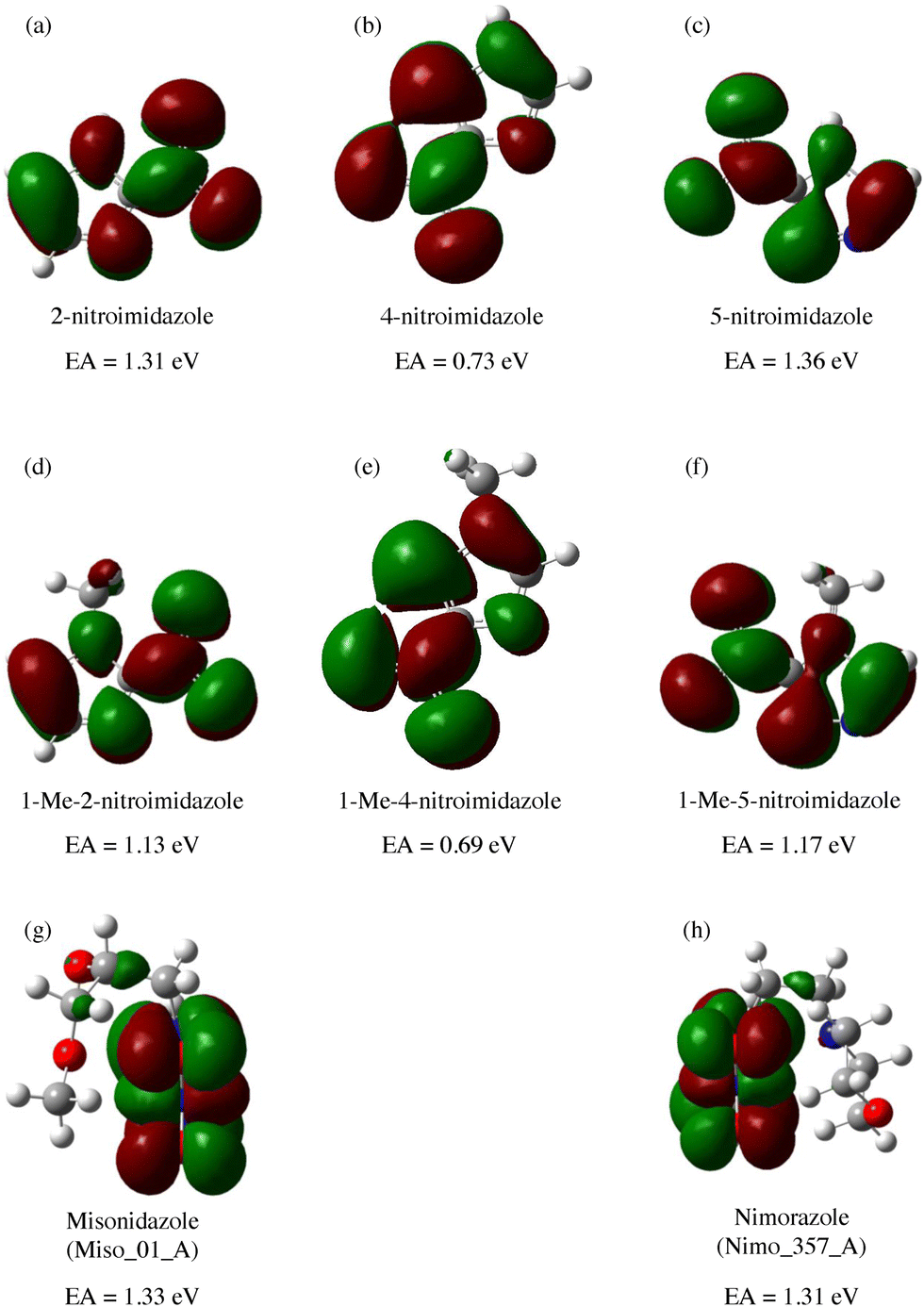 | ||
| Fig. 8 M062x/6-311+G(d,p) optimized structures of radical anions and their SOMOs reproduced from the work of Feketeova et al.183 with permissions from Elsevier, copyright 2014. We can see that in these compounds, the electron is always localized over the imidazole ring and while the nitro group position shifts the SOMO energy levels, the other substituents have practically no influence. Single occupied molecular orbital SOMO, forms upon electron attachment into the lowest unoccupied orbital (LUMO) of the neutral precursor molecule. The effects of the substituents can be expected for attachment at higher energies due to the often observed additive effect of electron attachment.184,185 | ||
In all nitroimidazoles, the most intense DEA channel is the release of neutral OH. The second, still exothermic, channel is NO release and the third is hydrogen release. The nitro group is strongly bound to the ring and therefore cross section for the nitro C–N bond cleavage via both NO2− or (M-NO2)− channels is observed in a shape resonance around 3 eV. An additional fragment appears in 2-nitroimidazole due to the release of a neutral water molecule. The calculated reaction barriers for this process are practically isoenergetic for 5- and 2-nitroimidazoles,186 and therefore exclusive observation of this channel for 2-nitroimidazole is quite surprising. Either the channel is closed in 5-nitro derivative due to the dynamical restraints or the anion product may differ from that predicted by calculation. In the first case, one can imagine a two-step process. In 2-nitroimidazole oxygen interacts with hydrogen at position 1 and position 2 to form water, in 5 nitroimidazoles, upon the interaction with hydrogen at position 1, the formed OH and hydrogen at position 4 will be separated by the remaining NO group at position 5 of the molecule preventing the fragmentation. In the second case, e.g. ring opening isomerization of the formed anion may be energetically more favorable upon 1,5 dehydrogenation in contrast to 1,4 dehydrogenation, which can influence the reaction enthalpy as well as the lifetime of the formed anion with respect to the autodetachment.
Another interesting observation for nitroimidazoles is that methylation of N1 hydrogen suppresses the fragmentation of molecules below 2 eV.187,188 Methylation of hydrogen sites is quite a common approach used in fragmentation studies to assign dissociation sites.189–191 In the case of nitroimidazoles, however, the methylation on the N1 position closed all fragmentation at energies below 2 eV.187 This is clear for channels relying on N1–H dissociation such as OH release, but the influence on the NO or NO2− release is not straightforward. 2 processes have to be separated at these energies.
The first is π* resonance, identified theoretically.192 The lifetime of the resonance with respect to autodetachment is significantly lower upon methylation at the N1 site. The result is the disappearance of the resonance from fragmentation but also from the parent anion spectra. Autodetachment is faster than anion detection.
The second region of energies is below 1 eV, where strong VFRs result in the release of NO. This channel is closed upon methylation and a stable anion is formed, which can have several explanations. The first is the change in the molecular dipole moment and the size of the dipole-bound state. These effects on dipole-supported states were explained by Desfrançois and co-workers.193 A decrease of the dipole moment due to the methylation can result in a lowering of the affinity for the VFR.194 For small molecules important effect can have also an increase in the molecular size. For larger molecules, the molecular core potential is better shielded and the probability of electron attachment is lower. On the other side, the polarizability, also influencing the VFR formation, can be larger, and therefore, the interplay of these effects on VFRs should be carefully explored from one case to another. In the discussed case of methyl-4-nitroimidazole the dipole moment is huge, larger than that of the bare 4-nitroimidazole 8.50 D vs. 7.78 D and the molecules are relatively large, the addition of the methyl does not have a significant effect on molecular size. Probability for the formation of VFRs is therefore not influenced what is reflected in a strong signal of the molecular parent anion at near 0 eV energy.
The nitro group fragmentation via VFRs therefore must be influenced on the side of the outgoing – dissociative channel. Either it is the hydrogen migration upon DEA and NO release that can result in the formation of hydroxyl form of (M–NO)− anion instead of carboxyl form in 4-nitroimidazole but not in the methyl-4-nitroimidazole or methylation stabilizes the parent anion by a different mechanism, such as the ring opening. Additionally, all these molecules are studied upon sublimation. At such conditions, different propensities of molecules for dimerization can result in a surprising parent ion stabilization, particularly in the case of VFRs. This was observed e.g. in the case of pyruvic acid.195
Nitroimidazoles efficiently attach electrons also in the energy region of core excited resonances above 6 eV. However, practically no DEA fragmentation is observed at these high energies. In the study of a model compound 1-methyl-5-nitroimidazole, it was suggested that dissociation into two neutral fragments may occur at these energies, pointing out the importance of the studies of neutral dissociation products upon electron attachment.80 The total scattering cross section at low energies was complemented by differential and DEA as well as ionization cross sections in the follow-up publication.196 We would like to add that in complex environments the core excited resonances may play an important role irrespective of the dissociation as they can act as doorways for electron transfer (Fig. 2c) discussed in the introduction.
3.3 Metronidazole
The formation of negative ion intermediates of metronidazole during pulsed radiolysis was already identified in the 1970s by the ESR study of Willson.197 The fundamentals of LEE interactions with the molecule isolated in the gas phase and in the microhydrated environment were, however, explored only recently.198 Electron attachment spectroscopy of isolated and microhydrated molecules shows that irrespective of the environment the molecule forms a stable parent anion in agreement with the early pulsed radiolysis studies. Main DEA reaction channels include the release of NO2− and OH−. While the first channel is strongly quenched in the water environment, OH− may be still observed as a minor channel upon hydration.3.4 Nimorazole
Nimorazole interacts with low-energy electrons in two main energy ranges. Strong near 0 eV resonance results in the formation of a stable parent anion. Nimorazole has a large adiabatic as well as vertical electron affinities of 1.31 eV and 0.82 eV, respectively.183 The reported cross section of 3 × 10−18 cm2 indicates S-wave electron attachment. However, the bump at 0.2 eV in the parent anion yield measured in the same work and structure corresponding to the negative electron affinity of 0.4 eV in the electron transmission spectrum in collisions with K+ ions199 indicate that also VFRs are present, in line with the VFR feature in the electron transmission spectrum of imidazole.176 The adiabatic electron affinity is still not enough for imidazole ring opening or significant molecular rearrangement and the anion is primarily stabilized by intramolecular energy redistribution in the gas phase or intermolecular energy transfer in the solvent.76 At higher energies a rich fragmentation pattern can be observed via shape resonances in the 2 eV to 4 eV energy range. The only significant signal is, however, observed for NO2− anion.200The work of Meißner et al.76 is particularly important for demonstrating the caging effect of the environment on the DEA. In microhydrated water clusters, electron attaches to the same resonances of the nimorazole as in the isolated system. However, these resonances are no longer dissociative but lead to the formation of stable parent anions as shown in Fig. 9. With the rising number of water molecules, covering the nimorazole in the neutral precursor cluster, nimorazole parent and heterogeneous nimorazole-water cluster anions were reported for the same electron incident energies as observed for the dissociative shape resonances in the gas phase.
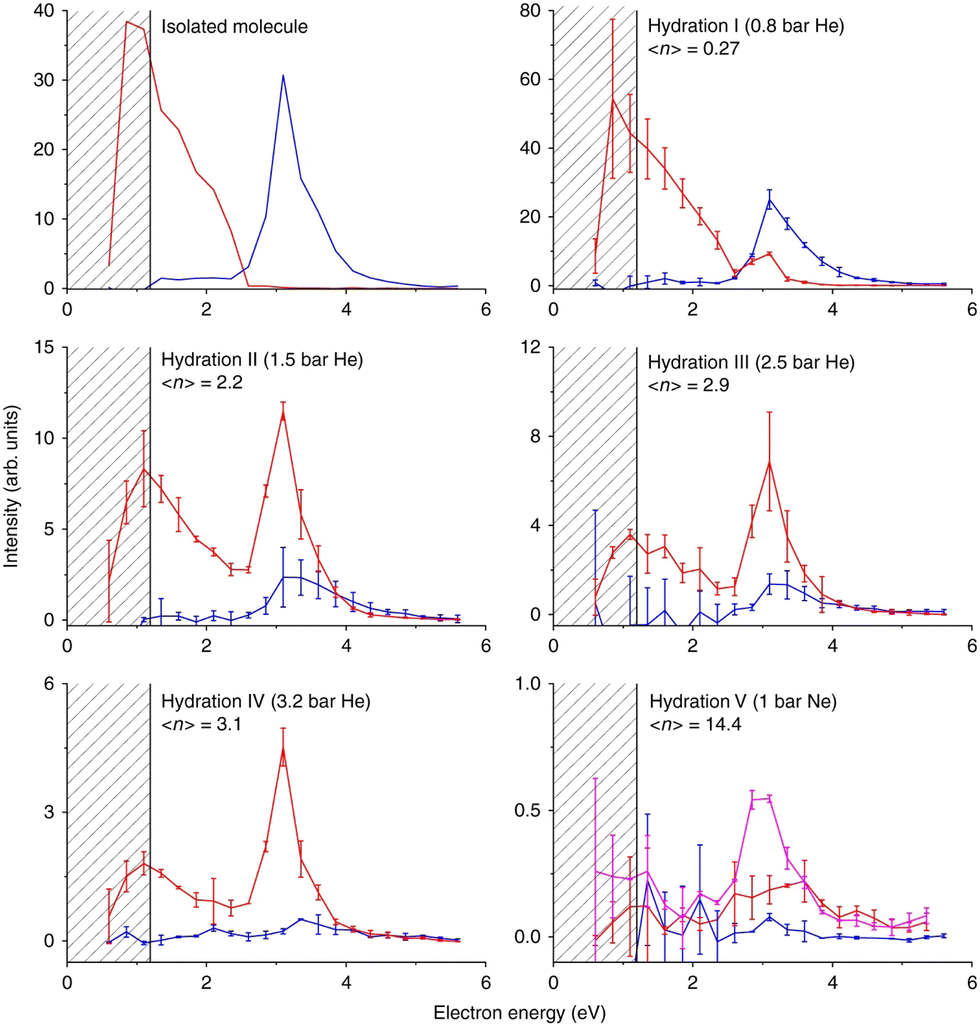 | ||
| Fig. 9 Caging of DEA fragments of nimorazole studied in the molecular beam of microhydrated molecules. Hydration (average number of water molecules attached to nimorazole neutral precursor) rises from top to bottom and from left to right. The red curve represents the ion yield of the parent anion M−, blue curve represents the ion yield of NO2− after the cluster interaction with LEEs at energies shown on the x-axis. Increasing hydration, the peak of NO2− at ∼3 eV disappears, while the M− signal appears at the same energy demonstrating the “caging effect” of the environment on the DEA reaction. A drop of the near 0 eV signal of the parent anion is caused by the fact that at higher hydration, the energy gained in the electron attachment is not enough to evaporate all water molecules from the cluster and M−.(H2O)n cluster anions are produced instead of M−. Reproduced from ref. 76 with permission from Springer Nature, copyright 2019. | ||
The stabilization of the nimorazole parent anion upon single electron reduction was recently confirmed by cyclic voltammetry in bulk water.201 However, also 4 electron reduction was reported resulting in the hydroxylamine derivative formation at specific conditions. Except for the confirmation of the relevance of cluster studies for the LEE-induced chemistry in the bulk, the work also expresses the need for extending the studies with microhydrated molecules to other solvents as well as towards the multiple electron reduction experiments.
3.5 Misonidazole
Pulsed radiolysis studies of misonidazole were performed in 1980's reporting a high rate constant for interaction with solvated electrons 2.5 × 1010 M−1 s−1.202 The products of such interaction as well as the interactions with LEE electrons in the (0–10 eV) can be understood based on the experiments in the gas phase203 and the microhydrated environment.204 Similar to imidazole, two energy ranges for electron attachment have been identified, where the 0 eV attachment leads to the formation of parent anion while 2–4 eV shape resonances lead to the variety of the fragment anions dominated by the NO2 bond cleavage. In contrast to nimorazole, MISO fragmentation upon interaction with LEEs seems not to be fully suppressed by hydration, but OH− channel remains open. This may be caused by the preferred hydration of the MISO over the hydroxyl group and predissociation as supported by theoretical calculations in the work.204 Computational modeling also reveals that TNI fragmentation via OH loss can be accompanied by significant rearrangement of the neutral counterpart forming ring structures and increasing the exothermicity of the process in solution.4 Organometallics
Most of the organometallic cancer chemotherapeutics (Fig. 10) are designed to bind covalently to the DNA or intercalate, resulting in inter- and intrastrand crosslings or secondary structure distortions. The most studied organometallics concerning chemo-radiation therapy are platinum compounds205 due to the initial success of cisplatin (CDDP). CDDP releases Cl atoms in a water environment becoming reactive towards DNA where it forms preferentially guanine adducts206 and intrastrand crosslinks with high susceptibility for further reaction to form DNA–protein207 or guanine–cytosine crosslinks.208 These interactions strongly interfere with the DNA repair mechanisms that are accepted as one of the leading explanations of the CDDP mechanism of action.209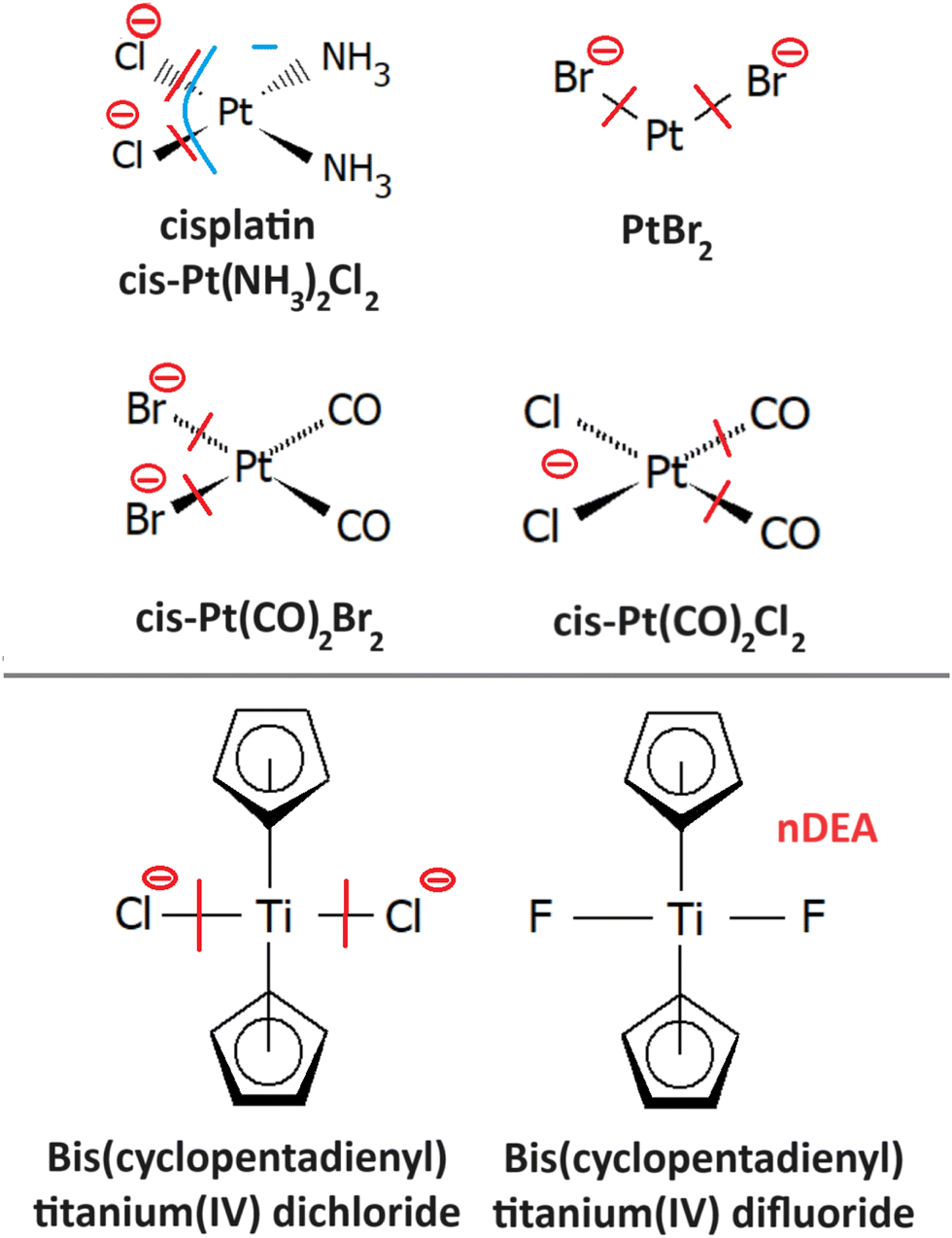 | ||
| Fig. 10 Radiosensitizers from the group of organometallics and model compounds, whose reactivity with LEEs is discussed in the text: cisplatin (4.1), PtBr2 (4.2); PtBr2 (CO)2 (4.3); titanocenes (4.4). The red line marks the bond broken in the most intense DEA reaction of the molecule identified for the isolated molecule, and the blue line the dissociation for a solvated molecule. The minus sign marks the anionic DEA fragment. | ||
Concerning the low-energy electrons, the most interest is in synergism observed for the CDDP and radiation therapy. The synergism can also be caused by the CDDP involvement in the DNA repair pathways in the biological stage of the tissue interaction with ionizing radiation. However, if the synergism occurs already during the physicochemical stage of the interaction, the tiny effects may result in significant radio-sensitization on the biological timescales.3 Therefore, the fundamental reactivity of low-energy electrons towards CDDP and its DNA adducts has been under investigation for more than 15 years. The first suggestions on the LEE involvement in CDDP synergism came from the works of Q. B. Lu and coworkers, demonstrating fast electron transfer from guanine base to CDDP within its single covalently bound DNA adduct. Such transfer can result in Cl− release having two possible sensitizing effects, increasing cross-linking efficiency for the Pt adduct and reactivity of the formed Cl− anion.210 Several follow-up works in the Q. B. Lu group were devoted to these mechanisms including suggestions for novel chemo-radio therapy combinations.210–212 In the L. Sanche group, the direct action of LEEs was investigated mainly in the drug-loaded DNA thin films in the surface experiments with ballistic low-energy electrons in vacuum77 and in the simulated environment.213 However, also a series of studies was performed in realistic conditions of plasmid DNA in buffer solutions, where the electron effects were disentangled by the suppression of the radical chemistry by scavengers. (e.g. ref. 214 and 215).
4.1 Cisplatin
The only gas phase study of electron attachment to isolated cisplatin is that of Kopyra.216 CDDP strongly attaches near 0 eV electrons resulting in a range of products in decreasing probability order Cl−, [Pt(NH3)2Cl]−, [Pt(NH3)2]−, Cl2−, and NH2−. Particularly the fact that a single electron can induce dissociation of both Cl to Pt bonds and form a reactive cross-linking intermediate [Pt(NH3)2]−. The mechanism of multiple bond cleavages can be operative also in cases when CDDP is already bound to the DNA. Additionally, the single dehalogenated fragment [Pt(NH3)2Cl]−, which is formed in the second most intense channel of DEA, is highly reactive towards DNA.217Most of the later studies focus on creating the links between LEE-induced processes and chemo-radiation therapy by studies of cisplatin-modified DNA. For example in the study of Bao et al., five monolayer DNA films were irradiated with and without cisplatin showing approximately doubling of the damage in the 3 eV to 19 eV range. Most important is the observation of a single electron induced double-strand breaks in the incoming electron energy range of 1.6 to 3.6 eV, which was not present in the unmodified DNA.218 The work of Rezaee shifts the LEE damage limit much lower, to 0.5 eV energies causing both single and double-strand breaks and enhancing the formation of DNA lesions.219 In a later work, absolute cross sections for the damages were determined for 5 monolayers of DNA irradiated in vacuum at resonance energies 4.6 eV and 9.6 eV. At 4.6 eV, 244 ± 42 × 10−15 damages per electron and molecule were reported, while at 9.6 eV, 359 ± 44 × 10−15 damages per electron and molecule.220 Clustered damages in DNA were reported in 5.6 eV to 10 eV range, while no clustered lesions are observed at LEE energies below 5 eV.221 This energy dependence, also shown at Fig. 11, demonstrates how important is to know the details of LEE interaction including the initial state of the TNI related to the energy of the incident electron as well as the evolution of the TNI.
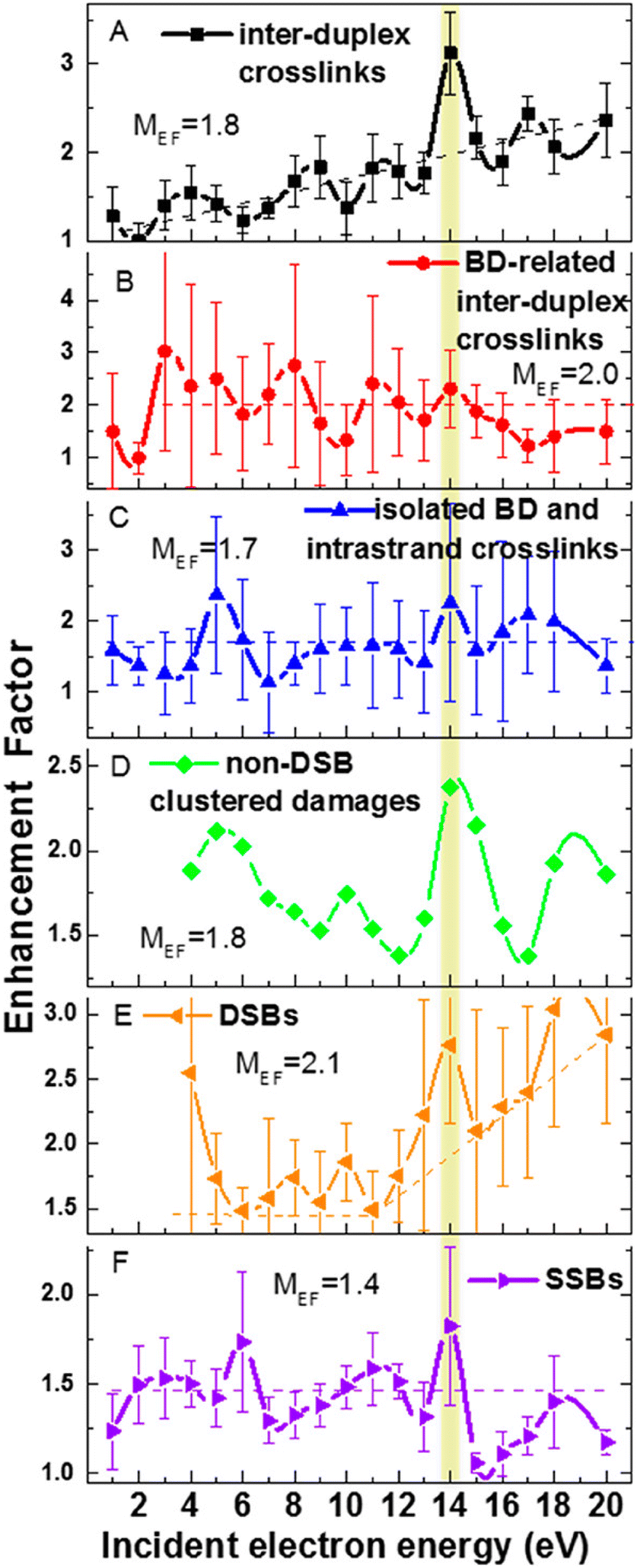 | ||
| Fig. 11 Enhancement factors for different types of DNA damages from the ratios of damages for cisplatin–DNA to those of unmodified DNA as a function of the energy of incident electron reproduced from the work of Dong et al.221 with permission from American Chemical Society, copyright 2020. We can see that cisplatin enhances all types of damages at all studied energies showing that there is a constant enhancement component introduced by cisplatin such as enhancement of LEE scavenging, enhancement of access sites or enhancement of the deposited energy. At the same time, we can see that at some energies the enhancements are more pronounced, demonstrating also resonant – LEE related contribution. | ||
An important set of experiments was performed also on DNA oligomers by Behmand and co-workers. In the first work,222 TTTTTGTGTTT and TTTTTTTGTTT oligomers loaded with CDDP were irradiated by gamma rays in solutions with EDTA, scavenging the OH radicals and enhancing the effects of solvated electrons. The work demonstrated the solvated electron damage at the CDDP site. In the follow-up work,223 HPLC MS was employed to elucidate more details on the mechanism of the damage. The process was described as an initial electron attachment to thymine followed by electron transfer to CDDP and base or cisplatin release. The same approach was used also in the third work on the topic,224 studying shorter GTG sequence demonstrating the catalytic effect of cisplatin on the reactivity with solvated electrons. CDDP presence increased the rate constant for interaction with solvated electrons from (7.4 × 109 mol−1 L s−1) for bare GTG oligomer to (2.23 × 1010 mol−1 L s−1) for GTG-CDDP complex. Similarly, a strong enhancement was observed also for double-stranded oligomers showing an increase of the reaction rate from 2 × 109 mol−1 L s−1 to 7 × 109 mol−1 L s−1. Additionally, the transformations of intra- to interstrand crosslinks were identified for CDDP-modified strands using gel electrophoresis.225
Despite a high number of studies in solution, the gas phase studies with Pt-based organometallics are scarce. The reason behind this is the weight of the Pt atom and the low decomposition temperature of organic ligands. If the molecule is not directly designed for sublimation, there is a high probability that it will thermally decompose before sublimation. Therefore, the gas phase experiments with these compounds are very complicated and as most of the experiments focus on the anion products, it is also hard to identify, if the attachment occurred to the parent anion or a part of the molecule formed during thermal decomposition. This requires either detailed studies of temperature dependencies or detection of neutral by-products as already mentioned several times, ideally in the coincidence regime. Few other organometallic compounds were therefore studied to better understand the chemistry of CDDP as well as the details of the chemo-radiation synergism. The examples are.
4.2 PtBr2
Radiosensitizing potential of this compound was identified in the work of Śmiałek et al.,226 by evaluating damage enhancement in plasmid DNA upon X-ray irradiation. The DEA reaction results exclusively in the formation of Br− anions.227 Three resonances were observed in the spectrum. The most intense signal at 0.4 eV was assigned to the impurity of HBr as a possible product of PtBr2 reactions with residual water in the inlet system since the strong temperature dependence of its signal did not correlate with the other observed features in the spectrum. It is worth noting that such behavior of near 0 eV cross section is not uncommon,228 even though the impurities are a significant issue in the electron attachment spectroscopy of halocarbons.229 Based on DFT calculations of reaction enthalpies, the signal peaking at 1.2 eV and 7 eV was assigned to DEA reactions leading to PtBr and Pt+Br neutral co-products.4.3 Pt(CO)2Br2,Pt(CO)2Cl2
Modification of organometallics by carbonyl groups represents a common way of increasing sublimation efficiency, mainly for applications in charged particle beam-induced deposition. The present study demonstrates that such modification can significantly influence also the DEA dynamics. For previously discussed PtBr2 and CDDP, the primary DEA channel is leading to the formation of halogen anion. Both carbonyls Pt(CO)2Br2230 and Pt(CO)2Cl2231 lose primary the CO ligands, despite the exothermicity of halogen anion formation channel. The provided explanation is based on the different orbitals available for electron attachment. While the LUMO of CDDP or PtBr2 has σ* antibonding character LUMO of the carbonyl compounds have π* character that additionally do not allow energy flow to the Pt-halogen vibration due to the symmetry restrictions. CO ligands can provide an effective energy sink via C–O stretches as shown for Fe(CO)5, prolonging TNI lifetime over several vibrational periods. As a result the PtBr2 and CDDP fastly dissociate in a direct non-ergodic process, while the dissociation of carbonyl molecules will be probably more ergodic. It would be interesting to theoretically explore the interplay using time-dependent molecular dynamics approaches.These studies of seemingly unrelated compounds bearing carbonyl ligands provide an important insight into the fundamental mechanisms of CDDP dissociation with possible consequences for the design of CDDP analogs. Most of the CDDP successor compounds suggested so far are complex molecules, where the direct dissociation upon single electron reduction can be quenched by energy dissipation into internal degrees of freedom. In other words, a significant portion of energy available upon single electron reduction is consumed by heating of the molecule rather than its fragmentation. Pt molecules, where such energy dissipation is minimized and direct bond cleavage is possible may be promising radiosensitizers. Indeed the high radiosensitizing potential of PtBr2 reported by Śmiałek and co-workers226 supports this hypothesis.
4.4 Bis(pentamethylcyclopentadienyl) titanium(IV) dichloride and difluoride
The idea behind the study232 is to separate LEE processes from ionization processes in realistic conditions by designing selective probes for such processes. Out of the two molecules, the dichloride efficiently dissociates by the release of Cl− while the difluoride anion signal upon electron attachment is dominated by the parent anion. At the same time, the ionization cross section is controlled by huge pentamethylcyclopentadienyl rings and it is similar for both molecules. Upon distribution into the environment, Cl− formation can be used as a selective probe for electron attachment and therefore contribute to the evaluation of absolute numbers for the reactions of secondary LEEs in radiation chemistry and chemo-radiation synergism.5 Other small molecules
LEE reactions of several other small molecule radiosnesitizers were explored, such as the ones shown in Fig. 12.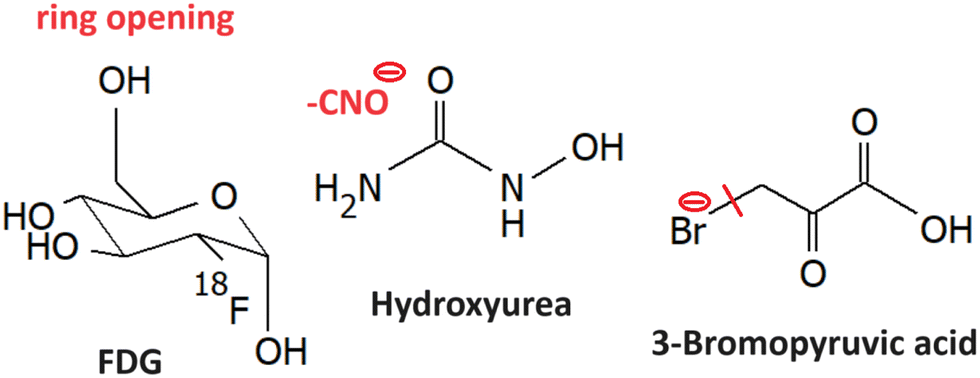 | ||
| Fig. 12 Other small molecule radiosensitizers discussed in the text: fluorodeoxyglucose (5.1), hydroxyurea2 (5.2) and 3-bromopyruvic acid (5.3). The red line marks the bond broken in the most intense DEA reaction of the molecule identified for the isolated molecule, and the blue line the dissociation for a solvated molecule. The minus sign marks the anionic DEA fragment. | ||
5.1 Fluorodeoxyglucose
Fluorodeoxyglucose, which is commonly used in tumor imaging in its F18 form is an example of a sugar with high reactivity towards LEEs. Electrons lower than 2 eV can cause the decomposition of FDG and it leads to 7 anions out of them, C3H3O2− has the highest yield. Dissociation of HF is less energetic demanding than dissociation of F−. Therefore, F− is forming only a minor DEA channel. However, the formed TNIs can decompose via a large number of dissociating channels including ring opening and reactions leading to the formation of neutral molecules, such as H2 O, H2, HF.2335.2 Hydroxyurea
Upon administration in living organisms, hydroxyurea efficiently creates nitric oxide in a set of one to three electron redox reactions.234 Nitric oxide inhibits ribonucleotide reductase enzyme, as discussed for nitroimidazoles, which is believed to be the primary mechanism of action.235 However, other reactive species can also be formed such as hydroxylamine or hydrogen peroxide, which are genotoxic.236 The need to understand multiple electron reduction as well as the involvement of electron affinic radiosensitizers in DNA repair was already mentioned in several places within the review.Hydroxyurea has a large dipole and therefore it can attach electrons via VFRs.237 However, the parent anion is unstable and fragments due to the high electron affinity of its components. The most intense anion reported is NCO−. The electron attachment spectrum shows three peaks at 0 eV, 0.1 eV, and 0.4 eV, which is not typical for the vibrational structure that should be in this region distributed approximately equidistantly. On the other side, Therefore an explanation for the structure was based on the possible attachment of the electron into three different conformers of the molecule that could be present in the gas phase. The process discussed e.g. for amino acid serine.238 Other fragments are produced via shape resonance peaking at ∼2 eV ((M–H)−, (M–OH)−, (M–H2O)−,CN−) and core excited resonance ∼6 eV ((M–H2O)−, NH2−, OH−, CN−). No hydroxylamine was observed upon LEE. It wil be definitely interesting to explore how hydration changes the hydroxyurea reaction dynamics.
5.3 3-Bromopyruvic acid
3-Bromopyruvic acid is similar to the discussed nitroimidazoles active in hypoxic cells. The believed mechanism is via inhibition of glycolytic pathway enzymes.239 These are more pronounced in tumors due to hypoxia. Under normoxia, the energy is gained via ATP phosphorylation. It is worth mentioning that electron transfer reactions play an important role in both hypoxic and normoxic glycolysis,240 which may have consequences for the secondary LEE effects on the cell viability. Electron attachment 3Br pyruvic acid is dominated by strong Br− ion yield at near 0 eV energies,241 which corresponds to the s-wave attachment and exothermic dissociation (ref. (Fig. 1a)). This is in strong contrast to pyruvic acid, where VFRs are observed with only a short lifetime with respect to autodetachment and therefore they are detected only upon anion stabilization in a three-body process.195 This strong enhancement of the electron attachment cross-section by Br substituent was proposed as one of the possible mechanisms of radiosensitization by 3-bromopyruvic acid.Another important observation is a strong shift in the electronic excited states of the anion due to the presence of bromine. While in pyruvic acid, the core excited resonances peak above ∼6 eV, in 3-bromopyruvic acid it is at under 5 eV.
6 Conclusions and perspective
A large number of radiosensitizing molecules has been studied concerning LEE interactions during recent years. Studies for DNA base components, nitroimidazoles, and organometallics already demonstrated the variety of interaction regimes with LEEs and suggested some important mechanisms that can contribute to the high efficiency of these compounds in concomitant chemo-radiotherapy. While DNA bases or platinum compounds decompose exothermically in processes that often cannot be prevented by energy transfer to the environment, nitro-azole decomposition is more energetically neutral and their anions often stabilize in the environment. DEA therefore does not provide the only explanation of the fundamental radiosensitizing mechanism by electron affinic radiosensitizers. The overview of so far suggested mechanisms is in Fig. 13 To provide real societal impact, these mechanisms need to be evaluated on more complex biophysical models.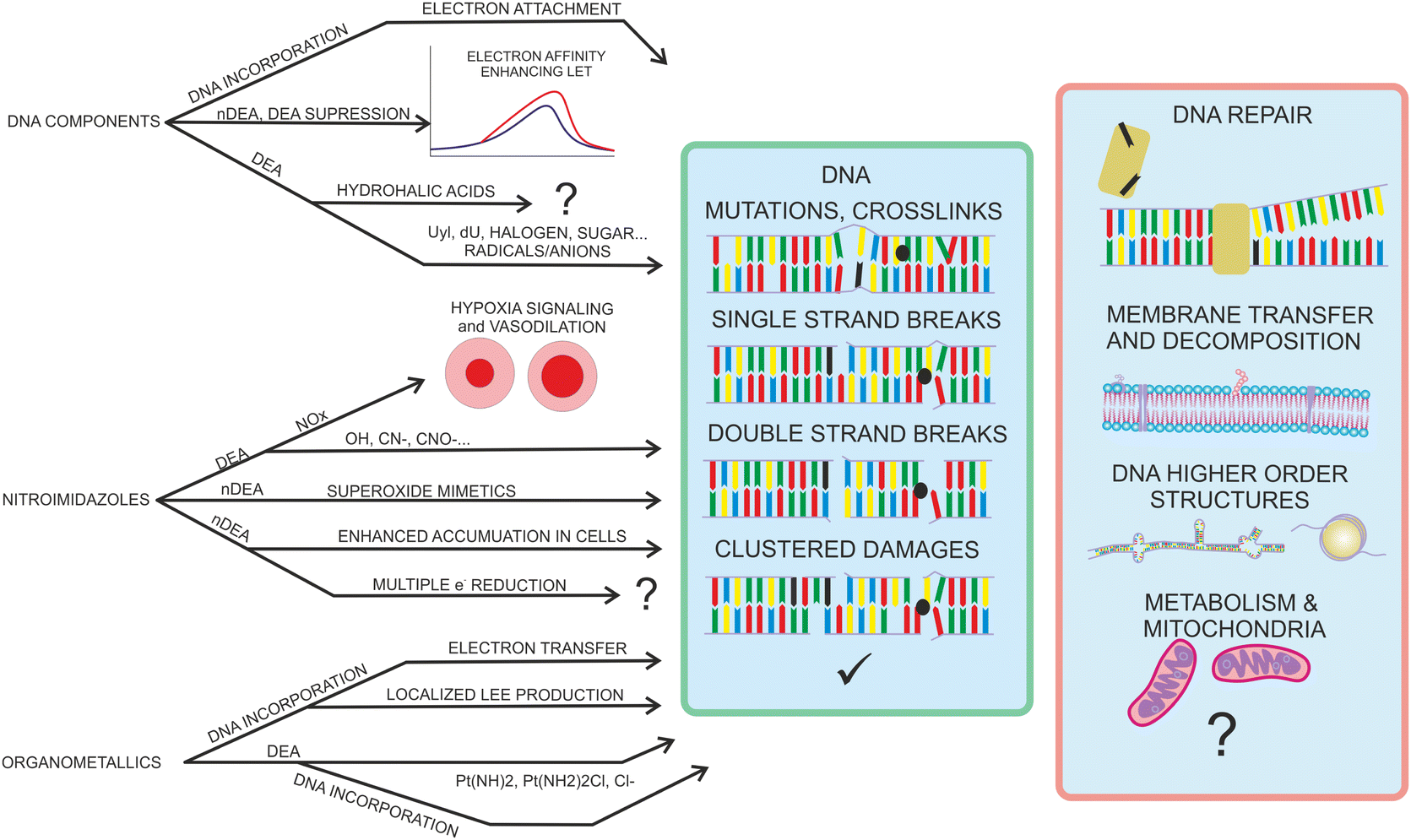 | ||
| Fig. 13 Different mechanisms proposed for radiosensitising and synergistic effects of LEEs with derivatives of DNA components, nitro azoles and organometallics. So far the research has focused on direct DNA sensitization towards the LEEs by enhancement of base excision, mutations, crosslinks, single and double-strand breaks, or clustered damages as depicted in the central green box of the image. The combined action of LEEs and the radiosensitizers on the other cellular mechanisms is practically unexplored. LEE based radiosnesitizers can form negative parent ions or reactive species that can influence repair enzymes, electron transfer chains through cellular membranes, interact with membrane lipids and proteins, modify the higher order structure of DNA or enter into mitochondrial and other cell metabolism processes. Exploration of these processes will require further extension of molecular physics experiments towards biochemistry and biophysics. | ||
For example, in the molecular physics community, enormous focus is given to DNA interactions and damage. This is natural since DNA damage is the most lethal form of cellular damage. On the other hand, life evolved in an environment with a relatively high radiation background, and therefore, various mechanisms exist in living organisms to mitigate radiation damage to DNA.242 Instead of focusing on DNA damage, as something expected by the organism, we should focus on the less expected changes in the cellular environment. When exploring the effects of the three types of molecules reviewed here, each of them is actively influencing the DNA repair processes. The role of LEEs in the repair process is practically unexplored. For that, a crucial point will be to involve the repair enzymes in more complex studies in a realistic environment. On the level of DNA in bulk solution, one can use time-resolved absorption spectroscopy243 or state-of-the-art X-ray crystallography.244,245 Another approach is to design complex architectures for single-molecule experiments.246 One such architecture, the DNA origami frame developed by Endo et al. was already demonstrated to be suitable for enzyme interaction studies.(e.g. ref. 247).
Further directions of complex biological pathways that may be explored concerning the evolution of the negative ions and radicals produced by secondary LEEs on chemical and biological timescales of radiation interaction with radiosensitizing compounds are shown in the red box of the Fig. 13. Except for the mentioned DNA repair processes these include effects on the cellular membrane or metabolism that are practically unexplored and will require in vitro biophysical and biochemistry studies.248 Another direction is the higher-order structure of DNA. In this direction, the telomeric sequences are in the focus at the moment.249,250
The suggested studies of biological consequences can provide guidelines for selecting relevant targets for LEE interactions on the molecular level. Only systematic experimental studies on the molecular level, which can be well complemented by computational modeling, can provide the level of understanding required for reaching the ultimate objective of all these efforts – providing the base for the rational design of novel chemo-radiotherapy agents. The basic needs that we identified are visualized in Fig. 14 and in this section, we will describe them in more detail.
 | ||
| Fig. 14 Suggested experimental research directions for exploration of LEE interactions with model radiosensitizers. Interplay between the needs and current state of the art can be visualized by a pyramid, where the importance decreases from the top to the bottom of the pyramid, while the present number of the studies is represented by the area of the corresponding level of the pyramid. | ||
6.1 Multiple electron reduction
Studies with isolated molecules or clusters are performed for singly charged species. Complex molecules can undergo multiple electron reductions, particularly in an environment, where the singly charged TNI can be stabilized by energy transfer to solvent. Multiply-charged anions in the gas phase attracted significant interest in 1990s and 2000s.251–254 Their preparation using the electrospray or sputtering technique is straightforward and state of the art time resolved photoelectron spectroscopy can already reveal intringuing details of state to state, oxidation or dissociation dynamics of these species.255–257Here we suggest that in addition to studies of these stable species, the field will profit from studies mimicking the multiple electron reduction and therefore performing step-by-step electron attachment, ideally in a time-resolved manner. While some information about the process can be revealed from voltametry201 the details of the dynamics can be revealed only by performing step-by-step attachment of electrons, similar to the pioneering experiment with C84 of Compton and coworkers.258
6.2 Absolute numbers for LEE formation and reaction rates
The number of works studying absolute numbers for LEE-induced reactions in complex biomolecular systems is low (see e.g. ref. 259) and the number of studies including radiosensitizers is even lower. Examples include surface experiments on DNA origami templates in vacuum,85,260,261 in controlled environment,213,220 or bulk.225 While the measurements of absolute cross sections for LEE interactions of isolated molecules is straightforward, the number of parameters influencing the reactivity in the more complex environment is high and needs to be carefully analysed.262The most promising approach seems to be pulsed radiolysis, where, however, the LEE reactions are generally induced by high-energy radiation. The disentangling of LEE contribution is not straightforward. Either the laser-induced electron transfer has to be used as in the seminal works of Q. B. Lu, or the environment has to be precisely controlled by modifying electron scavenger (such as oxygen), or radical scavenger (such as tris) content. (e.g. ref. 134, 222, 263 and 264). However, the experiments must be perfromed with care about the the LEE-induced chemistry of buffer components to avoid misinterpretations.101,265,266
Another possible approach can be quantitative electron-induced fluorescence.267 A properly designed fluorescent probe requiring singlet to triplet excitation by LEEs to induce fluorescence can be irradiated by LEEs in realistic environments e.g. in liquid jets.265,268
Two approaches to LEE-induced processes come from the field of aerosol research. In the first approach, a photoelectron is produced inside homogeneous or heterogeneous aerosol by a tunable wavelength light source and escapes the aerosol.269,270 Combination of this approach with biomolecular doping and advanced anion detection techniques may provide new insight into transient anion formation as well as dynamics. The second approach already significantly contributed to establish the links between the radiation chemistry in bulk and the formation of transient negative ions. In the experiment, a high-energy ionizing particle interacts with aerosols containing molecules of interest, and negative ions are analyzed using mass spectrometry.271 The technique was already applied to several biomolecular targets.272–274 Adaptation of these techniques for absolute data measurements would be beneficial.
The methods to prepare and study LEE-induced processes at biologically relevant conditions are the subject of recent reviews of Gao and co-workers275 or that of Alizadeh and Ptasinska.276
6.3 Environmental effects
The reviewed experiments are on a limited set of molecular systems that cannot reproduce the complex radiation chemistry in biological buffers. We need to experimentally explore a much larger amount of solvent, counter ions, or cellular components, such as DNA-peptide or lipid TNI formation, and their dynamics in various solvents.There is an ongoing discussion about shifts of the electron attachment resonances in the presence of a solvent.277–279 For some systems, the shifts can be identified using photoelectron spectroscopy.280,281 However, as reviewed here for radiosensitizers, the anions often undergo DEA, TNIs are pre-dissociated or they undergo isomerizations. In such cases, the stable anion states, which can be used for photoelectron spectroscopy, do not correspond to that of the anion forming in the vertical attachment process. For some systems, this can be solved by electron transfer photoelectron spectroscopy.31 However, the most straightforward approach to probe the energetics of the initial electron attachment states will be a combination of the state-of-the-art sources of neutral cluster beams with electron sources having high energy resolution.
Another important issue is the stability of the molecular anions in the solvent. The cluster experiments, with relatively large water clusters, demonstrated that the initial resonant states correspond to that of individual solvated molecules rather than to the electron attachment to the water cluster via polarization interaction (e.g.Fig. 9).76 However, there exists a theoretical prediction that in some systems the attachment or transfer of the electron to the solvent282–284 may be very effective. Except for the energy sink, the environment can provide an electron sink. In cluster beam experiments, we can probably explore the limits of the cluster size, at which the interaction with solvated molecules is completely shielded. Such information can be compared with the minimal molecular concentrations required to observe LEE effects in realistic environments. Important is to understand that while electron affinity is a characteristic of a single molecule, the potential well that traps the solvated electron requires the rearrangement of several molecules, and therefore even the adiabatically exothermic processes are often not barrierless. Anion lifetime and charge transfer to solvent studies using state of the art spectroscopy in gas phase or bulk can provide better insight into this interesting topic.
6.4 Neutral detection
The need for the studies of LEE processes in biologically relevant molecules was already well defined by Ptasinska et al.285 The detection of neutrals is important for the process of neutral dissociation upon electron impact as well as for dissociative electron attachment.1 In the first case, various techniques were developed historically from electron energy loss-based approaches used mainly for diatomics to electron-induced fluorescence measurements performing well at higher energies, where electronically excited fragments are forming.286–288 Interesting can be a combination of the fluorescence technique with coincidence measurement of the outgoing electron, since the outgoing electron will provide information about the initial dissociative state while photons the final state of the dissociated fragment.In the case of DEA process, except for the detection of neutral itself, as performed in the current state-of-the-art experiments,289–291 it will be beneficial to have the option to perform a detailed analysis of the neutral products. Only in such a way the complex rearrangement (e.g. ref. 204 or neutral molecule117) formation mechanisms could be unambiguously identified. Considering the low densities of the products formed upon DEA, this will probably require sensitive laser based techniques such as the resonance-enhanced multiphoton ionization, as employed e.g. in the electron-induced desorption studies of Lane and Orlando.292
6.5 DEA
The number of DEA studies with radiosensitizers is significant and some general trends in the dynamics were already overviewed in the present perspective. However, we are still missing systematic studies on larger sets of molecules.Particularly interesting could be studies of azole derivatives. While the field focuses on imidazoles, there are also other molecules with azole ring having radiosensitizing potential.293 They form specific stacking interactions and they are intensely used in the drug design due to their biomimetic nature as well as because azoles are the product of several click chemistry reactions294 widely used in the modern drug design.295 Except for the better understanding of the radiosensitizing properties and possibly giving guidelines for the rational design of chemo-radiotherapeutics, the ring opening attachment can provide a way for selective modification of bonds created by click chemistry.
Another possible field of interest are organometallic complexes. Practically, the only thoroughly explored molecule is CDDP, few attempts for resembling LEE processes in solution exist for the CDDP analogs.77,213,296 However, there are many other organometallic molecules with promising radiotherapy applications,297 particularly these are ruthenium arene complexes.263,298
The suggestions described in this part can be summarized in three directions. The first is the absolute numbers for the LEE-induced reactions that will allow unambiguous identification of the most relevant processes in chemo-radiation synergism. The second is the evolution of the TNI to understand not only the energetics of the processes but also the final products of LEE interaction in realistic environments upon electron transfer and multiple electron reduction. The third direction should focus on establishing more rigid connections between the initial LEE-induced processes occurring in the physico-chemical stage of ionizing radiation interaction and their biological consequences. For that, more active collaborations between physical chemists, biophysicists, and/or biochemists will be required.
Conflicts of interest
In accordance with our policy on. There are no conflicts to declare.Acknowledgements
We would like to thank Juraj Fedor and Leo Sala for reading the manuscript and providing valuable feedback. The work was supported by Czech Science Foundation project number 21-26601X (EXPRO) and COST Action CA18212 – Molecular Dynamics in the GAS phase (MD-GAS), supported by COST (European Cooperation in Science and Technology).Notes and references
- J. Bednar, Theoretical foundations of radiation chemistry, 1990 Search PubMed.
- E. Alizadeh and L. Sanche, Chem. Rev., 2012, 112, 5578–5602 CrossRef CAS PubMed.
- R. Schurmann, S. Vogel, K. Ebel and I. Bald, Chem. – Eur. J., 2018, 24, 10271–10279 CrossRef PubMed.
- C. Szmytkowski and P. Możejko, Eur. Phys. J. D, 2020, 74, 90 CrossRef CAS.
- R. D. White, D. Cocks, G. Boyle, M. Casey, N. Garland, D. Konovalov, B. Philippa, P. Stokes, J. de Urquijo, O. González-Magaña, R. P. McEachran, S. J. Buckman, M. J. Brunger, G. Garcia, S. Dujko and Z. L. Petrovic, Plasma Sources Sci. Technol., 2018, 27, 053001 CrossRef.
- H. Nikjoo, D. Emfietzoglou, T. Liamsuwan, R. Taleei, D. Liljequist and S. Uehara, Rep. Prog. Phys., 2016, 79, 116601 CrossRef CAS PubMed.
- S. A. Zein, M.-C. Bordage, Z. Francis, G. Macetti, A. Genoni, C. Dal Cappello, W.-G. Shin and S. Incerti, Nucl. Instrum. Methods Phys. Res., Sect. B, 2021, 488, 70–82 CrossRef CAS.
- T. Kai, A. Yokoya, M. Ukai, K. Fujii, T. Toigawa and R. Watanabe, Phys. Chem. Chem. Phys., 2018, 20, 2838–2844 RSC.
- S. M. Pimblott and J. A. LaVerne, Radiat. Phys. Chem., 2007, 76, 1244–1247 CrossRef CAS.
- T. P. R. Kumar, J. Kočišek, K. Bravaya and J. Fedor, Phys. Chem. Chem. Phys., 2020, 22, 518–524 RSC.
- D. Davis, V. P. Vysotskiy, Y. Sajeev and L. S. Cederbaum, Angew. Chem., Int. Ed., 2011, 50, 4119–4122 CrossRef CAS PubMed.
- D. Davis, V. P. Vysotskiy, Y. Sajeev and L. S. Cederbaum, Angew. Chem., Int. Ed., 2012, 51, 8003–8007 CrossRef CAS PubMed.
- C. S. Anstöter, G. Mensa-Bonsu, P. Nag, M. C. V. Ranković, T. P. R. Kumar, A. N. Boichenko, A. V. Bochenkova, J. Fedor and J. R. R. Verlet, Phys. Rev. Lett., 2020, 124, 203401 CrossRef PubMed.
- J. Med, t Sršeň, P. Slavíček, A. Domaracka, S. Indrajith, P. Rousseau, M. Fárník, J. Fedor and J. Kočišek, J. Phys. Chem. Lett., 2020, 11, 2482–2489 CrossRef CAS.
- T. Sommerfeld and M. C. Davis, J. Chem. Phys., 2018, 149, 084305 CrossRef PubMed.
- T. F. M. Luxford, J. Fedor and J. Kočišek, J. Chem. Phys., 2021, 154, 214303 CrossRef CAS PubMed.
- J. Forer, V. Kokoouline and T. Stoecklin, Phys. Rev. A, 2023, 107, 043117 CrossRef CAS.
- V. Chandrasekaran, A. Prabhakaran, B. Kafle, H. Rubinstein, O. Heber, M. Rappaport, Y. Toker and D. Zajfman, J. Chem. Phys., 2017, 146, 094302 CrossRef.
- I. I. Fabrikant and H. Hotop, Phys. Rev. A: At., Mol., Opt. Phys., 2001, 63, 022706 CrossRef.
- K. D. Jordan and F. Wang, Ann. Rev. Phys. Chem., 2003, 54, 367–396 CrossRef CAS PubMed.
- I. I. Fabrikant, H. Hotop and M. Allan, Phys. Rev. A: At., Mol., Opt. Phys., 2005, 71, 022712 CrossRef.
- M. Allan, M. Lacko, P. Papp, Š. Matejčík, M. Zlatar, I. I. Fabrikant, J. Kočišek and J. Fedor, Phys. Chem. Chem. Phys., 2018, 20, 11692–11701 RSC.
- Y.-R. Zhang, D.-F. Yuan, C.-H. Qian, G.-Z. Zhu and L.-S. Wang, J. Am. Chem. Soc., 2023, 145, 14952–14962 CrossRef CAS PubMed.
- D.-F. Yuan, Y. Liu, Y.-R. Zhang and L.-S. Wang, J. Am. Chem. Soc., 2023, 145, 5512–5522 CrossRef CAS PubMed.
- C.-H. Qian, G.-Z. Zhu and L.-S. Wang, J. Phys. Chem. Lett., 2019, 10, 6472–6477 CrossRef CAS PubMed.
- Y.-R. Zhang, D.-F. Yuan and L.-S. Wang, J. Phys. Chem. Lett., 2022, 13, 2124–2129 CrossRef CAS PubMed.
- C. Desfrançois, H. Abdoul-Carime and J.-P. Schermann, Int. J. Mod. Phys. B, 1996, 10, 1339–1395 CrossRef.
- D. H. Kang, J. Kim, H. J. Eun and S. K. Kim, J. Am. Chem. Soc., 2022, 144, 16077–16085 CrossRef CAS PubMed.
- T. Sommerfeld, J. Phys. Chem. A, 2004, 108, 9150–9154 CrossRef CAS.
- J. H. Hendricks, S. A. Lyapustina, H. L. de Clercq, J. T. Snodgrass and K. H. Bowen, J. Chem. Phys., 1996, 104, 7788–7791 CrossRef CAS.
- A. Kunin and D. M. Neumark, Phys. Chem. Chem. Phys., 2019, 21, 7239–7255 RSC.
- X. Li, K. H. Bowen, M. Haranczyk, R. A. Bachorz, K. Mazurkiewicz, J. Rak and M. Gutowski, J. Chem. Phys., 2007, 127, 174309 CrossRef PubMed.
- D. Chen and G. A. Gallup, J. Chem. Phys., 1990, 93, 8893–8901 CrossRef CAS.
- G. J. Schulz, Rev. Mod. Phys., 1973, 45, 423–486 CrossRef CAS.
- J. N. Bardsley, A. Herzenberg and F. Mandl, Proc. Phys. Soc., 1966, 89, 321 CrossRef CAS.
- T. P. Ragesh Kumar, P. Nag, M. Ranković, T. F. M. Luxford, J. Kočišek, Z. Mašín and J. Fedor, J. Phys. Chem. Lett., 2022, 13, 11136–11142 CrossRef CAS PubMed.
- E. M. de Oliveira, M. A. P. Lima, M. H. F. Bettega, S. D. Sanchez, R. F. da Costa and M. T. D. N. Varella, J. Chem. Phys., 2010, 132, 204301 CrossRef PubMed.
- M. Zawadzki, M. Čížek, K. Houfek, R. Čurík, M. Ferus, S. Civiš, J. Kočišek and J. Fedor, Phys. Rev. Lett., 2018, 121, 143402 CrossRef CAS PubMed.
- P. Nag, M. Tarana and J. Fedor, Phys. Rev. A, 2021, 103, 032830 CrossRef CAS.
- I. I. Fabrikant, S. Eden, N. J. Mason and J. Fedor, Recent Progress in Dissociative Electron Attachment: From Diatomics to Biomolecules, Academic Press, 2017, vol. 66, pp. 545–657 Search PubMed.
- S. A. Pshenichnyuk, N. L. Asfandiarov, A. S. Vorobev and Š. Matejčík, Physics-Uspekhi, 2022, 65, 163 CrossRef CAS.
- O. Ingolfsson, Low-Energy Electrons Fundamentals and Applications, Taylor and Francis, 2019, 434 Search PubMed.
- L. Christophorou and J. Olthoff, Fundamental Electron Interactions with Plasma Processing Gases, Fundamental Electron Interactions with Plasma Processing Gases, 2003 Search PubMed.
- E. Illenberger and J. Momigny, Electron Attachment Spectroscopy, Steinkopff, Heidelberg, 1992, pp. 264–298 Search PubMed.
- J. M. Herbert and M. P. Coons, Ann. Rev. Phys. Chem., 2017, 68, 447–472 CrossRef CAS PubMed.
- A. E. Bragg, J. R. R. Verlet, A. Kammrath, O. Cheshnovsky and D. M. Neumark, Science, 2004, 306, 669–671 CrossRef CAS PubMed.
- V. Svoboda, R. Michiels, A. C. LaForge, J. Med, F. Stienkemeier, P. Slavíček and H. J. Wörner, Sci. Adv., 2020, 6, eaaz0385 CrossRef CAS PubMed.
- F. Novelli, K. Chen, A. Buchmann, T. Ockelmann, C. Hoberg, T. Head-Gordon and M. Havenith, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2216480120 CrossRef CAS PubMed.
- J. Belloni, M. Mostafavi, T. Douki and M. Spotheim-Maurizot, Radiation chemistry from basics to applications in material and life sciences, Editions EDP Sciences, Les Ulis (France), France, 2008 Search PubMed.
- Q.-B. Lu, Mutat. Res., 2010, 704, 190–199 CAS.
- H. Zettergren, A. Domaracka, T. Schlathölter, P. Bolognesi, S. Díaz-Tendero, M. Abuda, S. Tosic, S. Maclot, P. Johnsson, A. Steber, D. Tikhonov, M. C. Castrovilli, L. Avaldi, S. Bari, A. R. Milosavljević, A. Palacios, S. Faraji, D. G. Piekarski, P. Rousseau, D. Ascenzi, C. Romanzin, E. Erdmann, M. Alcamí, J. Kopyra, P. Limão-Vieira, J. Kočišek, J. Fedor, S. Albertini, M. Gatchell, H. Cederquist, H. T. Schmidt, E. Gruber, L. H. Andersen, O. Heber, Y. Toker, K. Hansen, J. A. Noble, C. Jouvet, C. Kjær, S. B. Nielsen, E. Carrascosa, J. Bull, A. Candian and A. Petrignani, Eur. Phys. J. D, 2021, 75, 152 CrossRef CAS.
- J. Poštulka, P. Slavíček, J. Fedor, M. Fárník and J. Kočišek, J. Phys. Chem. B, 2017, 121, 8965–8974 CrossRef PubMed.
- J. Kočišek, A. Pysanenko, M. Fárník and J. Fedor, J. Phys. Chem. Lett., 2016, 7, 3401–3405 CrossRef PubMed.
- M. McAllister, N. Kazemigazestane, L. T. Henry, B. Gu, I. Fabrikant, G. A. Tribello and J. Kohanoff, J. Phys. Chem. B, 2019, 123, 1537–1544 CrossRef CAS PubMed.
- J. Schiedt, R. Weinkauf, D. Neumark and E. Schlag, Chem. Phys., 1998, 239, 511–524 CrossRef CAS.
- J. H. Hendricks, S. A. Lyapustina, H. L. de Clercq and K. H. Bowen, J. Chem. Phys., 1998, 108, 8–11 CrossRef CAS.
- L. Sala, B. Sedmidubská, I. Vinklárek, M. Fárník, R. Schürmann, I. Bald, J. Med, P. Slavicek and J. Kocisek, Phys. Chem. Chem. Phys., 2021, 23, 18173–18181 RSC.
- M. J. Brunger, Int. Rev. Phys. Chem., 2017, 36, 333–376 Search PubMed.
- P. Limão-Vieira, F. F. da Silva and G. G. Gómez-Tejedor, in Electron Transfer-Induced Fragmentation in (Bio)Molecules by Atom-Molecule Collisions, ed. G. García Gómez-Tejedor and M. C. Fuss, Springer Netherlands, Dordrecht, 2012, pp. 59–70 Search PubMed.
- A. M. Stanislav, A. Pshenichnyuk and A. S. Komolov, Int. Rev. Phys. Chem., 2018, 37, 125–170 Search PubMed.
- P. M. Cullis, G. D. D. Jones, M. C. R. Symons and J. S. Lea, Nature, 1987, 330, 773–774 CrossRef CAS PubMed.
- J. Simons, Acc. Chem. Res., 2006, 39, 772–779 CrossRef CAS PubMed.
- A. Kumar, D. Becker, A. Adhikary and M. D. Sevilla, Int. J. Mol. Sci., 2019, 20, 3998 CrossRef CAS PubMed.
- S. Ptasińska, S. Denifl, B. Mróz, M. Probst, V. Grill, E. Illenberger, P. Scheier and T. D. Märk, J. Chem. Phys., 2005, 123, 124302 CrossRef PubMed.
- M. Orzol, I. Martin, J. Kocisek, I. Dabkowska, J. Langer and E. Illenberger, Phys. Chem. Chem. Phys., 2007, 9, 3424–3431 RSC.
- D. S. Slaughter, T. Weber, A. Belkacem, C. S. Trevisan, R. R. Lucchese, C. W. McCurdy and T. N. Rescigno, Phys. Chem. Chem. Phys., 2020, 22, 13893–13902 RSC.
- V. Tadsare, S. Das, S. Gokhale, E. Krishnakumar and V. S. S. Prabhudesai, Atoms, 2022, 10, 98 CrossRef CAS.
- O. Ingólfsson, F. Weik and E. Illenberger, Int. J. Mass Spectrom. Ion Processes, 1996, 155, 1–68 CrossRef.
- H. Hotop, M.-W. Ruf, M. Allan and I. Fabrikant, Resonance and threshold phenomena in low-energy electron collisions with molecules and clusters, Academic Press, 2003, vol. 49, pp. 85–216 Search PubMed.
- J. Lengyel, J. Kočišek, M. Fárník and J. Fedor, J. Phys. Chem. C, 2016, 120, 7397–7402 CrossRef CAS.
- D. Mészáros, P. Papp and Š. Matejčík, Eur. Phys. J. D, 2023, 77, 62 CrossRef.
- M. McAllister, M. Smyth, B. Gu, G. A. Tribello and J. Kohanoff, J. Phys. Chem. Lett., 2015, 6, 3091–3097 CrossRef CAS PubMed.
- J. Kočišek, B. Sedmidubská, S. Indrajith, M. Fárník and J. Fedor, J. Phys. Chem. B, 2018, 122, 5212–5217 CrossRef PubMed.
- J. Ma, A. Kumar, Y. Muroya, S. Yamashita, T. Sakurai, S. A. Denisov, M. D. Sevilla, A. Adhikary, S. Seki and M. Mostafavi, Nat. Commun., 2019, 10, 102 CrossRef PubMed.
- T. F. M. Luxford, S. A. Pshenichnyuk, N. L. Asfandiarov, T. Perečko, M. Falk and J. Kočišek, Int. J. Mol. Sci., 2020, 21, 8173 CrossRef CAS PubMed.
- R. Meißner, J. Kočišek, L. Feketeová, J. Fedor, M. Fárník, P. Limão-Vieira, E. Illenberger and S. Denifl, Nat. Commun., 2019, 10, 2388 CrossRef PubMed.
- M. Rezaee, D. J. Hunting and L. Sanche, Int. J. Radiat. Oncol., Biol., Phys., 2013, 87, 847–853 CrossRef CAS PubMed.
- S. Zhao and R.-b Zhang, RSC Adv., 2016, 6, 83053–83059 RSC.
- L. Chomicz-Mañka, M. Zdrowowicz, F. Kasprzykowski, J. Rak, A. Zeigler, Y. Wang and K. Bowen, J. Phys. Chem. Lett., 2013, 4, 2853–2857 CrossRef.
- A. I. Lozano, F. Kossoski, F. Blanco, P. Limão-Vieira, M. T. N. Varella and G. García, J. Phys. Chem. Lett., 2022, 13, 7001–7008 CrossRef CAS PubMed.
- D. Nandi, E. Krishnakumar, A. Rosa, W. Schmidt and E. Illenberger, Chem. Phys. Lett., 2003, 373, 454–459 CrossRef CAS.
- F. Xiao, X. Luo, X. Fu and Y. Zheng, J. Phys. Chem. B, 2013, 117, 4893–4900 CrossRef CAS PubMed.
- N. Bhagavan and C.-E. Ha, Essentials of Medical Biochemistry, Academic Press, San Diego, 2nd edn, 2015, pp. 381–400 Search PubMed.
- Z. Li, P. Cloutier, L. Sanche and J. R. Wagner, J. Phys. Chem. B, 2011, 115, 13668–13673 CrossRef CAS PubMed.
- J. Rackwitz, M. Ranković, A. Milosavljevic and I. Bald, Eur. Phys. J. D, 2017, 71, 32 CrossRef.
- T. Kimura, K. Kawai, S. Tojo and T. Majima, J. Org. Chem., 2004, 69, 1169–1173 CrossRef CAS PubMed.
- L. Sala, H. Lyshchuk, J. Šáchová, D. Chvátil and J. Kočišek, J. Phys. Chem. Lett., 2022, 13, 3922–3928 CrossRef CAS PubMed.
- C. von Sonntag, The Chemical Basis of Radiation Biology: Radiobiology, Taylor Francis, London, 1987 Search PubMed.
- S. Makurat, L. Chomicz-Mańka and J. Rak, ChemPhysChem, 2016, 2572–2578 CrossRef CAS PubMed.
- M.-E. Dextraze, J. R. Wagner and D. J. Hunting, Biochemistry, 2007, 46, 9089–9097 CrossRef CAS PubMed.
- H. Yang and M. W. Wong, J. Am. Chem. Soc., 2013, 135, 5808–5818 CrossRef CAS PubMed.
- I. Dabkowska, J. Rak, M. Gutowski, J. M. Nilles, S. T. Stokes, D. Radisic and K. H. Bowen Jr., Phys. Chem. Chem. Phys., 2004, 6, 4351–4357 RSC.
- B. Gu, M. Smyth and J. Kohanoff, Phys. Chem. Chem. Phys., 2014, 16, 24350–24358 RSC.
- X. Wang, H. Liao, W. Liu, Y. Shao, Y. Zheng and L. Sanche, J. Phys. Chem. Lett., 2023, 14, 5674–5680 CrossRef CAS PubMed.
- P. Verma, J. Narayanan S J and A. K. Dutta, J. Phys. Chem. A, 2023, 127, 2215–2227 CrossRef CAS PubMed.
- J. Huang, A. Suma, M. Cui, G. Grundmeier, V. Carnevale, Y. Zhang, C. Kielar and A. Keller, Small Struct., 2020, 1, 2000038 CrossRef.
- E. Gloaguen, M. Mons, K. Schwing and M. Gerhards, Chem. Rev., 2020, 120, 12490–12562 CrossRef CAS PubMed.
- M. Ahmed and O. Kostko, Phys. Chem. Chem. Phys., 2020, 22, 2713–2737 RSC.
- M. Wieczór, P. Wityk, J. Czub, L. Chomicz and J. Rak, Chem. Phys. Lett., 2014, 595–596, 133–137 CrossRef.
- P. Spisz, M. Zdrowowicz, W. Kozak, L. Chomicz-Mańka, K. Falkiewicz, S. Makurat, A. Sikorski, D. Wyrzykowski, J. Rak, E. Arthur-Baidoo, P. Ziegler, M. Costa and S. Denifl, J. Phys. Chem. B, 2020, 5600–5613 CrossRef CAS PubMed.
- A. Ribar, S. E. Huber, M. A. Śmiałek, K. Tanzer, M. Neustetter, R. Schürmann, I. Bald and S. Denifl, Phys. Chem. Chem. Phys., 2018, 20, 5578–5585 RSC.
- F. Kossoski and M. T. N. Varella, Phys. Chem. Chem. Phys., 2015, 17, 17271–17278 RSC.
- P. Storoniak, H. Wang, Y. J. Ko, X. Li, S. T. Stokes, S. Eustis, K. H. Bowen and J. Rak, in Valence Anions of DNA-Related Systems in the Gas Phase: Computational and Anion Photoelectron Spectroscopy Studies, ed. J. Leszczynski and M. K. Shukla, Springer US, Boston, MA, 2014, pp. 323–392 Search PubMed.
- R. Abouaf, J. Pommier and H. Dunet, Int. J. Mass Spectrom., 2003, 226, 397–403 CrossRef CAS.
- R. Abouaf, J. Pommier and H. Dunet, Chem. Phys. Lett., 2003, 381, 486–494 CrossRef CAS.
- A. M. Scheer, K. Aflatooni, G. A. Gallup and P. D. Burrow, Phys. Rev. Lett., 2004, 92, 068102 CrossRef CAS PubMed.
- S. J. Blanksby and G. B. Ellison, Acc. Chem. Res., 2003, 36, 255–263 CrossRef CAS PubMed.
- J. C. Rienstra-Kiracofe, G. S. Tschumper, H. F. Schaefer, S. Nandi and G. B. Ellison, Chem. Rev., 2002, 102, 231–282 CrossRef CAS PubMed.
- C. Ning and Y. Lu, J. Phys. Chem. Ref. Data, 2022, 51, 021502 CrossRef CAS.
- X. Li, L. Sanche and M. D. Sevilla, J. Phys. Chem. A, 2002, 106, 11248–11253 CrossRef CAS PubMed.
- S. Denifl, S. Matejcik, B. Gstir, G. Hanel, M. Probst, P. Scheier and T. D. Märk, J. Chem. Phys., 2003, 118, 4107–4114 CrossRef CAS.
- W. Wang, M. Marshall, E. Collins, S. Marquez, C. Mu, K. H. Bowen and X. Zhang, Nat. Commun., 2019, 10, 1170 CrossRef PubMed.
- S. Klaiman and L. S. Cederbaum, Angew. Chem., Int. Ed., 2015, 54, 10470–10473 CrossRef CAS PubMed.
- H.-T. Liu, C.-G. Ning, D.-L. Huang and L.-S. Wang, Angew. Chem., Int. Ed., 2014, 53, 2464–2468 CrossRef CAS PubMed.
- F. Izadi, A. Szczyrba, M. Datta, O. Ciupak, S. Demkowicz, J. Rak and S. Denifl, Int. J. Mol. Sci., 2023, 24, 8706 CrossRef CAS PubMed.
- B. Ómarsson, E. H. Bjarnason, O. Ingólfsson, S. Haughey and T. A. Field, Chem. Phys. Lett., 2012, 539–540, 7–10 CrossRef.
- E. Arthur-Baidoo, G. Schöpfer, M. Ončák, L. Chomicz-Mańka, J. Rak and S. Denifl, Int. J. Mol. Sci., 2022, 23, 8325 CrossRef CAS PubMed.
- M. Cipriani and O. Ingólfsson, Radiat. Phys. Chem., 2023, 202, 110544 CrossRef CAS.
- L. M. Cornetta, T. J. Martinez and M. T. N. Varella, Phys. Chem. Chem. Phys., 2022, 24, 6845–6855 RSC.
- J. W. J. D. Zimbrick and L. Myers, Int. J. Radiat. Oncol., Biol., Phys., 1969, 16, 505–523 CAS.
- J. D. Zimbrick, J. F. Ward and L. S. Myers, Jr, Int. J. Radiat. Biol., 1969, 16, 525–534 CAS.
- K. Westphal, J. Wiczk, J. Miloch, G. Kciuk, K. Bobrowski and J. Rak, Org. Biomol. Chem., 2015, 13, 10362–10369 RSC.
- H. Abdoul-Carime, S. Gohlke and E. Illenberger, Phys. Chem. Chem. Phys., 2004, 6, 161–164 RSC.
- C.-R. Wang and Q.-B. Lu, Angew. Chem., Int. Ed., 2007, 46, 6316–6320 CrossRef CAS PubMed.
- C. Beach, A. F. Fuciarelli and J. D. Zimbrick, Radiat. Res., 1994, 137, 385–393 CrossRef CAS PubMed.
- J. Narayanan S J, D. Tripathi, P. Verma, A. Adhikary and A. K. Dutta, ACS Omega, 2023, 8, 10669–10689 CrossRef CAS PubMed.
- L. Cupellini, P. Wityk, B. Mennucci and J. Rak, Phys. Chem. Chem. Phys., 2019, 21, 4387–4393 RSC.
- P. Schyman, R. B. Zhang, L. A. Eriksson and A. Laaksonen, Phys. Chem. Chem. Phys., 2007, 9, 5975–5979 RSC.
- J. Rak, L. Chomicz-Mańka, J. Wiczk, K. Westphal, M. Zdrowowicz, P. Wityk, M. Żyndul, S. Makurat and Ł. Golon, J. Phys. Chem. B, 2015, 119, 8227–8238 CrossRef CAS PubMed.
- X. Li, M. D. Sevilla and L. Sanche, J. Am. Chem. Soc., 2003, 125, 8916–8920 CrossRef CAS PubMed.
- A. Paul and D. Nandi, Phys. Chem. Chem. Phys., 2022, 24, 21020–21029 RSC.
- J. Kopyra, H. Abdoul-Carime, F. Kossoski and M. T. N. Varella, Phys. Chem. Chem. Phys., 2014, 16, 25054–25061 RSC.
- J. Kopyra and H. Abdoul-Carime, J. Chem. Phys., 2016, 144, 034306 CrossRef PubMed.
- K. P. Prasanthkumar, J. R. Alvarez-Idaboy, P. V. Kumar, B. G. Singh and K. I. Priyadarsini, Phys. Chem. Chem. Phys., 2016, 18, 28781–28790 RSC.
- E. Arthur-baidoo, K. Falkiewicz, L. Chomiczâ-mańka, A. Czaja, S. Demkowicz, K. Biernacki, W. Kozak, J. Rak and S. Denifl, Int. J. Mol. Sci., 2021, 22, 1–13 Search PubMed.
- F. Meng, J. Mol. Struct.: THEOCHEM, 2007, 806, 159–164 CrossRef CAS.
- M. Sosnowska, S. Makurat, M. Zdrowowicz and J. Rak, J. Phys. Chem. B, 2017, 121, 6139–6147 CrossRef CAS PubMed.
- T. Trabelsi, O. Yazidi, J. S. Francisco, R. Linguerri and M. Hochlaf, J. Chem. Phys., 2015, 143, 164301 CrossRef CAS PubMed.
- P. Spisz, M. Zdrowowicz, W. Kozak, L. Chomicz-Mańka, K. Falkiewicz, S. Makurat, A. Sikorski, D. Wyrzykowski, J. Rak, E. Arthur-Baidoo, P. Ziegler, M. S. Rodrigues Costa and S. Denifl, J. Phys. Chem. B, 2020, 124, 5600–5613 CrossRef CAS PubMed.
- R. Abouaf, S. Ptasinska and D. Teillet-Billy, Chem. Phys. Lett., 2008, 455, 169–173 CrossRef CAS.
- S. Ptasińska, E. Alizadeh, P. Sulzer, R. Abouaf, N. J. Mason, T. D. Märk and P. Scheier, Int. J. Mass Spectrom., 2008, 277, 291–295 CrossRef.
- S. J. Kogikoski, A. Dutta and I. Bald, ACS Nano, 2021, 15, 20562–20573 CrossRef CAS PubMed.
- R. Schürmann and I. Bald, Nanoscale, 2017, 9, 1951–1955 RSC.
- A. Dutta, R. Schürmann, S. J. Kogikoski, N. S. Mueller, S. Reich and I. Bald, ACS Catal., 2021, 11, 8370–8381 CrossRef CAS PubMed.
- N. L. Asfandiarov, S. A. Pshenichnyuk, R. G. Rakhmeyev, R. F. Tuktarov, N. L. Zaitsev, A. S. Vorobev, J. Kočišek, J. Fedor and A. Modelli, J. Chem. Phys., 2019, 150, 114304 CrossRef CAS PubMed.
- N. Asfandiarov, M. Muftakhov, R. Rakhmeev, A. Safronov, A. Markova and S. Pshenichnyuk, J. Electron Spectrosc. Related Phenomena, 2022, 256, 147178 CrossRef CAS.
- N. L. Asfandiarov, M. V. Muftakhov and S. A. Pshenichnyuk, J. Chem. Phys., 2023, 158, 194305 CrossRef CAS PubMed.
- N. L. Asfandiarov, M. V. Muftakhov, S. A. Pshenichnyuk, R. G. Rakhmeev, A. M. Safronov, A. V. Markova, A. S. Vorobev, T. F. M. Luxford, J. Kočišek and J. Fedor, J. Chem. Phys., 2021, 155, 244302 CrossRef CAS PubMed.
- K. Falkiewicz, W. Kozak, M. Zdrowowicz, P. Spisz, L. Chomicz-Mańka, M. Torchala and J. Rak, J. Phys. Chem. B, 2023, 2565–2574 CrossRef CAS PubMed.
- H. Abdoul-Carime, P. Limão-Vieira, S. Gohlke, I. Petrushko, N. J. Mason and E. Illenberger, Chem. Phys. Lett., 2004, 393, 442–447 CrossRef CAS.
- S. Denifl, P. Candori, S. Ptasińska, P. Limão-Vieira, V. Grill, T. D. Märk and P. Scheier, Eur. Phys. J. D, 2005, 35, 391–398 CrossRef CAS.
- C.-R. Wang, A. Hu and Q.-B. Lu, J. Chem. Phys., 2006, 124, 241102 CrossRef PubMed.
- J. Ma, T. Bahry, S. A. Denisov, A. Adhikary and M. Mostafavi, J. Phys. Chem. A, 2021, 125, 7967–7975 CrossRef CAS PubMed.
- M. Saqib, E. Arthur-Baidoo, F. Izadi, A. Szczyrba, M. Datta, S. Demkowicz, J. Rak and S. Denifl, J. Phys. Chem. Lett., 2023, 127, 8948–8955 CrossRef PubMed.
- M. Durante, R. Orecchia and J. S. Loeffler, Nat. Rev. Clin. Oncol., 2017, 14, 483–495 CrossRef PubMed.
- J. Kopyra, A. Keller and I. Bald, RSC Adv., 2014, 4, 6825–6829 RSC.
- C. Chatgilialoglu, Reactivity of Nucleic Acid Sugar Radicals, John Wiley Sons, Ltd, 2009, ch. 4, pp. 99–133 Search PubMed.
- A. Kumar and M. D. Sevilla, Chem. Rev., 2010, 110, 7002–7023 CrossRef CAS PubMed.
- J. Gu, Y. Xie and H. F. Schaefer III, ChemPhysChem, 2006, 7, 1885–1887 CrossRef CAS PubMed.
- K. Khistyaev, A. Golan, K. B. Bravaya, N. Orms, A. I. Krylov and M. Ahmed, J. Phys. Chem. A, 2013, 117, 6789–6797 CrossRef CAS PubMed.
- B. Boudaïffa, Science, 2000, 287, 1658–1660 CrossRef PubMed.
- S. Ptasińska, S. Denifl, P. Scheier and T. D. Märk, J. Chem. Phys., 2004, 120, 8505–8511 CrossRef PubMed.
- I. Bald, I. Dabkowska and E. Illenberger, Angew. Chem., Int. Ed., 2008, 47, 8518–8520 CrossRef CAS PubMed.
- H. Czapinska, M. Winiewska-Szajewska, A. Szymaniec-Rutkowska, A. Piasecka, M. Bochtler and J. Poznański, J. Phys. Chem. B, 2021, 125, 2491–2503 CrossRef CAS PubMed.
- G. E. Adams, Radiat. Res., 1992, 132, 129–139 CrossRef CAS PubMed.
- P. Wardman, Clin. Oncol., 2007, 19, 397–417 CrossRef CAS PubMed.
- B. T. Oronsky, S. J. Knox and J. J. Scicinski, Transl. Oncol., 2012, 5, 66–71 CrossRef PubMed.
- D. A. Wink, Y. Vodovotz, J. Laval, F. Laval, M. W. Dewhirst and J. B. Mitchell, Carcinogenesis, 1998, 19, 711–721 CrossRef CAS PubMed.
- M. Lepoivre, F. Fieschi, J. Coves, L. Thelander and M. Fontecave, Biochem. Biophys. Res. Commun., 1991, 179, 442–448 CrossRef CAS PubMed.
- R. Willson, W. Cramp and R. Ings, Int. J. Radiat. Biol. Relat. Stud. Phys., Chem. Med., 1974, 26, 557–569 CrossRef CAS PubMed.
- M. R. Horsman, L. S. Mortensen, J. B. Petersen, M. Busk and J. Overgaard, Nat. Rev. Clin. Oncol., 2012, 9, 674–687 CrossRef CAS PubMed.
- K. Nepali, H.-Y. Lee and J.-P. Liou, J. Med. Chem., 2019, 62, 2851–2893 CrossRef CAS PubMed.
- N. Boechat, A. S. Carvalho, K. Salomão, S. L. D. Castro, C. F. Araujo-Lima, F. V. Mello, I. Felzenszwalb, C. A. Aiub, T. R. Conde and H. P. Zamith, et al. , Mem. Inst. Oswaldo Cruz, 2015, 110, 492–499 CrossRef CAS PubMed.
- A. C. M. Von Trompowsky, T. R. Conde, R. C. Lemos, B. M. C. Quaresma, M. C. S. Pitombeira, A. S. de Carvalho, N. Boechat, K. Salomão, S. L. de Castro and H. P. D. S. Zamith, Mem. Inst. Oswaldo Cruz, 2019, 114, e190017 CrossRef PubMed.
- D. Christen, J. H. Griffiths and J. Sheridan, Zeitschrift für Naturforschung A, 1981, 36, 1378–1385 CrossRef.
- S. A. Pshenichnyuk, I. I. Fabrikant, A. Modelli, S. Ptasinska and A. S. Komolov, J. Phys.: Conf. Ser., 2020, 1412, 212003 CrossRef.
- F. Kossoski and M. H. Bettega, J. Chem. Phys., 2013, 138, 234311 CrossRef CAS PubMed.
- A. Ribar, K. Fink, Z. Li, S. Ptasińska, I. Carmichael, L. Feketeová and S. Denifl, Phys. Chem. Chem. Phys., 2017, 19, 6406–6415 RSC.
- T. Jani, P. C. Vinodkumar and M. Vinodkumar, J. Phys. Chem. A, 2023, 127, 4996–5004 CrossRef CAS PubMed.
- A. Modelli and P. D. Burrow, J. Phys. Chem. A, 2004, 108, 5721–5726 CrossRef CAS.
- Z. Li, I. Carmichael and S. Ptasińska, Phys. Chem. Chem. Phys., 2018, 20, 18271–18278 RSC.
- A. A. Wallace, Y. Dauletyarov and A. Sanov, J. Phys. Chem. A, 2021, 125, 317–326 CrossRef CAS PubMed.
- L. Feketeová, A. L. Albright, B. S. Sørensen, M. R. Horsman, J. White, R. A. OHair and N. Bassler, Int. J. Mass Spectrom., 2014, 365–366, 56–63 CrossRef.
- V. S. Prabhudesai, A. H. Kelkar, D. Nandi and E. Krishnakumar, Phys. Rev. Lett., 2005, 95, 143202 CrossRef PubMed.
- M. Zawadzki, T. F. M. Luxford and J. Kočišek, J. Phys. Chem. A, 2020, 124, 9427–9435 CrossRef CAS PubMed.
- A. Ribar, K. Fink, M. Probst, S. E. Huber, L. Feketeová and S. Denifl, Chem. – Eur. J., 2017, 23, 12892–12899 CrossRef CAS PubMed.
- K. Tanzer, L. Feketeová, B. Puschnigg, P. Scheier, E. Illenberger and S. Denifl, Angew. Chem., Int. Ed., 2014, 53, 12240–12243 CrossRef CAS PubMed.
- K. Tanzer, L. Feketeová, B. Puschnigg, P. Scheier, E. Illenberger and S. Denifl, J. Phys. Chem. A, 2015, 119, 6668–6675 CrossRef CAS PubMed.
- J. Kopyra, K. K. Kopyra, H. Abdoul-Carime and D. Branowska, J. Chem. Phys., 2018, 148, 234301 CrossRef PubMed.
- D. Barreiro-Lage, C. Nicolafrancesco, J. Kočišek, A. Luna, J. Kopyra, M. Alcamí, B. A. Huber, F. Martín, A. Domaracka, P. Rousseau and S. Díaz-Tendero, Phys. Chem. Chem. Phys., 2022, 24, 941–954 RSC.
- Y. V. Vasil'ev, B. J. Figard, V. G. Voinov, D. F. Barofsky and M. L. Deinzer, J. Am. Chem. Soc., 2006, 128, 5506–5515 CrossRef PubMed.
- F. Kossoski and M. T. D. N. Varella, J. Chem. Phys., 2017, 147, 164310 CrossRef CAS PubMed.
- C. Desfrançois, V. Périquet, S. Carles, J. P. Schermann, D. M. A. Smith and L. Adamowicz, J. Chem. Phys., 1999, 110, 4309–4314 CrossRef.
- D. M. A. Smith, J. Smets, Y. Elkadi and L. Adamowicz, J. Phys. Chem. A, 1997, 101, 8123–8127 CrossRef CAS.
- M. Zawadzki, M. Ranković, J. Kočišek and J. Fedor, Phys. Chem. Chem. Phys., 2018, 20, 6838–6844 RSC.
- A. I. Lozano, L. Álvarez, A. García-Abenza, C. Guerra, F. Kossoski, J. Rosado, F. Blanco, J. C. Oller, M. Hasan, M. Centurion, T. Weber, D. S. Slaughter, D. M. Mootheril, A. Dorn, S. Kumar, P. Limão-Vieira, R. Colmenares and G. García, Int. J. Mol. Sci., 2023, 24, 12182 CrossRef CAS PubMed.
- R. Willson, B. Gilbert, P. Marshall and R. Norman, Int. J. Radiat. Biol. Relat. Stud. Phys., Chem. Med., 1974, 26, 427–434 CrossRef CAS PubMed.
- C. Lochmann, T. F. Luxford, S. Makurat, A. Pysanenko, J. Kočišek, J. Rak and S. Denifl, Pharmaceuticals, 2022, 15, 1–12 CrossRef PubMed.
- S. Kumar, I. Ben Chouikha, B. Kerkeni, G. García and P. Limão-Vieira, Molecules, 2022, 27, 4134 CrossRef CAS PubMed.
- R. Meißner, L. Feketeová, A. Bayer, P. Limão-Vieira and S. Denifl, J. Chem. Phys., 2021, 154, 074306 CrossRef PubMed.
- C. L. Brito, R. S. Lins, M. Bertotti, E. I. Ferreira and M. A. La-Scalea, Electrochim. Acta, 2022, 403, 139709 CrossRef CAS.
- E. Gattavecchia, D. Tonelli and P. Fuochi, Int. J. Radiat. Biol. Relat. Stud. Phys., Chem. Med., 1984, 45, 469–477 CrossRef CAS PubMed.
- R. Meiβner, L. Feketeová, E. Illenberger and S. Denifl, Int. J. Mol. Sci., 2019, 20, 3496 CrossRef PubMed.
- M. Ončák, R. Meißner, E. Arthur-Baidoo, S. Denifl, T. F. Luxford, A. Pysanenko, M. Fárník, J. Pinkas and J. Kočišek, Int. J. Mol. Sci., 2019, 20, 1–12 Search PubMed.
- T. C. Johnstone, J. J. Wilson and S. J. Lippard, Inorg. Chem., 2013, 52, 12234–12249 CrossRef CAS PubMed.
- J. Reedijk, Inorg. Chim. Acta, 2016, 452, 268–272 CrossRef CAS.
- K. Chválová, V. Brabec and J. Kašpárková, Nucleic Acids Res., 2007, 35, 1812–1821 CrossRef PubMed.
- V. Brabec and M. Leng, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 5345–5349 CrossRef CAS PubMed.
- A. Eastman, The Mechanism of Action of Cisplatin: From Adducts to Apoptosis, John Wiley Sons, Ltd, 1999, ch. 4, pp. 111–134 Search PubMed.
- Q. B. Lu, J. Med. Chem., 2007, 50, 2601–2604 CrossRef CAS PubMed.
- Q. Zhang and Q. Lu, Sci. Rep., 2021, 11, 788 CrossRef CAS PubMed.
- T. Luo, J. Yu, J. Nguyen, C.-R. Wang, R. G. Bristow, D. A. Jaffray, X. Z. Zhou, K. P. Lu and Q.-B. Lu, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 10175–10180 CrossRef CAS PubMed.
- E. Alizadeh and L. Sanche, Eur. Phys. J. D, 2014, 68, 97 CrossRef.
- M. Rezaee, L. Sanche and D. J. Hunting, Radiat. Res., 2013, 179, 323–331 CrossRef CAS PubMed.
- S. Kouass Sahbani, M. Rezaee, P. Cloutier, L. Sanche and D. Hunting, Chem.-Biol. Interact., 2014, 217, 9–18 CrossRef CAS PubMed.
- J. Kopyra, C. Koenig-Lehmann, I. Bald and E. Illenberger, Angew. Chem., Int. Ed., 2009, 48, 7904–7907 CrossRef CAS PubMed.
- H. Y. Chen, H. F. Chen, C. L. Kao, P. Y. Yang and S. C. Hsu, Phys. Chem. Chem. Phys., 2014, 16, 19290–19297 RSC.
- Q. Bao, Y. Chen, Y. Zheng and L. Sanche, J. Phys. Chem. C, 2014, 118, 15516–15524 CrossRef CAS PubMed.
- M. Rezaee, E. Alizadeh, P. Cloutier, D. J. Hunting and L. Sanche, ChemMedChem, 2014, 9, 1145–1149 CrossRef CAS PubMed.
- Y. Dong, L. Zhou, Q. Tian, Y. Zheng and L. Sanche, J. Phys. Chem. C, 2017, 121, 17505–17513 CrossRef CAS.
- Y. Dong, Y. Wang, P. Zhuang, X. Fu, Y. Zheng and L. Sanche, J. Phys. Chem. B, 2020, 124, 3315–3325 CrossRef CAS PubMed.
- B. Behmand, P. Cloutier, S. Girouard, J. R. Wagner, L. Sanche and D. J. Hunting, J. Phys. Chem. B, 2013, 117, 15994–15999 CrossRef CAS PubMed.
- B. Behmand, J. R. Wagner, L. Sanche and D. J. Hunting, J. Phys. Chem. B, 2014, 118, 4803–4808 CrossRef CAS PubMed.
- B. Behmand, J. L. Marignier, M. Mostafavi, J. R. Wagner, D. J. Hunting and L. Sanche, J. Phys. Chem. B, 2015, 119, 9496–9500 CrossRef CAS PubMed.
- B. Behmand, A. M. Noronha, C. J. Wilds, J. L. Marignier, M. Mostafavi, J. R. Wagner, D. J. Hunting and L. Sanche, J. Radiat. Res., 2020, 61, 343–351 CrossRef CAS PubMed.
- M. A. Śmiałek, S. Ptasińska, J. Gow, S. V. Hoffmann and N. J. Mason, Eur. Phys. J. D, 2015, 69, 121 CrossRef.
- K. Tanzer, A. Pelc, S. Huber, M. Śmiałek, P. Scheier, M. Probst and S. Denifl, Int. J. Mass Spectrom., 2014, 365–366, 152–156 CrossRef CAS.
- Š. Matejčık, V. Foltin, M. Stano and J. Skalný, Int. J. Mass Spectrom., 2003, 223–224, 9–19 CrossRef.
- Y. V. Vasilev, V. G. Voinov, D. F. Barofsky and M. L. Deinzer, Int. J. Mass Spectrom., 2008, 277, 142–150 CrossRef CAS.
- M. Cipriani, S. Svavarsson, F. F. da Silva, H. Lu, L. McElwee-White and O. Ingólfsson, Int. J. Mol. Sci., 2021, 22, 8984 CrossRef CAS PubMed.
- F. Ferreira da Silva, R. M. Thorman, R. Bjornsson, H. Lu, L. McElwee-White and O. Ingólfsson, Phys. Chem. Chem. Phys., 2020, 22, 6100–6108 RSC.
- J. Langer, M. Zawadzki, M. Fárník, J. Pinkas, J. Fedor and J. Kočišek, Eur. Phys. J. D, 2018, 72, 112 CrossRef.
- E. Arthur-Baidoo, M. Ončák and S. Denifl, J. Chem. Phys., 2022, 157, 074301 CrossRef CAS PubMed.
- S. King, Free Radical Biol. Med., 2004, 37, 737–744 CrossRef CAS PubMed.
- J. Yarbro, Semin. Oncol., 1992, 19, 1–10 CAS.
- K. Sakano, S. Oikawa, K. Hasegawa and S. Kawanishi, Jpn. J. Cancer Res., 2001, 92, 1166–1174 CrossRef CAS PubMed.
- S. E. Huber, M. A. Śmiałek, K. Tanzer and S. Denifl, J. Chem. Phys., 2016, 144, 224309 CrossRef CAS PubMed.
- J. Kočišek, P. Papp, P. Mach, Y. Vasil'ev, M. Deinzer and S. Matejcik, J. Phys. Chem. A, 2010, 114, 1677–1683 CrossRef PubMed.
- A. M. Weljie and F. R. Jirik, Int. J. Biochem. Cell Biol., 2011, 43, 981–989 CrossRef CAS PubMed.
- M. Bonora, S. Patergnani, A. Rimessi, E. De Marchi, J. M. Suski, A. Bononi, C. Giorgi, S. Marchi, S. Missiroli, F. Poletti, M. R. Wieckowski and P. Pinton, Purinergic Signalling, 2012, 8, 343–357 CrossRef CAS PubMed.
- F. FerreiradaSilva, M. T. doN. Varella, N. C. Jones, S. Vrøn-ningHoffmann, S. Denifl, I. Bald and J. Kopyra, Chem. – Eur. J., 2019, 25, 5498–5506 CrossRef CAS PubMed.
- T. J. Jorgensen, Strange Glow: The Story of Radiation, Princeton University Press, 2016 Search PubMed.
- K. Kobayashi, Chem. Rev., 2019, 119, 4413–4462 CrossRef CAS PubMed.
- M. Maestre-Reyna, P.-H. Wang, E. Nango, Y. Hosokawa, M. Saft, A. Furrer, C.-H. Yang, E. P. G. N. Putu, W.-J. Wu, H.-J. Emmerich, N. Caramello, S. Franz-Badur, C. Yang, S. Engilberge, M. Wranik, H. L. Glover, T. Weinert, H.-Y. Wu, C.-C. Lee, W.-C. Huang, K.-F. Huang, Y.-K. Chang, J.-H. Liao, J.-H. Weng, W. Gad, C.-W. Chang, A. H. Pang, K.-C. Yang, W.-T. Lin, Y.-C. Chang, D. Gashi, E. Beale, D. Ozerov, K. Nass, G. Knopp, P. J. M. Johnson, C. Cirelli, C. Milne, C. Bacellar, M. Sugahara, S. Owada, Y. Joti, A. Yamashita, R. Tanaka, T. Tanaka, F. Luo, K. Tono, W. Zarzycka, P. Müller, M. A. Alahmad, F. Bezold, V. Fuchs, P. Gnau, S. Kiontke, L. Korf, V. Reithofer, C. J. Rosner, E. M. Seiler, M. Watad, L. Werel, R. Spadaccini, J. Yamamoto, S. Iwata, D. Zhong, J. Standfuss, A. Royant, Y. Bessho, L.-O. Essen and M.-D. Tsai, Science, 2023, 382, eadd7795 CrossRef CAS PubMed.
- N.-E. Christou, V. Apostolopoulou, D. V. M. Melo, M. Ruppert, A. Fadini, A. Henkel, J. Sprenger, D. Oberthuer, S. Günther, A. Pateras, A. R. Mashhour, O. M. Yefanov, M. Galchenkova, P. Y. A. Reinke, V. Kremling, T. E. S. Scheer, E. R. Lange, P. Middendorf, R. Schubert, E. D. Zitter, K. Lumbao-Conradson, J. Herrmann, S. Rahighi, A. Kunavar, E. V. Beale, J. H. Beale, C. Cirelli, P. J. M. Johnson, F. Dworkowski, D. Ozerov, Q. Bertrand, M. Wranik, C. Bacellar, S. Bajt, S. Wakatsuki, J. A. Sellberg, N. Huse, D. Turk, H. N. Chapman and T. J. Lane, Science, 2023, 382, 1015–1020 CrossRef CAS PubMed.
- S. Kogikoski, J. Ameixa, A. Mostafa and I. Bald, Chem. Commun., 2023, 59, 4726–4741 RSC.
- X. Xing, S. Sato, N.-K. Wong, K. Hidaka, H. Sugiyama and M. Endo, Nucleic Acids Res., 2020, 48, 4041–4051 CrossRef CAS PubMed.
- J. Reindl, A. M. Abrantes, V. Ahire, O. Azimzadeh, S. Baatout, A. Baeyens, B. Baselet, V. Chauhan, F. Da Pieve, W. Delbart, C. P. Dobney, N. F. J. Edin, M. Falk, N. Foray, A. François, S. Frelon, U. S. Gaipl, A. G. Georgakilas, O. Guipaud, M. Hausmann, A. J. Michaelidesova, M. Kadhim, I. A. Marques, M. Milic, D. Mistry, S. Moertl, A. Montoro, E. Obrador, A. S. Pires, R. Quintens, N. Rajan, F. Rödel, P. Rogan, D. Savu, G. Schettino, K. Tabury, G. I. Terzoudi, S. Triantopoulou, K. Viktorsson and A.-S. Wozny, Molecular Radiation Biology, ed. S. Baatout, Springer International Publishing, Cham, 2023, pp. 83–189 Search PubMed.
- J. Rackwitz and I. Bald, Chem. – Eur. J., 2018, 24, 4680–4688 CrossRef CAS PubMed.
- W. Li, E. Mjekiqi, W. Douma, X. Wang, O. Kavatsyuk, R. Hoekstra, J.-C. Poully and T. Schlathölter, Chem. – Eur. J., 2019, 25, 16114–16119 CrossRef CAS PubMed.
- M. K. Scheller, R. N. Compton and L. S. Cederbaum, Science, 1995, 270, 1160–1166 CrossRef CAS.
- A. Dreuw and L. S. Cederbaum, Chem. Rev., 2002, 102, 181–200 CrossRef CAS PubMed.
- A. I. Boldyrev, M. Gutowski and J. Simons, Acc. Chem. Res., 1996, 29, 497–502 CrossRef CAS.
- X.-B. Wang and L.-S. Wang, Ann. Rev. Phys. Chem., 2009, 60, 105–126 CrossRef CAS PubMed.
- M. E. Castellani, D. Avagliano, L. González and J. R. R. Verlet, J. Phys. Chem. Lett., 2020, 11, 8195–8201 CrossRef CAS PubMed.
- M. E. Castellani, D. Avagliano and J. R. R. Verlet, J. Phys. Chem. A, 2021, 125, 3646–3652 CrossRef CAS PubMed.
- J. A. Gibbard and J. R. R. Verlet, J. Chem. Phys., 2023, 158, 154306 CrossRef CAS PubMed.
- R. N. Compton, A. A. Tuinman, C. E. Klots, M. R. Pederson and D. C. Patton, Phys. Rev. Lett., 1997, 78, 4367–4370 CrossRef CAS.
- M. Rezaee, R. P. Hill and D. A. Jaffray, Radiat. Res., 2017, 188, 123–143 CrossRef CAS PubMed.
- L. Zhou, W. Liu, N. Brodeur, P. Cloutier, Y. Zheng and L. Sanche, J. Chem. Phys., 2019, 150, 195101 CrossRef PubMed.
- R. Schürmann, T. Tsering, K. Tanzer, S. Denifl, S. V. K. Kumar and I. Bald, Angew. Chem., Int. Ed., 2017, 56, 10952–10955 CrossRef PubMed.
- Y. Zheng and L. Sanche, Appl. Phys. Rev., 2018, 5, 021302 Search PubMed.
- D. Reimitz, M. Davídková, O. Mestek, J. Pinkas and J. Kočišek, Radiat. Phys. Chem., 2017, 141, 229–234 CrossRef CAS.
- M. Zdrowowicz, P. Spisz, A. Hać, A. Herman-Antosiewicz and J. Rak, Int. J. Mol. Sci., 2022, 23, 1429 CrossRef CAS PubMed.
- P. Nag, M. Ranković, H. C. Schewe, J. Rakovský, L. Sala, J. Kočišek and J. Fedor, J. Phys. B: At. Mol. Phys., 2023, 56, 215201 CrossRef.
- L. Sala, J. Rakovský, A. Zerolová and J. Kočišek, J. Phys. B: At. Mol. Phys., 2023, 56, 185101 CrossRef.
- J. Országh, A. Ribar, M. Danko, D. Bodewits, T. Matejčík and W. Barszczewska, ChemPhysChem, 2022, 23, e202100705 CrossRef PubMed.
- Z. Chen, Z. Li, J. Hu and S. X. Tian, Acc. Chem. Res., 2022, 55, 3071–3079 CrossRef CAS PubMed.
- T. E. Gartmann, S. Hartweg, L. Ban, E. Chasovskikh, B. L. Yoder and R. Signorell, Phys. Chem. Chem. Phys., 2018, 20, 16364–16371 RSC.
- S. Amanatidis, B. L. Yoder and R. Signorell, J. Chem. Phys., 2017, 146, 224204 CrossRef PubMed.
- H. Tsuchida, T. Kai, K. Kitajima, Y. Matsuya, T. Majima and M. Saito, Eur. Phys. J. D, 2020, 74, 212 CrossRef CAS.
- K. Kitajima, H. Tsuchida, T. Majima and M. Saito, J. Chem. Phys., 2019, 150, 095102 CrossRef PubMed.
- T. Majima, S. Mizutani, Y. Mizunami, K. Kitajima, H. Tsuchida and M. Saito, J. Chem. Phys., 2020, 153, 224201 CrossRef CAS PubMed.
- K. Kitajima, T. Majima, T. Nishio, Y. Oonishi, S. Mizutani, J. ya Kohno, M. Saito and H. Tsuchida, Nucl. Instrum. Methods Phys. Res., Sect. B, 2018, 424, 10–16 CrossRef CAS.
- Y. Gao, Y. Zheng and L. Sanche, Int. J. Mol. Sci., 2021, 22, 7879 CrossRef CAS PubMed.
- E. Alizadeh, D. Chakraborty and S. Ptasińska, Biophys., 2022, 2, 475–497 CrossRef.
- A. Sieradzka and J. D. Gorfinkiel, J. Chem. Phys., 2017, 147, 034302 CrossRef PubMed.
- L. M. Cornetta, K. Coutinho and M. T. D. N. Varella, J. Chem. Phys., 2020, 152, 084301 CrossRef CAS PubMed.
- J. D. Gorfinkiel, J. Phys.: Conf. Ser., 2020, 1412, 052003 CrossRef CAS.
- A. Lietard, G. Mensa-Bonsu and J. R. R. Verlet, Nat. Chem., 2021, 13, 737–742 CrossRef CAS PubMed.
- G. A. Cooper, C. J. Clarke and J. R. R. Verlet, J. Am. Chem. Soc., 2023, 145, 1319–1326 CrossRef CAS PubMed.
- M. Smyth, J. Kohanoff and I. I. Fabrikant, J. Chem. Phys., 2014, 140, 184313 CrossRef CAS PubMed.
- I. Anusiewicz, P. Skurski and J. Simons, J. Phys. Chem. A, 2020, 124, 2064–2076 CrossRef CAS PubMed.
- M. Mukherjee, D. Tripathi and A. K. Dutta, J. Chem. Phys., 2020, 153, 044305 CrossRef CAS PubMed.
- S. Ptasinska, Atoms, 2021, 9, 77 CrossRef CAS.
- J. McConkey, C. Malone, P. Johnson, C. Winstead, V. McKoy and I. Kanik, Phys. Rep., 2008, 466, 1–103 CrossRef CAS.
- A. Ribar, M. Danko, J. Országh, F. Ferreira da Silva, I. Utke and Š. Matejčík, Eur. Phys. J. D, 2015, 69, 117 CrossRef.
- J. Országh, A. Ribar, M. Danko, D. Bodewits, T. Matejčík and W. Barszczewska, ChemPhysChem, 2022, 23, e202100705 CrossRef PubMed.
- H. Abdoul-Carime, F. Mounier, F. Charlieux and H. André, Rev. Sci. Instrum., 2023, 94, 045104 CrossRef CAS PubMed.
- F. Charlieux and H. Abdoul-Carime, ChemPhysChem, 2023, 24, e202200722 CrossRef CAS PubMed.
- Z. Li, A. R. Milosavljević, I. Carmichael and S. Ptasinska, Phys. Rev. Lett., 2017, 119, 053402 CrossRef PubMed.
- C. D. Lane and T. M. Orlando, J. Chem. Phys., 2006, 124, 164702 CrossRef PubMed.
- S. D. Brinkevich, A. Y. Maliborskii, I. A. Kapusto, R. L. Sverdlov, Y. V. Grigor'ev, O. A. Ivashkevich and O. I. Shadyro, High Energy Chemistry, 2019, 53, 147–154 CrossRef CAS.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS PubMed.
- X. Jiang, X. Hao, L. Jing, G. Wu, D. Kang, X. Liu and P. Zhan, Expert Opin. Drug Discovery, 2019, 14, 779–789 CrossRef CAS PubMed.
- M. Rezaee, D. J. Hunting and L. Sanche, Int. J. Radiat. Oncol. Biol. Phys., 2013, 87, 847–853 CrossRef CAS PubMed.
- M. R. Gill and K. A. Vallis, Chem. Soc. Rev., 2019, 48, 540–557 RSC.
- R. Carter, A. Westhorpe, M. J. Romero, A. Habtemariam, C. R. Gallevo, Y. Bark, N. Menezes, P. J. Sadler and R. A. Sharma, Sci. Rep., 2016, 6, 20596 CrossRef CAS PubMed.
| This journal is © the Owner Societies 2024 |