
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSensitizer-controlled photochemical reactivity via upconversion of red light†
Felix
Glaser
 and
Oliver S.
Wenger
*
and
Oliver S.
Wenger
*
Department of Chemistry, University of Basel, St. Johanns-Ring 19, 4056 Basel, Switzerland. E-mail: oliver.wenger@unibas.ch
First published on 1st December 2022
Abstract
By combining the energy input from two red photons, chemical reactions that would normally require blue or ultraviolet irradiation become accessible. Key advantages of this biphotonic excitation strategy are that red light usually penetrates deeper into complex reaction mixtures and causes less photo-damage than direct illumination in the blue or ultraviolet. Here, we demonstrate that the primary light-absorber of a dual photocatalytic system comprised of a transition metal-based photosensitizer and an organic co-catalyst can completely alter the reaction outcome. Photochemical reductions are achieved with a copper(I) complex in the presence of a sacrificial electron donor, whereas oxidative substrate activation occurs with an osmium(II) photosensitizer. Based on time-resolved laser spectroscopy, this changeover in photochemical reactivity is due to different underlying biphotonic mechanisms. Following triplet energy transfer from the osmium(II) photosensitizer to 9,10-dicyanoanthracene (DCA) and subsequent triplet–triplet annihilation upconversion, the fluorescent singlet excited state of DCA triggers oxidative substrate activation, which initiates the cis to trans isomerization of an olefin, a [2 + 2] cycloaddition, an aryl ether to ester rearrangement, and a Newman–Kwart rearrangement. This oxidative substrate activation stands in contrast to the reactivity with a copper(I) photosensitizer, where photoinduced electron transfer generates the DCA radical anion, which upon further excitation triggers reductive dehalogenations and detosylations. Our study provides the proof-of-concept for controlling the outcome of a red-light driven biphotonic reaction by altering the photosensitizer, and this seems relevant in the greater context of tailoring photochemical reactivities.
Introduction
Conventional photoreactions typically aim at one catalytic turnover per absorbed photon, but recently many new applications of multi-photon excitation strategies in photoredox catalysis were developed.1 In these cases, two (or more) photons are required per catalytic turnover, but thermodynamically unusually challenging reactions become accessible as a result of the combined energy input from multiple photons. Most systems investigated in this context operate on the basis of a single photocatalyst, for example involving consecutive photoinduced electron transfer (conPET),2–10 photoionization processes,11,12 structural in situ catalyst modifications,13–15 or light-driven catalyst recovery.16,17 The combination of a photoredox catalyst with a co-catalyst is less common,18,19 and combinations of two photoactive catalysts are yet very scarce.20,21,22,39Sensitized triplet–triplet annihilation upconversion (sTTA-UC) is an attractive approach to combining the energy input of multiple photons.1,23–25 In this case, the co-catalyst (the so-called annihilator) is not directly excited, but instead its lowest triplet excited state is formed via triplet–triplet energy transfer (TTET) from the primary photosensitizer and then the annihilator's fluorescent singlet excited state is populated by triplet–triplet annihilation. That singlet excited state can either activate the substrate directly, or it can be quenched by sacrificial electron donors to yield a radical anion, which subsequently engages with the substrate.24 Direct photo-excitation of the respective singlet excited state of the annihilator would typically require ultraviolet excitation, and consequently the main advantage of the sTTA-UC strategy is that much lower-energy input radiation can be employed. In the recent past, different upconversion systems relying on blue,24,26,27 green,28–30 red,31 or near-infrared excitation sources32–35 were developed for applications in photo(redox) catalysis. With rare exceptions,33 mainly reductive dehalogenations have been reported until now, typically in the presence of a sacrificial amine donor, sometimes complemented by a radical trapping reagent. This limited reaction scope stands in contrast to the very large diversity of light-driven reactions relying on monophotonic mechanisms.36 It seems surprising that almost exclusively reductive substrate activations have been investigated so far by multi-photonic excitation mechanisms, whereas only few oxidative transformations have been targeted.37,38
Recently, we reported a new strategy for reductive dehalogenations and detosylations based on red light irradiation (Fig. 1h) by combining [Cu(dap)2]+ (dap = 2,9-dianisyl-1,10-phenanthroline) together with 9,10-dicyanoanthracene (DCA).39 The involved mechanism resembled the Z-scheme of natural photosynthesis, and this represents a potentially widely applicable concept for multi-photonic catalysis. In this previous study, photoinduced electron transfer from [Cu(dap)2]+ generated DCA radical anions (DCA˙−), which engaged in reductive substrate activation upon further excitation. In the present study, we targeted oxidative substrate activations from the fluorescent singlet excited state of DCA (1*DCA), which has a very high oxidation potential of +1.99 V vs. SCE.40 There have been many previous reports on photocatalytic applications of DCA41–49 and related cyanoarenes,40 though usually involving direct ultraviolet or blue excitation. Here, we targeted the formation of 1*DCA via upconversion of red light, to accomplish chemical transformations requiring high oxidative potentials, contrasting our recent studies, in which excitation of DCA˙− led to thermodynamically challenging reductions under red irradiation.39 In other words, the goal was to completely reverse the photoredox reactivity of a biphotonic reaction system with DCA a key catalytic component.
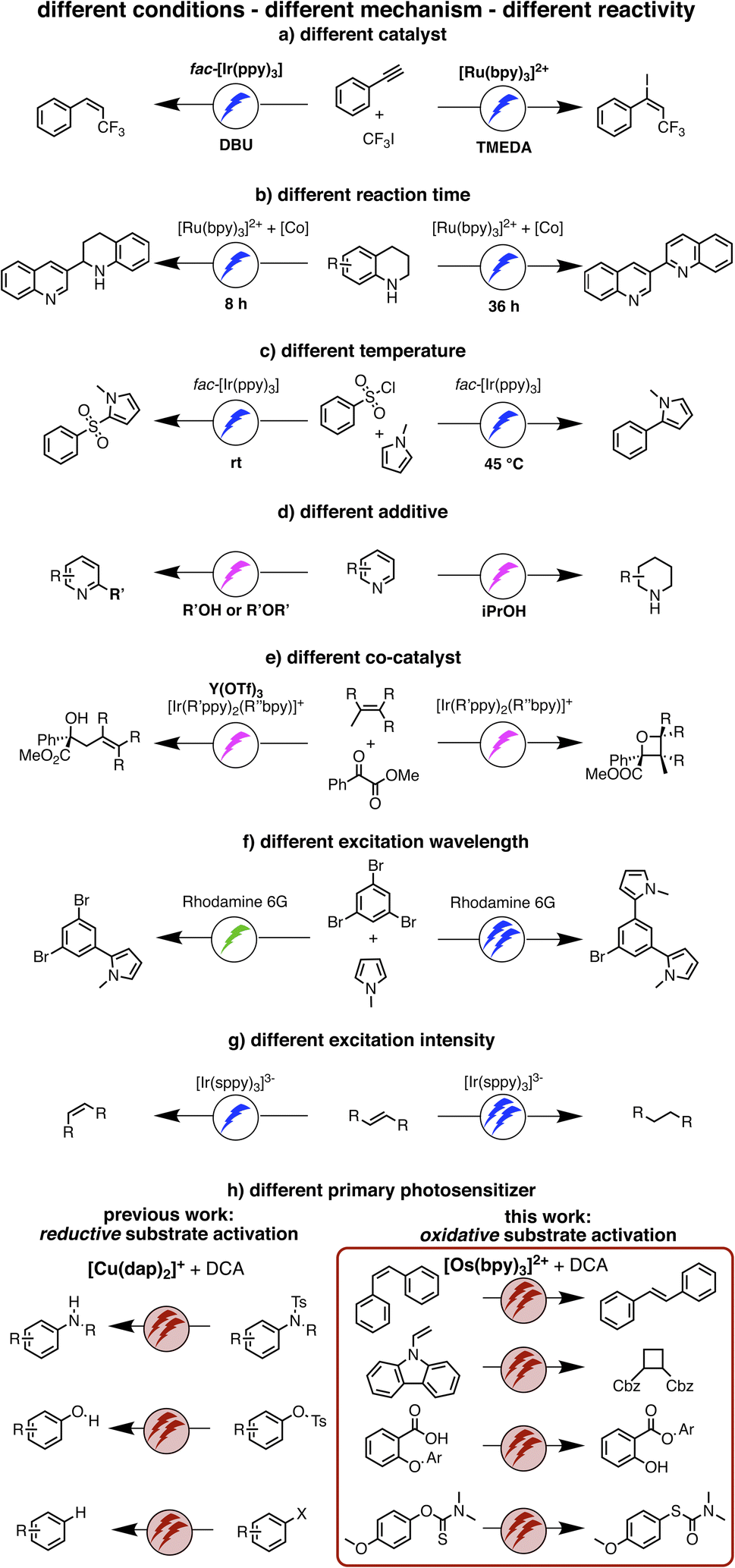 | ||
| Fig. 1 (a–g) Previously reported examples of light-driven reactions with changes of reaction conditions to achieve different photochemical reaction outcomes, mostly relying on monophotonic (blue or green) excitation.5,51–56 (h) Our work on reductive substrate activation39 and oxidative substrate activation with red light (this work). Depending on whether the primary photosensitizer is a CuI or an OsII polypyridine complex, different biphotonic reaction mechanisms are operative, and either very negative reduction potentials (left) or very positive oxidation potentials are accessible (right). | ||
The idea of controlling and switching the reaction outcome by changing the conditions in photo(redox) catalysis has obtained attention mostly for monophotonic mechanisms until now.50 For example, the change of the photocatalyst from [Ru(bpy)3]2+ to fac-[Ir(ppy)3] led to different products for the reaction of trifluoromethyl iodide with an alkyne (Fig. 1a).51 In other studies, the reaction conditions rather than the catalyst played a key role in controlling the change of the formed photoproducts, for example the reaction time (Figure 1b),52 the temperature (Figure 1c),53 the use of additives (Figure 1d),54 or co-catalysts (Fig. 1e).55 Furthermore, a change of the excitation wavelength (Fig. 1f)5 and the irradiation light intensity (Fig. 1g)56 have been found to alter the achievable photoreactions by switching between monophotonic and biphotonic excitation. However, apart from these two latter examples, divergent photochemical reactivities of biphotonic systems have been less in focus,12 presumably due to their inherently high overall complexity.
Against this background, we were curious whether we could revert the reactivity of the photosensitizer/DCA combination from the previously observed highly reducing behaviour (involving DCA˙−), to strong oxidizing reactivity (based on 1*DCA) as noted above, while keeping red light as a low-energy irradiation source. We envisioned that sTTA-UC would give access to the strongly oxidizing 1*DCA, which should be readily possible with red light given the low triplet energy of ∼1.8 eV of 3*DCA.57 Thus, instead of the previously exploited photoinduced electron transfer (PET) from [Cu(dap)2]+ to DCA,39 we targeted TTET as an initial elementary reaction step, and this required a different primary photosensitizer which replaces [Cu(dap)2]+. In other words, we aimed to actively change the mechanism of a biphotonic reaction by altering the photosensitizer (Fig. 1h). Detailed mechanistic insight into all light-driven reaction steps is the key to understanding observable photochemical reactivities,24,58–68 and here such understanding provides the basis for the targeted active reaction control.
The number of photocatalysts with suitable absorption properties in the red spectral range is somewhat limited,31,69–72 and we chose a well-known polypyridyl complex of osmium(II) from as class of robust sensitizers that have been used successfully for triplet–triplet annihilation previously.73–79 In combination with DCA, the change from [Cu(dap)2]+ to [Os(bpy)3]2+ as a primary light absorber completely alters the photochemical reactivity of the system. With the copper(I) sensitizer in the presence of a sacrificial electron donor, we observed the reductive activation of substrates requiring potentials below −2.0 V vs. SCE, whereas with the osmium(II) sensitizer the oxidative activation of substrates requiring potentials close to +2.0 V vs. SCE is achievable. The specific reactions explored herein are four different overall redox-neutral reactions that all rely on an initial light-induced substrate oxidation step initiated by 1*DCA (Fig. 1h).
Results and discussion
Triplet–triplet annihilation upconversion
In a first set of experiments we explored whether the combination of DCA and [Os(bpy)3]2+ is indeed suitable for our purposes. In principle, sTTA-UC is well established for many anthracene derivatives,76,80–83 and TTET from [Os(bpy)3]2+ to DCA is expected to be thermodynamically viable based on the relevant triplet energies.39,57 On the other hand, a more thorough analysis suggests that photoinduced electron transfer could in principle compete (detailed discussion in Section 4.2.2 of the ESI†), hence the anticipated TTET (and ensuing upconversion) reactivity cannot be taken for granted. Transient UV-vis absorption spectroscopy provides unambiguous evidence for 3*DCA as the only detectable photoproduct formed directly upon quenching of [Os(bpy)3]2+, hence TTET is clearly the dominant reaction pathway of excited 3*[Os(bpy)3]2+. In the following, the TTA upconversion properties were investigated in acetonitrile, acetone and dichloromethane. Less polar solvents were not considered due to anticipated challenges associated with photoinduced charge separation and enhanced exciplex formation (see below).84–91The choice of solvent can play an important role for the performance of an upconversion system,92–94 and in our case solubility issues had to be considered. In very polar solvents, the solubility of DCA decreases, while in more apolar solvents the charged osmium(II) complex becomes less soluble. The solubility of the DCA annihilator is the more important factor, due to the higher concentrations needed of this component in comparison to the sensitizer.
The rate constants for energy transfer from 3*[Os(bpy)3]2+ to DCA (kTTET) and for the triplet–triplet annihilation (kTTA) are both largely independent on the solvent (Table 1). Due to the abovementioned differences in solubility and different excited state properties of the osmium(II) sensitizer (e.g. the lifetime τ0 change from 94 ns in dichloromethane to 61 ns in acetonitrile), a direct comparison of the determined upconversion quantum yield (ΦsTTA-UC) in different solvents is not straightforward. However, a clear trend to better upconversion quantum yields in less polar solvents is found. Specifically, the respective quantum yield is ∼1.5% in dichloromethane (with respect to a theoretical maximum of 50%, Fig. 2a and b) whereas in acetone and acetonitrile values of ∼0.13% and ∼0.22%, respectively, are achievable (Table 1). The main reason for these discrepant ΦsTTA-UC values is most likely the substantially higher solubility of DCA in dichloromethane, leading to more efficient excited-state quenching of the osmium(II) sensitizer by TTET. Even in the case of dichloromethane with 3 mM of dissolved DCA (used for the determination of ΦsTTA-UC in Table 1), less than 50% of 3*[Os(bpy)3]2+ is quenched (kTTET·[DCA]/(kTTET·[DCA] + τ0−1) = (3 × 109 M−1 s−1 × 3 mM)/(3 × 109 M−1 s−1 × 3 mM + (94 ns)−1) ≈ 46%). Thus, the solubility of DCA limits the overall achievable upconversion quantum yield (further discussion in Section 4.3.3 of the ESI†). In the other investigated solvents, the DCA solubility is only ∼0.5 mM (Table 1), and consequently the resulting TTET efficiency is further diminished.
| Solvent | k TTET/M−1 s−1 | k TTA/M−1 s−1 | [DCA]/mM | λ ex/nm | Φ sTTA-UC /% | ΔEb/eV |
|---|---|---|---|---|---|---|
| a The data were recorded in de-aerated solvents. Details for the determination of the triplet–triplet energy transfer rate constant kTTET, the triplet–triplet annihilation rate constant kTTA, the sensitized triplet–triplet annihilation upconversion quantum yield ΦsTTA-UC and the apparent pseudo anti-Stokes shifts ΔE are in the ESI in Sections 4.2 and 4.3. b The pseudo anti-Stokes shift and the upconversion quantum yield were determined using the concentrations of DCA indicated in the table. The concentration of [Os(bpy)3](PF6)2 was different for excitation with a 635 nm cw laser (20 μM) and a 705 nm cw laser excitation (50 μM). | ||||||
| Acetonitrile | 4 × 109 | 6.9 × 109 | 0.5 | 635 | ∼0.22 | 0.88 |
| 705 | ∼0.10 | 1.06 | ||||
| Acetone | 4 × 109 | 6.4 × 109 | 0.5 | 635 | ∼0.13 | 0.88 |
| 705 | ∼0.15 | 1.08 | ||||
| Dichloromethane | 3 × 109 | 6.7 × 109 | 3 | 635 | ∼1.5 | 0.73 |
| 705 | ∼1.4 | 0.92 | ||||
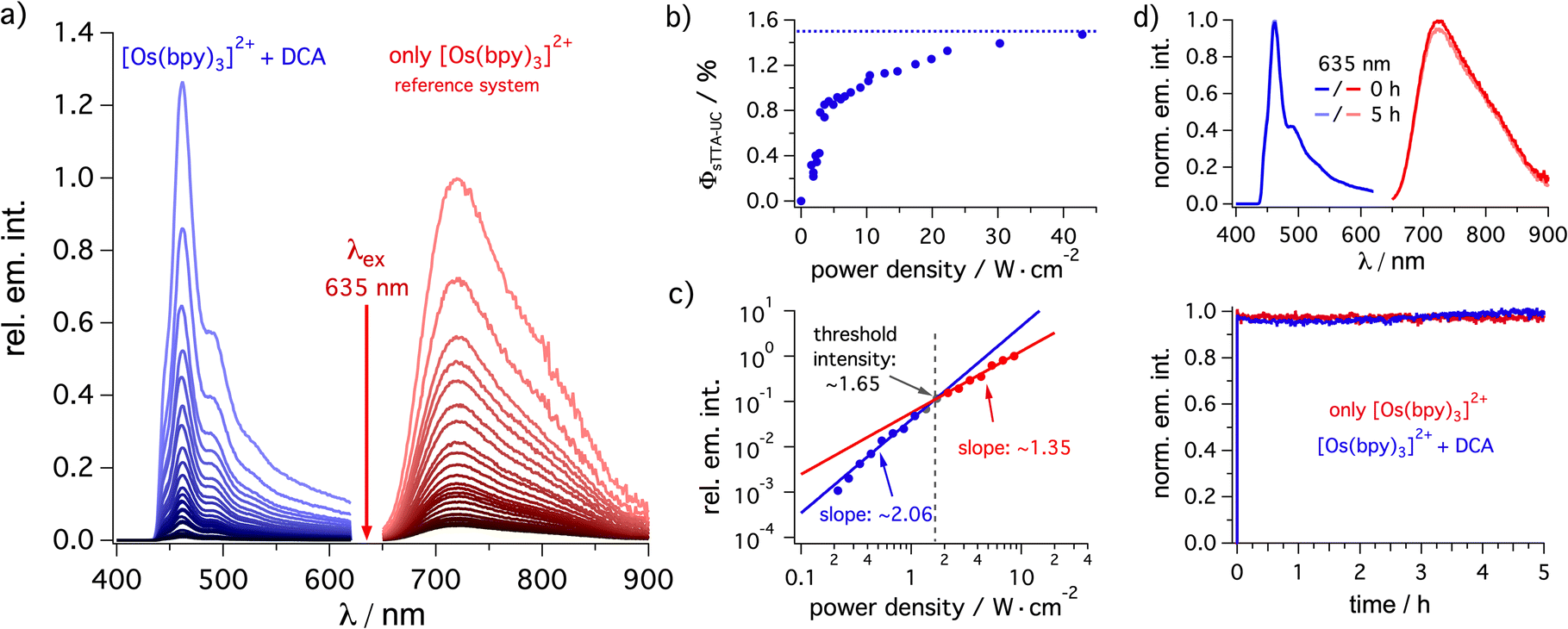 | ||
| Fig. 2 Triplet–triplet annihilation upconversion analysis. (a) Steady-state emission spectra of [Os(bpy)3](PF6)2 (20 μM) in de-aerated dichloromethane at 20 °C excited at 635 nm with a cw laser using different excitation densities in the absence of DCA (red, reference system) and upconverted delayed fluorescence spectra in the presence of DCA (3 mM) under otherwise identical conditions. (b) Upconversion quantum yield (ΦsTTA-IC) determination based on the data in (a) using the emission quantum yield of [Os(bpy)3](PF6)2 (2.7%, determined against [Ru(bpy)3]2+ in a separate experiment) as reference system. (c) Determination of the threshold for the changeover from the quadratic excitation power dependence to a nearly linear regime, based on the integrated upconversion fluorescence intensity of DCA (3 mM) sensitized by [Os(bpy)3](PF6)2 (20 μM) in de-aerated dichloromethane (ESI Section 4.3.2†). (d) Photostability of [Os(bpy)3](PF6)2 (50 μM) (red trace) and of the [Os(bpy)3](PF6)2 (50 μM)/DCA (3 mM) upconversion system (blue trace) under 635 nm cw laser excitation (450 mW). The upconverted emission was detected at 460 nm while the prompt sensitizer emission was detected at 720 nm (ESI Section 4.3.5†). The upper part of this panel contains the emission spectra of both solutions before and after 5 hours of irradiation. | ||
As expected for a biphotonic process, a quadratic dependence of the DCA upconversion fluorescence intensity on the excitation power density is found (Fig. 2c, blue). At elevated excitation power densities, the change towards an approximately linear regime with a slope of 1.35 on a double logarithmic scale (Fig. 2c, red) indicates that the so-called strong annihilation limit is reached. From the intersection between the (approximately) quadratic and linear fits, a threshold of ∼1.65 W cm−2 can be estimated. A reference experiment without annihilator revealed almost perfectly linear power dependence of the prompt osmium(II) photoluminescence with a slope of 0.97 (Section 4.3.2 in the ESI†).
In all three investigated solvents (weak) excimer emission is detectable under upconversion conditions, manifesting in an emission band extending up to ∼650 nm (ESI, Section 4.3.2†). In acetonitrile and acetone the highest-energy DCA fluorescence peak around 440 nm (corresponding to the 0–0 transition) is clearly detectable under upconversion conditions, whereas in dichloromethane this band is not detectable anymore due to inner filter effects caused by the higher concentration in this solvent (Fig. 2a).95 Thus, the delayed fluorescence band maximum in dichloromethane corresponds to the first vibrational progression member of the prompt DCA fluorescence and consequently, the apparent pseudo anti-Stokes shift for dichloromethane is 0.73 eV, while in acetone and acetonitrile the emission maximum is 0.88 eV higher in energy than the excitation wavelength (Table 1, see ESI Section 4.3.2† for details). Using excitation at 705 nm instead of 635 nm, similar upconversion quantum yields ΦsTTA-UC remain achievable (Table 1),34 but now pseudo anti-Stokes shifts over 1 eV result (Table 1). While systems with significantly better upconversion quantum yields are known,80,96–102 our apparent pseudo anti-Stokes shifts are close to current state of the art.31,76,94,98,101,103,104
Since the highest upconversion quantum yield was obtained in dichloromethane, the long-term photostability was studied in this specific solvent (Fig. 2d). Irradiation over 5 hours revealed that our photosensitizer/annihilator combination is very robust and essentially no loss in the upconversion intensity is detectable (blue trace). Unsurprisingly, the reference without annihilator revealed that the osmium(II)-based sensitizer is also very photostable (red trace). Even after 14 days of irradiating a reaction mixture with a red cw laser, the characteristic signals of the sensitizer as well as the annihilator remain easily detectable by 1H-NMR spectroscopy (ESI Section 4.8.5†). These measurements indicate a good photostability of our sensitizer/catalyst combination under long-term red irradiation. Overall, these properties seem useful for the application of the [Os(bpy)3]2+/DCA combination for photoredox catalysis via upconversion.
Cis–trans photo-isomerisation of stilbene
Isomerisation reactions of stilbenes sensitized by direct blue or UV excitation of DCA have long been known to proceed through a mechanism involving stilbene radical cations.87,105,109–114 The prototypical example is the photo-isomerisation of cis-stilbene (1) to trans-stilbene (1-P),105,109,111 and this reaction seemed promising for our proof-of-concept study involving upconversion of red light, particularly because this is a simple reaction for which comparatively fast substrate conversion can be anticipated on the basis of the previous UV irradiation studies.109 First reactions with 5 mol% of DCA under blue LED irradiation (440 nm, 40 W) in acetonitrile and dichloromethane resulted in conversions of over 75% within 1 hour (see ESI Table S1 and Fig. S1†). In dichloromethane ∼85% of 1-P are present in the photostationary state,107 while in acetonitrile the reactant is essentially completely converted to product.109For initial tests with red light irradiation in the presence of 1 mol% of [Os(bpy)3]2+, a collimated high-power 623 nm LED (min. 3.8 W) was used to drive the reaction on an NMR scale (Fig. 3, ESI Section 2.3†). In contrast to what is observed under blue irradiation above, the reaction progress depended strongly on the solvents. In acetonitrile-d3, only 18% product formation was observed after 2 hours while in dichloromethane-d2 61% of 1-P were detected after the same irradiation time (ESI, Fig. S1†). This finding is in line with the significantly higher triplet–triplet annihilation upconversion quantum yield in dichloromethane compared to acetonitrile. In the latter solvent, 96% formation of trans-stilbene required a much longer irradiation time of 40 hours (ESI, Table S2†).
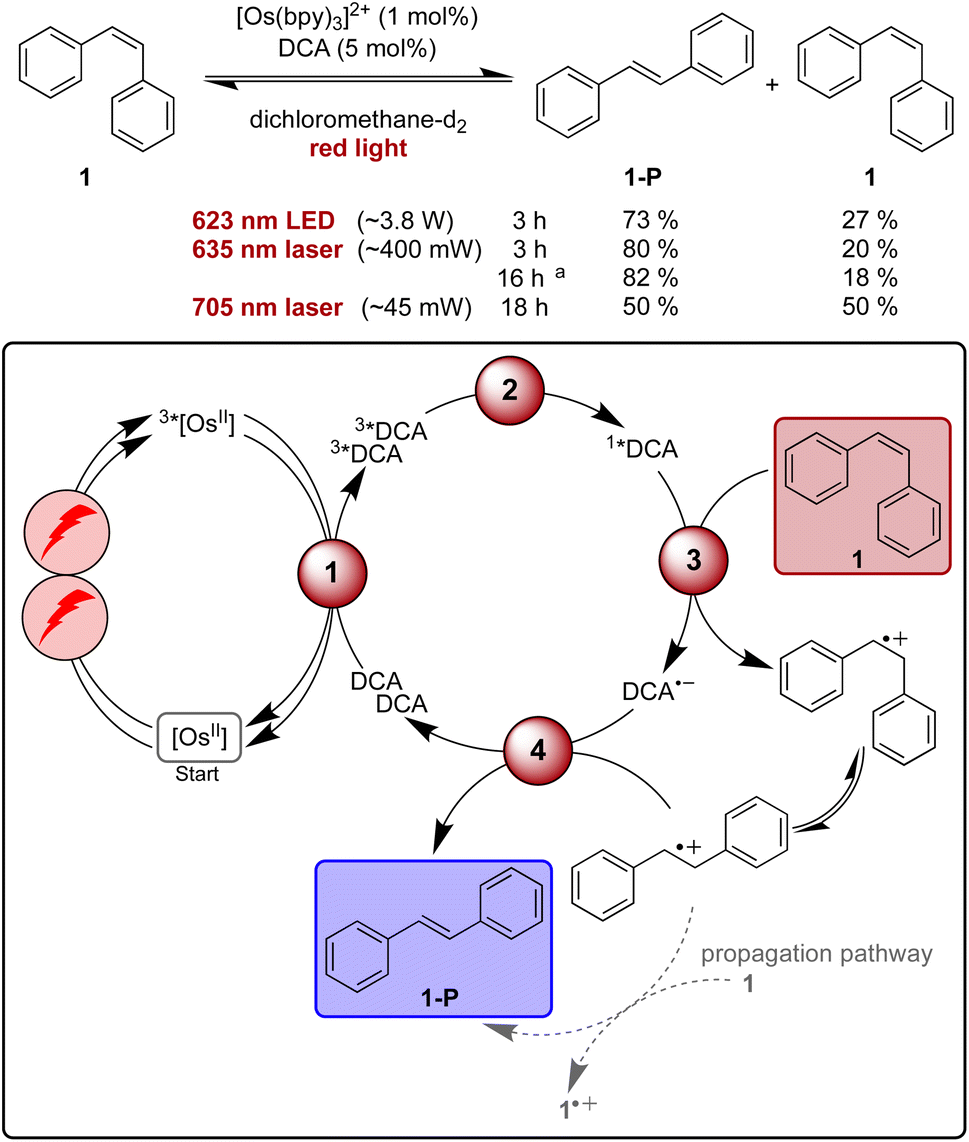 | ||
| Fig. 3 Light-driven isomerisation of cis-stilbene (1) under red-light irradiation along with a plausible mechanism involving triplet–triplet energy transfer (1), triplet–triplet annihilation upconversion (2), substrate activation (3), and catalyst recovery (4). Main steps of substrate activation adapted from a known monophotonic mechanism.105 A radical chain propagation pathway is plausible for this reaction (details in text and ESI Section 4.8.3†),106 and further intermediates (for example a transient dimer radical cation) can be involved.107–109 All yields were determined by 1H-NMR spectroscopy using 1,4-dioxane as internal standard. Further information is in Section 4.4 of the ESI.† | ||
As our spectroscopic investigations indicated a threshold intensity around 1.65 W cm−2 (Fig. 2c) for the changeover from quadratic to linear dependence of the upconversion process, excitation with a cw laser rather than a LED was tested in the next step. The cw laser power is lower (∼400 mW) compared to the LED (min. 3.8 W, further discussion in ESI Section 2.3.1†), yet the laser source gave a slightly better product yield (80% of 1-P) compared to 73% after 3 h of irradiation with the collimated LED for NMR-scale reactions (Fig. 3). Thus, the laser irradiation setup, which provides a higher power density, due to the more collimated nature of the laser beam, was used for further investigations. With prolonged irradiation times of 16 h, the loading of [Os(bpy)3]2+ could be lowered to 0.1 mol% (Fig. 3), resulting in a turnover number of over 1500 with respect to the photosensitizer.
Contrary to previous studies,109 following direct excitation of DCA at 405 nm in the absence of sensitizer and under otherwise identical conditions as for the sensitized catalysis, no clear evidence for a propagation pathway was found neither in acetonitrile (photochemical quantum yield ΦPC ∼0.13) nor in dichloromethane (ΦPC ∼0.03, ESI Section 4.8.3†) under the conditions used herein. An experiment with our upconversion system under red-light irradiation results in an only slightly smaller value for the quantum yield of cis- to trans-stilbene isomerization in dichloromethane (ΦPC ∼0.015), which is close to the triplet–triplet annihilation upconversion quantum yield in that solvent (Table 1). It does not seem plausible that every successful upconversion event leads to product, and therefore contributions from a radical chain mechanism under upconversion conditions seem likely (ESI Section 4.8.3†). When using a comparatively low-power 705 nm cw laser (∼45 mW), 50% of the substrate was converted to the trans-isomer after 18 hours of irradiation (Fig. 1). As for all other investigated light-driven transformations reported herein, no product formation was observed in the absence of light or any of the catalysts (ESI Tables S3–S8†).
Mechanistic investigations reveal that photoinduced electron transfer from cis-stilbene 1 to 1*DCA (1.46 × 1010 M−1 s−1 in dichloromethane) as well as from the trans-stilbene product 1-P to 1*DCA (1.54 × 1010 M−1 s−1) are both essentially diffusion-limited (the diffusion limit in dichloromethane at 20 °C is 1.5 × 1010 M−1 s−1).57,109 Direct substrate activation by triplet energy transfer from 3*[Os(bpy)3]2+, by reductive quenching of 3*[Os(bpy)3]2+ or by quenching of 3*DCA are not important for the observed reactivity in our system (ESI Section 4.4†). Consequently, the mechanism involving triplet–triplet annihilation upconversion presented in Fig. 3 (including direct oxidation of 1 as well as contributions from a radical chain propagation pathway) is suggested as the main contributor for product formation in this red-light driven isomerisation reaction.
Carbon–carbon bond formation via [2 + 2] cycloaddition
Functionalization of radical cations and [2 + 2] cycloaddition reactions catalysed by pyrylium salts or DCA as photocatalysts have been reported previously.42,87,116–119 One of the challenges associated with [2 + 2] cycloadditions of alkenes is the reversibility of the overall process, due to a possible (unwanted) ring-opening reaction of the formed product.116 Therefore, the use of a redox mediator can be advantageous to selectively activate the substrate whilst preventing the undesirable backward reaction of the cycloaddition product.116,120 9-Vinylcarbazole (2) has been found to undergo successful [2 + 2] cycloaddition sensitized by an organic dye in the absence of a redox mediator, and therefore this specific substrate was selected as model compound for our proof-of-principle studies.116Starting with direct blue excitation (440 nm LED, 40 W) of 1*DCA in acetone, 80% of product 2-P were formed in 0.5 hours (ESI Fig. S2 and Table S3†), whereas only 19% of 2-P (Table S3†) formed in dichloromethane under identical conditions. Consequently, acetone was used for the reaction under red cw laser irradiation, despite the markedly lower upconversion yield in this solvent (Table 1). Under 635 nm laser irradiation the reaction becomes significantly slower and 90 hours are needed to achieve 71% product formation (Fig. 4), presumably due to the low upconversion quantum yield in acetone. Based on the comparison of the quantum yield for the photochemical [2 + 2] cycloaddition reaction and ΦsTTA-UC, a radical chain propagation pathway is likely to contribute under red light irradiation (ESI Section 4.8.3†).106
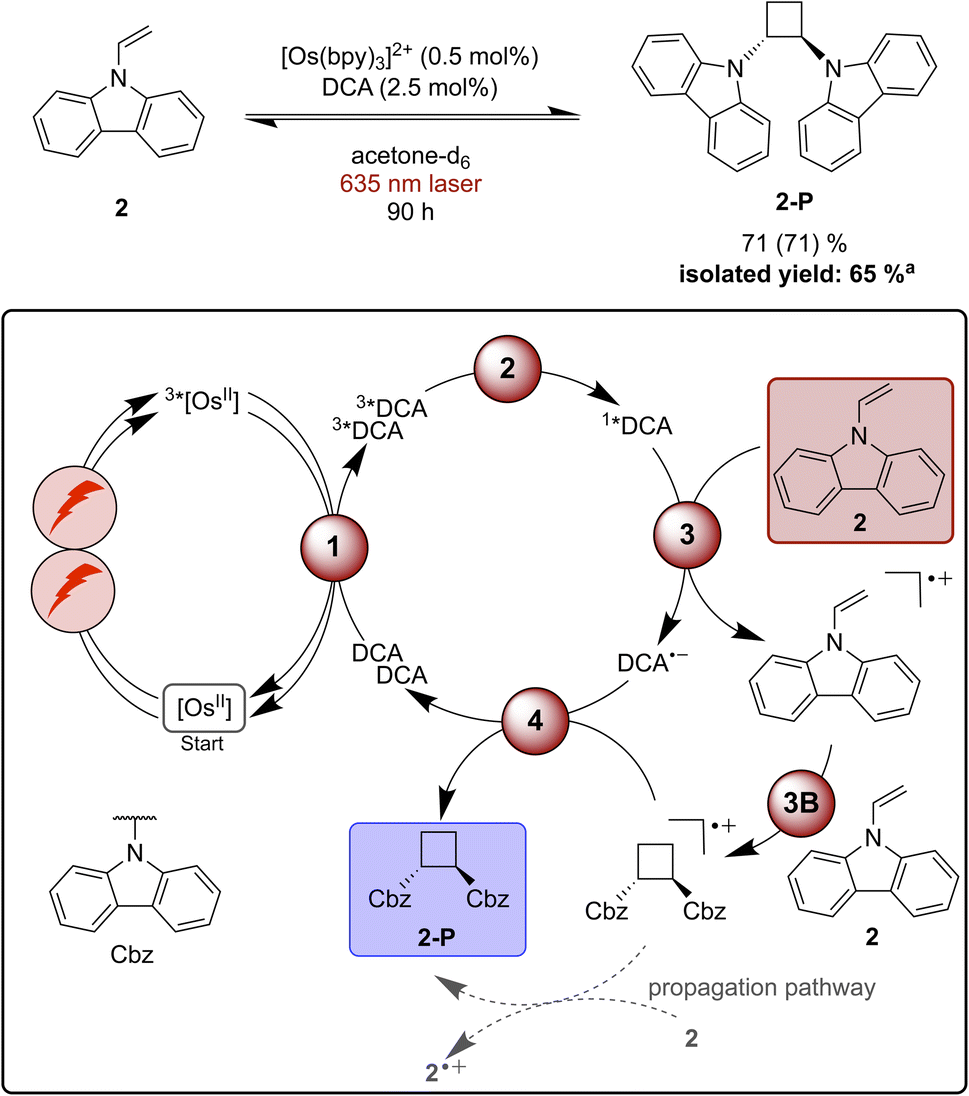 | ||
| Fig. 4 [2 + 2] Cycloaddition of vinylcarbazole (2) under red-light irradiation and a plausible mechanism involving triplet–triplet energy transfer (1), triplet–triplet annihilation upconversion (2), substrate activation (3), cycloaddition (3B), and catalyst recovery (4). Main steps of substrate activation adapted from a previously reported monophotonic mechanism.106,115,116 A radical chain propagation pathway is likely for this reaction (details in text and ESI Section 4.8.3†).106 The yield (conversion in parenthesis) was determined by 1H-NMR spectroscopy using 1,4-dioxane as internal standard. Further information is in Section 4.5 of the ESI.†aProduct isolation was performed on a 400 μmol scale of 2 under 623 nm LED irradiation. | ||
Unwanted quenching of triplet-excited DCA or direct 3MLCT quenching of the osmium(II) sensitizer by the substrate are inefficient based on spectroscopic investigations (ESI Section 4.5†). The proposed mechanism for the formation of product 2-P (Fig. 4) is similar to the stilbene isomerisation discussed above, except of course for the cycloaddition step between the radical cation and one equivalent of unreacted substrate (step 3B in Fig. 4).116,121
When using a 623 nm LED (min. 3.8 W) instead of the 635 nm cw laser, a larger volume can be irradiated, and the reaction can be performed better on somewhat enlarged scale (ESI Section 4.3†).120 Using 0.5 mol% sensitizer and 2.5 mol% of DCA in acetone, the reaction was performed on a 400 μmol scale and 2-P was isolated with a yield of 65% after 90 hours of irradiation. This example demonstrates that in principle upconversion with red light is useable for (small-scale) preparative reactions with (collimated) LED irradiation – a light source that is typically more available in synthetic laboratories than cw lasers.
Newman–Kwart rearrangement
Newman–Kwart rearrangements have previously been investigated by chemical,125 photochemical122,124 and electrochemical123 approaches involving single-electron substrate oxidation. This represents an attractive alternative way for this reaction to occur under mild conditions compared to thermal activation, which typically requires temperatures between 200 °C and 300 °C.126 Fast substrate oxidation with pyrylium salts and detailed mechanistic studies have been reported previously, providing a helpful basis for our investigations of red light driven Newman–Kwart rearrangement.122,124Initial experiments focusing on the direct formation of 1*DCA with blue light revealed that both naphthalene and TBAPF6 (tetra-n-butylammonium hexafluorophosphate) are necessary additives in dichloromethane to accomplish the Newman–Kwart rearrangement of substrate 3 in acceptable yields (see ESI Table S5† for details). Under [Os(bpy)3]2+ sensitized conditions with 635 nm laser irradiation the reaction became markedly slower, and after 20 hours a yield of 10% for the rearrangement product 3-P was found for a reaction on NMR scale. Essentially linear product formation as a function time was observed (ESI Fig. S3†), and after 14 days of irradiation 88% of rearranged product 3-P was obtained (Fig. 5). This indicates a very good long-term stability of our upconversion system, in line with the short-term investigations in Fig. 2d. A more potent irradiation source in combination with a flow setup would be desirable to accelerate this reaction,127 but the proof of principle is now made.
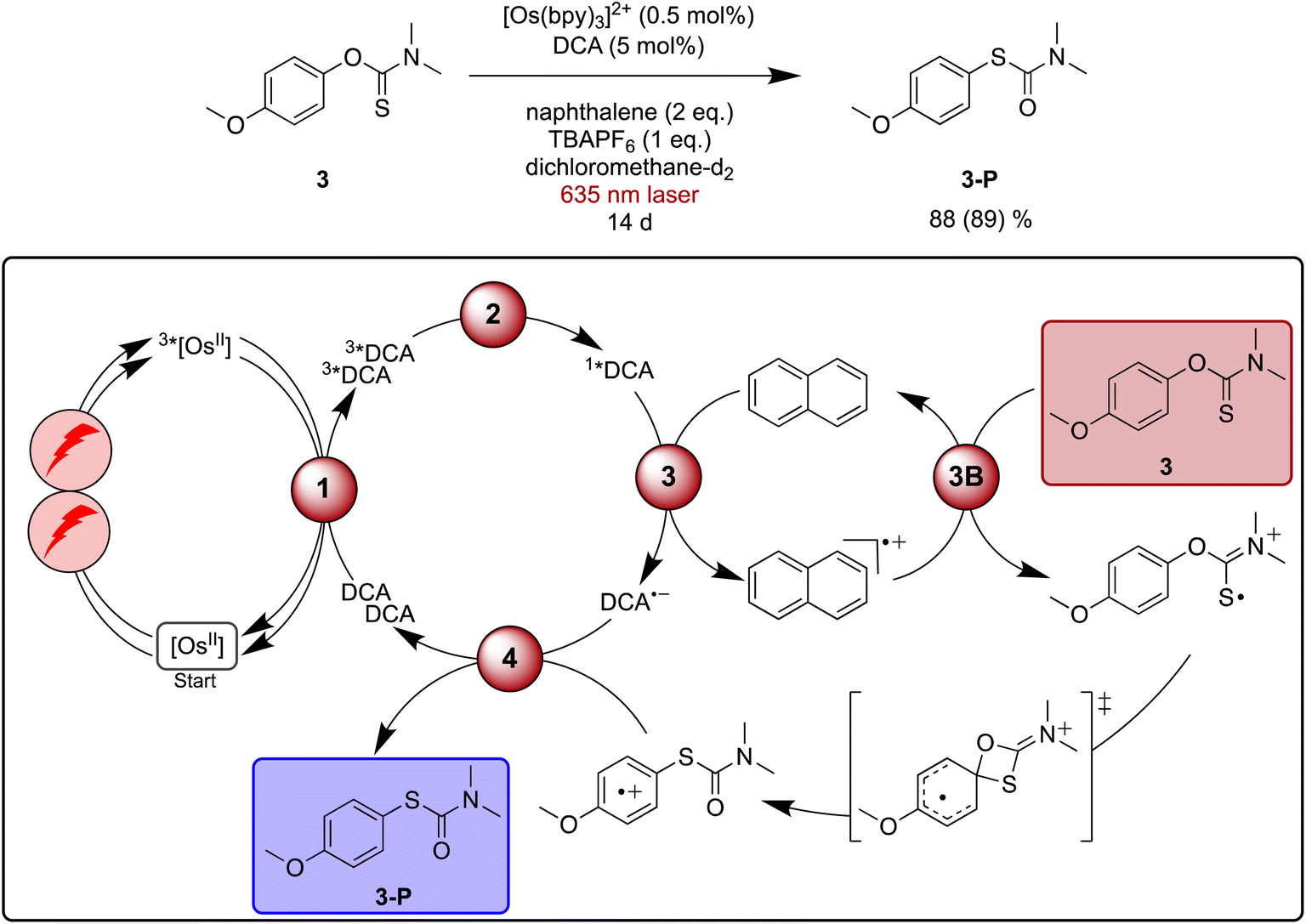 | ||
| Fig. 5 Light-driven Newman–Kwart rearrangement of substrate 3 under red-light irradiation, along with a plausible mechanism including triplet–triplet energy transfer (1), triplet–triplet annihilation upconversion (2), excited state quenching by naphthalene mediator (3), substrate activation (3B), and catalyst recovery (4). The main steps of substrate activation are analogous those postulated previously for a monophotonic mechanism.122,123 A possible radical chain propagation pathway and previously proposed off-cycle intermediates are omitted for simplicity.122–124 Yield (conversion in parenthesis) was determined by 1H-NMR spectroscopy against 1,4-dioxane as internal standard. Further information is in Section 4.6 of the ESI.† | ||
Pulsed laser experiments and UV-vis transient absorption spectroscopy under upconversion conditions in the presence of naphthalene provide no clear evidence for oxidized naphthalene and reduced DCA, most likely because the overall process is inefficient and because there are spectrally overlapping signals with 3*DCA. However, an unexpected ground-state absorption bleach appears between 450 and 700 nm in the presence of naphthalene. A comparison to spectro-electrochemical data indicates that this bleach signals the interim formation of [Os(bpy)3]3+ (ESI Section 4.8.4†), suggesting a cascade reactivity to form DCA˙− and Nap˙+ after TTA-UC, followed by subsequent electron transfer from [Os(bpy)3]2+ to Nap˙+. This latter step represents an unwanted deactivation pathway, which can serve as a plausible explanation for the slow reaction progress observed under red light driven upconversion conditions, in addition to the low sTTA-UC quantum yield. Based on the mechanistic insights gained here, it seems reasonable to assume that this deactivation pathway involving sensitizer oxidation generally contributes to the observable significantly prolonged reaction times when using upconversion from the red instead of direct blue DCA excitation.
Based on the abovementioned previous Newman–Kwart rearrangement studies involving single electron transfer,122–124 a simplified mechanism for our upconversion system including a naphthalene-mediated pathway is proposed in Fig. 5. Previous studies of related reactions in acetonitrile furthermore proposed a radical chain propagation pathway as well as off-cycle intermediates, which are not considered here for simplicity.122–124
Ether-to-ester rearrangement
As a final reaction example, the rearrangement of the aryl ether 4 to aryl ester 4-Pvia aryl migration was explored. This reaction has been previously reported with perylene diimide or acridinium salts as photocatalysts under blue irradiation in the presence of a sub-stoichiometric amount of base.128–130 The base is necessary to facilitate substrate oxidation, which in this case occurs from the deprotonated carboxylate (4—). Similar to what was found above for the Newman–Kwart rearrangement reaction, TBAPF6 helps accelerate product formation in dichloromethane upon direct excitation of DCA with blue light (Table S7†), and several different bases provided good results. However, with aliphatic amines such as 1,8-diazabicyclo(5.4.0)undec-7-ene (DBU, +1.24 V vs. SCE)131 or triethylamine (+0.69 V vs. SCE)132 unwanted reductive quenching of 1*DCA (+1.99 V vs. SCE),3,133 or even reductive quenching of 3*DCA can potentially occur.39 Consequently, 2,4,6-trimethylpyridine (tMePy, 2.25 V vs. SCE, ESI Section 4.1.4†) was chosen. Expectedly, the quenching of 1*DCA by tMePy is two orders below the diffusion limit (108 M−1 s−1), and our reaction optimisation experiments under blue irradiation (440 nm LED, 40 W) indicated that a stoichiometric amount of tMePy is beneficial (ESI Table S7†).Under irradiation with a 635 nm cw laser, the NMR scale rearrangement of ether 4 to 64% of ester 4-P required 140 h, while 81% of starting material was consumed. Isolation of the product on a slightly larger scale with a 623 nm LED resulted in a yield of 75% after the same reaction time.
Time-resolved experiments indicate that 1*DCA is quenched by 4— with a rate constant of 5 × 109 M−1 s−1, whereas no DCA fluorescence quenching is detectable without substrate deprotonation (ESI Section 4.7†). Control experiments monitoring the photoluminescence of [Os(bpy)3]2+ provide evidence for static quenching, manifesting in a shortened decay time independent of the concentration of 4—. This suggests that [Os(bpy)3]2+ and 4— aggregate, which possibly opens an unwanted deactivation pathway that could lead to diminished product formation under upconversion conditions compared to direct excitation of DCA with blue light. However, the addition of salt seems to counteract this deactivation by diminishing unwanted aggregation (ESI Section 4.7.2†). Control experiments confirm that DCA is an indispensable component for the red light driven reaction (Table S8†), hence photoexcited [Os(bpy)3]2+ seems to be unable to oxidize 4— in catalytically relevant amounts.
General mechanistic aspects
The four investigated proof-of-principle reactions reported above provide direct insight into what factors and processes affect the efficiency of biphotonic reactions in comparison to traditional monophotonic mechanisms. First, the optimal reaction conditions are not necessarily identical for both excitation strategies. This finding is illustrated by the photo-isomerisation of cis- to trans-stilbene, where the limited solubility and the resulting low triplet–triplet annihilation upconversion quantum yield clearly affect the overall reaction progress over time. While under blue light irradiation comparable reaction progress has been found in different solvents, under upconversion conditions significant differences in reaction progress are observed. Second, when two (photo)catalysts are present, additional (unwanted) deactivation steps can occur. Our time-resolved spectroscopic investigations of the naphthalene-mediated rearrangement of substrate 3, where the naphthalene radical cation intermediate was found to be partially deactivated in an unproductive pathway by electron transfer from the [Os(bpy)3]2+ sensitizer, illustrate this aspect. Third, as illustrated by the ether-to-ester rearrangement in Fig. 6, aggregation of a negatively charged substrate with the cationic photosensitizer is possible in apolar solvents, and the addition of salt can become important to minimize deactivation of the photosensitizer in an unwanted side reaction (Fig. 5 and 6). All of these aspects need to be considered in biphotonic mechanisms (Fig. 7), in addition to complications that are already present under monophotonic excitation conditions. Specifically, this includes unproductive pathways such as excimer and exciplex formation, as well as unproductive excited state deactivation by intersystem crossing to 3*DCA, or direct decay of 1*DCA to the ground state (ESI Sections 4.8.1 and 4.8.4†).134–136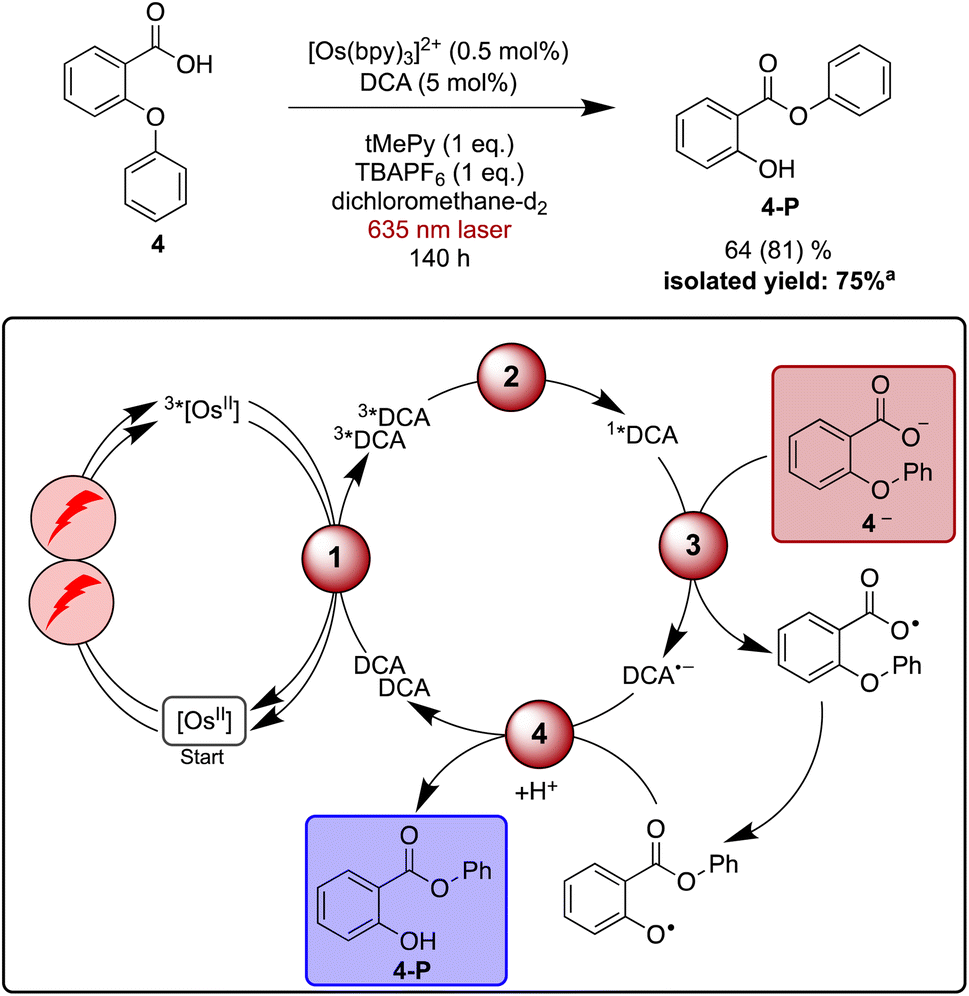 | ||
| Fig. 6 Red light-driven ether-to-ester rearrangement of substrate 4 along with a plausible mechanism including triplet–triplet energy transfer (1), triplet–triplet annihilation upconversion (2), substrate activation (3), and catalyst recovery (4). Main steps of substrate activation adapted from a previously published monophotonic mechanism.128 Yield (conversion in parenthesis) was determined by 1H-NMR spectroscopy using 1,4-dioxane as internal standard. Further information is in Section 4.7 of the ESI.†aProduct isolation was performed on a 250 μmol scale of 4 under 623 nm LED irradiation. | ||
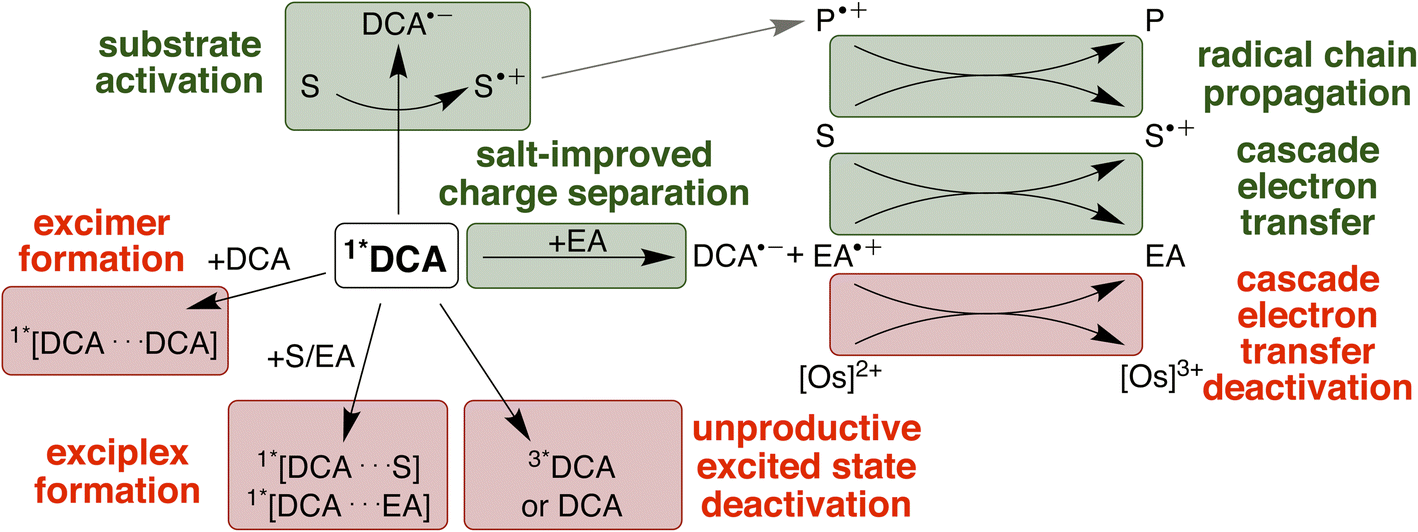 | ||
| Fig. 7 Summary of productive and unproductive pathways for the deactivation of 1*DCA after triplet–triplet annihilation upconversion. EA = electron acceptor/mediator; S = substrate; P = product. Favourable processes are coloured in green, unwanted processes in red. | ||
On the other hand, radical chain propagation pathways can contribute substantially to the overall reaction efficiency and seem to play important roles for at least two of the four investigated reactions herein. Furthermore, our studies suggest that improved cage escape yields to form solvent separated radical ions are achievable by the addition of salt, and the use of redox mediators for electron transfer cascade pathways is beneficial for some of the upconversion-driven photoreactions. Many of these effects can differ significantly based on exact conditions (solvent, additive, reaction type), and our work indicates that complementary UV-vis transient absorption and time-resolved luminescence spectroscopic investigations can provide particularly valuable insights when targeting biphotonic excitation strategies. Some of the challenges outlined above could potentially be tackled with higher annihilator concentrations78 (for example with modified DCA structures)49 or photosensitizers with longer lifetimes.80,98 Any enhancement of the sTTA-UC quantum yield would likely substantially improve the achievable photochemical quantum yield under red light irradiation in our system (ESI Section 4.8.3†). To counteract unwanted cascade deactivations, different catalysts137–139 or chemical environments8,21,61 might be promising. The main productive and unproductive pathways for the deactivation of 1*DCA are summarized in Fig. 7 (further discussion in ESI Section 4.8†).
Conclusions
Biphotonic excitation strategies for photoredox catalysis have received growing attention within the last few years, and many different mechanisms have been considered.1 Until now, most biphotonic reaction approaches rely on a single light absorber (acridinium dyes in Fig. 8a, DCA in Fig. 8c). Recently, we explored a reaction system comprised of two different light absorbers, where we observed photoinduced electron transfer from a CuI photosensitizer to DCA, and subsequent (secondary) excitation of DCA˙− led to thermodynamically challenging reductive dehalogenations and detosylations (Fig. 8e).39 In the present study, the use of an OsII photosensitizer leads to triplet–triplet energy transfer to DCA and ensuing upconversion, and the resulting highly oxidizing 1*DCA (instead of DCA˙−) induces the photochemical reactions of interest (Fig. 8f).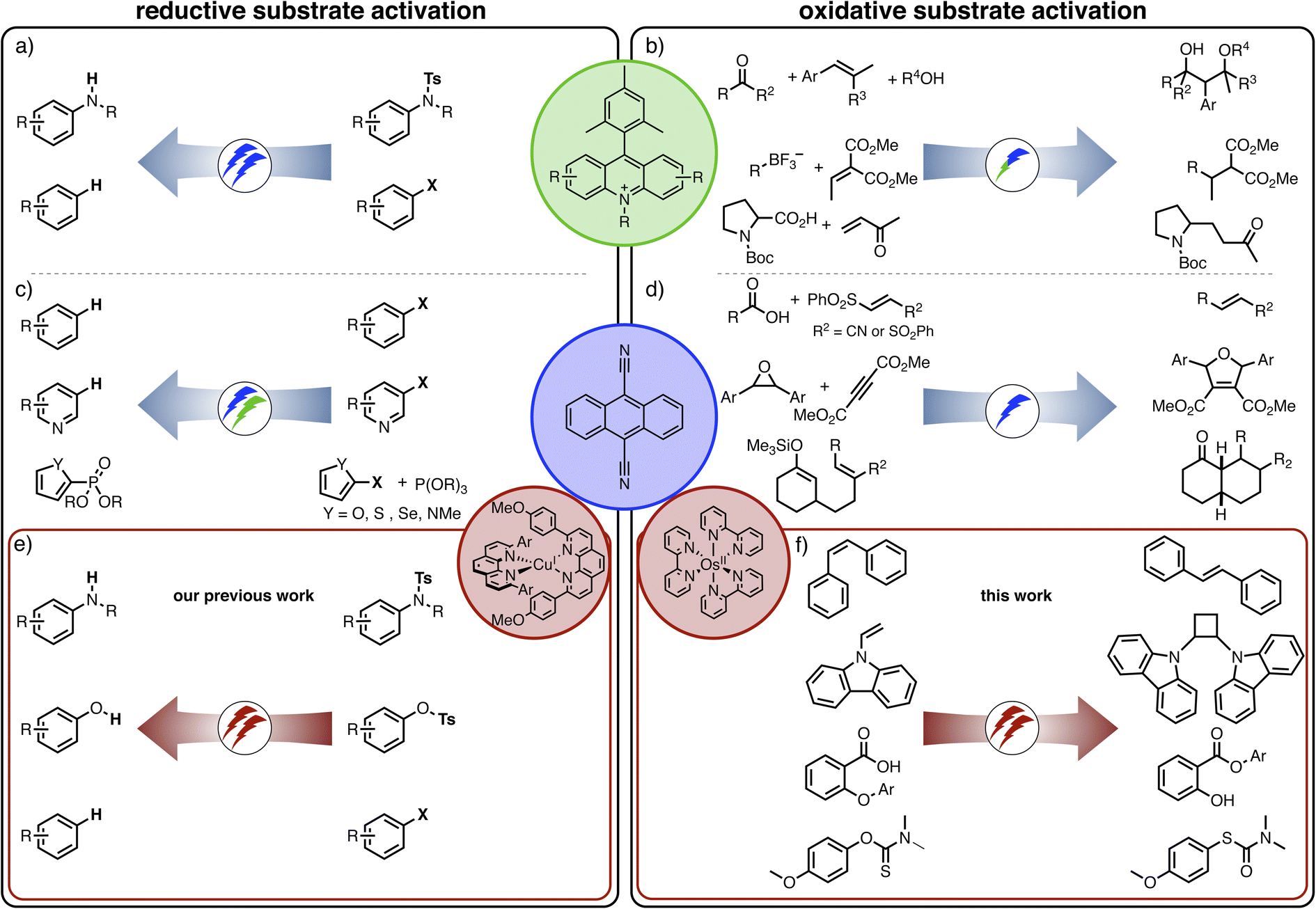 | ||
| Fig. 8 Selected examples of different approaches to oxidative and reductive substrate activation with acridinium dyes and direct (monophotonic) blue/green light (a and b),4,140–142 direct excitation of DCA in the blue or green spectral range (c and d),3,9,42,43,119 and two-catalyst systems operating under red light irradiation (e and f).39 Overall redox-neutral reactions requiring an initial substrate oxidation step have been chosen on the right-hand side of the figure. | ||
Conceptually, this changeover in photochemical reactivity (Fig. 8e and f) is related to the changeover between oxidative and reductive substrate activation with acridinium salts (Fig. 8a and b)4,143 under direct (monophotonic) blue excitation, or via consecutive photoinduced electron transfer (ConPET) using DCA3,40 or perylene diimide (PDI)40 under blue and green irradiation (Fig. 8c and d). Similar reactivity changes might become achievable with other systems, and the idea of investigating new photochemical mechanisms rather than developing new catalysts could become more important in the future.38 Increasing attention to elucidate mechanisms will be helpful in this context.24,58,59,62,68,80,144–151 Mechanistic studies seem particularly desirable for oxidative substrate activation, for several reasons. First, reductive dehalogenations often involve a fast and irreversible bond cleavage step,152 whereas oxidative substrate activations can be reversible reactions. Second, low cage escape yields for photogenerated radical pairs, unwanted back-electron transfer, and deactivation via electron transfer cascades are typically less important (or at least less in focus)146,153 for reductive transformations, but have to be considered carefully for oxidative transformations originating from singlet excited states (Fig. 7).38,49,116,154–156
The development of new red or near-infrared light absorbing catalysts could potentially give access to new photochemical applications with low-energy input light.31,79,120,151,157–164 Furthermore, recent interest in triplet–triplet annihilation upconversion will likely promote the development of more sophisticated systems with lower threshold irradiation intensities that permit more effective excitation by LEDs, higher photochemical quantum yields and larger pseudo anti-Stokes shifts,34,76,78,96,100,104,165 resulting in more efficient setups for applications in photo(redox) catalysis in the future.166,167 We hope that the concepts and proof-of-principle reactions presented herein can serve as useful inspiration for future catalytic applications with red and near-infrared light.9,23,163,168–170
Data availability
All experimental data, procedures for data analysis and pertinent data sets are provided in the ESI.† All electronic data is available on https://Zenodo.org.Author contributions
F. G. conceived the project, designed photochemical studies, carried out spectroscopic, synthetic and electrochemical work, analysed data and performed photocatalytic measurements; O. S. W. conceived the project and provided guidance. The manuscript was written through contributions of all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the Swiss National Science Foundation through grant number 200020_207329. Björn Pfund is acknowledged for helpful discussions.References
- F. Glaser, C. Kerzig and O. S. Wenger, Angew. Chem., Int. Ed., 2020, 59, 10266–10284 CrossRef CAS.
- I. Ghosh, T. Ghosh, J. I. Bardagi and B. König, Science, 2014, 346, 725–728 CrossRef CAS PubMed.
- M. Neumeier, D. Sampedro, M. Májek, V. A. de la Peña O'Shea, A. Jacobi von Wangelin and R. Pérez-Ruiz, Chem.–Eur. J., 2018, 24, 105–108 CrossRef CAS.
- I. A. MacKenzie, L. Wang, N. P. R. Onuska, O. F. Williams, K. Begam, A. M. Moran, B. D. Dunietz and D. A. Nicewicz, Nature, 2020, 580, 76–80 CrossRef CAS PubMed.
- I. Ghosh and B. König, Angew. Chem., Int. Ed., 2016, 55, 7676–7679 CrossRef CAS.
- R. Naumann, F. Lehmann and M. Goez, Angew. Chem., Int. Ed., 2018, 57, 1078–1081 CrossRef CAS PubMed.
- R. Naumann, C. Kerzig and M. Goez, Chem. Sci., 2017, 8, 7510–7520 RSC.
- M. Giedyk, R. Narobe, S. Weiß, D. Touraud, W. Kunz and B. König, Nat. Catal., 2020, 3, 40–47 CrossRef CAS.
- J. C. Herrera-Luna, D. D. Díaz, M. C. Jiménez and R. Pérez-Ruiz, ACS Appl. Mater. Interfaces, 2021, 13, 48784–48794 CrossRef CAS PubMed.
- J. Soika, C. McLaughlin, T. Neveselý, C. G. Daniliuc, J. J. Molloy and R. Gilmour, ACS Catal., 2022, 12, 10047–10056 CrossRef CAS.
- T. Kohlmann, R. Naumann, C. Kerzig and M. Goez, Photochem. Photobiol. Sci., 2017, 16, 1613–1622 CrossRef CAS PubMed.
- C. Kerzig, X. Guo and O. S. Wenger, J. Am. Chem. Soc., 2019, 141, 2122–2127 CrossRef CAS PubMed.
- J. P. Cole, D.-F. Chen, M. Kudisch, R. M. Pearson, C.-H. Lim and G. M. Miyake, J. Am. Chem. Soc., 2020, 142, 13573–13581 CrossRef CAS PubMed.
- H. Li, X. Tang, J. H. Pang, X. Wu, E. K. L. Yeow, J. Wu and S. Chiba, J. Am. Chem. Soc., 2021, 143, 481–487 CrossRef CAS.
- T. U. Connell, C. L. Fraser, M. L. Czyz, Z. M. Smith, D. J. Hayne, E. H. Doeven, J. Agugiaro, D. J. D. Wilson, J. L. Adcock, A. D. Scully, D. E. Gómez, N. W. Barnett, A. Polyzos and P. S. Francis, J. Am. Chem. Soc., 2019, 141, 17646–17658 CrossRef CAS PubMed.
- Y. Qiao, Q. Yang and E. J. Schelter, Angew. Chem., Int. Ed., 2018, 57, 10999–11003 CrossRef CAS.
- P. W. Antoni and M. M. Hansmann, J. Am. Chem. Soc., 2018, 140, 14823–14835 CrossRef PubMed.
- I. Ghosh, R. S. Shaikh and B. König, Angew. Chem., Int. Ed., 2017, 56, 8544–8549 CrossRef CAS PubMed.
- M. S. Coles, G. Quach, J. E. Beves and E. G. Moore, Angew. Chem., Int. Ed., 2020, 59, 9522–9526 CrossRef CAS.
- A. Hu, Y. Chen, J.-J. Guo, N. Yu, Q. An and Z. Zuo, J. Am. Chem. Soc., 2018, 140, 13580–13585 CrossRef CAS.
- C. Kerzig and M. Goez, Chem. Sci., 2016, 7, 3862–3868 RSC.
- A. Chinchole, M. A. Henriquez, D. Cortes-Arriagada, A. R. Cabrera and O. Reiser, ACS Catalysis, 2022, 12(21), 13549–13554 CrossRef CAS.
- R. Pérez-Ruiz, Top. Curr. Chem., 2022, 380, 23 CrossRef.
- F. Glaser, C. Kerzig and O. S. Wenger, Chem. Sci., 2021, 12, 9922–9933 RSC.
- J. Castellanos-Soriano, D. Álvarez-Gutiérrez, M. C. Jiménez and R. Pérez-Ruiz, Photochem. Photobiol. Sci., 2022, 21, 1175–1184 CrossRef CAS PubMed.
- M. Majek, U. Faltermeier, B. Dick, R. Pérez-Ruiz and A. Jacobi von Wangelin, Chem.–Eur. J., 2015, 21, 15496–15501 CrossRef CAS PubMed.
- T. J. B. Zähringer, J. A. Moghtader, M.-S. Bertrams, B. Roy, M. Uji, N. Yanai and C. Kerzig, Angew. Chem., Int. Ed., 2022, e202215340 Search PubMed.
- M. Häring, R. Pérez-Ruiz, A. J. Von Wangelin and D. D. Díaz, Chem. Commun., 2015, 51, 16848–16851 RSC.
- C. Kerzig and O. S. Wenger, Chem. Sci., 2018, 9, 6670–6678 RSC.
- S. Liu, H. Liu, L. Shen, Z. Xiao, Y. Hu, J. Zhou, X. Wang, Z. Liu, Z. Li and X. Li, Chem. Eng. J., 2022, 431, 133377 CrossRef CAS.
- J. B. Bilger, C. Kerzig, C. B. Larsen and O. S. Wenger, J. Am. Chem. Soc., 2021, 143, 1651–1663 CrossRef CAS.
- M. Freitag, N. Möller, A. Rühling, C. A. Strassert, B. J. Ravoo and F. Glorius, ChemPhotoChem, 2019, 3, 24–27 CrossRef CAS.
- B. D. Ravetz, A. B. Pun, E. M. Churchill, D. N. Congreve, T. Rovis and L. M. Campos, Nature, 2019, 565, 343–346 CrossRef CAS.
- L. Huang, W. Wu, Y. Li, K. Huang, L. Zeng, W. Lin and G. Han, J. Am. Chem. Soc., 2020, 142, 18460–18470 CrossRef CAS PubMed.
- L. Huang, L. Zeng, Y. Chen, N. Yu, L. Wang, K. Huang, Y. Zhao and G. Han, Nat. Commun., 2021, 12, 122 CrossRef CAS PubMed.
- C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS.
- D. Rombach and H. A. Wagenknecht, Angew. Chem., Int. Ed., 2020, 59, 300–303 CrossRef CAS PubMed.
- K. Targos, O. P. Williams and Z. K. Wickens, J. Am. Chem. Soc., 2021, 143, 4125–4132 CrossRef CAS PubMed.
- F. Glaser and O. S. Wenger, JACS Au, 2022, 2, 1488–1503 CrossRef CAS PubMed.
- N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS PubMed.
- C. Yang, J.-D. Yang, Y.-H. Li, X. Li and J.-P. Cheng, J. Org. Chem., 2016, 81, 12357–12363 CrossRef CAS.
- E. Alfonzo, F. S. Alfonso and A. B. Beeler, Org. Lett., 2017, 19, 2989–2992 CrossRef CAS.
- L. M. Kammer, B. Lipp and T. Opatz, J. Org. Chem., 2019, 84, 2379–2392 CrossRef CAS PubMed.
- Y. Matsui, T. Ikeda, Y. Takahashi, M. Kamata, M. Akagi, Y. Ohya, R. Fujino, H. Namai, E. Ohta, T. Ogaki, T. Miyashi, S. Tero-Kubota, K. Mizuno and H. Ikeda, Asian J. Org. Chem., 2017, 6, 458–468 CrossRef CAS.
- Z. L. Wang, J. Chen, Y. H. He and Z. Guan, J. Org. Chem., 2021, 86, 3741–3749 CrossRef CAS PubMed.
- H. T. Qin, X. Xu and F. Liu, ChemCatChem, 2017, 9, 1409–1412 CrossRef CAS.
- G. Pandey and R. Laha, Angew. Chem., Int. Ed., 2015, 54, 14875–14879 CrossRef CAS PubMed.
- P. A. Waske and J. Mattay, Tetrahedron, 2005, 61, 10321–10330 CrossRef CAS.
- M. J. P. Mandigma, J. Žurauskas, C. I. MacGregor, L. J. Edwards, A. Shahin, L. D'Heureuse, P. Yip, D. J. S. Birch, T. Gruber, J. Heilmann, M. P. John and J. P. Barham, Chem. Sci., 2022, 13, 1912–1924 RSC.
- Y. Sakakibara and K. Murakami, ACS Catal., 2022, 12, 1857–1878 CrossRef CAS.
- N. Iqbal, J. Jung, S. Park and E. J. Cho, Angew. Chem., 2014, 126, 549–552 CrossRef.
- Z. Jia, Q. Yang, L. Zhang and S. Luo, ACS Catal., 2019, 9, 3589–3594 CrossRef CAS.
- S. K. Pagire, A. Hossain and O. Reiser, Org. Lett., 2018, 20, 648–651 CrossRef CAS.
- T. McCallum, S. P. Pitre, M. Morin, J. C. Scaiano and L. Barriault, Chem. Sci., 2017, 8, 7412–7418 RSC.
- J. Zheng, X. Dong and T. P. Yoon, Org. Lett., 2020, 22, 6520–6525 CrossRef CAS.
- C. Kerzig and O. S. Wenger, Chem. Sci., 2019, 10, 11023–11029 RSC.
- M. Montalti, A. Credi, L. Prodi and M. T. Gandolfi, Handbook of Photochemistry, CRC Press, Third edn, 2006 Search PubMed.
- B. G. Stevenson, E. H. Spielvogel, E. A. Loiaconi, V. M. Wambua, R. V. Nakhamiyayev and J. R. Swierk, J. Am. Chem. Soc., 2021, 143, 8878–8885 CrossRef CAS.
- C. J. Zeman, S. Kim, F. Zhang and K. S. Schanze, J. Am. Chem. Soc., 2020, 142, 2204–2207 CrossRef CAS PubMed.
- N. A. Till, L. Tian, Z. Dong, G. D. Scholes and D. W. C. MacMillan, J. Am. Chem. Soc., 2020, 142, 15830–15841 CrossRef CAS.
- R. Fayad, S. Engl, E. O. Danilov, C. E. Hauke, O. Reiser and F. N. Castellano, J. Phys. Chem. Lett., 2020, 11, 5345–5349 CrossRef CAS PubMed.
- L. Tian, N. A. Till, B. Kudisch, D. W. C. MacMillan and G. D. Scholes, J. Am. Chem. Soc., 2020, 142, 4555–4559 CrossRef CAS PubMed.
- Y. Zhang, T. S. Lee, J. L. Petersen and C. Milsmann, J. Am. Chem. Soc., 2018, 140, 5934–5947 CrossRef CAS.
- J. P. Menzel, B. B. Noble, J. P. Blinco and C. Barner-Kowollik, Nat. Commun., 2021, 12, 1691 CrossRef CAS PubMed.
- S. Gisbertz, S. Reischauer and B. Pieber, Nat. Catal., 2020, 3, 611–620 CrossRef CAS.
- F. Glaser, C. B. Larsen, C. Kerzig and O. S. Wenger, Photochem. Photobiol. Sci., 2020, 19, 1035–1041 CrossRef CAS.
- T. Noël and E. Zysman-Colman, Chem Catal., 2022, 2, 468–476 CrossRef.
- L. Buzzetti, G. E. M. Crisenza and P. Melchiorre, Angew. Chem., Int. Ed., 2019, 58, 3730–3747 CrossRef CAS PubMed.
- K. Watanabe, N. Terao, I. Kii, R. Nakagawa, T. Niwa and T. Hosoya, Org. Lett., 2020, 22, 5434–5438 CrossRef CAS PubMed.
- H. Janeková, M. Russo, U. Ziegler and P. Štacko, Angew. Chem., 2022, 134, e202204391 CrossRef.
- C. Grundke, R. C. Silva, W. R. Kitzmann, K. Heinze, K. T. De Oliveira and T. Opatz, J. Org. Chem., 2022, 87, 5630–5642 CrossRef CAS.
- L. Mei, J. M. Veleta and T. L. Gianetti, J. Am. Chem. Soc., 2020, 142, 12056–12061 CrossRef CAS PubMed.
- Y. Sasaki, M. Oshikawa, P. Bharmoria, H. Kouno, A. Hayashi-Takagi, M. Sato, I. Ajioka, N. Yanai and N. Kimizuka, Angew. Chem., Int. Ed., 2019, 58, 17827–17833 CrossRef CAS.
- Y. Sasaki, S. Amemori, H. Kouno, N. Yanai and N. Kimizuka, J. Mater. Chem. C, 2017, 5, 5063–5067 RSC.
- D. Liu, Y. Zhao, Z. Wang, K. Xu and J. Zhao, Dalton Trans., 2018, 47, 8619–8628 RSC.
- Y. Wei, M. Zheng, L. Chen, X. Zhou and S. Liu, Dalton Trans., 2019, 48, 11763–11771 RSC.
- D. Wei, X. Li, L. Shen, Y. Ding, K. Liang and C. Xia, Org. Chem. Front., 2021, 8, 6364–6370 RSC.
- R. Haruki, Y. Sasaki, K. Masutani, N. Yanai and N. Kimizuka, Chem. Commun., 2020, 56, 7017–7020 RSC.
- Z. Yuan, J. He, Z. Mahmood, L. Xing, S. Ji, Y. Huo and H. L. Zhang, Dyes Pigm., 2022, 199, 110049 CrossRef CAS.
- C. Wang, F. Reichenauer, W. R. Kitzmann, C. Kerzig, K. Heinze and U. Resch-Genger, Angew. Chem., Int. Ed., 2022, 61, e202202238 CAS.
- S. Baluschev, T. Miteva, V. Yakutkin, G. Nelles, A. Yasuda and G. Wegner, Phys. Rev. Lett., 2006, 97, 7–9 CrossRef.
- A. Olesund, V. Gray, J. Mårtensson and B. Albinsson, J. Am. Chem. Soc., 2021, 143, 5745–5754 CrossRef CAS PubMed.
- D. Dzebo, K. Moth-Poulsen and B. Albinsson, Photochem. Photobiol. Sci., 2017, 16, 1327–1334 CrossRef CAS.
- R. C. Kanner and C. S. Foote, J. Am. Chem. Soc., 1992, 114, 678–681 CrossRef CAS.
- A. F. Olea, D. R. Worrall and F. Wilkinson, Photochem. Photobiol. Sci., 2003, 2, 212–217 CrossRef CAS PubMed.
- A. F. Olea, D. R. Worrall, F. Wilkinson, S. L. Williams and A. A. Abdel-Shafi, Phys. Chem. Chem. Phys., 2002, 4, 161–167 RSC.
- E. Hasegawa, K. Okada, H. Ikeda, Y. Yamashita and T. Mukai, J. Org. Chem., 1991, 56, 2170–2178 CrossRef CAS.
- S. L. P. Chang and D. I. Schuster, J. Phys. Chem., 1987, 91, 3644–3649 CrossRef CAS.
- Y. Takahashi, K. Wakamatsu, K. Kikuchi and T. Miyashi, J. Phys. Org. Chem., 1990, 3, 509–518 CrossRef CAS.
- M. Ottolenghi, C. R. Goldschmidt and R. Potashnik, J. Phys. Chem., 1971, 75, 1025–1031 CrossRef CAS.
- K. Kikuchi, M. Hoshi, T. Niwa, Y. Takahashi and T. Miyashi, J. Phys. Chem., 1991, 95, 38–42 CrossRef CAS.
- C. Ye, V. Gray, J. Mårtensson and K. Börjesson, J. Am. Chem. Soc., 2019, 141, 9578–9584 CrossRef CAS.
- Q. Zhou, M. Zhou, Y. Wei, X. Zhou, S. Liu, S. Zhang and B. Zhang, Phys. Chem. Chem. Phys., 2017, 19, 1516–1525 RSC.
- Y. Murakami, A. Motooka, R. Enomoto, K. Niimi, A. Kaiho and N. Kiyoyanagi, Phys. Chem. Chem. Phys., 2020, 22, 27134–27143 RSC.
- Y. Zhou, F. N. Castellano, T. W. Schmidt and K. Hanson, ACS Energy Lett., 2020, 5, 2322–2326 CrossRef CAS.
- A. Olesund, J. Johnsson, F. Edhborg, S. Ghasemi, K. Moth-Poulsen and B. Albinsson, J. Am. Chem. Soc., 2022, 144, 3706–3716 CrossRef CAS.
- X. Cao, B. Hu and P. Zhang, J. Phys. Chem. Lett., 2013, 4, 2334–2338 CrossRef CAS.
- N. Nishimura, V. Gray, J. R. Allardice, Z. Zhang, A. Pershin, D. Beljonne and A. Rao, ACS Mater. Lett., 2019, 1, 660–664 CrossRef CAS.
- M. Uji, N. Harada, N. Kimizuka, M. Saigo, K. Miyata, K. Onda and N. Yanai, J. Mater. Chem. C, 2022, 10, 4558–4562 RSC.
- W. Sun, A. Ronchi, T. Zhao, J. Han, A. Monguzzi and P. Duan, J. Mater. Chem. C, 2021, 9, 14201–14208 RSC.
- Y. Wei, Y. Li, M. Zheng, X. Zhou, Y. Zou and C. Yang, Adv. Opt. Mater., 2020, 8, 1902157 CrossRef CAS.
- Z. A. VanOrman and L. Nienhaus, Matter, 2021, 4, 2625–2626 CrossRef CAS.
- C. Fan, L. Wei, T. Niu, M. Rao, G. Cheng, J. J. Chruma, W. Wu and C. Yang, J. Am. Chem. Soc., 2019, 141, 15070–15077 CrossRef CAS PubMed.
- T. J. B. Zähringer, M.-S. Bertrams and C. Kerzig, J. Mater. Chem. C, 2022, 10, 4568–4573 RSC.
- F. D. Lewis, A. M. Bedell, R. E. Dykstra, J. E. Elbert, I. R. Gould and S. Farid, J. Am. Chem. Soc., 1990, 112, 8055–8064 CrossRef CAS.
- R. A. Crellin, M. C. Lambert and A. Ledwith, J. Chem. Soc. D, 1970, 682 RSC.
- Y. Kuriyama, H. Sakuragi, K. Tokumaru, Y. Yoshida and S. Tagawa, Bull. Chem. Soc. Jpn., 1993, 66, 1852–1855 CrossRef CAS.
- B. W. Zhang, Y. Kuriyama, Y. Cao and K. Tokumaru, Bull. Chem. Soc. Jpn., 1993, 66, 1859–1862 CrossRef CAS.
- F. D. Lewis, J. R. Petisce, J. D. Oxman and M. J. Nepras, J. Am. Chem. Soc., 1985, 107, 203–207 CrossRef CAS.
- T. Tamai, N. Ichinose, T. Tanaka, T. Sasuga, I. Hashida and K. Mizuno, J. Org. Chem., 1998, 63, 3204–3212 CrossRef CAS.
- F. D. Lewis, R. E. Dykstra, I. R. Gould and S. Farid, J. Phys. Chem., 1988, 92, 7042–7043 CrossRef CAS.
- K. Mizuno, N. Ichinose and Y. Otsuji, Chem. Lett., 1985, 14, 455–458 CrossRef.
- K. Mizuno, N. Ichinose and Y. Otsuji, J. Org. Chem., 1992, 57, 1855–1860 CrossRef CAS.
- Y. Kuriyama, T. Arai, H. Sakuragi and K. Tokumaru, Chem. Lett., 1992, 21, 879–882 CrossRef.
- S. Mckinley, J. V. Crawford and C. H. Wang, J. Org. Chem., 1966, 31, 1963–1964 CrossRef CAS.
- M. Riener and D. A. Nicewicz, Chem. Sci., 2013, 4, 2625 RSC.
- S. L. Mattes, H. R. Luss and S. Farid, J. Phys. Chem., 1983, 87, 4779–4781 CrossRef CAS.
- E. Hasegawa, T. Mukai and K. Yanagi, J. Org. Chem., 1989, 54, 2053–2058 CrossRef CAS.
- S. Hintz, J. Mattay, R. Van Eldik and W. F. Fu, Eur. J. Org. Chem., 1998, 1583–1596 CrossRef CAS.
- B. D. Ravetz, N. E. S. Tay, C. L. Joe, M. Sezen-Edmonds, M. A. Schmidt, Y. Tan, J. M. Janey, M. D. Eastgate and T. Rovis, ACS Cent. Sci., 2020, 6, 2053–2059 CrossRef CAS PubMed.
- M. A. Ischay, Z. Lu and T. P. Yoon, J. Am. Chem. Soc., 2010, 132, 8572–8574 CrossRef CAS PubMed.
- C. L. Cruz and D. A. Nicewicz, ACS Catal., 2019, 9, 3926–3935 CrossRef CAS.
- A. F. Roesel, M. Ugandi, N. T. T. Huyen, M. Májek, T. Broese, M. Roemelt and R. Francke, J. Org. Chem., 2020, 85, 8029–8044 CrossRef CAS PubMed.
- A. J. Perkowski, C. L. Cruz and D. A. Nicewicz, J. Am. Chem. Soc., 2015, 137, 15684–15687 CrossRef CAS.
- S. K. Pedersen, A. Ulfkjær, M. N. Newman, S. Yogarasa, A. U. Petersen, T. I. Sølling and M. Pittelkow, J. Org. Chem., 2018, 83, 12000–12006 CrossRef CAS.
- H. Kwart and E. R. Evans, J. Org. Chem., 1966, 31, 410–413 CrossRef CAS.
- L. Buglioni, F. Raymenants, A. Slattery, S. D. A. Zondag and T. Noël, Chem. Rev., 2022, 122, 2752–2906 CrossRef CAS PubMed.
- S.-F. Wang, X.-P. Cao and Y. Li, Angew. Chem., Int. Ed., 2017, 56, 13809–13813 CrossRef CAS.
- F.-F. Tan, X.-Y. He, W.-F. Tian and Y. Li, Nat. Commun., 2020, 11, 6126 CrossRef CAS.
- J. C. Gonzalez-Gomez, N. P. Ramirez, T. Lana-Villarreal and P. Bonete, Org. Biomol. Chem., 2017, 15, 9680–9684 RSC.
- E. Hasegawa, N. Izumiya, T. Fukuda, K. Nemoto, H. Iwamoto, S. Takizawa and S. Murata, Tetrahedron, 2016, 72, 7805–7812 CrossRef CAS.
- Y. Pellegrin and F. Odobel, C. R. Chim., 2017, 20, 283–295 CrossRef CAS.
- M. A. Kellett, D. G. Whitten, I. R. Gould and W. R. Bergmark, J. Am. Chem. Soc., 1991, 113, 358–359 CrossRef CAS.
- M. Itoh, J. Am. Chem. Soc., 1974, 96, 7390–7394 CrossRef CAS.
- E. A. Chandross and J. Fercuson, J. Chem. Phys., 1967, 47, 2557–2560 CrossRef CAS.
- I. R. Gould, R. H. Young, L. J. Mueller and S. Farid, J. Am. Chem. Soc., 1994, 116, 8176–8187 CrossRef CAS.
- P. Herr, F. Glaser, L. A. Büldt, C. B. Larsen and O. S. Wenger, J. Am. Chem. Soc., 2019, 141, 14394–14402 CrossRef CAS PubMed.
- L. A. Büldt, X. Guo, R. Vogel, A. Prescimone and O. S. Wenger, J. Am. Chem. Soc., 2017, 139, 985–992 CrossRef.
- C. E. McCusker and F. N. Castellano, Chem. Commun., 2013, 49, 3537–3539 RSC.
- L. Pitzer, F. Sandfort, F. Strieth-Kalthoff and F. Glorius, J. Am. Chem. Soc., 2017, 139, 13652–13655 CrossRef CAS.
- T. Chinzei, K. Miyazawa, Y. Yasu, T. Koike and M. Akita, RSC Adv., 2015, 5, 21297–21300 RSC.
- J. B. McManus, N. P. R. Onuska and D. A. Nicewicz, J. Am. Chem. Soc., 2018, 140, 9056–9060 CrossRef CAS.
- B. Zilate, C. Fischer and C. Sparr, Chem. Commun., 2020, 56, 1767–1775 RSC.
- A. J. Rieth, M. I. Gonzalez, B. Kudisch, M. Nava and D. G. Nocera, J. Am. Chem. Soc., 2021, 143, 14352–14359 CrossRef CAS PubMed.
- Y. Qin, R. Sun, N. P. Gianoulis and D. G. Nocera, J. Am. Chem. Soc., 2021, 143, 2005–2015 CrossRef CAS.
- A. Aydogan, R. E. Bangle, A. Cadranel, M. D. Turlington, D. T. Conroy, E. Cauët, M. L. Singleton, G. J. Meyer, R. N. Sampaio, B. Elias and L. Troian-Gautier, J. Am. Chem. Soc., 2021, 143, 15661–15673 CrossRef CAS PubMed.
- S. Rafiq, B. Fu, B. Kudisch and G. D. Scholes, Nat. Chem., 2021, 13, 70–76 CrossRef CAS PubMed.
- N. Kandoth, J. Pérez Hernández, E. Palomares and J. Lloret-Fillol, Sustainable Energy Fuels, 2021, 5, 638–665 RSC.
- K. Goliszewska, K. Rybicka-Jasińska, J. A. Clark, V. I. Vullev and D. Gryko, ACS Catal., 2020, 10, 5920–5927 CrossRef CAS.
- A. Aydogan, R. E. Bangle, S. De Kreijger, J. C. Dickenson, M. L. Singleton, E. Cauët, A. Cadranel, G. J. Meyer, B. Elias, R. N. Sampaio and L. Troian-Gautier, Catal. Sci. Technol., 2021, 11, 8037–8051 CAS.
- E. H. Spielvogel, B. G. Stevenson, M. J. Stringer, Y. Hu, L. A. Fredin and J. R. Swierk, J. Org. Chem., 2022, 87, 223–230 CrossRef CAS PubMed.
- L. Pause, M. Robert and J. M. Savéant, J. Am. Chem. Soc., 1999, 121, 7158–7159 CrossRef CAS.
- D. Y. Jeong, D. S. Lee, H. L. Lee, S. Nah, J. Y. Lee, E. J. Cho and Y. You, ACS Catal., 2022, 12, 6047–6059 CrossRef CAS.
- E. Vauthey, C. Högemann and X. Allonas, J. Phys. Chem. A, 1998, 102, 7362–7369 CrossRef CAS.
- E. Vauthey, P. Suppan and E. Haselbach, Helv. Chim. Acta, 1988, 71, 93–99 CrossRef CAS.
- T. Niwa, K. Kikuchi, N. Matsusita, M. Hayashi, T. Katagiri, Y. Takahashi and T. Miyashi, J. Phys. Chem., 1993, 97, 11960–11964 CrossRef CAS.
- S. L. Goldschmid, E. Bednářová, L. R. Beck, K. Xie, N. E. S. Tay, B. D. Ravetz, J. Li, C. L. Joe and T. Rovis, Synlett, 2022, 33, 247–258 CrossRef CAS.
- S. De Kreijger, O. Schott, L. Troian-Gautier, E. Cauët, G. S. Hanan and B. Elias, Inorg. Chem., 2022, 61, 5245–5254 CrossRef CAS PubMed.
- P. A. Scattergood, A. Sinopoli and P. I. P. Elliott, Coord. Chem. Rev., 2017, 350, 136–154 CrossRef CAS.
- Y. Sasaki, N. Yanai and N. Kimizuka, Inorg. Chem., 2022, 61, 5982–5990 CrossRef CAS.
- D. A. W. Ross, P. A. Scattergood, A. Babaei, A. Pertegás, H. J. Bolink and P. I. P. Elliott, Dalton Trans., 2016, 45, 7748–7757 RSC.
- S. A. E. Omar, P. A. Scattergood, L. K. McKenzie, C. Jones, N. J. Patmore, A. J. H. M. Meijer, J. A. Weinstein, C. R. Rice, H. E. Bryant and P. I. P. Elliott, Inorg. Chem., 2018, 57, 13201–13212 CrossRef CAS PubMed.
- A. R. Obah Kosso, N. Sellet, A. Baralle, M. Cormier and J.-P. Goddard, Chem. Sci., 2021, 12, 6964–6968 RSC.
- K. Rybicka-Jasińska, T. Wdowik, K. Łuczak, A. J. Wierzba, O. Drapała and D. Gryko, ACS Org. Inorg. Au, 2022, 2, 422–426 CrossRef.
- M. Yang, S. Sheykhi, Y. Zhang, C. Milsmann and F. N. Castellano, Chem. Sci., 2021, 12, 9069–9077 RSC.
- S. N. Sanders, M. K. Gangishetty, M. Y. Sfeir and D. N. Congreve, J. Am. Chem. Soc., 2019, 141, 9180–9184 CrossRef.
- S. N. Sanders, T. H. Schloemer, M. K. Gangishetty, D. Anderson, M. Seitz, A. O. Gallegos, R. C. Stokes and D. N. Congreve, Nature, 2022, 604, 474–478 CrossRef CAS.
- N. Sellet, M. Cormier and J.-P. Goddard, Org. Chem. Front., 2021, 8, 6783–6790 RSC.
- C. J. Imperiale, P. B. Green, M. Hasham and M. W. B. Wilson, Chem. Sci., 2021, 12, 14111–14120 RSC.
- J. Huang, J. Sun, Y. Wu and C. Turro, J. Am. Chem. Soc., 2021, 143, 1610–1617 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2sc05229f |
| This journal is © The Royal Society of Chemistry 2023 |