
Surface oxidation protection strategy of CoS2 by V2O5 for electrocatalytic hydrogen evolution reaction†‡
Jie
Wu§
ab,
Xuetao
Qin§
c,
Yu
Xia§
de,
Yuanyuan
Zhang
b,
Bin
Zhang
b,
Yunchen
Du
 b,
Hsing-Lin
Wang
*d,
Siwei
Li
*bf and
Ping
Xu
*b
b,
Hsing-Lin
Wang
*d,
Siwei
Li
*bf and
Ping
Xu
*b
aNational Engineering Laboratory for VOCs Pollution Control Technology and Equipment, School of Environment and Energy, South China University of Technology, Guangzhou 510640, China. E-mail: yhwj1105914174@foxmail.com
bMIIT Key Laboratory of Critical Materials Technology for New Energy Conversion and Storage, School of Chemistry and Chemical Engineering, Harbin Institute of Technology, Harbin 150001, China. E-mail: pxu@hit.edu.cn
cBeijing National Laboratory for Molecular Sciences, College of Chemistry and Molecular Engineering and College of Engineering, and BIC-ESAT, Peking University, Beijing 100871, China. E-mail: xuetaoqin@pku.edu.cn
dDepartment of Materials Science and Engineering, Southern University of Science and Technology, Shenzhen 518055, China. E-mail: wangxl3@sustech.edu.cn
eSchool of Physics and Astronomy, University of Birmingham, Edgbaston, Birmingham B152TT, UK. E-mail: 11856005@mail.sustech.edu.cn
fInstitute of Industrial Catalysis, School of Chemical Engineering and Technology, Xi’an Jiaotong University, Xi’an 710049, China. E-mail: lisiwei@xjtu.edu.cn
First published on 23rd December 2022
Abstract
Transition metal sulfides (TMSs) are promising electrocatalysts for hydrogen evolution reaction (HER), while TMSs usually suffer from inevitable surface oxidation in air, and the impact of the surface oxidation on their HER catalytic activity remains unclear. Herein, we demonstrate an effective strategy for reducing the surface oxidation degree of easily oxidized CoS2 by introducing glued vanadium pentoxide (V2O5) nanoclusters, taking advantage of the preferential adsorption and strong interaction between high-valence V and O2. Combining oxidation protection and elaborate oxidation control experiments reveal that reduced surface oxidation degree of CoS2 is conducive to affording promising HER catalytic performance, as the oxidized surface of CoS2 can hinder the dissociation of water and thus is harmful to the HER process. Direct evidence is provided that surface oxidation should be carefully considered for TMS-based HER catalysts. The present work not only develops a new strategy for protecting CoS2 from surface oxidation, but also provides deep insight into the impact of surface oxidation on the HER performance of transition metal compounds.
New conceptsTransition metal sulfides (TMSs) are important catalytic materials being widely used in the field of electrocatalysis, photocatalysis and thermal catalysis. Surface of TMSs can be easily oxidized under ambient conditions, however, the impact of the inevitable surface oxidation on the catalytic performance of TMSs has been always ignored. What's worse, efficient strategy for preventing TMSs from serious surface oxidation, crucial for academic and even industrial field, has not been developed. Herein, a novel strategy for preventing multiple TMSs from serious surface oxidation has been developed by introducing amorphous V2O5 clusters with close affinity to oxygen. Taking electrochemical hydrogen evolution reaction (HER) as a model reaction, the huge impact of the surface oxidation degree on the catalytic performance of TMSs has been shown clearly. We believe this work can not only arouse the attention on the surface oxidation of TMSs-based catalyst, but also provide a useful strategy for the surface protection of transition metal compounds including but not limited to TMSs. |
Introduction
Hydrogen evolution reaction (HER) is the cathodic reaction of water electrolysis, which is recognized as an important and sustainable approach to hydrogen production as clean fuels and chemical feedstocks.1,2 Pt-based materials exhibit excellent activity for HER, but their high cost and scarcity limit the wide-spread applications.3,4 Transition metal sulfides (TMSs) have been widely explored as a class of non-noble metal-based HER catalysts due to their unique physical and chemical properties.5,6 Both layered MS2 (e.g. MoS27 and WS28 and non-layered MxSy (e.g. CoS2,9 FeS210 and Ni3S2,11etc.) exhibit outstanding catalytic performance towards the HER, which are comparable or even superior to commercial Pt/C. Notably, multiple factors can impact the HER activity of TMSs, such as metal elements, ratios of metals and S atoms, vacancies, and electronic structures.12,13 However, the impact of inevitable surface oxidation of TMSs on their HER performance has been paid less attention.Like many TM compounds (e.g. carbides,14,15 nitrides16,17 and phosphides18,19), surface of TMSs can be easily oxidized under ambient conditions. In fact, the phenomenon that TMSs are susceptible to surface oxidation has been widely discovered and reported for almost all kinds of TMSs, though the degree of surface oxidation depends on the kinds of metal and crystalline phase.20–23 For example, surface oxidation degree of CoS2 calculated based on X-ray photoelectron spectroscopy (XPS) increases from ∼20% to ∼90% only after 8 days exposure to air at room temperature.24 In contrast, the surface oxidation degree of CoS2, with the same metallic element as CoS2, only increases from 10% to 20% under the same condition. Furthermore, the surface oxidation of MoS2 is even too slight to be tested by some well-known surface sensitive characterization techniques such as XPS, which can only be detected by scanning tunneling microscopy.20,24 To the best of our knowledge, most of the current works on this topic have not carefully considered the surface oxidation, and theoretical calculations based on the intrinsic TMS model are performed to explain the structure–performance relationship and catalytic mechanism. In our view, this is not rigorous for the easily oxidized TMSs (e.g. CoS2).
Since the importance of surface oxidation of TMSs has not been paid enough attention, efficient strategy for preventing TMSs from serious surface oxidation, crucial for academic and even industrial field, has not been developed. High-valence early transition metals (e.g. V) have close affinity to O due to the characteristic of strong metal–O bonds,25,26 and therefore can be used to stabilize the structure of the catalyst under the oxidative condition. Inspired by this strategy, herein we develop a new method for reducing the surface oxidation degree of easily oxidized CoS2 by introducing glued V2O5 nanoclusters (marked as CoS2–V2O5). The structure and surface oxidation degree of CoS2 have been investigated by using electron microscopic and spectroscopic characterizations. Moreover, the impact of the surface oxidation degree of CoS2 on the HER performance are explored through control oxidation experiments. This work provides a new method to protect the surface oxidation of TMS, and investigates the influence of the surface oxidation on the electrocatalytic HER performance of CoS2.
Results
The synthesis process of CoS2–V2O5 supported on carbon cloth (CC) is shown in Fig. 1. Firstly, CC supported Co zeolitic imidazolate frameworks (marked as Co MOFs) were immersed in Na3VO4 solution to get Co3V2O8–Co MOFs intermediate via ion exchange process (Fig. S1–S7, ESI‡). Subsequently, the Co3V2O8–Co MOFs was subjected to mild sulfidation at 400 °C under Ar atmosphere to obtain the CoS2–V2O5 catalyst. The Co3V2O8–Co MOFs intermediate with highly mixed Co and V elements may be a key factor for the generation of highly dispersed V2O5 nanoclusters around CoS2. For comparison, the CoS2 catalyst was synthesized via the same sulfidation treatment by directly using Co MOFs as the precursor (Fig. S8 and S9, ESI‡).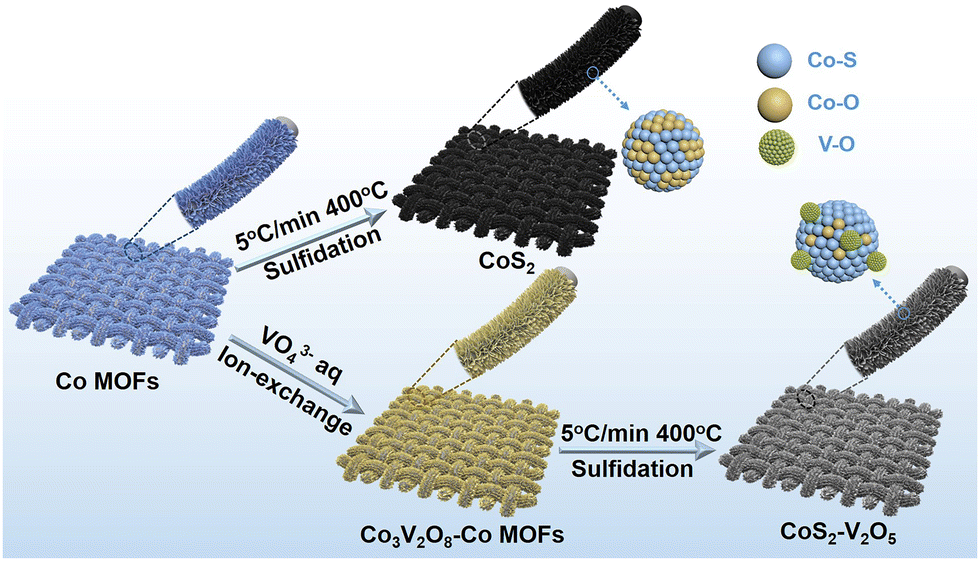 | ||
| Fig. 1 Schematic illustration of the synthesis strategy of CoS2 and CoS2–V2O5 nanoarrays supported on carbon cloth (CC). | ||
X-ray diffraction (XRD) was carried to study the structures of the as-synthesized samples (Fig. 2a). The XRD pattern of as-synthesized CoS2 matches well with the standard cubic CoS2 crystal phase (JCPDS No. 41-1471). However, for the V-incorporated CoS2 sample, only diffraction peaks assigned to CoS2 appear because V species cannot be detected by XRD due to low crystallinity and small particle size. The chemical composition of the V species was further investigated by using X-ray absorption fine structure (XAFS). The V K-edge X-ray absorption near-edge spectroscopy (XANES)of the as-synthesized sample is drastically similar to that of V2O5 (Fig. 2b), suggesting the formation of CoS2–V2O5. Moreover, the bonding structures of V species were carried out by extend X-ray absorption fine structure (EXAFS, Fig. 2c). There are only V–O (1.56 Å) and V–O–V (2.83 Å) scatterings in the V K-edge EXAFS spectra of CoS2–V2O5, while the V–S scattering (1.97 Å) is absent. This is a direct evidence for the existence of vanadium oxides in the as-synthesized CoS2–V2O5 heterostructure.
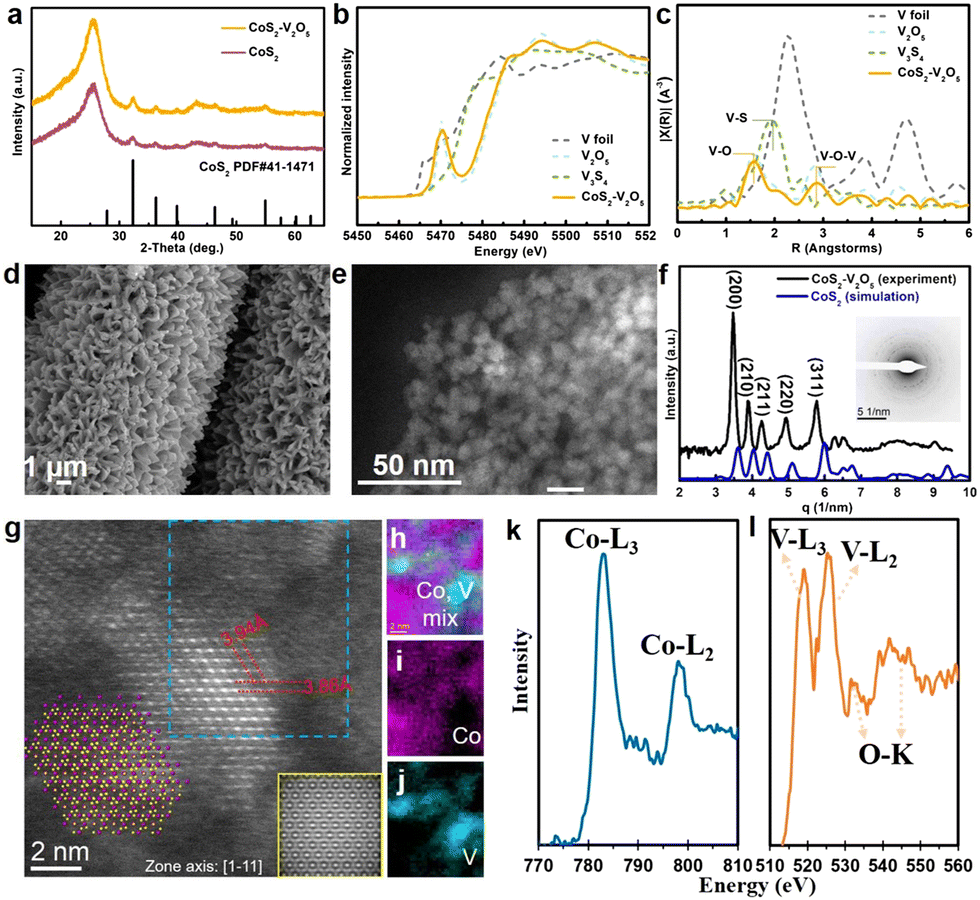 | ||
| Fig. 2 Structural analysis of CoS2–V2O5. (a) XRD patterns of CoS2 and CoS2–V2O5, (b) V K-edge XANES of CoS2–V2O5, V foil, V2O5 and V3S4, (c) V K-edge EXAFS of CoS2–V2O5, V foil, V2O5 and V3S4, (d) SEM image of CoS2–V2O5 nanoarrays loaded on CC, (e) HAADF-STEM image of CoS2–V2O5 particles, (f) radial intensity profile of CoS2–V2O5 (black line) and the simulated radial intensity profile of CoS2 (blue line), inset shows the SAED of CoS2–V2O5, (g) atomic HAADF-STEM image of CoS2–V2O5 along with the zone axis [1−11] direction (inset shows the atomic model of CoS2), (h) cobalt and vanadium colormix, (i) cobalt mapping, (j) vanadium mapping, EELS spectra of k cobalt L edge, (l) oxygen K edge, and vanadium L edge acquired from the blue rectangular region in (g). | ||
The morphology and crystallinity of the CoS2–V2O5 heterostructure were further investigated by electron microscopy. The scanning electron microscopy (SEM) shows the similar nanoarray morphology to that of the Co MOFs precursor and Co3V2O8–Co MOFs intermediate (Fig. 2d and Fig. S1, ESI‡). Upon closer observation by the high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) image in Fig. 2e, the nanoarray grown on the carbon cloth consists of CoS2–V2O5 nanoparticles that are about 8 nm in size. Notably, the integrated radial intensity profile (Fig. 2f) extracted from the selected area electron diffraction (SAED) corresponding to Fig. 2e is similar to the simulated radial intensity profile of CoS2, where no peak can be attributed to V2O5, which indicates that the V2O5 is amorphous in nature.
Aberration corrected STEM and corresponding electron energy loss spectrum (EELS) were further employed to characterize the structure of CoS2–V2O5. The atomic resolution image of CoS2 is presented in Fig. 2g with the zone axis [1−11]. The measured lattice spacings of 3.94 Å and 3.86 Å indicate (110) and (0−11) planes with an angle of 120°. The simulated HAADF-STEM image of CoS2 (110) facets with a d-spacing of 3.92 Å is presented at the bottom right corner of Fig. 2g, matching well with the atomic model of CoS2 with (110) planes (inset in Fig. 2g).27 More importantly, an area marked by the blue dashed rectangle in the STEM image was scanned for collecting the EEL spectra of V and Co elements to map the distribution of Co and V. As shown in Fig. 2h–j, Co and V elements were separated in space. Moreover, the low magnification mappings (Fig. S10, ESI‡) can also indicate that V2O5 was adhered on the surface of CoS2 nanoparticles, with the size about 1–2 nm. As the amorphous V2O5 has been confirmed according to the previous characterizations, V2O5 nanoclusters (1–2 nm) are glued to the crystalline CoS2 nanoparticles. The valence of V and Co were also analyzed from the EEL spectra (Fig. 2k–l). The peak intensity of V-L3 (518.8 eV) is lower than that of V-L2 (526.1 eV) with a ratio of L3/L2 = 0.7, indicating the valence of vanadium oxides in the as-synthesized sample is +5,28 consistent to the XANES result. The calculated Co-L3/L2 ratio of 3 reveals that the valence of Co in the CoS2 is a mixture of +2 and +3.29
Based on the above characterizations, the as-prepared CoS2–V2O5 heterostructure is composed of crystalline CoS2 nanoparticles around with amorphous V2O5 nanoclusters. The homogeneously distribution of V2O5 and CoS2 at nanoscale is a key to the surface oxidation protection due to the formation of abundant CoS2–V2O5 interfaces.
To demonstrate the surface oxidation protection effect of V2O5 nanoclusters, the surface oxidation degree of the CoS2 and CoS2–V2O5 samples is investigated by using spectroscopic characterizations and control experiments (Fig. 3). Raman spectrum of CoS2 sample (Fig. 3a) shows peaks centered at ∼487, 529, 600 and 698 cm−1 corresponding to the Eg, F2g, F2g and A1g modes of Co–O species, respectively,30 whereas the peak centered at 382 cm−1 is identified as the Ag mode of Co–S species.31 The appearance of the Co–O species in the Raman spectrum have been reported to result from the surface oxidation of CoS2 in air.32 As seen from the Raman spectrum, the as-reported HER performance for CoS2 has been heavily affected by the spontaneous surface oxidation, which may not reflect its catalytic activity. In contrast, the peaks assigned to Co–O species disappear in the Raman spectrum of the CoS2–V2O5 sample. Instead, two peaks for V–O species (centered at 743 and 806 cm−1) emerge along with the peaks for Co–S species.33,34 This result implies that the surface oxidation degree of CoS2 is significantly reduced upon the introduction of V2O5 nanoclusters.
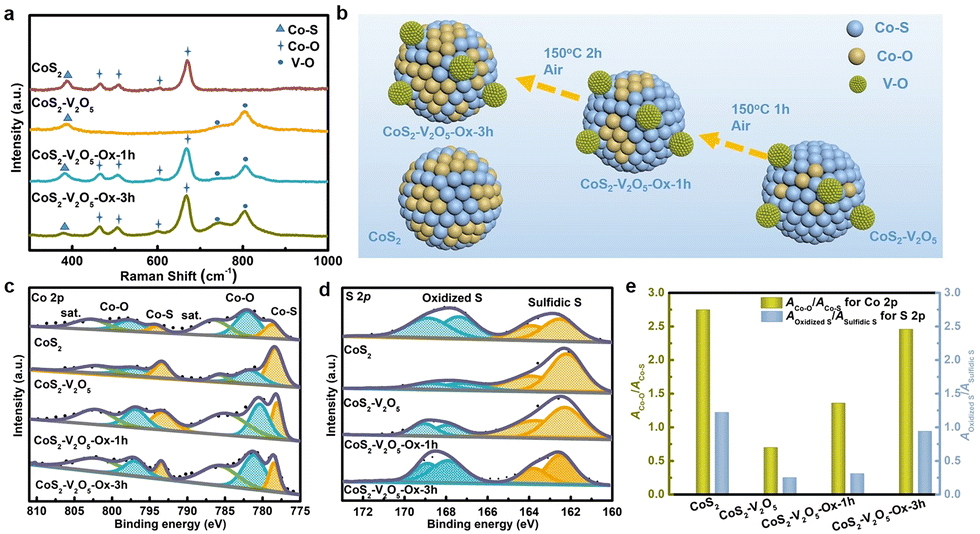 | ||
Fig. 3 (a) Raman spectra of CoS2, CoS2–V2O5, CoS2–V2O5-Ox-1h and CoS2–V2O5-Ox-3h, in which Ox means a control oxidation process in Air at 150 °C, (b) schematic illustration of surface oxidation phenomenon of CoS2, comparison of (c) Co 2p and (d) S 2p XPS spectra of CoS2, CoS2–V2O5, CoS2–V2O5-Ox-1h and CoS2–V2O5-Ox-3h, and (e) the ratios of ACo–O![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ACo–S and AOxidized S:ASulfidic S, in which A represents the peak area in the XPS spectra. :ACo–S and AOxidized S:ASulfidic S, in which A represents the peak area in the XPS spectra. | ||
Even though the CoS2–V2O5 and CoS2 catalysts have different surface oxidation degree, it is not enough to compare their HER performance to show the impact of surface oxidation. Therefore, we carried out a control oxidation experiments for the CoS2–V2O5 (Fig. 3b) to obtain more control groups with different surface oxidation degree. Specifically, the CoS2–V2O5 was oxidized in air at 150 °C for different time periods to tune the surface oxidation degree of CoS2, which were marked as CoS2–V2O5-Ox-1h and CoS2–V2O5-Ox-3h, respectively. XRD patterns and SEM images of the control groups (Fig. S8 and S9, ESI‡) tell that the crystalline structures and morphologies of these oxidized samples almost keep unchanged. However, as seen in Fig. 3a, the four peaks assigned to Co–O peaks appear clearly in the Raman spectra of CoS2–V2O5-Ox-1h and CoS2–V2O5-Ox-3h, indicating that controlled surface oxidation of CoS2 has been successfully realized.
XPS was further employed to investigate the surface oxidation degree of the above four samples. There are only Co, V (absent for the CoS2 sample), O, S, C, and N elements in the survey spectra, indicating the purity of the as-synthesized samples (Fig. S11, ESI‡). In the V 2p spectra of the CoS2–V2O5 and the oxidized samples, the peaks centered at 517.5 and 524.8 eV are assigned to V(+ 5) species (Fig. S12, ESI‡).35 The Co 2p spectra of the samples can be divided into three couples of peaks (Fig. 3c). Specifically, the peaks centered at 778.7 and 794 eV are assigned to the Co–S species, whereas the peaks at 782, 797.6, 786.5 and 803 eV are assigned to the Co–O species and the corresponding satellite peaks.36 For the CoS2 sample, the peak intensity of Co–O is relatively higher than that of Co–S, indicating the serious surface oxidation of CoS2. The area ratio of Co–O and Co–S (ACo–O:ACo–S), which can reflect the surface oxidation degree of CoS2, is calculated to be 2.75 for CoS2. For CoS2–V2O5, both Co–S and Co–O species can be detected in the Co 2p spectrum, because XPS is more sensitive to the surface species than Raman spectroscopy. The intensity of Co–O is much lower than that of Co–S and the value of ACo–O:ACo–S decreases sharply to 0.7, a strong proof that the surface oxidation of CoS2 is effectively reduced with the introduction of V2O5 nanoclusters. Furthermore, the peaks of Co–S for CoS2–V2O5 are located at 778.18 and 793.08 eV, which are negatively shifted compared with CoS2, suggesting the construction of CoS2–V2O5 heterojunction structure. When the CoS2–V2O5 is oxidized in air, the peak intensity of Co–O increases, while the peak intensity of Co–S decreases. The ACo–O:ACo–S are calculated to be 1.36 and 2.46 for the CoS2–V2O5-Ox-1h and CoS2–V2O5-Ox-3h, respectively.
The above analyses based on the Co 2p spectra can be further supported by the S 2p spectra of these samples (Fig. 3d). In brief, both oxidized S and sulfidic S species can be found in all the S 2p spectra of CoS2, CoS2–V2O5, CoS2–V2O5-Ox-1h and CoS2–V2O5-Ox-3h, and the ratio of AOxidized S:ASulfidic S is calculated to be 1.22, 0.25, 0.31 and 0.94, respectively, following the same trend as ACo–O:ACo–S (Fig. 3e).
Three major conclusions about the surface oxidation of CoS2 can be drawn according to the above Raman and XPS results. 1) As reported, CoS2 suffers from serious surface oxidation in air. 2) Construction of the CoS2–V2O5 hybrid materials can significantly reduce the surface oxidation degree of CoS2. 3) The control oxidation experiment of CoS2–V2O5 can tune the surface oxidation degree, which is helpful to understand the effect of surface oxidation on the HER performance of CoS2.
The electrocatalytic performance of CC, commercial Pt/C, CoS2, CoS2–V2O5 as well as the control samples towards the HER was tested in alkaline media (1 M KOH). The reference electrode is calibrated experimentally (Fig. S13, ESI‡).37 As shown in the iR-corrected linear sweep voltammetry (LSV, Fig. 4a) curves, bare CC shows very poor catalytic performance towards HER. The CoS2 exhibits much better catalytic performance towards HER, requiring an overpotential of 272 mV to deliver a current density of 10 mA cm−2. Significantly, once the CoS2–V2O5 heterostructure is fabricated, the required overpotential is sharply reduced to 128 mV at 10 mA cm−2. Moreover, CoS2–V2O5 affords a mass activity of 0.71 A g−1 at an overpotential of 100 mV (Fig. S14, ESI‡), which is much higher than that of CoS2 (0.18 A g−1). Interestingly, when the CoS2–V2O5 samples are oxidized for 1 h and 3 h, the HER performances drop gradually, requiring an overpotential of 166 and 196 mV at 10 mA cm−2, respectively.
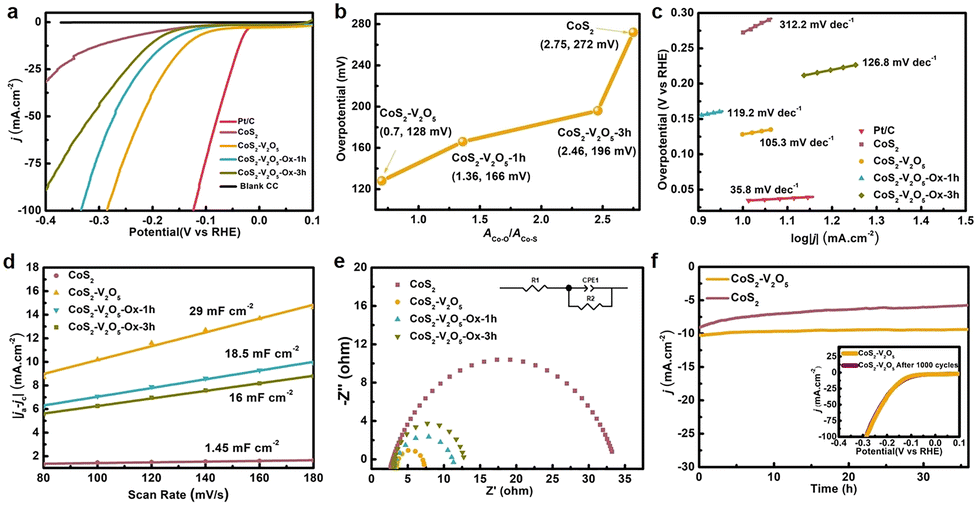 | ||
| Fig. 4 HER performance of the as-prepared electrocatalysts in 1 M KOH solution. (a) LSV curves after iR correction in comparison to Pt/C, (b) the relationship between ACo–O/ACo–S and the overpotential at 10 mA cm−2, (c) Tafel plots, (d) Cdl extracting from the CV curves, (e) EIS spectra (inset shows the equivalent circuit used to simulate the Nyquist plots), and (f) long-time stability of CoS2–V2O5 for 36 h (inset exhibits the LSV curves before and after 1000 CV cycles test). | ||
To further understand the impact of surface oxidation on the HER performance of CoS2-based catalysts, we plot the overpotential of the above CoS2–V2O5 and CoS2 catalysts as a function of the ACo–O:ACo–S from XPS (Fig. 4b). For the CoS2–V2O5 and CoS2–V2O5-Ox samples, the overpotential increases almost linearly with surface oxidation degree from 128 to 196 mV, suggesting the surface oxidation degree is the dominant role for the different HER performance of these samples. The overpotential increases sharply for the CoS2 catalyst (272 mV), implying that the V2O5 nanoclusters in the heterostructure may also contributes to the catalytic process. In sum, the lower surface oxidation degree of CoS2, the better HER performance.
Furthermore, the HER kinetics of the CoS2-based catalysts are informed from the corresponding Tafel plots in Fig. 4c. The Tafel slope of CoS2–V2O5 is 105.3 mV dec−1, which is smaller than that of CoS2 (312.2 mV dec−1), CoS2–V2O5-Ox-1h (119.2 mV dec−1) and CoS2–V2O5-Ox-3h (126.8 mV dec−1), indicating the facilitated reaction kinetics during HER process over CoS2–V2O5. Additionally, electrochemical double layer capacitance (Cdl) is obtained from the cyclic voltammetry (CV) curves at different scan rates in the non-faradaic potential range (Fig. 4d and Fig. S15, ESI‡), which is an important parameter to derive the electrochemically active surface area (ECSA).38 As the construction of CoS2–V2O5 heterojunction structure, which is considered to effectively improve the capacitance performance39,40 and as the more catalytical active sites (Co–S) are maintained due to the protection of V2O5, the CoS2–V2O5 owns the highest ECSA (725 cm2, Fig. S16, ESI‡). Thus, the CoS2–V2O5 owns the highest Cdl (29 mF cm−2) according to the equation ECSA = Cdl/Cs, in which Cdl is positively correlated to ECSA. As shown in Fig. S17 (ESI‡), we also conducted the turn over frequency (TOF) of CoS2 and CoS2–V2O5. The TOF values of CoS2 and CoS2–V2O5 are 0.13 s−1 and 0.08 s−1 at the overpotential of 150 mV, indicating the better intrinsic catalytic activity of CoS2–V2O5, which is in accordance with Cdl and ECSA results. Moreover, as has been reported, the interface between CoS2 and V2O5 can accelerate the kinetics of HER process and promote electron transport and finally reduce the charge transfer resistance.41 Thus, the electrochemical impedance spectroscopy (EIS) for these samples suggests that the charge transfer resistance for CoS2–V2O5 (3.86 Ω) is much smaller than that of CoS2 (31.24 Ω), CoS2–V2O5-Ox-1h (9.87 Ω) and CoS2–V2O5-Ox-3h (8.46 Ω) (Fig. 4e), demonstrating the enhanced charge transfer efficiency and improved reaction kinetics of CoS2–V2O5 during the HER process. And CoS2–V2O5 catalyst is also demonstrated to have an excellent mass transport property (Fig. S18, ESI‡). Besides, the HER Faraday efficiency (FE) of CoS2 and CoS2–V2O5 were performed under the current of 10 mA cm−2 for 6000 s, which were 98.1% and 98%, indicating the high selectivity during the HER process for both catalysts (Fig. S19, ESI‡).
The poor stability is a main drawback for CoS2-based HER electrocatalyst. The chronoamperometric i–t curves at 10 mA cm−2 of CoS2 and CoS2–V2O5 were conducted for 36 h (Fig. 4f). There is significant degradation of current density of CoS2, whereas the i–t curve of CoS2–V2O5 keeps stable. It means the lower surface oxidation degree may also help to improve the stability of CoS2-based HER catalyst. Moreover, the LSV curve of CoS2–V2O5 after 1000 CV cycles basically coincides with the initial one (Fig. 4f, inset). Both experiments indicate the excellent long-term stability of the CoS2–V2O5 catalyst towards HER. The excellent stability of CoS2–V2O5 can be attributed to the well-maintained morphology and maintained Co–S surface, which is confirmed by using SEM, TEM and XPS (Fig. S20 and S21, ESI‡). We have also carried out Co K-edge XANES and EXAFS of CoS2 and CoS2–V2O5 after HER tests to understand the different stability of these two catalysts. Both XANES and EXAFS (Fig. S22, ESI‡) show CoS2 converts to cobalt (hydr)oxides, which is consistent to the reported literatures.23 In contrast, the Co–S and Co–S–V bonds in EXAFS clearly show that CoS2 in the CoS2–V2O5 catalyst retain stable after HER test, which is also supported by XPS result (Fig. S21, ESI‡). The V2O5 clusters can not only reduce the surface oxidation degree of CoS2 in air, but also keep it stable during HER by avoiding the bulk oxidation. It is the structural stability of CoS2–V2O5 that leads to the outstanding stability and long-term durability of this catalyst.).
Density functional theory (DFT) calculations were carried out to understand why the CoS2 with lower surface oxidation degree exhibits better catalytic performance for HER. As shown in Fig. 5a–c, CoS2 (100) and O modified CoS2 (100) (marked as CoS2–O) were selected as the models for simplification according to XRD results. Moreover, V2O5 clusters on CoS2 (100) are built to represent the CoS2–V2O5 sample. Generally, the elemental HER steps in alkaline solution include H2O adsorption, H2O activation, H desorption and OH desorption.42,43 The chemisorption models of reaction intermediates adsorbed on the (100) surface of CoS2 (*H2O, *OH–H and *OH) are displayed concretely in Fig. S23 (ESI‡). The standard free energy (ΔG) diagrams for CoS2 CoS2–O and CoS2–V2O5 of HER reaction steps are shown in Fig. 5d. For CoS2, the Gibbs free energies of H2O adsorption and H2O dissociation are 0.37 and 0.67 eV, respectively, while the following steps are spontaneous. It can be learned that H2O dissociation is the rate determining step (RDS) for CoS2. With the CoS2 surface modified by O, although the H2O adsorption is effectively accelerated (ΔG = −0.05 eV), the energy barrier for H2O dissociation is even higher, resulting in a much higher ΔGRDS of 1.56 eV. The above results clearly show that the surface oxidation of CoS2 is harmful to the HER process due to the hindering of H2O dissociation.
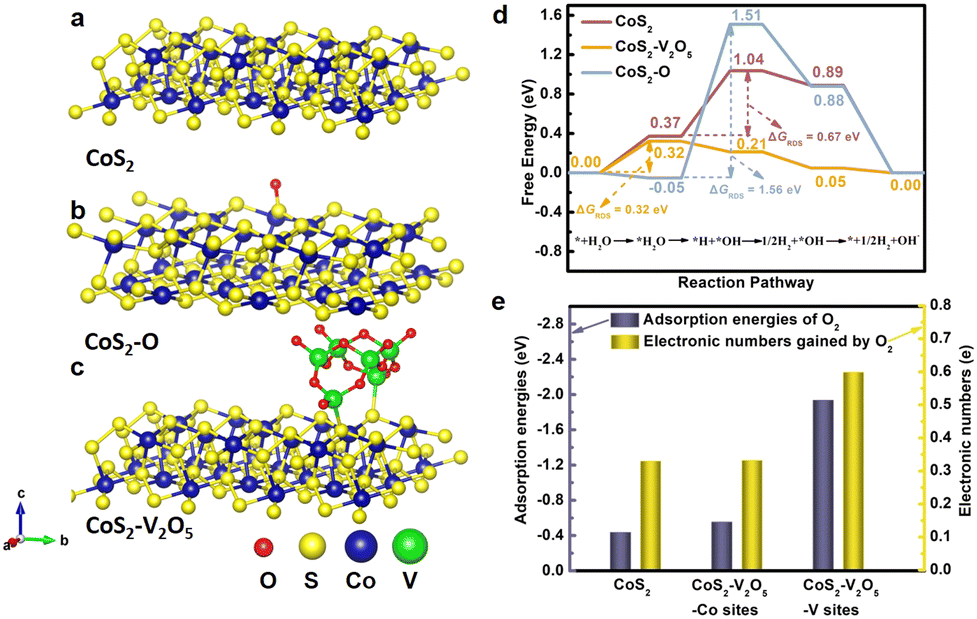 | ||
| Fig. 5 (a–c) Crystal structures of CoS2, CoS2–O and CoS2–V2O5, (d) comparison of standard Gibbs free energies at the rate determining step of CoS2, CoS2–O and CoS2–V2O5 during the HER process, and (e) the adsorption energies of O2 molecules at metal sites (Co sites and V sites) and the electronic numbers gained by O2 molecules. | ||
We further investigate the synergetic effect of V2O5 and CoS2 through DFT calculations. The contribution of V2O5 to HER is highlighted by comparing CoS2 and CoS2–V2O5 models (the red and yellow line, Fig. R6, ESI‡). The introduction of V2O5 will not bring an obvious change for H2O adsorption, but it significantly accelerates the H2O dissociation, leading to a spontaneous process (ΔG = −0.11 eV). As a result, the H2O adsorption step becomes the RDS for CoS2–V2O5, with a ΔGRDS of 0.32 eV. The ΔGRDS of CoS2–V2O5 is lower than that of CoS2, which means CoS2–V2O5 possesses better catalytic activity for HER than CoS2 from a theoretical point of view, matching well with the experimental results. Based on the theoretical calculations, the homogeneously distributed V2O5 can facilitate the dissociation of water and further the whole HER process. Therefore, the synergetic effect of V2O5 and CoS2 include two parts. On one hand, V2O5 can reduce the surface oxidation of CoS2 in air and the bulk oxidation of CoS2 during HER, leading to exposed and stable Co–S sites for HER. On the other hand, V2O5 also contributes to the HER by facilitating the dissociation of water and further the whole HER process.
Another important issue is why the V2O5 nanoclusters can protect CoS2 from surface oxidation. To answer this question, it is reasonable to investigate the adsorption and interaction between O2 and the CoS2 or CoS2–V2O5 catalyst. V2O5 clusters on CoS2 (100) were built to represent the CoS2–V2O5 sample (Fig. 5c) and multiple theoretical methods were adopted. On one hand, the adsorption energy of O2 molecule (EO2) on the CoS2 (Co sites) and CoS2–V2O5 (Co sites and V sites) was calculated (Fig. 5e, blue bar). EO2 of Co site in CoS2 is calculated to be −0.436 eV, meaning the adsorption of O2 is spontaneously. By contrast, EO2 of Co sites in the CoS2–V2O5 is slightly reduced to −0.553 eV, which may result from the electronic interaction between the CoS2 and V2O5 species. Significantly, EO2 of V sites in the CoS2–V2O5 reaches −1.935 eV, much lower than that of Co sites. The results clearly indicate that O2 molecule preferentially adsorbs on V sites rather than Co sites in the CoS2–V2O5. On the other hand, Bader charge analysis was applied to explore the charge transfer process between metal sites and adsorbed O2 molecules (Fig. 5e, yellow bar). The number of the transferred electron is 0.330 for Co sites in CoS2 and 0.332 and 0.559 for Co sites and V sites in CoS2–V2O5. The more transferred electron, the stronger interaction between O2 and metal sites. According to the above calculations, O2 molecules in air can preferentially adsorb on V2O5 rather than CoS2 and interact strongly with the V sites rather than Co sites in the CoS2–V2O5 catalyst. As a result, the V2O5 nanoclusters protect the CoS2 nanoparticles from serious surface oxidation.
Conclusions
In this work, we demonstrate a novel surface oxidation protection method for CoS2 by introducing V2O5 nanoclusters, and systematically investigate the impact of surface oxidation on the HER performance. XAFS, HADDF-STEM and EELS demonstrate that amorphous V2O5 nanoclusters homogeneously glue to CoS2 nanoparticles thanks to the MOFs-derived synthetic method, leading to significantly reduced surface oxidation degree of CoS2 and excellent stability of CoS2–V2O5 during HER by avoiding the bulk oxidation. The preferential adsorption and strong interaction between O2 and V2O5 clusters help to protect CoS2 from serious surface oxidation. As a result, CoS2–V2O5 delivers a superior HER performance in alkaline media, requiring a low overpotential of 128 mV to deliver a current density of 10 mA cm−2, which is much better than that of CoS2 (272 mV). Through the control oxidation experiments of CoS2–V2O5, it is demonstrated that the lower surface oxidation degree of CoS2, the better HER performance. DFT calculations indicate that the surface oxidation of CoS2 can hinder the dissociation of water, and the introduction of V2O5 can effectively facilitate the dissociation of water and further improve the HER process. This work shows the crucial impact of the surface oxidation of TMSs on their HER performance and provides a new idea for surface oxidation protection of CoS2 that may be expanded to other TMSs and transition metal compounds.Experimental section
Materials
Cobalt(II) nitrate hexahydrate (CoNO3·6H2O), dimethyl imidazole (C4H6N2), sodium orthovanadate (Na3VO4), sulfur powder (S), potassium hydroxide (KOH), iridium dioxide (IrO2) and Nafion were all were purchased from Aladdin Industrial Corporation. Pt sheet was purchased from Aldrich. And deionized water (DI), absolute ethanol (C2H5OH). All chemicals were used directly without any purification treatment.Methods
![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) . The software read the coordinates of Co and S atoms from the generated atomic model, creating the simulated image along with the direction of [1−11]. We used Renishaw in Via micro Raman spectroscopy system (laser wavelength: 532 nm) to record the Raman spectra. X-ray photoelectron spectra (XPS) were received by operating PHI 5700 ESCA system, in which an Al Kα radiation was used as a source (hν = 1486.6 eV). The Fourier transform infrared spectroscopy (FT-IR) was received by operating with Tensor-27, Germany, Bruker system. The X-ray absorption fine structure spectra V K-edge were collected at 44A beamline of National Synchrotron Radiation Research Center (NSRRC) Taiwan.44,45
. The software read the coordinates of Co and S atoms from the generated atomic model, creating the simulated image along with the direction of [1−11]. We used Renishaw in Via micro Raman spectroscopy system (laser wavelength: 532 nm) to record the Raman spectra. X-ray photoelectron spectra (XPS) were received by operating PHI 5700 ESCA system, in which an Al Kα radiation was used as a source (hν = 1486.6 eV). The Fourier transform infrared spectroscopy (FT-IR) was received by operating with Tensor-27, Germany, Bruker system. The X-ray absorption fine structure spectra V K-edge were collected at 44A beamline of National Synchrotron Radiation Research Center (NSRRC) Taiwan.44,45
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the financial support from Natural Science Foundation of China (No. 21871065, and 22071038), China Postdoctoral Science Foundation Grant (No. 2020M670894), Heilongjiang Touyan Team (HITTY-20190033), and Interdisciplinary Research Foundation of HIT (IR2021205). Prof. Li acknowledges the financial support from the “Young Talent Support Plan” of Xi’an Jiaotong University (HG6J024), High-Level Innovation and Entrepreneurship Talent Project of Qinchuangyuan (QCYRCXM-2022-123) and “Young Talent Lift Plan” of Xi’an city (095920221352). We would like to express our great gratitude to the supercomputing center in Wuhan University.References
- B. Zhu, R. Zou and Q. Xu, Adv. Energy Mater., 2018, 8, 1801193 CrossRef.
- J. Zhu, L. Hu, P. Zhao, L. Lee and K. Wong, Chem. Rev., 2020, 120, 851–918 CrossRef CAS PubMed.
- D. Liu, X. Li, S. Chen, H. Yan, C. Wang, C. Wu, Y. Halleem, S. Duan, J. Lu, B. Ge, P. Ajayan, Y. Luo, J. Jiang and L. Song, Nat. Energy, 2019, 4, 512–518 CrossRef CAS.
- E. Kemppainen, A. Bodin, B. Sebok, T. Pedersen, B. Seger, B. Mei, D. Bae, P. Vesborg, J. Halme, O. Hansen, P. Lund and I. Chorkendorff, Energy Environ. Sci., 2015, 8, 2991–2999 RSC.
- Y. Guo, T. Park, J. Yi, J. Henzie, J. Kim, Z. Wang, B. Jiang, Y. Bando, Y. Sugahara, J. Tang and Y. Yamauchi, Adv. Mater., 2019, 31, e1807134 CrossRef PubMed.
- H. Sun, Z. Yan, F. Liu, W. Xu, F. Cheng and J. Chen, Adv. Mater., 2020, 32, e1806326 CrossRef PubMed.
- Y. Yin, J. Han, Y. Zhang, X. Zhang, P. Xu, Q. Yuan, L. Samad, X. Wang, Y. Wang, Z. Zhang, P. Zhang, X. Cao, B. Song and S. Jin, J. Am. Chem. Soc., 2016, 138, 7965–7972 CrossRef CAS PubMed.
- L. Cheng, W. Huang, Q. Gong, C. Liu, Z. Liu, Y. Li and H. Dai, Angew. Chem., Int. Ed., 2014, 126, 7994–7997 CrossRef.
- S. Peng, L. Li, X. Han, W. Sun, M. Srinivasan, S. Mhaisalkar, F. Cheng, Q. Yan, J. Chen and S. Ramakrishna, Angew. Chem., Int. Ed., 2014, 126, 12802–12807 CrossRef.
- Y. Chen, S. Xu, Y. Li, R. Jacob, Y. Kuang, B. Liu, Y. Wang, G. Pastel, L. Salamanca, M. Zachariah and L. Hu, Adv. Energy Mater., 2017, 7, 1700482 CrossRef.
- L. Feng, G. Yu, Y. Wu, G. Li, H. Li, Y. Sun, T. Asefa, W. Chen and X. Zou, J. Am. Chem. Soc., 2015, 137, 14023–14026 CrossRef CAS PubMed.
- C. Xie, D. Yan, H. Li, S. Du, W. Chen, Y. Wang, Y. Zou, R. Chen and S. Wang, ACS Catal., 2020, 10, 11082–11098 CrossRef CAS.
- T. Wang, H. Xie, M. Chen, A. Aloia, J. Cho, G. Wu and Q. Li, Nano Energy, 2017, 42, 69–89 CrossRef CAS.
- C. Yang, H. Zhao, Y. Hou and D. Ma, J. Am. Chem. Soc., 2012, 134, 15814–15821 CrossRef CAS PubMed.
- S. Li, R. Cao, M. Xu, Y. Deng, L. Lin, S. Yao, X. Liang, M. Peng, Z. Gao, Y. Ge, J. Liu, W. Li, W. Zhou and D. Ma, Natl. Sci. Rev., 2022, 9, nwab026 CrossRef CAS PubMed.
- X. Yang, J. Nash, J. Anibal, M. Dunwell, S. Kattel, E. Stavitski, K. Attenkofer, J. Chen, Y. Yan and B. Xu, J. Am. Chem. Soc., 2018, 140, 13387–13391 CrossRef CAS PubMed.
- Y. Liu, D. Tian, A. Biswas, Z. Xie, S. Hwang, J. Lee, H. Meng and J. Chen, Angew. Chem., Int. Ed., 2020, 59, 11345–11348 CrossRef CAS PubMed.
- Z. Wu, X. Li, W. Liu, Y. Zhong, Q. Gan, X. Li and H. Wang, ACS Catal., 2017, 7, 4026–4032 CrossRef CAS.
- Y. Li, S. Li, J. Hu, Y. Zhang, Y. Du, X. Han, X. Liu and P. Xu, J. Energy Chem., 2021, 53, 1–8 CrossRef.
- J. Peto, T. Ollar, P. Vancso, Z. Popov, G. Magda, G. Dobrik, C. Hwang, P. Sorokin and L. Tapaszto, Nat. Chem., 2018, 10, 1246–1251 CrossRef CAS PubMed.
- H. Liu, Z. Liu, F. Wang and L. Feng, Chem. Eng. J., 2020, 397, 125507 CrossRef CAS.
- Z. Yu, Y. Xie, B. Xie, C. Cao, Z. Zhang, H. Huo, Z. Jiang, Q. Pan, G. Yin and J. Wang, Energy Storage Mater., 2020, 25, 416–425 CrossRef.
- W. Liu, E. Hu, H. Jiang, Y. Xiang, Z. Weng, M. Li, Q. Fan, X. Yu, E. Altman and H. Wang, Nat. Commun., 2016, 7, 10771 CrossRef CAS PubMed.
- Z. Wu, L. Huang, H. Liu, M. Li and H. Wang, Nano Res., 2020, 14, 2264–2267 CrossRef.
- H. Sun, C. Tung, Y. Qiu, W. Zhang, Q. Wang, Z. Li, J. Tang, H. Chen, C. Wang and H. Chen, J. Am. Chem. Soc., 2022, 144, 1174–1186 CrossRef CAS PubMed.
- S. Li, C. Xi, Y. Jin, D. Wu, J. Wang, T. Liu, H. Wang, C. Dong, H. Liu, S. Kulinich and X. Du, ACS Energy Lett., 2019, 4, 1823–1829 CrossRef CAS.
- D. He, Z. Li and J. Yuan, Micron, 2015, 74, 47–53 CrossRef CAS PubMed.
- S. Kalavathi, S. Amirthapandian, S. Chandra, P. Sahu and K. Sahu, J. Phys.: Condens. Matter, 2014, 26, 015601 CrossRef CAS PubMed.
- Z. Wang and Y. Jiang, Micron, 2000, 31, 571–580 CrossRef CAS PubMed.
- Z. Chen, L. Cai, X. Yang, C. Kronawitter, L. Guo, S. Shen and B. Koel, ACS Catal., 2018, 8, 1238–1247 CrossRef CAS.
- S. Lyapin, A. Utyuzh, A. Petrova, A. Novikov, T. Lograsso and S. Stishov, J. Phys.: Condens. Matter, 2014, 26, 396001 CrossRef CAS PubMed.
- J. Li, Z. Xia, M. Zhang, S. Zhang, J. Li, Y. Ma and Y. Qu, J. Mater. Chem. A, 2019, 7, 17775–17781 RSC.
- P. Shvets, O. Dikaya, K. Maksimova and A. Goikhman, J. Raman Spectrosc., 2019, 50, 1226–1244 CrossRef CAS.
- P. Vilanova, J. Hernánde, A. Landa and F. Agulló, J. Alloys Compd., 2016, 661, 122–125 CrossRef.
- S. Zhang, L. Zhang, G. Xu, X. Zhang and A. Zhao, New J. Chem., 2020, 44, 10918–10923 RSC.
- X. Wang, X. Zhong, Z. Zha, G. He, Z. Miao, H. Lei, Q. Luo, R. Zhang, Z. Liu and L. Cheng, Appl. Mater. Today, 2020, 18, 100464 CrossRef.
- S. Niu, S. Li, Y. Du, X. Han and P. Xu, ACS Energy Lett., 2020, 5, 1083–1087 CrossRef CAS.
- J. Wu, Z. Yu, Y. Zhang, S. Niu, J. Zhao, S. Li and P. Xu, Small, 2021, 17, e2105150 CrossRef PubMed.
- R. Hu, Y. Liao, H. Qiao, J. Li, K. Wang, Z. Huang and X. Qi, Ceram. Interfaces, 2022, 48, 23498–23503 CrossRef CAS.
- X. Wang, H. Li, H. Li, S. Lin, W. Ding, X. Zhu, Z. Sheng, H. Wang, X. Zhu and Y. Sun, Adv. Funct. Mater., 2020, 30, 1910302 Search PubMed.
- Y. Lin, K. Sun, S. Liu, X. Chen, Y. Cheng, W. Cheong, Z. Chen, L. Zheng, J. Zhang, X. Li, Y. Pan and C. Chen, Adv. Energy Mater., 2019, 9, 1901213 CrossRef.
- L. Yu, I. Mishra, Y. Xie, H. Zhou, J. Sun, J. Zhou, Y. Ni, D. Luo, F. Yu, Y. Yu, S. Chen and Z. Ren, Nano Energy, 2018, 53, 492–500 CrossRef CAS.
- W. Xu, N. Apodaca, H. Wang, L. Yan, G. Chen, M. Zhou, D. Ding, P. Choudhury and H. Luo, ACS Catal., 2019, 9, 5074–5083 CrossRef CAS.
- Z. Yu, H. Shan, Y. Zhong, X. Zhang and G. Hong, ACS Energy Lett., 2022, 7, 3151–3176 CrossRef CAS.
- Z. Yu, X. Zhang, C. Fu, H. Wang, M. Chen, G. Yin, H. Huo and J. Wang, Adv. Energy Mater., 2021, 11, 2003250 CrossRef CAS.
- G. Kress and J. Furthmuller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef.
- G. Kress and J. Furthmuller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169 CrossRef PubMed.
- J. Perdew, K. Burke and M. Emzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758 CrossRef CAS.
- P. Blochl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953 CrossRef PubMed.
Footnotes |
| † Dedicated to the 120th anniversary of Southeast University. |
| ‡ Electronic supplementary information (ESI) available: Fig. S1–S23. See DOI: https://doi.org/10.1039/d2nh00431c |
| § These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |