
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThermosensitive biomaterial gels with chemical permeation enhancers for enhanced microneedle delivery of naltrexone for managing opioid and alcohol dependency†
Kevin V.
Tobin
a and
Nicole K.
Brogden
 *ab
*ab
aDepartment of Pharmaceutical Sciences and Experimental Therapeutics, The University of Iowa College of Pharmacy, Iowa City, IA 52242, USA. E-mail: nicole-brogden@uiowa.edu
bDepartment of Dermatology, The University of Iowa Carver College of Medicine, Iowa City, IA 52242, USA
First published on 17th July 2023
Abstract
Naltrexone (NTX) can be transdermally delivered using microneedles (MN) to treat opioid and alcohol misuse disorders, but delivery is blunted by rapid in vivo micropore closure. Poloxamer (P407), a thermosensitive biocompatible hydrogel, sustains NTX delivery through MN-treated skin by generating a drug depot within the micropores. Optimizing P407 formulations could maintain sustained delivery after micropore closure while reducing required patch sizes, which would be more discreet and preferred by most patients. Here we developed NTX-loaded P407 gels with chemical permeation enhancers (CPEs) and used these novel formulations alongside MN treatment to enhance NTX permeation, utilizing parallel micropore and intact skin transport pathways. We analyzed physicochemical and rheological properties of CPE-loaded P407 formulations and selected formulations with DMSO and benzyl alcohol for further study. In vitro permeation tests demonstrated more consistent and sustained NTX delivery through MN-treated porcine skin from 16% P407 formulations vs. aqueous solutions. P407 with 1% benzyl alcohol and 10% DMSO significantly, P < 0.05, increased flux through MN-treated skin vs. formulations with benzyl alcohol alone. This formulation would require a smaller size patch than previously used to deliver NTX in humans, with half the NTX concentration. This is the first time poloxamer biomaterials have been used in combination with CPEs to improve MN-assisted transdermal delivery of an opioid antagonist. Here we have demonstrated that P407 in combination with CPEs effectively sustains NTX delivery in MN-treated skin while requiring less NTX than previously needed to meet clinical goals.
Introduction
For people who suffer from opioid or alcohol misuse disorders, naltrexone HCl (NTX) is a μ-opioid receptor antagonist that can reduce and suppress cravings when used alongside counseling and other behavioral therapies.1 These disorders require lifelong treatment, which calls for regimens that are easy to adhere to – ensuring the lowest chance of a patient prematurely discontinuing treatment. NTX is available as daily oral tablets or a monthly depot injection, both of which have challenges for chronic dosing scenarios.2 These challenges include extensive and highly variable first pass metabolism (5–40%) and hepatotoxicity associated with the oral dosage form, and the high cost and inconvenience of the injectable depot. Transdermal NTX delivery could provide distinct advantages, including more consistent plasma concentrations and potential for self-administration. Despite clear benefits of transdermal delivery, NTX does not possess the ideal physicochemical properties to passively permeate through the lipophilic stratum corneum, the outermost skin layer and primary barrier to dermal drug absorption. However, NTX has enough hydrophilicity to partition well into the aqueous environment of the dermis.Because of its physicochemical properties, NTX is an excellent candidate for transdermal delivery with microneedles (MNs). Solid MNs are small projections ranging from 100–1000 μm long that can be applied to the skin and then removed, creating aqueous micropores. With this pretreatment approach, a drug formulation can be applied over the MN-treated area (often in the form of a patch or semisolid formulation), allowing drug molecules to diffuse into the micropores and enter systemic circulation from the blood supply in the dermis. The hydrophilic micropores provide a direct pathway through the stratum corneum and into the lower skin layers.
Studies using solid MNs as a pretreatment approach have shown promising results for delivering NTX in humans.3 However, one challenge that still exists with MN-assisted NTX delivery is the skin's ability to heal the micropores in a very short period of time. Under an occlusive drug patch the micropores may close by 48 h, which would prevent further transdermal delivery (this has also been observed in NTX pharmacokinetics studies in animals and humans).3–7 Once-weekly dosing of a transdermal patch would be ideal, therefore it is important to identify novel formulation approaches that can overcome this challenge.
Poloxamers are biocompatible, thermosensitive polymers with well-known properties for controlling drug delivery rates, and they are FDA-approved as inactive ingredients in many preparations (including topical products). These biomaterials are liquids at lower temperature and form viscous gels as temperature increases. We have recently demonstrated the feasibility of using poloxamers to control NTX delivery through MN-treated skin, through creation of a local skin depot in the micropores (poloxamers applied as liquids can enter the micropores and quickly form a rate-controlling gel network).8In vivo this would allow continued NTX delivery even after the micropores close at the skin surface. While the poloxamers allow NTX to enter the hydrophilic micropores, there is still a large area of intact skin around the micropores that could also be potentially used for NTX delivery with an optimized formulation.
In the current work we investigated the combination of thermosensitive poloxamers with chemical permeation enhancers (CPEs) as a novel approach to enhance MN-assisted NTX delivery. Our objectives were to characterize physicochemical and rheological properties of poloxamer 407 (P407) formulations containing various CPEs, and quantify effects of formulation excipients on permeation enhancement of NTX through MN-treated skin ex vivo.
Results and discussion
Transdermal NTX delivery could provide a more patient-friendly treatment option for alcohol and opioid misuse. The convenience of a transdermal patch, along with more consistent plasma concentrations, could effectively suppress cravings throughout lifelong treatment, potentially introducing a lower chance of relapse. While solid MNs may provide a new route for NTX delivery that is more convenient, the challenge of rapid in vivo micropore closure significantly blunts the delivery window. In the present work, we targeted this delivery challenge from two perspectives: (1) creating a P407 depot in the micropores for controlled delivery after micropores close, and (2) enhancing NTX delivery through the hydrophobic intact skin surrounding the micropores. Here we report the effect of select CPEs on NTX solubility, P407 gelation properties, and permeation of NTX through intact and MN-treated skin. A schematic of the treatment approach can be seen in Fig. 1.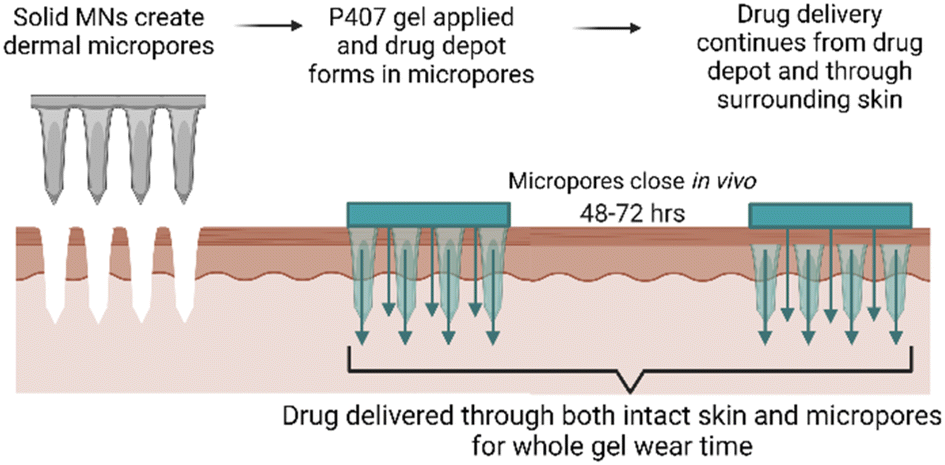 | ||
| Fig. 1 Schematic of proposed poloxamer formulation application with microneedle pre-treatment. The formulation remains on the skin even after the micropores have closed, which permits ongoing drug delivery from a drug depot in the micropores as well as delivery through the surrounding intact skin. Figure made with Biorender.com. | ||
Visualization of P407 migration into micropores
P407 was fluorescently labeled with 5-DTAF to visually confirm that P407 can quickly migrate into micropores, despite being delivered as a viscous liquid that gels almost immediately on contact with the skin. The labeling reaction reached ∼20% completion; this somewhat low reaction efficiency is consistent with previous reports using similar methods9 and is beneficial for limiting non-specific fluorescence in the images. Chromatograms of reaction mixtures before and after dialysis showed complete removal of the peak designated as free 5-DTAF, indicating that dialysis effectively removed unreacted 5-DTAF (data not shown). After skin sectioning, the micropores were imaged using confocal microscopy (Fig. 2). Within 5 min, almost no 5-DTAF can be seen within the skin for either 5-DTAF solution or labeled P407. After 30 min of skin contact, both 5-DTAF solution and labeled P407 have migrated into the micropores. This suggests that despite the quick gelation of P407 formulations at the skin surface, P407 does migrate into micropores – allowing for the local creation of a gel depot within the skin. These results are in line with previously published permeation studies of P407 applied to MN-treated skin that showed sustained drug delivery after formulation removal from the skin surface,8 and the findings were attributed to P407 gelling within the micropores. This also agrees with previous studies showing that fluorescent probes can be delivered into micropores from P407 gels.14,15 Videos demonstrating the ability for P407 labeled with 5-DTAF to migrate into a micropore (depicted by 3D visualization of confocal z-stacks), and intact skin controls, can be found in the ESI.†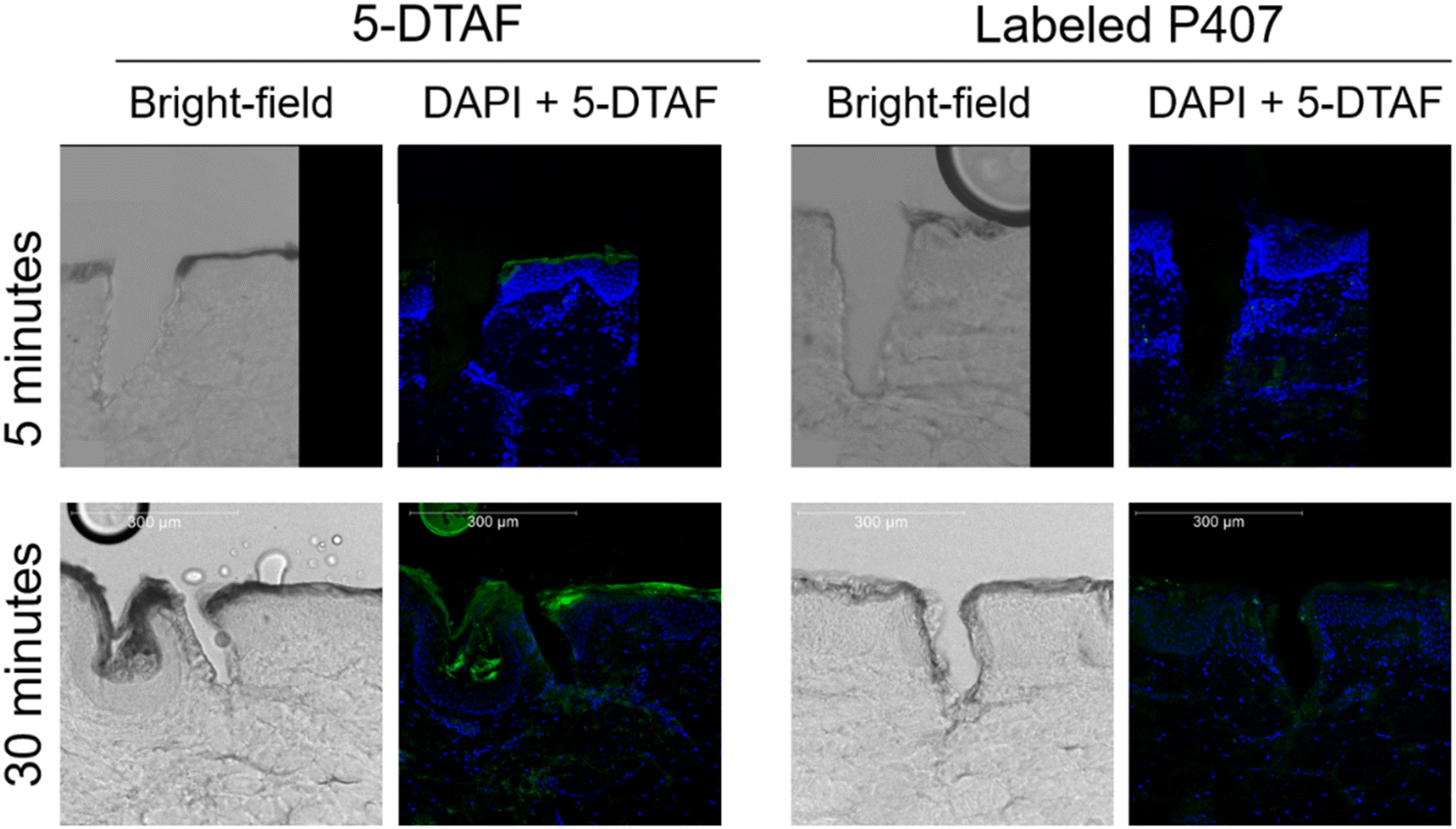 | ||
| Fig. 2 Brightfield and fluorescent images of micropores in skin. Maximum projection from a z-stack taken on a confocal microscope. Blue is DAPI staining and green is 5-DTAF. | ||
Solubility of NTX in P407 formulations
Solubility studies were performed to identify potential formulations for skin permeation studies. We selected eight CPEs for these studies, from the sulfoxide, alcohol, polyol, and fatty acid ester classes; all solubility results can be seen in Fig. 3. The overall goal was to identify formulations with increased NTX solubility, as increased drug concentration can enhance permeation through MN-treated skin.16,17 NTX flux through micropores created by MNs is also pH dependent, benefiting from decreased pH and higher solubility. NTX flux is maximized near the skin acid mantle pH of 5, and for this reason acetate buffer was chosen as the buffering agent in all P407 solutions (±CPE).18 The acetate pKa of 4.76 is very close to our desired pH of 5.0 and acetate does not damage skin integrity,17,19 making this a good choice for the P407 formulations.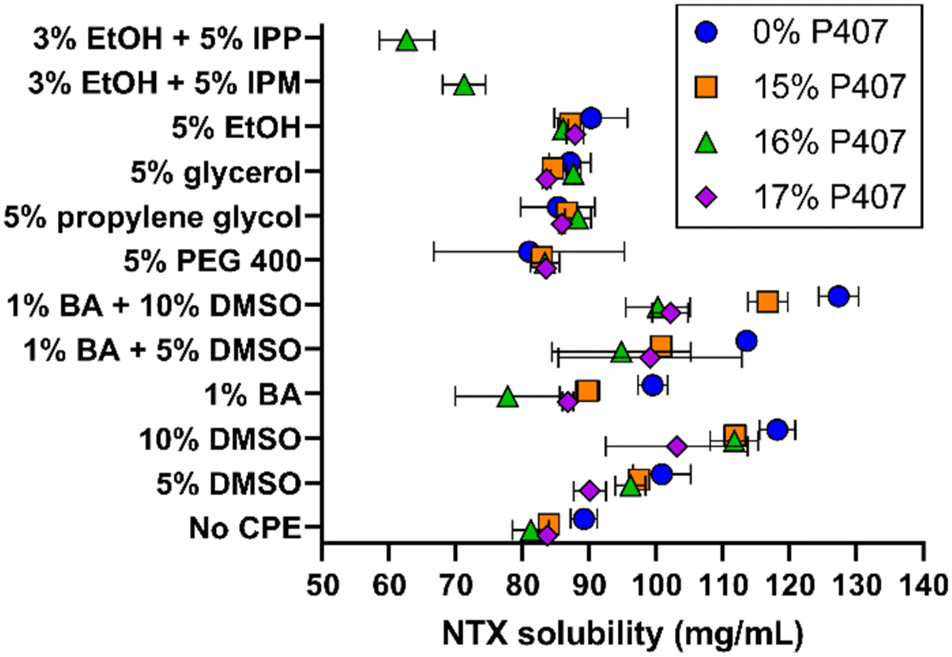 | ||
| Fig. 3 NTX solubility at 20 °C in 15–17% P407 formulations with chemical permeation enhancers (n = 3, mean ± SD). These P407 concentrations were selected because of the likelihood of gel formation at physiologic skin temperature and for comparison with prior studies of NTX in P407. BA = benzyl alcohol, CPE = chemical permeation enhancer, DMSO = dimethyl sulfoxide, EtOH = ethanol, IPP = isopropyl palmitate, IPM = isopropyl myristate, PEG 400 = polyethylene glycol (MW 400). | ||
NTX solubility in pH 5.0 acetate buffer at 20 °C was 89.17 ± 2.01 mg mL−1, which was significantly higher than NTX solubility of 79.21 ± 1.45 mg mL−1 in water (data not shown), P < 0.01. NTX exhibits a pKa of 8.38 at 20 °C because of protonation of the tertiary amine.20 The approximate percent ionization increases from 99.7 to 99.9% when the formulation is buffered at pH 5.0 with acetate. This slight change in ionization may contribute to the increased NTX solubility, and it is also likely that NTX-acetate salts formed and improved the solubility.17
Addition of P407 with no CPE significantly reduced NTX solubility (vs. acetate buffer aqueous solution) to 83.93 ± 1.17, 81.20 ± 2.72, and 83.73 ± 0.76 mg mL−1 for 15%, 16%, and 17% P407, respectively, P < 0.05. The inverse relationship of decreased NTX solubility with increasing P407 is likely attributed to lower water content per volume in the formulation. Despite decreased NTX solubility in P407 compared to aqueous solution, in several cases this was compensated for with the addition of CPEs alone or in combination, as described below. In contrast, NTX solubility in formulations containing 15–17% P407 were not significantly different compared to aqueous solution (without P407) with 5% of PEG 400, propylene glycol, glycerol, or ethanol, P > 0.05. The mean NTX solubilities across the 15–17% P407 formulations were 83.22 ± 1.21 mg mL−1 (5% PEG 400), 86.91 ± 1.47 mg mL−1 (5% propylene glycol), 85.23 ± 2.03 mg mL−1 (5% glycerol), and 87.01 ± 1.10 mg mL−1 (5% ethanol). NTX solubility was significantly higher in P407 formulations containing 5% propylene glycol, 5% glycerol, and 5% ethanol compared to no CPE, P < 0.05, though it was a small increase of 3–4 mg mL−1.
Ethanol (3%) was required in formulations containing isopropyl myristate (IPM) and isopropyl palmitate (IPP) to stabilize the emulsion produced when adding these oily compounds into aqueous P407 solutions. The IPM emulsions in 16% P407 were stable for at least 72 h, while IPM in aqueous solution (no P407) would completely separate within minutes. Because of this immediate phase separation, NTX solubility was not measured in 0% P407. In 16% P407, 5% IPP and 5% IPM lowered NTX solubility to 62.62 ± 4.10 and 71.21 ± 3.21 mg mL−1, respectively. This was significantly lower than in 16% P407 without CPE, P < 0.05, likely due to the oily consistency of these fatty ester acids which is unfavorable for NTX at this pH.
Dimethyl sulfoxide (DMSO), 5%, increased NTX solubility in 0, 15%, and 16% P407 when compared to formulations without CPEs at the same P407 concentration, P < 0.01, and 10% DMSO increased NTX solubility in all P407 formulations (P < 0.05). This increase ranged for 6.3 to 14.94 mg mL−1 in 5% DMSO and 19.3 to 30.5 mg mL−1 in 10% DMSO. Due to this notable solubility increase, DMSO formulations were selected for further testing for gelation properties. Benzyl alcohol (1%) significantly increased NTX solubility in the absence of P407, P < 0.05, but did not significantly change solubility in formulations with P407, P > 0.05. The effect of DMSO and benzyl alcohol in combination was also tested. The combination of 5 or 10% DMSO and 1% benzyl alcohol synergistically increased NTX solubility in formulations with no P407, vs. DMSO or benzyl alcohol alone, P < 0.05. This synergistic effect was not observed for P407 formulations, P > 0.05.
While benzyl alcohol alone or with DMSO did not significantly increase NTX solubility in P407 formulations, benzyl alcohol was still selected for further testing in permeation studies for many reasons. It is FDA approved for use in cosmetic, personal care, and skincare products, as well as drug products (including for topical and transdermal delivery). It has been used previously as a preservative (at the presently studied concentration) in NTX formulations for in vivo MN delivery.3,21,22 Additionally, benzyl alcohol is an antioxidant and this may help limit degradation of poloxamer,23 which could be useful for long-term formulation stability. Therefore, 1% benzyl alcohol, 5% and 10% DMSO, and their combinations were chosen for permeation studies. As an additional comparator, permeation studies were performed with 5% IPM (+3% ethanol) formulations, because IPM has more oily properties (compared to benzyl alcohol and DMSO).
Variability in NTX solubility overall was higher in samples that had gelation temperatures near 20 °C (the temperature at which solubility studies were conducted). Examples of formulations that fit this included 1% benzyl alcohol in 16% P407, 1% benzyl alcohol with 5% DMSO in 16% and 17% P407, and 10% DMSO in 17% P407 (at 20 °C each of these did not follow overall trends, in addition to showing high variability).
Gelation temperature of formulations with P407 and CPEs
The thermogelling properties of poloxamer formulations arise from the unique triblock copolymer structure of polypropylene oxide and polyethylene oxide. With increasing temperature the hydrophobic center dehydrates, folding in on itself and generating micelles. Once enough micelles are generated and the critical volume fraction is surpassed, the tails of these micelles overlap and generate a rigid gel.24,25 These thermogelation properties present a distinct opportunity to form a localized NTX depot in the micropores created by MN application, which can provide sustained NTX delivery over time.8 The ability of poloxamers to form an in situ drug depot in the skin has been reported previously,14,15 but these prior studies primarily focused on the micropore environment. However, there is a large area of intact skin surrounding the micropores that can also be utilized for drug delivery, and it would be a missed opportunity to not simultaneously exploit this pathway. However, this does present a unique challenge because the requirements for delivery through intact skin are much different than those for delivery through micropores; we used CPEs as a novel approach to this dichotomy.The temperature at which gelation occurs is highly relevant because a critical attribute of a desired transdermal NTX product is for gelation to occur at or before skin surface temperature (∼32 °C). Gelation at this temperature would ensure that the P407 gel will provide sustained release properties. It is well known that formulation additives can alter gelation properties of poloxamer formulations,25,28–30 and thus it was essential to evaluate the gelation temperatures of the P407 gels loaded with NTX ± CPEs. Here we used rheological techniques to compare gelation temperatures between formulations and identify those suitable for permeation studies.
The gelation temperatures of P407 formulations tested in permeation studies with CPEs can be seen in Fig. 4. The NTX concentration was held constant at 7.5% in all formulations; this concentration was selected based on the solubility studies. As expected, increasing P407 concentration decreased the gelation temperature for all formulations (the inverse relationship between gelation temperature and P407 concentration is well established).26–28 Gelation temperatures for the 15%, 16%, and 17% P407 (all made in pH 5.0 acetate buffer) were 29.21 ± 1.09, 28.60 ± 1.88, and 23.78 ± 0.35 °C, respectively. The presence of NTX generally increased gelation temperature in the absence of CPEs but had inconsistent effects on gelation temperature in the presence of CPEs. Ultimately, however, NTX did not change the gelation temperature significantly in absence of CPEs, P > 0.05.
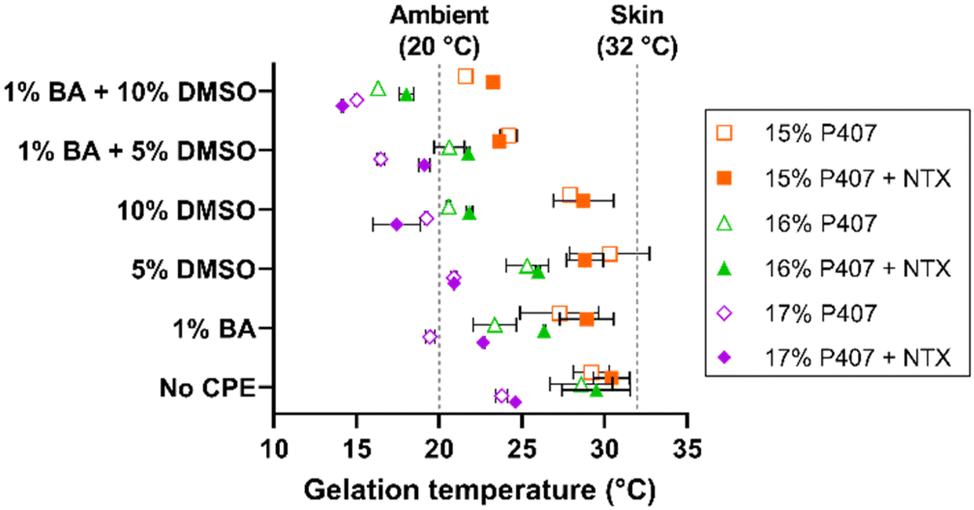 | ||
| Fig. 4 Gelation temperatures for 15, 16, and 17% w/w P407 formulations with (filled symbols) and without (open symbols) 7.5% NTX and/or chemical permeation enhancers (n = 3, mean ± SD). Some error bars are too small to be shown; data were nudged along the y axis so that they would not overlap. BA = benzyl alcohol, CPE = chemical permeation enhancer, DMSO = dimethyl sulfoxide. | ||
The gelation temperature of 15%, 16%, and 17% P407 formulations ± NTX, 1% benzyl alcohol, and 5% or 10% DMSO were also measured to select the formulations most suitable for transdermal application. The gelation temperatures for 16% P407 with 7.5% NTX were 29.5 ± 2.1 (0% CPE), 26.4 ± 0.1 (1% benzyl alcohol), 26.0 ± 0.1 (5% DMSO), and 21.8 ± 0.1 °C (1% benzyl alcohol + 5% DMSO). The same trends were seen for the 15% and 17% P407 formulations, as shown in Fig. 3. On increasing DMSO content from 5% to 10%, the gelation temperature decreased by ∼3–4 °C to 18.0 ± 0.5 °C and 21.8 ± 0.2 °C with and without 1% benzyl alcohol, respectively. Overall, the presence of benzyl alcohol and/or DMSO lowered gelation temperature compared to formulations without CPE (DMSO has been previously shown to decrease gelation temperature of P407 formulations31,32). The overall trend of decreasing gelation temperature observed upon addition of CPEs could be attributed to water molecules having more affinity for CPEs; this would result in fewer free water molecules to interact with poloxamer, increasing the need to generate micelles.26
Because 16% P407 formulations with CPEs had gelation temperatures that were generally within the desired range between ambient and skin temperature, 16% P407 formulations were selected for skin permeation experiments.
Formulation viscosity
To determine the impact of P407 and CPEs on gel characteristics, viscosity was measured for 15%, 16%, and 17% P407 formulations with 7.5% NTX at 32 °C, Fig. 5. At 32 °C the P407 formulations (15–17% P407) exhibited non-Newtonian shear thinning behavior, meaning that viscosity decreased as the applied shear increased. This is typical for polymers and has been attributed to higher molecular weight polymers disentangling and aligning with the direction of flow, causing proportionally less drag at higher shear rates.33 At a shear rate of 100 s−1 the viscosity of NTX-containing poloxamer formulations increased with increasing P407 concentration: 0.78 ± 0.29, 1.32 ± 0.77, and 2.35 ± 0.02 Pa s for 15, 16, and 17% P407, respectively. This positive relationship of increased viscosity with increased poloxamer concentration is consistent with previous studies and can be attributed to more molecules being available to catch on each other, resisting flow of the solution.26,27 We also measured the viscosity of 16% P407, 7.5% NTX formulations containing 5% or 10% DMSO, 1% benzyl alcohol, or their combination, since these preparations were selected for permeation studies based on solubility and gelation temperatures. The viscosity of these formulations at 100 s−1 was not significantly different from 0% CPE, P > 0.05, ranging from 0.83 to 2.57 Pa s. It is possible that the CPE concentrations were too low to have any notable impact on the macroviscosity.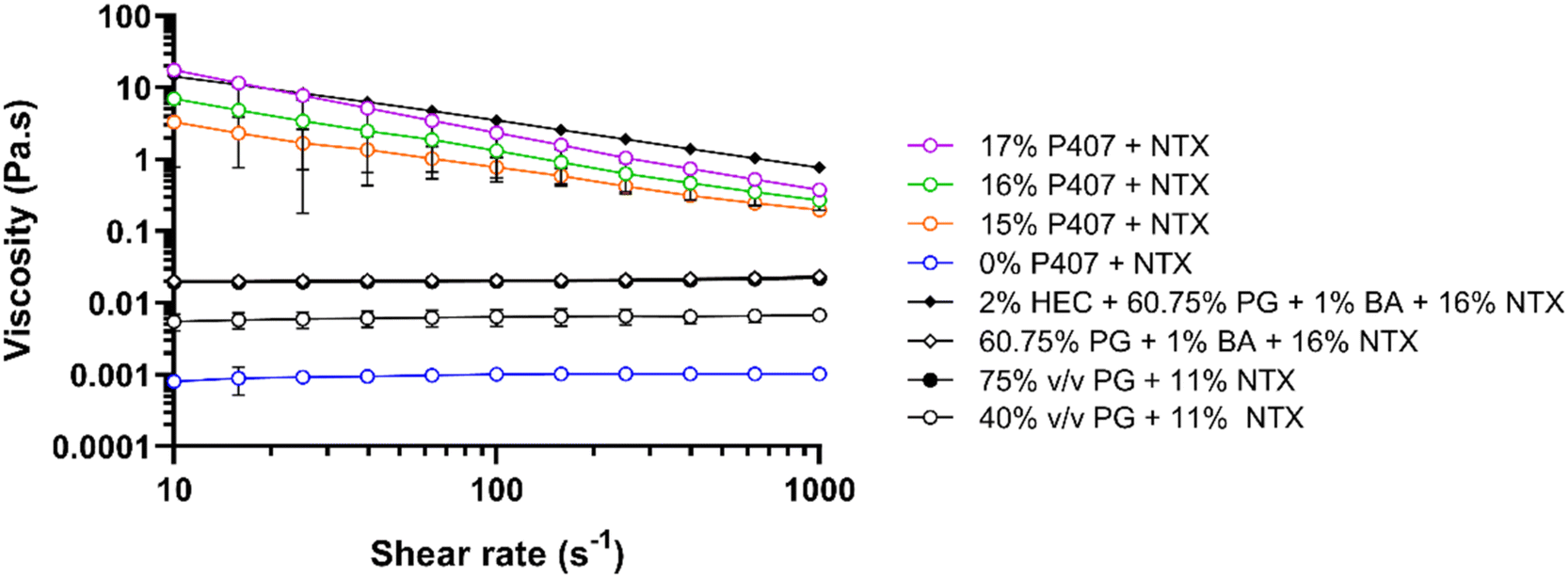 | ||
| Fig. 5 Viscosity at 32 °C vs. shear rate of 10–1000 s−1 for 40% or 75% propylene glycol solutions, 2% hydroxyethyl cellulose gels, or 16% P407 gels containing various NTX concentrations. Gels contained 7.5% NTX unless otherwise stated. All percentages are w/w unless otherwise stated. PG and HEC formulations were included for comparison of P407 gels with NTX gels used in prior studies. n = 3, mean ± SD. BA = benzyl alcohol, HEC = hydroxyethyl cellulose, NTX = naltrexone, PG = propylene glycol. | ||
In addition to the P407 formulations, we also measured viscosity of the gel used in the first-in-human study of MN-assisted NTX transdermal delivery. The viscosity of this formulation was compared with the viscosity of our P407 preparations as an additional variable that may impact differences in permeation between formulations. This gel contained 2% hydroxyethyl cellulose, 60.75% propylene glycol, 1% benzyl alcohol, and 16% NTX.3 Like P407 formulations, the viscosity of this hydroxyethyl cellulose formulation showed non-Newtonian behavior at 32 °C. At 100 s−1 and 32 °C the viscosity was 3.51 ± 0.24 Pa s, which is significantly higher than all 16% P407 formulations with 7.5% NTX, with or without CPEs, at this temperature and shear rate, P < 0.05. This suggests that NTX may permeate faster through MN-treated skin from the 16% P407 formulations.
The viscosity of aqueous solutions containing 40 or 75% propylene glycol with 11% NTX and no gelling agent at 32 °C were also measured to compare with viscosities reported by Milewski et al. These previously studied NTX-loaded propylene glycol solutions displayed a strong influence of formulation viscosity on NTX permeation through MN-treated skin.12 At 32 °C, the propylene glycol formulations exhibited Newtonian fluid behavior, meaning that viscosity remained constant across all shear rates. Viscosity at 100 s−1 was 6.39 ± 0.00 and 19.98 ± 0.00 mPa s for 40% and 75% propylene glycol, respectively. This was slightly higher, though not significantly different, when compared to the prior measurements of 4.01 ± 0.01 and 14.40 ± 0.00 mPa s for 40% and 75% propylene glycol with 11% NTX.12
At 32 °C, 15–17% P407 and 2% hydroxyethyl cellulose formulations were much more viscous than all aqueous solutions without hydroxyethyl cellulose or P407. The notably increased viscosity of P407 and hydroxyethyl cellulose formulations (vs. solutions) is expected because at 32 °C all of these formulations are macroscopically in a gelled state with inhibited flow, while solutions with propylene glycol are still liquid and flow more easily. The viscosity of the P407 formulations may contribute to the sustained in vitro NTX release from P407 gels that has been previously reported,8 which is an ideal characteristic for the present goals and clinical condition.
In vitro NTX permeation from P407 formulations
There are two major routes of drug transport to consider with a MN pretreatment approach: micropores (a hydrophilic environment) and intact skin surrounding the micropores (a hydrophobic environment). CPEs may impact intact skin permeability by affecting drug skin partitioning, solubility in a formulation, or disorganizing the structure of skin lipids. Due to the very small area of the micropores (<1% of total area covered by the formulation),34 the surrounding intact skin may contribute significantly to absorption depending on the drug molecule and formulation. Here we aimed to study whether CPEs in combination with MN treatment would synergistically increase NTX permeation. To evaluate the impact of select CPEs and their combinations on transdermal NTX delivery, in vitro skin permeation studies were performed using excised porcine skin treated with solid MNs. All novel P407 formulations tested in permeation studies contained 7.5% NTX to minimize effects of changes in chemical activity on the permeation through skin, and 0.5 mL was the application volume for all conditions. NTX flux through skin was calculated from the 56–72 h data on the cumulative permeation vs. time curve (representative formulations shown in Fig. 6). All flux data can be seen in Fig. 7.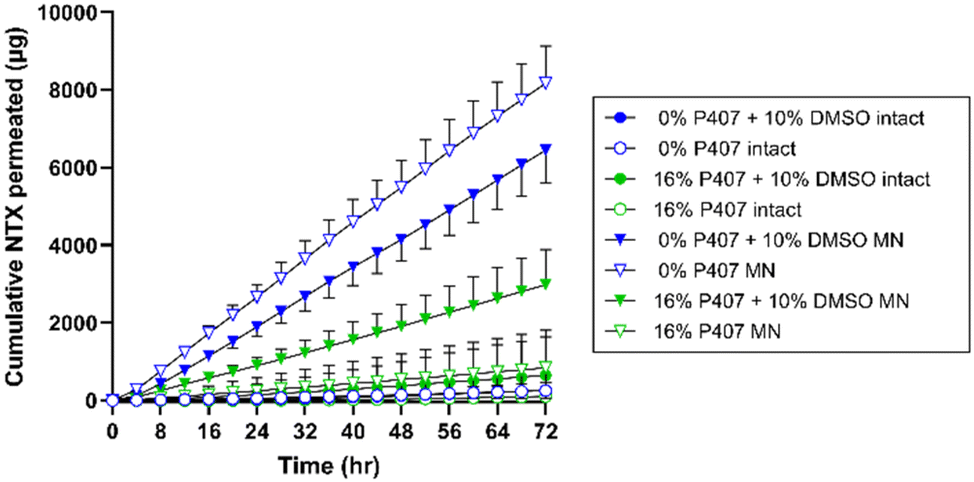 | ||
| Fig. 6 Representative profiles of cumulative NTX permeated through intact (circles) or MN-treated (triangles) porcine skin from formulations containing 0% or 16% P407 ± 10% DMSO. n = 3–8, mean ± SD. BA = benzyl alcohol, DMSO = dimethyl sulfoxide, MN = microneedle. | ||
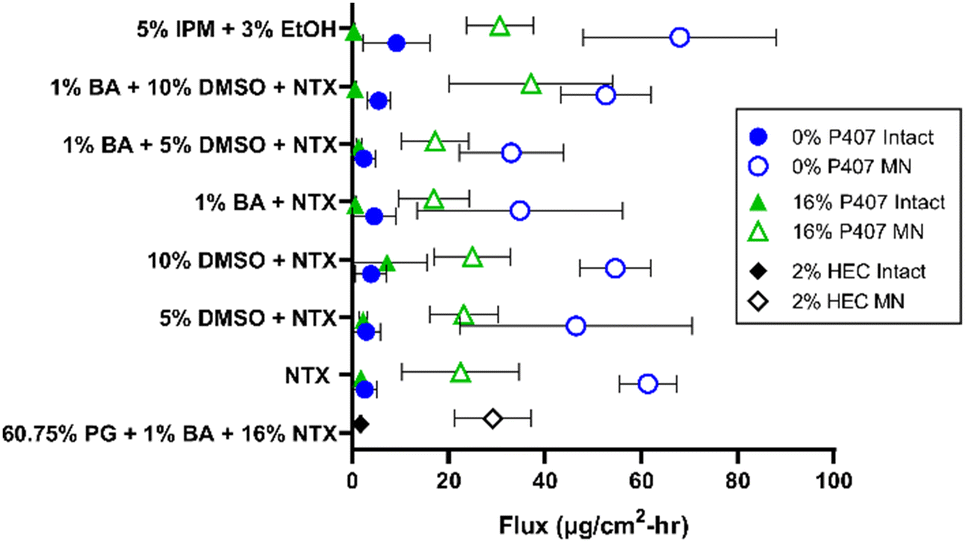 | ||
| Fig. 7 Terminal flux over 56–72 h through intact or MN-treated skin from different formulations with CPEs (n = 3–8, mean ± SD). All formulations contained 7.5% NTX except for the 2% hydroxyethyl cellulose formulations which contained 16% NTX. BA = benzyl alcohol, CPE = chemical permeation enhancer, DMSO = dimethyl sulfoxide, EtOH = ethanol, HEC = hydroxyethyl cellulose, IPM = isopropyl myristate, MN = microneedle, PG = propylene glycol. | ||
Effect of MN treatment on skin barrier and permeation
Transepidermal water loss (TEWL) was measured to verify that intact skin samples had an appropriate skin barrier, and confirm that micropore formation impaired the barrier. Lower TEWL values confirm intact barrier function, and TEWL significantly increases upon micropore formation (as water loss increases through the epidermal micropores).35,36 On average the TEWL values of intact skin were 9.93 ± 3.92 g m−2 h−1, while MN-treated skin had significantly higher TEWL values of 33.27 ± 2.93 g m−2 h−1, P < 0.01. These TEWL measurements confirmed that MN treatment significantly inhibited skin barrier properties.NTX flux was significantly higher through MN-treated (vs. intact) skin for all formulations, P < 0.05, except for the 0% P407/1% benzyl alcohol and the 16% P407/1% benzyl alcohol/10% DMSO preparations. This significant flux increase after MN application is expected, since MN treatment disrupts the skin barrier function and increases permeability of hydrophilic drugs.18 Because the P407 formulations were buffered in acetate, NTX is at a higher ionization percentage and is therefore more highly favors the aqueous micropore environment. In addition, CPEs such as DMSO, fatty acids, and alcohols may preferentially enhance permeation of hydrophilic molecules.37
A flux enhancement ratio (ER) was calculated to quantify the ability of micropores to physically enhance permeation (vs. intact skin). All formulations had an enhancement ratio > 1, confirming that MNs successfully enhanced permeation regardless of formulation. However, the extent of this enhancement differed between formulations. For instance, the ER for 0% P407 was lower for formulations with 1% benzyl alcohol: the ER did not surpass 14-fold in these cases, compared to ERs as high as 23-fold when benzyl alcohol was not present. The opposite was true for 16% P407: the enhancement reached as high as 60-fold in the presence of benzyl alcohol vs. at most 12-fold without. This suggests that when benzyl alcohol is used in P407 formulations it may promote NTX permeation through the micropores, and/or it may decrease the permeation through intact skin. The former could be due to benzyl alcohol having a slightly hydrophobic nature (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P 1.1),38 which is not preferable for NTX, promoting its partitioning into the aqueous epidermal micropores. The latter could be due to benzyl alcohol interacting with the P407 rather than the skin barrier; this is supported by previous reports of lowered dexamethasone permeation through intact skin when formulated with P407 and benzyl alcohol.39 A balance of these two properties is highly likely.
P 1.1),38 which is not preferable for NTX, promoting its partitioning into the aqueous epidermal micropores. The latter could be due to benzyl alcohol interacting with the P407 rather than the skin barrier; this is supported by previous reports of lowered dexamethasone permeation through intact skin when formulated with P407 and benzyl alcohol.39 A balance of these two properties is highly likely.
The highest flux enhancement from any formulation was achieved with 5% IPM (with 3% ethanol) in 16% P407, in which an ER of 95-fold between intact and MN-treated flux was observed. This could be due to NTX preferring the aqueous micropore channels to the oilier formulation. The flux enhancement for this formulation could have been aided by the ethanol in the formulation (used to stabilize the emulsion), but permeation from formulations with ethanol alone was not tested so this impact could not be directly determined.
Effect of P407 on NTX permeation and flux
Flux through intact skin from 16% P407 gels was not significantly different than from aqueous solution without P407 at the same CPE concentrations (P > 0.05). For example, NTX flux through intact skin from 16% P407 (with 7.5% NTX and no CPE) was 1.84 ± 1.76 μg cm−2 h−1, which was not significantly different than the flux of 2.63 ± 2.44 μg cm−2 h−1 from the same aqueous solution containing no P407; P > 0.05. Since these values are not significantly different, this likely means that NTX partitioning into the stratum corneum is the rate-limiting step in permeation through intact skin (not the release of NTX from the gel matrix).As a comparator condition for a CPE with different physicochemical properties, we performed permeation studies with formulations containing 5% IPM. Previous reports have shown IPM enhancement of naloxone permeation through intact skin (another opioid receptor antagonist with similar structure to NTX but slightly different physicochemical properties),40 also making this a noteworthy comparator. As shown in Fig. 7, NTX flux through intact skin was higher for 5% IPM in 0% P407 (10.87 ± 9.06 μg cm−2 h−1) than all other formulations on intact skin including 5% IPM in 16% P407 (0.32 ± 0.25 μg cm−2 h−1). The variability of NTX flux from 5% IPM in solution (no P407) was also much larger than all other formulations on intact skin, which could be attributed to IPM being an oily compound with a logP of 7.341 (unfavorable for NTX at this pH) and this formulation showing immediate phase separation. IPM can interact with the lipid domains in the skin and cause fluidity, which likely explains its permeation enhancement for intact skin.11,42 While in solution, IPM may be able to interact more with the stratum corneum because of the absence of competing interaction from micelles, further increasing its permeation enhancement. In contrast, in the P407 formulations there is an extra step of IPM release from the gel structure before it can interact with the skin, which may have lowered its effectiveness as a CPE. It is possible that ethanol could be providing some further permeation enhancement from this formulation, but we did not run additional control permeation studies with just ethanol as the CPE to rule this out.
In comparison, NTX flux through MN-treated skin from 16% P407 was lower than from aqueous solution. This difference was significant for formulations with no CPE (22.5 ± 12.17 vs. 61.37 ± 5.93 μg cm−2 h−1), 10% DMSO (25.00 ± 7.89 vs. 54.62 ± 7.42 μg cm−2 h−1), and 1% benzyl alcohol + 5% DMSO (17.25 ± 6.98 vs. 33.01 ± 10.78 μg cm−2 h−1); P < 0.05. This suggests that P407 can better sustain delivery of NTX through MN-treated skin (vs. aqueous solution which has no sustained release properties). This corroborates previous permeation studies of drug permeation through MN-treated skin from formulations with and without P407.14 These data also support some dependency of flux on viscosity, as lower viscosity aqueous solutions (without P407) produced higher flux through MN-treated skin than the higher viscosity 16% P407 gels.
As seen in Fig. 6 and 7, higher cumulative permeation and flux through MN-treated skin were obtained from the aqueous solutions compared to the same formulations with 16% P407. The decreased flux resulting from addition of P407 may at first seem counterproductive to the overall goal. However, there are several reasons why gelled P407 formulations are better suited in this situation. For a transdermal product that will require ongoing contact with the skin for treatment of a chronic condition such as opioid or alcohol misuse disorders, an aqueous solution is not ideal. First, the high rate of delivery would lead to dose depletion more quickly from the aqueous solution. This would ultimately lead to inconsistent dosing over the whole patch wear time as the diffusion gradient decreases with dose depletion (flux is directly dependent on the concentration gradient between formulation and skin). Second, aqueous solution does not have any sustained release properties, which is critical to the current indication, and is provided by the P407 gels. Third, for ongoing delivery, the solution would need to be in prolonged direct contact with the skin underneath a patch. Even slight stretching, bumping, or sliding of the patch could cause the formulation to leak out around the patch. This would ultimately lead to inconsistent dosing, and the decreased pharmaceutical elegance of such a product would also be undesirable to patients. Last, the aqueous solution would not have the ability to extend drug delivery past the point of micropore closure; that is, once the micropores have closed in vivo, delivery of NTX from the aqueous solution would decrease abruptly. Unfortunately, this is not something that can be modeled in excised skin permeation studies. In the context of in vivo micropore closure, NTX permeation from the aqueous solution would likely cease between 48–72 h, while the P407 gel depot formed in the micropores would allow continued delivery even after micropores closed at the skin surface. In the current study we did not simulate micropore closure. However, in previous studies when a P407 formulation is removed from the skin surface at 48 h and continued NTX permeation is measured, the depot effect of the P407 can be observed.8
Thus, while the P407 significantly reduced permeation and flux compared to aqueous solutions, the gelled formulation offers many other practical advantages that are more appropriate for transdermal products. This includes the ability to extend drug delivery by generating a gel depot in the micropores, which may somewhat counteract the limitation of rapid micropore closure.
Effect of CPE on flux
All comparisons of flux from P407 formulations ± DMSO and benzyl alcohol can be seen in Table 1. In formulations with 16% P407, only 5% IPM produced significantly higher flux through MN-treated skin compared to 16% P407 without CPE, P < 0.01 (data not shown). Despite the ability of DMSO to interact with stratum corneum proteins and lipids,11,44 increasing DMSO concentration in our formulations did not uniformly increase flux through intact or MN-treated skin in the no P407 conditions. DMSO 10% with or without benzyl alcohol generally increased flux through intact skin, whereas 5% DMSO with or without benzyl alcohol had mixed effects or lowered the flux. This suggests that at the present concentrations, these CPEs are not impacting the stratum corneum structure enough to consistently enhance permeation of the hydrophilic NTX molecule. The concentrations of DMSO studied here are lower than previous studies that have shown permeation enhancement with DMSO up to 60%.11,44 Additional, DMSO can decrease the release rate of hydrophilic drugs from formulations.31 Slower release from the P407 formulation could have counteracted any permeation enhancement benefits from the DMSO. Some researchers suggest applying CPEs to the skin for two hours before applying a drug formulation.43 While this may be more effective for disorganizing the stratum corneum lipids, this extra step would be inconvenient for patients and would not be suitable for our intended application.| 1% BA | 5% DMSO | 10% DMSO | 1% BA + 5% DMSO | 1% BA + 10% DMSO | ||
|---|---|---|---|---|---|---|
| BA = benzyl alcohol; CPE = chemical permeation enhancer; DMSO = dimethyl sulfoxide, MN = microneedle. | ||||||
| Intact skin | No P407 | ↑ | ↔ | ↑ | ↔ | ↑ |
| 16% P407 | ↓ | ↑ | ↑ | ↓ | ↓ | |
| MN | No P407 | ↓ | ↓ | ↔ | ↓ | ↔ |
| 16% P407 | ↓ | ↔ | ↔ | ↓ | ↑ | |
In contrast, increasing the DMSO concentration from 5 to 10% with 1% benzyl alcohol in aqueous solution without P407 significantly increased flux through MN-treated skin, P < 0.05. Additionally, increasing the DMSO concentration from 0 to 10% or from 5 to 10% significantly increased NTX flux through MN-treated skin from 16% P407 formulations with 1% benzyl alcohol; P < 0.05. This suggests that a threshold had been surpassed at which a high enough DMSO concentration was present to achieve permeation enhancement when combined with the micropore channel pathway. This is consistent with previous studies showing increased permeation when DMSO concentration was increased from 5 to 10%.45 This effect was only seen with 1% benzyl alcohol in the present studies, which suggest that benzyl alcohol and DMSO may act synergistically to increase NTX permeation (this could relate to the well-known membrane fluidizing properties of benzyl alcohol).46
Variability in flux measurements appeared to be formulation dependent. Flux through intact skin was much less variable overall vs. MN-treated skin. This is likely due to the highly effective stratum corneum barrier restricting permeation of the hydrophilic NTX molecule. Regardless of formulation, NTX will have poor permeation through intact skin (even with CPEs). In contrast, MN treatment removes the physical permeation barrier, and any variability in NTX release form the formulation itself will likely manifest as variability in permeation. Flux through MN-treated skin from 16% P407 showed generally lower variability compared to solutions with no P407. The amount of variability was also more consistent between formulations, which suggests that P407 can better control NTX delivery compared to aqueous solutions. When benzyl alcohol or DMSO were the only CPE in solution (without P407), the flux variability was very large; interestingly, variability was notably reduced when 1% benzyl alcohol was added in combination with DMSO at 5 or 10%.
In vivo considerations for MN NTX delivery
The first pharmacokinetic study using the solid MN pretreatment approach in humans used NTX as the model compound, with a formulation containing 16% NTX, 2% hydroxyethyl cellulose, 60.75% propylene glycol, and 1% benzyl alcohol.3 In our present study we also measured NTX permeation from this gel so that it could be compared with the present P407 formulations. NTX flux from our 16% P407 (no CPE) formulation through MN-treated skin was not significantly different from the 2% hydroxyethyl cellulose gel used in the prior studies, P > 0.05. This suggests that P407 formulations may deliver NTX with similar efficiency through MN-treated skin while using significantly less drug overall, as the present formulations had a nearly 50% reduction in NTX concentration compared to the previous pharmacokinetic study (7.5% vs. 16% NTX, respectively). This would reduce the cost of product manufacturing and could ultimately also reduce cost to the consumer, which further enhances the patient-centered approach that a transdermal NTX product could offer. Interestingly, the viscosity of 2% hydroxyethyl cellulose gels at 32 °C was higher than 16% P407 gels. This may have contributed to the higher NTX flux from the poloxamer gels (22.5 ± 12.2 μg cm−2 h−1) vs. flux reported by Wermeling et al. (14.7 ± 4.9 μg cm−2 h−1) for the hydroxyethyl cellulose gel;3 this further supports the idea that flux is at least partly viscosity dependent.In the prior pharmacokinetic study, a total patch size of ∼28 cm2 was required to achieve a therapeutic plasma NTX concentration of 2 ng mL−1 (this concentration provides sufficient opioid receptor blockade, reduces cravings, and may reduce the chance of relapse47,48). The 28 cm2 patch size was based on the following equation:
| Apatch = (Cl·Css)/Jss |
| No P407 | 16% P407 | |
|---|---|---|
| BA = benzyl alcohol; CPE = chemical permeation enhancer; DMSO = dimethyl sulfoxide. | ||
| No CPE | 7 | 19 |
| 1% BA | 12 | 25 |
| 5% DMSO | 9 | 18 |
| 10% DMSO | 8 | 17 |
| 1% BA + 5% DMSO | 13 | 24 |
| 1% BA + 10% DMSO | 8 | 11 |
Using this approach to estimate patch size, it is assumed that the NTX-loaded P407 gel is in continuous contact with the skin even after the micropores close. The addition of CPEs (specifically, DMSO and benzyl alcohol) to our formulations allows continued NTX delivery through the intact skin surrounding the micropores. This would allow a patient to simply apply one gel-loaded patch over the micropores and not need to remove or change that patch until the end of the specified wear period (the long-term goal is 7 days). While the estimated patch size for our formulation is smaller than previously used and is in the typical range of transdermal patch sizes, it may be beneficial for patient discretion to further minimize the patch size for therapeutic delivery.
Limitations and future studies
Presently, we have demonstrated the potential for permeation optimization using formulation excipients and solid MNs as a pretreatment. In addition to the formulation, properties of the geometry of the MN themselves (length, number of micropores, or MN material) can impact both the extent of permeation and the in vivo micropore closure time.8,51,52 Here we used MNs of 600 μm length, which is similar to the 620 μm length used by Wermeling et al.3 However, MN lengths of up to 800 μm have been safely used in humans and this would present another variable for improving NTX permeation.53,54 Here we also used the two-step process in which solid MNs serve as a skin pretreatment to create micropores (an approach that has been used successfully in prior NTX MN studies in vivo). While somewhat less convenient overall, this approach permits greater control over both the MN geometry and the formulation, as these variables can be adjusted separately to achieve specific goals. This approach has been successfully used with poloxamer gels to sustain delivery of hydrophilic compounds for up to 96 h in vitro.8,15 However, a design in which polymeric MNs were used could also be of benefit and may provide greater ease of use, though it may be limiting in the dose that can be applied. Strategically optimizing the combined effects of MN characteristics and CPE combinations could further drive down the patch size, which would be more discreet and preferred by most patients.The studies here were all performed in vitro, and micropore closure is not observed in ex vivo skin samples, and the gradual process of micropore closure cannot be readily modeled in vitro. For this reason, in vivo pharmacokinetic studies will be necessary in animals and/or humans. Prior in vivo studies of MN-assisted NTX delivery have been conducted in guinea pigs, minipigs, and humans to establish strong correlations between in vitro predictions and in vivo plasma concentrations.3,34 While the purpose of the present work was focused on the in vitro development and characterization of NTX-loaded poloxamer formulations, in vivo studies will help demonstrate sustained NTX from the formulations developed here, and confirm if the formulations maintain NTX plasma concentrations within the therapeutic window for a prolonged time. In vivo studies in animals or humans would also help quantify NTX permeation both before and after the aqueous micropores close in the skin, as the impact of the CPEs could shift as the primary transport pathway changes (intact skin vs. micropores).
CPEs can cause some skin irritation, and the irritation potential of the presently tested formulations on living skin was not evaluated (and we did not specifically quantify mass of CPE that permeated through the skin). However, all CPE concentrations presently tested were below concentrations previously shown to produce minimal irritation,55,56 though this will still need to be evaluated in vivo.
Last, there are many mechanisms by which these CPEs can enhance drug permeation,11,43 but the specific methods by which these CPEs acted were not investigated (though some hypothesized mechanisms were proposed). Determining which mechanism of enhancement is most suitable for delivery with MN treatment may be of interest, both for NTX and for other drugs with similar properties.
Conclusions
Opioid and alcohol misuse disorders are increasingly being recognized as chronic conditions, and transdermal NTX delivery could provide a more patient-friendly option for at-home treatment. Here we have demonstrated that poloxamer biomaterials (specifically P407) in combination with CPEs effectively sustains NTX delivery in MN-treated skin while requiring less than half of the NTX concentration previously required to meet clinical goals.Materials and methods
Materials
Naltrexone hydrochloride (NTX) was obtained from Mallinckrodt Pharmaceuticals (Webster Groves, MO, USA). P407 was obtained from Anatrace (Maumee, OH, USA). HEPES, methanol, and sodium bicarbonate were obtained from Research Products International (Mt Prospect, IL, USA). Sodium hydroxide (1 N), o-phosphoric acid, polyethylene glycol (PEG, MW 400), and methanol Optima® were obtained from Fisher Chemical (Lenexa, KS, USA). Acetic acid, octanesulfonate sodium, Hank's balanced salts, gentamicin sulfate, dimethyl sulfoxide (DMSO), glycerol, propylene glycol, benzyl alcohol, isopropyl myristate, oleic acid, HPLC grade water, and HPLC grade acetonitrile were obtained from Sigma Aldrich (St Louis, MO, USA). Ethanol was obtained from Decon Laboratories (King of Prussia, PA, USA). Isopropyl palmitate was obtained from Alfa Aesar (Haverhill, MA, USA). Hydroxyethyl cellulose (750 kDa) was obtained from Ashland (Wilmington, DE, USA). Solid stainless steel MN arrays of 600 μm length (56 projections, 200 μm width, 75 μm thickness, 1.3 mm inter-needle spacing) were purchased from Tech Etch (Plymouth, MA, USA). (5-(4,6-Dichlorotriazinyl)aminofluorescein) (5-DTAF) was obtained from Thermo Fisher.Visualizing P407 migration into micropores
To demonstrate the ability of P407 molecules to enter micropores generated using solid MNs, P407 was fluorescently labeled in aqueous medium with 5-DTAF according to previous reports.9 Stock solution of 6% w/v P407 and 20 mg mL−1 5-DTAF were prepared in 0.1 M sodium bicarbonate (pH 9.3) and DMSO, respectively. The 5-DTAF stock solution was diluted to 5 mg mL−1 in 0.1 M sodium bicarbonate (pH 9.3) before mixing with P407 solution such that the molar ratio of 5-DTAF to P407 was 2:1. The reaction was protected from light and stirred at room temperature for 12 h. Reaction products were isolated from free 5-DTAF by dialyzing in 2 L of DI water for 24 h (with dialysate changes at 2 and 4 h). Isolated product was frozen at −80 °C for 2 h before being lyophilized under 0.021 mbar for 24 hours. HEPES buffer (pH 8.0) was added to the lyophilized product to achieve 160 mg mL−1 P407.
One MN array (800 μm length) was applied to full thickness excised porcine skin to create 50 micropores. This MN length was slightly longer than the MNs used for permeation studies, but would ensure that micropores would be clear and easy to locate on processed samples (micropore depth has been previously shown to only be ∼25% as deep as the actual MN length because of the impact of skin elasticity).10 To stain micropores for visual confirmation while cryotoming, gentian violet was applied over the entire MN-treated area and was removed from the skin surface after 30 s with ethanol wipes. The skin sample was warmed to 32 °C on a manifold and 10 μL of either labeled P407 or 5-DTAF solution at the same concentration was applied to skin for 5 or 30 min. Excess formulation was gently removed from the skin surface with a Kimwipe® before being rinsed in HEPES buffer (pH 8.0) for 5 min to remove access dye attached to skin. Skin samples were dried by blotting with a Kimwipe® immediately before flash freezing in liquid nitrogen. The skin samples were sectioned into 50 μm thick slices and adhered to a microscope slide, followed by a rinse with HEPES buffer (pH 8.0) before being mounted with a DAPI stain. Confocal microscopy was used for visualization with excitation at 200 nm (for DAPI) and 495 nm (for 5-DTAF).
Formulation preparation with CPEs
Eight CPEs were used in these studies: DMSO, glycerol, propylene glycol, polyethylene glycol (PEG 400), benzyl alcohol, ethanol, isopropyl myristate (IPM), and isopropyl palmitate (IPP). These potential CPEs were selected because they cover a wide range of chemical classes with different proposed mechanisms of action.11The required mass of P407, NTX, and CPE(s) were added to a single glass vial and balanced with acetate buffer to maintain a final pH of 5.0. Final concentrations were as follows: 15–17% P407, 7.5% w/w NTX (based on solubility studies), and 1–10% CPE. After combining all components, the vials were rotated overnight at 4 °C (all P407 formulations were liquid at this temperature) to fully dissolve P407. Formulations were stored at 4 °C for no longer than one week and rotated at room temperature for ∼1–3 hours on the day of use to dissolve NTX.
Rheological characterization
The viscosity of 15–17% P407 formulations containing 7.5% NTX in acetate buffer was measured to better understand how poloxamer concentration affects formulation viscosity. The viscosity of formulations used in permeation experiments were also measured to relate flux and viscosity. The formulation viscosities were also compared to solutions containing 40–75% propylene glycol with 11% NTX, in order to benchmark against previous studies that have shown the impact of formulation viscosity on NTX skin permeability.12 An ARES-G2 rotational rheometer (TA Instruments, New Castle, DE, USA) was used, outfitted with a Peltier plate and 40 mm diameter, 1-degree stainless steel cone, and a truncation gap set to 23 μm. Viscosity was measured as shear rate increased logarithmically from 1 to 100 s−1. All formulations were evaluated at 32 °C to maintain the temperature that the formulation would reach while on the skin surface after application.Gelation temperature was measured for all formulations, with the goal of further studying the formulations that transition to a gel within the range of 20 to 32 °C. Fifty mm diameter stainless steel parallel plates with a 0.5 mm gap were used on the rheometer, equipped with a solvent trap to prevent evaporation. Temperature ramps were performed by continuously measuring the storage modulus (G′) as the temperature was increased 1 °C per minute from 15 °C to 40 °C. The gelation temperature was defined as the temperature at which the G′ was halfway between the G′ of the liquid and the G′ of the fully formed gel. Amplitude sweeps were performed on all gels at 37 °C with a 1 Hz frequency by increasing strain from 0.01 to 100% to determine the linear viscoelastic region; identifying this region for each gel confirmed that the 0.2% strain used in oscillatory experiments would not cause structural breakdown of the gel network.
In vitro permeation testing (IVPT)
Permeation studies were completed in excised porcine skin to quantify the impact of CPEs on NTX permeation from P407 formulations. The permeation was measured using in-line diffusion cells with 1.77 cm2 diffusion area (Permegear, Hellertown, PA, USA). Dorsal skin from Yucatan miniature pigs was obtained from Sinclair Bio Resources, LLC (Auxvasse, MO, USA) and stored at −80 °C until use. Skin samples were dermatomed to 1 mm thickness (Nouvag, Model TCM3000BL, Goldach, Switzerland) and cut to the appropriate size to cover the entire diffusion area. Skin samples were pretreated with MN arrays consisting of 56 stainless steel projections of 600 μm length. This MN length was used because previous reports have demonstrated that 600 μm length MNs are suitable for P407 delivery of NTX.8 To create a total of 112 non-overlapping micropores, the MN array was applied to the skin using gentle thumb pressure for 15 s, removed and rotated 45°, and then reapplied to the skin a second time. Intact skin (not MN-treated) samples served as controls. Prepared skin samples were mounted in the diffusion cells and maintained at 32 °C; receiver solution (HEPES buffer with Hank's balanced salts and 0.1 mM gentamicin sulfate, pH 7.4) was warmed to 37 °C and pumped at 1.5 mL h−1 to maintain sink conditions. After skin samples equilibrated for 30 min, transepidermal water loss measurements were performed on select samples to confirm the barrier integrity of intact skin and confirm that MN treatment breached the skin barrier. Experiments were initiated by applying 0.5 ml of donor; receiver samples were collected every 4 h for up to 72 h. NTX concentration was quantified via HPLC. n = 3–8 replicates were performed for each formulation.High performance liquid chromatography (HPLC) assays
5-DTAF reaction and isolation efficiency was analyzed using reverse-phase HPLC on a Shimadzu Prominence i-Series LC-2030 Plus system with UV detector (Shimadzu, Torrance, CA, USA), outfitted with a C-18 column (5 μm particle size, 100 Å pore size, 150 mm length, 4.6 mm inner diameter) (Phenomenex®, Torrance, CA, USA). The mobile phase, based on Gaun et al.,13 consisted of 40:60 acetonitrile:water from 0 to 3 min, gradually increased acetonitrile from 40:60 to 99:1 acetonitrile:water from 3 to 12 min, then equilibrated back to 40:60 acetonitrile:water for 8 min before injecting the next sample. Fluorescence intensity was measured by exciting at 592 nm and detecting at 516 nm.9 An injection volume of 10 μL was used for all samples.
NTX concentration was quantified using the same HPLC system and column. The mobile phase consisted of 18:82 acetonitrile:aqueous buffer (0.065% w/v octanesulfonate sodium and 0.13% v/v o-phosphoric acid, pH 2.1) at a flow rate of 1 mL min−1, run time of 10 min, and 10 μL injection volume. NTX absorption was detected at 280 nm.
Data analysis
Cumulative mass of NTX permeated was plotted vs. time and the terminal flux was measured over 56–72 h. All data were collected in at least triplicate for statistical analysis (n = 3–8). One-way ANOVA with Tukey's multiple comparison was used for comparisons of solubility and gelation temperatures. Individual flux values were compared using a two-tailed Student's t-test with a check for equal variance using an F-test in Excel®. Summary statistics were reported for all measures using counts and percentages for categorical variables and using medians and inter-quartile ranges for continuous variables. The generalized linear modeling (GLM) framework was used to assess the univariate relationships between the primary predictors of interest (P407, DMSO, benzyl alcohol, NTX) and outcomes (flux) using a log link due to the right-skewed nature of the three outcomes. Then bivariate GLMs with an interaction effect were fit for each primary predictor-outcome pairing where skin condition (intact vs. MN-treated) was introduced as the second covariate. Estimates for mean ratios, 95% confidence intervals, and p-values were obtained from the models to assess the effect of predictors and compare whether this was altered by the skin status. Corrections for multiple comparisons were not employed due to the exploratory nature of the analysis. R version 4.2.2 was used for all GLM analyses. Data are reported as mean ± SD and P < 0.05 was considered statistically significant.Author contributions
KVT: conceptualization, methodology, investigation, formal analysis, writing – original draft, editing and review. NKB: conceptualization, funding acquisition, formal analysis, writing – original draft, editing and review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Institutes of Health award 1R35GM124551. We would also like to acknowledge Patrick Ten Eyck and Linder Wendt for their assistance with statistical analyses.References
- A. N. Edinoff, C. A. Nix, C. V. Orellana, S. M. StPierre, E. A. Crane, B. T. Bulloch, E. M. Cornett, R. L. Kozinn, A. M. Kaye and K. S. Murnane, Neurol. Int., 2021, 14, 49–61 CrossRef PubMed.
- S. Racha, M. Buresh and M. Fingerhood, Psychiatr. Clin. North Am., 2022, 45(3), 335–346 CrossRef PubMed.
- D. P. Wermeling, S. L. Banks, D. A. Hudson, H. S. Gill, J. Gupta, M. R. Prausnitz and A. L. Stinchcomb, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 2058–2063 CrossRef CAS PubMed.
- H. Kalluri and A. K. Banga, Pharm. Res., 2011, 28, 82–94 CrossRef CAS PubMed.
- P. Ghosh, N. K. Brogden and A. L. Stinchcomb, J. Pharm. Sci., 2014, 103, 652–660 CrossRef CAS PubMed.
- D. A. Muller, J. Henricson, S. B. Baker, T. Togö, C. M. Jayashi Flores, P. A. Lemaire, A. Forster and C. D. Anderson, Sci. Rep., 2020, 10, 18468 CrossRef CAS PubMed.
- A. T. Ogunjimi, C. Lawson, J. Carr, K. K. Patel, N. Ferguson and N. K. Brogden, Skin Pharmacol. Physiol., 2021, 34, 214–228 CrossRef CAS PubMed.
- K. V. Tobin, J. Fiegel and N. K. Brogden, Polymers, 2021, 13, 933 CrossRef CAS PubMed.
- F. Ahmed, P. Alexandridis and S. Neelamegham, Langmuir, 2001, 17, 537–546 CrossRef CAS.
- X. Gao and N. K. Brogden, J. Pharm. Sci., 2019, 108, 3695–3703 CrossRef CAS PubMed.
- D. Kaushik, P. Batheja, B. Kilfoyle, V. Rai and B. Michniak-Kohn, Expert Opin. Drug Delivery, 2008, 5, 517–529 CrossRef CAS PubMed.
- M. Milewski and A. L. Stinchcomb, Pharm. Res., 2011, 28, 124–134 CrossRef CAS PubMed.
- B. Guan, A. Ali, H. Peng, W. Hu, L. R. Markely, S. Estes and S. Prajapati, Anal. Methods, 2016, 8, 2812–2819 RSC.
- A. Sivaraman and A. K. Banga, Drug Delivery Transl. Res., 2017, 7, 16–26 CrossRef CAS PubMed.
- S. Khan, M. U. Minhas, I. A. Tekko, R. F. Donnelly and R. R. S. Thakur, Drug Delivery Transl. Res., 2019, 9, 764–782 CrossRef CAS PubMed.
- P. Ghosh, R. R. Pinninti, D. C. Hammell, K. S. Paudel and A. L. Stinchcomb, J. Pharm. Sci., 2013, 102, 1458–1467 CrossRef CAS PubMed.
- M. Milewski, R. R. Pinninti and A. L. Stinchcomb, J. Pharm. Sci., 2012, 101, 2777–2786 CrossRef CAS PubMed.
- P. Ghosh, N. K. Brogden and A. L. Stinchcomb, Mol. Pharm., 2013, 10, 2331–2339 CrossRef CAS PubMed.
- M. Milewski, T. R. Yerramreddy, P. Ghosh, P. A. Crooks and A. L. Stinchcomb, J. Controlled Release, 2010, 146, 37–44 CrossRef CAS PubMed.
- J. J. Kaufman, N. M. Semo and W. S. Koski, J. Med. Chem., 1975, 18, 647–655 CrossRef CAS PubMed.
- N. K. Brogden, S. L. Banks, L. J. Crofford and A. L. Stinchcomb, Pharm. Res., 2013, 30, 1947–1955 CrossRef CAS PubMed.
- S. L. Banks, K. S. Paudel, N. K. Brogden, C. D. Loftin and A. L. Stinchcomb, Pharm. Res., 2011, 28, 1211–1219 CrossRef CAS PubMed.
- E. P. Chung, A. R. Wells, M. M. Kiamco and K. P. Leung, AAPS PharmSciTech, 2020, 21, 265 CrossRef CAS PubMed.
- A. M. Bodratti and P. Alexandridis, J. Funct. Biomater., 2018, 9(1), 11 CrossRef PubMed.
- G. Dumortier, J. L. Grossiord, F. Agnely and J. C. Chaumeil, Pharm. Res., 2006, 23, 2709–2728 CrossRef CAS PubMed.
- E. Ricci, M. V. L. B. Bentley, M. Farah, R. Bretas and J. M. Marchetti, Eur. J. Pharm. Sci., 2002, 17, 161–167 CrossRef CAS PubMed.
- K. Edsman, J. Carlfors and R. Petersson, Eur. J. Pharm. Sci., 1998, 6, 105–112 CrossRef CAS PubMed.
- J. Gilbert, C. Washington, M. Davies and J. Hadgraft, Int. J. Pharm., 1987, 40, 93–99 CrossRef CAS.
- M. V. L. Bentley, J. M. Marchetti, N. Ricardo, Z. Ali-Abi and J. H. Collett, Int. J. Pharm., 1999, 193, 49–55 CrossRef CAS PubMed.
- M. Vadnere, G. Amidon, S. Lindenbaum and J. L. Haslam, Int. J. Pharm., 1984, 22, 207–218 CrossRef CAS.
- T. Ur-Rehman, S. Tavelin and G. Gröbner, Int. J. Pharm., 2010, 394, 92–98 CrossRef CAS PubMed.
- R. R. Ford, P. H. Gilbert, R. Gillilan, Q. Huang, R. Donnelly, K. K. Qian, D. P. Allen, N. J. Wagner and Y. Liu, J. Pharm. Sci., 2023, 112, 731–739 CrossRef CAS PubMed.
- S. Förster, M. Konrad and P. Lindner, Phys. Rev. Lett., 2005, 94, 017803 CrossRef PubMed.
- M. Milewski, K. S. Paudel, N. K. Brogden, P. Ghosh, S. L. Banks, D. C. Hammell and A. L. Stinchcomb, Mol. Pharm., 2013, 10, 3745–3757 CrossRef CAS PubMed.
- J. Russo, J. Fiegel and N. K. Brogden, Pharmaceutics, 2020, 12, 1214 CrossRef CAS PubMed.
- F. Verbaan, S. Bal, D. Van den Berg, W. Groenink, H. Verpoorten, R. Lüttge and J. Bouwstra, J. Controlled Release, 2007, 117, 238–245 CrossRef CAS PubMed.
- V. Rai, I. Ghosh, S. Bose, S. Silva, P. Chandra and B. Michniak-Kohn, J. Drug Delivery Sci. Technol., 2010, 20, 75–88 CrossRef CAS.
- A. M. Jörgensen, J. D. Friedl, R. Wibel, J. Chamieh, H. Cottet and A. Bernkop-Schnürch, Mol. Pharm., 2020, 17, 3236–3245 CrossRef PubMed.
- W. Li, J. J. Hartsock, C. Dai and A. N. Salt, Otol. Neurotol., 2018, 39, 639–647 CrossRef PubMed.
- B. J. Aungst, N. J. Rogers and E. Shefter, Int. J. Pharm., 1986, 33, 225–234 CrossRef CAS.
- V. M. Cardoso, A. G. R. Solano, M. A. F. Prado and E. de Aguiar Nunan, J. Pharm. Biomed. Anal., 2006, 42, 630–634 CrossRef CAS PubMed.
- S. B. de Souza Ferreira and M. L. Bruschi, J. Mol. Liq., 2021, 332, 115856 CrossRef CAS.
- O. Pillai, V. Nair and R. Panchagnula, Int. J. Pharm., 2004, 269, 109–120 CrossRef CAS PubMed.
- A. C. Williams and B. W. Barry, Adv. Drug Delivery Rev., 2012, 64, 128–137 CrossRef.
- B. Kumar, S. Jain and S. Prajapati, Int. J. Drug Delivery, 2011, 3(1), 83–94 CrossRef CAS.
- R. Regev, Y. G. Assaraf and G. D. Eytan, Eur. J. Biochem., 1999, 259(1–2), 18–24 CrossRef CAS PubMed.
- G. K. Hulse, H. T. Ngo and R. J. Tait, Biol. Psychiatry, 2010, 68, 296–302 CrossRef CAS PubMed.
- D. Sudakin, J. Med. Toxicol., 2016, 12, 71–75 CrossRef CAS PubMed.
- A. M. DeVeaugh-Geiss, L. H. Chen, M. L. Kotler, L. R. Ramsay and M. J. Durcan, Clin. Ther., 2010, 32, 1140–1148 CrossRef CAS PubMed.
- P. I. Hair, G. M. Keating and K. McKeage, Drugs, 2008, 68, 2001–2009 CrossRef CAS PubMed.
- M. N. Kelchen, K. J. Siefers, C. C. Converse, M. J. Farley, G. O. Holdren and N. K. Brogden, J. Controlled Release, 2016, 225, 294–300 CrossRef CAS PubMed.
- J. Gupta, H. S. Gill, S. N. Andrews and M. R. Prausnitz, J. Controlled Release, 2011, 154, 148–155 CrossRef CAS PubMed.
- A. T. Ogunjimi, J. Carr, C. Lawson, N. Ferguson and N. K. Brogden, Sci. Rep., 2020, 10, 18963 CrossRef PubMed.
- N. K. Brogden, M. Milewski, P. Ghosh, L. Hardi, L. J. Crofford and A. L. Stinchcomb, J. Controlled Release, 2012, 163, 220–229 CrossRef CAS PubMed.
- M. Myristate, J. Am. Coll. Toxicol., 1982, 1, 55–80 CrossRef.
- U. T. Lashmar, J. Hadgraft and N. Thomas, J. Pharm. Pharmacol., 1989, 41, 118–121 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3bm00972f |
| This journal is © The Royal Society of Chemistry 2023 |